
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Genet., 03 April 2019
Sec. Genetics of Aging
Volume 10 - 2019 | https://doi.org/10.3389/fgene.2019.00303
This article is part of the Research TopicProceedings of the International Conference on Interventions to Extend Healthspan and LifespanView all 8 articles
 Wolfgang Wagner1,2*
Wolfgang Wagner1,2*Replicative senescence of cells in vitro is often considered as counterpart for aging of the organism in vivo. In fact, both processes are associated with functional decay and similar molecular modifications. On epigenetic level, replicative senescence and aging evoke characteristic modifications in the DNA methylation (DNAm) pattern, but at different sites in the genome. Various epigenetic signatures, which are often referred to as epigenetic clocks, provide useful biomarkers: Senescence-associated epigenetic modifications can be used for quality control of cell preparations or to elucidate effects of culture conditions on the state of cellular aging. Age-associated epigenetic modifications hold high expectations to determine chronological age in forensics or to identify parameters that impact on biological aging. Despite these differences, there are some striking similarities between senescence- and age-associated DNAm, such as complete rejuvenation during reprogramming into induced pluripotent stem cells (iPSCs). It is yet unclear what makes epigenetic clocks tick, but there is evidence that the underlying mechanisms of both processes are related to similar modifications in the histone code or higher order chromatin. Replicative senescence therefore appears to be a suitable model system to gain better insight into how organismal aging might be governed epigenetically.
Primary cells, which are taken directly from living tissues, can only be culture expanded for a limited number of cell divisions before entering irreversible proliferation arrest (Campisi and d’Adda di Fagagna, 2007). Since the first description of this phenomenon by Hayflick and Moorhead (1961) it has been discussed if replicative senescence is merely a cell culture artifact, or if it is directly related to aging of the organism. In fact, both processes are associated – and potentially caused – by telomere attrition (Harley et al., 1990; Allsopp et al., 1992). Furthermore, accumulating DNA damage and functional decline of mitochondria may contribute to metabolic dysfunction (Sun et al., 2016; McHugh and Gil, 2018). Additional commonalities of senescence and aging include alteration of cellular morphology, metabolic changes, loss of differentiation potential, activation of the p53/p21CIP1 and p16INK4A/pRb signaling pathways, increased senescence-associated β-galactosidase activity (SA-β-gal), formation of senescence-associated heterochromatic foci (SAHF), and the senescence-associated secretory phenotype (SASP) (Lopez-Otin et al., 2013). Cellular senescence is even considered as a hallmark of aging, as senescent cells accumulate in aged tissues (Lopez-Otin et al., 2013), but it remains to be proven if the underlying mechanisms for senescence and aging are directly related (Campisi, 2013).
The phenomenon of cellular senescence is quite diverse. Decreasing proliferation of cells in culture is not only evoked by long-term expansion, but it can also be triggered, e.g., by irradiation or oncogene induced senescence (Campisi and d’Adda di Fagagna, 2007). While these different types of cellular senescence result in similar functional and morphologic changes on cellular level, they can clearly be distinguished by molecular means. Unless stated otherwise, I will particularly focus on replicative senescence in this mini review. Furthermore, I apologize that due to space constrains for this format it will not be possible to give credit to all the important studies in this field and I will particularly discuss the possible link between DNA methylation (DNAm) changes in aging and replicative senescence.
There is a growing perception that culture expansion toward replicative senescence and aging are both reflected by specific modifications in the DNAm pattern, which might indicate that they are evoked by epigenetic processes (Wagner et al., 2016). DNAm plays a crucial role in mammalian development (Meissner et al., 2008). It occurs predominantly in the context of cytosine-guanine dinucleotides – so called CpG sites. The methylation pattern of CpG sites is maintained and modified by DNA methyltransferases (DNMTs) (Liao et al., 2015). On the other hand, demethylation can be mediated indirectly by ten-eleven translocation (TET) family enzymes, which oxidize 5-methylcytosine into 5-hydroxymethylcytosine that can either be passively depleted through DNA replication or actively reverted to cytosine by iterative oxidation and thymine DNA glycosylase (TDG)-mediated base excision repair (Kohli and Zhang, 2013). However, it is largely unclear how DNAm changes are regulated at specific sites in the genome. Furthermore, the functional relevance of specific DNAm changes remains largely elusive. In 2010, our group has demonstrated that aging as well as senescence are reflected by DNAm changes at specific CpG sites (Bork et al., 2010). Since then, many other groups have fine-tuned epigenetic signatures that can be used to track these processes independently and for different purposes (Figure 1), demonstrating that replicative senescence and aging are epigenetically distinct (Wagner et al., 2016; Kabacik et al., 2018).
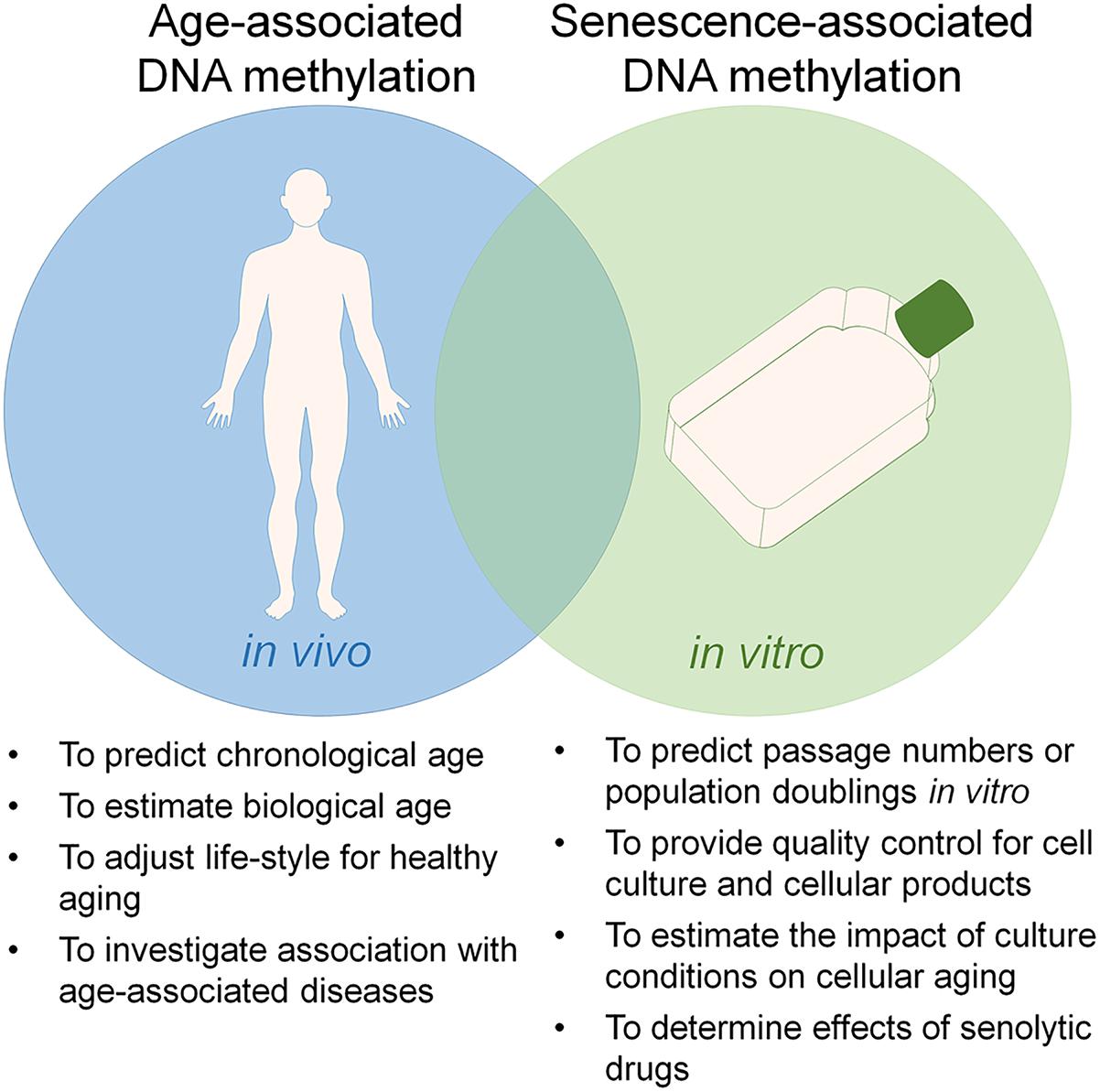
Figure 1. Utility of epigenetic clocks for aging and senescence. Aging and replicative senescence are both reflected by highly reproducible DNA methylation (DNAm) changes and there is a significant overlap between these epigenetic modifications. On the other hand, the two processes can be tracked independently by epigenetic signatures, which can be utilized for different applications.
Almost 30 years ago, it has been suggested that cell culture is associated with a continuous loss in the general DNAm level, and that this loss might be related to the number of cell divisions (Catania and Fairweather, 1991). These authors speculated that the progressive loss of DNAm could generate a multi-step cell division “clock” which underlies the Hayflick phenomenon. The today available global DNAm profiles reveal a more complex picture. Whole-genome single-nucleotide bisulfite sequencing has demonstrated widespread DNA hypomethylation and focal hypermethylation upon replicative senescence (Cruickshanks et al., 2013). In fact, a relatively large proportion of the genome reveals reproducible and highly significant changes of DNAm during culture expansion, particularly in developmental genes (Koch et al., 2013). Furthermore, chromatin conformation analysis demonstrated that replicative senescence is associated with an unidirectional loss in local chromatin connectivity, suggesting that senescence is an endpoint of the continuous nuclear remodeling process during differentiation (Chandra et al., 2015; Criscione et al., 2016). Yet, senescence-associated DNAm changes are relatively consistent across different cell types (Franzen et al., 2018; Zirkel et al., 2018).
In our previous work, we have utilized DNAm profiles of mesenchymal stromal cells (MSCs) and fibroblasts, which at the time were analyzed with 27k Illumina BeadChip arrays, to select six CpGs that revealed almost linear hyper- or hypo-methylation with subsequent passages (Koch et al., 2012). Thus, DNAm levels at these six CpGs can be used to estimate the number of passages during in vitro expansion or the number of cumulative population doublings (cPDs) (Koch and Wagner, 2013). This relatively small Epigenetic Senescence Signature facilitates a targeted analysis by pyrosequencing in a fast and cost-effective manner. Tracking of the state of senescence is of particular importance for MSCs, which raise high hopes in regenerative medicine and are currently tested in a multitude of clinical trials (Wagner and Ho, 2007). In comparison to other biomarkers for senescence, such as telomere attrition (Bernadotte et al., 2016) or staining of SA-β-gal (Itahana et al., 2007), the DNAm changes provide a more quantitative measure. Notably, gamma irradiation of MSCs resulted in typical senescence-associated changes in morphology, gene expression, and loss of differentiation potential, but in contrast to replicative senescence it did not evoke any significant DNAm changes (Koch et al., 2013). This exemplifies that the different types of senescence – e.g., oncogene induced senescence, DNA damage induced senescence, or replicative senescence – seem to be triggered by different mechanisms (Koch et al., 2013; Lowe et al., 2016; Kabacik et al., 2018).
Already in 1973 it was described that aging is reflected by a global decrease in the 5-methylcytosine content of various tissues, and this led to the assumption that DNAm may regulate gene activity and somehow trigger the process of aging (Vanyushin et al., 1973) – a hypothesis that is still valid. Presently, it is estimated that almost one third of the CpG sites reveal age-associated DNAm changes, of which 60% become hypomethylated and 40% hypermethylated upon aging (Johansson et al., 2013). Overall, the age-associated DNAm changes are similar across different tissues, while they are certainly influenced by the very different epigenetic makeup of different cell types (Teschendorff et al., 2010; Koch and Wagner, 2011).
The first epigenetic clocks to estimate donor age were again derived from the 27k Illumina Bead Chips (Bocklandt et al., 2011; Koch and Wagner, 2011). These signatures were further improved in the advent of more available datasets and refined bioinformatics approaches (Hannum et al., 2013; Horvath, 2013; Weidner et al., 2014). For example, Hannum et al. (2013) used DNAm profiles of 656 whole blood samples of donors aged 19–101 and integrated 71 CpG sites into a multivariable linear regression model enabling age-predictions with an error of 4.9 years in an independent validation dataset of blood samples. In analogy, we derived similar epigenetic aging signatures for blood (Weidner et al., 2014; Lin et al., 2016). Horvath established a very robust multi-tissue predictor by an elastic net regression model that was trained on 7,844 samples from 82 datasets (including 51 different tissues and cell types) (Horvath, 2013). This model is based on 353 age-associated CpG sites and facilitates good precision of age-estimation in various tissue types. The relatively high precisions of age-predictions hold high hopes in forensics to estimate the donor age of blood traces or of people with allegedly unknown age. Furthermore, epigenetic age-predictions do not only correlate with chronological age, they are also indicative for life expectancy (Marioni et al., 2015, 2018; Field et al., 2018). Particularly specific hypomethylated CpGs seem to correlate with all-cause mortality (Lin et al., 2016). This indicates that epigenetic age-predictors may be trained to rather correlate with biological age (Levine et al., 2018).
Epigenetic age-predictions are less precise for cells in culture, due to the above mentioned impact of in vitro expansion on the epigenetic makeup (Horvath, 2013; Frobel et al., 2014; Sheng et al., 2018). There is a moderate correlation between age-associated and senescence-associated DNAm changes (Bork et al., 2010; Pasumarthy et al., 2017) and hence it is not surprising that several epigenetic clocks for aging are also increasing with higher passage numbers (Horvath et al., 2018). However, as mentioned above, replicative senescence and aging are overall rather reflected by independent CpGs, indicating that the different types of epigenetic clocks might be modulated independently.
It is possible to rejuvenate cells by reprogramming into induced pluripotent stem cells (iPSCs). While in pluripotent state, iPSCs can be passaged virtually infinitively without any signs of cellular aging. Furthermore, iPSCs derived from elderly organisms can give rise to young organisms that pursue normal aging, as for example demonstrated in mice (Boland et al., 2009). Also on molecular level iPSCs seem to be fully rejuvenated: their telomeres become elongated and other age-related molecular parameters are reset upon reprogramming (Marion et al., 2009). Notably, age-associated as well as senescence-associated DNAm patterns are completely reversed in iPSCs (Horvath, 2013; Koch et al., 2013; Weidner et al., 2014). This epigenetic switch apparently occurs simultaneous with DNAm changes in pluripotency associated CpGs, indicating that the epigenetic rejuvenation belongs to the fundamental changes in reprogramming (Franzen et al., 2018). However, it has also been demonstrated that the loss of cell-type specific gene expression, e.g., for fibroblast lineage, follows different kinetics, which may suggest that there might be a safe time window for rejuvenation without complete erasure of somatic identity (Olova et al., 2018).
Upon re-differentiation of iPSCs toward other cell types the senescence-associated DNAm patterns are continuously reacquired as observed during culture expansion of primary cells (Frobel et al., 2014). In contrast, the estimates for epigenetic age remain overall rejuvenated in the iPSC-derived cells and are only very slowly accelerated upon differentiation (Frobel et al., 2014). Direct conversion of cells, e.g., into induced neurons (iNs), at least initially retains age-associated transcriptomic and epigenetic signatures (Mertens et al., 2015; Huh et al., 2016). Furthermore, direct conversion into induced neuronal stem cells (iNSCs) maintained some of the age-related DNAm patterns, which further erode upon culture expansion (Sheng et al., 2018). This epigenetic rejuvenation of iPSC and iPSC-derived cells needs to be taken into account when studying age-related diseases – to better address such research questions there is a need to identify ways to artificially accelerate epigenetic aging clocks.
To investigate if overexpression of the catalytic subunit of the human telomerase (TERT) would be enough to stop aging, we have immortalized fibroblasts and MSCs with TERT. While this facilitated elongation of telomeres and long-term expansion without any signs of replicative senescence, it did not reset senescence-associated DNAm changes – in fact, these epigenetic modifications were even further acquired during culture expansion of immortalized lines (Koch et al., 2013). This was more recently validated also for age-associated DNAm changes (Kabacik et al., 2018; Lu et al., 2018). Furthermore, age-associated DNAm changes were not generally accelerated in telomeropathies, such as dyskeratosis congenita (Weidner et al., 2016). Thus, TERT and telomere length have apparently no immediate impact on the epigenetic clock, but this may be further analyzed in the future.
So far, it is largely unclear how the very complex DNAm patterns can be modulated during development by predominantly two de novo DNA methyltransferases: DNMT3A and DNMT3B. We have recently demonstrated that different splice variants of DNMT3A have transcript specific effects on the DNAm pattern (Bozic et al., 2018), but it remains unclear how these enzymes are guided to specific sites in the genome. It is conceivable that this process is mediated by other DNA-binding proteins or long non-coding RNAs. Various transcription factor motifs have been associated with senescence-associated DNAm changes, including binding sites for early growth response protein 1 (EGR1), activating enhancer-binding protein 2 (TFAP2A), protein C-ets-1 (ETS1), neuroblastoma MYC oncogene (MYCN), and aryl hydrocarbon receptor nuclear translocator (ARNT) (Hänzelmann et al., 2015; Pasumarthy et al., 2017). Furthermore, the long non-coding RNA HOTAIR was suggested to target such differentially methylated regions, potentially by triple helix formation (Kalwa et al., 2016).
On the other hand, there is also evidence that the differentially methylated regions of epigenetic clocks are not directly mediated. We have used bisulfite barcoded amplicon sequencing (BBA-seq) to compare senescence-associated DNAm in different subpopulations of MSCs. Notably, in clonally derived subpopulations, the DNAm levels of neighboring CpGs differed extensively, indicating that these genomic regions are not synchronously modified during senescence (Franzen et al., 2017). In a more recent study, we have analyzed if senescence-associated DNAm changes are strand-specific by BBA-seq of hairpin-linked DNA molecules. In fact, many CpG dyads at these sites became only methylated on either the forward or the reverse strand. This hemimethylation was conserved over many passages, indicating that it was not due to insufficient maintenance of DNAm (Franzen et al., 2018). Circular chromatin conformation capture (4C) of senescence-associated CpGs indicated that there is no specific interaction of these genomic regions with other regions that undergo senescence-associated DNAm changes (Franzen et al., 2018). Furthermore, functional annotation of age-associated CpGs showed enrichment in CCCTC-binding factor (CTCF), which is relevant for the 3D organization of the genome (Day et al., 2013; Wang et al., 2018). It appears that the changes in DNAm partly overlap with changing histone modifications upon aging (Johansson et al., 2013). These findings suggest that senescence-associated and age-associated DNAm are not regulated in a targeted manner but rather evoked by other chromatin modifications.
It is still controversially discussed if aging is governed by a purposeful program or if it rather resembles an accumulation of stochastic, accidental events (Hayflick, 2007). While several authors argued that aging cannot be programmed (Blagosklonny, 2013), there is also evidence that it may be evolutionary purposeful for the species (Longo et al., 2005; Mitteldorf, 2016; Horvath and Raj, 2018). Aging necessitates a regular generation cycle, which supports better adaptation to environmental changes. In analogy, replicative senescence has been suggested to simply resemble an artificial process caused by the cell culture conditions, or to reflect a beneficial organized process that is somewhat related to aging. It has also been postulated that replicative senescence acts as a safeguard for malignant transformation (Campisi, 2000). On the other hand, there seems to be a significant overlap of DNAm changes in senescence and cancer (Cruickshanks et al., 2013), indicating that senescence might even promote malignant transformation. Furthermore, fibroblasts surrounding the tumor are known drivers of tumor growth by providing a permissive environment (Campisi, 2005) and hence senescence of neighboring cells might also be relevant. However, despite global similarities there are locally distinct DNAm changes in senescence versus malignant transformation (Xie et al., 2018). Overall, the precision of epigenetic aging clocks is very low in cancer, which might be due to the fact that cancer cells capture only the specific epigenetic state of the tumor-initiating cell (Lin and Wagner, 2015; Eipel et al., 2019). Most types of malignancies occur in the elderly and it may hence be speculated, that the chromatin reorganization upon aging supports specific mutations – age-associated DNAm changes would then rather resemble a trigger than a safeguard for malignant transformation (Wagner et al., 2015).
Epigenetic clocks for aging and senescence are certainly ticking independently, but the striking similarities indicate that the processes are regulated by similar means. There is evidence that the epigenetic modifications are not governed by targeting of regulatory complexes but that the DNAm changes may rather be orchestrated indirectly by other types of chromatin organization. Studying of senescence-associated changes in vitro has several strategic advantages for aging research: Effects of relevant genes or drugs can be systematically screened in a high throughput manner and with large compound libraries. It appears to be plausible that such findings can then be extrapolated to understand how to modulate age-associated epigenetic modifications – and possibly the process of aging.
The author confirms being the sole contributor of this work and has approved it for publication.
This work was supported by the Else Kröner-Fresenius-Stiftung (2014_A193), Interdisciplinary Center for Clinical Research within the faculty of Medicine at the RWTH Aachen University (O3-3), Deutsche Forschungsgemeinschaft (DFG; WA 1706/8-1 and WA1706/11-1), Deutsche Krebshilfe (TRACK-AML), and Bundesministerium für Bildung und Forschung (VIP+ Epi-Blood-Count).
WW is cofounder of Cygenia GmbH (www.cygenia.com), which can provide service for Epigenetic Senescence Signatures and Epigenetic Aging Signatures to other scientists.
Allsopp, R. C., Vaziri, H., Patterson, C., Goldstein, S., Younglai, E. V., Futcher, A. B., et al. (1992). Telomere length predicts replicative capacity of human fibroblasts. Proc. Natl. Acad. Sci. U.S.A. 89, 10114–10118. doi: 10.1073/pnas.89.21.10114
Bernadotte, A., Mikhelson, V. M., and Spivak, I. M. (2016). Markers of cellular senescence. Telomere shortening as a marker of cellular senescence. Aging 8, 3–11. doi: 10.18632/aging.100871
Blagosklonny, M. V. (2013). Aging is not programmed: genetic pseudo-program is a shadow of developmental growth. Cell Cycle 12, 3736–3742. doi: 10.4161/cc.27188
Bocklandt, S., Lin, W., Sehl, M. E., Sanchez, F. J., Sinsheimer, J. S., Horvath, S., et al. (2011). Epigenetic predictor of age. PLoS One 6:e14821. doi: 10.1371/journal.pone.0014821
Boland, M. J., Hazen, J. L., Nazor, K. L., Rodriguez, A. R., Gifford, W., Martin, G., et al. (2009). Adult mice generated from induced pluripotent stem cells. Nature 461, 91–94. doi: 10.1038/nature08310
Bork, S., Pfister, S., Witt, H., Horn, P., Korn, B., Ho, A. D., et al. (2010). DNA methylation pattern changes upon long-term culture and aging of human mesenchymal stromal cells. Aging Cell 9, 54–63. doi: 10.1111/j.1474-9726.2009.00535.x
Bozic, T., Frobel, J., Raic, A., Ticconi, F., Kuo, C. C., Heilmann-Heimbach, S., et al. (2018). Variants of DNMT3A cause transcript-specific DNA methylation patterns and affect hematopoiesis. Life Sci. Alliance 1:e201800153. doi: 10.26508/lsa.201800153
Campisi, J. (2005). Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell 120, 513–522. doi: 10.1016/j.cell.2005.02.003
Campisi, J. (2013). Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 75, 685–705. doi: 10.1146/annurev-physiol-030212-183653
Campisi, J., and d’Adda di Fagagna, F. (2007). Cellular senescence: when bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 8, 729–740. doi: 10.1038/nrm2233
Catania, J., and Fairweather, D. S. (1991). DNA methylation and cellular ageing. Mutat. Res. 256, 283–293. doi: 10.1016/0921-8734(91)90019-8
Chandra, T., Ewels, P. A., Schoenfelder, S., Furlan-Magaril, M., Wingett, S. W., Kirschner, K., et al. (2015). Global reorganization of the nuclear landscape in senescent cells. Cell Rep. 10, 471–483. doi: 10.1016/j.celrep.2014.12.055
Criscione, S. W., De Cecco, M., Siranosian, B., Zhang, Y., Kreiling, J. A., Sedivy, J. M., et al. (2016). Reorganization of chromosome architecture in replicative cellular senescence. Sci. Adv. 2:e1500882. doi: 10.1126/sciadv.1500882
Cruickshanks, H. A., McBryan, T., Nelson, D. M., Vanderkraats, N. D., Shah, P. P., van, T. J., et al. (2013). Senescent cells harbour features of the cancer epigenome. Nat. Cell Biol. 15, 1495–1506. doi: 10.1038/ncb2879
Day, K., Waite, L. L., Thalacker-Mercer, A., West, A., Bamman, M. M., Brooks, J. D., et al. (2013). Differential DNA methylation with age displays both common and dynamic features across human tissues that are influenced by CpG landscape. Genome Biol. 14:R102. doi: 10.1186/gb-2013-14-9-r102
Eipel, M., Božiæ, T., Mies, A., Beier, F., Jost, E., Brümmendorf, T. H., et al. (2019). Tracking of myeloid malignancies by targeted analysis of successive DNA methylation at neighboring CG dinucleotides. Haematologica doi: 10.3324/haematol.2018.209734[Epub ahead of print].
Field, A. E., Robertson, N. A., Wang, T., Havas, A., Ideker, T., and Adams, P. D. (2018). DNA methylation clocks in aging: categories, causes, and consequences. Mol. Cell 71, 882–895. doi: 10.1016/j.molcel.2018.08.008
Franzen, J., Georgomanolis, T., Selich, A., Stoeger, R., Brant, L., Fernandez-Rebollo, E., et al. (2018). DNA methylation patterns of replicative senescence are strand-specific and reflect changes in chromatin conformation. bioRxiv [Preprint]. doi: 10.1101/445114
Franzen, J., Zirkel, A., Blake, J., Rath, B., Benes, V., Papantonis, A., et al. (2017). Senescence-associated DNA methylation is stochastically acquired in subpopulations of mesenchymal stem cells. Aging Cell 16:183. doi: 10.1111/acel.12544
Frobel, J., Hemeda, H., Lenz, M., Abagnale, G., Joussen, S., Denecke, B., et al. (2014). Epigenetic rejuvenation of mesenchymal stromal cells derived from induced pluripotent stem cells. Stem Cell Rep. 3, 414–422. doi: 10.1016/j.stemcr.2014.07.003
Hannum, G., Guinney, J., Zhao, L., Zhang, L., Hughes, G., Sadda, S., et al. (2013). Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 49, 359-367. doi: 10.1016/j.molcel.2012.10.016
Hänzelmann, S., Beier, F., Gusmao, E. G., Koch, C. M., Hummel, S., Charapitsa, I., et al. (2015). Replicative senescence is associated with nuclear reorganization and with DNA methylation at specific transcription factor binding sites. Clin. Epigen. 4:19. doi: 10.1186/s13148-015-0057-5
Harley, C. B., Futcher, A. B., and Greider, C. W. (1990). Telomeres shorten during ageing of human fibroblasts. Nature 345, 458–460. doi: 10.1038/345458a0
Hayflick, L. (2007). Biological aging is no longer an unsolved problem. Ann. N. Y. Acad. Sci. 1100, 1–13. doi: 10.1196/annals.1395.001
Hayflick, L., and Moorhead, P. (1961). The serial cultivation of human diploid cell strains. Exp. Cell Res. 25, 585–621. doi: 10.1016/0014-4827(61)90192-6
Horvath, S. (2013). DNA methylation age of human tissues and cell types. Genome Biol. 14:R115. doi: 10.1186/gb-2013-14-10-r115
Horvath, S., Oshima, J., Martin, G. M., Lu, A. T., Quach, A., Cohen, H., et al. (2018). Epigenetic clock for skin and blood cells applied to hutchinson gilford progeria syndrome and ex vivo studies. Aging 10, 1758–1775. doi: 10.18632/aging.101508
Horvath, S., and Raj, K. (2018). DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 19, 371–384. doi: 10.1038/s41576-018-0004-3
Huh, C. J., Zhang, B., Victor, M. B., Dahiya, S., Batista, L. F., Horvath, S., et al. (2016). Maintenance of age in human neurons generated by microRNA-based neuronal conversion of fibroblasts. Elife 5:e18648. doi: 10.7554/eLife.18648
Itahana, K., Campisi, J., and Dimri, G. P. (2007). Methods to detect biomarkers of cellular senescence: the senescence-associated beta-galactosidase assay. Methods Mol. Biol. 371, 21–31. doi: 10.1007/978-1-59745-361-5_3
Johansson, A., Enroth, S., and Gyllensten, U. (2013). Continuous aging of the human DNA methylome throughout the human lifespan. PLoS One 8:e67378. doi: 10.1371/journal.pone.0067378
Kabacik, S., Horvath, S., Cohen, H., and Raj, K. (2018). Epigenetic ageing is distinct from senescence-mediated ageing and is not prevented by telomerase expression. Aging 10, 2800–2815. doi: 10.18632/aging.101588
Kalwa, M., Hanzelmann, S., Otto, S., Kuo, C. C., Franzen, J., Joussen, S., et al. (2016). The lncRNA HOTAIR impacts on mesenchymal stem cells via triple helix formation. Nucleic Acids Res. 44, 10631–10643. doi: 10.1093/nar/gkw802
Koch, C. M., Joussen, S., Schellenberg, A., Lin, Q., Zenke, M., and Wagner, W. (2012). Monitoring of cellular senescence by DNA-Methylation at specific CpG sites. Aging Cell 11, 366–369. doi: 10.1111/j.1474-9726.2011.00784.x
Koch, C. M., Reck, K., Shao, K., Lin, Q., Joussen, S., Ziegler, P., et al. (2013). Pluripotent stem cells escape from senescence-associated DNA methylation changes. Genome Res. 23, 248–259. doi: 10.1101/gr.141945.112
Koch, C. M., and Wagner, W. (2011). Epigenetic-aging-signature to determine age in different tissues. Aging 3, 1018–1027. doi: 10.18632/aging.100395
Koch, C. M., and Wagner, W. (2013). Epigenetic biomarker to determine replicative senescence of cultured cells. Methods Mol. Biol. 1048, 309–321. doi: 10.1007/978-1-62703-556-9_20
Kohli, R. M., and Zhang, Y. (2013). TET enzymes, TDG and the dynamics of DNA demethylation. Nature 502, 472–479. doi: 10.1038/nature12750
Levine, M. E., Lu, A. T., Quach, A., Chen, B. H., Assimes, T. L., Bandinelli, S., et al. (2018). An epigenetic biomarker of aging for lifespan and healthspan. Aging 10, 573–591. doi: 10.18632/aging.101414
Liao, J., Karnik, R., Gu, H., Ziller, M. J., Clement, K., Tsankov, A. M., et al. (2015). Targeted disruption of DNMT1, DNMT3A and DNMT3B in human embryonic stem cells. Nat. Genet. 47, 469–478. doi: 10.1038/ng.3258
Lin, Q., and Wagner, W. (2015). Epigenetic aging signatures are coherently modified in cancer. PLoS Genet. 11:e1005334. doi: 10.1371/journal.pgen.1005334
Lin, Q., Weidner, C. I., Costa, I. G., Marioni, R. E., Ferreira, M. R., Deary, I. J., et al. (2016). DNA methylation levels at individual age-associated CpG sites can be indicative for life expectancy. Aging 8, 394–401. doi: 10.18632/aging.100908
Longo, V. D., Mitteldorf, J., and Skulachev, V. P. (2005). Programmed and altruistic ageing. Nat. Rev. Genet. 6, 866–872. doi: 10.1038/nrg1706
Lopez-Otin, C., Blasco, M. A., Partridge, L., Serrano, M., and Kroemer, G. (2013). The hallmarks of aging. Cell 153, 1194–1217. doi: 10.1016/j.cell.2013.05.039
Lowe, D., Horvath, S., and Raj, K. (2016). Epigenetic clock analyses of cellular senescence and ageing. Oncotarget 7, 8524–8531. doi: 10.18632/oncotarget.7383
Lu, A. T., Xue, L., Salfati, E. L., Chen, B. H., Ferrucci, L., Levy, D., et al. (2018). GWAS of epigenetic aging rates in blood reveals a critical role for TERT. Nat. Commun. 9:387. doi: 10.1038/s41467-017-02697-5
Marion, R. M., Strati, K., Li, H., Tejera, A., Schoeftner, S., Ortega, S., et al. (2009). Telomeres acquire embryonic stem cell characteristics in induced pluripotent stem cells. Cell Stem Cell 4, 141–154. doi: 10.1016/j.stem.2008.12.010
Marioni, R. E., Belsky, D. W., Deary, I. J., and Wagner, W. (2018). Association of facial ageing with DNA methylation and epigenetic age predictions. Clin. Epigen. 10:140. doi: 10.1186/s13148-018-0572-2
Marioni, R. E., Shah, S., McRae, A. F., Chen, B. H., Colicino, E., Harris, S. E., et al. (2015). DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol. 16:25. doi: 10.1186/s13059-015-0584-6
McHugh, D., and Gil, J. (2018). Senescence and aging: causes, consequences, and therapeutic avenues. J. Cell Biol. 217, 65–77. doi: 10.1083/jcb.201708092
Meissner, A., Mikkelsen, T. S., Gu, H., Wernig, M., Hanna, J., Sivachenko, A., et al. (2008). Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454, 766–770. doi: 10.1038/nature07107
Mertens, J., Paquola, A. C. M., Ku, M., Hatch, E., Bohnke, L., Ladjevardi, S., et al. (2015). Directly reprogrammed human neurons retain aging-associated transcriptomic signatures and reveal age-related nucleocytoplasmic defects. Cell Stem Cell 17, 705–718. doi: 10.1016/j.stem.2015.09.001
Mitteldorf, J. (2016). An epigenetic clock controls aging. Biogerontology 17, 257–265. doi: 10.1007/s10522-015-9617-5
Olova, N., Simpson, D. J., Marioni, R. E., and Chandra, T. (2018). Partial reprogramming induces a steady decline in epigenetic age before loss of somatic identity. Aging Cell 18:e12877. doi: 10.1111/acel.12877
Pasumarthy, K. K., Doni Jayavelu, N., Kilpinen, L., Andrus, C., Battle, S. L., Korhonen, M., et al. (2017). Methylome analysis of human bone marrow MSCs reveals extensive age- and culture-induced changes at distal regulatory elements. Stem Cell Rep. 9, 999–1015. doi: 10.1016/j.stemcr.2017.07.018
Sheng, C., Jungverdorben, J., Wiethoff, H., Lin, Q., Flitsch, L. J., Eckert, D., et al. (2018). A stably self-renewing adult blood-derived induced neural stem cell exhibiting patternability and epigenetic rejuvenation. Nat. Commun. 9:4047. doi: 10.1038/s41467-018-06398-5
Sun, N., Youle, R. J., and Finkel, T. (2016). The mitochondrial basis of aging. Mol. Cell 61, 654–666. doi: 10.1016/j.molcel.2016.01.028
Teschendorff, A. E., Menon, U., Gentry-Maharaj, A., Ramus, S. J., Weisenberger, D. J., Shen, H., et al. (2010). Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 20, 440–446. doi: 10.1101/gr.103606.109
Vanyushin, B. F., Nemirovsky, L. E., Klimenko, V. V., Vasiliev, V. K., and Belozersky, A. N. (1973). The 5-methylcytosine in DNA of rats. Tissue and age specificity and the changes induced by hydrocortisone and other agents. Gerontologia 19, 138–152. doi: 10.1159/000211967
Wagner, W., Fernandez-Rebollo, E., and Frobel, J. (2016). DNA-methylation changes in replicative senescence and aging: two sides of the same coin? Epigenomics 8, 1–3. doi: 10.2217/epi.15.100
Wagner, W., and Ho, A. D. (2007). Mesenchymal stem cell preparations–comparing apples and oranges. Stem Cell Rev. 3, 239-248. doi: 10.1007/s12015-007-9001-1
Wagner, W., Weidner, C. I., and Lin, Q. (2015). Do age-associated DNA methylation changes increase the risk of malignant transformation? Bioessays 37, 20–24. doi: 10.1002/bies.201400063
Wang, Y., Karlsson, R., Lampa, E., Zhang, Q., Hedman, A. K., Almgren, M., et al. (2018). Epigenetic influences on aging: a longitudinal genome-wide methylation study in old Swedish twins. Epigenetics 13, 975–987. doi: 10.1080/15592294.2018.1526028
Weidner, C. I., Lin, Q., Birkhofer, C., Gerstenmaier, U., Kaifie, A., Kirschner, M., et al. (2016). DNA methylation in PRDM8 is indicative for dyskeratosis congenita. Oncotarget 7, 10765–10772. doi: 10.18632/oncotarget.7458
Weidner, C. I., Lin, Q., Koch, C. M., Eisele, L., Beier, F., Ziegler, P., et al. (2014). Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 15:R24. doi: 10.1186/gb-2014-15-2-r24
Xie, W., Kagiampakis, I., Pan, L., Zhang, Y. W., Murphy, L., Tao, Y., et al. (2018). DNA methylation patterns separate senescence from transformation potential and indicate cancer risk. Cancer Cell 33, 309.e305–321.e305. doi: 10.1016/j.ccell.2018.01.008
Keywords: DNA methylation, epigenetic, senescence, aging, iPSC, quality control, senolytic drugs, biomarker
Citation: Wagner W (2019) The Link Between Epigenetic Clocks for Aging and Senescence. Front. Genet. 10:303. doi: 10.3389/fgene.2019.00303
Received: 10 January 2019; Accepted: 19 March 2019;
Published: 03 April 2019.
Edited by:
Alexey Moskalev, Institute of Biology, Komi Scientific Center (RAS), RussiaReviewed by:
Ionel Sandovici, University of Cambridge, United KingdomCopyright © 2019 Wagner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wolfgang Wagner, d3dhZ25lckB1a2FhY2hlbi5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.