 Sigrid Le Clerc1
Sigrid Le Clerc1 Sophie Limou
Sophie Limou Jean-François Zagury
Jean-François Zagury
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet. , 13 September 2019
Sec. Applied Genetic Epidemiology
Volume 10 - 2019 | https://doi.org/10.3389/fgene.2019.00799
This article is part of the Research Topic Host Genetics in Viral Pathogenesis and Controls View all 22 articles
In this review, we present the main large-scale experimental studies that have been performed in the HIV/AIDS field. These “omics” studies are based on several technologies including genotyping, RNA interference, and transcriptome or epigenome analysis. Due to the direct connection with disease evolution, there has been a large focus on genotyping cohorts of well-characterized patients through genome-wide association studies (GWASs), but there have also been several invitro studies such as small interfering RNA (siRNA) interference or transcriptome analyses of HIV-1–infected cells. After describing the major results obtained with these omics technologies—including some with a high relevance for HIV-1 treatment—we discuss the next steps that the community needs to embrace in order to derive new actionable therapeutic or diagnostic targets. Only integrative approaches that combine all big data results and consider their complex interactions will allow us to capture the global picture of HIV molecular pathogenesis. This novel challenge will require large collaborative efforts and represents a huge open field for innovative bioinformatics approaches.
HIV remains a major global public health issue, having claimed more than 35 million lives so far. In 2017, 940,000 people died from HIV-related causes globally (Global AIDS Update, 2016). The active anti-retroviral therapies are efficient and have saved many lives but still present multiple caveats: need for high compliance, permanent treatment, and unwanted side effects and complications (Li et al., 2017). Developing alternative and simple solutions such as immunoprophylactic or immunotherapeutic options remains a public health priority. In line with this, a better understanding of the molecular etiology of disease progression is essential.
Due to the impact of the disease in the Western world, HIV research has been the subject of intense efforts for the past 35 years and has helped in promoting several new research technologies, in particular with high-throughput studies from the “omics” era.
In this review, we will present large-scale studies based on various technologies that have been undertaken to tackle HIV molecular etiology and their main results. These large-scale studies encompass mainly genomics with genome-wide association studies (GWASs) based on genotyping chips and with exome sequencing, transcriptomic studies from SIV and HIV patient cells, and small interfering RNA (siRNA) studies in sensitive cell lines. We will also briefly describe the results obtained from proteomics and epigenomics screenings.
There are three successive stages in HIV infection: the acute primary infection, the asymptomatic stage and symptomatic HIV infection, and acquired immunodeficiency syndrome (AIDS). Depending on the individual, AIDS, the most advanced stage of the infection course, can occur within a few months to several years after HIV infection, with an average of around 8 years in the Western world. This stage has been defined by the Center for Disease Control (CDC) either as a drop of CD4 T-cell count below 200/mm3 or as the appearance of opportunistic infections or some cancers (Center for Disease Control and Prevention, 1992). Quite early, AIDS cohorts were enrolled and prospectively followed, and it became apparent that this infection was exhibiting a considerable phenotypic heterogeneity at different levels: virus acquisition, disease progression, viral load control, and response to treatment (Langlade-Demoyen et al., 1994; Ludlam et al., 1985; Fowke et al., 1996; Pantaleo and Fauci, 1996; Hetherington et al., 2002; Mallal et al., 2002; Saksena et al., 2007). For instance, some individuals, called long-term non-progressors, are infected but never progress to AIDS; the elite controllers have never exhibited any detectable viral load; and the rapid progressors reach the AIDS stage within a few months following their infection. This phenotypic variability may be attributed to a complex interplay between viral, environmental, and host genetic factors that could be investigated through several types of large-scale studies or omics.
If the CD4 cell count was the major marker to follow HIV-1 infection and immune deficiency in patients in the early 1980s, the progress of molecular biology techniques has made it possible to measure precisely HIV-1 viral load in the blood (i.e., the number of viral particles present in each ml of serum) by the late 1990s. Together with the CD4 cell count, this marker has become very useful to evaluate the status of an infected patient, either a low viral load suggesting a good control of HIV-1 infection or a high viral load suggesting a progressive infection at an early stage of infection or an uncontrolled infection at a late disease stage (AIDS). Most cohorts focus on viral load outcomes (e.g., viral control, viral load at set point), but (slow and rapid) progression phenotypes have also been defined based on the CD4 counts (e.g., the GRIV cohort).
Host genes associated with various phenotypes have been extensively explored since the mid-1990s. The concept is as follows: if a particular phenotype (for instance, elite control) is statistically associated with the presence or absence of a genetic variant, the corresponding gene or its product may be involved in the molecular mechanisms of viral infection/dissemination. Genetic association studies can thus provide new clues on the molecular mechanisms of infection and disease progression and, in the long run, identify new targets for the development of new therapeutic or diagnostic strategies. Initial studies have focused on candidate genes such as HLA (Kaslow et al., 1996; Carrington et al., 1999; Hendel et al., 1999), and a large number of host genetic associations with HIV outcomes have been identified. The main confirmed association was a 32-base-pair deletion in the CCR5 gene (CCR5-Δ32) (Dean et al., 1996; Liu et al., 1996; Samson et al., 1996; Rappaport et al., 1997), but other variants closely located in the CCR5 promoter (Martin et al., 1998; McDermott et al., 1998) or in the nearby CCR2 gene (Smith et al., 1997) were also influential. This deletion led to the expression of a truncated and non-functional cell surface CCR5 protein that happened to be the major HIV-1 entry co-receptor (Alkhatib et al., 1996; Deng et al., 1996; Liu et al., 1996). Other candidate gene studies pointed to immunity-related genes (e.g., KIR, IL10, IFNγ) and genes encoding HIV restriction factors (e.g., CCL5, APOBEC3G, CUL5) but were only partially replicated according to the phenotype and cohort tested. The functional interpretation for most of these variants is yet to be discovered, but the detailed account of each candidate gene is beyond the scope of this article and has been covered by previous enlightening reviews (Fellay, 2009; An and Winkler, 2010). Overall, most of the candidate gene associations displayed small to modest effect sizes and, combined all together, account for a small fraction of the phenotype variability (O’Brien and Nelson, 2004).
It was only in 2007 that the first large-scale genetic association studies or GWASs have been published with the seminal publication by Fellay et al. that focused mainly on viral load at set point as the main phenotype of interest (Fellay et al., 2007). These large-scale genomic studies have relied on genotyping chips targeting simultaneously hundreds of thousands to millions of specific genetic markers called single nucleotide polymorphisms (SNPs), the most frequent polymorphisms in the human genome, that can be rapidly and easily genotyped. In contrast to the candidate gene strategy, this approach measures and analyzes gene variants across the whole human genome in an effort to identify common genetic risk factors in the population without any biological hypothesis apriori. Since the 2007 publication, GWASs have taken the place of candidate gene studies in AIDS. More than 20 GWASs focusing on various phenotypes and cohorts have been published, and Table 1 summarizes these studies with their main characteristics (see also (Limou and Zagury, 2013)): date of the publication, origin of the cohort, size of the cohort, phenotype(s) of interest, genotyping chip type, associated SNP(s), best P-value, possible causing gene(s) involved, and publication reference number.
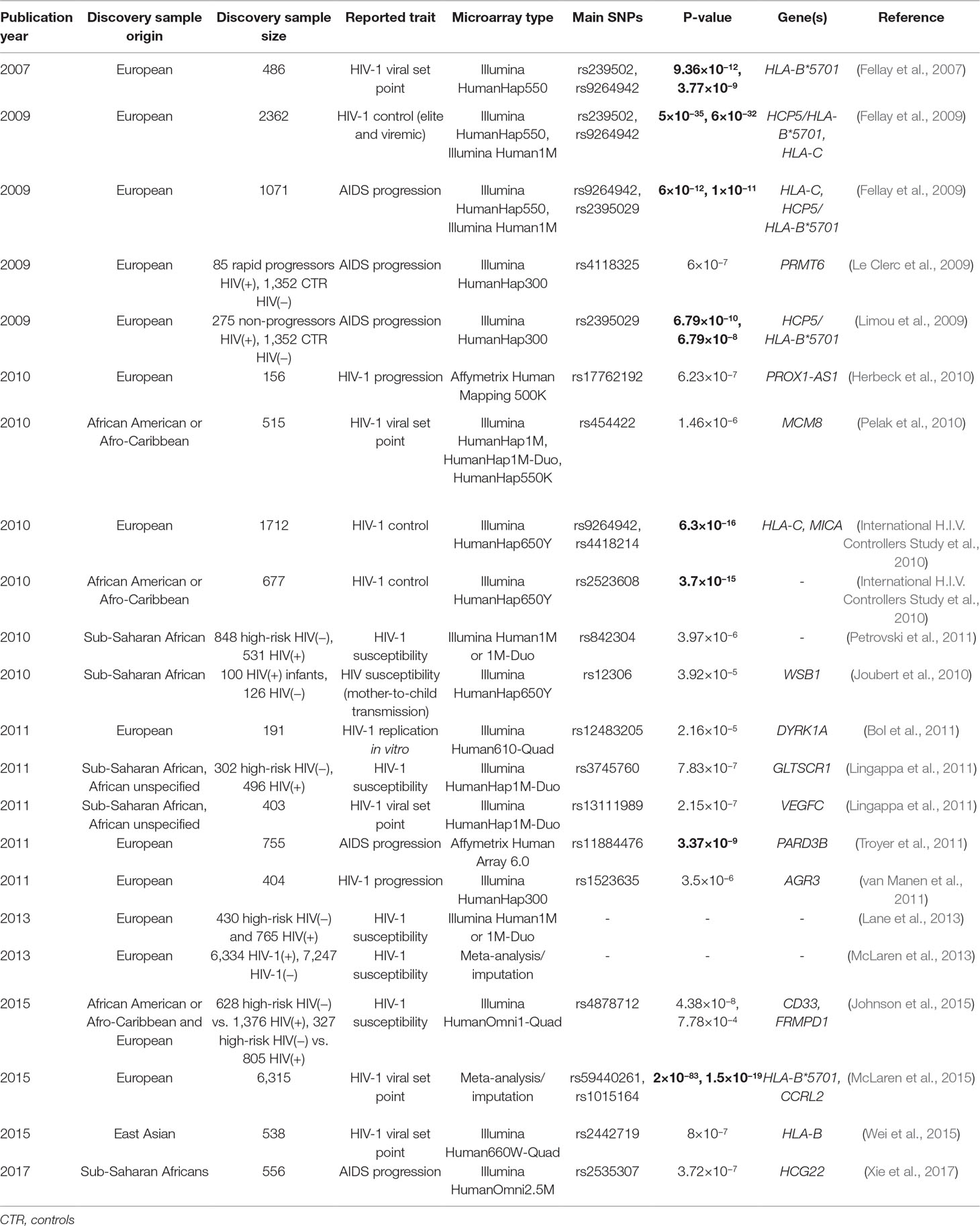
Table 1 GWASs published in the AIDS field since 2007. 5×10−8 is the standard threshold for considering an association as significant in a GWAS, and the significant ones in the table are in bold.
The following major conclusions can be underlined from these GWA studies:
1. One signal was repeatedly replicated in several cohorts (of European descent) both for the viral load phenotype and also for the non-progression phenotype: rs2395029, an SNP in the HCP5 gene within the MHC region, which was described in nearly full linkage disequilibrium (LD) with the HLA-B*5701 allele (Fellay et al., 2007; Dalmasso et al., 2008; Limou et al., 2009; McLaren et al., 2013; McLaren et al., 2017). This HLA allele could be critical for the destruction of infected cells via a yet-unidentified CD8 T-cell epitope, but we cannot exclude a role for other polymorphisms in the HLA region and possibly outside of the class I HLA genes, with relevant genes highlighted through LD such as MICB and TNF (Kulkarni et al., 2011). Similarly, in African Americans, the HLA-B*5703 allele was demonstrated to be important for viral control (Pelak et al., 2010; McLaren et al., 2012; McLaren et al., 2017). Beyond B*57 alleles, HLA seems to play a major role in viral control, as another SNP located 35 kb upstream of HLA-C was also identified in Europeans (Limou et al., 2009; McLaren et al., 2017). This −35 kb SNP was correlated with higher HLA-C cell surface expression (Thomas et al., 2009), which can be regulated through microRNA (miRNA) binding (Kulkarni et al., 2011) and through the binding of the Oct1 transcription factor (Vince et al., 2016).
2. In 2011, the HIV/AIDS research community conducted a massive effort to bring together their genomic data in order to gain statistical power of detection. This collaborative effort, named the International Consortium for the Genetics of HIV-1 (ICGH), gathered up to 6,300 HIV-infected individuals and 7,200 HIV-uninfected controls who were genotyped on various Illumina or Affymetrix platforms. A first publication emerged from this consortium in 2013; it focused on HIV-1 acquisition and confirmed the major role of CCR5-Delta32 in resistance to infection but did not identify any additional common genetic variant (McLaren et al., 2013). A second study was published 2 years later on viral control and disease progression and confirmed the complementary roles of CCR5 locus variants and of HLA genetic variants coding for amino acids in the peptide binding grooves of HLA-A and HLA-B (McLaren et al., 2015). In spite of the large sample size, no other locus reached genome-wide significance. These reports conclude that future genetic studies should target other classes of genetic variants (e.g., low or rare frequency), non-European populations, and well-defined homogeneous phenotypes (McLaren et al., 2015).
3. In addition, the numerous GWASs have identified novel candidate genes, which all deserve further exploration, notably the ones that reached genome-wide significance (see Table 1). In particular, a genetic variant within the CXCR6 gene was associated with long-term non-progression in four independent cohorts (Limou et al., 2010). This signal was only identified in individuals without sustained viral control, which explains why it was not highlighted in the consortium that mainly focused on viral control. Finally, the absence of replication for many other signals presented in Table 1 does not discount their scientific interest but complicates their biological interpretation.
GWASs rely on common SNPs (typically >1% in the population) and hardly take into account the possible effect of rare variants or other classes of genetic polymorphisms such as indels or copy number variants that can also significantly impact disease outcome. To investigate the impact of such variants, we first focused on low-frequency SNPs (<5%) in our progression GWASs and identified the gene RICH2 associated with non-progression, which interacts with BST-2, a major known HIV restriction factor (Le Clerc et al., 2011). Later, several studies based on next-generation sequencing (NGS) have emerged. Due to the high cost of such studies and to maximize statistical power of detection, these screenings have targeted coding variants (exome) and patients with very specific and extreme disease outcome. To our knowledge, only one publication has emerged from these studies (McLaren et al., 2017), which focused on 1,327 subjects, many of whom were elite and viral controllers. In spite of the significant number of patients studied, only variants in the HLA region came out, and this study suggested that exonic variants with large effect sizes are unlikely to have a major contribution to host control of HIV infection (McLaren et al., 2017).
Systematic inactivation of gene expression through siRNA and small hairpin RNA (shRNA) offers a unique chance to identify host genes required for HIV replication. In large-scale studies, authors used siRNAs (Brass et al., 2008; Konig et al., 2008; Zhou et al., 2008) or shRNAs (Yeung et al., 2009) to silence invitro most known genes one by one in HIV permissive cell lines. These screenings identified 273 (Brass et al., 2008), 213 (Konig et al., 2008), 311 (Zhou et al., 2008), and 252 (Yeung et al., 2009) HIV host dependency factors, respectively, for a total of 842 putative candidates. However, the overlap between the different studies was very low, suggesting low reproducibility and/or high false positive, which might not be surprising considering the different experimental models (cell lines, HIV strains, and measurement modes of HIV replication). Overall, these studies still provide an interesting list of candidate cellular factors and pathways potentially implicated in HIV-1 replication that could be considered as relevant targets for drug development.
The development of the CRISPR–Cas9 technology to screen each gene with a library of single-guided RNA offers a greater sensitivity and specificity than interference based-RNA (Wang et al., 2014). A recent report used this technology to screen a CD4 T-cell line and identified five host factors required for HIV replication, including CD4, C-C motif chemokine receptor 5 (CCR5), and activated leukocyte cell adhesion molecule (ALCAM (Park et al., 2017). These factors were further validated in primary human CD4 T-cells and therefore represent major candidates for a therapeutic intervention.
The first descriptions of transcriptome analysis by DNA microarrays were in cancer in 2002 (Pomeroy et al., 2002; van ‘t Veer et al., 2002). In AIDS, the first large-scale transcriptomic study (4,600 transcripts) was published in 2003 (van ‘t Wout et al., 2003). This study analyzed gene expression in HIV-infected CD4 T-cell lines at different time points and revealed the inhibition of genes involved in cell division, transcription, translation, splicing, and also cholesterol biosynthesis (van ‘t Wout et al., 2003). An exon transcriptome microarray analysis of purified HIV-infected cells revealed host cell factors required for viral replication and alternative splicing events (Imbeault et al., 2012). A bioinformatic analysis of HIV-resistant activated CD4 T-cells (due to CD3/CD28 antibodies’ co-stimulation) highlighted a few dozen genes critical for resistance or permissivity (Xu et al., 2013).
Several microarray studies focused on non-human primate models, such as cynomolgus monkeys (Bosinger et al., 2004) and African green monkeys (non-pathogenic model) vs. rhesus macaques (pathogenic model) (Jacquelin et al., 2009). These reports mainly identified a major role for IFN-stimulated genes, as well as a differential expression of some innate genes (such as LPS receptors CD14 and TLR4) and some apoptosis-related genes (Bosinger et al., 2004).
Finally, numerous transcriptome studies explored differential gene expression in HIV-infected individuals. A first report in 2005 claimed to have found (Ockenhouse et al., 2005) a 10-gene signature for HIV-1 serostatus and a 6-gene signature for subjects experiencing a CD4+ T-cell decrease (Ockenhouse et al., 2005). The genes identified were primarily linked with immune response and apoptosis, mitochondrial function, and RNA binding (downregulated in subjects with better prognosis) (Ockenhouse et al., 2005). A study focusing on HIV-1–resistant individuals (Huang et al., 2011) found a set of 185 HIV-1 resistance genes, suggesting a major role for nef in disease pathogenesis, and among them pointed out 29 potential targets for AIDS prevention or therapy (Huang et al., 2011). By comparing the complementary DNA (cDNA) profiles of CD3+ T-cells in long-term non-progressors vs. medium progressors (Salgado et al., 2011), 325 genes appeared over-expressed in regular progressors (from DNA replication, cell cycle, and DNA damage pathways), vs. 136 over-expressed genes in long-term non-progressors (from cytokine–cytokine receptor interaction and negative control of apoptosis pathways) (Salgado et al., 2011). The transcriptome comparison of CD4+ T-cells and CD8+ T-cells from rapid progressors, viremic non-progressors, and elite controllers showed a lower expression of IFN-stimulated genes and an upregulation of CASP1, CD38, LAG3, TNFSF13B, SOCS1, and EEF1D genes in viremic non-progressors (Rotger et al., 2011). Finally, a transcriptomic screening also targeted miRNA expression profiles in peripheral blood mononuclear cell (PBMC) from rapid and chronic progressors and identified five downregulated miRNAs in rapid progressors that all converged to the apoptosis pathway (Zhang et al., 2013).
Some proteomic studies have also been performed, but they were not very reproducible, as indicated in a recent review by Donnelly and Ciborowski (2016). To our knowledge, few epigenomic studies have been published to date in HIV/AIDS. One Korean group performed two chromatin immunoprecipitation sequencing (ChIP-seq) analyses in HIV latently infected CD4 T-cell lines to investigate the impact of H3K4me3 and H3K9ac histone modifications on latency. They revealed several potential candidate genes, including NFIX, tumor necrosis factor (TNF) receptor association factor 4 (TRAF4), and cell cycle regulating genes such as CDKN1A (p21) and CCND2 (Park et al., 2014; Kim et al., 2017). Finally, the blood DNA methylation signatures of HIV-infected and uninfected subjects were compared through an epigenome-wide association study (EWAS), which highlighted a down-methylation of NLC5 promoter in HIV-infected subjects (Zhang et al., 2016). This host gene encodes a key regulator of class I HLA gene expression and confirms the major role of the MHC locus in HIV viral control. Interestingly, NLC5 promoter and additional MHC clusters also appeared differentially methylated in HIV–Hepatitis C virus (HCV) co-infected subjects (Zhang et al., 2017), emphasizing the importance of inflammation-related genes in the course of HIV infection. Overall, these studies are promising and underline the need for additional large-scale epigenetic studies in order to better capture the breadth of host–HIV complex interactions.
In this review, we have presented numerous large-scale genomic and transcriptomic analyses that have taken place in the AIDS field, which are the consequences of the progress in molecular biology and biochemistry technologies. One can see that a huge research effort has been dedicated to genetic association studies, and this is logical since this experimental approach deals with real invivo data, i.e., cohorts of patients and HIV-1 infection invivo. Nevertheless, it was slightly surprising to observe that the main signals found by GWAS, in the HLA and CCR5 loci, had already been identified by previous candidate gene approaches. This apparent limitation could be explained by the yet-unidentified role of other polymorphisms such as copy number variations (CNVs) or interacting gene variants. It could also be explained by the statistical constraints (such as stringent multiple testing corrections) that limit the use of genetic association data (numerous false negatives) and the overall low number of samples at stake (a few thousands) compared to other human diseases such as diabetes or obesity (hundreds of thousands) (Shungin et al., 2015; Fuchsberger et al., 2016). In light of the available biological information provided by the other large-scale studies such as transcriptomic or functional genomic studies presented in this review, it appears important to reanalyze the genomic data by integrating biological information in order to enhance the genetic association results. For instance, our group has successfully implemented such approaches by pre-selecting relevant SNPs defined either by their low frequency (Le Clerc et al., 2011) or by their functional impact as potential expression quantitative trait loci (eQTLs) (Spadoni et al., 2015). More generally, there are several methods for data integration, the first one being to cross-check the results obtained by one method through another, for instance, using GWAS to identify SNPs with low P-values, even non-significant, and then using transcriptomics to pick genes that are differentially expressed in a relevant cell type or tissue. By combining two (or more) methods, researchers can zoom in on specific genes of high interest. This has been implemented with the development of PrediScan (Gamazon et al., 2015). Another example of cross-checking is the results obtained by metabolome analysis and GWAS in which the researchers have found that metabolites present at high levels in the blood of some subjects are highly correlated with specific variants present in the genes of enzymes involved in their metabolism (Illig et al., 2010). Other methods of data integration rely on rescuing genes by correlating signals not only at the gene level but also at the pathway level: for instance, one can suspect that if a gene X in a biological pathway is important for a clinical phenotype, the genes present upstream in the biological pathway may impact this gene X expression and, as a consequence, also become targets of interest. One will thus have to look for cross-checks at the level of pathways (Chen et al., 2011). Importantly, it is essential for data integration to perform all these cross-checks in a smart and automated manner. Finally, more sophisticated statistical approaches have recently emerged outside of the HIV field, such as the Bayesian method for data integration (Kichaev et al., 2014; Pickrell, 2014; Finucane et al., 2015; Yang et al., 2017; International Multiple Sclerosis Genetics Consortium, 2019). These new methods are yet to be implemented in the relatively small HIV/AIDS cohorts but might reveal novel underlying physiopathological mechanisms.
With the massive research effort to fight AIDS, this has been a true field of experimentation and development for novel technologies. A first challenge is now the cross-usage of all this information gathered from so many large-scale studies, to transform this “gold mine” into diagnostic or therapy strategies to fight AIDS, and the same integration of omics big data should of course take place also for other human diseases. This systems biology challenge has not yet been met. A second challenge is to pursue the exploration of alternative technologies such as epigenomics or proteomics to derive more understanding of HIV-1 molecular pathogenesis. We hope that the AIDS field will remain a “cultural” leader for research progress in order to fully understand the molecular mechanisms at stake in HIV-1 infection and AIDS and allow the rationale development of diagnostic and therapeutic strategies to finally tackle the HIV-1 virus.
SLC, SL, and J-FZ conceived this review, performed the bibliography search, and wrote it in a collective manner.
The authors have had their work funded from several sources. This review was directly funded by their employer: Conservatoire National des Arts et Métiers et Ecole Centrale de Nantes.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Alkhatib, G., Combadiere, C., Broder, C. C., Feng, Y., Kennedy, P. E., Murphy, P. M., et al. (1996). CC CKR5: a RANTES, MIP-1alpha, MIP-1beta receptor as a fusion cofactor for macrophage-tropic HIV-1. Science 272 (5270), 1955–1958. doi: 10.1126/science.272.5270.1955
An, P., Winkler, C. A. (2010). Host genes associated with HIV/AIDS: advances in gene discovery. Trends Genet. 26 (3), 119–131. doi: 10.1016/j.tig.2010.01.002
Bol, S. M., Moerland, P. D., Limou, S., van Remmerden, Y., Coulonges, C., van Manen, D., et al. (2011). Genome-wide association study identifies single nucleotide polymorphism in DYRK1A associated with replication of HIV-1 in monocyte-derived macrophages. PLoS One 6 (2), e17190. doi: 10.1371/journal.pone.0017190
Bosinger, S. E., Hosiawa, K. A., Cameron, M. J., Persad, D., Ran, L., Xu, L., et al. (2004). Gene expression profiling of host response in models of acute HIV infection. J. Immunol. 173 (11), 6858–6863. doi: 10.4049/jimmunol.173.11.6858
Brass, A. L., Dykxhoorn, D. M., Benita, Y., Yan, N., Engelman, A., Xavier, R. J., et al. (2008). Identification of host proteins required for HIV infection through a functional genomic screen. Science 319 (5865), 921–926. doi: 10.1126/science.1152725
Carrington, M., Nelson, G. W., Martin, M. P., Kissner, T., Vlahov, D., Goedert, J. J., et al. (1999). HLA and HIV-1: heterozygote advantage and B*35–Cw*04 disadvantage. Science 283 (5408), 1748–1752. doi: 10.1126/science.283.5408.1748
Center for Disease Control and Prevention (1992). 1993 revised classification system for HIV infection and expanded surveillance case definition for AIDS among adolescents and adults. MMWR Recomm. Rep. 41 (RR-17), 1–19.
Chen, M., Cho, J., Zhao, H. (2011). Incorporating biological pathways via a Markov random field model in genome-wide association studies. PLoS Genet. 7 (4), e1001353. doi: 10.1371/journal.pgen.1001353
Dalmasso, C., Carpentier, W., Meyer, L., Rouzioux, C., Goujard, C., Chaix, M. L., et al. (2008). Distinct genetic loci control plasma HIV-RNA and cellular HIV-DNA levels in HIV-1 infection: the ANRS genome wide association 01 study. PLoS One 3 (12), e3907. doi: 10.1371/journal.pone.0003907
Dean, M., Carrington, M., Winkler, C., Huttley, G. A., Smith, M. W., Allikmets, R., et al. (1996). Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. hemophilia growth and development study, multicenter AIDS cohort study, multicenter hemophilia cohort study, san francisco city cohort, ALIVE study. Science 273 (5283), 1856 1862. doi: 10.1126/science.273.5283.1856
Deng, H., Liu, R., Ellmeier, W., Choe, S., Unutmaz, D., Burkhart, M., et al. (1996). Identification of a major co-receptor for primary isolates of HIV-1. Nature 381 (6584), 661–666. doi: 10.1038/381661a0
Donnelly, M. R., Ciborowski, P. (2016). Proteomics, biomarkers, and HIV 1: a current perspective. Proteomics Clin. Appl. 10 (2), 110–125. doi: 10.1002/prca.201500002
Fellay, J. (2009). Host genetics influences on HIV type-1 disease. Antivir. Ther. 14 (6), 731–738. doi: 10.3851/IMP1253
Fellay, J., Ge, D., Shianna, K. V., Colombo, S., Ledergerber, B., Cirulli, E. T., et al. (2009). Common genetic variation and the control of HIV-1 in humans. PLoS Genet. 5 (12), e1000791. doi: 10.1371/journal.pgen.1000791
Fellay, J., Shianna, K. V., Ge, D., Colombo, S., Ledergerber, B., Weale, M., et al. (2007). A whole-genome association study of major determinants for host control of HIV-1. Science 317 (5840), 944–947. doi: 10.1126/science.1143767
Finucane, H. K., Bulik-Sullivan, B., Gusev, A., Trynka, G., Reshef, Y., Loh, P. R., et al. (2015). Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47 (11), 1228–1235. doi: 10.1038/ng.3404
Fowke, K. R., Nagelkerke, N. J., Kimani, J., Simonsen, J. N., Anzala, A. O., Bwayo, J. J., et al. (1996). Resistance to HIV-1 infection among persistently seronegative prostitutes in Nairobi, Kenya. Lancet 348 (9038), 1347–1351. doi: 10.1016/S0140-6736(95)12269-2
Fuchsberger, C., Flannick, J., Teslovich, T. M., Mahajan, A., Agarwala, V., Gaulton, K. J., et al. (2016). The genetic architecture of type 2 diabetes. Nature 536 (7614), 41–47. doi: 10.1038/nature18642
Gamazon, E. R., Wheeler, H. E., Shah, K. P., Mozaffari, S. V., Aquino-Michaels, K., Carroll, R. J., et al. (2015). A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet. 47 (9), 1091–1098. doi: 10.1038/ng.3367
Global AIDS Updated. (2016). UNAIDS. http://www.unaids.org/en/resources/documents/2016/Global-AIDS-update-2016.
Hendel, H., Caillat-Zucman, S., Lebuanec, H., Carrington, M., O’Brien, S., Andrieu, J. M., et al. (1999). New class I and II HLA alleles strongly associated with opposite patterns of progression to AIDS. J. Immunol. 162 (11), 6942–6946.
Herbeck, J. T., Gottlieb, G. S., Winkler, C. A., Nelson, G. W., An, P., Maust, B. S., et al. (2010). Multistage genomewide association study identifies a locus at 1q41 associated with rate of HIV-1 disease progression to clinical AIDS. J. Infect. Dis. 201 (4), 618–626. doi: 10.1086/649842
Hetherington, S., Hughes, A. R., Mosteller, M., Shortino, D., Baker, K. L., Spreen, W., et al. (2002). Genetic variations in HLA-B region and hypersensitivity reactions to abacavir. Lancet 359 (9312), 1121–1122. doi: 10.1016/S0140-6736(02)08158-8
Huang, T., Xu, Z., Chen, L., Cai, Y. D., Kong, X. (2011). Computational analysis of HIV-1 resistance based on gene expression profiles and the virus–host interaction network. PLoS One 6 (3), e17291. doi: 10.1371/journal.pone.0017291
Illig, T., Gieger, C., Zhai, G., Romisch-Margl, W., Wang-Sattler, R., Prehn, C., et al. (2010). A genome-wide perspective of genetic variation in human metabolism. Nat. Genet. 42 (2), 137–141. doi: 10.1038/ng.507
Imbeault, M., Giguere, K., Ouellet, M., Tremblay, M. J. (2012). Exon level transcriptomic profiling of HIV-1–infected CD4(+) T cells reveals virus-induced genes and host environment favorable for viral replication. PLoS Pathog. 8 (8), e1002861. doi: 10.1371/journal.ppat.1002861
International H.I.V. Controllers Study, Pereyra, F., Jia, X., McLaren, P. J., Telenti, A., de Bakker, P. I., et al. (2010). The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science 330 (6010), 1551–1557. doi: 10.1126/science.1195271
International Multiple Sclerosis Genetics Consortium (2019). A systems biology approach uncovers cell-specific gene regulatory effects of genetic associations in multiple sclerosis. Nat. Commun. 10 (1), 2236. doi: 10.1038/s41467-019-09773-y
Jacquelin, B., Mayau, V., Targat, B., Liovat, A. S., Kunkel, D., Petitjean, G., et al. (2009). Nonpathogenic SIV infection of African green monkeys induces a strong but rapidly controlled type I IFN response. J. Clin. Invest. 119 (12), 3544–3555. doi: 10.1172/JCI40093
Johnson, E. O., Hancock, D. B., Gaddis, N. C., Levy, J. L., Page, G., Novak, S. P., et al. (2015). Novel genetic locus implicated for HIV-1 acquisition with putative regulatory links to HIV replication and infectivity: a genome-wide association study. PLoS One 10 (3), e0118149. doi: 10.1371/journal.pone.0118149
Joubert, B. R., Lange, E. M., Franceschini, N., Mwapasa, V., North, K. E., Meshnick, S. R., et al. (2010). A whole genome association study of mother-to-child transmission of HIV in Malawi. Genome Med. 2 (3), 17. doi: 10.1186/gm138
Kaslow, R. A., Carrington, M., Apple, R., Park, L., Munoz, A., Saah, A. J., et al. (1996). Influence of combinations of human major histocompatibility complex genes on the course of HIV-1 infection. Nat. Med. 2 (4), 405–411. doi: 10.1038/nm0496-405
Kichaev, G., Yang, W. Y., Lindstrom, S., Hormozdiari, F., Eskin, E., Price, A. L., et al. (2014). Integrating functional data to prioritize causal variants in statistical fine-mapping studies. PLoS Genet. 10 (10), e1004722. doi: 10.1371/journal.pgen.1004722
Kim, K. C., Lee, S., Son, J., Shin, Y., Yoon, C. H., Kang, C., et al. (2017). Identification of novel genes associated with HIV-1 latency by analysis of histone modifications. Hum. Genomics 11 (1), 9. doi: 10.1186/s40246-017-0105-7
Konig, R., Zhou, Y., Elleder, D., Diamond, T. L., Bonamy, G. M., Irelan, J. T., et al. (2008). Global analysis of host–pathogen interactions that regulate early-stage HIV-1 replication. Cell 135 (1), 49–60. doi: 10.1016/j.cell.2008.07.032
Kulkarni, S., Savan, R., Qi, Y., Gao, X., Yuki, Y., Bass, S. E., et al. (2011). Differential microRNA regulation of HLA-C expression and its association with HIV control. Nature 472 (7344), 495–498. doi: 10.1038/nature09914
Lane, J., McLaren, P. J., Dorrell, L., Shianna, K. V., Stemke, A., Pelak, K., et al. (2013). A genome-wide association study of resistance to HIV infection in highly exposed uninfected individuals with hemophilia A. Hum. Mol. Genet. 22 (9), 1903–1910. doi: 10.1093/hmg/ddt033
Langlade-Demoyen, P., Ngo-Giang-Huong, N., Ferchal, F., Oksenhendler, E. (1994). Human immunodeficiency virus (HIV) nef-specific cytotoxic T lymphocytes in noninfected heterosexual contact of HIV-infected patients. J. Clin. Invest. 93 (3), 1293–1297. doi: 10.1172/JCI117085
Le Clerc, S., Coulonges, C., Delaneau, O., Van Manen, D., Herbeck, J. T., Limou, S., et al. (2011). Screening low-frequency SNPS from genome-wide association study reveals a new risk allele for progression to AIDS. J. Acquir. Immune. Defic. Syndr. 56 (3), 279–284. doi: 10.1097/QAI.0b013e318204982b
Le Clerc, S., Limou, S., Coulonges, C., Carpentier, W., Dina, C., Taing, L., et al. (2009). Genomewide association study of a rapid progression cohort identifies new susceptibility alleles for AIDS (ANRS Genomewide Association Study 03). J. Infect. Dis. 200 (8), 1194–1201. doi: 10.1086/605892
Li, H., Marley, G., Ma, W., Wei, C., Lackey, M., Ma, Q., et al. (2017). The role of ARV associated adverse drug reactions in influencing adherence among HIV-infected individuals: a systematic review and qualitative meta-synthesis. AIDS Behav. 21 (2), 341–351. doi: 10.1007/s10461-016-1545-0
Limou, S., Coulonges, C., Herbeck, J. T., van Manen, D., An, P., Le Clerc, S., et al. (2010). Multiple-cohort genetic association study reveals CXCR6 as a new chemokine receptor involved in long-term nonprogression to AIDS. J. Infect Dis. 202 (6), 908–915. doi: 10.1086/655782
Limou, S., Le Clerc, S., Coulonges, C., Carpentier, W., Dina, C., Delaneau, O., et al. (2009). Genomewide association study of an AIDS-nonprogression cohort emphasizes the role played by HLA genes (ANRS Genomewide Association Study 02). J. Infect. Dis. 199 (3), 419–426. doi: 10.1086/596067
Limou, S., Zagury, J. F. (2013). Immunogenetics: genome-wide association of non-progressive HIV and viral load control: HLA genes and beyond. Front. Immunol. 4, 118. doi: 10.3389/fimmu.2013.00118
Lingappa, J. R., Petrovski, S., Kahle, E., Fellay, J., Shianna, K., McElrath, M. J., et al. (2011). Genomewide association study for determinants of HIV-1 acquisition and viral set point in HIV-1 serodiscordant couples with quantified virus exposure. PLoS One 6 (12), e28632. doi: 10.1371/journal.pone.0028632
Liu, R., Paxton, W. A., Choe, S., Ceradini, D., Martin, S. R., Horuk, R., et al. (1996). Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 86 (3), 367–377. doi: 10.1016/S0092-8674(00)80110-5
Ludlam, C. A., Tucker, J., Steel, C. M., Tedder, R. S., Cheingsong-Popov, R., Weiss, R. A., et al. (1985). Human T-lymphotropic virus type III (HTLV-III) infection in seronegative haemophiliacs after transfusion of factor VIII. Lancet 2 (8449), 233–236. doi: 10.1016/S0140-6736(85)90288-0
Mallal, S., Nolan, D., Witt, C., Masel, G., Martin, A. M., Moore, C., et al. (2002). Association between presence of HLA-B*5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir. Lancet 359 (9308), 727–732. doi: 10.1016/S0140-6736(02)07873-X
Martin, M. P., Dean, M., Smith, M. W., Winkler, C., Gerrard, B., Michael, N. L., et al. (1998). Genetic acceleration of AIDS progression by a promoter variant of CCR5. Science 282 (5395), 1907–1911. doi: 10.1126/science.282.5395.1907
McDermott, D. H., Zimmerman, P. A., Guignard, F., Kleeberger, C. A., Leitman, S. F., Murphy, P. M. (1998). CCR5 promoter polymorphism and HIV-1 disease progression. Multicenter AIDS Cohort Study (MACS). Lancet 352 (9131), 866–870. doi: 10.1016/S0140-6736(98)04158-0
McLaren, P. J., Coulonges, C., Bartha, I., Lenz, T. L., Deutsch, A. J., Bashirova, A., et al. (2015). Polymorphisms of large effect explain the majority of the host genetic contribution to variation of HIV-1 virus load. Proc. Natl. Acad. Sci. U. S. A. 112 (47), 14658–14663. doi: 10.1073/pnas.1514867112
McLaren, P. J., Coulonges, C., Ripke, S., van den Berg, L., Buchbinder, S., Carrington, M., et al. (2013). Association study of common genetic variants and HIV-1 acquisition in 6,300 infected cases and 7,200 controls. PLoS Pathog. 9 (7), e1003515. doi: 10.1371/journal.ppat.1003515
McLaren, P. J., Pulit, S. L., Gurdasani, D., Bartha, I., Shea, P. R., Pomilla, C., et al. (2017). Evaluating the impact of functional genetic variation on HIV-1 control. J. Infect. Dis. 216 (9), 1063–1069. doi: 10.1093/infdis/jix470
McLaren, P. J., Ripke, S., Pelak, K., Weintrob, A. C., Patsopoulos, N. A., Jia, X., et al. (2012). Fine-mapping classical HLA variation associated with durable host control of HIV-1 infection in African Americans. Hum. Mol. Genet. 21 (19), 4334–4347. doi: 10.1093/hmg/dds226
O’Brien, S. J., Nelson, G. W. (2004). Human genes that limit AIDS. Nat. Genet. 36 (6), 565–574. doi: 10.1038/ng1369
Ockenhouse, C. F., Bernstein, W. B., Wang, Z., Vahey, M. T. (2005). Functional genomic relationships in HIV-1 disease revealed by gene-expression profiling of primary human peripheral blood mononuclear cells. J. Infect. Dis. 191 (12), 2064–2074. doi: 10.1086/430321
Pantaleo, G., Fauci, A. S. (1996). Immunopathogenesis of HIV infection. Annu. Rev. Microbiol. 50, 825–854. doi: 10.1146/annurev.micro.50.1.825
Park, J., Lim, C. H., Ham, S., Kim, S. S., Choi, B. S., Roh, T. Y. (2014). Genome-wide analysis of histone modifications in latently HIV-1 infected T cells. AIDS 28 (12), 1719–1728. doi: 10.1097/QAD.0000000000000309
Park, R. J., Wang, T., Koundakjian, D., Hultquist, J. F., Lamothe-Molina, P., Monel, B., et al. (2017). A genome-wide CRISPR screen identifies a restricted set of HIV host dependency factors. Nat. Genet. 49 (2), 193–203. doi: 10.1038/ng.3741
Pelak, K., Goldstein, D. B., Walley, N. M., Fellay, J., Ge, D., Shianna, K. V., et al. (2010). Host determinants of HIV-1 control in African Americans. J. Infect. Dis. 201 (8), 1141–1149. doi: 10.1086/651382
Petrovski, S., Fellay, J., Shianna, K. V., Carpenetti, N., Kumwenda, J., Kamanga, G., et al. (2011). Common human genetic variants and HIV-1 susceptibility: a genome-wide survey in a homogeneous African population. AIDS 25 (4), 513–518. doi: 10.1097/QAD.0b013e328343817b
Pickrell, J. K. (2014). Joint analysis of functional genomic data and genome-wide association studies of 18 human traits. Am. J. Hum. Genet. 94 (4), 559–573. doi: 10.1016/j.ajhg.2014.03.004
Pomeroy, S. L., Tamayo, P., Gaasenbeek, M., Sturla, L. M., Angelo, M., McLaughlin, M. E., et al. (2002). Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature 415 (6870), 436–442. doi: 10.1038/415436a
Rappaport, J., Cho, Y. Y., Hendel, H., Schwartz, E. J., Schachter, F., Zagury, J. F. (1997). 32 bp CCR-5 gene deletion and resistance to fast progression in HIV-1 infected heterozygotes. Lancet 349 (9056), 922–923. doi: 10.1016/S0140-6736(05)62697-9
Rotger, M., Dalmau, J., Rauch, A., McLaren, P., Bosinger, S. E., Martinez, R., et al. (2011). Comparative transcriptomics of extreme phenotypes of human HIV-1 infection and SIV infection in sooty mangabey and rhesus macaque. J. Clin. Invest. 121 (6), 2391–2400. doi: 10.1172/JCI45235
Saksena, N. K., Rodes, B., Wang, B., Soriano, V. (2007). Elite HIV controllers: myth or reality? AIDS Rev. 9 (4), 195–207.
Salgado, M., Lopez-Romero, P., Callejas, S., Lopez, M., Labarga, P., Dopazo, A., et al. (2011). Characterization of host genetic expression patterns in HIV-infected individuals with divergent disease progression. Virology 411 (1), 103–112. doi: 10.1016/j.virol.2010.12.037
Samson, M., Libert, F., Doranz, B. J., Rucker, J., Liesnard, C., Farber, C. M., et al. (1996). Resistance to HIV-1 infection in Caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 382 (6593), 722–725. doi: 10.1038/382722a0
Shungin, D., Winkler, T. W., Croteau-Chonka, D. C., Ferreira, T., Locke, A. E., Magi, R., et al. (2015). New genetic loci link adipose and insulin biology to body fat distribution. Nature 518 (7538), 187–196. doi: 10.1038/nature14132
Smith, M. W., Dean, M., Carrington, M., Winkler, C., Huttley, G. A., Lomb, D. A., et al. (1997). Contrasting genetic influence of CCR2 and CCR5 variants on HIV-1 infection and disease progression. Hemophilia Growth and Development Study (HGDS), Multicenter AIDS Cohort Study (MACS), Multicenter Hemophilia Cohort Study (MHCS), San Francisco City Cohort (SFCC), ALIVE Study. Science 277 (5328), 959–965. doi: 10.1126/science.277.5328.959
Spadoni, J. L., Rucart, P., Le Clerc, S., van Manen, D., Coulonges, C., Ulveling, D., et al. (2015). Identification of genes whose expression profile is associated with non-progression towards AIDS using eQTLs. PLoS One 10 (9), e0136989. doi: 10.1371/journal.pone.0136989
Thomas, R., Apps, R., Qi, Y., Gao, X., Male, V., HUigin, C., et al. (2009). HLA-C cell surface expression and control of HIV/AIDS correlate with a variant upstream of HLA-C. Nat. Genet. 41 (12), 1290–1294. doi: 10.1038/ng.486
Troyer, J. L., Nelson, G. W., Lautenberger, J. A., Chinn, L., McIntosh, C., Johnson, R. C., et al. (2011). Genome-wide association study implicates PARD3B-based AIDS restriction. J. Infect. Dis. 203 (10), 1491–1502. doi: 10.1093/infdis/jir046
van ‘t Veer, L. J., Dai, H., de Vijver, M. J., He, Y. D., Hart, A. A., Mao, M., et al. (2002). Gene expression profiling predicts clinical outcome of breast cancer. Nature 415 (6871), 530–536. doi: 10.1038/415530a
van ‘t Wout, A. B., Lehrman, G. K., Mikheeva, S. A., O’Keeffe, G. C., Katze, M. G., Bumgarner, R. E., et al. (2003). Cellular gene expression upon human immunodeficiency virus type 1 infection of CD4(+)-T-cell lines. J. Virol. 77 (2), 1392–1402. doi: 10.1128/JVI.77.2.1392-1402.2003
van Manen, D., Delaneau, O., Kootstra, N. A., Boeser-Nunnink, B. D., Limou, S., Bol, S. M., et al. (2011). Genome-wide association scan in HIV-1–infected individuals identifying variants influencing disease course. PLoS One 6 (7), e22208. doi: 10.1371/journal.pone.0022208
Vince, N., Li, H., Ramsuran, V., Naranbhai, V., Duh, F. M., Fairfax, B. P., et al. (2016). HLA-C level is regulated by a polymorphic Oct1 binding site in the HLA-C promoter region. Am. J. Hum. Genet. 99 (6), 1353–1358. doi: 10.1016/j.ajhg.2016.09.023
Wang, T., Wei, J. J., Sabatini, D. M., Lander, E. S. (2014). Genetic screens in human cells using the CRISPR–Cas9 system. Science 343 (6166), 80–84. doi: 10.1126/science.1246981
Wei, Z., Liu, Y., Xu, H., Tang, K., Wu, H., Lu, L., et al. (2015). Genome-wide association studies of HIV-1 host control in ethnically diverse Chinese populations. Sci. Rep. 5, 10879. doi: 10.1038/srep10879
Xie, W., Agniel, D., Shevchenko, A., Malov, S. V., Svitin, A., Cherkasov, N., et al. (2017). Genome-wide analyses reveal gene influence on HIV disease progression and HIV-1C acquisition in Southern Africa. AIDS Res. Hum. Retroviruses 33 (6), 597–609. doi: 10.1089/aid.2016.0017
Xu, W. W., Han, M. J., Chen, D., Chen, L., Guo, Y., Willden, A., et al. (2013). Genome-wide search for the genes accountable for the induced resistance to HIV-1 infection in activated CD4+ T cells: apparent transcriptional signatures, co-expression networks and possible cellular processes. BMC Med. Genomics 6, 15. doi: 10.1186/1755-8794-6-15
Yang, J., Fritsche, L. G., Zhou, X., Abecasis, G., International Age-Related Macular Degeneration Genomics Consortium (2017). A scalable Bayesian method for integrating functional information in genome-wide association studies. Am. J. Hum. Genet. 101 (3), 404–416. doi: 10.1016/j.ajhg.2017.08.002
Yeung, M. L., Houzet, L., Yedavalli, V. S., Jeang, K. T. (2009). A genome-wide short hairpin RNA screening of Jurkat T-cells for human proteins contributing to productive HIV-1 replication. J. Biol. Chem. 284 (29), 19463–19473. doi: 10.1074/jbc.M109.010033
Zhang, X., Hu, Y., Justice, A. C., Li, B., Wang, Z., Zhao, H., et al. (2017). DNA methylation signatures of illicit drug injection and hepatitis C are associated with HIV frailty. Nat. Commun. 8 (1), 2243. doi: 10.1038/s41467-017-02326-1
Zhang, X., Justice, A. C., Hu, Y., Wang, Z., Zhao, H., Wang, G., et al. (2016). Epigenome-wide differential DNA methylation between HIV-infected and uninfected individuals. Epigenetics 11 (10), 750–760. doi: 10.1080/15592294.2016.1221569
Zhang, Z. N., Xu, J. J., Fu, Y. J., Liu, J., Jiang, Y. J., Cui, H. L., et al. (2013). Transcriptomic analysis of peripheral blood mononuclear cells in rapid progressors in early HIV infection identifies a signature closely correlated with disease progression. Clin. Chem. 59 (8), 1175–1186. doi: 10.1373/clinchem.2012.197335
Keywords: HIV, genome-wide association study, genomics, omics, big data
Citation: Le Clerc S, Limou S and Zagury J-F (2019) Large-Scale “OMICS” Studies to Explore the Physiopatholgy of HIV-1 Infection. Front. Genet. 10:799. doi: 10.3389/fgene.2019.00799
Received: 30 January 2019; Accepted: 30 July 2019;
Published: 13 September 2019.
Edited by:
Dana C. Crawford, Case Western Reserve University, United StatesReviewed by:
Paul J. McLaren, Public Health Agency of Canada (PHAC), CanadaCopyright © 2019 Le Clerc, Limou and Zagury. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sophie Limou, c29waGllLmxpbW91QHVuaXYtbmFudGVzLmZy; Jean-François Zagury, emFndXJ5QGNuYW0uZnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.