
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 10 July 2019
Sec. Genetics of Common and Rare Diseases
Volume 10 - 2019 | https://doi.org/10.3389/fgene.2019.00605
This article is part of the Research Topic Technologies for Prenatal Diagnosis and Assessment of Genetic Disorders View all 12 articles
 Qiongdan Wang1,2†
Qiongdan Wang1,2† Zhenwei Liu3†Zhongdong Lin4Ru Zhang2Yutian Lu1,2Weijue Su5Feng Li4Xi Xu1,2Mengyun Tu1,2Yongliang Lou2,6*Junzhao Zhao5*
Zhenwei Liu3†Zhongdong Lin4Ru Zhang2Yutian Lu1,2Weijue Su5Feng Li4Xi Xu1,2Mengyun Tu1,2Yongliang Lou2,6*Junzhao Zhao5* Xiaoqun Zheng1,2,6*
Xiaoqun Zheng1,2,6*Infantile spasm (IS) is an early-onset epileptic encephalopathy that usually presents with hypsarrhythmia on an electroencephalogram with developmental impairment or regression. In this study, whole-exome sequencing was performed to detect potential pathogenic de novo mutations, and finally we identified a novel damaging de novo mutation in SEMA5A and a compound heterozygous mutation in CLTCL1 in three sporadic trios with IS. The expression profiling of SEMA5A in the human brain showed that it was mainly highly expressed in the cerebral cortex, during the early brain development stage (8 to 9 post-conception weeks and 0 to 5 months after birth). In addition, we identified a close protein-protein interaction network between SEMA5A and candidate genes associated with epilepsy, autism spectrum disorder (ASD) or intellectual disability. Gene enrichment and function analysis demonstrated that genes interacting with SEMA5A were significantly enriched in several brain regions across early fetal development, including the cortex, cerebellum, striatum and thalamus (q < 0.05), and were involved in axonal, neuronal and synapse-associated processes. Furthermore, SEMA5A and its interacting genes were associated with ASD, epilepsy syndrome and developmental disorders of mental health. Our results provide insightful information indicating that SEMA5A may contribute to the development of the brain and is associated with IS. However, further genetic studies are still needed to evaluate the role of SEMA5A in IS to definitively establish the role of SEMA5A in this disorder.
Epileptic encephalopathies (EEs) are a group of complex brain disorders characterized by intractable and early-onset epilepsy with or without developmental delays, which are highly genetically heterogeneous (Berg et al., 2010; Scheffer et al., 2017). Infantile spasms (IS, also known as West syndrome) are considered a subset of EEs that are characterized by the early onset of epileptic spasms, typically in the first year of life, and are always accompanied by a hypsarrhythmia pattern on the electroencephalogram (EEG) and developmental impairment (Nabbout and Dulac, 2003; Pavone et al., 2014). Moreover, EEs are caused by a variety of etiologies that are not yet fully understood. However, recent studies have provided evidence that genetic factors have a tremendous impact on the pathogenesis of EEs (McTague et al., 2016; Shbarou and Mikati, 2016). The discovery of candidate genes will further our understanding of the mechanisms underlying epileptogenesis.
The development of next-generation sequencing techniques, such as whole-exome sequencing (WES), has greatly facilitated gene discovery in EEs, and these approaches have been widely used to detect pathogenic mutations (Dimassi et al., 2016; Jin et al., 2018). Furthermore, recent genetic studies have used trio exome sequencing to confirm that many pathogenetic de novo mutations (DNMs) are critical genetic components in the pathogenesis underlying EEs (Claes et al., 2001; Carvill et al., 2013; Epi4K Consortium and Epilepsy Phenome/Genome Project, 2013). DNMs are the most damaging form of rare genetic mutations and occur mainly in the germline (Veltman and Brunner, 2012). Most genes with pathogenetic DNMs in EE encode voltage-gated ion channels or receptors associated with neurotransmitter, including sodium channels, potassium channels, GABA receptors, glutamate receptors and NMDA receptors (Fukata and Fukata, 2017; He et al., 2019), and are often involved in a variety of functional pathways related to neuronal excitability or synaptic and neuronal connectivity (Pardo et al., 2014).
Axon guidance proteins, including semaphorins, ephrins, slits, repulsive guidance molecules, and netrins, can act as attractants or repellents during axon branching, synapse formation and plasticity (Shen and Cowan, 2010; Van Battum et al., 2015). In the central nervous system, semaphorins play a role in synaptic plasticity including the regulation of synaptic structures and synaptic transmission (Pasterkamp and Giger, 2009). Moreover, both NMDA and AMPA receptors participate in synaptic plasticity (Song and Huganir, 2002; Lau and Zukin, 2007). Strikingly, DNMs have been identified in the GRIN1 and GRIN2B genes, which encode the proteins that form the subunits of the NMDA receptor, in individuals with West syndrome and a severe developmental delay (Epi4K Consortium and Epilepsy Phenome/Genome Project, 2013; Lemke et al., 2014). Because appropriate brain function and mental health rely on the accurate regulation of the central nervous system synapse density, an imbalance between excitatory and inhibitory synaptic transmission is partly responsible for neurodevelopmental disorders such as schizophrenia and autism spectrum disorder (ASD) characterized by impairments in social interactions and restricted behaviors and interests (Penzes et al., 2011).
In this study, we performed WES on three individuals with IS as well as their unaffected parents and identified a novel damaging DNM in SEMA5A. As a member of the semaphorin gene family with bifunctional axon guidance activities, the discovery of a mutation in SEMA5A may further strengthen the role of axon guidance proteins in EEs. In addition, we also detected a DNM in PLEKHG4B and a compound heterozygous mutation in CLTCL1 in patients with IS.
Three probands with IS and their healthy parents were enrolled in the study from the Second Affiliated Hospital and Yuying Children’s Hospital of Wenzhou Medical University, after study approval was provided by the Hospital Ethics Committee. Moreover, written informed consent of all participants was obtained from their parents or guardians at the time of recruitment. All probands were referred with typical seizure presentation and were diagnosed with IS by an experienced pediatric neurologist.
Peripheral blood (2 ml) was drawn from the three affected probands and their unaffected family members. Genomic DNA was isolated using a QIAGEN DNeasy Blood & Tissue Kit (Qiagen, Valencia, CA, USA) from the peripheral blood of each included individual. The DNA was then subjected to an additional quality and quantity evaluation step using a NanoDrop 2000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA). Subsequently, exome-coding DNA was captured with an Agilent SureSelect Human All Exon v6 Kit (Agilent Technologies, Santa Clara, CA, USA), and the libraries were sequenced on an Illumina HiSeq2000 sequencer (San Diego, CA, USA), which produced 150-bp paired-end reads.
All raw sequencing data obtained from these three trios were analyzed in a similar manner according to a customized bioinformatics pipeline (Wang et al., 2013). After quality filtering was performed on the raw sequence data using the Trim Galore program, the cleaned sequences were aligned to the human reference genome (GRCH37/hg19) using Burrows-Wheeler Aligner (BWA). Picard was performed to realign the reads and remove any reads that were duplicated or mapped to multiple genome locations. Variant and genotype calling were performed using the Genome Analysis Toolkit (GATK) (McKenna et al., 2010), and DNMs were detected by two software tools [ForestDNM (Michaelson et al., 2012) and mirTrios (Li et al., 2015)].
ANNOVAR was used to annotate all called variants. The minor allele frequency (MAF) of the detected sequence variants was estimated in various publicly available databases, including ExAC, UK10K, dbSNP147, 1000 Genomes, and ESP6500. If a detected variant was presented with a MAF > 0.1% in any database, it was eliminated. Subsequently, the effects of the detected variants were predicted according to SIFT (https://sift.bii.a-star.edu.sg/, a variant with SIFT score < 0.05 predicted damaging), VEST3 (https://karchinlab.org/apps/appVest.html, a variant with VEST3 score ≥ 0.5 indicated damaging), Polyphen2 (http://genetics.bwh.harvard.edu/pph2/, a variant with a score of 0.909 to 1.0 indicated probable damage, while those with scores between 0.0 to 0.446 meant benign), and GERP++ (http://mendel.stanford.edu/SidowLab/downloads/gerp/, a variant with a GERP++ score ≥ 2 indicated conserved). All potential damaging DNMs were visualized by Splicing Viewer (Liu et al., 2012) and confirmed by Sanger sequencing.
The RNA sequencing data for human tissue-derived SEMA5A were obtained from the human protein atlas (HPA) (https://www.proteinatlas.org). In addition, data were obtained from the human brain transcriptome at HBT (http://hbatlas.org/) to evaluate the spatial and temporal expression pattern of SEMA5A in human brain tissues. Furthermore, a digital atlas of gene expression patterns in mouse tissues at embryonic day 14.5 was selected and assessed in GenePaiant (www.genepaint.org). Expression patterns were defined by non-radioactive in situ hybridization (ISH), and selected images were annotated in detail.
A critical assessment and integration of protein–protein interaction (PPI) information was obtained from the STRING database to create the PPI networks. Then, we selected genes that interacted with SEMA5A and that had interaction scores greater than 200. Specific expression analysis (SEA) was performed on a developed online tool (http://genetics.wustl.edu/jdlab/csea-tool-2/) to explore whether SEMA5A and the identified interacting genes were highly enriched in a particular human brain region. In addition, R package clusterProfiler and DOSE were used to further explore the functions of the interacting genes in the PPI network. There were 43 SEMA5A-interacting genes that were shared by neuropsychiatric disorders and used to construct the interconnected PPI network. Random simulations of 100,000 permutations for genes and connections were performed to verify the non-random nature of the networks. Moreover, fisher’s exact test was used to evaluate the enrichment of 43 genes in the postsynaptic density (PSD) proteins (Collins et al., 2006) and genes under evolutionary constraint (Samocha et al., 2014).
To detect candidate pathogenic DNMs, we recruited three trios with IS for WES. After low-quality reads and adapters were removed, approximately 5.2–11.03 GB of clean reads were obtained for each individual. All samples passed the quality control measures, and more than 99% of the qualified bases were aligned to the human reference genome (hg19) with an average sequencing depth of 74.91-fold. It is important to note that a mean of 87.49% of the target regions was covered at 20× and that more than 90% of the exonic regions were sequenced at 10× coverage (Supplementary Table S1). Following alignment and variant annotation, we detected a total of five de novo SNVs and three de novo indel mutations in coding regions. We confirmed that two DNMs and a compound heterozygous mutation were present among the three trios. In addition, all of the identified mutations were confirmed by Sanger sequencing, and the deleteriousness of mutations were predicted by SIFT, VEST3, Polyphen2, and GERP++.
Patient 1, who was born at term following an uneventful pregnancy to healthy non-consanguineous parents with no pertinent family history, had seizure symptom onset at the age of 7 months with the EEG displaying hypsarrhythmia (Figure 1A), a characteristic feature of IS. Additionally, the magnetic resonance imaging (MRI) of patient 1 at 7 months old, revealed brain dysplasia and agenesis of the corpus callosum (Figures 1B, C). The patient carried one missense mutation (c.1201C > T) in the SEMA5A gene that was identified by WES and confirmed as a DNM by Sanger sequencing (Figure 2A). At the structural level, the de novo missense mutation caused an arginine to cysteine substitution at amino acid 401 in the Sema domain [p.(Arg401Cys)], a conserved domain containing 450 amino acids (Figure 2B). The Sema domain serves as a repulsive guidance cue and might prevent axons from branching into surrounding myotome regions (Hilario et al., 2009). Then we predicted the structural model of the SEMA5A and there was no hydrogen bond between Arg-401 of SEMA5A and other residues. However, Cys-401 forms three hydrogen bonds with Phe-402, Asp-425, and Gln-292, respectively (Supplementary Figure S1). Hence, the substitution of arginine by cystine at amino acid 401 of SEMA5A may affect the structural stability.
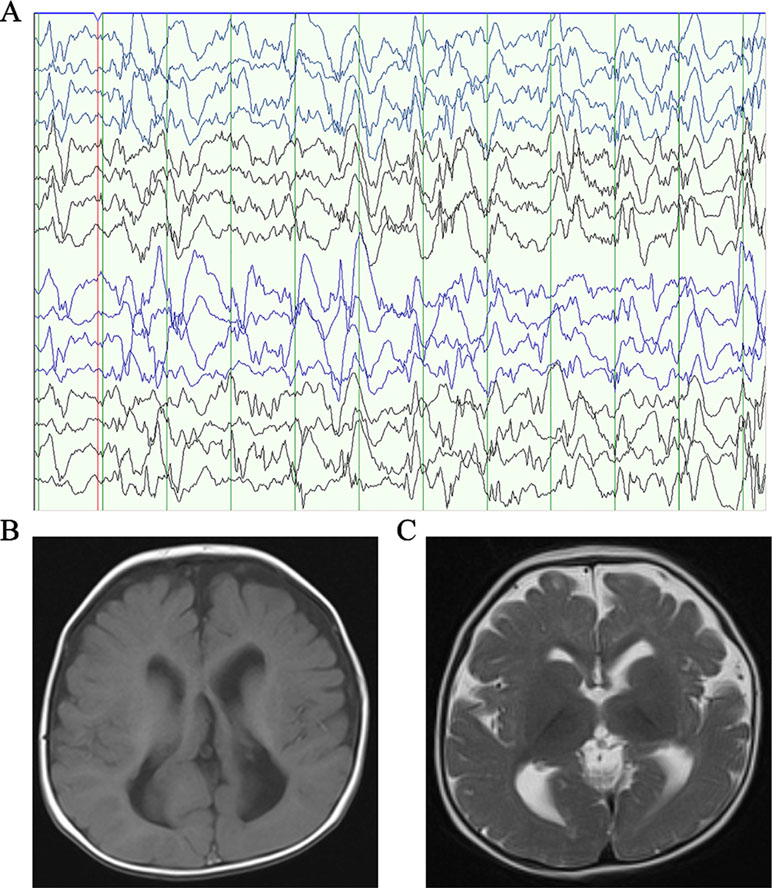
Figure 1 Clinical presentation. (A) The electroencephalogram obtained in patient 1 when he was 7 months old. (B) Axial T1-weighted brain MRI obtained in patient 1 when he was 7 months old. (C) Axial T2-weighted brain MRI obtained in patient 1 when he was 7 months old.
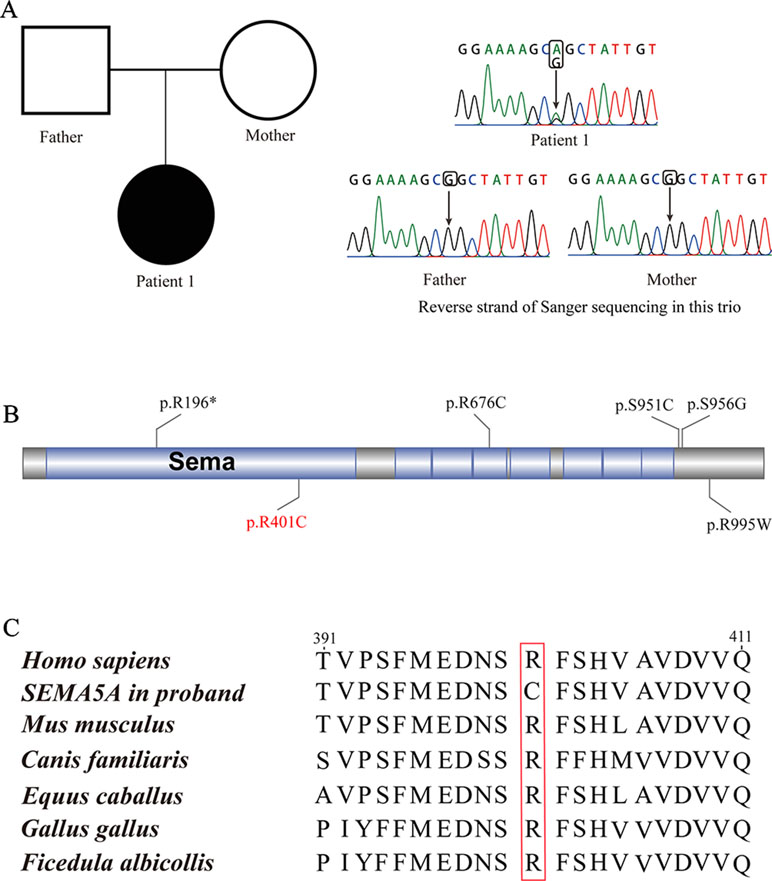
Figure 2 De novo mutations (DNMs) in SEMA5A. (A) Sanger sequencing of patient 1. The filled symbol indicates the affected individual. (B) Protein schematic of SEMA5A. Red indicates the DNM identified in this study, and black indicates mutations detected in ASD. (C) The conservation of the DNM in SEMA5A among various vertebrates.
In addition, we found that the identified DNM is evolutionarily conserved across various vertebrates (Figure 2C). Moreover, in silico prediction programs (SIFT, VEST3, Polyphen2 and GERP++) indicated that the de novo missense mutation in SEMA5A identified in this study is likely to be damaging and conserved in the protein (Table 1). Furthermore, the missense Z score of SEMA5A is 1.6794, which means that it tended to be intolerant of functional genetic variation (Lek et al., 2016). It should be mentioned that five different mutations, including a de novo missense mutation [c.2852C > G, p.(Ser951Cys)], a nonsense mutation [c.586C > T p.(Arg196*)] and three missense mutations, have been found in SEMA5A in individuals diagnosed with ASD (Figure 2B) (Iossifov et al., 2014; D’Gama et al., 2015; Mosca-Boidron et al., 2016). Additionally, SEMA5A has not been reported to be involved in epilepsy and is absent in any gene database related to epilepsy (Ran et al., 2015).
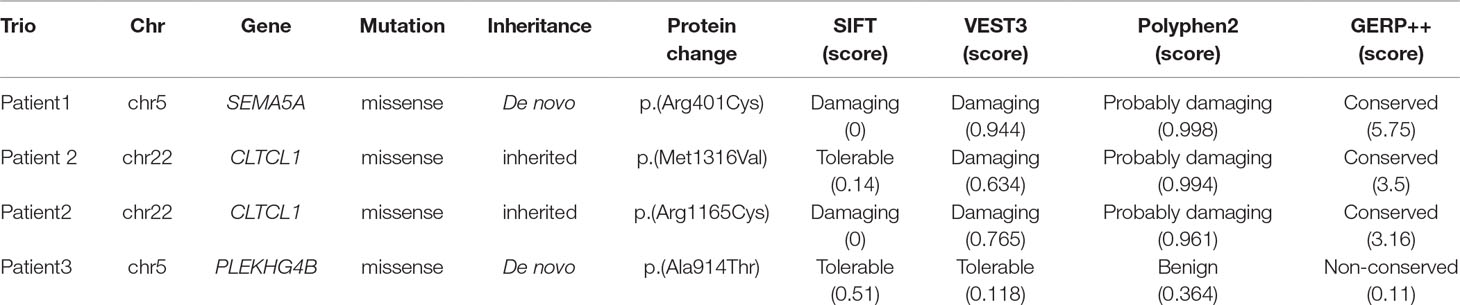
Table 1 Mutations detected by WES in this study in patients with IS.
We additionally identified a compound heterozygous mutation in CLTCL1 in patient 2 (Supplementary Figure S2A). In this patient, one missense mutation was inherited from the mother [c.3946A > G, p.(Met1316Val)], and was predicted to be damaging and conserved by VEST3, Polyphen2 and GERP++. Another missense mutation [c.3493C > T, p.(Arg1165Cys)] that was inherited from the father was predicted by four prediction tools (SIFT, VEST3, Polyphen2, and GERP++) to be damaging and conserved (Supplementary Figure S2B, Table 1). Both mutations were located in a highly conserved domain (Supplementary Figure S2C).
In patient 3, we identified a missense mutation in PLEKHG4B [c.2740G > A, p.(Ala914Thr)] that was regarded as a DNM and confirmed by Sanger sequencing. Nevertheless, the SIFT, VEST3 and Polyphen2 tools predicted that this mutation is tolerable and benign, and GERP++ predicted that it is non-conserved. Therefore, it was not considered as a possible candidate mutation in further analyses.
Considering that IS can give rise to severe cognitive and behavioral impairments, we attempted to determine the role of SEMA5A in the development of brain tissues by evaluating its expression pattern. RNA sequencing data were available for 36 tissues in the HPA database and indicated that SEMA5A acts in a variety of tissues. It is important to note that SEMA5A is highly expressed in the cerebral cortex, in which it showed its fourth-highest expression level in the 36 tissues (Figure 3A). To affirm this finding and to gain a higher-resolution spatiotemporal view of SEMA5A expression in the human brain, we analyzed RNA-seq data across various brain regions and developmental stages in HBT. The results showed that SEMA5A expression is extensively scattered across different developmental periods and regions of the human brain. Interestingly, SEMA5A is preferentially highly expressed in 11 areas of the neocortex, as well as several other brain regions such as the hippocampus (HIP), amygdala (AMY), and the striatum (STR), during early embryonic development (8 to 9 post-conception weeks, Figure 3B). In addition, SEMA5A was also highly expressed in almost all human brain tissues at 0 to 5 months after birth, which also represents an important stage of human brain development (Figure 3B).
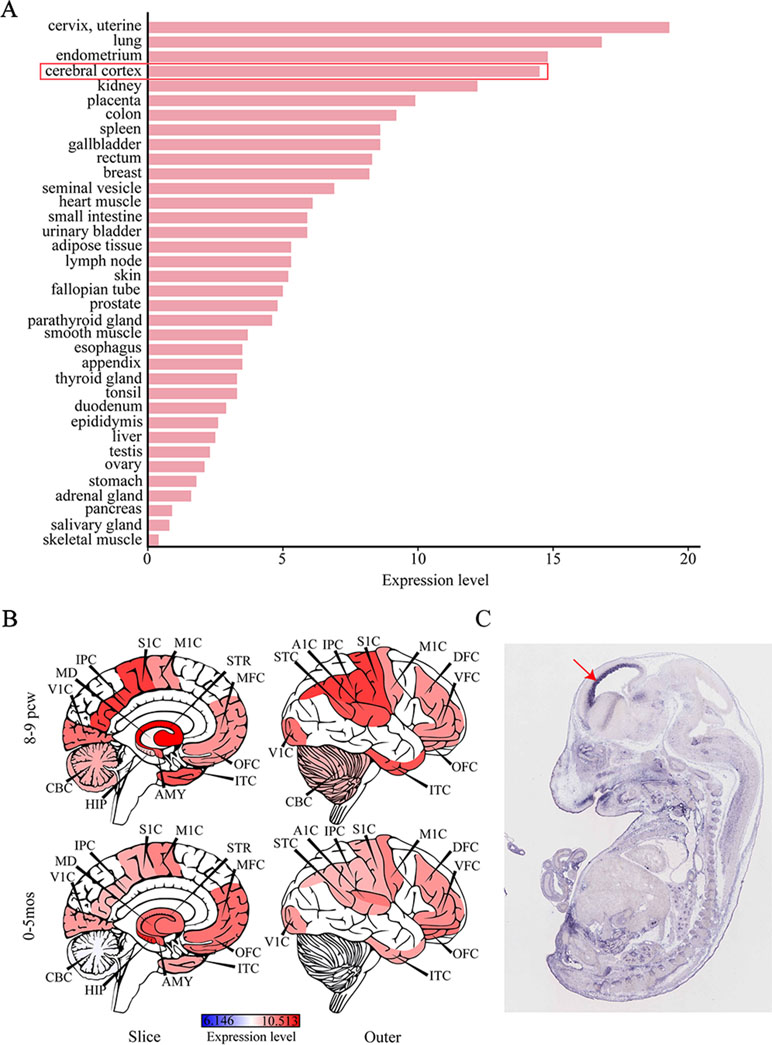
Figure 3 The expression profile of SEMA5A. (A) The expression of SEMA5A in 36 tissues in the human protein atlas (HPA) database. (B) Spatiotemporal expression profile of SEMA5A in HBT. A1C, primary auditory cortex; AMY, amygdala; CBC, cerebellar cortex; DFC, dorsolateral prefrontal cortex; HIP, hippocampus; IPC, posterior inferior parietal cortex; ITC, inferior temporal cortex; M1C, primary motor cortex; MD, mediodorsal nucleus of the thalamus; MFC, medial prefrontal cortex; OFC, orbital prefrontal cortex; S1C, primary somatosensory cortex; STC, posterior superior temporal cortex; STR, striatum; V1C, primary visual cortex; VFC, ventrolateral prefrontal cortex. (C) The expression of SEMA5A in a mouse embryo at embryonic day 14.5 was analyzed by situ hybridization. The arrow indicates the neocortex.
Furthermore, we investigated the spatiotemporal expression pattern of SEMA5A in the mouse embryo at embryonic day 14.5, on images of ISH from GenePaint. Consistent with the findings mentioned above, it showed that SEMA5A was strongly expressed in the neocortex of the embryonic mouse brain (Figure 3C). The expression profiles of SEMA5A in the brain indicates that this gene may play an essential role in the development of the brain and that dysfunctional SEMA5A may lead to human neurodevelopmental disorders such as EEs.
To gain insight into the biological function of SEMA5A and those genes that interact with SEMA5A, we extracted genes with interaction scores greater than 200 from the STRING database, and then constructed a PPI network. In the resulting PPI network, we found 196 nodes, among which 43 genes were known candidate genes in neurodevelopmental disorders. These 43 genes included 16 ASD candidate genes, six epilepsy candidate genes, six intellectual disability (ID) candidate genes, three candidate genes shared by ASD and epilepsy, nine shared by ASD and ID, one shared by ID and epilepsy and three shared by ASD, ID and epilepsy (Figure 4A). To evaluate the interconnectivity among these 43 genes, we performed an interconnected PPI analysis and observed a non-random interaction connectivity with 100,000 permutation tests (q = 1 × 10−5 for genes, q = 1 × 10−5 for connections). Moreover, we discovered that these genes were significantly enriched in PSD (q = 7.50 × 10−6) and genes under evolutionary constraint (q = 2.04 × 10−5) (Supplementary Figure S3 and S4).
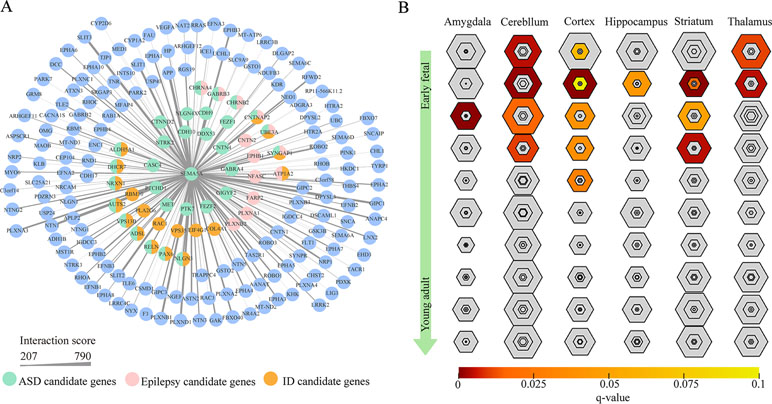
Figure 4 Protein–protein interaction (PPI) network and specific expression analysis (SEA) enrichment analysis. (A) PPI network of genes found to interact with SEMA5A in the STRING database and to have interaction scores greater than 200. The nodes represent genes, and edges represent the intersection between gene pairs. The thickness of an edge denotes the interaction scores between gene pairs. (B) SEA of genes in the PPI network. Each hexagon shows different stringency thresholds, with colors indicating q values.
Furthermore, when we performed a specific spatiotemporal expression analysis to further explore the expression patterns of the interacting genes, as expected, we observed that SEMA5A and its interacting genes were significantly enriched in the cortex, cerebellum, striatum and thalamus from early fetal to early infancy periods (q < 0.05) (Figure 4B). Next, a Gene Ontology (GO) enrichment analysis was performed to determine whether the genes in the PPI network were enriched in neurodevelopment-associated GO terms. The results showed that SEMA5A and its interacting genes were significantly enriched in axon-, neuron- and synapse-associated GO terms, such as axon development (q = 2.05 × 10−49), axon guidance (q = 5.22 × 10−39), the regulation of neuron projection development (q = 4.86 × 10−31), and synapse organization (q = 2.73 × 10−19) (Figure 5A). Moreover, it is important to note that genes in the PPI network were relevant to ASD (q = 1.88 × 10−13), developmental disorders of mental health (q = 2.48 × 10−12), Parkinson’s disease (q = 1.61 × 10−6) and epilepsy syndrome (q = 8.92 × 10−5) based on Disease Ontology (DO) analysis (Figure 5B).
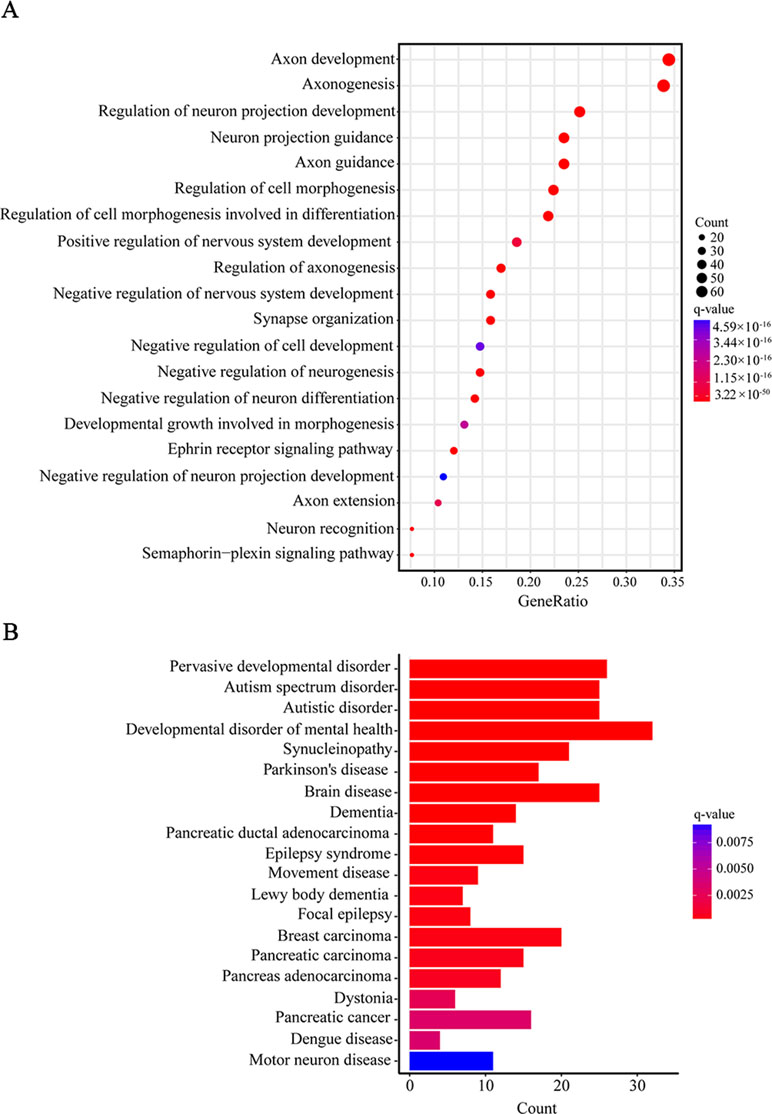
Figure 5 Gene Ontology (GO) and Disease Ontology (DO) enrichment analyses. (A) The top 20 enriched GO terms of biological process. Circle size indicates the number of genes enriched in each term. Color saturation represents the significance level. (B) The top 20 terms in the DO enrichment analysis. The x-axis shows the number of genes enriched in each term. The q-value of each term is indicated by color according to the legend.
In this study, we performed WES on three trios and presented evidence showing that a de novo missense mutation in SEMA5A was likely associated with IS. Considering the potential role of recessive inheritance in EEs, we also tried to detect homozygous inherited SNVs or indel mutations in other genes in our samples. However, there were no candidate damaging compound mutations or homozygous mutations in all patients except a compound heterozygous mutation in CLTCL1 in patient 2. SEMA5A belongs to the semaphorin gene family, the members of which serve as canonical axon guidance proteins and function in pathological conditions of the nervous system (Yaron and Zheng, 2007). It is now clear that semaphorins and their receptors play many crucial roles in the development of neural circuits (Pasterkamp, 2012), including roles in neuronal migration (Hernandez-Miranda et al., 2011), axon bundling (Van Battum et al., 2015), axon pruning (Pasterkamp, 2012) and synaptic transmission (Sahay et al., 2005; Carrillo et al., 2010). Moreover, some alterations in the expression of axon guidance proteins, such as semaphorins and ephrins, have been observed in animal models of epilepsy (Barnes et al., 2003; Xia et al., 2013). It is generally accepted that semaphorins that can act as axon guidance proteins are involved in a variety of neurological diseases, including ASD (McFadden and Minshew, 2013), epilepsy (Xia et al., 2013), Parkinson’s disease (Lin et al., 2009) and Alzheimer’s disease (Good et al., 2004).
Moreover, semaphorins are sensitive to electrical activity and experience and form a large family consisting of eight classes, among which class 5 semaphorins (SEMA5A and SEMA5B) play essential roles in the functions of the developing nervous system (Mann et al., 2007). SEMA5A, as an integral membrane protein, acts as a bifunctional guidance cue that could be directly implicated in both attractive and inhibitory processes during axon development (Kantor et al., 2004). Furthermore, Sema5A was shown to be essential for the proper development of fasciculus retroflexus in a rat model (Kantor et al., 2004). A separate study showed that Sema5A functions as a negative regulator of synaptogenesis during early brain development and synaptic transmission (Duan et al., 2014).
Cumulative evidence obtained from various sources shows that SEMA5A is a transmembrane protein that has been identified as an autism susceptibility gene in humans according to a genome-wide association study (Weiss et al., 2009), cDNA microarray technology (Melin et al., 2006) and expression quantitative trait locus mapping (Cheng et al., 2013). Notably, a de novo microdeletion in SEMA5A was identified in a patient with ASD and ID (Mosca-Boidron et al., 2016). It is worth noting that ASD and EEs often occur together and share some common genetic etiologies (Srivastava and Sahin, 2017). Abnormal synaptic plasticity may represent a common pathophysiological mechanism in ASD and epilepsy that can lead to an imbalance between excitatory and inhibitory neurotransmission in the developing brain (Brooks-Kayal, 2010). NRP2 is a receptor for both SEMA3C and SEMA3F, each of which plays an important role in axon guidance in the peripheral and central nervous systems (Chen et al., 2000) in addition to synaptic plasticity (Lee et al., 2012). Positional and functional evidence has been indicated that polymorphisms and mutations in NRP2 may be associated with autism (Wu et al., 2007). For example, NRP2 knockout mice were found to have a lowered seizure threshold and were more sensitive to chemical challenges aimed at inducing epileptogenesis (Gant et al., 2009). Notably, several studies have demonstrated that SEMA3C and SEMA3F, which are homologous to SEMA5A, are candidate genes for EEs (Barnes et al., 2003; Mefford et al., 2011). The patient carrying damaging DNM in SEMA5A in this study was diagnosed with IS at the age of only 7 months, though there was no behaviors or phenotypes related to ASD based on clinical diagnosis, which may be due to the very young age of the patient. Therefore, it is necessary to continue examinations or follow-up surveys for this patient as they become older, to evaluate whether ASD develops.
CLTCL1, a clathrin heavy chain protein in humans, is associated with the neuromuscular system (Towler et al., 2004), and neuropeptide degradation and secretion during neuronal development (Nahorski et al., 2018). The homozygous R125C mutation in CLTCL1, inherited from heterozygous parents, was predicted to be damaging and has been identified in patients with autism (Chahrour et al., 2012). A mutation in CLTCL1 located on chromosome 22q11.2 has also been associated with susceptibility to schizophrenia (Karayiorgou et al., 2010; Chahrour et al., 2012). DiGeorge syndrome is usually associated with the deletion of chromosome 22q11.2, which is linked with cognitive impairments, susceptibility to schizophrenia and neuroanatomical changes (Karayiorgou et al., 2010). It is worth noting that an interruption in the CLTCL gene observed in a patient with DiGeorge syndrome was found to contribute to the patient’s phenotype, including a seizure disorder, ID and facial dysmorphia (Holmes et al., 1997). However, whether the compound mutation in CLTCL identified in this study was associated with IS still required further genetic evidence or functional studies to support it.
To our knowledge, this is a novel DNM of SEMA5A found in an individual with IS. Our results provide genetic and functional evidence showing that SEMA5A plays a significant role in the development of the brain and might be involved in several neurodevelopmental disorders, such as EE and ASD. The discovery of damaging mutations in SEMA5A provides further evidence supporting the role of axon guidance proteins in the pathogenesis and causes underlying EE. However, to definitively establish the role of SEMA5A in IS, further genetic function studies are needed.
This study was carried out with the approval provided by the Second Affiliated Hospital and Yuying Children’s Hospital of Wenzhou Medical University. Written informed consent of all participants was collected from their parents or guardians at the time of recruitment for participation in the study and publication of the study.
QW and ZWL contributed to the drafting and revision of the manuscript, data acquisition, and analysis. ZDL, RZ, YL, and FL contributed to individual recruitment, data acquisition, and data analysis. XX, MT, and WS contributed to data acquisition and manuscript revision. XZ, JZ, and YL contributed to study concept and design, critical review, and manuscript revision.
This study was supported by the Zhejiang Provincial Natural Science Foundation of China (Grant No. LY18C060007).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.00605/full#supplementary-material
Barnes, G., Puranam, R. S., Luo, Y., McNamara, J. O. (2003). Temporal specific patterns of semaphorin gene expression in rat brain after kainic acid-induced status epilepticus. Hippocampus 13, 1–20. doi: 10.1002/hipo.10041
Berg, A. T., Berkovic, S. F., Brodie, M. J., Buchhalter, J., Cross, J. H., van Emde Boas, W., et al. (2010). Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005-2009. Epilepsia 51, 676–685. doi: 10.1111/j.1528-1167.2010.02522.x
Brooks-Kayal, A. (2010). Epilepsy and autism spectrum disorders: are there common developmental mechanisms? Brain Dev. 32, 731–738. doi: 10.1016/j.braindev.2010.04.010
Carrillo, R. A., Olsen, D. P., Yoon, K. S., Keshishian, H. (2010). Presynaptic activity and CaMKII modulate retrograde semaphorin signaling and synaptic refinement. Neuron 68, 32–44. doi: 10.1016/j.neuron.2010.09.005
Carvill, G. L., Heavin, S. B., Yendle, S. C., McMahon, J. M., O’Roak, B. J., Cook, J., et al. (2013). Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat. Genet. 45, 825–830. doi: 10.1038/ng.2646
Chahrour, M. H., Yu, T. W., Lim, E. T., Ataman, B., Coulter, M. E., Hill, R. S., et al. (2012). Whole-exome sequencing and homozygosity analysis implicate depolarization-regulated neuronal genes in autism. PLoS Genet. 8, e1002635. doi: 10.1371/journal.pgen.1002635
Chen, H., Bagri, A., Zupicich, J. A., Zou, Y., Stoeckli, E., Pleasure, S. J., et al. (2000). Neuropilin-2 regulates the development of selective cranial and sensory nerves and hippocampal mossy fiber projections. Neuron 25, 43–56. doi: 10.1016/S0896-6273(00)80870-3
Cheng, Y., Quinn, J. F., Weiss, L. A. (2013). An eQTL mapping approach reveals that rare variants in the SEMA5A regulatory network impact autism risk. Hum. Mol. Genet. 22, 2960–2972. doi: 10.1093/hmg/ddt150
Claes, L., Del-Favero, J., Ceulemans, B., Lagae, L., Van Broeckhoven, C., De Jonghe, P. (2001). De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am. J. Hum. Genet. 68, 1327–1332. doi: 10.1086/320609
Collins, M. O., Husi, H., Yu, L., Brandon, J. M., Anderson, C. N., Blackstock, W. P., et al. (2006). Molecular characterization and comparison of the components and multiprotein complexes in the postsynaptic proteome. J. Neurochem. 97 Suppl 1, 16–23. doi: 10.1111/j.1471-4159.2005.03507.x
D’Gama, A. M., Pochareddy, S., Li, M., Jamuar, S. S., Reiff, R. E., Lam, A. N., et al. (2015). Targeted DNA sequencing from autism spectrum disorder brains implicates multiple genetic mechanisms. Neuron 88, 910–917. doi: 10.1016/j.neuron.2015.11.009
Dimassi, S., Labalme, A., Ville, D., Calender, A., Mignot, C., Boutry-Kryza, N., et al. (2016). Whole-exome sequencing improves the diagnosis yield in sporadic infantile spasm syndrome. Clin. Genet. 89, 198–204. doi: 10.1111/cge.12636
Duan, Y., Wang, S. H., Song, J., Mironova, Y., Ming, G. L., Kolodkin, A. L., et al. (2014). Semaphorin 5A inhibits synaptogenesis in early postnatal- and adult-born hippocampal dentate granule cells. eLife 3. doi: 10.7554/eLife.04390
Epi4K Consortium, Epilepsy Phenome/Genome Project. (2013). De novo mutations in epileptic encephalopathies. Nature 501, 217–221. doi: 10.1038/nature12439
Fukata, Y., Fukata, M. (2017). Epilepsy and synaptic proteins. Curr. Opin. Neurobiol. 45, 1–8. doi: 10.1016/j.conb.2017.02.001
Gant, J. C., Thibault, O., Blalock, E. M., Yang, J., Bachstetter, A., Kotick, J., et al. (2009). Decreased number of interneurons and increased seizures in neuropilin 2 deficient mice: implications for autism and epilepsy. Epilepsia 50, 629–645. doi: 10.1111/j.1528-1167.2008.01725.x
Good, P. F., Alapat, D., Hsu, A., Chu, C., Perl, D., Wen, X., et al. (2004). A role for semaphorin 3A signaling in the degeneration of hippocampal neurons during Alzheimer’s disease. J. Neurochem. 91, 716–736. doi: 10.1111/j.1471-4159.2004.02766.x
He, N., Lin, Z. J., Wang, J., Wei, F., Meng, H., Liu, X. R., et al. (2019). Evaluating the pathogenic potential of genes with de novo variants in epileptic encephalopathies. Genet. Med. 21, 17–27. doi: 10.1038/s41436-018-0011-y
Hernandez-Miranda, L. R., Cariboni, A., Faux, C., Ruhrberg, C., Cho, J. H., Cloutier, J. F., et al. (2011). Robo1 regulates semaphorin signaling to guide the migration of cortical interneurons through the ventral forebrain. J. Neurosci. 31, 6174–6187. doi: 10.1523/JNEUROSCI.5464-10.2011
Hilario, J. D., Rodino-Klapac, L. R., Wang, C., Beattie, C. E. (2009). Semaphorin 5A is a bifunctional axon guidance cue for axial motoneurons in vivo. Dev. Biol. 326, 190–200. doi: 10.1016/j.ydbio.2008.11.007
Holmes, S. E., Riazi, M. A., Gong, W., McDermid, H. E., Sellinger, B. T., Hua, A., et al. (1997). Disruption of the clathrin heavy chain-like gene (CLTCL) associated with features of DGS/VCFS: a balanced (21;22)(p12;q11) translocation. Hum. Mol. Genet. 6, 357–367. doi: 10.1093/hmg/6.3.357
Iossifov, I., O’Roak, B. J., Sanders, S. J., Ronemus, M., Krumm, N., Levy, D., et al. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221. doi: 10.1038/nature13908
Jin, Z. B., Li, Z., Liu, Z., Jiang, Y., Cai, X. B., Wu, J. (2018). Identification of de novo germline mutations and causal genes for sporadic diseases using trio-based whole-exome/genome sequencing. Biol. Rev. Camb. Philos. Soc. 93, 1014–1031. doi: 10.1111/brv.12383
Kantor, D. B., Chivatakarn, O., Peer, K. L., Oster, S. F., Inatani, M., Hansen, M. J., et al. (2004). Semaphorin 5A is a bifunctional axon guidance cue regulated by heparan and chondroitin sulfate proteoglycans. Neuron 44, 961–975. doi: 10.1016/j.neuron.2004.12.002
Karayiorgou, M., Simon, T. J., Gogos, J. A. (2010). 22q11.2 microdeletions: linking DNA structural variation to brain dysfunction and schizophrenia. Nat. Rev. Neurosci. 11, 402–416. doi: 10.1038/nrn2841
Lau, C. G., Zukin, R. S. (2007). NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat. Rev. Neurosci. 8, 413–426. doi: 10.1038/nrn2153
Lee, K., Kim, J. H., Kwon, O. B., An, K., Ryu, J., Cho, K., et al. (2012). An activity-regulated microRNA, miR-188, controls dendritic plasticity and synaptic transmission by downregulating neuropilin-2. J. Neurosci. 32, 5678–5687. doi: 10.1523/JNEUROSCI.6471-11.2012
Lek, M., Karczewski, K. J., Minikel, E. V., Samocha, K. E., Banks, E., Fennell, T., et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. doi: 10.1038/nature19057
Lemke, J. R., Hendrickx, R., Geider, K., Laube, B., Schwake, M., Harvey, R. J., et al. (2014). GRIN2B mutations in West syndrome and intellectual disability with focal epilepsy. Ann. Neurol. 75, 147–154. doi: 10.1002/ana.24073
Li, J., Jiang, Y., Wang, T., Chen, H., Xie, Q., Shao, Q., et al. (2015). mirTrios: an integrated pipeline for detection of de novo and rare inherited mutations from trios-based next-generation sequencing. J. Med. Genet. 52, 275–281. doi: 10.1136/jmedgenet-2014-102656
Lin, L., Lesnick, T. G., Maraganore, D. M., Isacson, O. (2009). Axon guidance and synaptic maintenance: preclinical markers for neurodegenerative disease and therapeutics. Trends Neurosci. 32, 142–149. doi: 10.1016/j.tins.2008.11.006
Liu, Q., Chen, C., Shen, E., Zhao, F., Sun, Z., Wu, J. (2012). Detection, annotation and visualization of alternative splicing from RNA-Seq data with SplicingViewer. Genomics 99, 178–182. doi: 10.1016/j.ygeno.2011.12.003
Mann, F., Chauvet, S., Rougon, G. (2007). Semaphorins in development and adult brain: implication for neurological diseases. Prog. Neurobiol. 82, 57–79. doi: 10.1016/j.pneurobio.2007.02.011
McFadden, K., Minshew, N. J. (2013). Evidence for dysregulation of axonal growth and guidance in the etiology of ASD. Front. Hum. Neurosci. 7, 671. doi: 10.3389/fnhum.2013.00671
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
McTague, A., Howell, K. B., Cross, J. H., Kurian, M. A., Scheffer, I. E. (2016). The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. 15, 304–316. doi: 10.1016/S1474-4422(15)00250-1
Mefford, H. C., Yendle, S. C., Hsu, C., Cook, J., Geraghty, E., McMahon, J. M., et al. (2011). Rare copy number variants are an important cause of epileptic encephalopathies. Ann. Neurol. 70, 974–985. doi: 10.1002/ana.22645
Melin, M., Carlsson, B., Anckarsater, H., Rastam, M., Betancur, C., Isaksson, A., et al. (2006). Constitutional downregulation of SEMA5A expression in autism. Neuropsychobiology 54, 64–69. doi: 10.1159/000096040
Michaelson, J. J., Shi, Y., Gujral, M., Zheng, H., Malhotra, D., Jin, X., et al. (2012). Whole-genome sequencing in autism identifies hot spots for de novo germline mutation. Cell 151, 1431–1442. doi: 10.1016/j.cell.2012.11.019
Mosca-Boidron, A. L., Gueneau, L., Huguet, G., Goldenberg, A., Henry, C., Gigot, N., et al. (2016). A de novo microdeletion of SEMA5A in a boy with autism spectrum disorder and intellectual disability. Eur. J. Hum. Genet. 24, 838–843. doi: 10.1038/ejhg.2015.211
Nabbout, R., Dulac, O. (2003). Epileptic encephalopathies: a brief overview. J. Clin. Neurophysiol. 20, 393–397. doi: 10.1097/00004691-200311000-00002
Nahorski, M. S., Borner, G. H. H., Shaikh, S. S., Davies, A. K., Al-Gazali, L., Antrobus, R., et al. (2018). Clathrin heavy chain 22 contributes to the control of neuropeptide degradation and secretion during neuronal development. Sci. Rep. 8, 2340. doi: 10.1038/s41598-018-19980-0
Pardo, C. A., Nabbout, R., Galanopoulou, A. S. (2014). Mechanisms of epileptogenesis in pediatric epileptic syndromes: Rasmussen encephalitis, infantile spasms, and febrile infection-related epilepsy syndrome (FIRES). Neurotherapeutics 11, 297–310. doi: 10.1007/s13311-014-0265-2
Pasterkamp, R. J. (2012). Getting neural circuits into shape with semaphorins. Nat. Rev. Neurosci. 13, 605–618. doi: 10.1038/nrn3302
Pasterkamp, R. J., Giger, R. J. (2009). Semaphorin function in neural plasticity and disease. Curr. Opin. Neurobiol. 19, 263–274. doi: 10.1016/j.conb.2009.06.001
Pavone, P., Striano, P., Falsaperla, R., Pavone, L., Ruggieri, M. (2014). Infantile spasms syndrome, West syndrome and related phenotypes: what we know in 2013. Brain Dev. 36, 739–751. doi: 10.1016/j.braindev.2013.10.008
Penzes, P., Cahill, M. E., Jones, K. A., VanLeeuwen, J. E., Woolfrey, K. M. (2011). Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 14, 285–293. doi: 10.1038/nn.2741
Ran, X., Li, J., Shao, Q., Chen, H., Lin, Z., Sun, Z. S., et al. (2015). EpilepsyGene: a genetic resource for genes and mutations related to epilepsy. Nucleic Acids Res. 43, D893–D899. doi: 10.1093/nar/gku943
Sahay, A., Kim, C. H., Sepkuty, J. P., Cho, E., Huganir, R. L., Ginty, D. D., et al. (2005). Secreted semaphorins modulate synaptic transmission in the adult hippocampus. J. Neurosci. 25, 3613–3620. doi: 10.1523/JNEUROSCI.5255-04.2005
Samocha, K. E., Robinson, E. B., Sanders, S. J., Stevens, C., Sabo, A., McGrath, L. M., et al. (2014). A framework for the interpretation of de novo mutation in human disease. Nat. Genet. 46, 944–950. doi: 10.1038/ng.3050
Scheffer, I. E., Berkovic, S., Capovilla, G., Connolly, M. B., French, J., Guilhoto, L., et al. (2017). ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia 58, 512–521. doi: 10.1111/epi.13709
Shbarou, R., Mikati, M. A. (2016). The expanding clinical spectrum of genetic pediatric epileptic encephalopathies. Semin. Pediatr. Neurol. 23, 134–142. doi: 10.1016/j.spen.2016.06.002
Shen, K., Cowan, C. W. (2010). Guidance molecules in synapse formation and plasticity. Cold Spring Harb. Perspect. Biol. 2, a001842. doi: 10.1101/cshperspect.a001842
Song, I., Huganir, R. L. (2002). Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci. 25, 578–588. doi: 10.1016/S0166-2236(02)02270-1
Srivastava, S., Sahin, M. (2017). Autism spectrum disorder and epileptic encephalopathy: common causes, many questions. J. Neurodev. Disord. 9, 23. doi: 10.1186/s11689-017-9202-0
Towler, M. C., Gleeson, P. A., Hoshino, S., Rahkila, P., Manalo, V., Ohkoshi, N., et al. (2004). Clathrin isoform CHC22, a component of neuromuscular and myotendinous junctions, binds sorting nexin 5 and has increased expression during myogenesis and muscle regeneration. Mol. Biol. Cell 15, 3181–3195. doi: 10.1091/mbc.e04-03-0249
Van Battum, E. Y., Brignani, S., Pasterkamp, R. J. (2015). Axon guidance proteins in neurological disorders. Lancet Neurol. 14, 532–546. doi: 10.1016/S1474-4422(14)70257-1
Veltman, J. A., Brunner, H. G. (2012). De novo mutations in human genetic disease. Nat. Rev. Genet. 13, 565–575. doi: 10.1038/nrg3241
Wang, T., Liu, Q., Li, X., Wang, X., Li, J., Zhu, X., et al. (2013). RRBS-analyser: a comprehensive web server for reduced representation bisulfite sequencing data analysis. Hum. Mutat. 34, 1606–1610. doi: 10.1002/humu.22444
Weiss, L. A., Arking, D. E., The Gene Discovery Project of Johns Hopkins & the Autism Consortium (2009). A genome-wide linkage and association scan reveals novel loci for autism. Nature 461, 802–808. doi: 10.1038/nature08490
Wu, S., Yue, W., Jia, M., Ruan, Y., Lu, T., Gong, X., et al. (2007). Association of the neuropilin-2 (NRP2) gene polymorphisms with autism in Chinese Han population. Am. J. Med. Genet. B Neuropsychiatr. Genet. 144B, 492–495. doi: 10.1002/ajmg.b.30495
Xia, Y., Luo, C., Dai, S., Yao, D. (2013). Increased EphA/ephrinA expression in hippocampus of pilocarpine treated mouse. Epilepsy Res. 105, 20–29. doi: 10.1016/j.eplepsyres.2013.01.001
Keywords: epileptic encephalopathy, infantile spasms, de novo mutations, SEMA5A, whole-exome sequencing
Citation: Wang Q, Liu Z, Lin Z, Zhang R, Lu Y, Su W, Li F, Xu X, Tu M, Lou Y, Zhao J and Zheng X (2019) De Novo Germline Mutations in SEMA5A Associated With Infantile Spasms. Front. Genet. 10:605. doi: 10.3389/fgene.2019.00605
Received: 04 December 2018; Accepted: 07 June 2019;
Published: 10 July 2019.
Edited by:
Fan Jin, Zhejiang University, ChinaReviewed by:
Yueqiu Tan, Central South University, ChinaCopyright © 2019 Wang, Liu, Lin, Zhang, Lu, Su, Li, Xu, Tu, Lou, Zhao and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoqun Zheng, anN6aGVuZ3hxQDE2My5jb20=; Junzhao Zhao, ei5qb3ljZTA4QDE2My5jb20=; Yongliang Lou, bG91eW9uZ2xpYW5nMjAxM0AxNjMuY29t
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.