 Emanuele Micaglio1
Emanuele Micaglio1 Michelle M. Monasky1
Michelle M. Monasky1 Giuseppe Ciconte1
Giuseppe Ciconte1 Gabriele Vicedomini1
Gabriele Vicedomini1 Manuel Conti1Valerio Mecarocci1Luigi Giannelli1Federica Giordano1Alberto Pollina1Massimo Saviano1Paolo R. Pozzi1
Manuel Conti1Valerio Mecarocci1Luigi Giannelli1Federica Giordano1Alberto Pollina1Massimo Saviano1Paolo R. Pozzi1 Chiara Di Resta2,3
Chiara Di Resta2,3 Sara Benedetti4Maurizio Ferrari2,3,4Vincenzo Santinelli1
Sara Benedetti4Maurizio Ferrari2,3,4Vincenzo Santinelli1 Carlo Pappone1*
Carlo Pappone1*- 1Arrhythmology Department, IRCCS Policlinico San Donato, Milan, Italy
- 2Genomic Unit for the Diagnosis of Human Pathologies, Division of Genetics and Cellular Biology, IRCCS San Raffaele Hospital, Milan, Italy
- 3Vita-Salute San Raffaele University, Milan, Italy
- 4Laboratory of Clinical Molecular Biology and Cytogenetics, IRCCS San Raffaele Hospital, Milan, Italy
In this case report, we characterize a novel inherited frameshift mutation c.4700_4701del (p.Phe1567Cysfs*221) in a single copy of the SCN5A gene and its association with Brugada syndrome (BrS). The proband experienced a life-threatening ventricular arrhythmia successfully treated with DC-shock and he also suffered from supraventricular tachycardia. Ajmaline test confirmed the BrS diagnosis. No other mutation nor low frequency variants in the other 23 analyzed genes were detected. The same mutation was found in the father and sister, who were both diagnosed with BrS. We hypothesize that this mutation could be responsible for BrS and potentially linked to supraventricular tachycardias. Further studies are needed to confirm this observation and to assess the clinical relevance of this mutation, in terms of risk-stratification.
Introduction
The Brugada syndrome (BrS) is characterized by a coved-type ST-segment elevation in the right precordial leads on the electrocardiogram (ECG) and an increased risk of sudden cardiac death (SCD). Diagnosis requires the presence of a type 1 BrS pattern on the ECG that occurs either spontaneously or after administration of a sodium channel blocking agent, such as ajmaline (Antzelevitch et al., 2016), which reveals the type 1 pattern. The arrhythmias that occur in BrS can be eliminated using transcatheter radiofrequency (RF) ablation of the arrhythmogenic substrate (AS) that is usually found in the epicardial surface of the right ventricle (RV). Ajmaline may be used to fully visualize the extent of the AS to improve the success of the procedure.
Brugada syndrome is inherited as an autosomal dominant condition with incomplete penetrance. The most commonly associated genetic mutations are found in the SCN5A gene, thought to be responsible for about 15–30% of BrS cases (Kapplinger et al., 2010). However, no genetic mutations of any kind are detected in the majority of BrS patients, perhaps due to both locus heterogeneity and large genomic rearrangements only detectable with other genetic analyses. The need exists to further understand the genetics of BrS, especially for the high rate of asymptomatic individuals who could undergo pre-symptomatic non-invasive genetic testing and, if mutated, be recommended for a specific arrhythmologic follow-up.
Case Presentation
Written informed consent of human subjects included in this case report was obtained for their participation in the study and for publication of this case report. The procedures employed were reviewed and approved by the local Ethics Committee. The proband is a 28-year-old male with a history of recurrent syncope episodes. A 12-lead ECG performed at the age of 27 years demonstrated a type 3 BrS pattern. After a couple of weeks, the proband underwent a 12-lead ECG that demonstrated atrioventricular nodal reentrant tachycardia (AVNRT). One month later, he was admitted to the emergency room elsewhere because of a life-threatening ventricular arrhythmia successfully treated with DC-shock. After sinus rhythm restoration, a type 1 BrS pattern was noticed. The patient underwent an electrophysiological study (EPS) and was found to be inducible. Therefore, he received a subcutaneous ICD.
At this stage he came to our attention. All the performed clinical tests (metabolic assessment, EEG, anion-cation concentration, hormonal functions) were negative. Next, an ajmaline test confirmed BrS. Thereafter, he underwent catheter ablation of AVNRT, and the AS responsible for BrS (Figure 1). The EPS and ajmaline test were repeated after ablation and were both negative. A control echocardiogram showed no pericardial effusion.
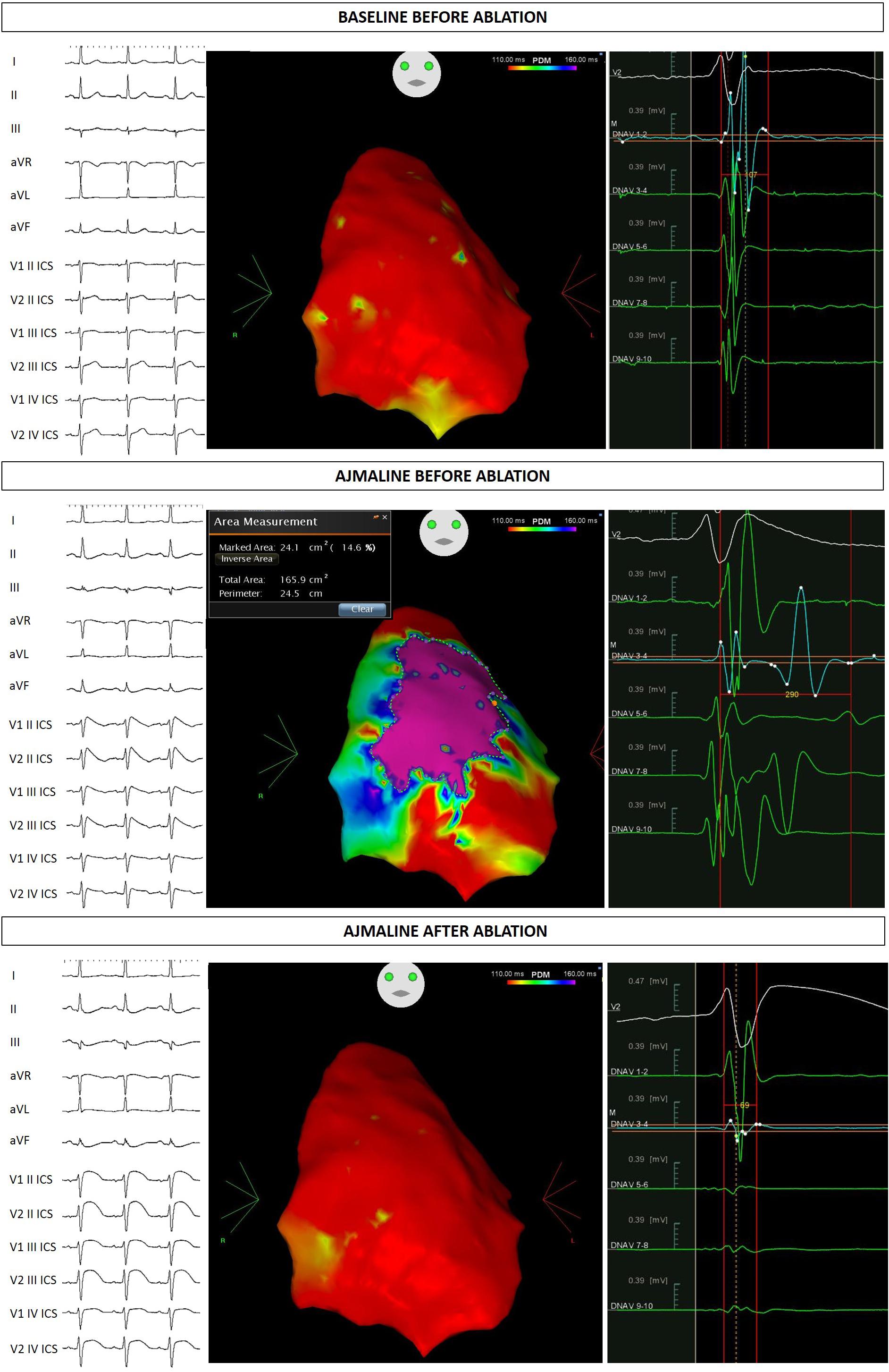
Figure 1. Proband ECG and potential duration map at baseline, after ajmaline administration, and after ablation.
Genetic Studies
After careful genetic counseling, we decided to perform a genetic analysis of genomic DNA extracted from saliva and study 24 genes known to be causative of BrS (ABCC9, AKAP9, CACNA1C, CACNA2D1, CACNB2, DSG2, GPD1L, HCN4, KCND2, KCND3, KCNE3, KCNE5, KCNH2, KCNJ8, MOG1, PKP2, RANGRF, SCN5A, SCN10A, SCN1B, SCN2B, SCN3B, SEMA3A, TRPM4). This analysis demonstrated that the proband carries the novel mutation c.4700_4701del (p.Phe1567Cysfs*221) in a single copy of the SCN5A gene (LOVD1). No other mutation nor low frequency variants in the other 23 analyzed genes were detected. The sequence variation can be found in Figure 2.
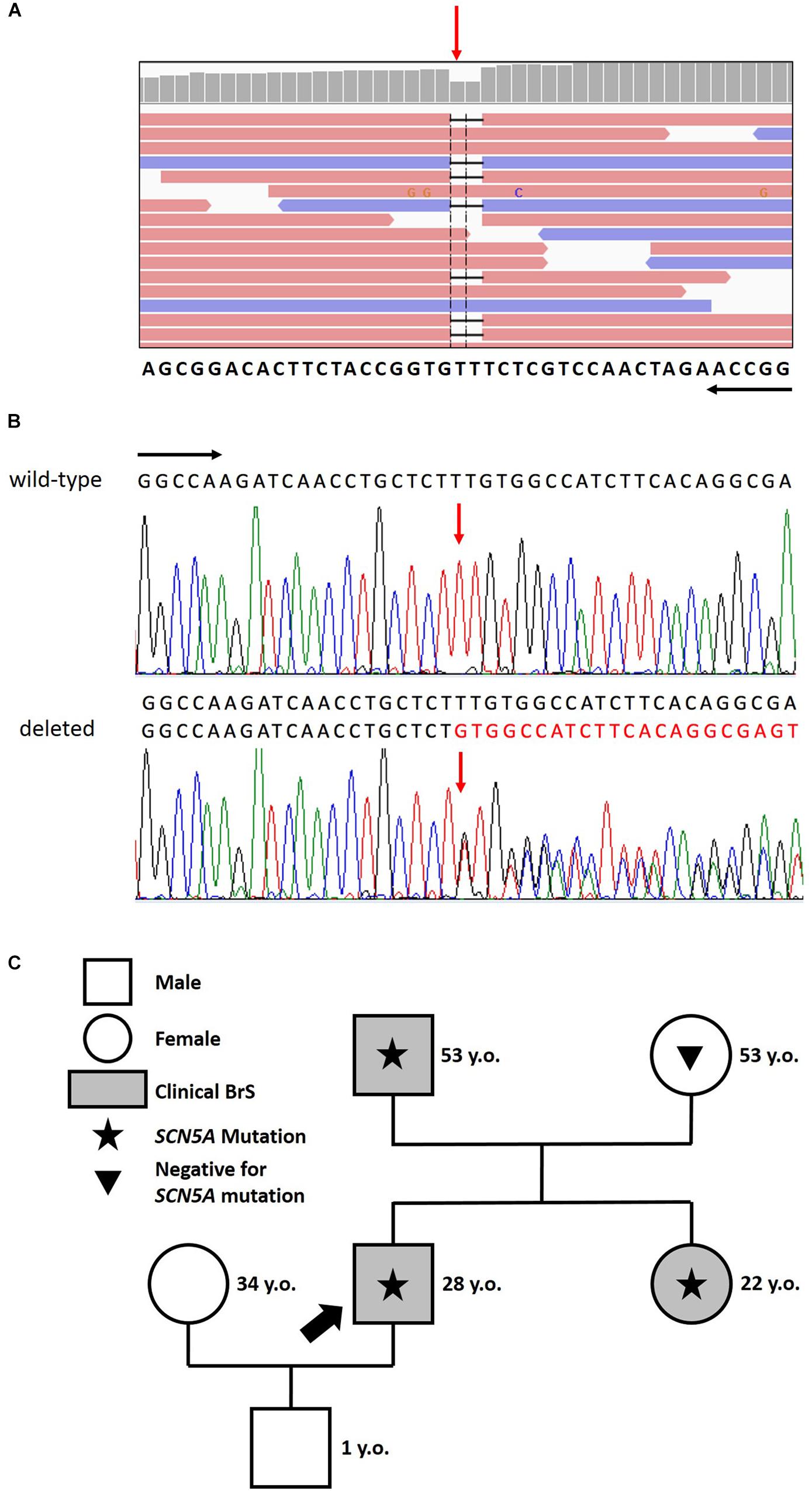
Figure 2. (A) Identification of the heterozygous c.4700_4701del deletion by next generation sequencing. NGS paired-end reads loaded in the IGV genome browser. The gene sequence is in the reverse orientation on the chromosome. (B) Identification of the heterozygous c.4700_4701del deletion by sanger sequencing. Sanger sequencing confirmation of the deletion compared with the wild-type sequence. The wt (black) and mutated (red) sequences are reported. The position of the two bases deleted is indicated by the red arrow. Black arrows indicate the direction of the gene. (C) Family pedigree. Proband identified with an arrow. Square: male; Circle: female; Shaded: clinically affected by Brugada syndrome; Star: molecularly confirmed SCN5A mutation: Triangle: genetically tested and negative for SCN5A mutation.
Assessment of Family Members
The proband’s father is an otherwise healthy 53-year-old man who suffered from atrial fibrillation since he was 23 years old. At the age of about forty, he was diagnosed with arterial hypertension. Consequently, several 12-lead ECGs were performed, but none raised the suspicion of BrS. A positive ajmaline test confirmed a BrS diagnosis. The patient was inducible during EPS (Figure 3). Thus, the patient underwent an ICD implantation, AS ablation (Figure 3), and was referred for genetic counseling. Genetic testing revealed the variant c.4700_4701del (p.Phe1567Cysfs*221) in a single copy of the SCN5A gene (Figure 2).
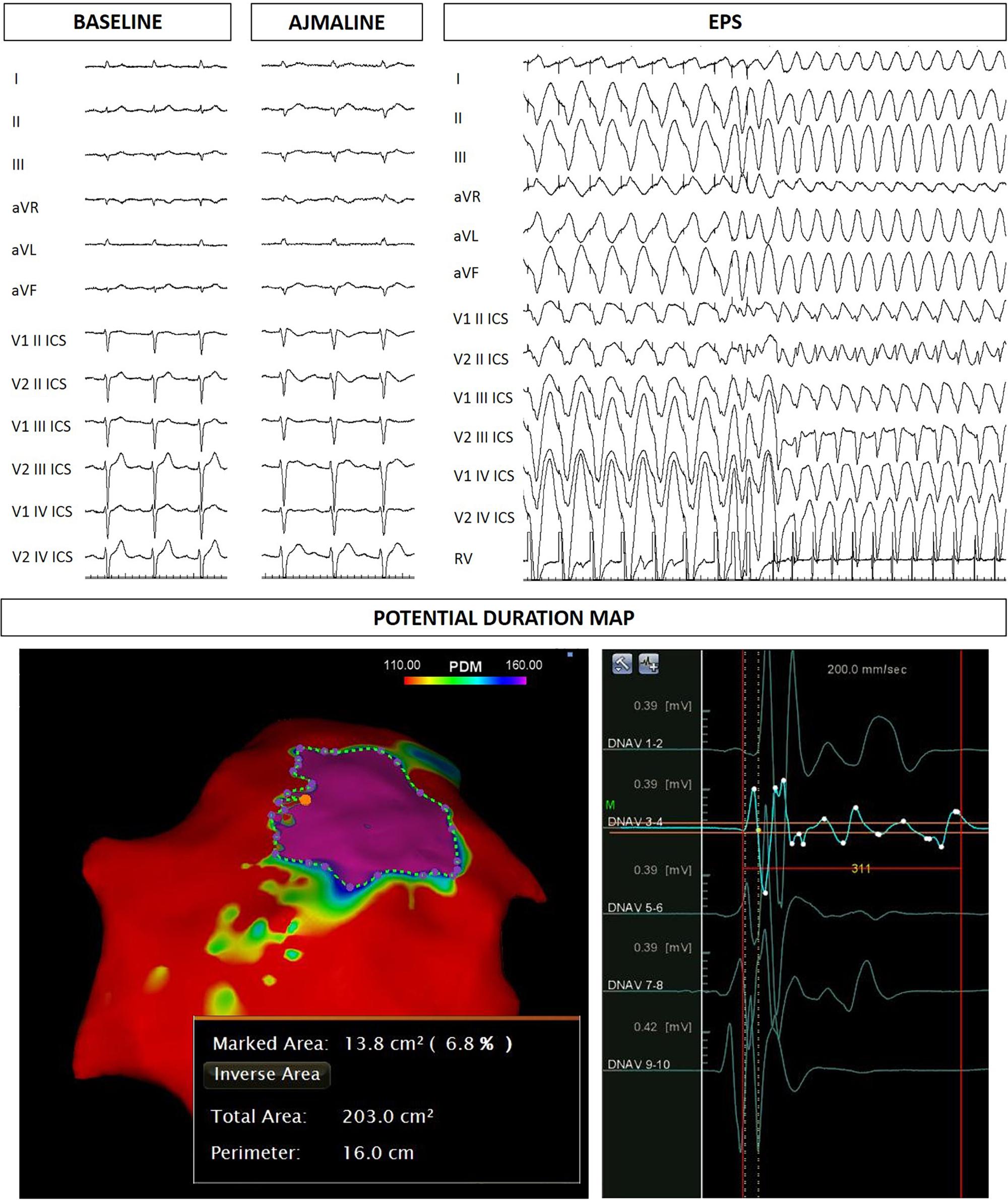
Figure 3. Father of the proband: ECG at baseline, after ajmaline administration, VT/VF inducibility during EPS. Epicardial arrhythmogenic substrate.
The proband’s 22-year-old sister experienced syncope after gastroenteritis at the age of 20 years. She also experienced recurrent unexplained syncope episodes. Consequently, several 12-lead ECGs were performed, but none raised the suspicion of BrS. A positive ajmaline test confirmed a BrS diagnosis. For these reasons she received ICD. AS ablation was performed (Figure 4). Genetic testing revealed the variant c.4700_4701del (p.Phe1567Cysfs*221) in a single copy of the SCN5A gene.
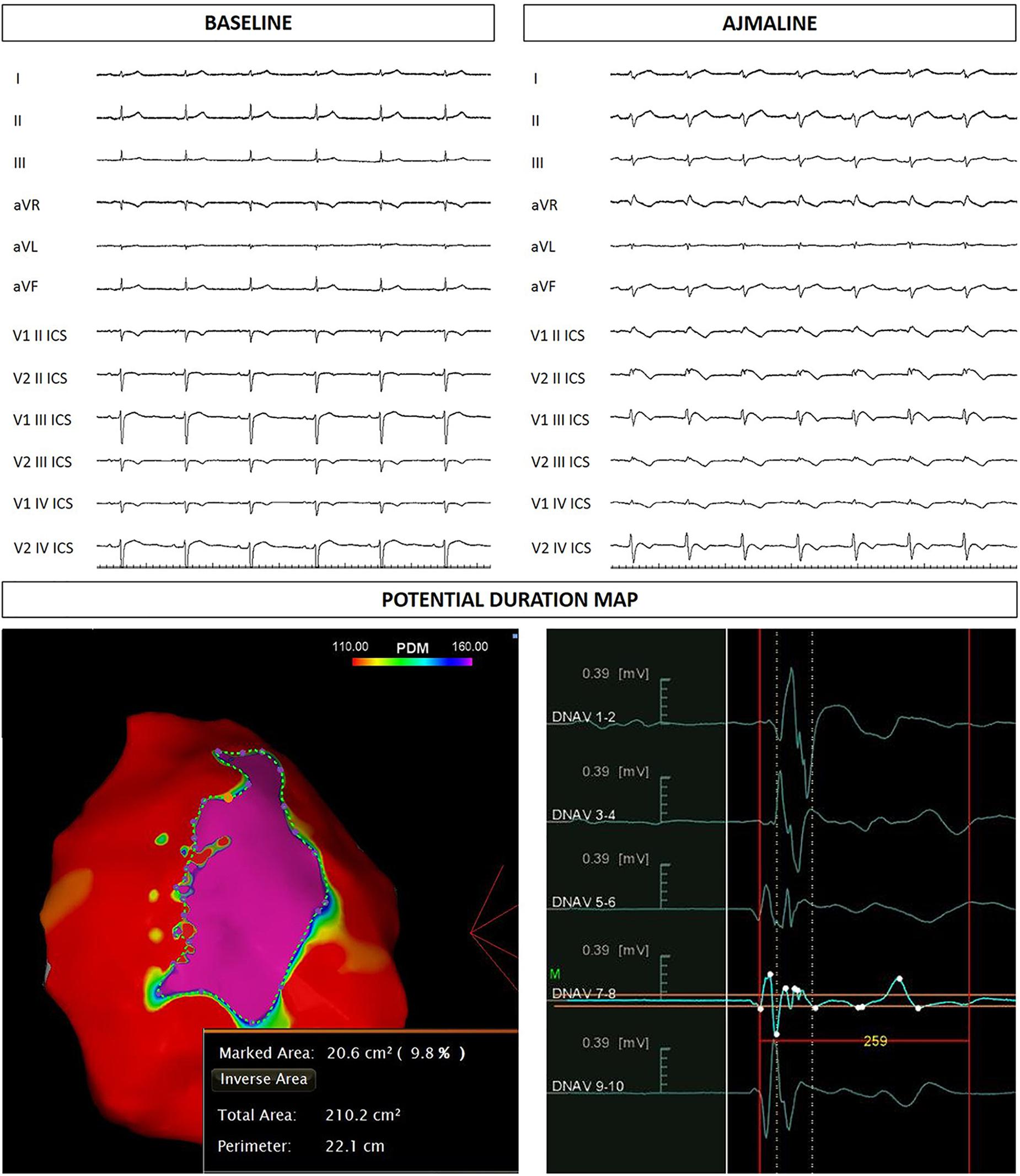
Figure 4. Sister of the proband: ECG at baseline and after ajmaline administration. Epicardial arrhythmogenic substrate.
The proband’s mother does not carry the familial variant in the SCN5A gene and any kind of consanguinity with her husband was excluded.
Discussion
Many studies have described single copy SCN5A frameshift mutations in BrS (Probst et al., 2009; Dolz-Gaiton et al., 2013). It is known that SCN5A frameshift mutations can cause BrS alone or in compound heterozygosity with a missense mutation (Barajas-Martinez et al., 2013). Our hypothesis is that the novel frameshift mutation herein described is responsible for the proband’s clinical picture characterized by a complex arrhythmic phenotype.
Brugada syndrome clinical expression varies from totally asymptomatic to life-threatening arrhythmias. BrS patients harboring a compound heterozygous mutation of the SCN5A gene seem to be affected by the worst clinical picture (Sacilotto et al., 2017). However, a possible rescue effect due to the different contributions of two mutations in the same gene has been reported (Nunez et al., 2013).
A relationship between syncopal episodes and pediatric arrhythmias was known even before the discovery of BrS (Romano et al., 1963). Further studies have highlighted the frequency of supraventricular arrhythmias in patients affected by BrS (Liu et al., 2015). A clinical study involving 46 BrS patients (39 male and 7 female) showed that 5 (3 male and 2 female) exhibited supraventricular tachycardias (Liu et al., 2015). Another clinical study (Hasdemir, 2016) emphasized the prevalence of atrial arrhythmias in BrS patients. In fact, mutations in the SCN5A gene have been found responsible for atrial fibrillation, Long QT syndrome type 3, sick sinus syndrome type 2, idiopathic ventricular fibrillation, and heart block type 1A (Olson et al., 2005). While atrial fibrillation is the most common arrhythmia to overlap with BrS, cases of supraventricular tachycardia have been described (Milanesi et al., 2015). An article by Hasdemir et al. (2015) studied 96 patients with AVNRT screened for BrS. The result was that 26 out of 96 patients exhibited a BrS type 1 pattern after ajmaline challenge. Genetic testing was then performed on these patients exhibiting a dual phenotype, revealing mutations in genes known or suspected to be responsible for sodium channel loss-of-function (Hasdemir et al., 2015). In fact, several familial forms of AVNRT have been reported (Hayes et al., 2004; Namgung et al., 2012). Thus, the overlap between AVNRT and BrS may well be underappreciated.
SCN5A mutations have been described as causative of a variety of pathologies. SCN5A missense mutations can be associated with BrS or atrial fibrillation, while frameshift mutations can result in pathologies including BrS and AVNRT (Chen et al., 1998). While it is certain that SCN5A mutations can cause BrS, it is less clear whether the SCN5A mutation described herein could be responsible for the other phenotypes. Variants in genes not tested for in our patients have been associated with AVNRT (Andreasen et al., 2018). Thus, we cannot rule out the existence of AVNRT-causing variants that were not detected in this study. Additionally, we cannot exclude mitochondrial DNA variations between the proband and his sister that may account for variations in phenotype, as mitochondrial and nuclear genomes may cross-regulate each other (Kim et al., 2018). Regardless, the novel mutation herein described should be further investigated as a cause of BrS and possibly the other pathologies presented. Finally, BrS arrhythmogenic substrate ablation is a novel approach for this disease and long-term follow-up data are not yet available.
Concluding Remarks
In this report, we documented for the first time the novel inherited frameshift mutation c.4700_4701del (p.Phe1567Cysfs*221) in a single copy of the SCN5A gene and its association with BrS. Further studies are needed to confirm this observation and to assess the clinical relevance of this mutation, in terms of risk-stratification.
Ethics Statement
Informed consent of human subjects included in this case series report was obtained. The procedures employed were reviewed and approved by the local Ethics Committee.
Author Contributions
EM, GC, GV, MC, VM, LG, FG, AP, MS, PP, CD, SB, MF, VS, and CP collected the data. EM and MM wrote the manuscript. All authors interpreted the results, critically reviewed the manuscript, and approved the final version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was partially supported by Ricerca Corrente funding from Italian Ministry of Health to IRCCS Policlinico San Donato. The authors thank the members of the family for their help and participation in the study.
Footnotes
References
Andreasen, L., Ahlberg, G., Tang, C., Andreasen, C., Hartmann, J. P., Tfelt-Hansen, J., et al. (2018). Next-generation sequencing of AV nodal reentrant tachycardia patients identifies broad spectrum of variants in ion channel genes. Eur. J. Hum. Genet. 26, 660–668. doi: 10.1038/s41431-017-0092-0
Antzelevitch, C., Yan, G. X., Ackerman, M. J., Borggrefe, M., Corrado, D., Guo, J., et al. (2016). J-Wave syndromes expert consensus conference report: emerging concepts and gaps in knowledge. Heart Rhythm 13:e295–e324. doi: 10.1016/j.hrthm.2016.05.024
Barajas-Martinez, H., Hu, D., and Antzelevitch, C. (2013). [Genetic and molecular basis for sodium channel-mediated Brugada syndrome]. Arch. Cardiol. Mex. 83, 295–302. doi: 10.1016/j.acmx.2013.10.001
Chen, Q., Kirsch, G. E., Zhang, D., Brugada, R., Brugada, J., Brugada, P., et al. (1998). Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 392, 293–296. doi: 10.1038/32675
Dolz-Gaiton, P., Nunez, M., Nunez, L., Barana, A., Amoros, I., Matamoros, M., et al. (2013). Functional characterization of a novel frameshift mutation in the C-terminus of the Nav1.5 channel underlying a Brugada syndrome with variable expression in a Spanish family. PLoS One 8:e81493. doi: 10.1371/journal.pone.0081493
Hasdemir, C. (2016). Atrial arrhythmias in inherited arrhythmogenic disorders. J. Arrhythm 32, 366–372. doi: 10.1016/j.joa.2015.11.007
Hasdemir, C., Payzin, S., Kocabas, U., Sahin, H., Yildirim, N., Alp, A., et al. (2015). High prevalence of concealed Brugada syndrome in patients with atrioventricular nodal reentrant tachycardia. Heart Rhythm 12, 1584–1594. doi: 10.1016/j.hrthm.2015.03.015
Hayes, J. J., Sharma, P. P., Smith, P. N., and Vidaillet, H. J. (2004). Familial atrioventricular nodal reentry tachycardia. Pacing Clin. Electrophysiol. 27, 73–76. doi: 10.1111/j.1540-8159.2004.00388.x
Kapplinger, J. D., Tester, D. J., Alders, M., Benito, B., Berthet, M., Brugada, J., et al. (2010). An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm 7, 33–46. doi: 10.1016/j.hrthm.2009.09.069
Kim, K. H., Son, J. M., Benayoun, B. A., and Lee, C. (2018). The mitochondrial-encoded peptide MOTS-c translocates to the nucleus to regulate nuclear gene expression in response to metabolic stress. Cell Metab. 28, 516–524.e7. doi: 10.1016/j.cmet.2018.06.008
Liu, B., Guo, C., Fang, D., and Guo, J. (2015). Clinical observations of supraventricular arrhythmias in patients with brugada syndrome. Int. J. Clin. Exp. Med. 8, 14520–14526.
Milanesi, R., Bucchi, A., and Baruscotti, M. (2015). The genetic basis for inherited forms of sinoatrial dysfunction and atrioventricular node dysfunction. J. Interv. Card Electrophysiol. 43, 121–134. doi: 10.1007/s10840-015-9998-z
Namgung, J., Kwak, J. J., Choe, H., Kwon, S. U., Doh, J. H., Lee, S. Y., et al. (2012). Familial occurrence of atrioventricular nodal reentrant tachycardia in a mother and her son. Korean Circ. J. 42, 718–721. doi: 10.4070/kcj.2012.42.10.718
Nunez, L., Barana, A., Amoros, I., De La Fuente, M. G., Dolz-Gaiton, P., Gomez, R., et al. (2013). p.D1690N Nav1.5 rescues p.G1748D mutation gating defects in a compound heterozygous Brugada syndrome patient. Heart Rhythm 10, 264–272. doi: 10.1016/j.hrthm.2012.10.025
Olson, T. M., Michels, V. V., Ballew, J. D., Reyna, S. P., Karst, M. L., Herron, K. J., et al. (2005). Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA 293, 447–454.
Probst, V., Wilde, A. A., Barc, J., Sacher, F., Babuty, D., Mabo, P., et al. (2009). SCN5A mutations and the role of genetic background in the pathophysiology of Brugada syndrome. Circ. Cardiovasc. Genet. 2, 552–557. doi: 10.1161/CIRCGENETICS.109.853374
Romano, C., Gemme, G., and Pongiglione, R. (1963). [Rare cardiac arrythmias of the pediatric age. II. syncopal attacks due to paroxysmal ventricular fibrillation. (presentation of 1st case in italian pediatric literature)]. Clin. Pediatr. 45, 656–683.
Keywords: Brugada syndrome, sudden cardiac death, genetic testing, mutation, SCN5A, sodium channel, arrhythmia, atrial fibrillation
Citation: Micaglio E, Monasky MM, Ciconte G, Vicedomini G, Conti M, Mecarocci V, Giannelli L, Giordano F, Pollina A, Saviano M, Pozzi PR, Di Resta C, Benedetti S, Ferrari M, Santinelli V and Pappone C (2019) Novel SCN5A Frameshift Mutation in Brugada Syndrome Associated With Complex Arrhythmic Phenotype. Front. Genet. 10:547. doi: 10.3389/fgene.2019.00547
Received: 30 July 2018; Accepted: 23 May 2019;
Published: 06 June 2019.
Edited by:
Prashant Kumar Verma, All India Institute of Medical Sciences Rishikesh, IndiaReviewed by:
Nelson L. S. Tang, The Chinese University of Hong Kong, ChinaGiannis G. Baltogiannis, Vrije Universiteit Brussel, Belgium
Copyright © 2019 Micaglio, Monasky, Ciconte, Vicedomini, Conti, Mecarocci, Giannelli, Giordano, Pollina, Saviano, Pozzi, Di Resta, Benedetti, Ferrari, Santinelli and Pappone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carlo Pappone, Y2FybG8ucGFwcG9uZUBhZi1hYmxhdGlvbi5vcmc=