
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 28 March 2019
Sec. Livestock Genomics
Volume 10 - 2019 | https://doi.org/10.3389/fgene.2019.00274
This article is part of the Research TopicAnimal Genetics and Diseases: Advances in Farming and Livestock SystemsView all 16 articles
 Zhong Xu1
Zhong Xu1 Hao Sun1
Hao Sun1 Zhe Zhang1
Zhe Zhang1 Qingbo Zhao1
Qingbo Zhao1 Babatunde Shittu Olasege1
Babatunde Shittu Olasege1 Qiumeng Li1
Qiumeng Li1 Yang Yue1
Yang Yue1 Peipei Ma1Xiangzhe Zhang1
Peipei Ma1Xiangzhe Zhang1 Qishan Wang1*
Qishan Wang1* Yuchun Pan1,2*
Yuchun Pan1,2*Jinhua pig, a well-known Chinese indigenous breed, has evolved as a pig breed with excellent meat quality, greater disease resistance, and higher prolificacy. The reduction in the number of Jinhua pigs over the past years has raised concerns about inbreeding. Runs of homozygosity (ROH) along the genome have been applied to quantify individual autozygosity to improve the understanding of inbreeding depression and identify genes associated with traits of interest. Here, we investigated the occurrence and distribution of ROH using next-generation sequencing data to characterize autozygosity in 202 Jinhua pigs, as well as to identify the genomic regions with high ROH frequencies within individuals. The average inbreeding coefficient, based on ROH longer than 1 Mb, was 0.168 ± 0.052. In total, 18,690 ROH were identified in all individuals, among which shorter segments (1–5 Mb) predominated. Individual ROH autosome coverage ranged from 5.32 to 29.14% in the Jinhua population. On average, approximately 16.8% of the whole genome was covered by ROH segments, with the lowest coverage on SSC11 and the highest coverage on SSC17. A total of 824 SNPs (about 0.5%) and 11 ROH island regions were identified (occurring in over 45% of the samples). Genes associated with reproduction (HOXA3, HOXA7, HOXA10, and HOXA11), meat quality (MYOD1, LPIN3, and CTNNBL1), appetite (NUCB2) and disease resistance traits (MUC4, MUC13, MUC20, LMLN, ITGB5, HEG1, SLC12A8, and MYLK) were identified in ROH islands. Moreover, several quantitative trait loci for ham weight and ham fat thickness were detected. Genes in ROH islands suggested, at least partially, a selection for economic traits and environmental adaptation, and should be subject of future investigation. These findings contribute to the understanding of the effects of environmental and artificial selection in shaping the distribution of functional variants in the pig genome.
Autozygosity refers to homozygosity in which the two alleles are identical by descent (IBD). It can result from several different phenomena, such as genetic drift, consanguineous matings, population bottleneck, as well as natural and artificial selection (Curik et al., 2014). Jinhua pig, as a valuable natural resource, is a well-known indigenous breed in eastern China that has evolved as a pig breed with excellent meat quality, greater disease resistance, higher prolificacy and greater adaptability to hot and humid climate (Gao et al., 2014). Due to their superior meat quality, Jinhua pigs have been used for the production of a famous ham brand called Jinhua Ham, which is a famous ham in China (Miao et al., 2009). The number of Jinhua pigs has been decreasing in the last two decades as a result of large import of Western pig breeds to improve leanness rate of pork (China National Commission of Animal Genetic Resources, 2011). A deficient control of inbreeding may lead to a reduction of the genetic variability and therefore of the effective population size (Ne), a key parameter that influences the conservation planning and determines the rate of change in the composition of a population caused by genetic drift (Charlesworth, 2009). In addition, inbreeding may also increase the frequency of autozygosity for deleterious alleles with the consequent reduction in individual performance (Ouborg et al., 2010). For these reasons, there is a growing interest in characterizing and understanding inbreeding and autozygosity in Jinhua pigs. This would help to better preserve the genetic diversity and allow long-term viability of breeding programs of this breed.
Runs of homozygosity (ROH) are contiguous homozygous segments of the genome where the two haplotypes inherited from the parents are identical (Gibson et al., 2006). The development of high-density single nucleotide polymorphism (SNP) markers to scan the genome for ROH has been proposed as a proxy for the detection of genomic regions where a reduction in heterozygosity has occurred (Howrigan et al., 2011). Nowadays, whole genome inbreeding estimated from ROH is considered as a powerful method to distinguish between recent and ancient inbreeding (Keller et al., 2011). As the expected length of a ROH is equal to 1/2G Morgan, where G is the number of generations since the common ancestor (Thompson, 2013), the number of generations can be inferred from the length and frequency of ROH (Howrigan et al., 2011). The autozygosity, based on ROH, can help to improve the understanding of inbreeding depression of a trait (Keller et al., 2011) and also help to identify genes associated with traits of economic interest present in these ROH island regions (Purfield et al., 2017). In addition, given the stochastic nature of recombination, the occurrence of ROH is not randomly distributed across the genome, and ROH islands across a large number of samples may be the result of selective pressure (Zavarez et al., 2015). Recently, ROH has been used to explore signatures of selection in cattle (Peripolli et al., 2018), chicken (Marchesi et al., 2018), and sheep (Mastrangelo et al., 2017), but less commonly in pig, especially Chinese indigenous pigs such as the Jinhua pigs.
In this study, we investigated the occurrence and distribution of ROH in a sample of 202 Jinhua pigs, in order to characterize genome-wide autozygosity levels and to detect potential ROH islands that may provide insights into past events of selection in this population. In addition, other parameters to address the levels of genetic variability, including Ne and different measures of inbreeding from pedigree and genomic information, were also investigated. For that, we used genotyping by genome reducing and sequencing (GGRS) (Chen et al., 2013), which was successfully applied to evaluate genetic diversity in Chinese indigenous pig breeds in the Taihu region (Wang et al., 2015).
Ear tissue samples were collected from 202 Jinhua pigs (189 females and 13 males) from conservation pig farms in Zhejiang province. Those pigs were born between 2014 and 2017, with an average depth of about four generations. A commercial kit (Lifefeng Biotech, Co., Ltd., Shanghai, China) was used to extract genomic DNA, and verified the integrity and purity of DNA by agarose gel electrophoresis and the A260/280 ratio. The Genomic DNA samples were genotyped using the GGRS protocol (Chen et al., 2013). Quality control (QC) of ∼1.4 billion raw reads were performed using NGS QC Toolkit v2.3 (Patel and Jain, 2012). In this study, we mapped the clean sequencing reads to the latest released pig reference genome (Sscrofa11.1) using BWA (Li and Durbin, 2010). SNP calling was performed using SAMTOOLS v0.1.19 and the missing genotypes were imputed using BEAGLE (Howie et al., 2009; Li et al., 2009). Additional quality controls were applied following Xiao et al. (2017). These included a minimum number of samples genotyped > 30%, a calling quality > 20 (99% accuracy), and a minor allele frequency (MAF) ≥ 5%. SNPs mapped to sex chromosomes were excluded from the analyses.
To determine novel variants in our sequence data, we compared the identified SNPs with the dbSNP data (Build 1521). These SNPs were annotated according to the Ensembl pig gene annotation set (Ensembl release 922) as previously reported by Wang et al. (2015).
Observed (Ho) and expected heterozygosity (He) were estimated using PLINK v1.07 (Purcell et al., 2007). The Ne was estimated using SNeP v1.1 (Barbato et al., 2015). This approach simultaneously estimated historical effective population size based on the relationship between LD, Ne, and recombination rate:
where Ne(t) is the effective population size t generations ago, calculated as t = (2f (ct))-1; ct is the recombination rate for a specific physical distance between SNPs, which was estimated using Sved and Feldman (1973); f is the Haldane mapping function built between recombination rate and genetic distance measured by Morgan; is the LD value corrected for sample size and α is a correction for the occurrence of mutations.
Linkage disequilibrium between SNP pairs was estimated using PLINK v1.07 (Purcell et al., 2007). Haplotype blocks were obtained with a confidence intervals algorithm and with the software Haploview (Barrett, 2009), which was also used to visualize haplotype patterns.
Runs of homozygosity were identified for each individual using PLINK v1.07 (Purcell et al., 2007). The default parameter –homozyg were used to define ROH (Peripolli et al., 2018) and the following criteria were chosen: (1) a sliding window of 50 SNPs across the genome; (2) one heterozygous and five missing calls were allowed per window to account for genotyping error; (3) the minimum number of consecutive SNPs included in a ROH was set to 100; (4) to exclude short ROH that was derived from strong LD, the minimum length for a ROH was set to 1 Mb (Purfield et al., 2012); (5) the required minimum SNP density to define a ROH was 1 SNP per 50 kb. Considering an approximate genetic distance of 1 cM each 1 Mb (Zanella et al., 2016), a minimum ROH length of 1 Mb was expected to capture inbreeding up to 50 ancestral generations.
Different types of inbreeding coefficients were estimated based on pedigree and genomic information. Pedigree-based inbreeding coefficients (FPED) for all pigs were estimated using R package “pedigree.”
Genomic inbreeding for each animal was estimated from ROH (FROH), as the ratio of the total length of genome covered by ROH to the total length of the genome covered by SNPs or sequences, as proposed by McQuillan et al. (2008):
in which LROH is the total length of an individual’s ROH and Lauto is the length of the autosomal genome covered by the SNPs, which was 2.26 Gb in our study. For each animal, four ROH estimates were calculated based on lengths from sequence data as the proportion of its genome: ROH > 10 Mb (FROH > 10 Mb), 5–10 Mb (FROH5-10 Mb), 1–5 Mb (FROH1-5 Mb), and ROH > 1 Mb (FROH_all), corresponding to 5 generations, 5 to 10 generations, 10 to 50 generations, and 50 generations, respectively.
In addition, three SNP-based estimates of inbreeding coefficients were calculated using the option –ibc from the GCTA software (Yang et al., 2011): the first estimator, FSNP1, was based on the variance of the additive genotypes (VanRaden, 2008); the FSNP2 estimate was calculated based on the homozygous excess; the third estimator, FSNP3, was calculated based on the correlation between uniting gametes (Wright, 1922). The formulae are as follows:
Where Yi is the number of the reference allele copies for the i-th SNP, pi is the frequency of this allele in the sample and hi = 2pi (1-pi), and n is the total number of SNPs. Note that these coefficients were corrected by the allele frequencies of the current population and they can take negative values (Yang et al., 2013), while FPED and FROH ranged from 0 to 1. The inbreeding coefficients obtained by the eight methods were compared using Pearson’s correlation.
Genomic regions with reduced genetic diversity can be found in ROH islands, and high homozygosity around the ROH islands that might harbor targets of positive selection and are under strong selective pressure (Pemberton et al., 2012). To identify the genomic overlapping ROH regions, we calculated the proportion of the occurrences of a SNP in ROH by counting the number of times the SNP was detected in those ROH across individuals, and this was plotted against the position of the SNP along the chromosome. The genomic regions most commonly associated with ROH were identified by selecting the top 0.5% of the SNPs most commonly observed in ROH (Pemberton et al., 2012). A series of adjacent SNPs, merged to constitute ROH islands and genes within each ROH island, were further extracted using the BIOMART package (Durinck et al., 2005). To further analyze the functions of identified genes, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway and Gene Ontology (GO) enrichment analyses were performed using DAVID 6.83. Only terms with a p-value less than 0.05 were considered as significant and listed.
To compare the selection signatures obtained from ROH, the integrated haplotype score (iHS) test was performed within Jinhua pigs. The iHS is a measure of the amount of extended haplotype homozygosity at a given SNP, designed to use phased genotypes to identify putative regions of recent or ongoing positive selection in genomes (Voight et al., 2006). The haplotype was phased using fastPHASE with default parameters (Scheet and Stephens, 2006). The derived haplotypes were then analyzed using the rehh v2.0 R package (Gautier et al., 2017) as previously reported by Bertolini et al. (2018). The iHS score was computed for each autosomal SNP, and values obtained were standardized so that they followed a standard normal distribution. To calculate the p-value at the genomic level, the scores for each SNP were transformed as piHS = - log10 [1 - 2|Φ (iHS) - 0.5|], where Φ(x) represents the Gaussian cumulative distribution function and piHS is the two sided p-value associated with the neutral hypothesis of no selection (Gautier et al., 2017). Corresponding to the threshold of 0.5% for ROH islands, the | iHS| scores higher than 2.81 (p < 0.005) were considered as putative signatures of selection (Cardoso et al., 2018). In this study, significant iHS signals were reported only for ROH islands.
A total of 166,661 informative SNPs satisfying the quality filters were obtained, 26,458 of which were identified as unreported in the pig SNP database of NCBI. The SNP density was about 1 SNP per 13.6 kb and they were equally distributed on each chromosome, with the exception of some isolated regions (Figure 1). According to the Ensembl pig gene annotation set (Ensembl release 92), 81,753 SNPs were mapped to gene regions, of which 8,092 were mapped as exons, and 4,481 were mapped as UTRs.
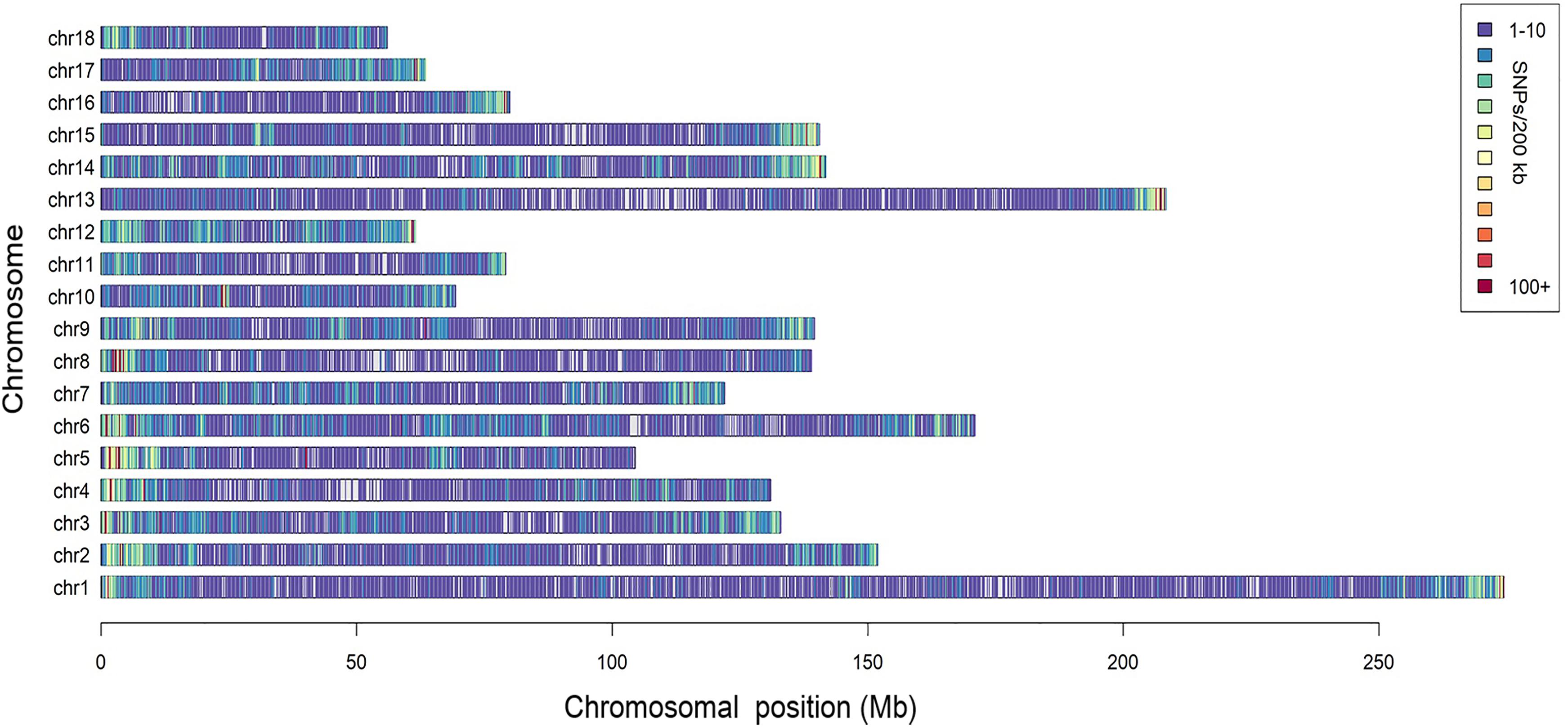
Figure 1. Distribution of the SNPs across the chromosomes. The x-axis denotes the chromosomal position (Mb), and the y-axis represents the chromosomes. The number of the SNPs present in each 200 kb genome block is expressed via colors.
The average observed (Ho) and expected (He) heterozygosities in this Jinhua pig population were 0.312 ± 0.070 and 0.429 ± 0.057, respectively. The value of He was slightly higher than that reported by Chen et al. (2018) for Jinhua pig breed in Zhejiang province. The earliest legend regarding the Jinhua pigs may be traced back to approximately 1600 years ago (China National Commission of Animal Genetic Resources, 2011). Considering a generation interval of 1.5 years, the 1000 generations correspond to Jinhua populations 1500 years ago, approximately. The Ne was estimated from five to 1000 generations ago in our study. The results show that Ne has decreased through time, at a faster rate at 1,000 to 970 generations ago (Supplementary Figure S1). This finding could be explained by the domestication bottleneck caused by human-driven artificial selection approximately 1500 years ago. The effective population size in the last five generations was about 88 and was about 3018 in the 1000th generation. This value was larger than that reported by Xiao et al. (2017) for pig breeds in the Taihu region of China (ranging from 47 to 71) using the same method, therefore, this may indicate higher genetic diversity in Jinhua pigs.
The average inbreeding coefficients estimated using different approaches are shown in Table 1. Incomplete pedigree failed to capture the influence of relatedness among founders from the base population, thus, the levels of inbreeding based on pedigree were expected to be lower than levels of inbreeding based on ROH and SNP-by-SNP (Table 1). The average FROH based on larger segments (0.041–0.053) was closer to FPED (0.01) than the average FSNP. This was to some extent expected, given that the pedigree depth (about four generations) is in agreement with larger ROH segments. In addition, these two coefficients vary in the same range (0–1), while SNP-by-SNP coefficients estimated here can take lower or larger values. These low average FROH values of inbreeding suggest that recent inbreeding was low. However, according to SNP-by-SNP based coefficients, which reflect deviations of the observed inbreeding from the expected values in the current population, recent inbreeding seems to be considerably higher (0.262) than that based on pedigree and ROH.
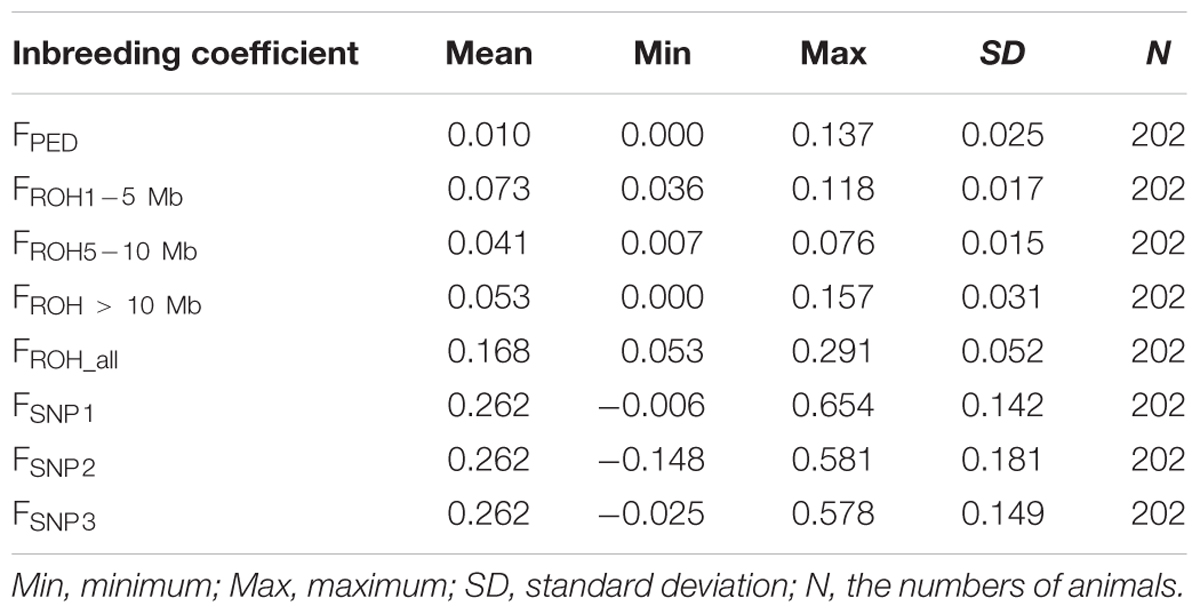
Table 1. Descriptive statistics of the inbreeding coefficient based on pedigree (FPED), ROH (FROH1-5 Mb, FROH5-10 Mb, FROH> 10 Mb, FROH_all) and SNP-by-SNP (FSNP1, FSNP2, FSNP3).
In contrast, the correlations between FPED with all the genomic coefficients was low (from -0.009 to 0.053, Table 2), which may be indicative of a lack of power of FPED to determine relatedness among founders from the base population (Visscher et al., 2006). These low correlations may also be affected by a poor and incomplete pedigree recording, as the base population assumed for FPED, based on long ROH and FSNP was within the range of the last four generations. Correlations between genomic coefficients, based in ROH and in SNP-by-SNP approaches were considerably higher (from 0.218 to 0.698), increasing between coefficients computed from the same source of information, as expected (i.e., ROH segments or SNP-by-SNP information), thus suggesting that genotype-based estimates provide greater accuracy on relatedness as supported by previous studies (Purfield et al., 2012; Zanella et al., 2016).
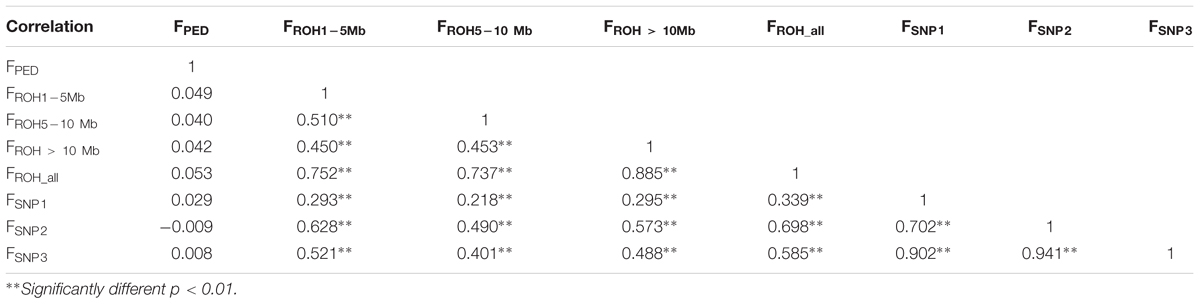
Table 2. Correlation coefficients (lower panel) between pedigree-based inbreeding coefficients (FPED), four inbreeding coefficients based on different ROH lengths (FROH1-5 Mb, FROH5-10 Mb, FROH > 10 Mb, and FROH_all) and three inbreeding coefficients based on SNP-by-SNP (FSNP1, FSNP2, and FSNP3).
Among the four inbreeding coefficients based on different ROH lengths, FROH_all (FROH> 1 Mb) had higher correlations with FSNP1, FSNP2, and FSNP3. A similar trend was also reported by Purfield et al. (2017) while studying six commercial meat sheep breeds. Among the three inbreeding coefficients based on SNP-by-SNP, FSNP2 had higher correlations with FROH1-5 Mb, FROH5-10 Mb, FROH > 10Mb and all FROH_all. These results corroborate previous results observed in cattle (Zhang et al., 2015a; Mastrangelo et al., 2016; Purfield et al., 2017). Similarly, Zhang et al. (2015a) also found that FSNP2 based on excess of homozygosity correlated relatively highly with FROH detected from 50k and sequence data. This trend may be due to the fact that both FROH and FSNP2 directly reflect homozygosity on the genome (Brito et al., 2017). FSNP2 (to some extent) capture all of the homozygosity, whereas, the FROH uses only ROH. Furthermore, the moderate to high correlations between FROH and the three other estimates of genomic inbreeding (FSNP1, FSNP2, and FSNP3) suggested that the proportion of the genome in ROH can be used as an accurate estimate of individual inbreeding levels (Purfield et al., 2012; Peripolli et al., 2018).
The abundance and genomic distribution of ROH provide efficient information about the demographic history of livestock species (Bosse et al., 2012). In total, 18,690 ROH were identified in 202 individuals. The mean ROH length was 4.11 Mb and the longest segment, found in chromosome SSC1 had 72.45 Mb (2,237 SNPs). The distribution of ROH according to length is shown in Figure 2A. The descriptive statistics of ROH number and length by classes is given in Table 3. The total ROH number for Jinhua pigs was composed mostly of a high number of shorter segments (1–5 Mb), which accounted for approximately 77% of all ROH detected, and contributed about 43% of the cumulative ROH length. In contrast, larger ROH (>10 Mb), which were only 8% of all ROH, still covered about 32% of the total ROH length. These results revealed that both ancient (up to 50 generations ago) and recent (within the last five generations) inbreeding have had an impact on the genome of the Jinhua pig population.
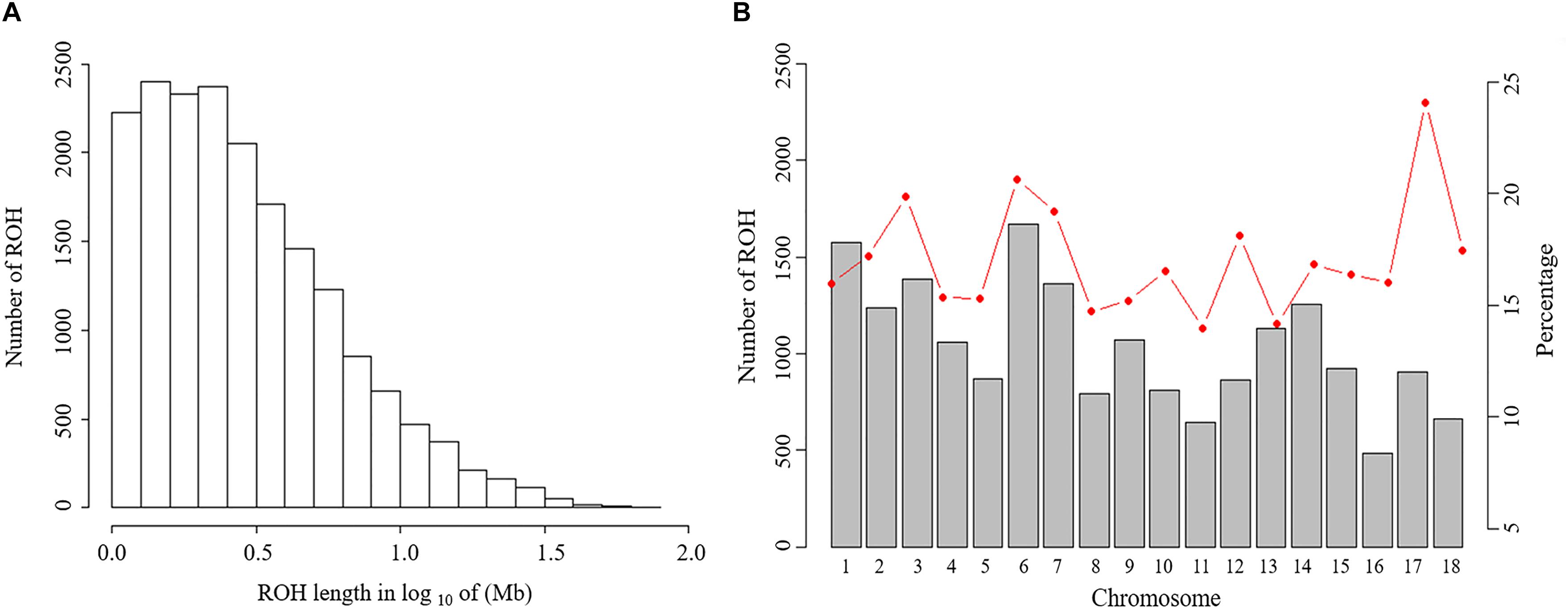
Figure 2. Distribution of the runs of homozygosity (ROH). (A) Distribution of ROH in different lengths (Mb). The values of length in Mb were transformed in log10. (B) Number of ROH longer than 1 Mb per chromosome (bars) and average percentage of each chromosome covered by ROH (red line).

Table 3. Descriptive statistics of runs of homozygosity (ROH) number and length (in Mb) by ROH length class (ROH 1-5 Mb, ROH 5-10 Mb, ROH > 10 Mb and total).
For individuals, the relationship between total number of ROH and total length of the genome covered by ROH showed considerable variation among animals (Supplementary Figure S2). Individual ROH autosome coverage ranged from 5.32% (120.52 Mb) to 29.14% (659.87 Mb) in the Jinhua population. Similar distributions were also observed in other livestock species, such as sheep (Mastrangelo et al., 2017) and cattle (Peripolli et al., 2018).
For chromosomes, the number of ROH per chromosome and the percentage of chromosomes covered by ROH are shown in Figure 2B. The highest number of ROH per chromosome was on SSC6 (1,672 segments), whereas the lowest was on SSC16 (485 segments). On average, approximately 16.8% of the whole genome was under ROH segments, with the lowest coverage shown by SSC11 (14.0%) and the highest coverage of ROHs was on SSC17 (24.1%).
Twenty seven percent of SNPs were comprised in ROH in at least 20% of individuals, thus suggesting that candidate autozygosity regions are present in this population. This finding was similar to that reported by Ferencakovic et al. (2013). The most frequent SNP detected in ROH (131 occurrences, 64.9%) mapped at ∼36 Mb in SSC3, according to the updated reference genome (Sscrofa11.1), although no genes have been currently mapped in this position, suggesting regulatory regions may be involved.
To identify the genomic regions that were most commonly associated with ROH in all individuals, the top 0.5% of SNPs with the highest occurrences (occurring in over 45% of the samples) in a ROH were considered as candidate SNPs (Figure 3). A total of 11 ROH island regions were identified, and the length of these regions ranged from 90 bp on SSC10 to 3.62 Mb on SSC3 (Table 4). The SNPs within these regions showed significantly higher linkage disequilibrium levels than the estimates obtained for the entire chromosome (Supplementary Table S1). On SSC8, we found the longest ROH cold-spot of 194 contiguous SNPs (12.56 Mb) that were not part of a ROH region in any of the individuals, thus suggesting a high heterozygosity region. This region might be produced by high recombination rates, or harboring loci with heterozygous advantage and under selection favoring high haplotype diversity. These results are in good agreement with low LD levels determined in these regions (Supplementary Figure S3) (Barrett, 2009). In the same way, ROH islands showed high levels of LD, as expected (Supplementary Figure S4).
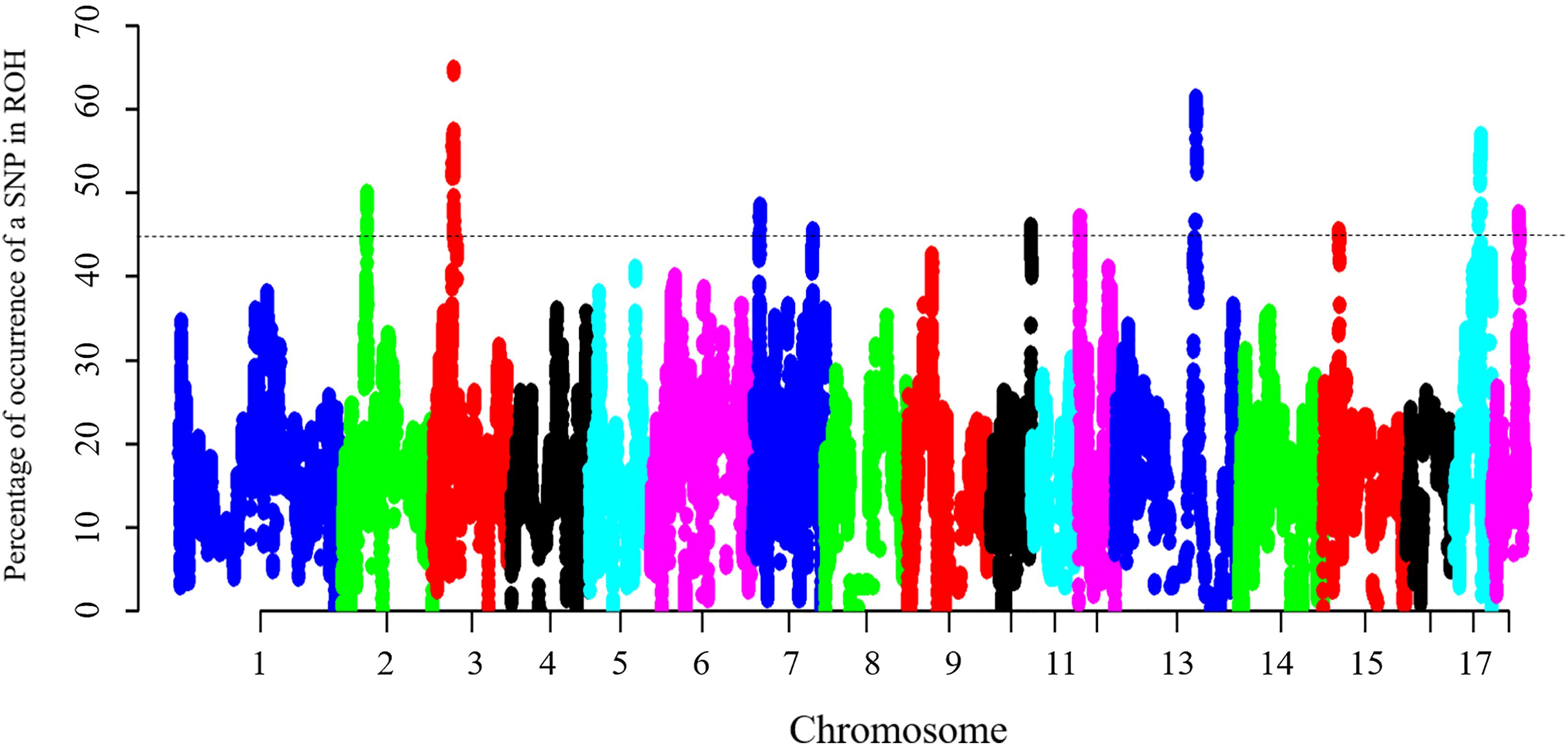
Figure 3. Manhattan plot of incidence of each SNP in the ROH across individuals. The dashed line represents the 45% threshold.
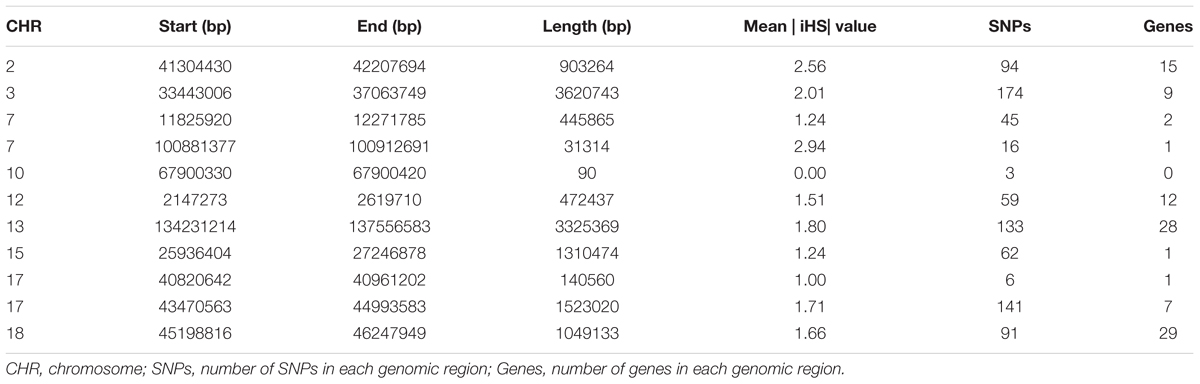
Table 4. List of genomic regions of extended homozygosity detected in Jinhua pigs.
Chromosome position, start and end position of ROH, ROH length, number of SNPs, and number of genes within the genomic regions of extended homozygosity are reported in Table 4. We found that some SNPs in ROH occurred in poor gene content regions. Some identified regions, such as that on SSC15, contained only one annotated gene, although it is longer than 1.3 Mb, either because the annotation of pig reference genome is still incomplete, or the genomic region is positioned in a non-coding region. A total number of 105 genes inside the ROH islands were analyzed using GO enrichment analysis. Supplementary Table S2 provides the chromosome position, start and end, gene name and Ensembl Gene ID for 105 genes. Supplementary Table S3 shows the significant GO terms and KEGG pathways; most of the genes were involved in metabolic pathways and biosynthetic process.
We also checked if the ROH islands overlapped with putative selection signatures in pigs in other literatures. We found that a ROH island at SSC7: 100881377–100912691 overlapped with gene ADCK1, involved in phosphate metabolism, which was in the selection signature region between Berkshire and Korean native pig breeds (Edea and Kim, 2014). A ROH island on SSC2 partially overlapped with a selection region for intramuscular fat and backfat thickness in two Duroc populations, which spanned three genes (ABCC8, MYOD1, and PIK3C2A) (Kim et al., 2015). The MYOD1 gene in this region was also detected in the selection signatures between Jinhua pig group and European breeds group (Li et al., 2016).
In this paper, we focused on some of the most relevant genes within ROH that showed associations with several specific traits related to livestock breeding. Several candidate genes relating to reproduction traits were identified, such as the HOXA genes cluster: HOXA3, HOXA7, HOXA10, and HOXA11 on SSC18, which affects embryo implantation and prolificacy traits (Bagot et al., 2000; Gao et al., 2010; Wu et al., 2013); ROPN1, involved in litter size trait in pigs (Lan et al., 2012); and HNRNPA2B1, which plays key roles in the preimplantation of pig embryo during elongation (Wilson et al., 2000). Some genes associated with specific traits related to meat quality were detected: MYOD1, which affects muscle fiber characteristics, the loin eye area and back fat thickness (Lee et al., 2012; Cepica et al., 2013); LPIN3, one member of lipin gene family associated with back-fat thickness in pigs (He et al., 2009), which are the important regulators in fat-tailed sheep with active lipid metabolism (Jiao et al., 2016); CTNNBL1, associated with porcine fat deposition and backfat traits (Yin et al., 2012). One gene was involved in appetite: NUCB2, which plays an important role in whole-body energy homeostasis and body weight at puberty by regulation of appetite (Lents et al., 2013). Most of genes we detected were involved in disease resistance traits: MUC4, MUC13, MUC20, LMLN, ITGB5, HEG1, SLC12A8, and MYLK on SSC13, were potential candidate genes for controlling the expression of the enterotoxigenic Escherichia coli (ETEC) with F4 fimbriae (F4ac) receptor (Huang et al., 2008; Jacobsen et al., 2010; Rampoldi et al., 2011; Fu et al., 2012; Ren et al., 2012). These genes played key roles in resistance to diarrhea by defending the attachment and adhesion of ETEC to porcine jejunal cells and in maintaining the epithelial barrier as well as immunity function (Zhou et al., 2013). Many studies have revealed that resistance to ETEC F4ac adhesion in pigs can be inherited as an autosomal recessive trait, and the pigs with homozygous genotype were usually resistant to ETEC F4ac (Rampoldi et al., 2011). The regional climate of the Jinhua pig is mainly subtropical with a weather condition that is hot and very humid, which is capable of inducing diarrhea especially during summer (Lyutskanov, 2011). Several researches have also shown that Jinhua pigs were more resistant to ETEC F4ac (Yan et al., 2009; Gao et al., 2014). These genes on SSC13 that display autozygosity in the Jinhua pigs may be linked to selection in response to hot and humid climate, as a result of local adaptation.
The Pig quantitative trait loci (QTL) database4 lists several QTL for reproduction, meat quality and immunity traits that overlapped these ROH islands (Quintanilla et al., 2011; Verardo et al., 2015; Zhang et al., 2016). In particular, some QTLs related to ham traits have already been reported: on SSC2, Cepica et al. (2013) identified significant QTL for ham weight (ID = 28220), Harmegnies et al. (2006) reported QTL for ham fat thickness (ID = 3938, ID = 3960, and ID = 3968); on SSC3, Choi et al. (2011) detected highly significant QTL for ham weight (ID = 21357), Stratil et al. (2006) reported QTL for ham meat weight (ID = 3102) and ham weight (ID = 3104).
In summary, the results show that the genomic regions that display autozygosity in the Jinhua pig breed are related to important production traits under selection, and possibly also help improve their adaptability to survive in hot and very humid environments.
Results from the iHS test revealed that this coefficient had an average value of 0.77, with a maximum value of 4.69 on SSC7, thus indicating that the iHS values were not uniform across the genome. Figure 4 shows the genome-wide distribution of | iHS| values. The plots suggest evidence of selective forces in different regions of the genome. To compare these two methods, the occurrence of a SNP in a ROH was correlated with the SNP | iHS| value (Supplementary Figure S5). Significant moderate correlations were found between the iHS selection signature method and percentage of occurrence of a SNP in a ROH (Pearson’s correlation coefficient = 0.25, <0.0001). The average | iHS| values were also calculated in each ROH islands (Table 4). The result showed that mean | iHS| values of SNPs in each ROH islands (except one on SSC10) were higher than that across the genome (0.77) (Table 4).
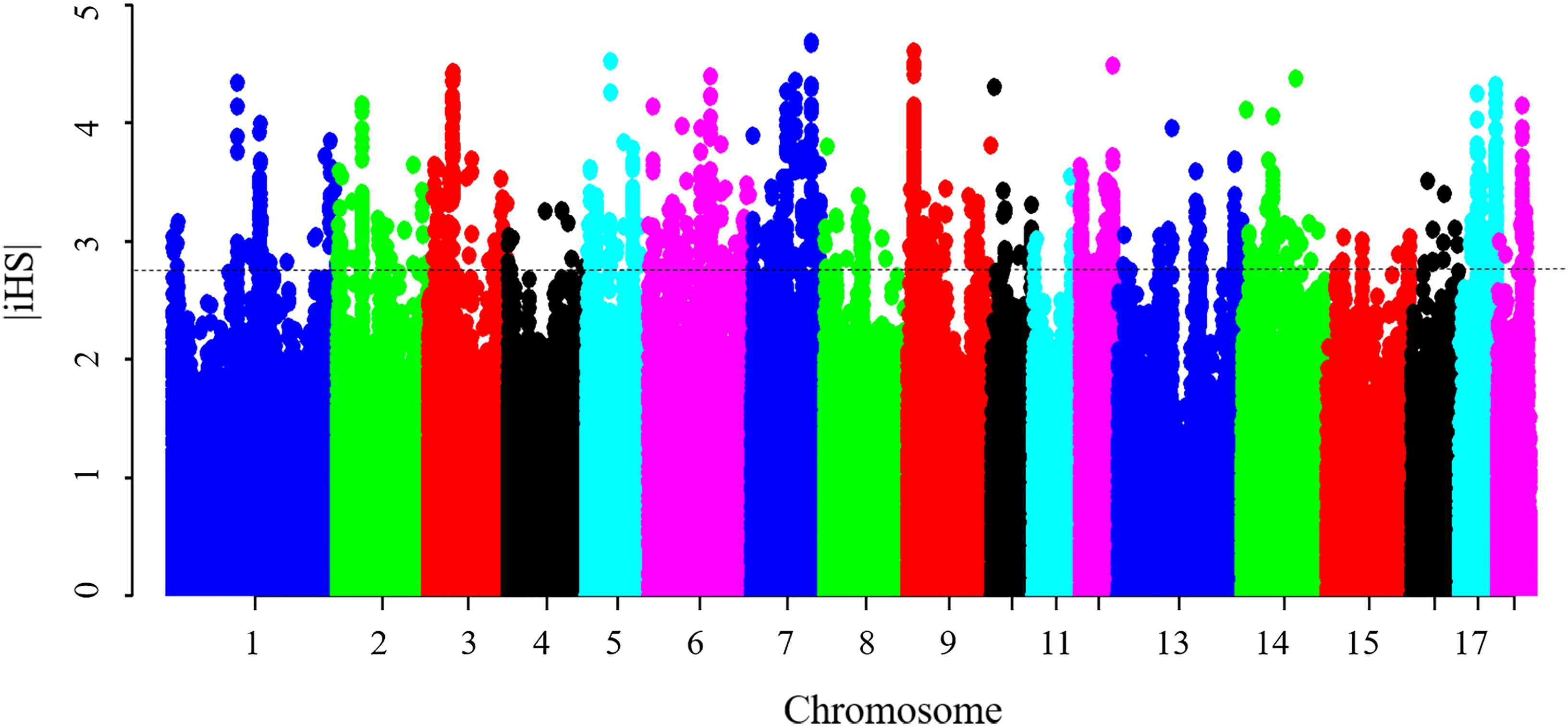
Figure 4. Genome-wide distribution of selection signatures detected by iHS. The dashed line represents the threshold levels of 0.5% (| iHS| = 2.81).
There were 1,535 SNPs with p-value < 0.005 that harbored signatures of selection, 92 of which were found in ROH islands (Supplementary Table S4). A total of 42 candidate genes were found to overlap with these regions (Supplementary Table S5). It includes several genes mentioned above, such as MYOD1, ADCK1, LPIN3, ITGB5, NUCB2, PIK3C2A, and ABCC8. These genes obtained by the two methods should be given more consideration in further studies. The significant correlation between the iHS selection signature method and the percentage of occurrence of SNP in a ROH, in the present study and elsewhere (Zhang et al., 2015b), supports the hypothesis that the observed ROH islands are not only as a result of demography, but could also be due to selection.
To our knowledge, this is the first study to describe the occurrence and distribution of ROH in the genome of Jinhua pigs. Autozygosity levels varied largely in this population, which has experienced both recent and historical inbreeding events. We have shown that, despite the low to moderate inbreeding levels in most animals, there were individuals with high inbreeding coefficients, indicating the need to account for inbreeding when planning mating strategy. Several genes within ROH islands are associated with adaptive and economic traits and should be the subject of future investigation. These findings may contribute to the understanding of the effects of environmental and artificial selection in shaping the distribution of functional variants in pig genome.
All BAM data were deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA). 202 samples are available under the Bioproject No. PRJNA525747.
All experimental procedures were approved by the Institutional Animal Care and Use Committee of Shanghai Jiao Tong University, and all methods involved pigs were in accordance with the agreement of Institutional Animal Care and Use Committee of Shanghai Jiao Tong University (Contract No. 2011–0033).
YP, QW, and ZX designed the experiments. ZX, HS, QZ, XZ, QL, and YY performed the experiments. ZX, ZZ, and PM developed some of the analysis software. ZX wrote the manuscript with the help of BO. All authors read and approved the final manuscript.
This work was supported by Zhejiang Province agriculture (livestock) varieties breeding Key Technology R&D Program (Grant No. 2016C02054-2), National Natural Science Foundation of China (Grant No. 31872976), and National Natural Science Foundation of China (Grant No. U1402266).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.00274/full#supplementary-material
Bagot, C. N., Troy, P. J., and Taylor, H. S. (2000). Alteration of maternal Hoxa10 expression by in vivo gene transfection affects implantation. Gene Ther. 7, 1378–1384. doi: 10.1038/sj.gt.3301245
Barbato, M., Orozco-terWengel, P., Tapio, M., and Bruford, M. W. (2015). SNeP: a tool to estimate trends in recent effective population size trajectories using genome-wide SNP data. Front. Genet. 6:109. doi: 10.3389/fgene.2015.00109
Barrett, J. C. (2009). Haploview: visualization and analysis of SNP genotype data. Cold Spring Harb. Protoc. 2009:pdb.ip71. doi: 10.1101/pdb.ip71
Bertolini, F., Servin, B., Talenti, A., Rochat, E., Kim, E. S., Oget, C., et al. (2018). Signatures of selection and environmental adaptation across the goat genome post-domestication. Genet. Sel. Evol. 50:57. doi: 10.1186/s12711-018-0421-y
Bosse, M., Megens, H. J., Madsen, O., Paudel, Y., Frantz, L. A., Schook, L. B., et al. (2012). Regions of homozygosity in the porcine genome: consequence of demography and the recombination landscape. PLoS Genet. 8:e1003100. doi: 10.1371/journal.pgen.1003100
Brito, L. F., Kijas, J. W., Ventura, R. V., Sargolzaei, M., Porto-Neto, L. R., Cánovas, A., et al. (2017). Genetic diversity and signatures of selection in various goat breeds revealed by genome-wide SNP markers. BMC Genomics 18:229. doi: 10.1186/s12863-015-0220-1
Cardoso, D. F., Albuquerque, L. G., Reimer, C., Qanbari, S., Erbe, M., Nascimento, A. V., et al. (2018). Genome-wide scan reveals population stratification and footprints of recent selection in Nelore cattle. Genet. Sel. Evol. 50, 22. doi: 10.1186/s12711-018-0381-2
Cepica, S., Zambonelli, P., Weisz, F., Bigi, M., Knoll, A., Vykoukalova, Z., et al. (2013). Association mapping of quantitative trait loci for carcass and meat quality traits at the central part of chromosome 2 in Italian Large White pigs. Meat Sci. 95, 368–375. doi: 10.1016/j.meatsci.2013.05.002
Charlesworth, B. (2009). Effective population size and patterns of molecular evolution and variation. Nat. Rev. Genet. 10:195. doi: 10.1038/nrg2526
Chen, J., Peng, J., Xiao, Q., Pan, Y., Zhang, X., Lo, L. J., et al. (2018). The genetic diversity and population structures of indigenous pig breeds in Zhejiang Province revealed by GGRS sequencing. Anim. Genet. 49, 36–42. doi: 10.1111/age.12625
Chen, Q., Ma, Y., Yang, Y., Chen, Z., Liao, R., Xie, X., et al. (2013). Genotyping by genome reducing and sequencing for outbred animals. PLoS One 8:e67500. doi: 10.1371/journal.pone.0067500
China National Commission of Animal Genetic Resources (2011). Animal Genetic Resources in China Pigs. Beijing: Chinese Agriculture Press.
Choi, I., Steibel, J. P., Bates, R. O., Raney, N. E., Rumph, J. M., and Ernst, C. W. (2011). Identification of carcass and meat quality QTL in an F2 Duroc ( Pietrain pig resource population using different least-squares analysis models. Front. Genet. 2:18. doi: 10.3389/fgene.2011.00018
Curik, I., Ferencakovic, M., and Solkner, J. (2014). Inbreeding and runs of homozygosity: a possible solution to an old problem. Livest. Sci. 166, 26–34. doi: 10.1016/j.livsci.2014.05.034
Durinck, S., Moreau, Y., Kasprzyk, A., Davis, S., De Moor, B., Brazma, A., et al. (2005). BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics 21, 3439–3440. doi: 10.1093/bioinformatics/bti525
Edea, Z., and Kim, K. S. (2014). A whole genomic scan to detect selection signatures between Berkshire and Korean native pig breeds. J. Anim. Sci. Technol. 56:23. doi: 10.1186/2055-0391-56-23
Ferencakovic, M., Solkner, J., and Curik, I. (2013). Estimating autozygosity from high-throughput information: effects of SNP density and genotyping errors. Genet. Sel. Evol. 45:42. doi: 10.1186/1297-9686-45-42
Fu, W. X., Liu, Y., Lu, X., Niu, X. Y., Ding, X. D., Liu, J. F., et al. (2012). A genome-wide association study identifies two novel promising candidate genes affecting Escherichia coli F4ab/F4ac susceptibility in swine. PLoS One 7:e32127. doi: 10.1371/journal.pone.0032127
Gao, Y., Hyttel, P., and Hall, V. J. (2010). Regulation of H3K27me3 and H3K4me3 during early porcine embryonic development. Mol. Reprod. Dev. 77, 540–549. doi: 10.1002/mrd.21180
Gao, Y., Rong, Y., Wang, Y., Xiong, H., Huang, X., Han, F., et al. (2014). Expression pattern of porcine antimicrobial peptide PR-39 and its induction by enterotoxigenic Escherichia coli (ETEC) F4ac. Vet. Immunol. Immunopathol. 160, 260–265. doi: 10.1016/j.vetimm.2014.05.012
Gautier, M., Klassmann, A., and Vitalis, R. (2017). rehh 2.0: a reimplementation of the R package rehh to detect positive selection from haplotype structure. Mol. Ecol. Resour. 17, 78–90. doi: 10.1111/1755-0998.12634
Gibson, J., Morton, N. E., and Collins, A. (2006). Extended tracts of homozygosity in outbred human populations. Hum. Mol. Genet. 15, 789–795. doi: 10.1093/hmg/ddi493
Harmegnies, N., Davin, F., De Smet, S., Buys, N., Georges, M., and Coppieters, W. (2006). Results of a whole-genome quantitative trait locus scan for growth, carcass composition and meat quality in a porcine four-way cross. Anim. Genet. 37, 543–553. doi: 10.1111/j.1365-2052.2006.01523.x
He, X., Xu, X., and Liu, B. (2009). Molecular characterization, chromosomal localization and association analysis with back-fat thickness of porcine LPIN2 and LPIN3. Mol. Biol. Rep. 36, 1819–1824. doi: 10.1007/s11033-008-9385-2
Howie, B. N., Donnelly, P., and Marchini, J. (2009). A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 5:e1000529. doi: 10.1371/journal.pgen.1000529
Howrigan, D. P., Simonson, M. A., and Keller, M. C. (2011). Detecting autozygosity through runs of homozygosity: a comparison of three autozygosity detection algorithms. BMC Genomics 12:460. doi: 10.1186/1471-2164-12-460
Huang, X., Ren, J., Yan, X., Peng, Q., Tang, H., Zhang, B., et al. (2008). Polymorphisms of three gene-derived STS on pig chromosome 13q41 are associated with susceptibility to enterotoxigenic Escherichia coli F4ab/ac in pigs. Sci. China C Life Sci. 51, 614–619. doi: 10.1007/s11427-008-0078-9
Jacobsen, M., Kracht, S. S., Esteso, G., Cirera, S., Edfors, I., Archibald, A. L., et al. (2010). Refined candidate region specified by haplotype sharing for Escherichia coli F4ab/F4ac susceptibility alleles in pigs. Anim. Genet. 41, 21–25. doi: 10.1111/j.1365-2052.2009.01952.x
Jiao, X. L., Jing, J. J., Qiao, L. Y., Liu, J. H., Li, L. A., Zhang, J., et al. (2016). Ontogenetic expression of Lpin2 and Lpin3 genes and their associations with traits in two breeds of chinese fat-tailed sheep. Asian Australas. J. Anim. Sci. 29, 333–342. doi: 10.5713/ajas.15.0467
Keller, M. C., Visscher, P. M., and Goddard, M. E. (2011). Quantification of inbreeding due to distant ancestors and its detection using dense single nucleotide polymorphism data. Genetics 189, 237–249. doi: 10.1534/genetics.111.130922
Kim, E. S., Ros-Freixedes, R., Pena, R. N., Baas, T. J., Estany, J., and Rothschild, M. F. (2015). Identification of signatures of selection for intramuscular fat and backfat thickness in two Duroc populations. J. Anim. Sci. 93, 3292–3302. doi: 10.2527/jas.2015-8879
Lan, J., Zhao, J., and Liu, Y. (2012). Molecular cloning, sequence characterization, polymorphism and association analysis of porcine ROPN1 gene. Mol. Biol. Rep. 39, 2739–2743. doi: 10.1007/s11033-011-1029-2
Lee, E. A., Kim, J. M., Lim, K. S., Ryu, Y. C., Jeon, W. M., and Hong, K. C. (2012). Effects of variation in porcine MYOD1 gene on muscle fiber characteristics, lean meat production, and meat quality traits. Meat. Sci. 92, 36–43. doi: 10.1016/j.meatsci.2012.03.018
Lents, C. A., Barb, C. R., Hausman, G. J., Nonneman, D., Heidorn, N. L., Cisse, R. S., et al. (2013). Effects of nesfatin-1 on food intake and LH secretion in prepubertal gilts and genomic association of the porcine NUCB2 gene with growth traits. Domest. Anim. Endocrinol. 45, 89–97. doi: 10.1016/j.domaniend.2013.06.002
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, Z., Chen, J., Wang, Z., Pan, Y., Wang, Q., Xu, N., et al. (2016). Detection of selection signatures of population-specific genomic regions selected during domestication process in Jinhua pigs. Anim. Genet. 47, 672–681. doi: 10.1111/age.12475
Lyutskanov, M. (2011). Epidemiological characteristics of post-weaning diarrhoea associated with toxin-producing Escherichia coli in large intensive pig farms. Trakia J. Sci. 9, 68–73.
Marchesi, J. A. P., Buzanskas, M. E., Cantao, M. E., Ibelli, A. M. G., Peixoto, J. O., Joaquim, L. B., et al. (2018). Relationship of runs of homozygosity with adaptive and production traits in a paternal broiler line. Animal 12, 1126–1134. doi: 10.1017/S1751731117002671
Mastrangelo, S., Tolone, M., Di Gerlando, R., Fontanesi, L., Sardina, M. T., and Portolano, B. (2016). Genomic inbreeding estimation in small populations: evaluation of runs of homozygosity in three local dairy cattle breeds. Animal 10, 746–754. doi: 10.1017/S1751731115002943
Mastrangelo, S., Tolone, M., Sardina, M. T., Sottile, G., Sutera, A. M., Di Gerlando, R., et al. (2017). Genome-wide scan for runs of homozygosity identifies potential candidate genes associated with local adaptation in Valle del Belice sheep. Genet. Sel. Evol. 49:84. doi: 10.1186/s12711-017-0360-z
McQuillan, R., Leutenegger, A. L., Abdel-Rahman, R., Franklin, C. S., Pericic, M., Barac-Lauc, L., et al. (2008). Runs of homozygosity in European populations. Am. J. Hum. Genet. 83, 359–372. doi: 10.1016/j.ajhg.2008.08.007
Miao, Z. G., Wang, L. J., Xu, Z. R., Huang, J. F., and Wang, Y. R. (2009). Developmental changes of carcass composition, meat quality and organs in the Jinhua pig and Landrace. Animal 3, 468–473. doi: 10.1017/S1751731108003613
Ouborg, N. J., Pertoldi, C., Loeschcke, V., Bijlsma, R., and Hedrick, P. W. (2010). Conservation genetics in transition to conservation genomics. Trends Genet. 26, 177–187. doi: 10.1016/j.tig.2010.01.001
Patel, R. K., and Jain, M. (2012). NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One 7:e30619. doi: 10.1371/journal.pone.0030619
Pemberton, T. J., Absher, D., Feldman, M. W., Myers, R. M., Rosenberg, N. A., and Li, J. Z. (2012). Genomic patterns of homozygosity in worldwide human populations. Am. J. Hum. Genet. 91, 275–292. doi: 10.1016/j.ajhg.2012.06.014
Peripolli, E., Stafuzza, N. B., Munari, D. P., Lima, A. L. F., Irgang, R., Machado, M. A., et al. (2018). Assessment of runs of homozygosity islands and estimates of genomic inbreeding in Gyr (Bos indicus) dairy cattle. BMC Genomics 19:34. doi: 10.1186/s12864-017-4365-3
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A., Bender, D., et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. doi: 10.1086/519795
Purfield, D. C., Berry, D. P., McParland, S., and Bradley, D. G. (2012). Runs of homozygosity and population history in cattle. BMC Genet. 13:70. doi: 10.1186/1471-2156-13-70
Purfield, D. C., McParland, S., Wall, E., and Berry, D. P. (2017). The distribution of runs of homozygosity and selection signatures in six commercial meat sheep breeds. PLoS One 12:e0176780. doi: 10.1371/journal.pone.0176780
Quintanilla, R., Pena, R. N., Gallardo, D., Canovas, A., Ramirez, O., Diaz, I., et al. (2011). Porcine intramuscular fat content and composition are regulated by quantitative trait loci with muscle-specific effects. J. Anim. Sci. 89, 2963–2971. doi: 10.2527/jas.2011-3974
Rampoldi, A., Jacobsen, M. J., Bertschinger, H. U., Joller, D., Burgi, E., Vogeli, P., et al. (2011). The receptor locus for Escherichia coli F4ab/F4ac in the pig maps distal to the MUC4-LMLN region. Mamm. Genome 22, 122–129. doi: 10.1007/s00335-010-9305-3
Ren, J., Yan, X., Ai, H., Zhang, Z., Huang, X., Ouyang, J., et al. (2012). Susceptibility towards enterotoxigenic Escherichia coli F4ac diarrhea is governed by the MUC13 gene in pigs. PLoS One 7:e44573. doi: 10.1371/journal.pone.0044573
Scheet, P., and Stephens, M. (2006). A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase. Am. J. Hum. Genet. 78, 629–644. doi: 10.1086/502802
Stratil, A., Van Poucke, M., Bartenschlager, H., Knoll, A., Yerle, M., Peelman, L. J., et al. (2006). Porcine OGN and ASPN: mapping, polymorphisms and use for quantitative trait loci identification for growth and carcass traits in a Meishan x Pietrain intercross. Anim. Genet. 37, 415–418. doi: 10.1111/j.1365-2052.2006.01480.x
Sved, J., and Feldman, M. (1973). Correlation and probability methods for one and two loci. Theor. Popul. Biol. 4, 129–132. doi: 10.1016/0040-5809(73)90008-7
Thompson, E. A. (2013). Identity by descent: variation in meiosis, across genomes, and in populations. Genetics 194, 301–326. doi: 10.1534/genetics.112.148825
VanRaden, P. M. (2008). Efficient methods to compute genomic predictions. J. Dairy Sci. 91, 4414–4423. doi: 10.3168/jds.2007-0980
Verardo, L. L., Silva, F. F., Varona, L., Resende, M. D., Bastiaansen, J. W., Lopes, P. S., et al. (2015). Bayesian GWAS and network analysis revealed new candidate genes for number of teats in pigs. J. Appl. Genet. 56, 123–132. doi: 10.1007/s13353-014-0240-y
Visscher, P. M., Medland, S. E., Ferreira, M. A., Morley, K. I., Zhu, G., Cornes, B. K., et al. (2006). Assumption-free estimation of heritability from genome-wide identity-by-descent sharing between full siblings. PLoS Genet. 2:e41. doi: 10.1371/journal.pgen.0020041
Voight, B. F., Kudaravalli, S., Wen, X., and Pritchard, J. K. (2006). A map of recent positive selection in the human genome. PLoS Biol. 4:e72. doi: 10.1371/journal.pbio.0040072
Wang, Z., Chen, Q., Yang, Y., Liao, R., Zhao, J., Zhang, Z., et al. (2015). Genetic diversity and population structure of six Chinese indigenous pig breeds in the Taihu Lake region revealed by sequencing data. Anim. Genet. 46, 697–701. doi: 10.1111/age.12349
Wilson, M. E., Sonstegard, T. S., Smith, T. P., Fahrenkrug, S. C., and Ford, S. P. (2000). Differential gene expression during elongation in the preimplantation pig embryo. Genesis 26, 9–14. doi: 10.1002/(SICI)1526-968X(200001)26:1<9::AID-GENE4>3.0.CO;2-1
Wright, S. (1922). Coefficients of inbreeding and relationship. Am. Nat. 56, 330–338. doi: 10.1086/279872
Wu, D., Song, D., Li, X., Yu, M., Li, C., and Zhao, S. (2013). Molecular characterization and identification of the E2/P4 response element in the porcine HOXA10 gene. Mol. Cell Biochem. 374, 213–222. doi: 10.1007/s11010-012-1522-5
Xiao, Q., Zhang, Z., Sun, H., Wang, Q., and Pan, Y. (2017). Pudong White pig: a unique genetic resource disclosed by sequencing data. Animal 11, 1117–1124. doi: 10.1017/S1751731116002494
Yan, X., Huang, X., Ren, J., Zou, Z., Yang, S., Ouyang, J., et al. (2009). Distribution of Escherichia coli F4 adhesion phenotypes in pigs of 15 Chinese and Western breeds and a White Duroc × Erhualian intercross. J. Med. Microbiol. 58, 1112–1117. doi: 10.1099/jmm.0.009803-0
Yang, J., Lee, S. H., Goddard, M. E., and Visscher, P. M. (2011). GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82. doi: 10.1016/j.ajhg.2010.11.011
Yang, J., Lee, S. H., Goddard, M. E., and Visscher, P. M. (2013). “Genome-wide complex trait analysis (GCTA): methods, data analyses, and interpretations,” in Genome-Wide Association Studies and Genomic Prediction, eds C. Gondro, J. van der Werf, and B. Hayes (Totowa, NJ: Humana Press), 215–236.
Yin, Q., Yang, H., Han, X., Fan, B., and Liu, B. (2012). Isolation, mapping, SNP detection and association with backfat traits of the porcine CTNNBL1 and DGAT2 genes. Mol. Biol. Rep. 39, 4485–4490. doi: 10.1007/s11033-011-1238-8
Zanella, R., Peixoto, J. O., Cardoso, F. F., Cardoso, L. L., Biegelmeyer, P., Cantao, M. E., et al. (2016). Genetic diversity analysis of two commercial breeds of pigs using genomic and pedigree data. Genet. Sel. Evol. 48:24. doi: 10.1186/s12711-016-0203-3
Zavarez, L. B., Utsunomiya, Y. T., Carmo, A. S., Neves, H. H., Carvalheiro, R., Ferencakovic, M., et al. (2015). Assessment of autozygosity in Nellore cows (Bos indicus) through high-density SNP genotypes. Front. Genet. 6:5. doi: 10.3389/fgene.2015.00005
Zhang, J., Chen, J. H., Liu, X. D., Wang, H. Y., Liu, X. L., Li, X. Y., et al. (2016). Genomewide association studies for hematological traits and T lymphocyte subpopulations in a Duroc x Erhualian F resource population. J. Anim. Sci. 94, 5028–5041. doi: 10.2527/jas.2016-0924
Zhang, Q., Calus, M. P., Guldbrandtsen, B., Lund, M. S., and Sahana, G. (2015a). Estimation of inbreeding using pedigree, 50k SNP chip genotypes and full sequence data in three cattle breeds. BMC Genet. 16:88. doi: 10.1186/s12863-015-0227-7
Zhang, Q., Guldbrandtsen, B., Bosse, M., Lund, M. S., and Sahana, G. (2015b). Runs of homozygosity and distribution of functional variants in the cattle genome. BMC Genomics 16:542. doi: 10.1186/s12864-015-1715-x
Keywords: pig, runs of homozygosity, inbreeding coefficients, selection, animal breeding
Citation: Xu Z, Sun H, Zhang Z, Zhao Q, Olasege BS, Li Q, Yue Y, Ma P, Zhang X, Wang Q and Pan Y (2019) Assessment of Autozygosity Derived From Runs of Homozygosity in Jinhua Pigs Disclosed by Sequencing Data. Front. Genet. 10:274. doi: 10.3389/fgene.2019.00274
Received: 20 June 2018; Accepted: 12 March 2019;
Published: 28 March 2019.
Edited by:
Andrea B. Doeschl-Wilson, Roslin Institute, University of Edinburgh, United KingdomReviewed by:
Maria Saura, Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria (INIA), SpainCopyright © 2019 Xu, Sun, Zhang, Zhao, Olasege, Li, Yue, Ma, Zhang, Wang and Pan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qishan Wang, d2FuZ3Fpc2hhbkBzanR1LmVkdS5jbg== Yuchun Pan, cGFueXVjaHVuMTk2M0BhbGl5dW4uY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.