 Yanru Wang1
Yanru Wang1 Yuanyuan Zheng1Dan Guo2Xinghui Zhang2Suling Guo3Taiyu Hui1Chang Yue1Jiaming Sun1Suping Guo1Zhixian Bai1Weidong Cai1Xinjiang Zhang1
Yuanyuan Zheng1Dan Guo2Xinghui Zhang2Suling Guo3Taiyu Hui1Chang Yue1Jiaming Sun1Suping Guo1Zhixian Bai1Weidong Cai1Xinjiang Zhang1 Yixing Fan1Zeying Wang1*Wenlin Bai1*
Yixing Fan1Zeying Wang1*Wenlin Bai1*- 1College of Animal Science & Veterinary Medicine, Shenyang Agricultural University, Shenyang, China
- 2Academy of Animal Husbandry Science of Liaoning Province, Liaoyang, China
- 3Prosperous Community, Huade, China
N6-methyladenosine (m6A) is the most common internal modification in mRNAs of all higher eukaryotes. Here we perform two high-throughput sequencing methods, m6A-modified RNA immunoprecipitation sequence (MeRIP-seq) and RNA sequence (RNA-seq) to identify key genes with m6A modification in cashmere fiber growth. A total of 9,085 m6A sites were differentially RNA m6A methylated as reported from by MeRIP-seq, including 7,170 upregulated and 1,915 downregulated. In addition, by comparing m6A-modified genes between the fine-type Liaoning cashmere goat (FT-LCG) and coarse-type Liaoning Cashmere Goat (CT-LCG) skin samples, we obtain 1,170 differentially expressed genes. In order to identify the differently methylated genes related to cashmere fiber growth, 19 genes were selected to validate by performing qRT-PCR in FT-LCG and CT-LCG. In addition, GO enrichment analysis shows that differently methylated genes are mainly involved in keratin filament and intermediate filament. These findings provide a theoretical basis for future research on the function of m6A modification during the growth of cashmere fiber.
Introduction
In the 1970s, scientists discovered m6A modification, which can occur on RNA adenine (A) such as mRNA and long non-coding RNA (Dubin and Taylor, 1975). Subsequent studies have shown that methylation of m6A exists in the mRNA of different prokaryotes, eukaryotes, and viruses (Desrosiers et al., 1974; Dubin and Taylor, 1975; Furuichi and Miura, 1975; Beemon and Keith, 1977). However, the clear function and mechanism of the m6A modification were largely unclear until recently. Until now, more than 150 types of posttranscriptional modifications have been identified in RNA among all living organisms (Boccaletto et al., 2018). m6A is a chemical modification that exists in a variety of RNA and has the highest abundance in mRNA (Yang et al., 2018).
Earlier studies on m6A mainly focused on the methylation modification at the mRNA 5'Cap, including the maintenance of mRNA stability, mRNA precursor shear, polyadenylation, mRNA transport, and translation initiation. The modification of 3' polyA contributes to nuclear transport, initiation of translation, and maintenance of structural stability of mRNA with polyA binding protein. In recent years, there have been more and more studies on internal modification of RNA, including N6 methyladenosine modification (m6A), N1 methyladenosine modification (m1A), methylcytosine modification (m5C), and pseudouridine modification (Ψ) (Weng et al., 2018). m6A is present transcriptome-wide in over 25% of all RNAs (Hibar et al., 2015), as the most common methylation modification in RNA, m6A is mainly concentrated in long exons, stop codons, and in 3′UTRs (Dominissini et al., 2012). As similar with DNA and histone methylation, m6A RNA methylation is also dynamic and reversible in mammalian cells and it has been proposed as another form of epigenetic regulation. In mammals, m6A modification occurs by a methyltransferase complex (writers) mainly consisting of METTL3, METTL14, WTAP, and other components. The demethylases (erasers) FTO and ALKBH5 can mediate the demethylation of m6A modification (Li et al., 2015; Meyer and Jaffrey, 2017).
A large amount of original work of m6A is still concentrated in human, mouse, and other model animals. Studies have shown that m6A methylation is associated with obesity (Ben-Haim et al., 2015). Lin indicated that m6A was dynamically regulated and played crucial roles to shape gene expression in spermatogonial stem cells development and during murine spermatogenesis (Lin et al., 2017). In Drosophila melanogaster, the m6A-deficient Drosophila melanogaster could survive, but could not fly and its fertility declined (Haussmann et al., 2016). Recent studies have demonstrated that the deletion of YTHDF2 resulted in the non-regulation of transcripts of related genes during oocyte maturation, leading to the infertility of specific YTHDF2 deficient female mice (Ivanova et al., 2017). Wang indicated that both m6A in total RNA and expression of METTL3 were significantly decreased in mouse hippocampus after traumatic brain injury (Wang et al., 2019). But there is no relevant reference on cashmere goats.
Cashmere goat is a kind of goat breed which mainly produces cashmere. It produces more cashmere and has better cashmere quality. Cashmere produced by the secondary hair follicles of cashmere goat is a shiny and comfortable natural fiber (Xiaolong et al., 2016; Bai et al., 2018). The fineness of cashmere is the most important factor affecting cashmere production. It is an important index to evaluate the quality of cashmere in many countries. The standard of cashmere classification in China is that fineness between 14.5 to 16.0 μm is fine type, between 16.0 to18.5 μm is coarse type (GB18267-2013). The fineness of cashmere has been determined as early as the development of hair follicles, the development of hair follicles has a certain impact on the fineness of cashmere. Therefore, in order to improve the quality of cashmere, the fineness of cashmere has become more and more popular (Bai et al., 2016; Zhang et al., 2018). Investigations indicated that many genes might widely participate in the growth regulation of cashmere fiber (Stenn and Paus, 2001; Liu et al., 2016). Many studies have identified some genes associated with the growth and properties of wool fibers in sheep and goats, such as DSG1, IGF-IR, KRTAPs, ILK, as well as the KRT and KRTAP genes (Rufaut et al., 1999; Bin et al., 2006; Yu et al., 2011; Yang et al., 2012; Liu et al., 2015). In the past few decades, the regulators of cashmere fiber growth have been studied at several levels, including genes with methylation characteristics (Bai et al., 2013; Fan et al., 2015; Bai et al., 2017). Given the indispensable function of RNA m6A modification in regulating gene express and involving in various bioprocesses, it is reasonable to speculate that regulation of m6A modification might also be associated with the cashmere fineness. Herein, we aim to find the m6A methylation modification landscape of differential genes in the skin of FT-LCG and CT-LCG. And some potential functions of RNA m6A methylation genes in cashmere growth will be explored in the future.
Materials and Methods
Sample Collection and Ethics Statement
We collected scapular skin samples from two groups Liaoning Cashmere Goats from Academy of Animal Husbandry Science of Liaoning Province. The two groups goats are three coarse type and three fine type adult female cashmere goats, which had the same feed conditions and growth environment. They are all 1.5 years old. The upper 1/3 of the right scapula along the mid-dorsal and mid-abdominal lines were taken from the lateral skin about 1 cm2, and immediately stored in liquid nitrogen until RNA isolation. In addition, three coarse and three fine type skin samples of Liaoning Cashmere Goat were collected for qRT-PCR. All experimental procedures used in this study were approved and conducted according to the guidelines by the Laboratory Animal Management Committee of Shenyang Agricultural University.
Experimental Procedure
Total RNA was extracted using Trizol reagent (Invitrogen, CA, USA) following the manufacturer's procedure. The total RNA quality and quantity were analysis of Bioanalyzer 2100 and RNA 6000 Nano LabChip Kit (Agilent, CA, USA) with RIN number >7.0. Approximately more than 200ug of total RNA was subjected to isolate Poly (A) mRNA with poly-T oligo attached magnetic beads (Invitrogen). Following purification, the poly(A) mRNA fractions is fragmented into 100-nt-long oligonucleotides using divalent cations under elevated temperature. Then the cleaved RNA fragments were subjected to incubated for 2h at 4°C; with m6A-specific antibody (No. 202003, Synaptic Systems, Germany) in IP buffer (50 mM Tris-HCl, 750 mM NaCl, and 0.5% Igepal CA-630) supplemented with BSA (0.5 μg/ μl). The mixture was then incubated with protein-A beads and eluted with elution buffer (1 × IP buffer and 6.7mM m6A). Eluted RNA was precipitated by 75% ethanol. Eluted m6A-containing fragments (IP) and untreated input control fragments are converted to final cDNA library in accordance with a strand-specific library preparation by dUTP method. The average insert size for the paired-end libraries was 100 ± 50 bp. And then we performed the paired-end 2 × 150 bp sequencing on an Illumina Novaseq™ 6000 platform at the LC-BIO Bio-tech ltd (Hangzhou, China) following the vendor's recommended protocol.
Bioinformatics Analysis Process
Firstly, Cutadapt and perl scripts in house were used to remove the reads that contained adaptor contamination, low quality bases, and undetermined bases (Martin, 2011). Then sequence quality was verified using fastp. We used HISAT2 to map reads to the genome of Capra_hircus_goat (Version: Capra_hircus_goat_NCBI) with default parameters (Kim et al., 2015). Mapped reads of IP and input libraries were provided for R package exomePeak, which identifies m6A peaks with bed or bam format that can be adapted for visualization on the UCSC genome browser or IGV software (http://www.igv.org/) (Meng et al., 2014). MEME and HOMER were used for de novo and known motif finding followed by localization of the motif with respect to peak summit by perl scripts in house (Bailey et al., 2009; Heinz et al., 2010). Called peaks were annotated by intersection with gene architecture using ChIPseeker (Yu et al., 2015). Then StringTie was used to perform expression level for all mRNAs from input libraries by calculating FPKM (FPKM = [total_exon_fragments/mapped_reads (millions) × exon_length (kB)]) (Pertea et al., 2015). The differentially expressed mRNAs were selected with log2 (fold change) > 1 or log2 (fold change) <−1 and P < 0.05 by R package edgeR (Robinson et al., 2010).
Quantitative Real-Time PCR Validation
We detected 19 differently m6A methylated genes for qRT-PCR. In order to validate the differentially methylated genes, total RNAs were synthesized directly to cDNA synthesis by an RT-PCR kit (Takara, Dalian, China). According to the manufacturer's instructions, Real-time PCR was performed using SYBR Green (TaKaRa Biotech, Dalian). The glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene was used as an internal control to normalize the expression level of genes. Three independent experiments were carried out on three CT-LCG and three FT-LCG skin samples. Nineteen pairs primers were designed by Primer 5 software and listed in the Supplementary Material, and all primers were spanning the distal ends of genes. The relative expression levels of differentially expressed genes were analyzed by the 2−ΔΔCt method in qPCR data. The data were indicated as the means ± SE (n = 3). All statistical analyses in the two groups were calculated using a t-test in SPSS statistical software (Version 22.0, Chicago, IL, USA), the difference was significant at P < 0.05.
Results and Analysis
Sequencing Sequence Statistics and Quality Control
The raw data from IP and input RNA-seq were firstly trimmed by Cutadapt and perl scripts in house to remove the adaptor and low quality data, and the clean reads were obtained. In the MeRIP-seq library, two groups of skin samples were obtained 65964582 and 68530268 raw data reads, 56434060 and 60393976 valid data reads, and the effective reads accounted for 72.94% and 66.49% respectively. In the RNA-seq library, three groups of skin samples were obtained 65680810 and 75067398 raw data reads, 57014334 and 64832184 valid data reads, and the effective reads accounted for 71.84% and 62.20% respectively. The results are shown in Table 1. In Table 1, Q20% represents proportion of bases with mass value ≥ 20 (sequencing error rate less than 0.01) and Q30% represents proportion of bases with quality value ≥ 30 (sequencing error rate less than 0.001).
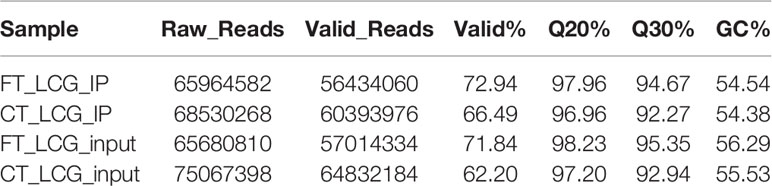
Table 1 Summary of reads quality control.
Mapping Reads to the Reference Genome
We used HISAT2 to map reads to the genome of Capra hircus goat (Version: Capra_hircus_goat_NCBI) with default parameters. By comparing reads with reference sequences, we can make detailed statistics of the alignment of sequencing data. In the m6A-seq library, the mapping ratio of valid data in cashmere goat skin IP samples FT-LCG and CT-LCG were 90.93% and 92.65%. In the RNA-seq library, the mapping ratio of valid reads in cashmere goat skin IP sample FT-LCG and CT-LCG were 90.48% and 92.14%. The ratio of unique mapped reads and the multi-mapped reads were shown in Table 2. According to the regional information of the reference genome, it can be defined as the alignment to exon, intron, and intergenic. Under normal circumstances, the percentage of sequencing sequence localization in the Exon region should be the highest. The results of this experiment showed that the ratio of IP samples and Input samples to exons of fine cashmere goat skin was 96.13% and 96.21%. The ratio of IP samples to exons of coarse cashmere goat skin was 96.25%, and the ratio of input samples was 96.01%. The results are shown in Figure 1.
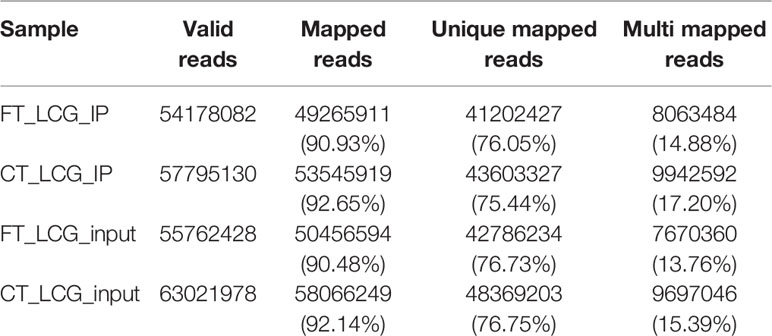
Table 2 Summary of reads mapping to the goat reference genome.
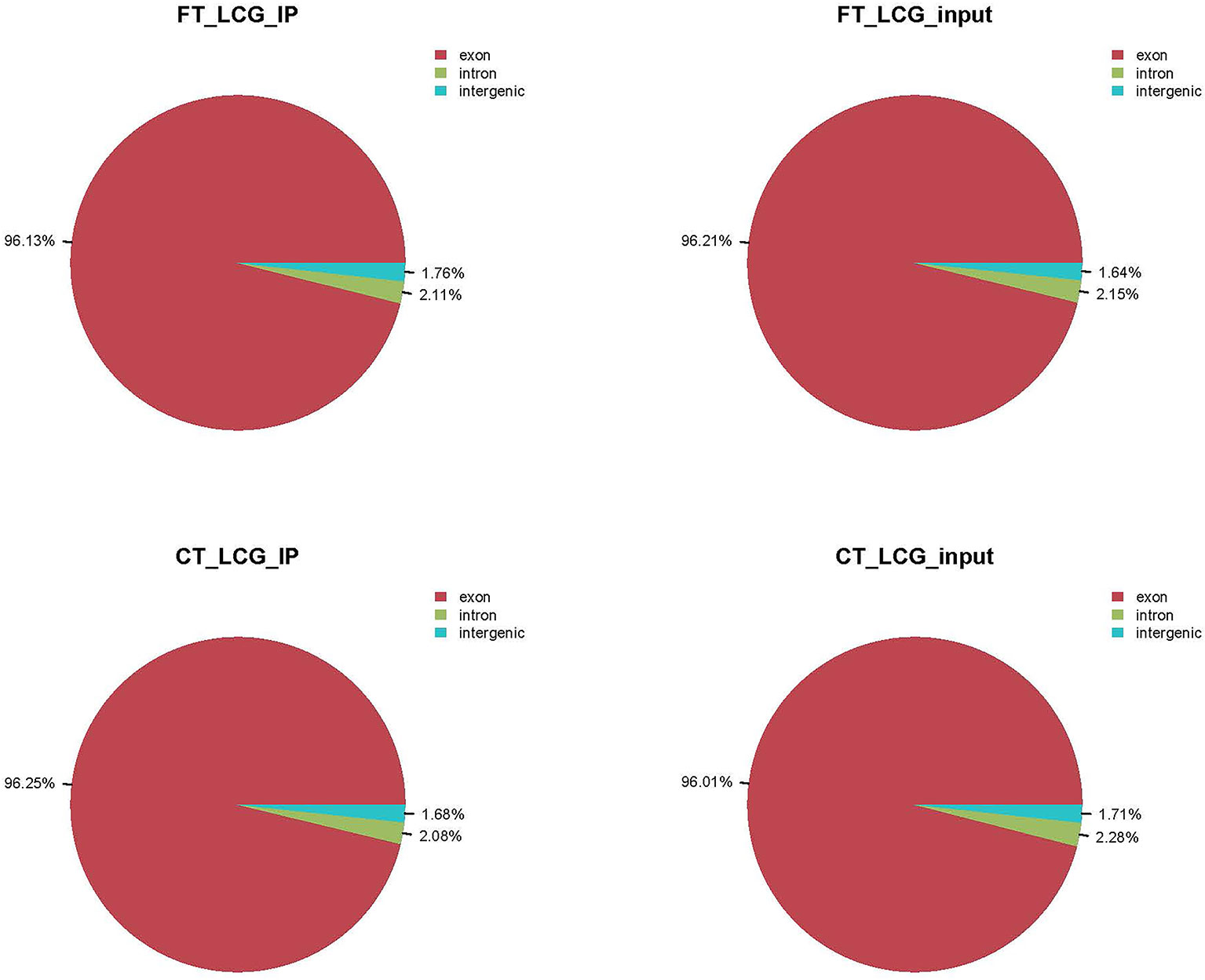
Figure 1 Refer to the genome to compare the regional distribution.
Identification of m6A Modification Sites and Differential Methylation Analysis
m6A modification sites were identified by using R package exomePeak, which performed peaks scanning in the whole gene range. Peak identification was based on MeRIP-seq and RNA-seq (Input) sequencing data. If p-value is less than 0.05, the regions less than the p-value are considered to be a peak. We combined all samples to demonstrate the enrichment of reads near TSS at the transcriptome initiation site of the gene. Peak distributions in the combined regions around TSS are shown in heatmap form (Figure 2A). Next, we use the ChIPseeker to annotate the difference peaks. At present, the default P < 0.05 is the screening threshold of differential peak. A total of 9,085 differential peaks were detected, of which 7,170 m6A peaks were up-regulated and 1,915 peaks were down-regulated. To analyze the distribution of m6A peaks in different regions of the transcript, we divided the transcript into four areas 5′ untranslated region (5'UTR), 3′UTR, 1st exon, and other exon (Figure 2B). The differential m6A peaks were mainly enriched in the 3′UTRs. And the top 20 differential m6A peaks were showed in the Table 3. Fold change <1 represents hypo-methylation and fold change ≥1 represents hyper-methylation in Table 3.
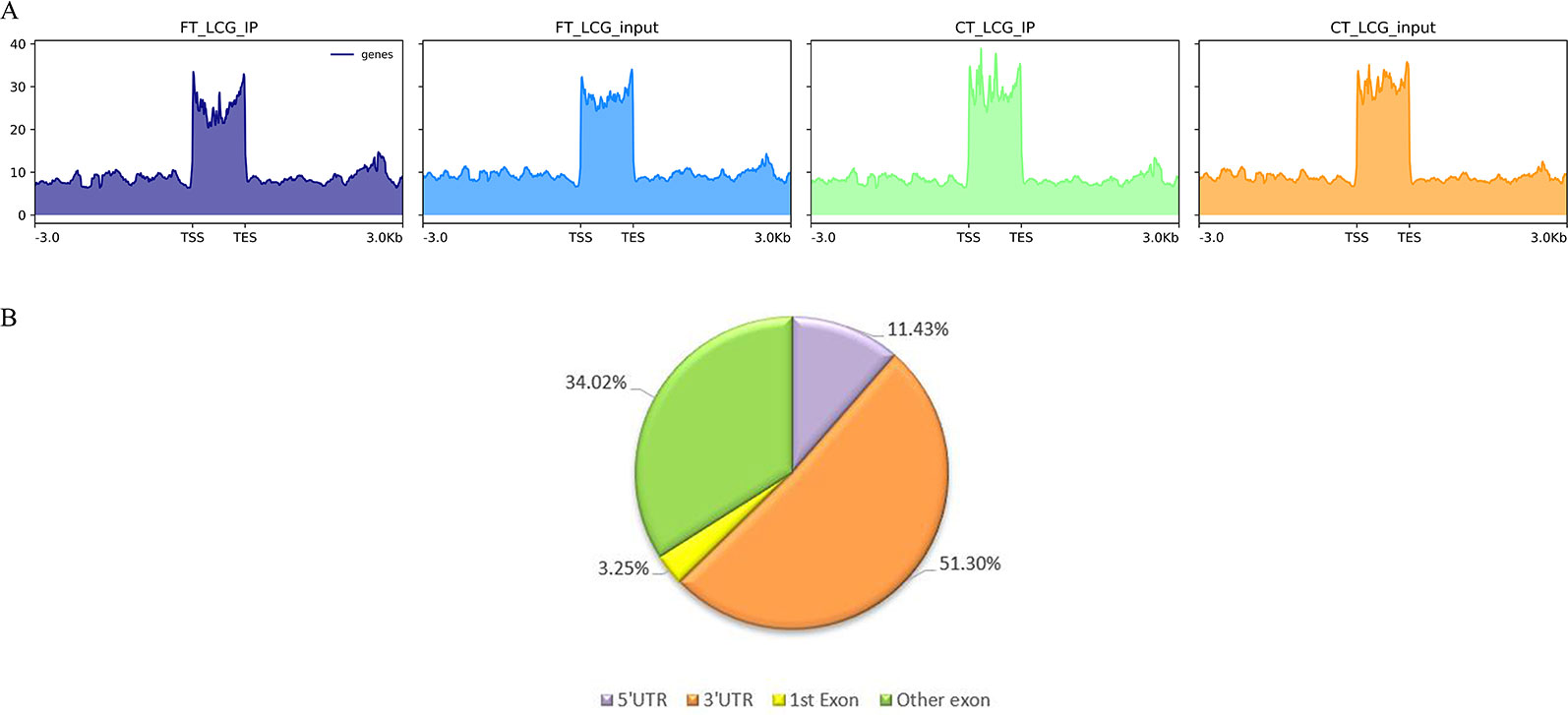
Figure 2 (A) The enrichment of reads near TSS at the transcriptome initiation site of the gene. (B) The distribution of differential peaks on gene functional elements.
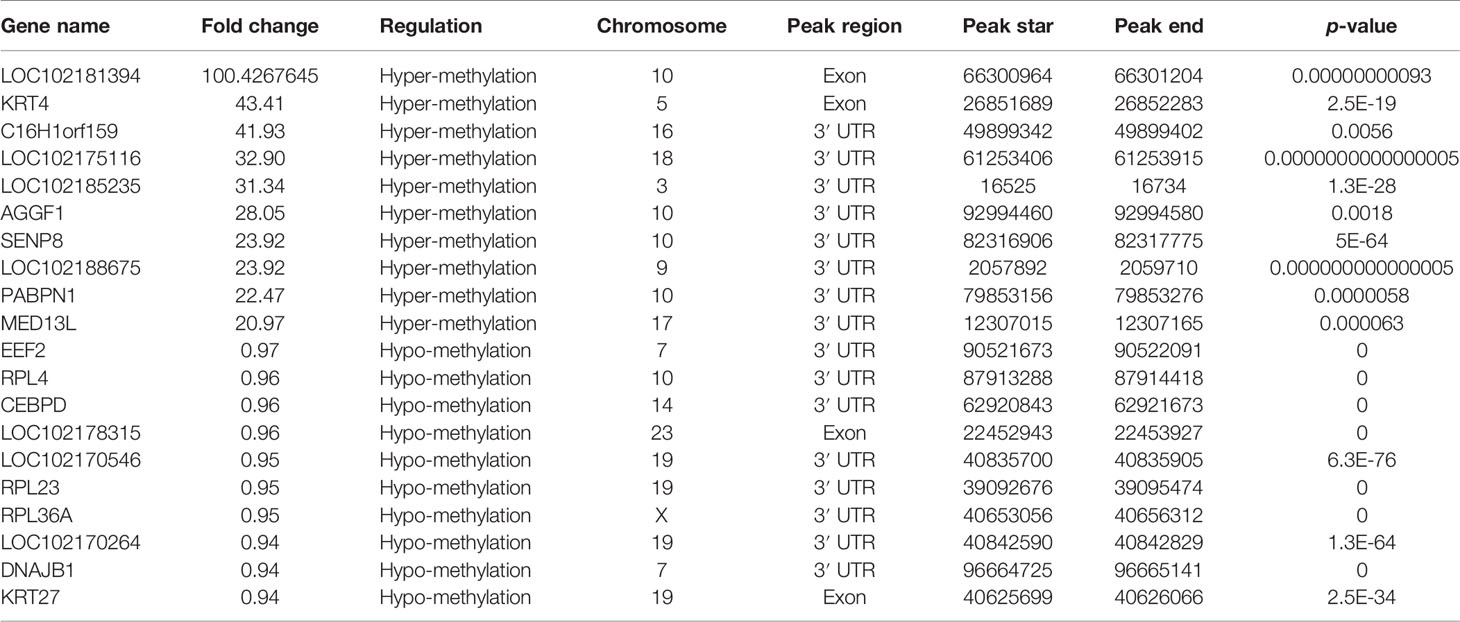
Table 3 The top 20 differently expressed m6A peaks.
Motif Analysis
RNA methylation and demethylation begin with the motif binding of various binding proteins to methylation sites. A motif is essentially a sequence pattern of nucleic acids with biological significance, and these RNA methylation related enzymes recognize and bind to these motifs, thereby affecting gene expression. Motif analysis software MEME was used to search for motifs with high credibility in the peak region, and the width, e-value, p-value of each motif, and its general position information in each peak sequence were obtained. We conducted motif prediction for samples and differential results. We sorted the predicted motif in the Figure 3. According to the p-value, and the smaller the p-value, the higher the rank. The most commonly reported RNA motif structures are RRACH (where R = A or G, H = A, C or U). The differential m6A peaks were characterized by the GAAGA motif, which is the first rank motif structures.
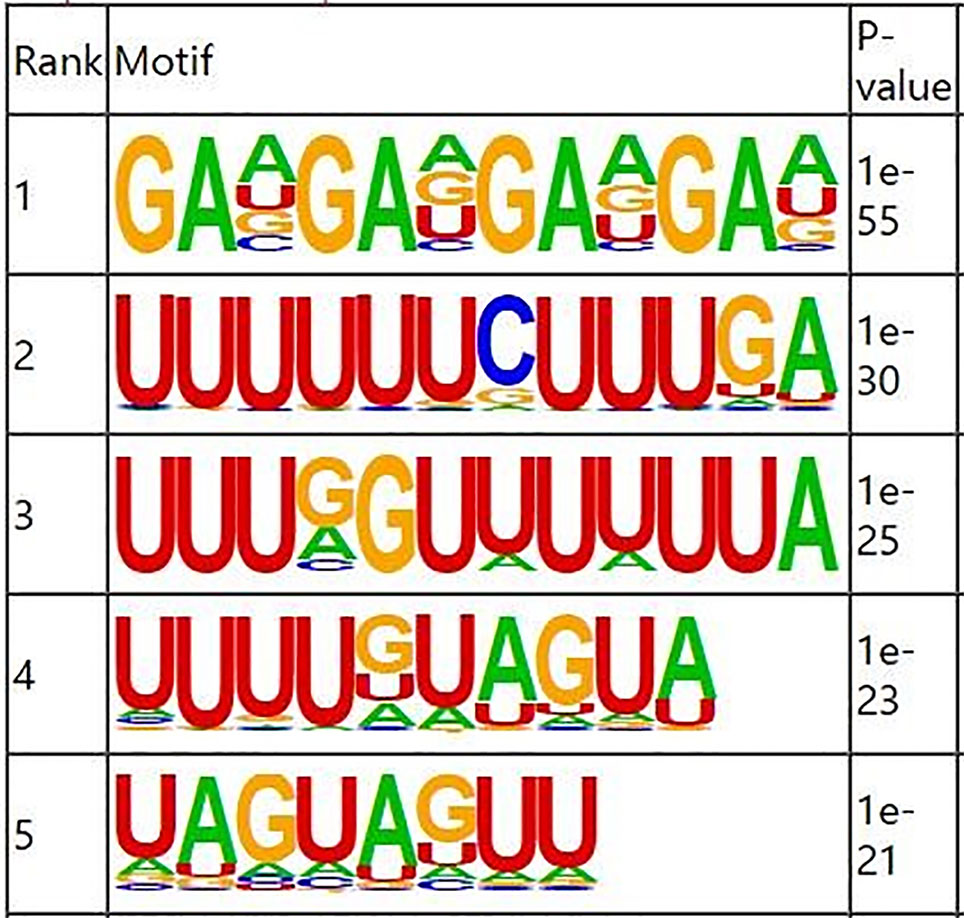
Figure 3 Sequence logo showing the top motifs enriched across differential m6A peaks identified from skin samples.
Overall Gene Outcome Analysis and Differential Genes Analysis
The level of gene expression is mainly measured by RPKM (reads per kilobase of exon model per million mapped reads) or FPKM (fragments per kilobase of exon model per million mapped reads). In this project, FPKM was used to calculate the expression abundance of known genes in different samples. FPKM represents the number of sequencing fragments contained in the sequence bases of every 1,000 transcripts per million sequencing bases. In short, the FPKM value can be understood as the amount of gene expression. Of the 18,619 genes we have identified, 1,170 were significantly different, while 17,449 were not. Among them, 527 differentially expressed genes were up-regulated and 643 down-regulated. The top 20 differently expressed genes are listed in Table 4. Gene expression box plot and gene expression density map are showed in Figures 4A, B. The overall distribution of differentially expressed genes can be understood by mapping volcanoes in Figure 4C. And the heatmap of the genes between FT-LCG and CT-LCG is showed in Figure 4D.
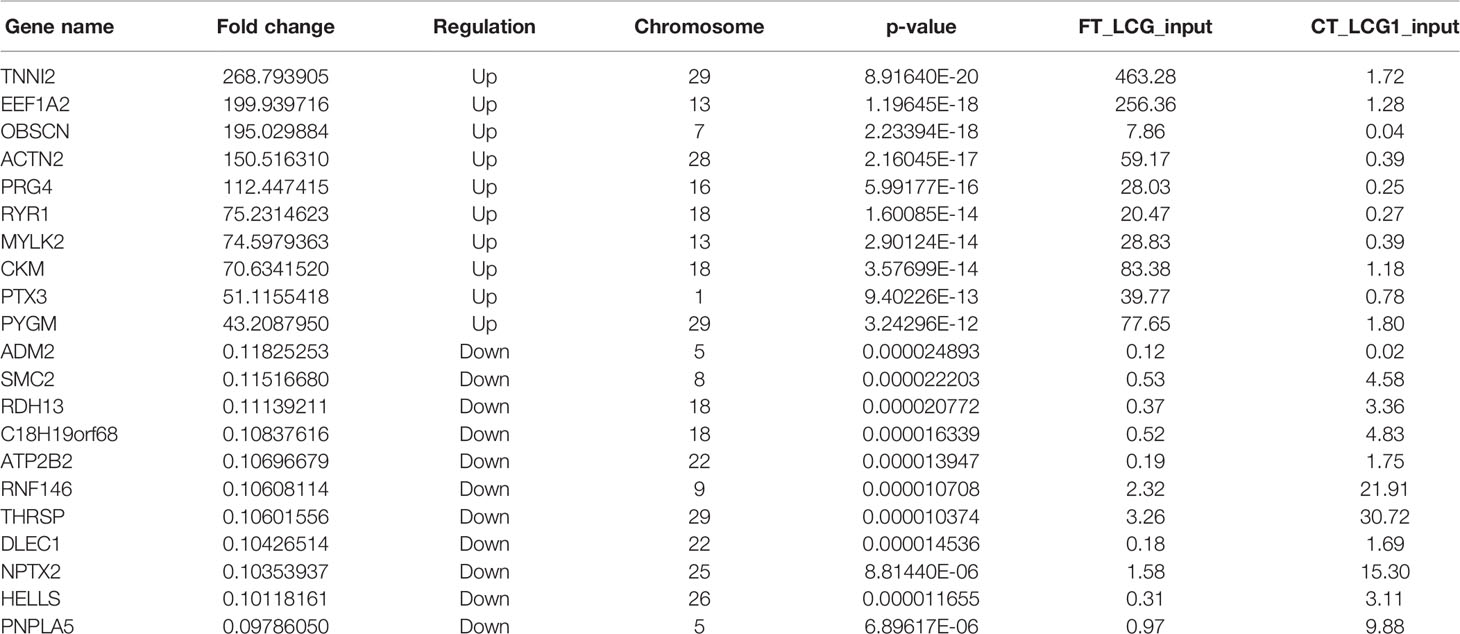
Table 4 The top 20 differentially expressed genes.
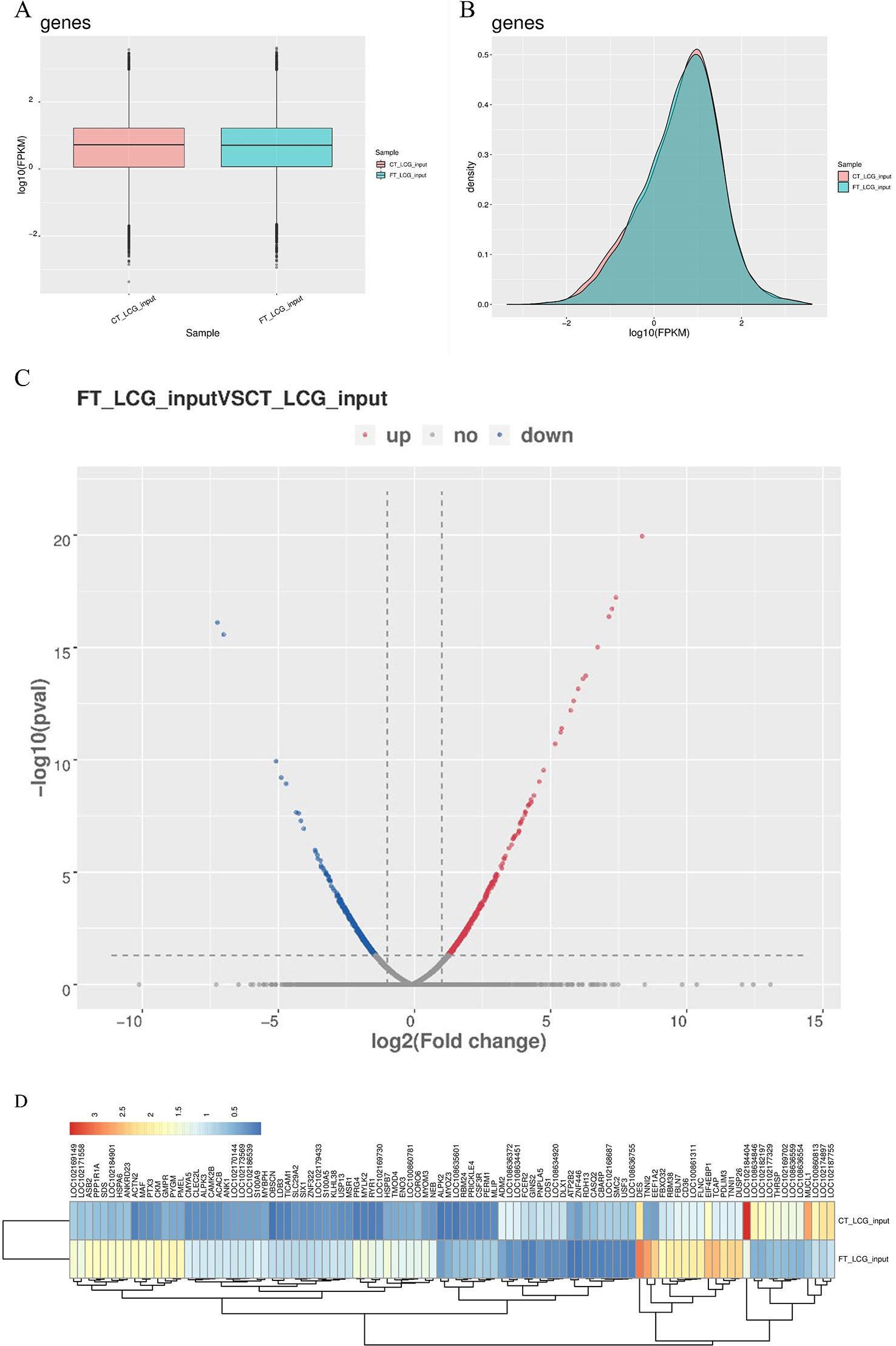
Figure 4 Overview of differentially expressed genes in goats skin samples. (A) Gene expression box plot. (B) Gene expression density map. (C) Volcanic map of differentially expressed genes. [log2(fold change) was used as the horizontal coordinate, and -log10(p-value) was used as the vertical coordinate. Volcanic map was drawn for all genes in the differentially expressed analysis. The abscissa represents the multiple change of gene expression in different samples. The ordinate represents the statistical significance of differences in gene expression level. Red represents significantly differentially expressed genes up-regulated, blue represents significantly differentially expressed genes down-regulated, and gray dots represent non-significantly differentially expressed genes.] (D) Heatmap of the genes of FT-LCG and CT-LCG.
Correlation Analysis Results of Differential Genes and Differential Peaks
In transcriptome sequencing, the results were up-regulated and down-regulated. In the same MeRIP-seq results, the up-regulated methylation of the gene itself can also be found according to the peak abundance changes. Here we conduct a correlation analysis of the contents of the two omics, comparing the transcriptional level with the methylation level. Based on these results, we screened out 19 candidate genes from 256 differentially differently methylated genes associated with the growth of the cashmere, which are listed in Table 5. The m6A regulation of these genes are upregulated, the gene regulation are downregulated, and the differences between the two samples are significant.
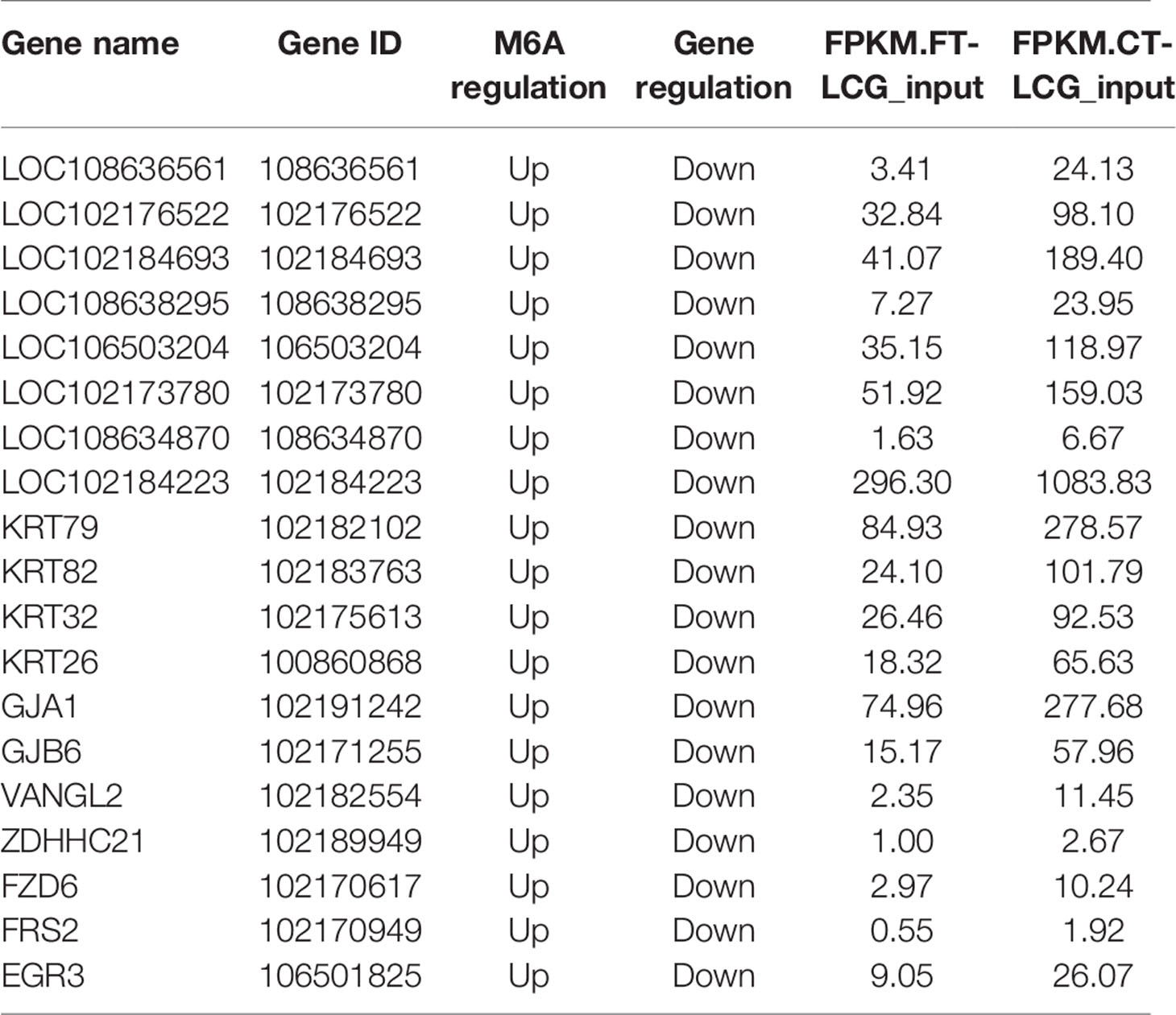
Table 5 Candidate m6A modified genes related to cashmere fineness.
GO Analysis and KEGG Pathway Analysis of Differently Methylated Genes
To explore the physiological and pathological significance of m6A modification, GO (http://www.geneontology.org/) and KEGG (http://www.kegg.jp/) pathway (P < 0.05) analysis were performed for differentially peaks. These peaks were enriched for 1,683 GO terms and 225 KEGG pathways. Top 25 in biological process, top 15 in cellular component and top 10 in molecular function are listed in Figure 5A. In the Figure 5B, Go analysis revealed that the differently methylated genes were significantly enriched in structural molecule activity, keratin filament, and intermediate filament. KEGG Pathway analysis showed that differently methylated genes were significantly associated with microRNA in cancer, PPAR signaling pathway, and protein digestion and absorption (Figure 5C).
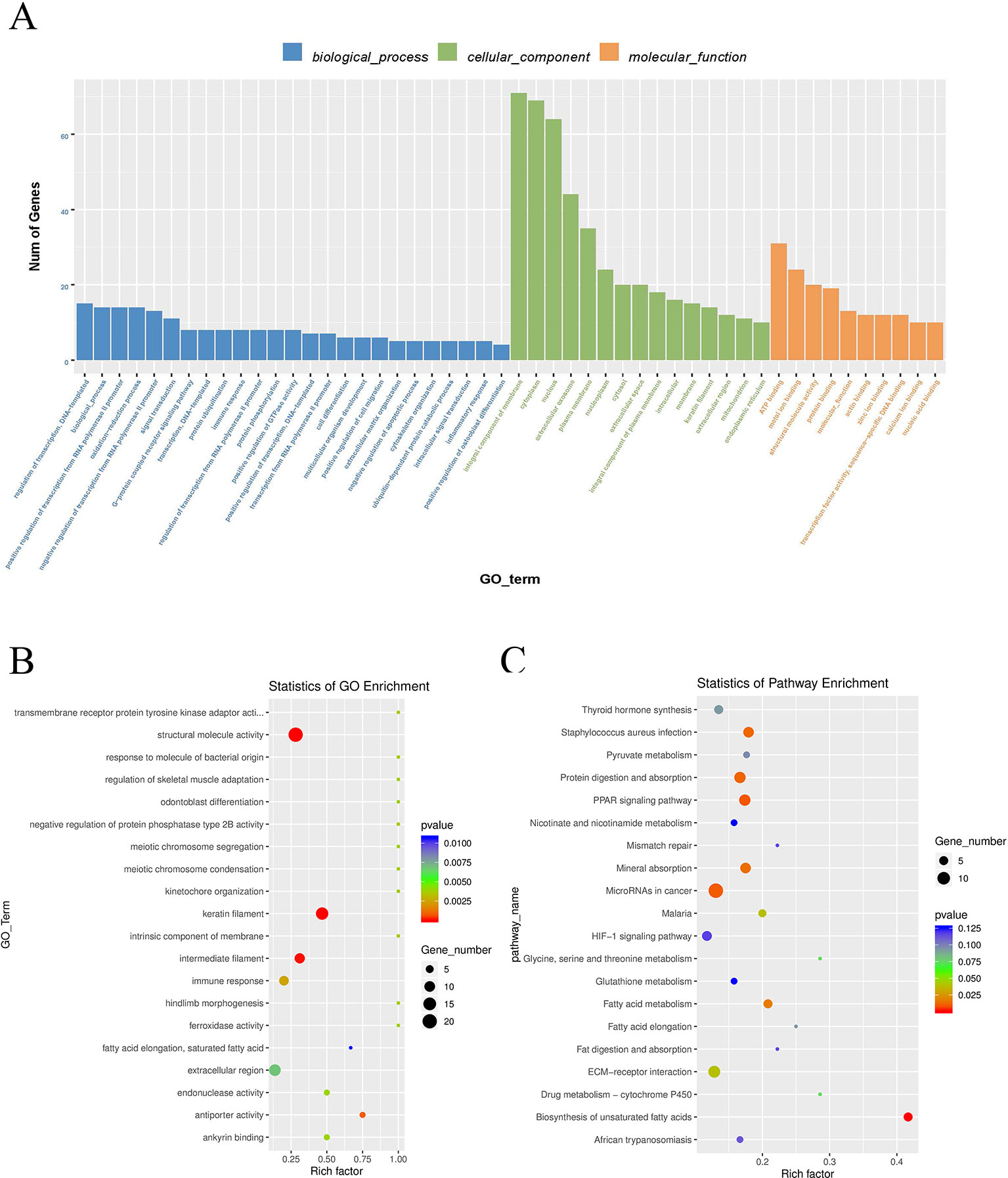
Figure 5 Gene ontology enrichment and KEGG pathway analysis of differential m6A peaks. (A) Major enrichment and meaningful GO terms of m6A peaks. (B) The top 20 significantly GO enrichment terms. (C) The top 20 enriched pathways of m6A peaks.
Validation of Differentially Expressed Genes by qPCR
To explore the potential role of m6A-modified genes and identify which genes are important to regulate cashmere fiber growth in LCG skin tissue, qRT-PCR was performed. RNA-seq results showed that the expression of differentially expressed genes in FT-LCG skin tissue was higher than that in CT-LCG. qRT-PCR confirmed that these m6A genes did exist in cashmere goat skin tissue. Except ZDHHC21, VANGL2, LOC108634870, FRS2, the variation trend of these genes was consistent with the results of RNA-seq. The results are showed in Figure 6.
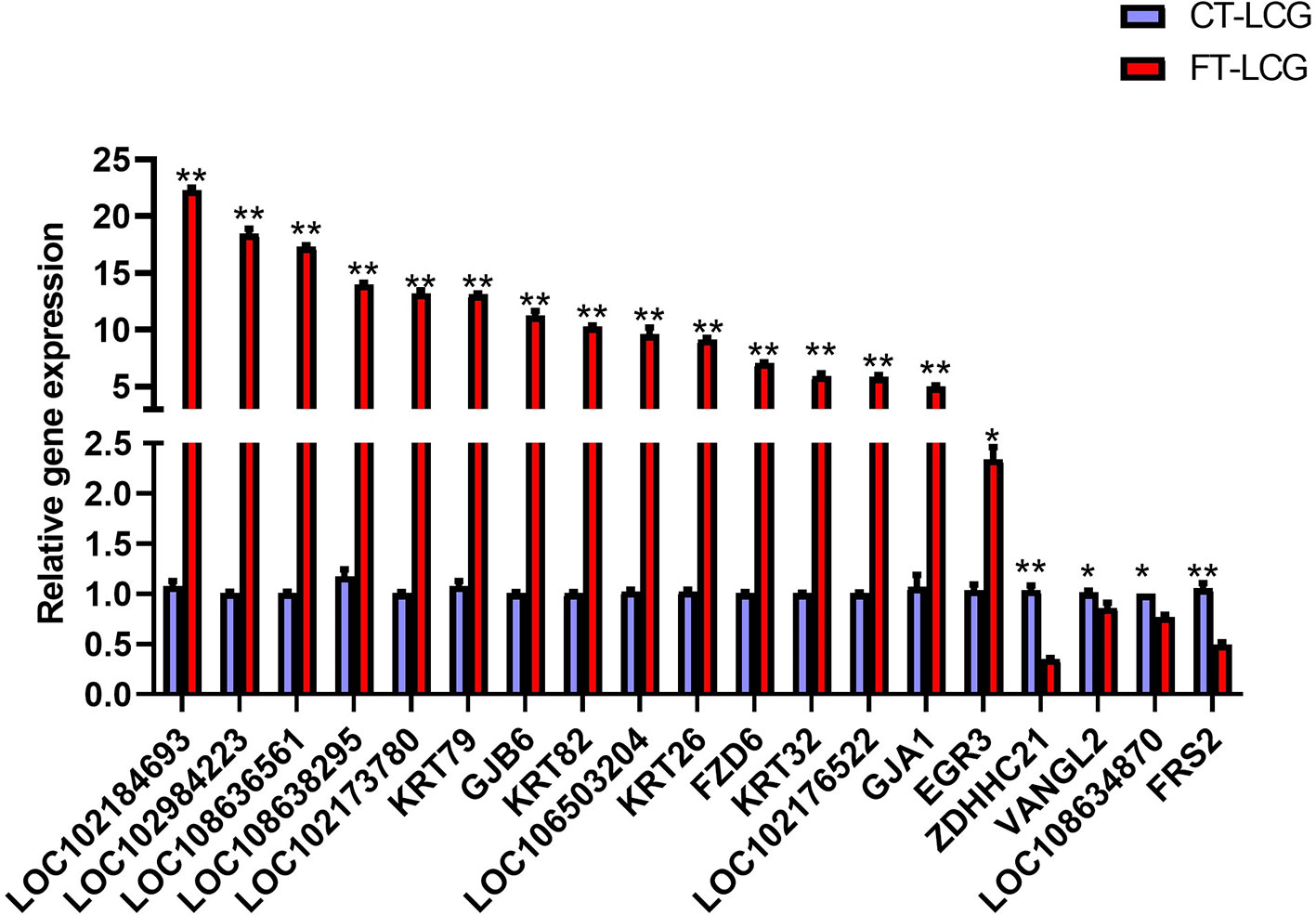
Figure 6 qPCR results of the 19 differentially m6A modified genes in FT-LCG and CT-LCG. The blue-purple column represents CT-LCG, the red column represents FT-LCG, ** represents P < 0.01, * represents P < 0.05.
Discussion
The same as DNA modifications, a series of reversible modification is present on RNA (Dominissini et al., 2012). Many m6A-related studies have been performed in many mammals, plant, and yeast, but no report has been published on m6A profiling in cashmere goats. Wang et al. identified that 922 m6A peaks, with 370 upregulated and 552 down regulated after traumatic brain injury (Wang et al., 2019). To our knowledge, this is the first comprehensive high-throughput study of RNA methylation in cashmere goat skin tissue. Our data showed that during the growth of cashmere, there are a large number of m6A methylation modification of genes in skin tissues. Further analysis showed that m6A modification may play an important role in cashmere growth by regulating gene expression level.
Herein, two sequencing libraries, namely m6A-seq library (IP) and RNA-seq library (Input), were constructed for high-throughput sequencing, and the results were analyzed with bioinformatics. Using MeRIP-seq method, we detected a large number of m6A methylation peaks in the transcriptome of cashmere goat skin tissue. In this study, 9,085 different peaks were detected, 7,170 m6A peaks were upregulated and 1,915 peaks were downregulated. It was reported that m6A peaks were mainly concentrated in long exons, stop codons, and in 3′UTRs (Dominissini et al., 2012). Our results also showed that these peaks were mainly concentrated in the 3′UTRs region. Based on the combined analysis of transcriptome and differential peaks, 19 differentially expressed genes containing RNA methylation modification were screened and all of them were related to cashmere growth. The FPKM of these genes in CT-LCG was higher than that in FT-LCG. According to published data, a consistent motif sequence “RRACH” is over-expressed in the m6A motif region. However, in our current data, another motif sequence, GAAGA, was enriched by MEME and HOMER software. The specific reasons are not clear, which need to be further studied. In addition, GO and KEGG pathway analysis were performed to deduce potential functions of m6A modified genes. Some pathways are known to play a vital role in the regulation of hair follicle development, such as Wnt, TGF-β, Notch, and MAPK which have 7, 6, 1, 13 differentially expressed genes, respectively. In this study, the m6A regulatory level of the differentially selected genes was negatively correlated with the transcriptional level in the cashmere goats' skin. Therefore, it can indicate that m6A could regulate mRNA degradation. The expression of differential genes in the skin tissue of CT-LCG were higher than that of FT-LCG in the RNA-seq result. Quantitative real-time PCR (qRT-PCR) confirmed that these differently methylated genes did exist in the skin tissue of cashmere goats.
As an important part of the skin-specific genetic network, EGR3 has been found to be related to the keratinocyte differentiation-related module and contribute to late epidermal differentiation (Kim et al., 2019). Keratin, as a structural protein of ectodermal cells, constitutes the hair skeleton, protects cells from mechanical damage, participates in cell signal transduction and apoptosis, and has functions of influencing hair diameter, participating in hair structural differentiation and skin appendage organ formation (Jin et al., 2011; Liffers et al., 2011). KRT family and KAP family are the main structural proteins of cashmere fiber, which determine the quality of cashmere fiber cluster (Rogers et al., 2004). Studies found that KAP1.1, KAP1.3, KAP8.1, KAP7.1, and KAP8.2 were related to cashmere fiber growth (Rogers et al., 2004; Jin et al., 2011; Plowman et al., 2012), we also identified these genes in our sequencing results. As an acidic type I keratin gene, KRT26 may be involved in the morphogenesis of hair follicles (Zhao et al., 2009). KRT32, and KRT82 mRNA were localized to cells forming the wool fiber and its cuticle in sheep. The study has shown that loss of ZDHHC21 function in hair loss mice results in defects in interfollicular epidermis, sebaceous gland hyperplasia, and delay in hair follicle differentiation (Pleasantine et al., 2009). LOC102176522, LOC108638295, LOC106503204, LOC102173780, LOC108634870, GJA1, GJB6, FRS2, and CTSK are also modified by m6A, but little research has been done on them, which may also be related to cashmere growth.
FZD6 is involved in regulating the Wnt signaling pathway. Previous studies have proved that Wnt signaling pathway plays a key role in maintaining hair follicle induction and forming new hair fibers in human and mouse data (Bai et al., 2017). The Wnt signaling pathway can regulate hair growth cycle and promote hair follicle differentiation (Andl et al., 2002). In addition, CAMK2G, FZD6, KANK3, LEF1, LYG2, MXRA8, and VANGL2 were enriched in the Wnt signaling pathway in our results. The classical Wnt signaling pathway participates in the biological process of skin and has the functions of regulating epithelial morphology, hair follicle development, and related cell differentiation (Andl et al., 2002). LEF1 is a downstream transcription factor of Wnt signaling pathway. Many experiments have shown that when Wnt signaling pathway is activated, LEF1 promotes hair follicle stem cells to differentiate into cells constituting hair follicle structure and plays an important role in hair follicle development (Ito et al., 2007). Other pathways related to cashmere growth, such as Notch, MAPK, and TGF-β signaling pathways, have m6A-modified genes enrichment. The role of most genes has not been reported in hair follicle growth and development. Notch signaling pathway can affect the formation of epidermal appendages. Notch signal is involved in the growth and development of various hair follicle cells. There were 13 differentially expressed genes were enriched in MAPK signaling pathway. It has been reported that the MAPK signaling pathway may be related to hair color formation (Schellenberger et al., 2011). MAPK signaling pathway plays an important role in regulating hair cycle and self-renewal of hair follicle stem cells (Akilli Öztürk et al., 2015). They are all modified by RNA N6-methylation, and they are involved in regulating these signaling pathways. It can be inferred indirectly that m6A may regulate these signaling pathways to some extent, which may regulate hair follicle development and differentiation.
In summary, this study analyzed the modification of m6A methylation in fine and coarse type Liaoning Cashmere Goat skin tissue. It was suggested that m6A methylation may play a role in regulating the expression of genes related to cashmere growth and cashmere fineness. This study suggests that KRT26, KRT32, KRT82, EGR3, and FZD6 are most likely to play a key role in regulating cashmere growth and fineness. In addition, the data obtained by high-throughput sequencing provide a basis for future research on the function of m6A methylation modification in cashmere growth process.
Data Availability Statement
The raw data has been made publically available. SRA accession: PRJNA591317.
Ethics Statement
The animal study was reviewed and approved by Shenyang Agricultural University.
Author Contributions
Data curation: YW. Formal analysis: YW, XJZ and YZ. Funding acquisition: ZW and WB. Methodology: SupG, ZW, and WB. Project administration: WB. Resources: DG and SulG. Software: YW and TH. Supervision: ZW and XHZ. Validation: YW, JS, CY, ZB and WC. Writing–original draft: YW. Writing–review and editing: YW, YF, ZW, and WB.
Funding
The work was supported financially by grants from the National Natural Science Foundation of China (No. 31802038, 31872325, 31672388), Key Project Foundation of Education Department of Liaoning Province, China (NO. LSNZD201606), Breeding project of new Liaoning cashmere goat “meat and meat dual-use” (No. 2017202005), Science and Technology Innovation Talent Support Foundation for Young and Middle-aged People of Shenyang City, China (RC170447).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.01318/full#supplementary-material
References
Akilli Öztürk, Ö., Pakula, H., Chmielowiec, J., Qi, J., Stein, S., Lan, L., et al. (2015). Gab1 and mapk signaling are essential in the hair cycle and fair follicle stem cell quiescence. Cell Rep. 13, 561–572. doi: 10.1016/j.celrep.2015.09.015
Andl, T., Reddy, S. T., Gaddapara, T., Millar, S. E. (2002). Wnt signals are required for the initiation of hair follicle development. Dev. Cell 2, 643–653. doi: 10.1016/s1534-5807(02)00167-3
Bai, W., Yin, R., Yin, R., Wang, J., Jiang, W., Luo, G., et al. (2013). IGF1 mrna splicing variants in liaoning cashmere goat: identification, characterization, and transcriptional patterns in skin and visceral organs. Anim. Biotechnol. 24, 13. doi: 10.1080/10495398.2012.750245
Bai, W. L., Dang, Y. L., Wang, J. J., Yin, R. H., Wang, Z. Y., Zhu, Y. B., et al. (2016). Molecular characterization, expression and methylation status analysis of BMP4 gene in skin tissue of Liaoning cashmere goat during hair follicle cycle. Genetica 144, 457–467. doi: 10.1007/s10709-016-9914-1
Bai, W. L., Wang, J. J., Yin, R. H., Dang, Y. L., Wang, Z. Y., Zhu, Y. B., et al. (2017). Molecular characterization ofhoxc8gene and methylation status analysis of its exon 1 associated with the length of cashmere fiber in liaoning cashmere goat. Genetica 145, 115–126. doi: 10.1007/s10709-017-9950-5
Bai, W. L., Zhao, S. J., Wang, Z. Y., Zhu, Y. B., Dang, Y. L., Cong, Y. Y., et al. (2018). LncRNAs in secondary hair follicle of cashmere goat: identification, expression, and their regulatory network in wnt signaling pathway. Anim. Biotechnol. 29, 1–13. doi: 10.1080/10495398.2017.1356731
Bailey, T. L., Boden, M., Buske, F. A., Frith, M., Grant, C. E., Clementi, L., et al. (2009). Meme suite: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208. doi: 10.1093/nar/gkp335
Beemon, K., Keith, J. (1977). Localization of N6-methyladenosine in the Rous sarcoma virus genome. J. Mol. Biol. 113, 165–179. doi: 10.1016/0022-2836(77)90047-x
Ben-Haim, M. S., Moshitch-Moshkovitz, S., Rechavi, G. (2015). FTO: linking m6A demethylation to adipogenesis. Cell Res. 25, 3–4. doi: 10.1038/cr.2014.162
Bin, J., Ji-Feng, X. I., Su-Yun, Z., Zong-Sheng, Z., Ru-Qian, Z., Jie, C. (2006). The developmental patterns of GH-R, IGF-1 and IGF-IR gene expression in sheep skin. Hereditas 28, 1078–1082. doi: 10.1360/yc-006-1078
Boccaletto, P., Machnicka, M. A., Purta, E., Piatkowski, P., Baginski, B., Wirecki, T. K., et al. (2018). MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. 46, D303–D307. doi: 10.1093/nar/gkx1030
Desrosiers, R., Friderici, K., Rottman, F. (1974). Identification of methylated nucleosides in messenger RNA from novikoff hepatoma cells. Proc. Natl. Acad. Sci. 71, 3971–3975. doi: 10.1073/pnas.71.10.3971
Dominissini, D., Moshitch-Moshkovitz, S., Schwartz, S., Salmon-Divon, M., Ungar, L., Osenberg, S., et al. (2012). Topology of the human and mouse m6a rna methylomes revealed by m6a-seq. Nature 485, 201–206. doi: 10.1038/nature11112
Dubin, D. T., Taylor, R. H. (1975). The methylation state of poly A-containing-messenger RNA from cultured hamster cells. Nucleic Acids Res. 2, 1653–1668. doi: 10.1093/nar/2.10.1653
Fan, Y. X., Wu, R. B., Qiao, X., Zhang, Y. J., Wang, R. J., Su, R., et al. (2015). Hair follicle transcriptome profiles during the transition from anagen to catagen in cashmere goat (Capra hircus). Genet. Mol. Res. Gmr 14, 17904. doi: 10.4238/2015.December.22.15
Furuichi, Y., Miura, K. I. (1975). A blocked structure at the 5'terminus of mRNA from cytoplasmic polyhedrosis virus. Nature 253, 374–375. doi: 10.1038/253374a0
Haussmann, I. U., Bodi, Z., Sanchez-Moran, E., Mongan, N. P., Archer, N., Fray, R. G., et al. (2016). m6A potentiates Sxl alternative pre-mRNA splicing for robust drosophila sex determination. Nature 540, 301–304. doi: 10.1038/nature20577
Heinz, S., Benner, C., Spann, N., Bertolino, E., Glass, C. K. (2010). Simple combinations of lineage-determining factors prime cis-regulatory elements required for macrophage and b-cell identities. Mol. Cell 38, 576–589. doi: 10.1016/j.molcel.2010.05.004
Hibar, D. P., Stein, J. L., Renteria, M. E., Arias-Vasquez, A., Desrivières, S., Jahanshad, N., et al. (2015). Common genetic variants influence human subcortical brain structures. Nature 520, 224–229. doi: 10.1038/nature14101
Ito, M., Yang, Z., Andl, T., Cui, C., Kim, N., Millar, S. E., et al. (2007). Wnt-dependent de novo hair follicle regeneration in adult mouse skin after wounding. Nature 447, 316–320. doi: 10.1038/nature05766
Ivanova, I., Much, C., Di, G. M., Azzi, C., Morgan, M., Moreira, P. N., et al. (2017). The RNA m(6)A reader YTHDF2 is essential for the post-transcriptional regulation of the maternal transcriptome and oocyte competence. Mol. Cell 67, 1059–1067.e4. doi: 10.1016/j.molcel.2017.08.003
Jin, M., Wang, L., Li, S., Xing, M. X., Zhang, X. (2011). Characterization and expression analysis of KAP7.1 KAP8.2 gene in Liaoning new -breeding cashmere goat hair follicle. Mol. Bio Rep. 38, 3023–3028. doi: 10.1007/s11033-010-9968-6
Kim, D., Langmead, B., Salzberg, S. L. (2015). Hisat: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Kim, K. H., Son, E. D., Kim, H. J., Lee, S. H., Bae, I. H., Lee, T. R. (2019). EGR3 is a late epidermal differentiation regulator that establishes the skin-specific gene network. J. Invest. Dermatol 139, 615–625. doi: 10.1016/j.jid.2018.09.019
Li, F., Kennedy, S., Hajian, T., Gibson, E., Seitova, A., Xu, C., et al. (2015). A radioactivity-based assay for screening human m6A-RNA methyltransferase, METTL3-METTL14 complex, and demethylase ALKBH5. J. Biomolecular Screening 21, 290–297. doi: 10.1177/1087057115623264
Liffers, S. T., Maghnouj, A., Munding, J. B., Jackstadt., R., Hahn, S. A. (2011). Keratin 23, a novel dpc4/smad4 target gene which binds 14-3-3. BMC Cancer 11, 137. doi: 10.1186/1471-2407-11-137
Lin, Z., Hsu, P. J., Xing, X., Fang, J., Lu, Z., Zou, Q., et al. (2017). Mettl3-/mettl14-mediated mRNA n6-methyladenosine modulates murine spermatogenesis. Cell Res. 27, 1216–1230. doi: 10.1038/cr.2017.117
Liu, G., Liu, R., Tang, X., Cao, J., Zhao, S., Yu, M. (2015). Expression profiling reveals genes involved in the regulation of wool follicle bulb regression and regeneration in sheep. Int. J. Mol. Sci. 16, 9152–9166. doi: 10.3390/ijms16059152
Liu, B., Gao, F., Guo, J., Wu, D., Hao, B., Li, ,. Y., et al. (2016). A microarray-based analysis reveals that a short photoperiod promotes hair growth in the arbas cashmere goat. PloS One 11, e0147124. doi: 10.1371/journal.pone.0147124
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet J. 17 (1), 1384. doi: 10.14806/ej.17.1.200
Meng, J., Lu, Z., Liu, H., Zhang, L., Zhang, S., Chen, Y., et al. (2014). A protocol for rna methylation differential analysis with merip-seq data and exomepeak r/bioconductor package. Methods 69, 274–281. doi: 10.1016/j.ymeth.2014.06.008
Meyer, K. D., Jaffrey, S. R. (2017). Rethinking m6A readers, writers, and erasers. Annu. Rev. Cell Dev. Biol. 33, 319. doi: 10.1146/annurev-cellbio-100616-060758
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T. C., Mendell, J. T., Salzberg, S. L. (2015). Stringtie enables improved reconstruction of a transcriptome from rna-seq reads. Nat. Biotechnol. 33, 290–295. doi: 10.1038/nbt.3122
Pleasantine, M., Lee, A. W. S., Yuko, F., Ryouhei, T., Masaki, F., Margaret, K., et al. (2009). Palmitoylation regulates epidermal homeostasis and hair follicle differentiation. PloS Genet. 5, e1000748. doi: 10.1371/journal.pgen.1000748
Plowman, J. E., Deb-Choudhury, S., Clerens, S., Thomas, A., Cornellison, C. D., Dyer, J. M. (2012). Unravelling the proteome of wool: towards markers of wool quality traits. J. Proteomics 75, 4315–4324. doi: 10.1016/j.jprot.2012.03.027
Robinson, M. D., McCarthy, D. J., Smyth, G. K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26 ((1)), 139–140. doi: 10.1093/bioinformatics/btp616
Rogers, M. A., Langbein, L., Winter, H., Beckmann, I., Schweizer, ,. J. (2004). Hair keratin associated proteins: characterization of a second high sulfur kap gene domain on human chromosome 211. J. Invest. Dermatol. 122, 147–158. doi: 10.1046/j.0022-202X.2003.22128.x
Rufaut, N. W., Pearson, A. J., Nixon, A. J., Wheeler, T. T., Wilkins, R. J. (1999). Identification of differentially expressed genes during a wool follicle growth cycle induced by prolactin. J. Invest. Dermatol. 113, 865–872. doi: 10.1046/j.1523-1747.1999.00775.x
Schellenberger, A. A., Horland, R., Rosowski, M., Paus, R., Lauster, R., Lindner, G. (2011). Cartilage oligomeric matrix protein (COMP) forms part of the connective tissue of normal human hair follicles. Exp. Dermatol. 20, 361–366. doi: 10.1111/j.1600-0625.2010.01217.x
Stenn, K. S., Paus, R. (2001). Controls of hair follicle cycling. Physiol. Rev. 81, 449–494. doi: 10.1152/physrev.2001.81.1.449
Wang, Y., Mao, J., Wang, X., Lin, Y., Hou, G., Zhu, J., et al. (2019). Genome-wide screening of altered m6A-tagged transcript profiles in the hippocampus after traumatic brain injury in mice. Epigenomics 11, 805–819. doi: 10.2217/epi-2019-0002
Weng, Y. L., Wang, X., An, R., Cassin, J., Visser, C., Liu, Y. Y., et al. (2018). Epitranscriptomic m6A regulation of axon regeneration in the adult mammalian nervous system. Neuron 97, 313. doi: 10.1016/j.neuron.2017.12.036
Xiaolong, W., Bei, C., Jiankui, Z., Haijing, Z., Yiyuan, N., Baohua, M., et al. (2016). Disruption of FGF5 in cashmere goats using CRISPR/CAS9 results in more secondary hair follicles and longer fibers. PloS One 11, e0164640. doi: 10.1371/journal.pone.0167322
Yang, J. B., Gan, S. Q., Yang, Y. L., Zhang, H. L., Shen, M. (2012). Cloning and expression in follicle anagen of ilk gene in sheep. Hereditas 34, 719–726. doi: 10.3724/SP.J.1005.2012.00719
Yang, Y., Hsu, P. J., Chen, Y. S., Yang, Y. G. (2018). Dynamic transcriptomic m6A decoration: writers, erasers, readers and functions in RNA metabolism. Cell Res. 28, 616–624. doi: 10.1038/s41422-018-0040-8
Yu, Z., Wildermoth, J. E., Wallace, O. A., Gordon, S. W., Maqbool, N. J., Maclean, P. H. (2011). Annotation of sheep keratin intermediate filament genes and their patterns of expression. Exp. Dermatol. 20, 582–588. doi: 10.1111/j.1600-0625
Yu, G., Wang, L. G., He, Q. Y. (2015). Chipseeker: an r/bioconductor package for chip peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383. doi: 10.1093/bioinformatics/btv145
Zhang, B., Chang, L., Lan, Y. X., Asif, N., Guan, F. L., Fu, D. K., et al. (2018). Genome-wide definition of selective sweeps reveals molecular evidence of trait-driven domestication among elite goat (Capra species) breeds for the production of dairy, cashmere, and meat. Gigascience 7, giy105. doi: 10.1093/gigascience/giy105
Keywords: MeRIP-seq, RNA-seq, m6A modification, cashmere fineness, m6A-modified genes
Citation: Wang Y, Zheng Y, Guo D, Zhang X, Guo S, Hui T, Yue C, Sun J, Guo S, Bai Z, Cai W, Zhang X, Fan Y, Wang Z and Bai W (2020) m6A Methylation Analysis of Differentially Expressed Genes in Skin Tissues of Coarse and Fine Type Liaoning Cashmere Goats. Front. Genet. 10:1318. doi: 10.3389/fgene.2019.01318
Received: 22 September 2019; Accepted: 03 December 2019;
Published: 22 January 2020.
Edited by:
Peter G. Zaphiropoulos, Karolinska Institutet (KI), SwedenReviewed by:
Jia Meng, Xi'an Jiaotong-Liverpool University, ChinaZhigang Wang, Inner Mongolia University, China
Zhuanjian Li, Henan Agricultural University, China
Copyright © 2020 Wang, Zheng, Guo, Zhang, Guo, Hui, Yue, Sun, Guo, Bai, Cai, Zhang, Fan, Wang and Bai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zeying Wang, d2FuZ3pleWluZzIwMTJAc3lhdS5lZHUuY24=; Wenlin Bai, YmFpd2VubGluQHN5YXUuZWR1LmNu