 Marcelo L. Ribeiro
Marcelo L. Ribeiro Diana Reyes-Garau
Diana Reyes-Garau Marc Armengol
Marc Armengol Miranda Fernández-Serrano
Miranda Fernández-Serrano Gaël Roué
Gaël Roué
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet., 16 October 2019
Sec. Epigenomics and Epigenetics
Volume 10 - 2019 | https://doi.org/10.3389/fgene.2019.00986
This article is part of the Research TopicEpigenetic Biomarker and Personalized Precision MedicineView all 33 articles
In the last 10 years, major advances have been made in the diagnosis and development of selective therapies for several blood cancers, including B-cell non-Hodgkin lymphoma (B-NHL), a heterogeneous group of malignancies arising from the mature B lymphocyte compartment. However, most of these entities remain incurable and current treatments are associated with variable efficacy, several adverse events, and frequent relapses. Thus, new diagnostic paradigms and novel therapeutic options are required to improve the prognosis of patients with B-NHL. With the recent deciphering of the mutational landscapes of B-cell disorders by high-throughput sequencing, it came out that different epigenetic deregulations might drive and/or promote B lymphomagenesis. Consistently, over the last decade, numerous epigenetic drugs (or epidrugs) have emerged in the clinical management of B-NHL patients. In this review, we will present an overview of the most relevant epidrugs tested and/or used so far for the treatment of different subtypes of B-NHL, from first-generation epigenetic therapies like histone acetyl transferases (HDACs) or DNA-methyl transferases (DNMTs) inhibitors to new agents showing selectivity for proteins that are mutated, translocated, and/or overexpressed in these diseases, including EZH2, BET, and PRMT. We will dissect the mechanisms of action of these epigenetic inhibitors, as well as the molecular processes underlying their lack of efficacy in refractory patients. This review will also provide a summary of the latest strategies being employed in preclinical and clinical settings, and will point out the most promising lines of investigation in the field.
At the origin of 4% of all cancers and more than 90% of the cases of lymphoma, B-NHLs comprise a heterogeneous group of lymphoid neoplasms. According to the last World Health Organization hematopoietic and lymphoid tumor classification, more than 40 distinct entities are categorized, according to a combination of morphological, immunophenotypic, genetic, and clinical features, having each entity its own clinical course and requiring specific treatments (Table 1) (Campo et al., 2011; Scott and Gascoyne, 2014; Swerdlow et al., 2016). Originated from either mature or immature B cells, B-NHLs are characterized by the proliferation of lymphocytes, mainly in lymphoid organs and in extranodal tissues. Their diversity can often be traced to a particular stage of differentiation, from the bone marrow where the normal precursor B cell is originated to secondary lymphoid tissues where B cells undergo multiple rounds of selection before their differentiation into plasma cells or memory B cells. During these processes, the VDJ heavy chain is formed, followed by VJ light-chain gene rearrangement, which allows the pre-B cells to express intracytoplasmic μ-heavy chains. Subsequently, immature immunoglobulin (Ig)-positive B cells are formed. Within the lymph node, and in contact with a determined antigen, naïve B cells can mature into IgM-secreting plasma cells or may proliferate into primary follicles to form germinal center (GC) centroblasts. Upon maturation, they further differentiate into centrocytes, which give place to memory B cells or plasma cells. Within the GC, somatic hypermutation in the Ig heavy or light chain variable region (IGHV or IGHL) genes leads to increased antigen affinity.
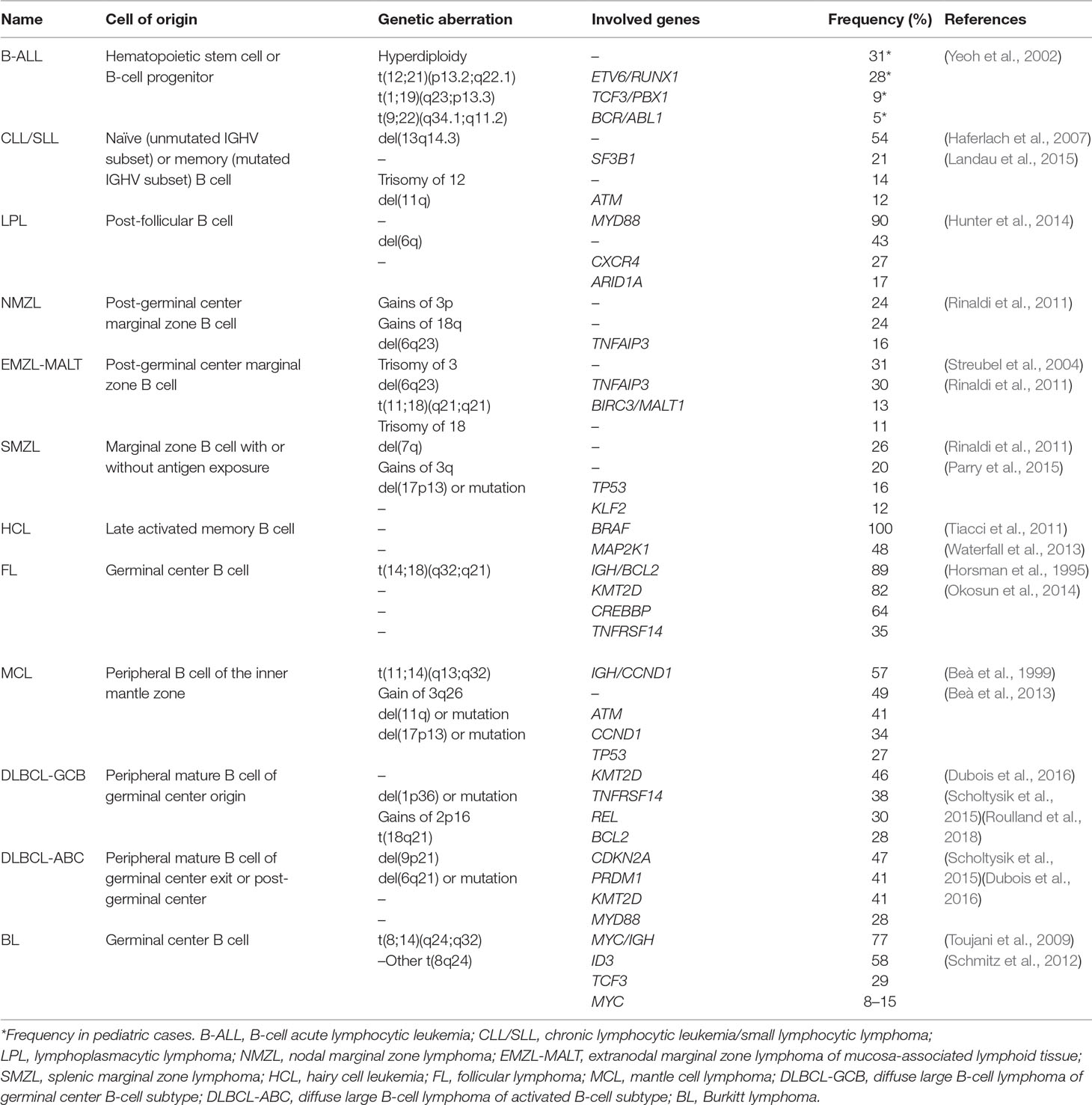
Table 1 Classification of B-cell non-Hodgkin lymphoma.
Although tightly regulated, the B-cell differentiation process and especially the antibody diversification phase can be accompanied by inherited events that may favor lymphomagenesis, such as chromosomal translocations, oncogene activation, and/or inactivating mutations in tumor suppressor genes. Infection by determined viruses, such as the Epstein–Barr virus, has also been involved in lymphomagenesis. The malignant counterparts of the early B-cell differentiation steps account for B lymphoblastic lymphomas, which harbor high similarity with B progenitor cells. On the other hand, mantle cell lymphomas (MCLs) and a subset of chronic lymphocytic leukemia (CLL) with unmutated IGHV are thought to derive from naive B cells and pre-GC mature B cells expressing the CD5 surface marker. Other GC-originated lymphomas, including follicular lymphoma (FL), Burkitt’s lymphoma (BL), a subset of diffuse large B-cell lymphoma (DLBCL), and Hodgkin’s lymphoma (HL), present mutations in IGHV gene. Additional entities, including marginal zone lymphoma (MZL), lymphoplasmacytic lymphoma, CLL with somatic IGHV mutation, another subset of DLBCL, and multiple myeloma (MM) correspond to post-GC cells. Each lymphoma subtype retains key features of their cell of origin as judged by the similarity of immunophenotype, histological appearance, and gene expression profiles (Seifert et al., 2013) (Table 1). The putative normal B-cell counterpart of each B-cell lymphoma is summarized in Figure 1.
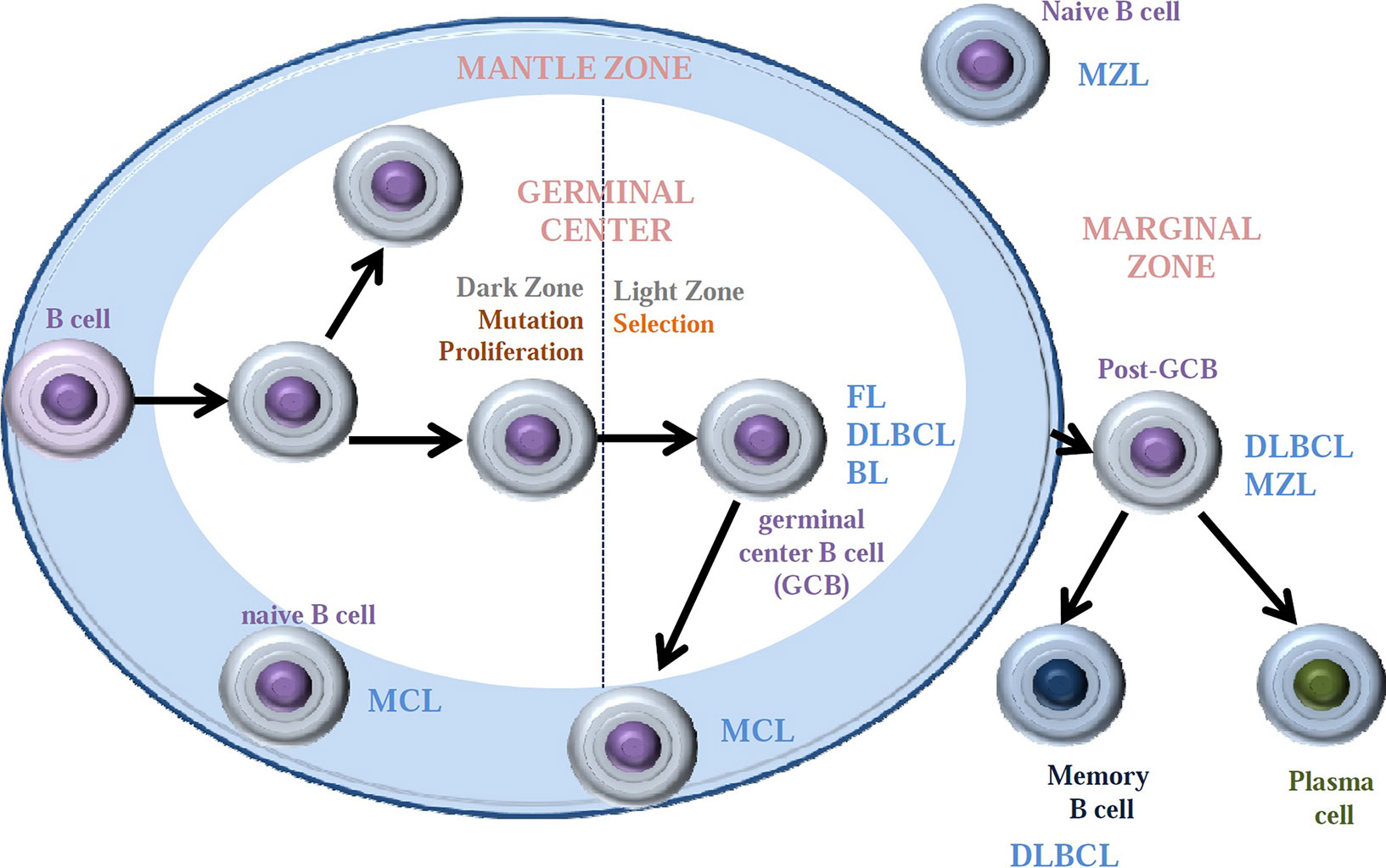
Figure 1 Major B-cell non-Hodgkin lymphoma subtypes arise from different cell of origin within the lymph node. Mantle cell lymphomas (MCL) arise from naive B cells or germinal center (GC) B cells found within the mantle zone. Marginal zone lymphomas initiate from naive B cells or GCB that have entered the marginal zone. GCB are the origin of follicular lymphomas (FL), Burkitt lymphoma (BL), and diffuse large B-cell lymphomas (DLBCL) when still in the germinal center. This last DLBCL appears to also form GCB within the marginal zone or from fully developed memory B cells.
In the last decade, loads of evidences have suggested an association between the frequent alterations in chromatin state and epigenetic regulators observed in B-NHL patients, and disease formation and progression.
Contrary to the general belief that only accumulations of DNA mutations might lead directly to the development of tumorigenic processes, it has been progressively reported a growing subset of epigenetic alterations lying at the basis of many malignancies, including those occurring in lymph nodes. Interestingly, in B-cell lymphomas, certain somatic mutations in chromatin-modifying enzymes account for several epigenetic alterations, suggesting that an aberrant epigenetic landscape in B-NHL may be a consequence of genetic alterations associated with a particular lymphoma subtype. For instance, deleterious and/or loss of function mutations in the histone acetyltransferase CREB binding protein (CREBBP) or the E1A binding protein 300 (EP330) have been reported in about 40% of DLBCL and FL patients as well as in other lymphoma subtypes (Morin et al., 2011; Pasqualucci et al., 2011b). Recurrent point mutations in the histone acetyl transferase (HAT) recruiting gene myocyte enhancer binding factor 2B (MEF2B) have been also described in 15% of FL and 13% of DLBCL patients with germinal center B cell (DLBCL-GCB) subtype (Morin et al., 2011). Although no mutations have been reported in the genes coding for histone deacetylases (HDACs), several members of this family like HDAC1, 2, and 6 can be overexpressed in DLBCL, in association with a decrease in the DNA accessibility to the transcription machinery (Marquard et al., 2009).
In addition to mutations in chromatin‐regulatory proteins, epigenetic modifications at chromatin level are also commonly observed in B-NHL as a result of profound changes in DNA methylation patterns. Indeed, while hypo- and hyper-DNA methylation status have been linked to the pathogenesis of several cancer subtypes, somatic mutations in epigenetic genes codifying for DNA methylation regulators have been particularly well associated to a repressed chromatin state and to malignant processes in B-NHL (Esteller et al., 2001; Hassler et al., 2013). Among the main reported alterations, activating mutations in enhancer of zeste homolog 2 (EZH2), a histone methyltransferase (HMT) gene, were found in 22% of DLBCL-GCB patients and 7% of FL patients (Morin et al., 2010). Further loss‐of‐function mutations were observed in the histone-Lysine N-Methyltransferase 2D (MLL2/KMT2D) gene in about 90% of FL and 30% of DLBCL patients (Morin et al., 2011; Pasqualucci et al., 2011b; Lohr et al., 2012). Concretely, MLL2 presents a defective SET domain when mutated by either truncation or frameshift mutations, leading to a reduced H3K4 methylation activity (Shilatifard, 2008; Morin et al., 2011; Pasqualucci et al., 2011b; Lohr et al., 2012).
Hence, B-NHL occurrence as a result of disruption in epigenetic mechanisms has generated a strong rationale to target epigenetic and chromatin regulators for drug discovery attempts. To address these alterations, several Food and Drug Administration (FDA)–approved epigenetic-modulating agents, whose clinical use has been mainly restrained so far to other hematological malignancies (Popovic et al., 2013), are now being made available for their evaluation in B-NHL. These agents include the HDAC inhibitors romidepsin (FK228, depsipeptide), vorinostat (suberanilohydroxamic acid, SAHA), panobinostat (LBH589), and belinostat (PXD101); the DNA methyltransferase (DNMT) inhibitors (hypomethylating agents, HMAs) azacitidine (5-azacytidine) and decitabine (5-aza-2′-deoxycytidine); and the isocitrate dehydrogenase (IDH) inhibitors enasidenib (AG-221) and ivosidenib (AG-120) (Table 2).
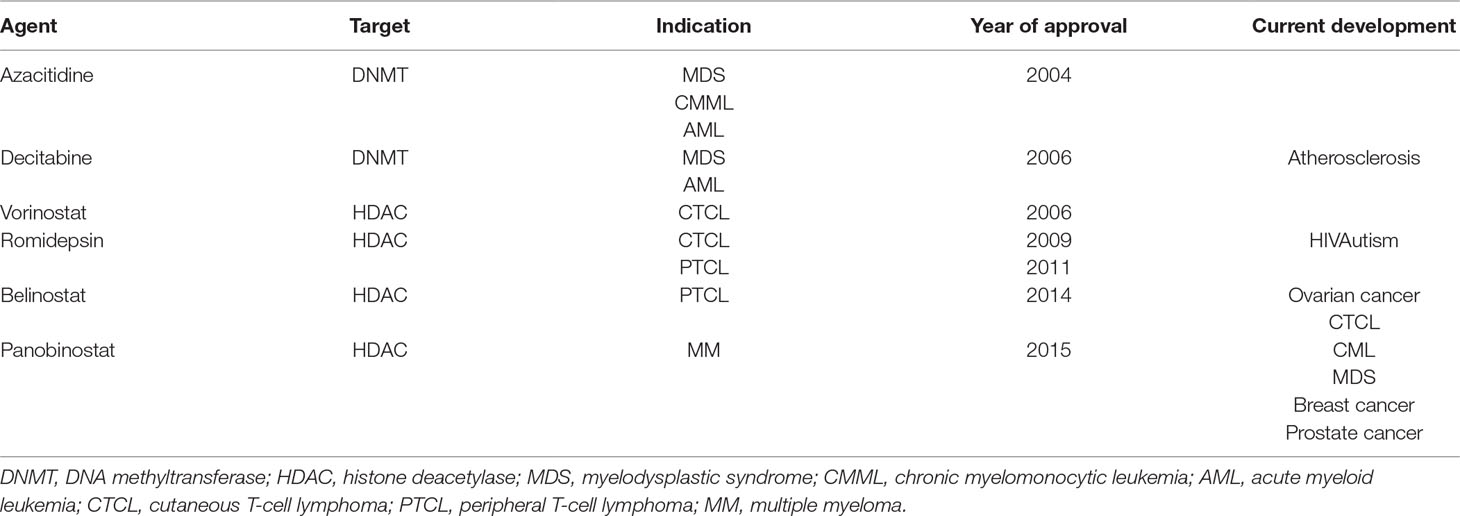
Table 2 FDA-approved epigenetic drugs for hematological malignancies.
DNA methylation is responsible for the control of gene expression and for maintaining genomic stability during embryogenesis and tissue differentiation (Meissner, 2010). This process is clonally inherited and preserved in daughter cells, and occurs through the inclusion of a methyl group at cytosine residues in CpG dinucleotides (Figure 2). It is carried out by the DNMTs, namely DNMT1 which primarily mediates maintenance methylation during cell division, and DNMT3A and 3B that regulate de novo DNA methylation (Belinsky et al., 2003; Hermann et al., 2004). DNA methylation is thought to have a significant role in the regulation of lymphoid compartment, as it has been demonstrated that differential recruitment of DNMT1, DNMT3A, and DNMT3B and consequent specific DNA methylation patterns are determined at early stages during lymphopoiesis and B-cell activation (Shaknovich et al., 2011; Lai et al., 2013).
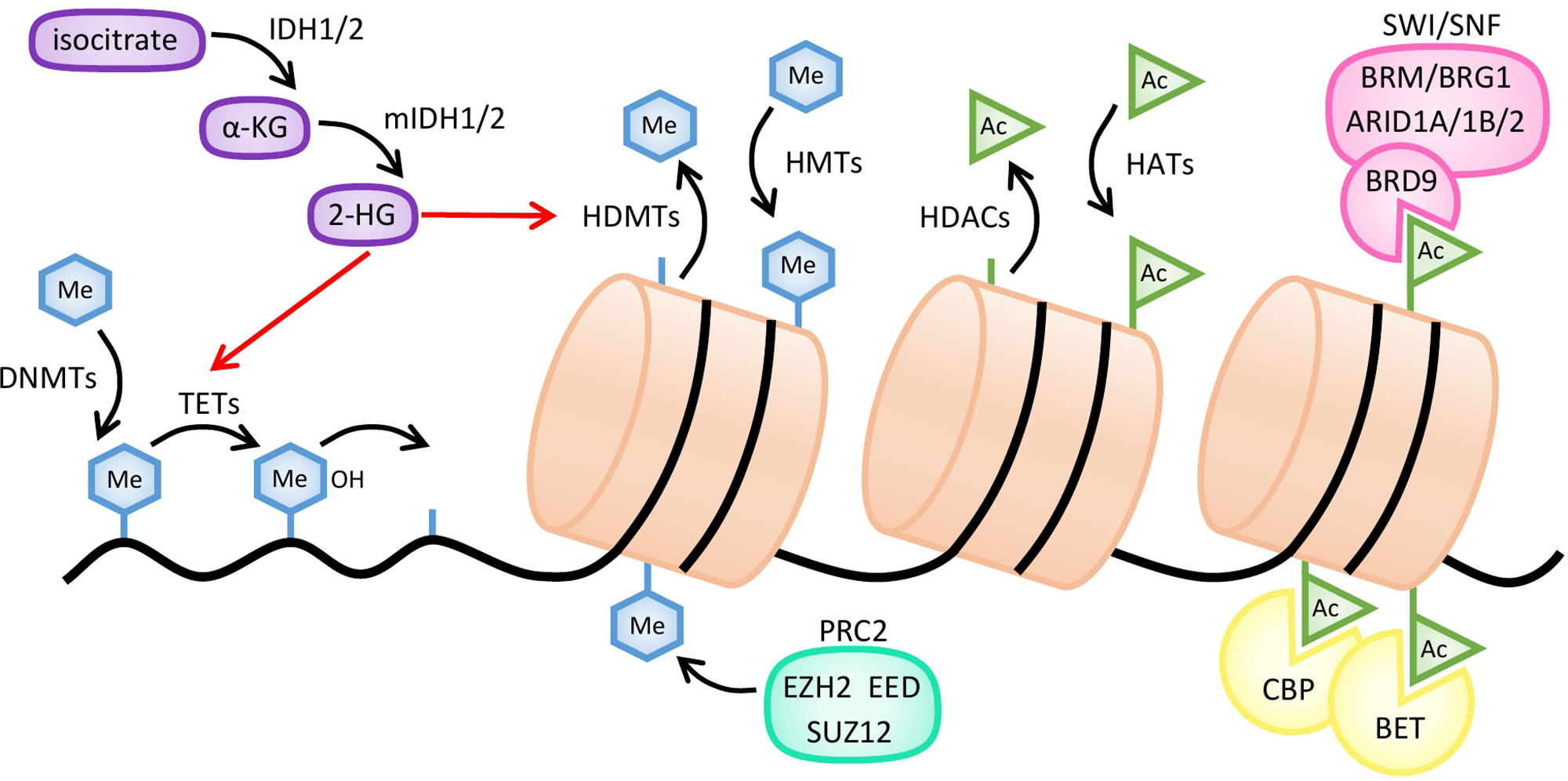
Figure 2 Mechanisms of action of common epigenetic enzymes. Histone methylation is regulated by histone methyltransferases (HMTs), such as the EZH2 subunit of polycomb repressive complex 2 (PRC2), and histone demethylases (HDMTs). DNA methylation is established by DNA methyltransferases (DNMTs) and reversed by several enzymes like TET hydroxymethylases. Demethylation of both histones and DNA is inhibited by 2-hydroxyglutarate (2-HG), produced from α-ketoglutarate (α-KG) by mutant forms of IDH1/2 enzymes (mIDH). Histone acetylation is regulated by histone acetyltransferases (HATs) and histone deacetylases (HDACs). Bromodomain-containing proteins, such as CREB-binding protein (CBP), BET, or the BRD9 subunit of the SWI/SNF complex, bind to acetylated residues of histones.
While on the one hand, DNA methylation is essential for cell homeostasis, on the other hand, disturbance in methylation pattern have been widely described in cancer. Changes in CpG methylation are indeed commonly associated with malignant transformation and tumor progression (Berdasco and Esteller, 2010). In addition, accumulating evidences suggest that aberrant epigenetic regulation, including DNA methylation, exerts an important role in regulating each cancer’s hallmarks (Flavahan et al., 2017). Illustrating this relationship in B-NHL, Shaknovich and collaborators demonstrated the relevance of DNA methylation in defining the molecular DLBCL subtypes (Shaknovich et al., 2010). It was further proposed that DNMT1 and DNMT 3B overexpression may play a role in malignant progression of these tumors (Amara et al., 2010) and also in BL neoplasm (Robaina et al., 2015). In line with this, the disruption of DNA methylation pattern is correlated with disease severity and patient survival in DLBCL and FL (De et al., 2013).
Considering that the majority of cancers, including B-NHL, harbor an altered DNA methylation pattern, and also taking into account the reversibility of this alteration, the idea to modulate the methylation machinery to restore a “normal” DNA methylation state has attracted great attention in cancer treatment (Azad et al., 2013). The first two DNA methylation epigenetic compounds (DNMTi) ratified by the FDA and the European Medicines Agency for cancer treatment, azacitidine and decitabine (Jones et al., 2016), were initially described as promising chemotherapeutic agents against myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML), although with moderate efficacy and high toxicity (Li et al., 1970; Vogler et al., 1976). In further trials, low-dose decitabine and azacitidine demonstrated to be effective in these patients, improving both the response and the overall survival (OS), leading to their further approval (Table 2 and Figure 3) (Silverman et al., 2002; Fenaux et al., 2009; Lübbert et al., 2016). In B-NHL patients, two phase I studies using decitabine have been completed so far, but the response to therapy and the effect on DNA methylation were moderate (Stewart et al., 2009; Blum et al., 2010). Currently, azacitidine and decitabine are being evaluated alone or in combination in approximately 10 active clinical trials involving relapsed/refractory R/R B-NHL patients (Table 3). Considering the preliminary data of these trials, it seems premature to conclude that DNMTis can be used as monotherapy in B-NHL.
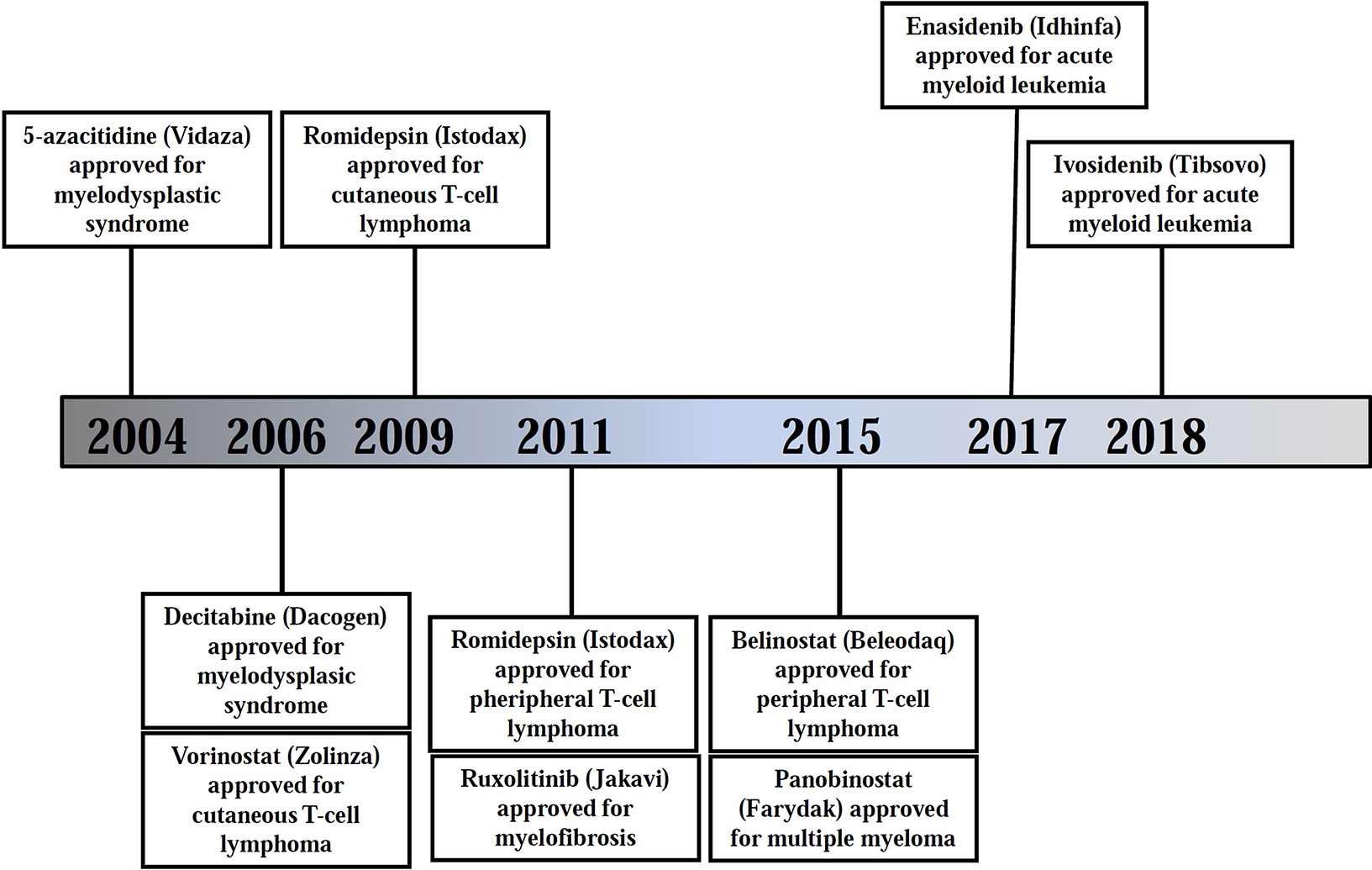
Figure 3 Timeline of FDA approvals of epigenetic-modulating therapies in hematological cancers. Source: https://www.accessdata.fda.gov/scripts/cder/daf/
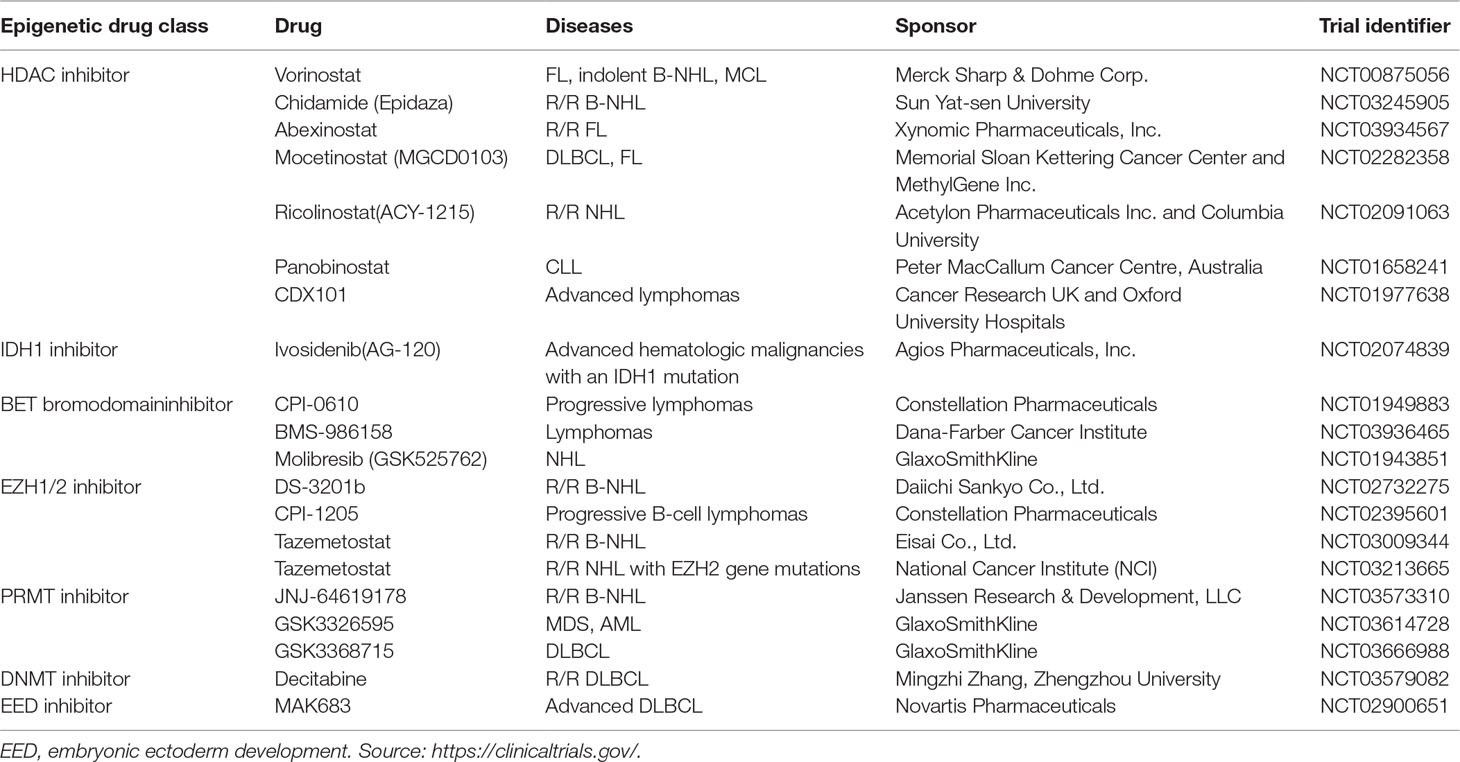
Table 3 Selected examples of epigenetic drugs under clinical evaluation in B-NHL patients as single agents.
Although the mechanism of action of DNMTi is not well understood, the activity of decitabine and azacitidine is known to involve their incorporation into the DNA of proliferating cells, followed by irreversible inhibition of DNMT1 enzymatic activity and the addressing of this latest to proteasomal degradation (Ghoshal et al., 2005; Juttermann et al., 2006). Accordingly, two main molecular effects have been described for DNMTi inhibitors: (1) a global demethylation of gene promoters (mainly tumor suppressor genes) and (2) the activation of immune system and the triggering of an anti-tumor immune response (Groudine et al., 1981; Almstedt et al., 2010; Goodyear et al., 2010; Chiappinelli et al., 2015;). As an illustration, in DLBCL it has been described that decitabine can reverse DNA methylation and restore expression of important cancer-related pathways in vitro and in vivo (Li et al., 2002; Clozel et al., 2013), although in other studies a less drastic and transient effect was observed (Karpf, 2004; McGarvey et al., 2006; Egger et al., 2007). Furthermore, DNMT inhibition is also linked to the demethylation of gene bodies, leading to oncogene downregulation (Wong et al., 2013; Yang et al., 2014).
Several new DNMTis have been developed in the last decade with potential activity in hematological malignancies. Among them, thioguanine (2-amino-1,7-dihydro-6H-purine-6-thione (6-tG)) has been approved by FDA to treat AML patients (Munshi et al., 2014). Its mechanism of action involves its incorporation into DNA, decrease in DNMT activity and DNA methylation, blockade of DNA and RNA synthesis, and ultimately cell death (Hogarth et al., 2008; Yuan et al., 2011; Flesner et al., 2014). Recently described as an experimental DNMTi, 5-fluoro-2′-deoxycytidine (FdCyd) is currently undergoing a phase I/II clinical trial in combination with other drugs (Kinders et al., 2011; Newman et al., 2015). Its mechanism of action involves the ability to block DNMT-dependent DNA methylation (Jones and Taylor, 1980; Beumer et al., 2008). 5,6-Dihydro-5-azacytidine is a reduced, hydrolytically stable form of 5-azacytidine nucleoside (Beisler et al., 1979). The mechanism of action is very similar to that described for azacytidine, with the advantage of a lower toxicity (Avramis et al., 1989). However, its evaluation in clinical settings revealed a reduced response rate and the rise of significant adverse effects (Samuels et al., 1998). Zebularine is another DNMTi, which has been previously described as tumor-selective inhibitor of DNMTs (Cheng et al., 2004). Although there are a lot of evidences, both in vitro and in vivo, indicating the potential of zebularine as a demethylating agent in a wide range of tumors (Agrawal et al., 2018), its poor bioavailability has prevented its introduction into clinical trials (Ben-Kasus et al., 2005). More recently, 4′-thio-2′-deoxycytidine (TdCyd) and its 5-aza analog, 5-aza-TdCyd, have been reported to downregulate DNMT1 and to exhibit anti-tumor activity in vitro and in human leukemia and lung cancer xenografts (Thottassery et al., 2014). Among these last molecules, TdCyd has already entered into phase I clinical evaluation (NCT02423057 and NCT03366116). Further molecules were developed with superior anti-tumoral efficacy and included guadecitabine (SGI-110), a second-generation DNMTi that harbors an improved DNA methylation inhibition in solid tumors both in vitro and in vivo (Chuang et al., 2010; Srivastava et al., 2015). A phase I clinical trial has provided promising results in patients with MDS and AML (Issa et al., 2015). Fluorocyclopentenylcytosine (RX-3117) is a cytidine analog that presents an anti-tumor activity in a large set of tumor cells and in vivo. Its mechanism of action is associated with an inhibition of DNMT1 (Choi et al., 2012). This agent is being evaluated in a phase II study with R/R pancreatic or advanced bladder cancer (NCT02030067).
EZH2 constitutes the catalytic subunit of the polycomb repressive complex 2 (PRC2). Its structure is composed by a SET domain, typical in chromatin-associated regulators of gene expression (Xiao et al., 2003). It catalyzes histone H3 lysine 27 tri-methylation (H3K27me3) and the subsequent formation of heterochromatic regions and downregulation of the nearby genes (Bracken and Helin, 2009; Ferrari et al., 2014) (Figure 2). In B lymphocytes, EZH2 becomes expressed and inhibited in a cyclic manner. First, in pre-B lymphocytes, induction of EZH2 expression is required for an optimal V(D)J recombination. Later on, during the migration to lymphoid tissues, it is downregulated until the GC reaction occurs, after which it becomes re-expressed to allow the silencing of the anti-proliferative genes cyclin-dependent kinase inhibitor 2A (CDKN2A) and cyclin-dependent kinase inhibitor 1A (CDKN1A1) and the pro-differentiation genes interferon regulatory factor 4 (IRF4) and PR domain zinc finger protein 1 (PRDM1/BLIMP1) during the somatic hypermutation and isotype switch processes. Finally, EZH2 becomes repressed when mature B cells leave the GC (Velichutina et al., 2010; Béguelin et al., 2013). Gain-of-function mutations in EZH2 have been reported in several solid tumors and hematological cancers. The consequence of those mutations in GC lymphocytes is the irreversible silencing of certain cell cycle checkpoint and plasma cell differentiation genes (Béguelin et al., 2013). The main gain-of-function mutation identified in DLBCL and FL patients includes a tyrosine deletion (Y641) at the EZH2 SET domain that increases the levels of H3K27me3, promoting a repressed state of cell differentiation and the repression of tumor suppressor genes (Morin et al., 2010; McCabe et al., 2012a). Similar effects have been described as a consequence of the A677G mutation in EZH2, which has been characterized in multiple human lymphoma cell lines. A change in the substrate preferences accounts for the aberrant H3K27me3 levels observed in cells bearing EZH2 mutant forms. Indeed, wt EZH2 displays preference for less methylated substrates whereas Y641 and A667G mutants prefer either substrates with higher methylation levels or show equal affinity for all three substrates (H3K73me0, me1, and me2) (McCabe et al., 2012a). Interestingly, these gain-of-function EZH2 variants expressed in GC B-cell lymphomas seem to synergize with BCL2 deregulation, favoring the progression of these malignancies (Béguelin et al., 2013). On the other hand, overexpression of wt EZH2 has been also reported in B-NHL (Van Kemenade et al., 2001; Visser and Gunster, 2001), with a positive correlation being observed between EZH2 transcript levels, tumor aggressiveness, and disease prognosis (Abd Al Kader et al., 2013). Taking into account these considerations, it looks reasonable that inhibiting EZH2 activity could result in a potential therapeutic strategy to treat B-NHL.
In this context, many efforts directed to develop highly selective EZH2 inhibitors have been made in the last decade. EZH2 activity was initially targeted by means of the carbocyclic adenosine analog 3-deazaneplanocin A (DZNep), an inhibitor of S-adenosylhomocysteine hydrolase. DZNep promotes a global increase in the levels of 5-adensylhomocystein and a further inhibition in the activity of many methyltransferases, including EZH2. Nevertheless, due to its mechanism of action, it resulted to be too unspecific as many other methyltransferases were similarly affected. In 2012, a small chemical compound named El1 with a good capacity to inhibit the Y641 mutant and wt EZH2 form was evaluated for the treatment of DLBCL. This compound was designed as a competitive inhibitor of the EZH2 methyl group donor S-adenosyl--methionine (SAM). Unlike DZNep, El1 showed a 10,000-fold selectivity for EZH2 over other HMTs and a 90-fold selectivity over EZH1 methyltransferase. This compound promoted a global decrease in methyl donor availability, leading to a lower global levels of H3K27me3 (Qi et al., 2012). Other subsequent compounds directed specifically against EZH2 are the dual EZH2/1 inhibitors UNC1999, with a potent capacity to suppress H3K27me3 and H3K27me2 levels and to inhibit proliferation of mixed lineage leukemia (MLL)-rearranged cells, and the OR-S1 and OR-S2 inhibitors, which were assessed for the treatment of DLBCL, AML, and MM (Konze et al., 2013; Honma et al., 2017). Later on, EPZ0005687 and GSK126, two selective and SAM-competitive EZH2 inhibitors with a higher inhibitory capacity for the mutant EZH2 form, were developed and tested in DLBCL and FL (McCabe et al., 2012b; Knutson et al., 2014). In 2014, GSK126 entered into phase I clinical trials with B-NHL and MM patients (NCT02082977) (Zeng et al., 2016; Yap et al., 2018), but unfortunately that study had to be discontinued as a consequence of insufficient therapeutic activity, evidencing the need to keep working in the improvement of those inhibitors. Also in 2014, CPI-360 and its more potent and stable analog, CPI-169, were reported to be effective EZH2 inhibitors for the treatment of several B-NHL subtypes (Vaswani et al., 2016). An improved version of these latest, CPI-1205, showed a higher oral bioavailability and was first tested in preclinical studies with xenograft mouse models generated from human B-NHL cell lines and further challenged in phase I trials for the treatment of DLBCL (NCT02395601).
Valemetostat (DS-3201) is another potent wild-type (wt) and mutant EZH1/2 inhibitor that demonstrated a strong anti-proliferative effect against NHL, DLBCL, and T-cell lymphoma (Maruyama et al., 2017). Currently, tazemetostat (EPZ‐6438), another SAM competitive inhibitor with a high affinity for the wt and the mutant EZH2 forms, is being evaluated in clinical studies to treat R/R B-NHL and MM patients (NCT03456726) (Knutson et al., 2014; Gulati et al., 2018), reaching an overall response rate of 38% in a phase I clinical trial (Italiano et al., 2018).
Despite first promising results, single-agent treatment with EZH2 inhibitors is in general slightly effective in aggressive lymphomas. Among the possible mechanism(s) of resistance, overactivation of the phosphatidylinositol 3-kinase (PI3K) and mitogen-activated protein kinase (MAPK) pathways has been identified in GSK126-resistant DLBCL cells (Bisserier and Wajapeyee, 2018). Thus, it looks reasonable to prioritize the discovery of new drug combination associating EZH2 inhibitors with other compounds targeting key signaling pathways in order to prevent and/or overcome the occurrence of EZH2i resistance in lymphoid neoplasm with mutated EZH2.
A conserved biological mechanism within all eukaryotic organisms, from yeast to higher mammals, is arginine methylation (Migliori et al., 2010). This post-translational modification is mediated by N-arginine methyltransferases (PRMTs), which catalyze the transfer of a methyl group, from SAM to the omega nitrogens found in terminus guanidine group of an arginine residue of the side chain. This transfer may occur in one or both nitrogens (Bedford and Clarke, 2009). Among the nine different members of the PRMT family (Schubert et al., 2003), PRMT1 is the major enzyme responsible for arginine methylation followed by PRMT5, according to the observation that PRMT1 and PRMT5 knockout mice die at an early stage during development whereas mice lacking any of the other seven PRMTs are fully viable (Hadjikyriacou et al., 2015). Protein modifications performed by PRMTs are traditionally related to important genetic processes such as DNA repair and gene transcription, among others. More recently, PRMT functions have been linked to carcinogenesis and metastasis, giving to these enzymes the status of potent therapeutic targets in a variety of cancers where they are overexpressed, including colon, breast, prostate, and lung cancers, neuroblastomas, leukemias, and B-cell lymphoma (Yoshimatsu et al., 2011).
Within this family, upregulation of PMRT1 and PRMT5 has been widely associated with hematological malignancies (Greenblatt et al., 2016; Smith et al., 2018). In particular, the expression and function of PMRT5 have been extensively examined during lymphomagenesis, as this enzyme is highly expressed in primary samples and cell lines from different leukemia and lymphoma subtypes, where it promotes the repression of tumor suppressors such as the retinoblastoma proteins. In these models, experimental studies have suggested that PRMT5 upregulation may be caused by overexpression of MYC and NOTCH oncogenes (Wang et al., 2008). In transformed DLBCL, the S-methyl-5’-thioadenosine phosphorylase (MTAP) gene encoding for a critical methionine metabolism enzyme is deleted due to its proximity to the tumor suppressor gene CDKN2A (Dreyling et al., 1998), and this phenomenon sensitizes cancer cells to PRMT5 inactivation (Marjon et al., 2016). A remarkable interplay has also been described between PRMT5 and the B cell lymphoma 6 (BCL6) oncogene during the lymphomagenesis in the GC (Lu et al., 2018), suggesting that pharmacological inhibition of arginine methylation could be of special interest in BCL6-driven lymphoma. Regarding PRMT1, an interesting interaction exists between this enzyme and EZH2 in DLBCL-GCB tumors. Indeed, recent works have reported an increase in PRMT1-related histone arginine methylation in DLBCL-GCB cells resistant to EZH2 inhibition, in association with BCL-2 overexpression and modulation of the B-cell receptor (BCR) downstream signaling, supporting the rational association of EZH2 and PRMT1 inhibitors in DLBCL patient samples (Goverdhan et al., 2017).
Among the multiple functional inhibitors that have been developed to target the different members of the family, PRMT1 and PRMT5 small molecule inhibitors have already shown great potential against B-NHL, either alone or upon their combination with other agents. As an illustration, promising results have been obtained with the specific PRMT5 inhibitor EPZ015666 (GSK3235025) when used as single agent in in vitro and in vivo models of MCL (Chan-Penebre et al., 2015).
By favoring an open chromatin state, histone acetylation allows numerous transcription factors to bind DNA and to activate gene expression. At the same time, acetylated histones increase DNA accessibility to transcriptional activators and counteract the function of transcriptional repressors (McClure et al., 2018). Acetylation of histones and non-histone proteins is regulated through a correct balance between HAT and HDAC activities. Among these enzymes, the most advanced subfamily is human HDACs, which have been classified into four classes according to their sequence homology, activity, and subcellular localization. HDACs 1, 2, 3, and 8 constitute class I. HDAC 4, 5, 6, 7, 9, and 10 belong to class II. Class III includes sirtuin 1 (SIRT1) and sirtuin 7 (SIRT7), two NAD-dependent structurally unrelated protein deacetylases (Minucci and Pelicci, 2006). Finally, class IV is represented by HDAC11. In contrast to class II HDACs which show a heterogeneous expression pattern, class I HDACs are found at particularly high levels in lymphoid cell lines and primary tumors, suggesting a predominant role of these latest in lymphomagenesis. Accordingly, the design of HDAC inhibitors (HDACis) in lymphoid malignancies has been mainly centered on this latest group of enzymes (Gloghini et al., 2009).
Several structurally distinct classes of HDACis have been developed. These molecules can be divided into five chemical groups: hydroxamic acids, cyclic peptides, electrophilic ketones, short-chain fatty acids, and benzamides. Pan-HDACis have the capacity to inhibit almost all HDACs with the exception of class III HDACs and include the hydroxamic acid derivatives vorinostat, givinostat (ITF2357), abexinostat, panobinostat, belinostat, and trichostatin A, the carboxylate sodium butyrate, and the cyclic peptide trapoxin (Bradner et al., 2010; Di Costanzo et al., 2014). Taking into account that HDACs can also modulate the function of several non-histone proteins regulating a number of physiological processes (Lane and Chabner, 2009), and that HDACs can simultaneously exert pro- and anti-leukemic activities (Heideman et al., 2013; Santoro et al., 2013), blocking individual HDACs with isotype-selective inhibitors specific for one or two classes of HDACs might represent a strategy of choice for the treatment of lymphoid tumors. In line with this, the isotype-selective HDACis include the benzamides entinostat (MS-275, SNDX-275) and mocetinostat (MGCD0103) (Fournel et al., 2008; Vannini et al., 2004), the hydroxamic acid derivative rocilinostat (ACY-1215) (Santo et al., 2012), and the cyclic peptide romidepsin, which show preference for HDAC1-6-8, HDAC6, and HDAC1-2, respectively (Lemoine and Younes, 2010). Several HDACis like vorinostat, mocetinostat, and entinostat can be administered orally; conversely, other agents like romidepsin are given intravenously (Batlevi et al., 2016; Mann et al., 2007; Younes et al., 2011; Holkova et al., 2017). By inhibiting the catalytic activity of their target HDAC(s), these compounds impair the formation of HDAC–substrate complexes, thus altering the transcriptomic pattern of the malignant cells as well as the activity of non‐histone proteins, ultimately leading to growth arrest, differentiation, and induction of apoptosis (Qiu et al., 2000). Of importance, when compared to their malignant counterparts, healthy tissues are generally unaffected by HDACis (Mai et al., 2005).
A number of preclinical studies have highlighted a role for HDACi therapy in a range of B-cell lymphoma, including DLBCL, HL, and BL, either alone or in combination with other epidrugs such as HMAs, with small molecule agents or with standard chemotherapeutics (Buglio et al., 2008; Kretzner et al., 2011; Kewitz et al., 2012; Ageberg et al., 2013; Klein et al., 2013; Rozati et al., 2016; Garrido Castro et al., 2018). Among these studies, the weak HDACi valproic acid was shown to overcome DLBCL cell resistance to the standard R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapeutic regimen (Ageberg et al., 2013). In preclinical models of DLBCL and MCL, panobinostat, belinostat, depsipeptide, and vorinostat were shown to evoke tumor growth arrest, differentiation, and/or apoptosis in vitro and/or in vivo, mediated by the accumulation of DNA damage upon PARP trapping (Valdez et al., 2018), G1 cell cycle arrest consequent to an increase in the expression of the cyclin-dependent kinase inhibitor p21, acetylation of histone H3 (Xue et al., 2016), or transcriptional activation of the BCL-2 family proapoptotic members BIM, BMF, and NOXA (Kalac et al., 2011; Xargay-Torrent et al., 2011).
Based on these preclinical studies, several HDACis have entered clinical trials under different modalities (monotherapies or in combination). Many of these trials have been conducted in DLBCLs, FLs, and HLs using HDACis, either alone or in combinatorial therapies (Watanabe et al., 2010; Stathis et al., 2011; Younes et al., 2012; Oki et al., 2013; Ogura et al., 2014; Chen et al., 2015; Morschhauser et al., 2015) (Table 2 and Figure 3). As monotherapy, HDACis have shown a wide range of response in lymphoma patients, varying from complete remissions (CRs) to no response. In the absence of biomarkers for prediction of clinical outcome, the molecular mechanisms of resistance are poorly understood. Vorinostat was the first proved in relapsed B-NHL patients, including FL, MZL, and MCL. In a phase II study including relapsed FL, non-FL indolent NHL and MCL patients, oral vorinostat showed low levels as a single agent, with the exception of FL, in which an overall response rate (ORR) of 47–49% (referring to the proportion of patients with tumor size reduction of a predefined amount and for a minimum time period) and a CR rate of 23% was observed (Kirschbaum et al., 2011; Ogura et al., 2014). This agent was also tolerated, but displayed limited activity in another phase II trial against R/R DLBCL, with only 1/18 patients presenting complete response (Crump et al., 2008).
With the pan-HDACis abexinostat and quisinostat, or the class-specific mocetinostat and entinostat, the response rates were quite variable (from 12% to 56%), and mostly dependent on the drug and on the lymphoma subtype. The most robust responses were obtained with abexinostat in FL patients (56% ORR). This latest drug showed a unique pharmacokinetic profile and an optimized oral dosing schedule that allowed for a superior anti-tumoral activity. In a recent phase II study with patients with R/R B-NHL or CLL, among the evaluable patients the ORR was 28%, with highest responses observed in FL patients (ORR 56%) and DLBCL (ORR 31%) (Ribrag et al., 2017). A phase II clinical trial with mocetinostat in patients with R/R DLBCL and FL showed promising results (Batlevi et al., 2017), whereas for entinostat only one B-NHL patient has been included in phase II trial; therefore, no conclusion can be made on its efficacy in this subgroup of patients (Kummar et al., 2007).
Similar to DNMTis, the effectiveness of the first-generation HDACis carries significant toxicity and is limited to hematopoietic malignancies, which makes them challenging to combine (Suraweera et al., 2018). It is believed that part of this toxicity may be related to the capacity of HDACis to alter directly the function of many non-histone proteins. Toxicity may also be due to widespread activity across HDAC isoforms; therefore, the focus of second-generation HDACi discovery was to enhance the discrimination over HDAC family members (Galli et al., 2010; Knipstein and Gore, 2011; Younes et al., 2011; Santo et al., 2012; Evens et al., 2016). In this context, targeting HDAC6 was associated to the upregulation of CD20 and consequent enhanced efficacy of anti-CD20 monoclonal antibody therapy (Bobrowicz et al., 2017). Also, tucidinostat (CS055/chidamide), the first oral subtype-selective HDACi, was approved for the treatment of refractory/relapsed PTCL by the China Food and Drug Administration. This compound inhibits HDAC1, HDAC2, HDAC3, and HDAC10, and has entered a phase II clinical trial as single-agent treatment for patients with R/R B-NHL (NCT03245905) based on preliminary evidences of clinical activity in DLBCL (Yang et al., 2018).
Another approach to maximize efficacy with manageable toxicity consists in developing dual inhibitors. In this field, CUDC-907, a novel first-in-class oral small molecule inhibitor of both HDAC (class I and II) and PI3K (class Iα, β, and δ), has demonstrated excellent levels of activity (55% ORR) and tolerability in DLBCL patients in a phase IA clinical trial (Younes et al., 2016). In a second phase IB trial, the drug has been tested in patients with R/R DLBCL and showed a response rate of 37%, with a higher effect in MYC-altered versus MYC-unaltered patients (Oki et al., 2017). As a result of these encouraging initial data, this agent is currently being evaluated in a phase II study including DLBCL patients, and also in a phase I trial involving pediatric patients with lymphomas (NCT02674750 and NCT02909777).
Among the post-translational modifiers with ability to orchestrate chromatin organization, bromodomain (BD)-containing proteins are readers of Ac-K residues at the N-terminal histone tails. They act as scaffolds that enable histone attachment to the chromatin and form active multi-protein transcription complexes, thereby modulating chromatin dynamics and ultimately diversifying gene expression (Filippakopoulos et al., 2012; Chaidos et al., 2015; Smith and Zhou, 2016). This family of proteins contains 46 members, comprising nuclear proteins with HAT or HMT activity, chromatin remodelers, helicases, transcription co-activators, and mediators or scaffold proteins. They are subdivided into eight subfamilies (I to VIII), based on their structure and sequence similarities. Subfamily II is the most studied one and includes the bromodomain-containing proteins mBRDT, BRD2, BRD3, and BRD4 (Padmanabhan et al., 2016). Besides the presence of two bromodomains (BD1 and BD2) that allow acetylated chromatin recognition, these proteins harbor an extra-terminal domain, which is responsible for protein–protein interactions. This bromodomain and extra-terminal (BET) subfamily has thus the capacity to act as protein adaptors facilitating the recruitment of chromatin remodelers and transcription factors for further initiation and elongation of transcription (Delmore et al., 2011; Chaidos et al., 2015; Padmanabhan et al., 2016). Several reports have highlighted the importance of the BET proteins action over DNA enhancers for the regulation of certain oncogenes expression (Lovén et al., 2013). Altogether, these studies make BET proteins attractive therapeutic targets in cancer.
As interfering with this family of proteins may serve as a strategy to address transcription irrespective of the presence of epigenetic mutations, BET proteins inhibitors have been a significant area of focus in the last decade, in cancer but also in inflammation, fibrosis, and heart diseases (Vakoc, 2015). Drug developmental studies have paid special attention to the Ac-K binding sites in the bromodomains, as these deep hydrophobic pockets with conserved asparagine and/or aspartate residues make BET proteins highly druggable (Cox et al., 2016). Indeed, the most common drug targeting approach in this family has been the development of small molecules that could block the lysine-binding pocket and disrupt the interactions between BDs and the Ac-K on chromatins (Smith and Zhou, 2016).
In 2005, a first bromodomain inhibitor developed by the Zhou laboratory, namely NP1, has the ability to target the BD of the P300/CBP-associated factor transcriptional coactivator (Zeng et al., 2005). This step was followed by the discovery in 2006 of MS7972, a weakly binding fragment specific for CREBBP-BD, hindering its binding to acetylated p53 (Sachchidanand et al., 2006). Among BET proteins, the first target considered to be druggable was BRD4, as a pioneering RNAi base unveiled its critical role in the maintenance of AML. In this study, authors found that BRD4-dependent transcriptional activity could be efficiently targeted by the pan-BET thieno-triazolo-1,4-diazepine (+)-JQ1 (Filippakopoulos et al., 2010; Zuber et al., 2011). This class of diazepine-based small molecule inhibitors, which also includes the benzodiazepine I-BET151 (GSK1210151A) (Dawson et al., 2011) and I-BET762 (GSK525762) (Mirguet et al., 2013) (NCT01943851), utilizes the methyltriazolo-diazepine ring system as the acetyl-mimetic. Further studies demonstrated that inhibition of BRD4 by (+)-JQ1 unveiled the MYC downregulation and, consequently, a genome-wide inhibition of its target genes (Filippakopoulos et al., 2010; Delmore et al., 2011). These results underlined significant preclinical activity of this inhibitor in MYC-driven B-NHL, including the aggressive, so-called “double hit” lymphoma (DHL), characterized by simultaneous oncogenic activation of MYC and/or BCL2/BCL6 (Johnson-Farley et al., 2015). Accordingly, (+)-JQ1 could increase survival of mice xenografted with MYC-driven lymphoma, including those ones bearing either TP53 deletions or intrinsic resistant to the topoisomerase II inhibitor etoposide (Hogg et al., 2016).
These promising results from (+)-JQ1 encouraged the development of BET inhibitors with similar chemical structure, including the BRD4 inhibitor CPI203 characterized by a higher bioavailability profile in mice (Normant et al., 2012; King et al., 2013). This agent displayed remarkable efficacy in different preclinical models of B-NHL, either as single agent or in combination with the BCL-2 antagonist venetoclax in DHLs (Esteve-Arenys et al., 2018), in DLBCL-ABC (Ceribelli et al., 2014) and in both ABC and GCB subtypes of DLBCL in combination with blockade of the CXCR4 chemokine receptor (Recasens-zorzo et al., 2018). In these studies, BRD4i activity was mainly related to the blockade of MYC transcriptional program. This is of special interest, as despite its central role in multiple hematological malignancies, including various subtypes of B-NHL, direct targeting of MYC was considered impossible until the demonstration that BET inhibition could regulate MYC activity in varied contexts, thanks to alleviation of BRD4 occupancy on MYC super-enhancers. Importantly, beside MYC, different anti-apoptotic proteins like BCL-2 and MCL-1 are also downregulated, either by direct transcription repression or as a downstream consequence of BRD4 antagonism (Vakoc, 2015). Unlike the expected general effects of BET inhibition in the elongation of transcription of several genes, changes in the expression of only a small subset of genes was observed in cultures and/or animals receiving this therapy, suggesting that bromodomain inhibitors might be suitable modulators of certain disease-associated genes. As an illustration, high levels of BRD4 co-localize in CLL cells with super-enhancer sites of genes and microRNAs belonging to the BCR-mediated signaling pathway with possible tumor-initiating activity, including miR-21, miR-15, TCL1, IL21R, and IL4R. Accordingly, in a mouse model of CLL, exposure to the BET inhibitor PLX51107 promoted an expression downmodulation of several tumor-associated genes, followed by consistent reduction in tumor burden (Ozer et al., 2018).
According to these promising results, in the last years a number of clinical leads have entered into trials for the treatment of hematological patients. Nevertheless, several side effects have been reported including some bone marrow and gastrointestinal toxicity that has forced to dose discontinuation or reduction. Nowadays, 18 BET inhibitors are being assessed in clinical trials either as single agents or in combination with other compounds (Table 4). While the data from various solid tumor trials look mitigated, several BETis including birabresib (OTX015, MK-8628), molibresib (GSK525762), RO6870810/TEN-010, and mivebresib (ABBV-075) have demonstrated remarkable clinical efficacy in myeloproliferative disorders, while other small molecule inhibitors such as PFI-1, BI-894999, FT-1101, INCB-54329, and CPI0610, a pharmacological derivative of CPI203, are currently undergoing human clinical trials in these patients (Table 3). Among these different molecules, molibresib has demonstrated an 18.5% ORR in various subtypes of NHLs including a CR in a DLBCL case (Dickinson et al., 2018). CPI0610 has also been evaluated in a phase I clinical trial (NCT01949883) in 64 R/R FL, DLBCL, or HL patients, showing leading to a complete remission in one FL case and in four DLBCL patients (Blum et al., 2018). In addition, the compound INCB057643 is currently being tested in a third phase I trial involving lymphoma patients, including some FL and DLBCL cases. In this evaluation trial, a CR has been achieved in one FL case whereas in two other patients, the disease has been stabilized (Forero-Torres et al., 2017). In the dose-escalation, open-label, phase I study with OTX015, a 47% complete remission was reported in 17 DLBCL cases; however, objective responses were seen in only three DLBCL patients and clinical activity in other six B-NHL patients (NCT01713582) (Amorim et al., 2016). More recently, the BETis molibresib, CC-90010, and INCB054329 are being challenged in clinical trials including various hematological malignancies (NCT02431260, NCT01943851, and NCT03220347), but no data have been released so far.
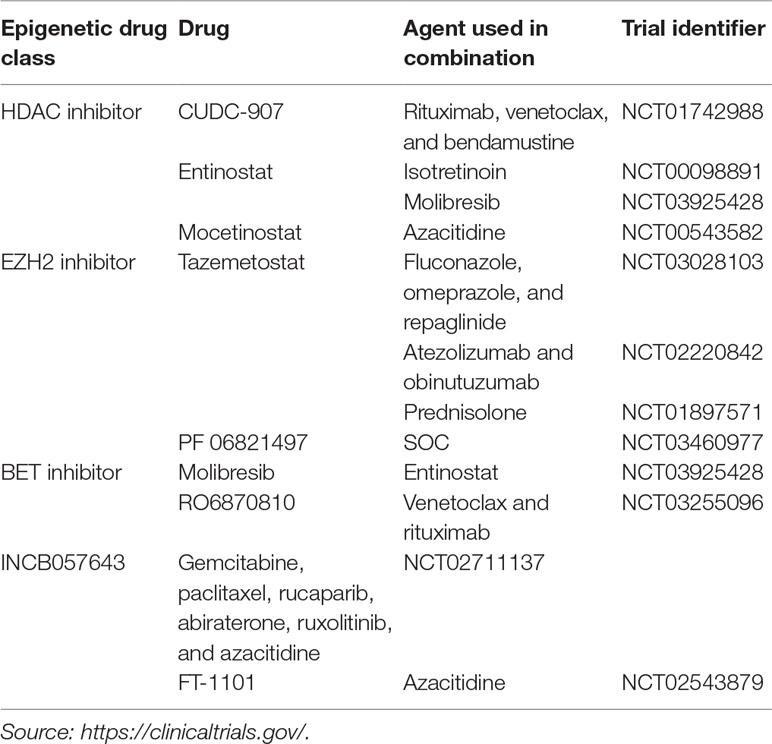
Table 4 Drug combinations with non-approved epigenetic agents in B-NHL.
Although at the moment most of the tested compounds aimed at inhibiting BET bromodomains are pan-BET inhibitors, many efforts are being focused in targeting BET proteins in a more specific and novel way. These new approaches include ABBV-744 (which targets bromodomain-containing protein II) (Sheppard et al., 2018), the bivalent BET inhibitors AZD5153 and MT1 (a JQ1-derived BETi) (Rhyasen et al., 2016; Tanaka et al., 2016), and the so-called BET-PROTACs (QCA570, dBET6, BETd-260, and ARV-771) that drive BET proteins to their degradation by proteolysis-targeted chimera (Raina et al., 2016; Winter et al., 2017; Qin et al., 2018 ). These molecules have shown both to promote apoptosis in MCL-derived cells resistant to the first-in-class Bruton’s kinase (BTK) inhibitor ibrutinib as well as to increase survival compared to OTX015-treated MCL xenografts (Sun et al., 2018). Although promising results have been reported for this new generation of BET-targeting agents in preclinical studies, their therapeutic window when moving to clinical trials has still to be evaluated.
As previously mentioned, chromatin modifications can regulate several important features of cell function. Among these modifications, histone lysine acetylation is generally associated with activation of gene expression (Shahbazian and Grunstein, 2007). HAT enzymes can deposit acetyl marks on histones and modify chromatin structure. Such marks are also recognized by bromodomains, thus adding a second level of regulation of the transcription process (Kouzarides, 2007). The transcriptional co-activators CBP/p300 are highly homologous, multifunctional proteins that encode a single bromodomain each and possess HAT activity (Chen and Li, 2011; Delvecchio et al., 2013). CBP/p300 act as transcriptional co-factors, involved in the regulation of several biological processes (Dancy and Cole, 2015). Animal studies have shown that CBP and p300 are required for the generation and activity of normal hematopoietic stem cells as well as for adult hematopoietic stem cell maintenance and function (Chan et al., 2011; Rebel et al., 2002). Consequently, CBP ablation has a direct impact on the quiescence, apoptosis, and self-renewal of adult hematopoietic stem cells (Chan et al., 2011) and CBP/p300 have a tumor suppressor role in mice models (Kung et al., 2000; Kang-Decker et al., 2004; Chan et al., 2011). This role of CBP and p300 as tumor suppressors has been also observed in B-NHL, where its inactivating mutation is a common event in FL and DLBCL, providing a rationale for employing drugs with the capacity to modulate acetylation and deacetylation processes in these tumors (Cerchietti et al., 2010; Mullighan et al., 2011; Pasqualucci et al., 2011a).
The SWItch/Sucrose Non-Fermentable (SWI/SNF) complex was initially discovered in yeast. It is composed by polypeptides associated with a subset of proteins codified by the SWI1, SWI2, SNF2, SWI3, SWI5, and SWI6 genes (Pazin and Kadonaga, 1997). This complex regulates gene transcription by altering DNA–nucleosome interactions at expenses of ATP consumption, thus facilitating or impeding the accession of the transcription machinery at concrete genomic regions (Workman and Kingston, 2002). Several studies have reported its capacity to repair nucleotide excisions and DNA double-strand breaks by homologous recombination (Chai et al., 2005). The mammalian analog of the SWI/SNF complex (mSWI/SNF) is the BRG1-Associated Factors (BAF) complex. It comprised approximately 11 subunits encoded by 19 distinct genes assembled in different combinations according to its specific molecular mechanism of action, and in a concrete genomic region. Two of the BAF components are the human Brahma (hBRM, also SMARCA2) and the Brahma-related gene 1 (BRG1, also SMARCA4). These proteins are ATPase subunits (Khavari et al., 1993) and either one or the other constitute the core component of the BAF complex. They contain BDs within their structure that recognize and contact acetyl groups present in histone proteins (Wang et al., 1996). Although they share similarities in their domain composition, they interact with different families of transcription factors what confers to them specific functions in the BAF complex (Kadam and Emerson, 2003).
BRG1 has been reported to be the most frequently mutated protein of the BAF complex in cancer. Classically, it has been described as a tumor suppressor gene as inactivating mutations of its protein have been found in numerous solid tumors like breast, lung, gastric, bladder, colon, ovarian cancers, and melanomas (Atlas et al., 2012; Hodis et al., 2012; Jelinic et al., 2014), but also in determined B-NHL subtype like DLBCL and MCL (Cuadros et al., 2017). Concretely, these loss-of-function mutations lead to the upregulation of the pro-survival gene BCL2L1 in MCL, conferring to this malignancy primary resistance to treatment or eventually relapse after dual exposure to ibutrinib and venetoclax (Agarwal et al., 2019). Other studies described BRG1 as a potent oncogene, since its function was required for AML progression in mice, through its binding to MYC enhancer region and consequent aberrant expression of this second oncogene (Shi et al., 2013; Buscarlet et al., 2014).
Beside BRG1, several BRG-/BRM-associated factors (BAF subunits) participate in tumoral progression. Two of these subunits, namely the AT-Rich Interaction Domain 1A (ARID1A/BAF250A) and its homologous ARID1B/BAF250B, contain domains capable of recognizing and binding to AT-enriched genomic regions and C terminus region, stimulating the activation of transcription in a glucocorticoid receptor-dependent manner. The presence of each of them in the complex is mutually exclusive, suggesting specific roles at concrete genomic regions (Wang et al., 2004).
Mutations that truncate the ARID1A sequence and promote its degradation have been widely characterized in endometrial carcinoma (Kandoth et al., 2013), colon cancer (Atlas et al., 2012), stomach cancer (Wang et al., 2011), bladder cancer (Gui et al., 2011), neuroblastoma (Sausen et al., 2013), and pancreatic or hepatocellular carcinoma (Biankin et al., 2012; Fujimoto et al., 2012), evidencing the role of this protein in preventing tumoral progression. Similar to the mutations reported for ARID1A, truncating mutations have also been identified for ARID1B although in a lesser frequency and most of them associated with neurodevelopmental disorders (Santen et al., 2012) or neuroblastomas (Lee et al., 2017). ARID1B knockdown has been reported to destabilize the SWI/SNF complex and inhibit cell proliferation in both ARID1A-mutant cancer cell lines and primary tumor cells, suggesting that this protein could constitute an interesting therapeutic target for the treatment of ARID1A-mutant tumors (Helming et al., 2014).
The enzyme isocitrate dehydrogenase (IDH) catalyzes the conversion of isocitrate into α-ketoglutarate (α-KG) by oxidative decarboxylation using NADP+ as a cofactor. The IDH1 isomer is located in the cytosol and the peroxisomes, whereas IDH2 is found in the mitochondria. IDH enzymes play an important role in the tricarboxylic (TCA) or Krebs’ cycle, but are also related with other cellular functions such as the regulation of redox balance (Dang et al., 2016; Dang and Su, 2017). Mutations in IDH genes are most commonly found in the R132 codon of IDH1 and the R172 and R140 codons of IDH2, which correspond to evolutionarily conserved residues in the enzyme active site which is critical for substrate binding. Mutant forms of IDH have much lower catalytic activity and are associated with metabolic alterations. More importantly, mutant IDH enzymes gain neomorphic activity as they convert α-KG into 2-hydroxyglutarate (2-HG). Under homeostatic conditions, 2-HG is only produced by errors in catalysis and it is maintained at low levels due to the action of 2-HG-hydroxigenases (2-HGHD). Unlike in bacteria and plants, 2-HG has no known physiological function in mammals (Dang and Su, 2017). 2-HG is structurally similar to α-KG and acts as a competitive inhibitor, blocking the activity of α-KG-dependent dioxygenases. This group of enzymes includes the TET family of hydroxylases, which participate in DNA demethylation, and the JMJ domain-containing histone demethylases (Dang and Su, 2017). The consequent aberrant hypermethylation of both DNA and histones has been associated to a blockade in differentiation in hematopoietic cells (Figueroa et al., 2010; Losman et al., 2013), hepatocytes (Saha et al., 2014), and mesenchymal stem cells (Jin et al., 2015), among other cell types.
Homozygous missense mutations in both IDH1 or IDH2 have been described in several cancer types, including glioma, cholangiocarcinoma, and hematological tumors, such as AML and MDS (Dang et al., 2016). Although infrequent, mutations have also been found in lymphoid malignancies like angioimmunoblastic T-cell lymphomas (Cairns et al., 2012) and acute lymphocytic leukemia, both in pediatric (Andersson et al., 2011; Tang et al., 2012) and adult cases (Kang et al., 2009; Abbas et al., 2010; Zhang et al., 2012). Dysregulation of the IDH pathway has also been reported in CLL, as leukemic B cells from these patients show overexpression of IDH1 and lower levels of IDH2 when compared to healthy B cells (Van Damme et al., 2016).
Two IDH inhibitors have been recently approved by the FDA for the treatment of R/R AML in adults. Enasidenib (AG-221) targets IDH2 with R172S, R172K, and R140Q mutations, whereas ivosidenib (AG-120) targets IDH1 with susceptible mutations, such as R132H and R132C (Han et al., 2019). Other non-approved IDH inhibitors are currently in clinical trials involving patients with advanced hematological cancers. Among these molecules, AG-881 is a pan-inhibitor of both IDH1 and IDH2 that can penetrate the blood–brain barrier, while IDH305, FT-2102, and BAY-1436032 are IDH1-specific inhibitors (Dang et al., 2016; Montalban-Bravo and DiNardo, 2018). At the preclinical level, the pharmacological IDH2 inhibitor AGI-6780 displayed synergistic cytotoxicity in MCL and BL cell lines in combination with the proteasome inhibitor carfilzomib, mediated by the blockade of tricarboxylic acid cycle and the decrease in ATP levels, as a consequence of enhanced IDH2 enzymatic inhibition (Bergaggio et al., 2019). Thus, although activating mutations of IDH genes are rare in B-NHL, there may be some room to evaluate, alone or in combination with standard chemotherapy, some of the molecules exhibiting clinical activity in non-lymphoid patients.
In recent years, thanks to the many works directed to characterize and get a better understanding of the human epigenome, it came out that more than 50% of the human cancers account for aberrant changes in chromatin organization at certain genomic regions, as a consequence of mutations in enzymes involved in the regulation of chromatin structure (You and Jones, 2012; The Cancer Genome Atlas Research Network, 2013). Changes in the activity of these chromatin modifiers can lead not only to the initiation of a tumor formation process but also to its progression, metastasis, development of drug resistances, and further relapse and/or escape from immune surveillance (Jones et al., 2016). Therapeutic modulation of such alterations can be achieved with chemical compounds that broadly affect the structure of the DNA such as DNMTis, histone HDACis, or BETis (Figure 4). While single-agent clinical trials with these compounds have been conducted with some success in MDS or R/R AML patients receiving azacitidine (Scott, 2016; Schuh et al., 2017) or in R/R FL, MZL, and MCL patients treated with vorinostat (Kirschbaum et al., 2011; Ogura et al., 2014), the association of these agents with other compounds has also been tested. As an example, the combinatorial treatment with vorinostat and the sirtuin inhibitor niacinamide was evaluated in R/R NHL and HL cases (NCT00691210) (Amengual et al., 2013), but it achieved a modest efficiency with an ORR below 50% (Olsen et al., 2007). Other examples include the combination of panobinostat with decitabine which displayed synergistic caspase-dependent cell death in DLBCL cells (Kalac et al., 2011) or the combination of romidepsin with the antimetabolite pralatrexate for the treatment of relapsed PTCL (Amengual et al., 2018).
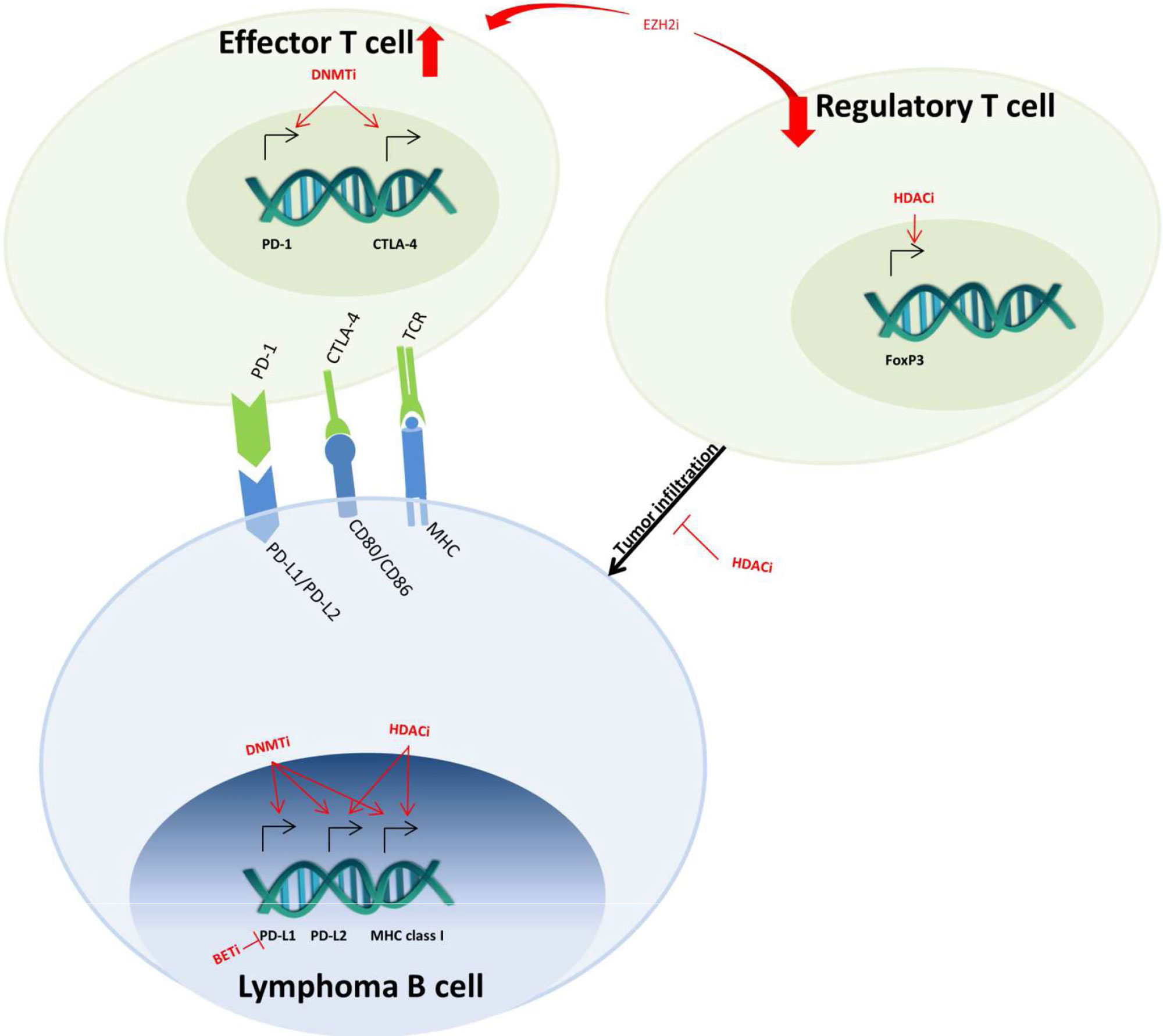
Figure 4 Epigenetic-targeted effects on immuno-oncology mechanisms. In lymphoma B cells, DNMT, HDAC, and BET inhibitors (DNMTi, HDACi, BETi) regulate the expression of MHC class I and PD-1 ligands (PD-L1 and PD-L2). In effector T cells, DNMTi also upregulates the expression of PD-1 and CTLA-4, which leads to T-cell exhaustion. The effects of HDACi on FoxP3 decrease the infiltration of regulatory T cells into the tumor. Lastly, EZH2i decreases the regulatory and increases effector T-cell population in the microenvironment.
A different therapeutic approach consists in targeting specifically certain chromatin regulatory proteins to achieve a more restricted effect in the transcription of a concrete subset of genes. Promising examples are the inhibition of the DOT1-like (DOT1L) histone H3K79 methyltransferase with pinometostat (EPZ-5676) in adults with MLL/KMT2A-driven leukemia (NCT02141828) (Stein et al., 2018) or inhibition of histone H3K4 and K9 demethylation by the lysine-specific demethylase 1 (LSD1) inhibitor seclidemstat, currently being assessed in clinical trials to treat refractory Ewing sarcomas (NCT03600649).
Combinations with chemical compounds that broadly affect an epigenetic mark and a specific inhibitor of a chromatin-modifying enzyme, such as the EZH2 inhibitor GSK126 and romidepsin, have also been assessed in preclinical studies with DLBCL-GCB cell lines, leading to synergistic tumor growth inhibition effects in mice (Lue et al., 2019). Another example of the strategies currently evaluated in clinical studies is the concomitant treatment of drug-resistant MM with panobinostat and bortezomib (NCT01083602) (Richardson et al., 2016).
Finally, and in concordance with the concept that acquired resistance to chemotherapy is tightly linked to changes in chromatin structure, many efforts have been made in identifying combinational strategies associating different types of cytotoxic drugs to small molecule regulators of chromatin modifiers. As an example, the dinitroazetidine derivative RRx-001 administered in combination with radiation, chemotherapy, or immunotherapies promotes the generation of reactive oxygen and nitrogen species, leading to the oxidation of the cysteines present at the catalytic sites of DNMTs and HDACs. This phenomenon entrains the inhibition in DNMT and HDAC enzymatic activities, with subsequent alterations in the chromatin structure. The therapeutic benefits of this compound have been assessed in phase II clinical trials both as a radio- and chemo-sensitizer, as well as a way to prone tumor response to conventional therapies (NCT02215512, NCT02452970, NCT02096341, NCT02871843) (Oronsky et al., 2017; Zhao et al., 2017).
The use of epigenetic agents combined with other anti-tumoral drugs may represent the future of epigenetic-targeted therapies (Figure 5). The rationale of such combinations would be, on the one hand, to benefit from the transcriptional effects of targeting epigenome. Indeed, growing evidences are showing that epigenetic therapy, using DNMTi or HDACi, in combination with conventional therapy or immunotherapy, might be an up-and-coming step toward the development of new and efficient cancer treatment strategies (Brahmer et al., 2012; Sharma and Allison 2015; Topalian et al., 2015; Issa et al., 2017). Accordingly, the acquired capacity of tumors to resist chemotherapy is related with changes in the cancer cell’s epigenome, which might affect directly the cell cycle and/or some key apoptosis regulators (Fodale et al., 2011).
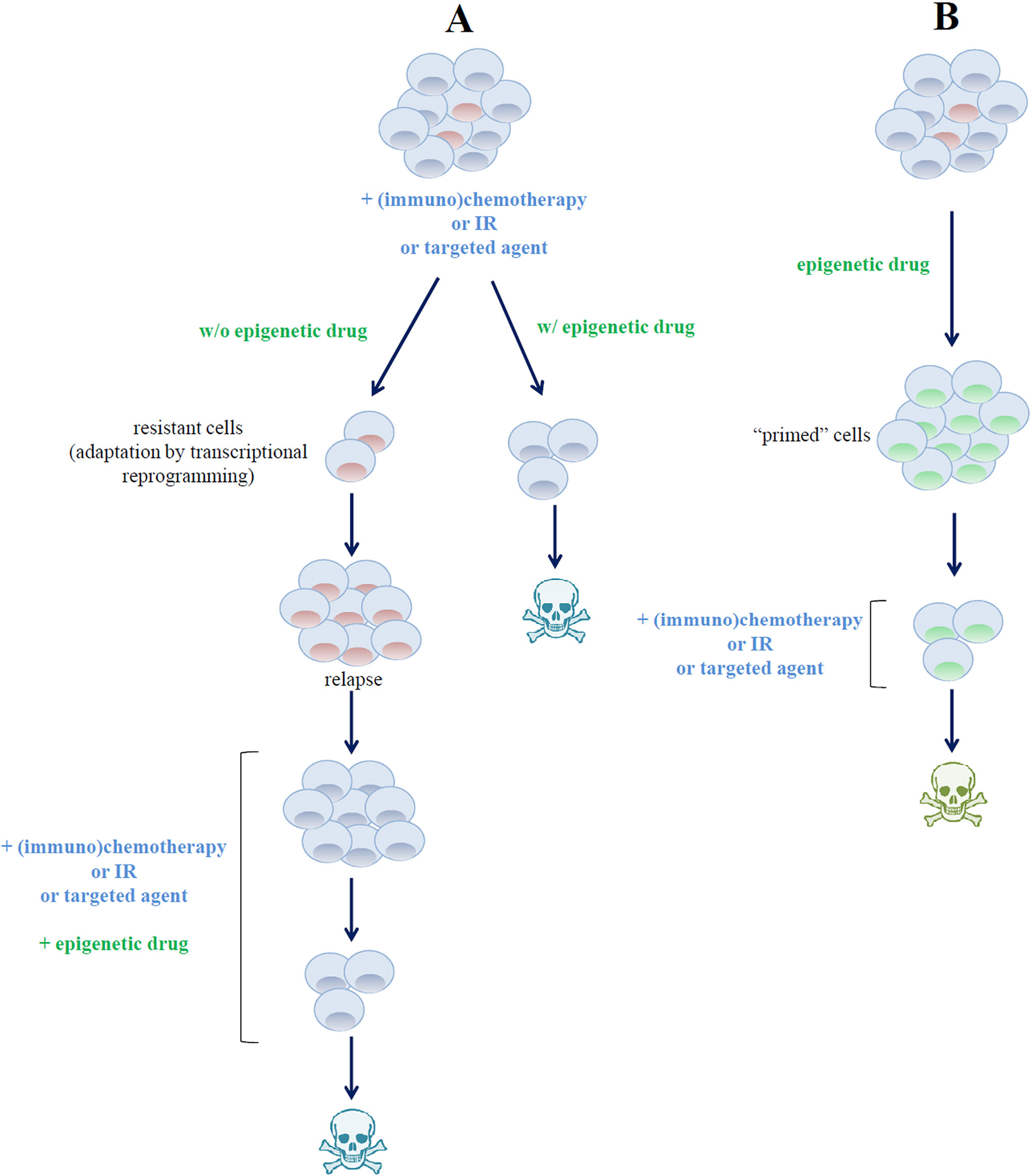
Figure 5 Different strategies for combining drugs targeting epigenetic regulators in B-cell lymphoma. (A) Epigenetic drugs can be used to overcome (left axis) or to circumvent (right axis) malignant B-cell resistance to targeted agents or to standard chemotherapeutic regimens. (B) Pre-exposure of tumor cells to epigenetic drugs induces profound modifications of cell transcriptional profile, thus priming them to the cytotoxic effect of chemotherapeutic and targeted agents.
In a phase I study, Clozel and collaborators proposed a new approach to beaten chemotherapy resistance in DLBCL patients. The authors demonstrated a high rate of complete remission when a 5-day exposure to azacitidine followed by treatment with R-CHOP was employed. Mechanistically, the treatment leads to the demethylation of the chemoresistance-associated gene SMAD1 and subsequent chemosensitization (Clozel et al., 2013). Based on these results, an ongoing phase I study using azacitidine combined with R-CHOP in therapy-naive DLBCL, grade 3B FL, or transformed FL patients is showing promising preliminary results (NCT02343536). Finally, the safety and tolerability of adding oral azacitidine to R-ICE therapy is being evaluated in R/R DLBCL patients (NCT03450343).
Regarding HDACi, in vitro studies have demonstrated that this class of agents can synergize with chemotherapy. Globally, these trials have had mixed heterogeneous results. Among the potently successful studies, in indolent B-NHL the vorinostat/rituximab combination exhibited a nice activity with an acceptable safety profile and durable responses (Chen et al., 2015). Ageberg and collaborators also showed that valproic acid sensitizes to CHOP and enhances the CHOP ability to induce apoptosis in DLBCL cell lines (Ageberg et al., 2013). Subsequently, it has been shown in a small set of DLBCL patients that the administration of valproate before R-CHOP treatment upregulated the CD20 levels and increased the efficacy of anti-CD20-based therapy (Damm et al., 2015). Recently, the VALFRID phase I trial (NCT01622439) showed that valproate when added to standard R-CHOP therapy is secure, tolerable, and increases OS in DLBCL patients (Drott et al., 2018). The efficacy of vorinostat combined with cyclophosphamide, etoposide, and prednisone (R-CVEP) was evaluated in aged patients with R/R DLBCL (NCT00667615); however, the R-CVEP association did not reach the criteria for cohort expansion (Straus et al., 2015). Similarly, the combination of vorinostat with R-CHOP was evaluated in the SWOG S0806 phase I/II trial (NCT00972478) without success in DLBCL patients (Persky et al., 2018). Panobinostat was tested in combination with conventional therapy and although the data from the clinical trial NCT01238692 suggested that as a single agent this drug induces a durable response in a subset of R/R DLBCL patients, its combination with rituximab did not improve the response rate (Assouline et al., 2016). Similarly, Barnes and collaborators observed that this combination was effective in a minority of DLBCL patients heavily pretreated (NTC01282476) (Barnes et al., 2018). The combination with immunomodulatory drug (IMiD) lenalidomide was assessed in a phase I/II clinical trial in patients with R/R HL (NCT01460940); however, the combination was not advantageous over single-agent treatment and raised relevant concerns regarding the toxicity (Maly et al., 2017). Finally, preclinical data have shown that belinostat exhibits synergistic cytotoxic activity in DLBCL cell lines when associated to the microtubule-interfering drug vincristine, mediated by the prevention of cell polyploidy (Havas et al., 2016).
Regarding EZH2 inhibitors, combinatorial treatments with tazemetostat and the anti-programmed death-ligand 1 (PDL1) antibody atezolizumab (NCT02220842), prednisone alone, or combined with other components of CHOP regimen are currently being evaluated in patients with refractory DLBCL (NCT02889523) (Gulati et al., 2018). Moreover, combinations with EZH2 inhibitors and inhibitors of the BCR signaling cascade such as ibrutinib, the spleen tyrosine kinase (SYK) inhibitor tamatinib, the mammalian target of rapamycin (mTOR) inhibitor everolimus, or MAPK inhibitor have also been challenged in pre-clinical models of DLBCL (Brach et al., 2017; Lue et al., 2017). Other therapeutic strategies currently assessed in pre-clinical studies for the treatment of MM consist in combining the inhibitor tazemetostat with IMiDs such as lenalidomide or pomalidomide (Dang et al., 2016), glucocorticoid receptor agonists (dexamethasone or prednisolone), proteasome inhibitors (bortezomib or ixazomib) (Drew et al., 2017), or HDACis (Issa et al., 2017).
Finally, in combination with the CDK4/6 inhibitor palbocilib, the BETi JQ1 has shown synergistic activity in MCL in vitro and in vivo (Sun et al., 2015). Another member of the CDK family, CDK9, is a core component of the assembly of the positive transcription elongation factor complex (P-TEFb), which is recruiting by BRD4. In relation with this, the BETi BI-894999 shows profound synergy with CDK9 inhibitors alvocidib and LDC000067 in both in vitro and in vivo models of hematological malignancies (Doroshow et al., 2017). Among other promising combinations, CPI203 combined with the proteasome inhibitor bortezomib or with lenalidomide was particularly efficient in aggressive bortezomib-resistant MCL tumors (Moros et al., 2014), and GS-5829 synergistically interacted with venetoclax or with BCR-interfering agents in preclinical models of DLBCL, MCL, and/or CLL (Bates et al., 2016; Kim et al., 2017).
Besides the well-known genomic changes, several epigenetic modifications that result in an altered chromatin state and alterations in the DNA methylation status have been described in lymphoma cells. In general, these alterations favor the malignant transformation and/or tumor progression. Among the mechanisms that may apply to several lymphoma entities, epigenetic activation of suppressors of lineage fidelity leads to downregulation of lineage-specific genes, while additional silencing of essential transcription factors through H3K27 trimethylation avoids the restoration of the cell type characteristic expression program. Therefore, there is undoubtedly an important clinical role for epigenetic drugs across the spectrum of lymphoid malignancies, including B-NHL.
In the last decade, the progresses in the awareness of epigenetic changes in lymphoma cells have paved the way for targeted therapy alternatives employing epigenetic drugs. Treatment approaches such as HDAC inhibition or DNMT blockade have shown remarkable activity in specific subsets of lymphoma patients who remained unresponsive to or relapsed after standard therapy. These drugs have already been added into routine use for patients with a particular lymphoma/leukemia subtype and are the most broadly studied now. However, the identification of biomarkers of clinical sensitivity/resistance to these agents is still needed in order to better identify those lymphoma patients suitable for treatment with these drugs, and for the design of rationally based targeted combination therapies. Although several epigenetic drugs can be successfully combined with standard chemotherapy, allowing to decrease the chemotherapy doses and to limit toxicities and adverse effects, co-administration of two epigenetic modulators like DNA hypomethylating agents and HDAC inhibitors, for example, can also show synergistic molecular effects, resulting in increased antitumor activity.
In the light of the large number of drugs currently in clinical development in B-NHL patients, selection of the most relevant targeted therapies will be extremely important to move the field ahead. Epigenetic drugs with more specific targets, such as EZH2 inhibitors or BRD4 inhibitors, but also the newer epigenetic agents like PRMT5 and IDH inhibitors, are also of great interest, as demonstrated by a particularly rapid translation from bench to bedside within the past 5 years.
Despite these considerable advances in epigenetic drug therapy in B-cell lymphoma, there is still some way to go before reaching a complete overview of the complex landscape of the epigenetic modifications occurring during the lymphomagenesis, and much work is still to be done to improve the rationale use of epigenetic drugs in lymphoma patients. According to the promising reports from several trials involving the newest agents and the most innovative drug combinations in B-NHL patients with relapse disease, it seems that we are entering a very exciting era for the field of epigenetics in lymphoma.
MR, DR, MA, MF and GR made a substantial contribution to all aspects of the preparation of this manuscript.
The authors received financial support from Fondo de Investigación Sanitaria PI15/00102 and PI18/01383, European Regional Development Fund (ERDF) “Una manera de hacer Europa” (to GR). The authors received fundings from TG Therapeutics and Celgene Corp to support researches unrelated to the present work. Funders were involved neither in the design, nor in the writing of this review.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbas, S., Lugthart, S., Kavelaars, F. G., Schelen, A., Koenders, J. E., Zeilemaker, A., et al. (2010). Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia: prevalence and prognostic value. Blood 116, 2122–2126. doi: 10.1182/blood-2009-11-250878
Abd Al Kader, L., Oka, T., Takata, K., Sun, X., Sato, H., Murakami, I., et al. (2013). In aggressive variants of non-Hodgkin lymphomas, Ezh2 is strongly expressed and polycomb repressive complex PRC1.4 dominates over PRC1.2. Virchows Arch. 463, 697–711. doi: 10.1007/s00428-013-1428-y
Agarwal, R., Chan, Y. C., Tam, C. S., Hunter, T., Vassiliadis, D., Teh, C. E., et al. (2019). Dynamic molecular monitoring reveals that SWI–SNF mutations mediate resistance to ibrutinib plus venetoclax in mantle cell lymphoma. Nat. Med. 25, 119–129. doi: 10.1038/s41591-018-0243-z
Ageberg, M., Rydström, K., Relander, T., Drott, K. (2013). The histone deacetylase inhibitor valproic acid sensitizes diffuse large B-cell lymphoma cell lines to CHOP-induced cell death. Am. J. Transl. Res. 5, 170–183.
Agrawal, K., Das, V., Vyas, P., Hajdúch, M. (2018). Nucleosidic DNA demethylating epigenetic drugs —A comprehensive review from discovery to clinic. Pharmacol. Ther. 188, 45–89. doi: 10.1016/j.pharmthera.2018.02.006
Almstedt, M., Blagitko-Dorfs, N., Duque-Afonso, J., Karbach, J., Pfeifer, D., Jäger, E., et al. (2010). The DNA demethylating agent 5-aza-2’2′-deoxycytidine induces expression of NY-ESO-1 and other cancer/testis antigens in myeloid leukemia cells. Leuk. Res. 34, 899–905. doi: 10.1016/j.leukres.2010.02.004
Amara, K., Ziadi, S., Hachana, M., Soltani, N., Korbi, S., Trimeche, M. (2010). DNA methyltransferase DNMT3b protein overexpression as a prognostic factor in patients with diffuse large B-cell lymphomas. Cancer Sci. 101, 1722–1730. doi: 10.1111/j.1349-7006.2010.01569.x
Amengual, J. E., Clark-Garvey, S., Kalac, M., Scotto, L., Marchi, E., Neylon, E., et al. (2013). Sirtuin and pan-class I/II deacetylase (DAC) inhibition is synergistic in preclinical models and clinical studies of lymphoma. Blood. 122, 2104–2113. doi: 10.1182/blood-2013-02-485441
Amengual, J. E., Lichtenstein, R., Lue, J., Sawas, A., Deng, C., Lichtenstein, E., et al. (2018). A phase 1 study of romidepsin and pralatrexate reveals marked activity in relapsed and refractory T-cell lymphoma. Blood. 131, 397–407. doi: 10.1182/blood-2017-09-806737
Amorim, S., Stathis, A., Gleeson, M., Iyengar, S., Magarotto, V., Leleu, X., et al. (2016). Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: a dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. 3, e196–e204. doi: 10.1016/S2352-3026(16)00021-1
Andersson, A. K., Miller, D. W., Lynch, J. A., Lemoff, A. S., Cai, Z., Pounds, S. B., et al. (2011). IDH1 and IDH2 mutations in pediatric acute leukemia. Leukemia 25, 1570–1577. doi: 10.1038/leu.2011.133
Assouline, S. E., Nielsen, T. H., Yu, S., Alcaide, M., Chong, L., MacDonald, D., et al. (2016). Phase 2 study of panobinostat with or without rituximab in relapsed diffuse large B-cell lymphoma. Blood. 128, 185–194. doi: 10.1182/blood-2016-02-699520
Atlas, T. C. G. N., Muzny, D., Bainbridge, M., Chang, K., Dinh, H., Drummond, J., et al. (2012). Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337. doi: 10.1038/nature11252
Avramis, V. I., Powell, W. C., Mecum, R. A. (1989). Cellular metabolism of 5,6-dihydro-5-azacytidine and its incorporation into DNA and RNA of human lymphoid cells CEM/O and CEM/dCk(-). (–). Cancer Chemother. Pharmacol. 24, 155–160. doi: 10.1007/BF00300235
Azad, N., Zahnow, C. A., Rudin, C. M., Baylin, S. B. (2013). The future of epigenetic therapy in solid tumours - —Lessons from the past. Nat. Rev. Clin. Oncol. 10, 256–266. doi: 10.1038/nrclinonc.2013.42
Barnes, J. A., Redd, R., Fisher, D. C., Hochberg, E. P., Takvorian, T., Neuberg, D., et al. (2018). Panobinostat in combination with rituximab in heavily pretreated diffuse large B-cell lymphoma: Results of a phase II study. Hematol. Oncol. 36, 633–637. doi: 10.1002/hon.2515
Bates, J., Kusam, S., Tannheimer, S., Clarke, A., Kenney, T., Breckenridge, D., et al. (2016). Combination of the BET inhibitor GS-5829 and a BCL2 inhibitor resulted in broader activity in DLBCL and MCL cell lines. Blood 128, 5104.
Batlevi, C. L., Crump, M., Andreadis, C., Rizzieri, D., Assouline, S. E., Fox, S., et al. (2017). A phase 2 study of mocetinostat, a histone deacetylase inhibitor, in relapsed or refractory lymphoma. Br. J. Haematol. 178, 434–441. doi: 10.1111/bjh.14698
Batlevi, C. L., Kasamon, Y., Bociek, R. G., Lee, P., Gore, L., Copeland, A., et al. (2016). ENGAGE- 501: Phase II study of entinostat (SNDX-275) in relapsed and refractory Hodgkin lymphoma. Haematologica 101, 968–975. doi: 10.3324/haematol.2016.142406
Beà, S., Ribas, M., Hernandez, J. M., Bosch, F., Pinyol, M., Hernández, L., et al. (1999). Increased number of chromosomal imbalances and high-level DNA amplifications in mantle cell lymphoma are associated with blastoid variants. Blood 93, 4365–4374.
Beà, S., Valdés-Mas, R., Navarro, A., Salaverria, I., Martín-Garcia, D., Jares, P., et al. (2013). Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc. Natl. Acad. Sci. U. S. A. 110,18250–18255. doi: 10.1073/pnas.1314608110
Bedford, M. T., Clarke, S. G. (2009). Protein arginine methylation in mammals: Who, what, and why. Mol. Cell 33, 1–13. doi: 10.1016/j.molcel.2008.12.013
Béguelin, W., Popovic, R., Teater, M., Jiang, Y., Bunting, K. L., Rosen, M., et al. (2013). EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 23, 677–692. doi: 10.1016/j.ccr.2013.04.011
Beisler, J. A., Abbasi, M. M., Driscoll, J. S. (1979). Synthesis and antitumor activity of 5-azacytosine arabinoside. J. Med. Chem. 22, 1230–1234. doi: 10.1021/jm00196a015
Belinsky, S. A., Klinge, D. M., Stidley, C. A., Issa, J. P., Herman, J. G., March, T. H., et al. (2003). Inhibition of DNA methylation and histone deacetylation prevents murine lung cancer. Cancer Res. 63, 7089–7093.
Ben-Kasus, T., Ben-Zvi, Z., Marquez, V. E., Kelley, J. A., Agbaria, R. (2005). Metabolic activation of zebularine, a novel DNA methylation inhibitor, in human bladder carcinoma cells. Biochem. Pharmacol. 70, 121–133. doi: 10.1016/j.bcp.2005.04.010
Berdasco, M., Esteller, M. (2010). Aberrant epigenetic landscape in cancer: How cellular identity goes awry. Dev. Cell. 19, 698–711. doi: 10.1016/j.devcel.2010.10.005
Bergaggio, E., Riganti, C., Garaffo, G., Vitale, N., Mereu, E., Bandini, C., et al. (2019). IDH2 inhibition enhances proteasome inhibitor responsiveness in hematological malignancies. Blood 133, 156 LP–15 167. doi: 10.1182/blood-2018-05-850826
Beumer, J. H., Parise, R. A., Newman, E. M., Doroshow, J. H., Synold, T. W., Lenz, H. J., et al. (2008). Concentrations of the DNA methyltransferase inhibitor 5-fluoro-2′- deoxycytidine (FdCyd) and its cytotoxic metabolites in plasma of patients treated with FdCyd and tetrahydrouridine (THU). Cancer Chemother. Pharmacol. 62, 363–368. doi: 10.1007/s00280-007-0603-8
Biankin, A. V., Waddell, N., Kassahn, K. S., Gingras, M. C., Muthuswamy, L. B., Johns, A. L., et al. (2012). Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 491, 399–405. doi: 10.1038/nature11547
Bisserier, M., Wajapeyee, N. (2018). Mechanisms of resistance to ezh2 inhibitors in diffuse large B-cell lymphomas. Blood. 131, 2125–2137. doi: 10.1182/blood-2017-08-804344
Blum, K. A., Abramson, J., Maris, M., Flinn, I., Goy, A., Mertz, J., et al. (2018). 41OA phase I study of CPI-0610, a bromodomain and extra terminal protein (BET) inhibitor in patients with relapsed or refractory lymphoma. Ann. Oncol. 29, A410. doi: 10.1093/annonc/mdy048
Blum, K. A., Liu, Z., Lucas, D. M., Chen, P., Xie, Z., Baiocchi, R., et al. (2010). Phase I trial of low dose decitabine targeting DNA hypermethylation in patients with chronic lymphocytic leukaemia and non-Hodgkin lymphoma: dose-limiting myelosuppression without evidence of DNA hypomethylation. Br. J. Haematol. 29, 410. doi: 10.1111/j.1365-2141.2010.08213.x
Bobrowicz, M., Dwojak, M., Pyrzynska, B., Stachura, J., Muchowicz, A., Berthel, E., et al. (2017). HDAC6 inhibition upregulates CD20 levels and increases the efficacy of anti-CD20 monoclonal antibodies. Blood 130, 1628–1638. doi: 10.1182/blood-2016-08-736066
Brach, D., Johnston-blackwellBlackwell, D., Drew, A., Lingaraj, T., Motwani, V., Warholic, N. M., et al. (2017). EZH2 inhibition by tazemetostat results in altered dependency on B-cell activation signaling in DLBCL 16. Mol. Cancer Ther. 2586-2597. doi: 10.1158/1535-7163.MCT-16-0840
Bracken, A. P., Helin, K. (2009). Polycomb group proteins: Navigators of lineage pathways led astray in cancer. Nat. Rev. Cancer 9, 773–784. doi: 10.1038/nrc2736
Bradner, J. E., West, N., Grachan, M. L., Greenberg, E. F., Haggarty, S. J., Warnow, T., et al. (2010). Chemical phylogenetics of histone deacetylases. Nat. Chem. Biol. 6, 238–243. doi: 10.1038/nchembio.313
Brahmer, J. R., Tykodi, S. S., Chow, L. Q. M., Hwu, W.-J., Topalian, S. L., Hwu, P., et al. (2012). Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 366, 2455–2465. doi: 10.1056/NEJMoa1200694
Buglio, D., Georgakis, G. V., Hanabuchi, S., Arima, K., Khaskhely, N. M., Liu, Y.-J., et al. (2008). Vorinostat inhibits STAT6-mediated TH2 cytokine and TARC production and induces cell death in Hodgkin lymphoma cell lines. Blood 112, 1424–1433. doi: 10.1182/blood-2008-01-133769
Buscarlet, M., Krasteva, V., Ho, L., Simon, C., Hébert, J., Wilhelm, B., et al. (2014). Essential role of BRG, the ATPase subunit of BAF chromatin remodeling complexes, in leukemia maintenance. Blood. 123, 1720–1728. doi: 10.1182/blood-2013-02-483495
Cairns, R. A., Iqbal, J., Lemonnier, F., Kucuk, C., De Leval, L., Jais, J.-P., et al. (2012). IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood 119, 1901–1903. doi: 10.1182/blood-2011-11-391748
Campo, E., Swerdlow, S. H., Harris, N. L., Pileri, S., Stein, H., Jaffe, E. S. (2011). The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood 117, 5019–5032. doi: 10.1182/blood-2011-01-293050
Cerchietti, L. C., Hatzi, K., Caldas-Lopes, E., Yang, S. N., Figueroa, M. E., Morin, R., et al. (2010). BCL6 repression of EP300 in human diffuse large B cell lymphoma cells provides a basis for rational combinatorial therapy. J. Clin. Invest. 120, 4569–4582. doi: 10.1172/JCI42869
Ceribelli, M., Kelly, P. N., Shaffer, A. L., Wright, G. W., Xiao, W., Yang, Y., et al. (2014). Blockade of oncogenic IκB kinase activity in diffuse large B-cell lymphoma by bromodomain and extraterminal domain protein inhibitors. Proc. Natl. Acad. Sci. U. S. A. 111, 11365–11370. doi: 10.1073/pnas.1411701111
Chai, B., Huang, J., Cairns, B. R., Laurent, B. C. (2005). Distinct roles for the RSC and Swi/Snf ATP-dependent chromatin remodelers in DNA double-strand break repair. Genes Dev. 19, 1656–1661. doi: 10.1101/gad.1273105
Chaidos, A., Caputo, V., Karadimitris, A. (2015). Inhibition of bromodomain and extra-terminal proteins (BET) as a potential therapeutic approach in haematological malignancies: Emerging preclinical and clinical evidence. Ther. Adv. Hematol. 6, 128–141. doi: 10.1177/2040620715576662
Chan-Penebre, E., Kuplast, K. G., Majer, C. R., Boriack-Sjodin, P. A., Wigle, T. J., Johnston, L. D., et al. (2015). A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat. Chem. Biol. 11, 432–437. doi: 10.1038/nchembio.1810
Chan, W.-I., Hannah, R. L., Dawson, M. A., Pridans, C., Foster, D., Joshi, A., et al. (2011). The transcriptional coactivator Cbp regulates self-renewal and differentiation in adult hematopoietic stem cells. Mol. Cell. Biol. 31, 5046–5060. doi: 10.1128/MCB.05830-11
Chen, J., Li, Q. (2011). Life and death of transcriptional co-activator p300. Epigenetics. 6, 957–961. doi: 10.4161/epi.6.8.16065
Chen, R., Frankel, P., Popplewell, L., Siddiqi, T., Ruel, N., Rotter, A., et al. (2015). A phase II study of vorinostat and rituximab for treatment of newly diagnosed and relapsed/refractory indolent non-Hodgkin lymphoma. Haematologica 100, 357–362. doi: 10.3324/haematol.2014.117473
Cheng, J. C., Yoo, C. B., Weisenberger, D. J., Chuang, J., Wozniak, C., Liang, G., et al. (2004). Preferential response of cancer cells to zebularine. Cancer Cell. 6, 151–158. doi: 10.1016/j.ccr.2004.06.023
Chiappinelli, K. B., Strissel, P. L., Desrichard, A., Li, H., Henke, C., Akman, B., et al. (2015). Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell. 162, 974–986. doi: 10.1016/j.cell.2015.07.011
Choi, W. J., Chung, H. J., Chandra, G., Alexander, V., Zhao, L. X., Lee, H. W., et al. (2012). Fluorocyclopentenyl-cytosine with broad spectrum and potent antitumor activity. J. Med. Chem. 55, 4521–4525. doi: 10.1021/jm3004009
Chuang, J. C., Warner, S. L., Vollmer, D., Vankayalapati, H., Redkar, S., Bearss, D. J., et al. (2010). S110, a 5-aza-2’2′-deoxycytidine-containing dinucleotide, is an effective DNA methylation inhibitor in vivo and can reduce tumor growth. Mol. Cancer Ther. 9, 1443–1450. doi: 10.1158/1535-7163.MCT-09-1048
Clozel, T., Yang, S. N., Elstrom, R. L., Tam, W., Martin, P., Kormaksson, M., et al. (2013). Mechanism-based epigenetic chemosensitization therapy of diffuse large B-cell lymphoma. Cancer Discov. 3, 1002–1019. doi: 10.1158/2159-8290.CD-13-0117
Cox, O. B., Krojer, T., Collins, P., Monteiro, O., Talon, R., Bradley, A., et al. (2016). A poised fragment library enables rapid synthetic expansion yielding the first reported inhibitors of PHIP(2), an atypical bromodomain. Chem. Sci. 7, 2322–2330. doi: 10.1039/C5SC03115J
Crump, M., Coiffier, B., Jacobsen, E. D., Sun, L., Ricker, J. L., Xie, H., et al. (2008). Phase II trial of oral vorinostat (suberoylanilide hydroxamic acid) in relapsed diffuse large-B-cell lymphoma. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 19, 964–969. doi: 10.1093/annonc/mdn031
Cuadros, M., Sánchez-Martín, V., Herrera, A., Baliñas, C., Martín-Padrón, J., Boyero, L., et al. (2017). BRG1 regulation by miR-155 in human leukemia and lymphoma cell lines. Clin. Transl. Oncol. 19, 1010–1017. doi: 10.1007/s12094-017-1633-2
Damm, J. K., Gordon, S., Ehinger, M., Jerkeman, M., Gullberg, U., Hultquist, A., et al. (2015). Pharmacologically relevant doses of valproate upregulate CD20 expression in three diffuse large B-cell lymphoma patients in vivo. Exp. Hematol. Oncol. 4, eColection. doi: 10.1186/2162-3619-4-4
Dancy, B. M., Cole, P. A. (2015). Protein lysine acetylation by p300/CBP. Chem. Rev. 115, 2419–2452. doi: 10.1021/cr500452k
Dang, L., Su, S.-S. M. (2017). Isocitrate dehydrogenase mutation and (R)-2-hydroxyglutarate: from basic discovery to therapeutics development. Annu. Rev. Biochem. 86, 305–331. doi: 10.1146/annurev-biochem-061516-044732
Dang, L., Yen, K. E., Attar, E. C. (2016). IDH mutations in cancer and progress toward development of targeted therapeutics. Ann. Oncol. 27, 599–608. doi: 10.1093/annonc/mdw013
Dawson, M. A., Prinjha, R. K., Dittmann, A., Giotopoulos, G., Bantscheff, M., Chan, W. I., et al. (2011). Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 478, 529–533. doi: 10.1038/nature10509
De, S., Shaknovich, R., Riester, M., Elemento, O., Geng, H., Kormaksson, M., et al. (2013). Aberration in DNA methylation in B-cell lymphomas has a complex origin and increases with disease severity. PLoS Genet. 9, e1003137. doi: 10.1371/journal.pgen.1003137
Delmore, J. E., Issa, G. C., Lemieux, M. E., Rahl, P. B., Shi, J., Jacobs, H. M., et al. (2011). BET bromodomain inhibition as as a therapeutic strategy to target c-Myc. Cell 146, 904–917. doi: 10.1016/j.cell.2011.08.017
Delvecchio, M., Gaucher, J., Aguilar-Gurrieri, C., Ortega, E., Panne, D. (2013). Structure of the p300 catalytic core and implications for chromatin targeting and HAT regulation. Nat. Struct. Mol. Biol. 20, 1040–1046. doi: 10.1038/nsmb.2642
Di Costanzo, A., Del Gaudio, N., Migliaccio, A., Altucci, L. (2014). Epigenetic drugs against cancer: an evolving landscape. Arch. Toxicol. 88, 1651–1668. doi: 10.1007/s00204-014-1315-6
Dickinson, M., Kamdar, M., Huntly, B. J., Larrea, C. F., Cordoba, R., Mateos, M.-V., et al. (2018). A phase I study of molibresib (GSK525762), a selective bromodomain (BRD) and extra terminal protein (BET) inhibitor: results from part 1 of a phase I/II open label single agent study in subjects with non-Hodgkin’s lymphoma (NHL). Blood 132, 1682–1682. doi: 10.1182/BLOOD-2018-99-117089
Doroshow, D. B., Eder, J. P., LoRusso, P. M. (2017). BET inhibitors: a novel epigenetic approach. Ann. Oncol. 28, 1776–1787. doi: 10.1093/annonc/mdx157
Drew, A. E., Motwani, V., Campbell, J. E., Tang, C., Smith, J. J., Chesworth, R., et al. (2017). Abstract 5060: activity of the EZH2 inhibitor tazemetostat as a monotherapy and in combination with multiple myeloma therapies in preclinical models. Cancer Res. 77, n5060. doi: 10.1158/1538-7445.AM2017-5060
Dreyling, M. H., Roulston, D., Bohlander, S. K., Vardiman, J., Olopade, O. I. (1998). Codeletion of CDKN2 and MTAP genes in a subset of non-Hodgkin’s lymphoma may be associated with histologic transformation from low-grade to diffuse large-cell lymphoma. Genes Chromosomes Cancer 22, 72–78. doi: 10.1002/(SICI)1098-2264(199805)22:1<72::AID-GCC10>3.3.CO;2-G
Drott, K., Hagberg, H., Papworth, K., Relander, T., Jerkeman, M. (2018). Valproate in combination with rituximab and CHOP as first-line therapy in diffuse large B-cell lymphoma (VALFRID). Blood Adv. 26, 1386–1392. doi: 10.1182/bloodadvances.2018019240
Dubois, S., Viailly, P., Mareschal, S., Bohers, E., Bertrand, P., Ruminy, P., et al. (2016). Next-generation sequencing in diffuse large B-cell lymphoma highlights molecular divergence and therapeutic opportunities : a LYSA study Clin. Cancer Res. 22, 2919–2929. doi: 10.1158/1078-0432.CCR-15-2305
Egger, G., Aparicio, A. M., Escobar, S. G., Jones, P. A. (2007). Inhibition of histone deacetylation does not block resilencing of p16 after 5-aza-2′-deoxycytidine treatment. Cancer Res. 67, 346–356. doi: 10.1158/0008-5472.CAN-06-2845
Esteller, M., Corn, P. G., Baylin, S. B., Herman, J. G. (2001). A gene hypermethylation profile of human cancer. Cancer Res. 61, 3225–3229.
Esteve-Arenys, A., Valero, J. G., Chamorro-Jorganes, A., Gonzalez, D., Rodriguez, V., Dlouhy, I., et al. (2018). The BET bromodomain inhibitor CPI203 overcomes resistance to ABT-199 (venetoclax) by downregulation of BFL-1/A1 in in vitro and in vivo models of MYC+/BCL2+ double hit lymphoma. Oncogene. 37, 1830–1844. doi: 10.1038/s41388-017-0111-1
Evens, A. M., Balasubramanian, S., Vose, J. M., Harb, W., Gordon, L. I., Langdon, R., et al. (2016). A phase I/II multicenter, open-label study of the oral histone deacetylase inhibitor abexinostat in relapsed/refractory lymphoma. Clin. Cancer Res. 22, 1059–1066. doi: 10.1158/1078-0432.CCR-15-0624
Fenaux, P., Mufti, G. J., Hellstrom-Lindberg, E., Santini, V., Finelli, C., Giagounidis, A., et al. (2009). Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: A randomised, open-label, phase III study. Lancet Oncol. 10, 223–232. doi: 10.1016/S1470-2045(09)70003-8
Ferrari, K. J., Scelfo, A., Jammula, S., Cuomo, A., Barozzi, I., Stützer, A., et al. (2014). Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Mol. Cell. 53, 49–62. doi: 10.1016/j.molcel.2013.10.030
Figueroa, M. E., Abdel-Wahab, O., Lu, C., Ward, P. S., Patel, J., Shih, A., et al. (2010). Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18, 553–567. doi: 10.1016/j.ccr.2010.11.015
Filippakopoulos, P., Picaud, S., Mangos, M., Keates, T., Lambert, J. P., Barsyte-Lovejoy, D., et al. (2012). Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 149, 214–231. doi: 10.1016/j.cell.2012.02.013
Filippakopoulos, P., Qi, J., Picaud, S., Shen, Y., Smith, W. B., Fedorov, O., et al. (2010). Selective inhibition of BET bromodomains. Nature. 468, 1067–1073. doi: 10.1038/nature09504
Flavahan, W. A., Gaskell, E., Bernstein, B. E. (2017). Epigenetic plasticity and the hallmarks of cancer. Science. 357, p.eaal2380. doi: 10.1126/science.aal2380
Flesner, B. K., Kumar, S. R., Bryan, J. N. (2014). 6-Thioguanine and zebularine down-regulate DNMT1 and globally demethylate canine malignant lymphoid cells. BMC Vet. Res. 10, 290. doi: 10.1186/s12917-014-0290-8
Fodale, V., Pierobon, M., Liotta, L., Petricoin, E. (2011). Mechanism of cell adaptation: When and how do cancer cells develop chemoresistance? Cancer J. 17, 89–95. doi: 10.1097/PPO.0b013e318212dd3d
Forero-Torres, A., Rosen, S., Smith, D. C., Lesser, G., Peguero, J., Gupta, S., et al. (2017). Preliminary results from an ongoing phase 1/2 Study of INCB057643, a bromodomain and extraterminal (BET) protein inhibitor, in patients (pts) with advanced malignancies. Blood 130, 4048.
Fournel, M., Bonfils, C., Hou, Y., Yan, P. T., Trachy-Bourget, M.-C., Kalita, A., et al. (2008). MGCD0103, a novel isotype-selective histone deacetylase inhibitor, has broad spectrum antitumor activity in vitro and in vivo. Mol. Cancer Ther. 7, 759–768. doi: 10.1158/1535-7163.MCT-07-2026
Fujimoto, A., Totoki, Y., Abe, T., Boroevich, K. A., Hosoda, F., Nguyen, H. H., et al. (2012). Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat. Genet. 44, 760–764. doi: 10.1038/ng.2291
Galli, M., Salmoiraghi, S., Golay, J., Gozzini, A., Crippa, C., Pescosta, N., et al. (2010). A phase II multiple dose clinical trial of histone deacetylase inhibitor ITF2357 in patients with relapsed or progressive multiple myeloma. Ann. Hematol. 89, 185–190. doi: 10.1007/s00277-009-0793-8
Garrido Castro, P., van Roon, E. H. J., Pinhanços, S. S., Trentin, L., Schneider, P., Kerstjens, M., et al. (2018). The HDAC inhibitor panobinostat (LBH589) exerts in vivo anti-leukaemic activity against MLL-rearranged acute lymphoblastic leukaemia and involves the RNF20/RNF40/WAC-H2B ubiquitination axis. Leukemia 32, 323–331. doi: 10.1038/leu.2017.216
Ghoshal, K., Datta, J., Majumder, S., Bai, S., Kutay, H., Motiwala, T., et al. (2005). 5-Aza-deoxycytidine induces selective degradation of DNA methyltransferase 1 by a proteasomal pathway that requires the KEN box, bromo-adjacent homology domain, and nuclear localization signal. Mol. Cell. Biol. 25, 4727–4741. doi: 10.1128/MCB.25.11.4727-4741.2005
Gloghini, A., Buglio, D., Khaskhely, N. M., Georgakis, G., Orlowski, R. Z., Neelapu, S. S., et al. (2009). Expression of histone deacetylases in lymphoma: implication for the development of selective inhibitors. Br. J. Haematol. 147, 515–525. doi: 10.1111/j.1365-2141.2009.07887.x
Goodyear, O., Agathanggelou, A., Novitzky-Basso, I., Siddique, S., McSkeane, T., Ryan, G., et al. (2010). Induction of a CD8+ T-cell response to the MAGE cancer testis antigen by combined treatment with azacitidine and sodium valproate in patients with acute myeloid leukemia and myelodysplasia. Blood. 116, 1908–1918. doi: 10.1182/blood-2009-11-249474
Goverdhan, A., Lee, H.-H., Havranek, O., Davis, R. E., Hung, M.-C., (2017). “Abstract 13: PRMT1 as a therapeutic target in diffuse large B-cell lymphoma,” in Epigenetics (American Association for Cancer Research), 13–13. doi: 10.1158/1557-3265.HEMMAL17-13
Greenblatt, S. M., Liu, F., Nimer, S. D. (2016). Arginine methyltransferases in normal and malignant hematopoiesis. Exp. Hematol. 44, 435–441. doi: 10.1016/j.exphem.2016.03.009
Groudine, M., Eisenman, R., Weintraub, H. (1981). Chromatin structure of endogenous retroviral genes and activation by an inhibitor of DNA methylation. Nature. 292, 311–317. doi: 10.1038/292311a0
Gui, Y., Guo, G., Huang, Y., Hu, X., Tang, A., Gao, S., et al. (2011). Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat. Genet. 44, 17–19. doi: 10.1038/ng.907
Gulati, N., Béguelin, W., Giulino-Roth, L. (2018). Enhancer of zeste homolog 2 (EZH2) inhibitors. Leuk. Lymphoma 59, 1574–1585. doi: 10.1080/10428194.2018.1430795
Hadjikyriacou, A., Yang, Y., Espejo, A., Bedford, M. T., Clarke, S. G. (2015). Unique features of human protein arginine methyltransferase 9 (PRMT9) and its substrate RNA splicing factor SF3B2. J. Biol. Chem. 290, 16723–16743. doi: 10.1074/jbc.M115.659433
Haferlach, C., Dicker, F., Schnittger, S., Kern, W., Haferlach, T. (2007). Comprehensive genetic characterization of CLL: a study on 506 cases analysed with chromosome banding analysis, interphase FISH, IgV(H) status and immunophenotyping. Leukemia 21, 2442–2451. doi: 10.1038/sj.leu.2404935
Han, M., Jia, L., Lv, W., Wang, L., Cui, W. (2019). Epigenetic enzyme mutations: role in tumorigenesis and molecular inhibitors. Front. Oncol. 9, 1–9. doi: 10.3389/fonc.2019.00194
Hassler, M. R., Schiefer, A.-I., Egger, G. (2013). Combating the epigenome: epigenetic drugs against non-Hodgkin’s lymphoma. Epigenomics 5, 397–415. doi: 10.2217/epi.13.39
Havas, A. P., Rodrigues, K. B., Bhakta, A., Demirjian, J. A., Hahn, S., Tran, J., et al. (2016). Belinostat and vincristine demonstrate mutually synergistic cytotoxicity associated with mitotic arrest and inhibition of polyploidy in a preclinical model of aggressive diffuse large B cell lymphoma. Cancer Biol. Ther. 17, 1240–1252. doi: 10.1080/15384047.2016.1250046
Heideman, M. R., Wilting, R. H., Yanover, E., Velds, A., de Jong, J., Kerkhoven, R. M., et al. (2013). Dosage-dependent tumor suppression by histone deacetylases 1 and 2 through regulation of c-Myc collaborating genes and p53 function. Blood 121, 2038–2050. doi: 10.1182/blood-2012-08-450916
Helming, K. C., Wang, X., Wilson, B. G., Vazquez, F., Haswell, J. R., Manchester, H. E., et al. (2014). ARID1B is a specific vulnerability in ARID1A-mutant cancers. Nat. Med. 20, 251–254. doi: 10.1038/nm.3480
Hermann, A., Goyal, R., Jeltsch, A. (2004). The Dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. J. Biol. Chem. 279, 48350–48359. doi: 10.1074/jbc.M403427200
Herviou, L., Kassambara, A., Boireau, S., Robert, N., Requirand, G., Vincent, L., et al. (2016). Targeting EZH2 in multiple myeloma could be promising for a subgroup of MM patients in combination with IMiDs. Blood. 128, 311.
Hodis, E., Watson, I. R., Kryukov, G. V., Arold, S. T., Imielinski, M., Theurillat, J. P., et al. (2012). A landscape of driver mutations in melanoma. Cell. 150, 251–266. doi: 10.1016/j.cell.2012.06.024
Hogarth, L. A., Redfern, C. P. F., Teodoridis, J. M., Hall, A. G., Anderson, H., Case, M. C., et al. (2008). The effect of thiopurine drugs on DNA methylation in relation to TPMT expression. Biochem. Pharmacol. 76, 1024–1035. doi: 10.1016/j.bcp.2008.07.026
Hogg, S. J., Newbold, A., Vervoort, S. J., Cluse, L. A., Martin, B. P., Gregory, G. P., et al. (2016). BET inhibition induces apoptosis in aggressive B-cell lymphoma via epigenetic regulation of BCL-2 family members. Mol. Cancer Ther. 15, 2030–2041. doi: 10.1158/1535-7163.MCT-15-0924
Holkova, B., Yazbeck, V., Kmieciak, M., Bose, P., Ma, S., Kimball, A., et al. (2017). A phase 1 study of bortezomib and romidepsin in patients with chronic lymphocytic leukemia/small lymphocytic lymphoma, indolent B-cell lymphoma, peripheral T-cell lymphoma, or cutaneous T-cell lymphoma. Leuk. Lymphoma 58, 1349–1357. doi: 10.1080/10428194.2016.1276287
Honma, D., Kanno, O., Watanabe, J., Kinoshita, J., Hirasawa, M., Nosaka, E., et al. (2017). Novel orally bioavailable EZH1/2 dual inhibitors with greater antitumor efficacy than an EZH2 selective inhibitor. Cancer Sci. 108, 2069–2078. doi: 10.1111/cas.13326
Horsman, D. E., Gascoyne, R. D., Coupland, R. W., Coldman, A. J., Adomat, S. A. (1995). Comparison of cytogenetic analysis, Southern analysis, and polymerase chain reaction for the detection of t(14; 18) in follicular lymphoma. Am. J. Clin. Pathol. 103, 472–478. doi: 10.1093/ajcp/103.4.472
Hunter, Z. R., Xu, L., Yang, G., Zhou, Y., Liu, X., Cao, Y., et al. (2014). The genomic landscape of Waldenström macroglobulinemia is characterized by highly recurring MYD88 and WHIM-like CXCR4 mutations, and small somatic deletions associated with B-cell lymphomagenesis. Blood 123, 1637–1646. doi: 10.1182/blood-2013-09-525808
Issa, J. P. J., Roboz, G., Rizzieri, D., Jabbour, E., Stock, W., O’Connell, C., et al. (2015). Safety and tolerability of guadecitabine (SGI-110) in patients with myelodysplastic syndrome and acute myeloid leukaemia: A multicentre, randomised, dose-escalation phase 1 study. Lancet Oncol. 16, 1099–1110. doi: 10.1016/S1470-2045(15)00038-8
Issa, M. E., Takhsha, F. S., Chirumamilla, C. S., Perez-Novo, C., Vanden Berghe, W., Cuendet, M. (2017). Epigenetic strategies to reverse drug resistance in heterogeneous multiple myeloma. Clin. Epigenetics. 9, 17. doi: 10.1186/s13148-017-0319-5
Italiano, A., Soria, J., Toulmonde, M., Michot, J., Lucchesi, C., Varga, A., et al. (2018). Articles Tazemetostat , an EZH2 inhibitor , in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours : a first-in-human, open-label , phase 1 study 2045, Lancet Oncol. 1–11. doi: 10.1016/S1470-2045(18)30145-1
Jelinic, P., Mueller, J. J., Olvera, N., Dao, F., Scott, S. N., Shah, R., et al. (2014). Recurrent SMARCA4 mutations in small cell carcinoma of the ovary. Nat. Genet. 46, 424–426. doi: 10.1038/ng.2922
Jin, Y., Elalaf, H., Watanabe, M., Tamaki, S., Hineno, S., Matsunaga, K., et al. (2015). Mutant IDH1 dysregulates the differentiation of mesenchymal stem cells in association with gene-specific histone modifications to cartilage- and bone-related genes. PLoS One 10, 1–15. doi: 10.1371/journal.pone.0131998
Johnson-Farley, N., Veliz, J., Bhagavathi, S., Bertino, J. R. (2015). ABT-199, a BH3 mimetic that specifically targets Bcl-2, enhances the antitumor activity of chemotherapy, bortezomib and JQ1 in "”double hit"” lymphoma cells. Leuk. Lymphoma 56, 2146–2152. doi: 10.3109/10428194.2014.981172
Jones, P. A., Issa, J. P. J., Baylin, S. (2016). Targeting the cancer epigenome for therapy. Nat. Rev. Genet. 17, 630–641. doi: 10.1038/nrg.2016.93
Jones, P. A., Taylor, S. M. (1980). Cellular differentiation, cytidine analogs and DNA methylation. Cell. 20, 85–93. doi: 10.1016/0092-8674(80)90237-8
Juttermann, R., Li, E., Jaenisch, R. (2006). Toxicity of 5-aza-2’2′-deoxycytidine to mammalian cells is mediated primarily by covalent trapping of DNA methyltransferase rather than DNA demethylation. Proc. Natl. Acad. Sci. 91, 11797–11801. doi: 10.1073/pnas.91.25.11797
Kadam, S., Emerson, B. M. (2003). Transcriptional specificity of human SWI/SNF BRG1 and BRM chromatin remodeling complexes. Mol. Cell. 1, 177–189. doi: 10.1016/S1097-2765(03)00034-0
Kalac, M., Scotto, L., Marchi, E., Amengual, J., Seshan, V. E., Bhagat, G., et al. (2011). HDAC inhibitors and decitabine are highly synergistic and associated with unique gene-expression and epigenetic profiles in models of DLBCL. Blood 118, 5506–5516. doi: 10.1182/blood-2011-02-336891
Kandoth, C., Schultz, N., Cherniack, A. D., Akbani, R., Liu, Y., Shen, H., et al. (2013). Integrated genomic characterization of endometrial carcinoma. Nature. 497, 67–73. doi: 10.1038/nature12113
Kang-Decker, N., Tong, C., Boussouar, F., Baker, D. J., Xu, W., Leontovich, A. A., et al. (2004). Loss of CBP causes T cell lymphomagenesis in synergy with p27 Kip1 insufficiency. Cancer Cell. 5, 177–189. doi: 10.1016/S1535-6108(04)00022-4
Kang, M. R., Kim, M. S., Oh, J. E., Kim, Y. R., Song, S. Y., Seo, S., et al. (2009). Mutational analysis of IDH1 codon 132 in glioblastomas and other common cancers. Int. J. Cancer 125, 353–355. doi: 10.1002/ijc.24379
Karpf, A. R. (2004). Limited gene activation in tumor and normal epithelial cells treated with the DNA methyltransferase Inhibitor 5-aza-2’2′-deoxycytidine. Mol. Pharmacol. 65, 18–27. doi: 10.1124/mol.65.1.18
Kewitz, S., Bernig, T., Staege, M. S. (2012). Histone deacetylase inhibition restores cisplatin sensitivity of Hodgkin’s lymphoma cells. Leuk. Res. 36, 773–778. doi: 10.1016/j.leukres.2012.02.021
Khavari, P. A., Peterson, C. L., Tamkun, J. W., Mendel, D. B., Crabtree, G. R. (1993). BRG1 contains a conserved domain of the SWI2/SNF2 family necessary for normal mitotic growth and transcription. Nature. 366, 170–174. doi: 10.1038/366170a0
Kim, E., Ten Hacken, E., Clarke, A., Keating, M. J., Wierda, W. G., Ferrajoli, A., et al. (2017). The clinical BET inhibitor, GS-5829, is active against chronic lymphocytic leukemia as single agent and in combination with B-cell receptor signaling inhibitors. Blood 130, 3844.
Kinders, R. J., Wang, L., Kummar, K. S., Balasubramanian, P., Zhu, W., Parchment, R. E., et al. (2011). Investigation of 5-fluorodeoxycytidine with tetrahydrouracil as a demethylation regimen in solid tumors. . Cancer Ther. 10, A106. doi: 10.1158/1535-7163.TARG-11-A106
King, B., Trimarchi, T., Reavie, L., Xu, L., Mullenders, J., Ntziachristos, P., et al. (2013). The ubiquitin ligase FBXW7 modulates leukemia-initiating cell activity by regulating MYC stability. Cell 153, 1552–1566. doi: 10.1016/j.cell.2013.05.041
Kirschbaum, M., Frankel, P., Popplewell, L., Zain, J., Delioukina, M., Pullarkat, V., et al. (2011). Phase II study of vorinostat for treatment of relapsed or refractory indolent non-Hodgkin’s lymphoma and mantle cell lymphoma. J. Clin. Oncol. 29, 1198–1203. doi: 10.1200/JCO.2010.32.1398
Klein, J. M., Henke, A., Sauer, M., Bessler, M., Reiners, K. S., Engert, A., et al. (2013). The histone deacetylase inhibitor LBH589 (panobinostat) modulates the crosstalk of lymphocytes with Hodgkin lymphoma cell lines. PLoS One 8, e79502. doi: 10.1371/journal.pone.0079502
Knipstein, J., Gore, L. (2011). Entinostat for treatment of solid tumors and hematologic malignancies. Expert Opin. Investig. Drugs 20, 1455–1467. doi: 10.1517/13543784.2011.613822
Knutson, S. K., Kawano, S., Minoshima, Y., Warholic, N. M., Huang, K.-C., Xiao, Y., et al. (2014). Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol. Cancer Ther. 13, 842–854. doi: 10.1158/1535-7163.MCT-13-0773
Konze, K. D., Ma, A., Li, F., Barsyte-Lovejoy, D., Parton, T., MacNevin, C. J., et al. (2013). An orally bioavailable chemical probe of the lysine methyltransferases EZH2 and EZH1. ACS Chem. Biol. 8, 1324–1334. doi: 10.1021/cb400133j
Kouzarides, T. (2007). Chromatin modifications and their function. Cell. 128, 693–705. doi: 10.1016/j.cell.2007.02.005
Kretzner, L., Scuto, A., Dino, P. M., Kowolik, C. M., Wu, J., Ventura, P., et al. (2011). Combining histone deacetylase inhibitor vorinostat with aurora kinase inhibitors enhances lymphoma cell killing with repression of c-Myc, hTERT, and microRNA levels. Cancer Res. 71, 3912–3920. doi: 10.1158/0008-5472.CAN-10-2259
Kummar, S., Gutierrez, M., Gardner, E. R., Donovan, E., Hwang, K., Chung, E. J., et al. (2007). Phase I trial of MS-275, a histone deacetylase inhibitor, administered weekly in refractory solid tumors and lymphoid malignancies. Clin. Cancer Res. 13, 5411–5417. doi: 10.1158/1078-0432.CCR-07-0791
Kung, A. L., Rebel, V. I., Bronson, R. T., Ch’ng, L. E., Sieff, C. A., Livingston, D. M., et al. (2000). Gene dose-dependent control of hematopoiesis and hematologic tumor suppression by CBP. Genes Dev. 14, 172–177.
Lai, A. Y., Mav, D., Shah, R., Grimm, S. A., Phadke, D., Hatzi, K., et al. (2013). DNA methylation profiling in human B cells reveals immune regulatory elements and epigenetic plasticity at Alu elements during B-cell activation. Genome Res. 12, 2030–2041. doi: 10.1101/gr.155473.113
Landau, D. A., Tausch, E., Taylor-Weiner, A. N., Stewart, C., Reiter, J. G., Bahlo, J., et al. (2015). Mutations driving CLL and their evolution in progression and relapse. Nature 526, 525–530. doi: 10.1038/nature15395
Lane, A. A., Chabner, B. A. (2009). Histone deacetylase inhibitors in cancer therapy. J. Clin. Oncol. 27, 5459–5468. doi: 10.1200/JCO.2009.22.1291
Lee, S. H., Kim, J.-S., Zheng, S., Huse, J. T., Bae, J. S., Lee, J. W., et al. (2017). ARID1B alterations identify aggressive tumors in neuroblastoma. Oncotarget. 8, 45943–45950. doi: 10.18632/oncotarget.17500
Lemoine, M., Younes, A. (2010). Histone deacetylase inhibitors in the treatment of lymphoma. Discov. Med. 10, 462–470.
Li, L. H., Olin, E. J., Buskirk, H. H., Reineke, L. M. (1970). Cytotoxicity and mode of action of 5-azacytidine on L1210 leukemia. Cancer Res. 30, 2760–2769.
Li, Y., Nagai, H., Ohno, T., Yuge, M., Hatano, S., Ito, E., et al. (2002). Aberrant DNA methylation of p57 KIP2 gene in the promoter region in lymphoid malignancies of B-cell phenotype. Blood. 100, 2572–2577. doi: 10.1182/blood-2001-11-0026
Lohr, J. G., Stojanov, P., Lawrence, M. S., Auclair, D., Chapuy, B., Sougnez, C., et al. (2012). Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc. Natl. Acad. Sci. U. S. A. 109, 3879–3884. doi: 10.1073/pnas.1121343109
Losman, J.-A., Looper, R., Koivunen, P., Lee, S., Schneider, R. K., McMahon, C., et al. (2013). (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science (80-. ). 339, 1621–1625. doi: 10.1126/science.1231677
Lovén, J., Hoke, H. A., Lin, C. Y., Lau, A., Orlando, D. A., Vakoc, C. R., et al. (2013). Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 153, 320–334. doi: 10.1016/j.cell.2013.03.036
Lu, X., Fernando, T. M., Lossos, C., Yusufova, N., Liu, F., Fontán, L., et al. (2018). PRMT5 interacts with the BCL6 oncoprotein and is required for germinal center formation and lymphoma cell survival. Blood 132, 2026–2039. doi: 10.1182/blood-2018-02-831438
Lübbert, M., Suciu, S., Hagemeijer, A., Rüter, B., Platzbecker, U., Giagounidis, A., et al. (2016). Decitabine improves progression-free survival in older high-risk MDS patients with multiple autosomal monosomies: results of a subgroup analysis of the randomized phase III study 06011 of the EORTC Leukemia cooperative group and german MDS study group. Ann. Hematol. 95, 191–199. doi: 10.1007/s00277-015-2547-0
Lue, J. K., Prabhu, S. A., Liu, Y., Gonzalez, Y., Verma, A., Mundi, P. S., et al. (2019). Precision targeting with EZH2 and HDAC inhibitors in epigenetically dysregulated lymphomas. Clin. Cancer Res. 25, 5271–5283. doi: 10.1158/1078-0432.CCR-18-3989
Lue, J. K., Prabhu, S. A., Liu, Y., Verma, A., Elemento, O., Amengual, J. E. (2017). Dual inhibition of EZH2 and HDAC is synergistic in EZH2 Dysregulated Lymphomas. Hematol. Oncol. 35, 254–255. doi: 10.1002/hon.2438_120
Mai, A., Massa, S., Rotili, D., Cerbara, I., Valente, S., Pezzi, R., et al. (2005). Histone deacetylation in epigenetics: an attractive target for anticancer therapy. Med. Res. Rev. 25, 261–309. doi: 10.1002/med.20024
Maly, J. J., Christian, B. A., Zhu, X., Wei, L., Sexton, J. L., Jaglowski, S. M., et al. (2017). A phase I/II trial of panobinostat in combination with lenalidomide in patients with relapsed or refractory Hodgkin lymphoma. Clin. Lymphoma Myeloma Leuk. 17, 347–353. doi: 10.1016/j.clml.2017.05.008
Mann, B. S., Johnson, J. R., Cohen, M. H., Justice, R., Pazdur, R. (2007). FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 12, 1247–1252. doi: 10.1634/theoncologist.12-10-1247
Marjon, K., Cameron, M. J., Quang, P., Clasquin, M. F., Mandley, E., Kunii, K., et al. (2016). MTAP deletions in cancer create vulnerability to targeting of the MAT2A/PRMT5/RIOK1 axis. Cell Rep. 15, 574–587. doi: 10.1016/j.celrep.2016.03.043
Marquard, L., Poulsen, C. B., Gjerdrum, L. M., de Nully Brown, P., Christensen, I. J., Jensen, P. B., et al. (2009). Histone deacetylase 1, 2, 6 and acetylated histone H4 in B- and T-cell lymphomas. Histopathology 54, 688–698. doi: 10.1111/j.1365-2559.2009.03290.x
Maruyama, D., Tobinai, K., Makita, S., Ishida, T., Kusumoto, S., Ishitsuka, K., et al. (2017). First-in-human study of the EZH1/2 dual inhibitor DS-3201b in patients with relapsed or refractory non-Hodgkin lymphomas — Preliminary results. Blood. 130, 4070.
McCabe, M. T., Graves, A. P., Ganji, G., Diaz, E., Halsey, W. S., Jiang, Y., et al. (2012a). Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc. Natl. Acad. Sci. U. S. A. 109, 2989–2994. doi: 10.1073/pnas.1116418109
McCabe, M. T., Ott, H. M., Ganji, G., Korenchuk, S., Thompson, C., Van Aller, G. S., et al. (2012b). EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 492, 108–112. doi: 10.1038/nature11606
McClure, J. J., Li, X., Chou, C. J. (2018). Advances and challenges of HDAC inhibitors in cancer therapeutics. Adv. Cancer Res. 183–211. doi: 10.1016/bs.acr.2018.02.006
McGarvey, K. M., Fahrner, J. A., Greene, E., Martens, J., Jenuwein, T., Baylin, S. B. (2006). Silenced tumor suppressor genes reactivated by DNA demethylation do not return to a fully euchromatic chromatin state. Cancer Res. 66, 3541–3549. doi: 10.1158/0008-5472.CAN-05-2481
Meissner, A. (2010). Epigenetic modifications in pluripotent and differentiated cells. Nat. Biotechnol. 28, 1079–1088. doi: 10.1038/nbt.1684
Migliori, V., Phalke, S., Bezzi, M., Guccione, E. (2010). Arginine/lysine-methyl/methyl switches: biochemical role of histone arginine methylation in transcriptional regulation. Epigenomics 2, 119–137. doi: 10.2217/epi.09.39
Minucci, S., Pelicci, P. G. (2006). Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 6, 38–51. doi: 10.1038/nrc1779
Mirguet, O., Gosmini, R., Toum, J., Clément, C. A., Barnathan, M., Brusq, J.-M., et al. (2013). Discovery of epigenetic regulator I-BET762: lead optimization to afford a clinical candidate inhibitor of the BET bromodomains. J. Med. Chem. 56, 7501–7515. doi: 10.1021/jm401088k
Montalban-Bravo, G., DiNardo, C. D. (2018). The role of IDH mutations in acute myeloid leukemia. Futur. Oncol. 14, 979–993. doi: 10.2217/fon-2017-0523
Morin, R. D., Johnson, N. A., Severson, T. M., Mungall, A. J., An, J., Goya, R., et al. (2010). Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 42, 181–185. doi: 10.1038/ng.518
Morin, R. D., Mendez-Lago, M., Mungall, A. J., Goya, R., Mungall, K. L., Corbett, R. D., et al. (2011). Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 476, 298–303. doi: 10.1038/nature10351
Moros, A., Rodríguez, V., Saborit-Villarroya, I., Montraveta, A., Balsas, P., Sandy, P., et al. (2014). Synergistic antitumor activity of lenalidomide with the BET bromodomain inhibitor CPI203 in bortezomib-resistant mantle cell lymphoma. Leukemia 28, 2049–2059. doi: 10.1038/leu.2014.106
Morschhauser, F., Terriou, L., Coiffier, B., Bachy, E., Varga, A., Kloos, I., et al. (2015). Phase 1 study of the oral histone deacetylase inhibitor abexinostat in patients with Hodgkin lymphoma, non-Hodgkin lymphoma, or chronic lymphocytic leukaemia. Invest. New Drugs 33, 423–431. doi: 10.1007/s10637-015-0206-x
Mullighan, C. G., Zhang, J., Kasper, L. H., Lerach, S., Payne-Turner, D., Phillips, L. A., et al. (2011). CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature. 471, 235–239. doi: 10.1038/nature09727
Munshi, P. N., Lubin, M., Bertino, J. R. (2014). 6-Thioguanine: a drug with unrealized potential for cancer therapy. Oncologist. 19, 760–765. doi: 10.1634/theoncologist.2014-0178
Newman, E., Morgan, R. J., Kummar, S., Beumer, J. H., Blanchard, M. S., Ruel, C., et al. (2015). A phase I, pharmacokinetic, and pharmacodynamic evaluation of the DNA methyltransferase inhibitor 5-fluoro-2’2′-deoxycytidine, administered with tetrahydrouridine. Cancer Chemother. Pharmacol. 75, 537–547.
Normant, E., Cummings, R., Bellon, S., Bailey, C., Albrecht, B., Hewitt, M., et al. (2012). “Abstract LB-237: in vitro and in vivo characterization of CPI-267203, a potent inhibitor of bromodomain-containing proteins,” in Experimental and molecular therapeutics (American Association for Cancer Research), LB–237-LB-237. doi: 10.1158/1538-7445.AM2012-LB-237
Ogura, M., Ando, K., Suzuki, T., Ishizawa, K., Oh, S. Y., Itoh, K., et al. (2014). A multicentre phase II study of vorinostat in patients with relapsed or refractory indolent B-cell non-Hodgkin lymphoma and mantle cell lymphoma. Br. J. Haematol. 165, 768–776. doi: 10.1111/bjh.12819
Oki, Y., Buglio, D., Fanale, M., Fayad, L., Copeland, A., Romaguera, J., et al. (2013). Phase I study of panobinostat plus everolimus in patients with relapsed or refractory lymphoma. Clin. Cancer Res. 19, 6882–6890. doi: 10.1158/1078-0432.CCR-13-1906
Oki, Y., Kelly, K. R., Flinn, I., Patel, M. R., Gharavi, R., Ma, A., et al. (2017). CUDC- 907 in relapsed/refractory diffuse large B-cell lymphoma, including patients with MYC-alterations: Results from an expanded phase I trial. Haematologica 102, 1923–1930. doi: 10.3324/haematol.2017.172882
Okosun, J., Bödör, C., Wang, J., Araf, S., Yang, C.-Y., Pan, C., et al. (2014). Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat Genet. 46, 176–181. doi: 10.1038/ng.2856
Olsen, E. A., Kim, Y. H., Kuzel, T. M., Pacheco, T. R., Foss, F. M., Parker, S., et al. (2007). Phase IIB multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J. Clin. Oncol. 25, 3109–3115. doi: 10.1200/JCO.2006.10.2434
Oronsky, B., Paulmurugan, R., Foygel, K., Scicinski, J., Knox, S. J., Peehl, D., et al. (2017). RRx-001: a systemically non-toxic M2-to-M1 macrophage stimulating and prosensitizing agent in phase II clinical trials. Expert Opin. Investig. Drugs. 26, 109–119. doi: 10.1080/13543784.2017.1268600
Ozer, H. G., El-Gamal, D., Powell, B., Hing, Z. A., Blachly, J. S., Harrington, B., et al. (2018). BRD4 profiling identifies critical chronic lymphocytic leukemia oncogenic circuits and reveals sensitivity to PLX51107, a novel structurally distinct BET inhibitor. Cancer Discov. 8, 458–477. doi: 10.1158/2159-8290.CD-17-0902
Padmanabhan, B., Mathur, S., Manjula, R., Tripathi, S. (2016). Bromodomain and extra-terminal (BET) family proteins: new therapeutic targets in major diseases. J. Biosci. 41, 295–311. doi: 10.1007/s12038-016-9600-6
Parry, M., Rose-Zerilli, M. J. J., Ljungström, V., Gibson, J., Wang, J., Walewska, R., et al. (2015). Genetics and prognostication in splenic marginal zone lymphoma: revelations from deep sequencing. Clin. Cancer Res. 21, 4174–4183. doi: 10.1158/1078-0432.CCR-14-2759
Pasqualucci, L., Dominguez-Sola, D., Chiarenza, A., Fabbri, G., Grunn, A., Trifonov, V., et al. (2011a). Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 471, 189–195. doi: 10.1038/nature09730
Pasqualucci, L., Trifonov, V., Fabbri, G., Ma, J., Rossi, D., Chiarenza, A., et al. (2011b). Analysis of the coding genome of diffuse large B-cell lymphoma. Nat. Genet. 43, 830–837. doi: 10.1038/ng.892
Pazin, M. J., Kadonaga, J. T. (1997). SWI2/SNF2 and related proteins: ATP-driven motors that disrupt protein- –DNA interactions? Cell. 88, 737–740. doi: 10.1016/S0092-8674(00)81918-2
Persky, D. O., Li, H., Rimsza, L. M., Barr, P. M., Popplewell, L. L., Bane, C. L., et al. (2018). A phase I/II trial of vorinostat (SAHA) in combination with rituximab-CHOP in patients with newly diagnosed advanced stage diffuse large B-cell lymphoma (DLBCL): SWOG S0806. Am. J. Hematol. 93, 486–493. doi: 10.1002/ajh.25010
Popovic, R., Shah, M. Y., Licht, J. D. (2013). Epigenetic therapy of hematological malignancies: Where are we now? Ther. Adv. Hematol. 4, 81–91. doi: 10.1177/2040620712466864
Qi, W., Chan, H., Teng, L., Li, L., Chuai, S., Zhang, R., et al. (2012). Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc. Natl. Acad. Sci. 109, 21360–21365. doi: 10.1073/pnas.1210371110
Qin, C., Hu, Y., Zhou, B., Fernandez-Salas, E., Yang, C.-Y., Liu, L., et al. (2018). Discovery of QCA570 as an exceptionally potent and efficacious proteolysis targeting chimera (PROTAC) degrader of the bromodomain and extra-terminal (BET) proteins capable of inducing complete and durable tumor regression. J. Med. Chem. 61, 6685–6704. doi: 10.1021/acs.jmedchem.8b00506
Qiu, L., Burgess, A., Fairlie, D. P., Leonard, H., Parsons, P. G., Gabrielli, B. G. (2000). Histone deacetylase inhibitors trigger a G2 checkpoint in normal cells that is defective in tumor cells. Mol. Biol. Cell 11, 2069–2083. doi: 10.1091/mbc.11.6.2069
Raina, K., Lu, J., Qian, Y., Altieri, M., Gordon, D., Rossi, A. M. K., et al.. (2016). PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. U. S. A. 113, 7124–7129. doi: 10.1073/pnas.1521738113
Rebel, V. I., Kung, A. L., Tanner, E. A., Yang, H., Bronson, R. T., Livingston, D. M. (2002). Distinct roles for CREB-binding protein and p300 in hematopoietic stem cell self-renewal. Proc. Natl. Acad. Sci. 99, 14789–14794. doi: 10.1073/pnas.232568499
Recasens-zorzo, C., Cardesa-salzmann, T., Petazzi, P., Ros-blanco, L., Esteve-arenys, A., Clot, G., et al. (2018). Pharmacological modulation of CXCR4 cooperates with BET bromodomain inhibition in diffuse large B-cell lymphoma. Haematologica 104, 778–788. doi: 10.3324/haematol.2017.180505
Rhyasen, G. W., Hattersley, M. M., Yao, Y., Dulak, A., Wang, W., Petteruti, P., et al. (2016). AZD5153: a novel bivalent BET bromodomain inhibitor highly active against hematologic malignancies. Mol. Cancer Ther. 15, 2563–2574. doi: 10.1158/1535-7163.MCT-16-0141
Ribrag, V., Kim, W. S., Bouabdallah, R., Lim, S. T., Coiffier, B., Illes, A., et al. (2017). Safety and efficacy of abexinostat, a pan-histone deacetylase inhibitor, in non-Hodgkin lymphoma and chronic lymphocytic leukemia: Results of a phase II study. Haematologica 102, 903–909. doi: 10.3324/haematol.2016.154377
Richardson, P. G., Hungria, V. T. M., Yoon, S. S., Beksac, M., Dimopoulos, M. A., Elghandour, A., et al. (2016). Panobinostat plus bortezomib and dexamethasone in previously treated multiple myeloma: Outcomes by prior treatment. Blood. 15, 2563–2574. doi: 10.1182/blood-2015-09-665018
Rinaldi, A., Mian, M., Chigrinova, E., Arcaini, L., Bhagat, G., Novak, U., et al.(2011). Genome-wide DNA profiling of marginal zone lymphomas identifies subtype-specific lesions with an impact on the clinical outcome. Blood 117, 1595–1604. doi: 10.1182/blood-2010-01-264275
Robaina, M. C., Mazzoccoli, L., Arruda, V. O., Reis, F. R., de, S., Apa, A. G., et al. (2015). Deregulation of DNMT1, DNMT3B and miR-29s in Burkitt lymphoma suggests novel contribution for disease pathogenesis. Exp. Mol. Pathol. 98, 200–207. doi: 10.1016/j.yexmp.2015.03.006
Roulland, S., Kasbekar, M., Young, R. M., Shaffer, A. L., Hodson, D. J., Xiao, W., et al. (2018). Genetics and pathogenesis of diffuse large B-cell lymphoma. N. Engl. Med. 378, 1396–1407. doi: 10.1056/NEJMoa1801445
Rozati, S., Cheng, P. F., Widmer, D. S., Fujii, K., Levesque, M. P., Dummer, R. (2016). Romidepsin and azacitidine synergize in their epigenetic modulatory effects to induce apoptosis in CTCL. Clin. Cancer Res. 22, 2020–2031. doi: 10.1158/1078-0432.CCR-15-1435
Sachchidanand, R-S, L., Yan, S., Mutjaba, S., Liu, W. J., Zeng, L., et al. (2006). Target structure-based discovery of small molecules that block human p53 and CREB binding protein association. Chem. Biol. 13, 81–90. doi: 10.1016/j.chembiol.2005.10.014
Saha, S. K., Parachoniak, C. A., Ghanta, K. S., Fitamant, J., Ross, K. N., Najem, M. S., et al. (2014). Mutant IDH inhibits HNF-4α to block hepatocyte differentiation and promote biliary cancer. Nature 513, 110–152. doi: 10.1038/nature13441
Samuels, B. L., Herndon, J. E., Harmon, D. C., Carey, R., Aisner, J., Corson, J. M., et al. (1998). Dihydro-5-azacytidine and cisplatin in the treatment of malignant mesothelioma: a phase II study by the Cancer and Leukemia Group B. Cancer. 82, 1578–1584. doi: 10.1002/(SICI)1097-0142(19980415)82:8<1578::AID-CNCR21>3.0.CO;2-0
Santen, G. W. E., Aten, E., Sun, Y., Almomani, R., Gilissen, C., Nielsen, M., et al. (2012). Mutations in SWI/SNF chromatin remodeling complex gene ARID1B cause Coffin-Siris syndrome. Nat. Genet. 44, 379–380. doi: 10.1038/ng.2217
Santo, L., Hideshima, T., Kung, A. L., Tseng, J.-C., Tamang, D., Yang, M., et al. (2012). Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood 119, 2579–2589. doi: 10.1182/blood-2011-10-387365
Santoro, F., Botrugno, O. A., Dal Zuffo, R., Pallavicini, I., Matthews, G. M., Cluse, L., et al. (2013). A dual role for Hdac1: oncosuppressor in tumorigenesis, oncogene in tumor maintenance. Blood 121, 3459–3468. doi: 10.1182/blood-2012-10-461988
Sausen, M., Leary, R. J., Jones, S., Wu, J., Reynolds, C. P., Liu, X., et al. (2013). Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat. Genet. 45, 12–17. doi: 10.1038/ng.2493
Schmitz, R., Young, R. M., Ceribelli, M., Jhavar, S., Xiao, W., Zhang, M., et al. (2012). Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 490, 116–120. doi: 10.1038/nature11378
Scholtysik, R., Kreuz, M., Hummel, M., Rosolowski, M., Szczepanowski, M., Klapper, W., et al. (2015). Characterization of genomic imbalances in diffuse large B-cell lymphoma by detailed SNP-chip analysis. Int. J. Cancer 136, 1033–1042. doi: 10.1002/ijc.29072
Schubert, H. L., Blumenthal, R. M., Cheng, X. (2003). Many paths to methyltransfer: a chronicle of convergence. Trends Biochem. Sci. 28, 329–335. doi: 10.1016/S0968-0004(03)00090-2
Schuh, A. C., Döhner, H., Pleyer, L., Seymour, J. F., Fenaux, P., Dombret, H. (2017). Azacitidine in adult patients with acute myeloid leukemia. Crit. Rev. Oncol. Hematol. 116, 159–177. doi: 10.1016/j.critrevonc.2017.05.010
Scott, D. W., Gascoyne, R. D. (2014). The tumour microenvironment in B cell lymphomas. Nat. Rev. Cancer 14, 517–534. doi: 10.1038/nrc3774
Scott, L. J. (2016). Azacitidine: A review in myelodysplastic syndromes and acute myeloid leukaemia. Drugs. 14, 517–534. doi: 10.1007/s40265-016-0585-0
Seifert, M., Scholtysik, R., Küppers, R., (2013). “Origin and pathogenesis of B cell lymphomas,” in Methods Mol. Biol. (Clifton, N.J.), 1–25. doi: 10.1007/978-1-62703-269-8_1
Shahbazian, M. D., Grunstein, M. (2007). Functions of site-specific histone acetylation and deacetylation. Annu. Rev. Biochem. 76, 75–100. doi: 10.1146/annurev.biochem.76.052705.162114
Shaknovich, R., Cerchietti, L., Tsikitas, L., Kormaksson, M., De, S., Figueroa, M. E., et al. (2011). DNA methyltransferase 1 and DNA methylation patterning contribute to germinal center B-cell differentiation. Blood. 118, 3559–3569. doi: 10.1182/blood-2011-06-357996
Shaknovich, R., Geng, H., Johnson, N. A., Tsikitas, L., Cerchietti, L., Greally, J. M., et al. (2010). DNA methylation signatures define molecular subtypes of diffuse large B-cell lymphoma. Blood. 116, e81–89. doi: 10.1182/blood-2010-05-285320
Sharma, P., Allison, J. P. (2015). Immune checkpoint targeting in cancer therapy: Toward combination strategies with curative potential. Cell. 161, 205–214. doi: 10.1016/j.cell.2015.03.030
Sheppard, G. S., Wang, L., Fidanze, S. D., Hasvold, L. A., Liu, D., Pratt, J. K., et al. (2018). “Abstract 931: Discovery of ABBV-744, a first-in-class highly BDII-selective BET bromodomain inhibitor,” in Cancer chemistry (American Association for Cancer Research), 931–931. doi: 10.1158/1538-7445.AM2018-931
Shi, J., Whyte, W. A., Zepeda-Mendoza, C. J., Milazzo, J. P., Shen, C., Roe, J. S., et al. (2013). Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev. 27, 2648–2662. doi: 10.1101/gad.232710.113
Shilatifard, A. (2008). Molecular implementation and physiological roles for histone H3 lysine 4 (H3K4) methylation. Curr. Opin. Cell Biol. 20, 341–348. doi: 10.1016/j.ceb.2008.03.019
Silverman, L. R., Demakos, E. P., Peterson, B. L., Kornblith, A. B., Holland, J. C., Odchimar-Reissig, R., et al. (2002). Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: A study of the cancer and leukemia group B. J. Clin. Oncol. 20, 2429–2440. doi: 10.1200/JCO.2002.04.117
Smith, E., Zhou, W., Shindiapina, P., Sif, S., Li, C., Baiocchi, R. A. (2018). Recent advances in targeting protein arginine methyltransferase enzymes in cancer therapy. Expert Opin. Ther. Targets 22, 527–545. doi: 10.1080/14728222.2018.1474203
Smith, S. G., Zhou, M.-M. (2016). The bromodomain: A new target in emerging epigenetic medicine. ACS Chem. Biol. 11, 598–608. doi: 10.1021/acschembio.5b00831
Srivastava, P., Paluch, B. E., Matsuzaki, J., James, S. R., Collamat-Lai, G., Taverna, P., et al. (2015). Immunomodulatory action of the DNA methyltransferase inhibitor SGI-110 in epithelial ovarian cancer cells and xenografts. Epigenetics. 10, 237–246. doi: 10.1080/15592294.2015.1017198
Stathis, A., Hotte, S. J., Chen, E. X., Hirte, H. W., Oza, A. M., Moretto, P., et al. (2011). Phase I study of decitabine in combination with vorinostat in patients with advanced solid tumors and non-Hodgkin’s lymphomas. Clin. Cancer Res. 17, 1582–1590. doi: 10.1158/1078-0432.CCR-10-1893
Stein, E. M., Garcia-Manero, G., Rizzieri, D. A., Tibes, R., Berdeja, J. G., Savona, M. R., et al. (2018). The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood. 131, 2661–2669. doi: 10.1182/blood-2017-12-818948
Stewart, D. J., Issa, J. P., Kurzrock, R., Nunez, M. I., Jelinek, J., Hong, D., et al. (2009). Decitabine effect on tumor global DNA methylation and other parameters in a phase I trial in refractory solid tumors and lymphomas. Clin. Cancer Res. 15, 3881–3888. doi: 10.1158/1078-0432.CCR-08-2196
Straus, D. J., Hamlin, P. A., Matasar, M. J., Lia Palomba, M., Drullinsky, P. R., Zelenetz, A. D., et al.. (2015). Phase I/II trial of vorinostat with rituximab, cyclophosphamide, etoposide and prednisone as palliative treatment for elderly patients with relapsed or refractory diffuse large B-cell lymphoma not eligible for autologous stem cell transplantation. Br. J. Haematol. 168, 663–670. doi: 10.1111/bjh.13195
Streubel, B., Simonitsch-Klupp, I., Müllauer, L., Lamprecht, A., Huber, D., Siebert, R., et al. (2004). Variable frequencies of MALT lymphoma-associated genetic aberrations in MALT lymphomas of different sites. Leukemia 18, 1722–1726. doi: 10.1038/sj.leu.2403501
Sun, B., Fiskus, W., Qian, Y., Rajapakshe, K., Raina, K., Coleman, K. G., et al. (2018). BET protein proteolysis targeting chimera (PROTAC) exerts potent lethal activity against mantle cell lymphoma cells. Leukemia 32, 343–352. doi: 10.1038/leu.2017.207
Sun, B., Shah, B., Fiskus, W., Qi, J., Rajapakshe, K., Coarfa, C., et al. (2015). Synergistic activity of BET protein antagonist-based combinations in mantle cell lymphoma cells sensitive or resistant to ibrutinib. Blood 126, 1565–1574. doi: 10.1182/blood-2015-04-639542
Suraweera, A., O’Byrne, K. J., Richard, D. J. (2018). Combination therapy with histone deacetylase inhibitors (HDACi) for the Treatment of cancer: achieving the full therapeutic potential of HDACi. Front. Oncol. 8, 92. doi: 10.3389/fonc.2018.00092
Swerdlow, S. H., Campo, E., Pileri, S. A., Harris, N. L., Stein, H., Siebert, R., et al. (2016). The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 127, 2375–2390. doi: 10.1182/blood-2016-01-643569
Tanaka, M., Roberts, J. M., Seo, H.-S., Souza, A., Paulk, J., Scott, T. G., et al. (2016). Design and characterization of bivalent BET inhibitors. Nat. Chem. Biol. 12, 1089–1096. doi: 10.1038/nchembio.2209
Tang, J.-Y., Chang, C.-C., Lin, P.-C., Chang, J.-G. (2012). Isocitrate dehydrogenase mutation hot spots in acute lymphoblastic leukemia and oral cancer. Kaohsiung J. Med. Sci. 28, 138–144. doi: 10.1016/j.kjms.2011.10.023
The Cancer Genome Atlas Research Network (2013). The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 45, 1113–1120. doi: 10.1038/ng.2764
Thottassery, J. V., Sambandam, V., Allan, P. W., Maddry, J. A., Maxuitenko, Y. Y., Tiwari, K., et al. (2014). Novel DNA methyltransferase-1 (DNMT1) depleting anticancer nucleosides, 4′-thio-2′-deoxycytidine and 5-aza-4′-thio-2′- deoxycytidine. Cancer Chemother. Pharmacol. 74, 291–302. doi: 10.1007/s00280-014-2503-z
Tiacci, E., Trifonov, V., Schiavoni, G., Holmes, A., Kern, W., Martelli, M. P., et al. (2011). BRAF mutations in hairy-cell leukemia. N. Engl. J. Med. 364, 2305–2315. doi: 10.1056/NEJMoa1014209
Topalian, S. L., Drake, C. G., Pardoll, D. M. (2015). Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 27, 450–461. doi: 10.1016/j.ccell.2015.03.001
Toujani, S., Dessen, P., Ithzar, N., Danglot, G., Richon, C., Vassetzky, Y., et al. (2009). High resolution genome-wide analysis of chromosomal alterations in Burkitt’s lymphoma. PLoS One 4, e7089. doi: 10.1371/journal.pone.0007089
Vakoc, C. R. (2015). “BET bromodomain inhibition as a therapeutic approach in hematological malignancies,” in Histone recognition. Ed. Zhou, M. M. (Heidelberg, Germany: Springer), 185–198. doi: 10.1007/978-3-319-18102-8_9
Valdez, B. C., Li, Y., Murray, D., Liu, Y., Nieto, Y., Champlin, R. E., et al. (2018). Combination of a hypomethylating agent and inhibitors of PARP and HDAC traps PARP1 and DNMT1 to chromatin, acetylates DNA repair proteins, down-regulates NuRD and induces apoptosis in human leukemia and lymphoma cells. Oncotarget 9, 3908–3921. doi: 10.18632/oncotarget.23386
Van Damme, M., Crompot, E., Meuleman, N., Maerevoet, M., Mineur, P., Bron, D., et al. (2016). Characterization of TET and IDH gene expression in chronic lymphocytic leukemia: comparison with normal B cells and prognostic significance. Clin. Epigenetics 8, 1–11. doi: 10.1186/s13148-016-0298-y
Van Kemenade, F. J., Raaphorst, F. M., Blokzijl, T., Fieret, E., Hamer, K. M., Satijn, D. P. E., et al. (2001). Coexpression of BMI-1 and EZH2 polycomb-group proteins is associated with cycling cells and degree of malignancy in B-cell non-Hodgkin lymphoma. Blood. 97, 3896–3901. doi: 10.1182/blood.V97.12.3896
Vannini, A., Volpari, C., Filocamo, G., Casavola, E. C., Brunetti, M., Renzoni, D., et al. (2004). Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc. Natl. Acad. Sci. U. S. A. 101, 15064–15069. doi: 10.1073/pnas.0404603101
Vaswani, R. G., Gehling, V. S., Dakin, L. A., Cook, A. S., Nasveschuk, C. G., Duplessis, M., et al. (2016). Identification of (R)-N-((4-Methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1-(1-(1-(2,2,2-trifluoroethyl)piperidin-4-yl)ethyl)-1H-indole-3-carboxamide (CPI-1205), a potent and selective inhibitor of histone methyltransferase EZH2, Suitabl. J. Med. Chem. 59, 9928–9941. doi: 10.1021/acs.jmedchem.6b01315
Velichutina, I., Shaknovich, R., Geng, H., Johnson, N. A., Gascoyne, R. D., Melnick, A. M., et al. (2010). EZH2-mediated epigenetic silencing in germinal center B cells contributes to proliferation and lymphomagenesis. Blood. 116, 5247–5255. doi: 10.1182/blood-2010-04-280149
Visser, H. P. J., Gunster, M. J. (2001). “The Polycomb group protein EZH2 is upregulated in proliferating, cultured human mantle cell lymphoma - Visser - 2001,” in Br. J. of Haematol. Mar; 112 (4), 950–958. (Wiley Online Library. Br. J). doi: 10.1046/j.1365-2141.2001.02641.x
Vogler, W. R., Miller, D. S., Keller, J. W. (1976). 5 Azacytidine (NSC 102816): A new drug for the treatment of myeloblastic leukemia. Blood. 48, 331–337.
Wang, K., Kan, J., Yuen, S. T., Shi, S. T., Chu, K. M., Law, S., et al. (2011). Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat. Genet. 43, 1219–1223. doi: 10.1038/ng.982
Wang, L., Pal, S., Sif, S. (2008). Protein arginine methyltransferase 5 suppresses the transcription of the RB family of tumor suppressors in leukemia and lymphoma cells. Mol. Cell. Biol. 28, 6262–6277. doi: 10.1128/MCB.00923-08
Wang, W., Côté, J., Xue, Y., Zhou, S., Khavari, P. A., Biggar, S. R., et al. (1996). Purification and biochemical heterogeneity of the mammalian SWI—SNF complex. EMBO J. 15, 5370–5382. doi: 10.1002/j.1460-2075.1996.tb00921.x
Wang, X., Nagl, N. G., Wilsker, D., Van Scoy, M., Pacchione, S., Yaciuk, P., et al. (2004). Two related ARID family proteins are alternative subunits of human SWI/SNF complexes. Biochem. J. 383, 319–325. doi: 10.1042/BJ20040524
Watanabe, T., Kato, H., Kobayashi, Y., Yamasaki, S., Morita-Hoshi, Y., Yokoyama, H., et al. (2010). Potential efficacy of the oral histone deacetylase inhibitor vorinostat in a phase I trial in follicular and mantle cell lymphoma. Cancer Sci. 101, 196–200. doi: 10.1111/j.1349-7006.2009.01360.x
Waterfall, J. J., Arons, E., Walker, R. L., Pineda, M., Roth, L., Killian, J. K., et al. (2013). High prevalence of MAP2K1 mutations in variant and IGHV4-34-expressing hairy-cell leukemias. Nat. Genet. 46, 8–10. doi: 10.1038/ng.2828
Winter, G. E., Mayer, A., Buckley, D. L., Erb, M. A., Roderick, J. E., Vittori, S., et al. (2017). BET bromodomain proteins function as master transcription elongation factors independent of CDK9 recruitment. Mol. Cell 67, 5–18.e19. doi: 10.1016/j.molcel.2017.06.004
Wong, Y. F., Jakt, L. M., Nishikawa, S. I. (2013). Prolonged treatment with DNMT inhibitors Induces distinct effects in promoters and gene-bodies. PLoS One. 8, e71099. doi: 10.1371/journal.pone.0071099
Workman, J. L., Kingston, R. E. (2002). Alteration of nucleosome structure as a mechanism of transcriptional regulation. Annu. Rev. Biochem. 67, 545–579. doi: 10.1146/annurev.biochem.67.1.545
Xargay-Torrent, S., López-Guerra, M., Saborit-Villarroya, I., Rosich, L., Campo, E., Roué, G., et al. (2011). Vorinostat-induced apoptosis in mantle cell lymphoma is mediated by acetylation of proapoptotic BH3-only gene promoters. Clin. Cancer Res. 17, 3956–3968. doi: 10.1158/1078-0432.CCR-10-3412
Xiao, B., Wilson, J. R., Gamblin, S. J. (2003). SET domains and histone methylation. Curr. Opin. Struct. Biol. 13, 699–705. doi: 10.1016/j.sbi.2003.10.003
Xue, K., Gu, J. J., Zhang, Q., Mavis, C., Hernandez-Ilizaliturri, F. J., Czuczman, M. S., et al. (2016). Vorinostat, a histone deacetylase (HDAC) inhibitor, promotes cell cycle arrest and re-sensitizes rituximab- and chemo-resistant lymphoma cells to chemotherapy agents. J. Cancer Res. Clin. Oncol. 142, 379–387. doi: 10.1007/s00432-015-2026-y
Yang, D., Zhang, W.-P., Yang, J.-M., He, M.-X., Cheng, C., Chen, J. (2018). Secondary skin involvement in gastric diffuse large B-cell lymphoma treated with chidamide: A case report. Medicine (Baltimore) 97, e13093. doi: 10.1097/MD.0000000000013093
Yang, X., Han, H., DeCarvalho, D. D., Lay, F. D., Jones, P. A., Liang, G. (2014). Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell. 26, 577–590. doi: 10.1016/j.ccr.2014.07.028
Yap, T. A., Johnson, P. W. M., Winter, J., Leonard, J., Giulino-Roth, L., Horner, T., et al.. (2018). A phase I, open-label study of GSK2816126, an enhancer of zeste homolog 2 (EZH2) inhibitor, in patients with relapsed/refractory diffuse large B-cell lymphoma (DLBCL), transformed follicular lymphoma (tFL), other non-Hodgkin’s lymphomas (NHL), multiple my. J. Clin. Oncol. 34, TPS2595. doi: 10.1200/jco.2016.34.15_suppl.tps2595
Yeoh, E.-J., Ross, M. E., Shurtleff, S. A., Williams, W. K., Patel, D., Mahfouz, R., et al.. (2002). Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell 1, 133–143. doi: 10.1016/S1535-6108(02)00032-6
Yoshimatsu, M., Toyokawa, G., Hayami, S., Unoki, M., Tsunoda, T., Field, H. I., et al. (2011). Dysregulation of PRMT1 and PRMT6, type I arginine methyltransferases, is involved in various types of human cancers. Int. J. Cancer 128, 562–573. doi: 10.1002/ijc.25366
You, J. S., Jones, P. A. (2012). Cancer genetics and epigenetics: Two sides of the same coin? Cancer Cell. 22, 9–20. doi: 10.1016/j.ccr.2012.06.008
Younes, A., Berdeja, J. G., Patel, M. R., Flinn, I., Gerecitano, J. F., Neelapu, S. S., et al. (2016). Safety, tolerability, and preliminary activity of CUDC-907, a first-in-class, oral, dual inhibitor of HDAC and PI3K, in patients with relapsed or refractory lymphoma or multiple myeloma: An open-label, dose-escalation, phase 1 trial. Lancet Oncol. 17, 622–631. doi: 10.1016/S1470-2045(15)00584-7
Younes, A., Oki, Y., Bociek, R. G., Kuruvilla, J., Fanale, M., Neelapu, S., et al. (2011). Mocetinostat for relapsed classical Hodgkin’s lymphoma: An open-label, single-arm, phase 2 trial. Lancet Oncol. 12, 1222–1228. doi: 10.1016/S1470-2045(11)70265-0
Younes, A., Sureda, A., Ben-Yehuda, D., Zinzani, P. L., Ong, T.-C., Prince, H. M., et al. (2012). Panobinostat in patients with relapsed/refractory Hodgkin’s lymphoma after autologous stem-cell transplantation: Results of a phase II study. J. Clin. Oncol. 30, 2197–2203. doi: 10.1200/JCO.2011.38.1350
Yuan, B., Zhang, J., Wang, H., Xiong, L., Cai, Q., Wang, T., et al. (2011). 6-Thioguanine reactivates epigenetically silenced genes in acute lymphoblastic leukemia cells by facilitating proteasome-mediated degradation of DNMT. Cancer Res. 71, 1904–1911. doi: 10.1158/0008-5472.CAN-10-3430
Zeng, D., Liu, M., Pan, J. (2016). Blocking EZH2 methylation transferase activity by GSK126 decreases stem cell-like myeloma cells. Oncotarget. 8, 3396–3411. doi: 10.18632/oncotarget.13773
Zeng, L., Li, J., Muller, M., Yan, S., Mujtaba, S., Pan, C., et al. (2005). Selective small molecules blocking HIV-1 Tat and coactivator PCAF association. J. Am. Chem. Soc. 127, 2376–2377. doi: 10.1021/ja044885g
Zhang, Y., Wei, H., Tang, K., Lin, D., Zhang, C., Mi, Y., et al. (2012). Mutation analysis of isocitrate dehydrogenase in acute lymphoblastic leukemia. Genet. Test. Mol. Biomarkers 16, 991–995. doi: 10.1089/gtmb.2011.0323
Zhao, H., Ning, S., Nolley, R., Scicinski, J., Oronsky, B., Knox, S. J., et al. (2017). The immunomodulatory anticancer agent, RRx-001, induces an interferon response through epigenetic induction of viral mimicry. Clin. Epigenetics. 9, 4. doi: 10.1186/s13148-017-0312-z
Keywords: B-cell lymphoma, DNMT, EZH2, HDAC, PRMT inhibitor, BET bromodomain inhibitor (BETi), combination therapy
Citation: Ribeiro ML, Reyes-Garau D, Armengol M, Fernández-Serrano M and Roué G (2019) Recent Advances in the Targeting of Epigenetic Regulators in B-Cell Non-Hodgkin Lymphoma. Front. Genet. 10:986. doi: 10.3389/fgene.2019.00986
Received: 11 June 2019; Accepted: 17 September 2019;
Published: 16 October 2019.
Edited by:
Jiucun Wang, Fudan University, ChinaReviewed by:
Naoko Hattori, National Cancer Center Research Institute (Japan), JapanCopyright © 2019 Ribeiro, Reyes-Garau, Armengol, Fernández-Serrano and Roué. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marcelo L. Ribeiro, bWxyaWJlaXJvQHZoaW8ubmV0; Gaël Roué, Z3JvdWVAdmhlYnJvbi5uZXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.