
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 11 December 2018
Sec. Genetics of Common and Rare Diseases
Volume 9 - 2018 | https://doi.org/10.3389/fgene.2018.00612
This article is part of the Research Topic Model Organisms: A Precious Resource for Understanding of the Molecular Mechanisms Underlying Human Physiology and Disease View all 14 articles
 Manuela Sollazzo*†
Manuela Sollazzo*† China Genchi†
China Genchi† Simona Paglia
Simona Paglia Simone Di Giacomo
Simone Di Giacomo Annalisa Pession
Annalisa Pession Dario de Biase
Dario de Biase Daniela Grifoni*
Daniela Grifoni*The term “field cancerisation” describes the formation of tissue sub-areas highly susceptible to multifocal tumourigenesis. In the earlier stages of cancer, cells may indeed display a series of molecular alterations that allow them to proliferate faster, eventually occupying discrete tissue regions with irrelevant morphological anomalies. This behaviour recalls cell competition, a process based on a reciprocal fitness comparison: when cells with a growth advantage arise in a tissue, they are able to commit wild-type neighbours to death and to proliferate at their expense. It is known that cells expressing high MYC levels behave as super-competitors, able to kill and replace less performant adjacent cells; given MYC upregulation in most human cancers, MYC-mediated cell competition is likely to pioneer field cancerisation. Here we show that MYC overexpression in a sub-territory of the larval wing epithelium of Drosophila is sufficient to trigger a number of cellular responses specific to mammalian pre-malignant tissues. Moreover, following induction of different second mutations, high MYC-expressing epithelia were found to be susceptible to multifocal growth, a hallmark of mammalian pre-cancerous fields. In summary, our study identified an early molecular alteration implicated in field cancerisation and established a genetically amenable model which may help study the molecular basis of early carcinogenesis.
The molecular events underlying cancer initiation are largely unknown. It is commonly accepted that most cancers are monoclonal in origin, evolving from a single cell whose lineage accumulates in time multiple molecular insults (Michor et al., 2004; Vogelstein et al., 2013; Feinberg et al., 2016). In particular, driver mutations, which provide cells with a growth advantage and are positively selected during lineage evolution, are generally associated with clonal expansion and are frequently found in pre-malignant lesions (Maley et al., 2004; Lawrence et al., 2014; Curtius et al., 2017). In the 1950s, Slaughter introduced the concept of “field cancerisation”: while studying oral cancers, he observed that they recurred more frequently adjacent to a resected tumour (Slaughter et al., 1953). Therefore, field cancerisation was defined as the process leading to the formation of a tissue sub-territory which, despite a normal appearance, bears a series of alterations that make cells more susceptible to malignant transformation than wild-type neighbours, giving rise to multifocal cancers (Wodarz et al., 2004). Successive studies, also fostered by the development of post-genomic technologies (Metzker, 2010), have demonstrated that this phenomenon is not specific to the oral mucosa, being rather a common feature of epithelial organs (Braakhuis et al., 2003; Dakubo et al., 2007; Nonn et al., 2009; Zeki et al., 2011; Jakubek et al., 2016; Lu et al., 2016; Park et al., 2016; Abdalla et al., 2017; Castven et al., 2017).
Although the interest in deciphering cancer’s molecular signature is obvious, it may be quite difficult to understand what mutations favour and maintain the malignant phenotype: a surprising number of driver mutations is indeed present in pre-cancerous tissues, also in those that are not likely to evolve into a frank malignancy (Hofstad et al., 1996; Martincorena et al., 2015; Kato et al., 2016). This suggests that several alterations are evolutionarily neutral and do not impact cell’s phenotype, maybe depending on their temporal occurrence (de Bruin et al., 2014), the tissue context (Galandiuk et al., 2012; Gagneur et al., 2013; Vermeulen et al., 2013) and the genetic background (Chandler et al., 2013). In human tissues, a number of genetic alterations have been associated with field cancerisation (Papadimitrakopoulou et al., 1996; Braakhuis et al., 2002; Santos-Garcia et al., 2005; Haaland et al., 2009; Trujillo et al., 2011; Mohan and Jagannathan, 2014), and genetic/genomic instability (Ellsworth et al., 2004; Zaky et al., 2008; Giaretti et al., 2012), mitochondrial defects (McDonald et al., 2008; Maggrah et al., 2013; Parr et al., 2013), production of reactive oxygen species (ROS) (Bongers et al., 1995; Jaloszynski et al., 2003; Chan et al., 2017), increased expression of proliferation and apoptosis markers (Birchall et al., 1997; Bascones-Martinez et al., 2013) and epigenetic modifications (Grady, 2005; Lee et al., 2011; Kamiyama et al., 2012; Luo et al., 2014) are also repeatedly found in regions adjacent to malignant tumours from a variety of organs. Whatever the cause of these modifications, from DNA replication errors to mutagenic injuries, the ongoing pre-cancerous field will most likely be composed of a number of genetically different clones, with the fittest one expected to colonise the entire territory over time (Driessens et al., 2012). This process of selection based on fitness comparison is a distinctive trait of cell competition (CC), a phenomenon first observed and characterised in Drosophila (Morata and Ripoll, 1975), and then demonstrated to be conserved in mammals (Penzo-Mendez and Stanger, 2014; Di Gregorio et al., 2016).
Competitive interactions are typically triggered when cells with different proliferation rates are found in close proximity: the fittest cells (winners) commit less fit neighbours (losers) to death and overgrow to replace them in the tissue (Levayer and Moreno, 2013, 2016; Tamori and Deng, 2013; Tsuboi et al., 2018). A number of molecules and signalling pathways have to date been found to play a role in CC (Moreno et al., 2002; Tyler et al., 2007; Vincent et al., 2011; Rodrigues et al., 2012; Akai et al., 2018): among these, the MYC protein was shown to be the most powerful inducer of CC (named in this case MYC-Mediated Cell Competition, MMCC) from Drosophila to mammals (Johnston, 2014), paving the way to studies that found this process implicated in a number of seemingly distant contexts, from organ development (de la Cova et al., 2004; Moreno and Basler, 2004; Claveria et al., 2013; Sancho et al., 2013; Villa del Campo et al., 2014; Villa Del Campo et al., 2016) to tissue regeneration (Oertel et al., 2006; Gogna et al., 2015; Rosen et al., 2015; Villa Del Campo et al., 2016; Shakiba and Zandstra, 2017), cell stemness (Rhiner et al., 2009; Diaz-Diaz et al., 2017) and cancer (Froldi et al., 2010; Ziosi et al., 2010; Eichenlaub et al., 2016; Suijkerbuijk et al., 2016). Of note, we and others recently demonstrated that MMCC is also active in human cancer cells (Patel et al., 2016; Di Giacomo et al., 2017). MYC upregulation is sufficient as to transform cells into super-competitors (Moreno and Basler, 2004), able to kill and replace suboptimal neighbours, and this capability has opened to speculations about a possible role for MMCC in field cancerisation (Rhiner and Moreno, 2009; Johnston, 2014). MYC family proteins are long investigated for their essential functions in cell physiology and in cancer (Stine et al., 2015); the Drosophila genome bears a single locus (diminutive, dm) encoding the MYC protein, which exerts the same functions as the mammalian orthologues (Gallant, 2013). MYC overexpression in wild-type cells may provoke a series of contradictory responses: on the one hand, it supports cell growth by accelerating biosynthesis, cell metabolism and cell cycle (Evan and Littlewood, 1993; Grewal et al., 2005; Meyer and Penn, 2008); on the other hand, it promotes potentially harmful reactions such as ROS production and genetic instability (Vafa et al., 2002; Greer et al., 2013; Kuzyk and Mai, 2014), and increases propensity to apoptotic cell death (Montero et al., 2008; McMahon, 2014). Cancer cells upregulating MYC are contrariwise protected from untimely death, primarily due to relevant changes in metabolic pathways leading to MYC addiction (Gabay et al., 2014). MYC seems thus to elicit in normal cells a number of biological responses similar to those found in mammalian pre-cancerous fields (Mohan and Jagannathan, 2014). Moreover, MYC upregulation is an early event in human prostate cancer (Gurel et al., 2008), and MYC overexpression is sufficient to transform luminal epithelial cells into pre-malignant derivatives in the mouse prostatic gland (Kim et al., 2009; Iwata et al., 2010). MYC upregulation has also been observed in cytologically normal bronchial epithelial cells of mice with pre-neoplastic lung squamous cell carcinoma lesions (Xiong et al., 2017), and it was reported to initiate gastric tumourigenesis following Hippo pathway deregulation in the pyloric stem cell (Choi et al., 2018). These observations led us to speculate that high MYC levels may be sufficient for an epithelial tissue to become responsive to the effect of second mutations that would otherwise be irrelevant when occurring in a wild-type epithelium.
In Drosophila, the tumour suppressor genes (TSGs) are historically subdivided into two classes, called “hyperplastic” and “neoplastic” according to the mutant phenotype (Hariharan and Bilder, 2006), most of which have in time been found to encode different components of the Hippo pathway (Grusche et al., 2010), a highly conserved signalling cascade central in cell growth and organ size modulation (Halder and Johnson, 2011). Broadly speaking, loss-of-function (LOF) mutants of these hyperplastic TSGs (fat, ft; dachsous, ds; expanded, ex; warts, wts; and hippo, hpo) show a substantial overgrowth of the larval epithelial organs, called imaginal discs (Aldaz and Escudero, 2010), and premature death at the pupal stage (Hariharan and Bilder, 2006), whereas LOF mutants of neoplastic TSGs do not survive beyond embryogenesis (Menut et al., 2007). An exception is made for scribble (scrib), discs large (dlg), and lethal giant larvae (lgl) neoplastic mutants which, given the abundant maternal transcript released into the zygote, survive up to the end of the larval life, showing abnormal growth of the imaginal discs with a complete loss of the epithelial structure (Bilder et al., 2000; Bilder, 2004). In case single mutant cells are created in a wild-type background through clonal analysis techniques (del Valle et al., 2012), those bearing hyperplastic LOF mutations survive and overgrow in the target tissue (Xu et al., 1995; Buratovich and Bryant, 1997; Udan et al., 2003; Maitra et al., 2006), whereas those bearing neoplastic LOF mutations are usually eliminated during development (Agrawal et al., 1995; Enomoto and Igaki, 2011). We and others demonstrated these opposite behaviours are dictated by MMCC: while hyperplastic mutant cells upregulate MYC and behave like winners in the wild-type tissue, killing the surrounding neighbours and growing at their expense (Neto-Silva et al., 2010; Ziosi et al., 2010), neoplastic mutant cells do not upregulate MYC and behave like losers in the context, being themselves out-competed by adjacent wild-type cells (Froldi et al., 2010; Menendez et al., 2010). We previously showed that a MYC-overexpressing background strengthens the super-competitive behaviour of ft, ds and ex mutant clones, which were found to kill the surrounding cells with increased efficiency and to grow more rapidly, although it did not provide mutant cells with the capability to evolve into a malignant mass (Ziosi et al., 2010).
Here we expanded on previous work by first identifying in Drosophila MYC-overexpressing epithelial organs a series of morphological and molecular markers typically found in human pre-cancerous fields. Moreover, we investigated the impact of a MYC-overexpressing background on the cellular phenotypes consequent to mutations in neoplastic TSGs, showing it is in this case sufficient to make mutant cells able to initiate multifocal malignant transformation, a peculiar trait of human pre-neoplastic fields.
The following fly lines were used in the study, built using stocks obtained from the Bloomington Drosophila Stock Center, Indiana: w; UAS-GFP(Bl-6874); hh-Gal4(Bl-67046) – yw, PI3K92ECAAX(Bl-25908) – w; Ubi-GFPnls, FRT40A(Bl-5629)/CyO; hh-Gal4(Bl-67046)/TM6b – w; l(2)gl4 P(neo-FRT)40A(Bl-36289)/ In(2-3)Gla,Bc; UAS-HAdm/TM6b – w; Rab52P(neo-FRT) 40A(Bl-42702)/In(2-3)Gla,Bc; UAS-HAdm/TM6b. UAS-HAdm on III is a gift of P. Bellosta. Plain genotypes are given for each experiment in the figure legends. For all experiments, flies were kept at 25°C. Larvae were heat-shocked once at 48 ± 4 h AEL in a water bath at 37°C for 10 min and dissected after additional 72 h development.
Frozen or fresh larvae were prepared for immunofluorescence by standard methods. The following antibodies and dilutions were used: mouse α-MYC (1:5, P. Bellosta); rabbit α-Lgl (1:400, D. Strand); rabbit α-active Caspase 3 (1:100, Cell Signalling Technologies); rabbit α-aPKCζ (1:200, Santa Cruz Biotechnology); rabbit α-pAKT (1:100, Cell Signaling Technologies); rabbit α-PH3 (1:100, Upstate Technology); mouse α-γH2Av (1:30, DSHB); mouse α-dIAP1 (1:100, B. A. Hay); rabbit α-Pc (1:400, Santa Cruz Biotechnology); mouse α-En (1:50, DSHB). Alexa Fluor 555 goat α-mouse and α-rabbit (1:500, Invitrogen) and DyLight 649-conjugated goat α-mouse and α-rabbit (1:750, Jackson ImmunoResearch Laboratories) were used as secondary antibodies. Samples were analysed with a Leica TSC SP2 laser confocal microscope and entire images were processed with Adobe Photoshop software or ImageJ free software from NIH. All the images represent a single confocal stack unless otherwise specified. Image magnification is 400× unless otherwise specified.
Larvae were dissected in PBS1X and carcasses were incubated for 30 min at room temperature in PBS1X – DHE (Dihydroethidium, Invitrogen Molecular Probes) at a final concentration of 30 μM in gentle shaking before fixation. Wing discs were immediately imaged under a Nikon 90i wide-field fluorescence microscope.
For the experiments shown in Figures 3–7, the number of wing discs analysed was 15÷25 from different larvae for each sample. For each experiment, the data presented are the average of three biological replicates. Multifocality was assessed on a total of 346 wing discs for l(2)gl4 clones (see Figures 9, 10), and on a total of 146 wing discs for Rab52 (see Figure 11). For the experiments shown in Figures 2, 8, 13, the number of discs analysed is indicated. Mean Fluorescence Intensity (MFI) (Figures 2, 5–7), clone area (Figures 8, 13) and positive signals (Figures 3, 4) were calculated by ImageJ free software (NIH) on images captured with a Nikon 90i wide-field fluorescence microscope at a magnification of 200×. All measurements have been taken inside the yellowish area highlighted in Figure 1A. P-values were as follows: ∗∗p ≤ 0.01 and ∗∗∗p ≤ 0.001. Mean, SEM and the t-Student test p-value were calculated by using GraphPad Prism software, San Diego, CA, United States.
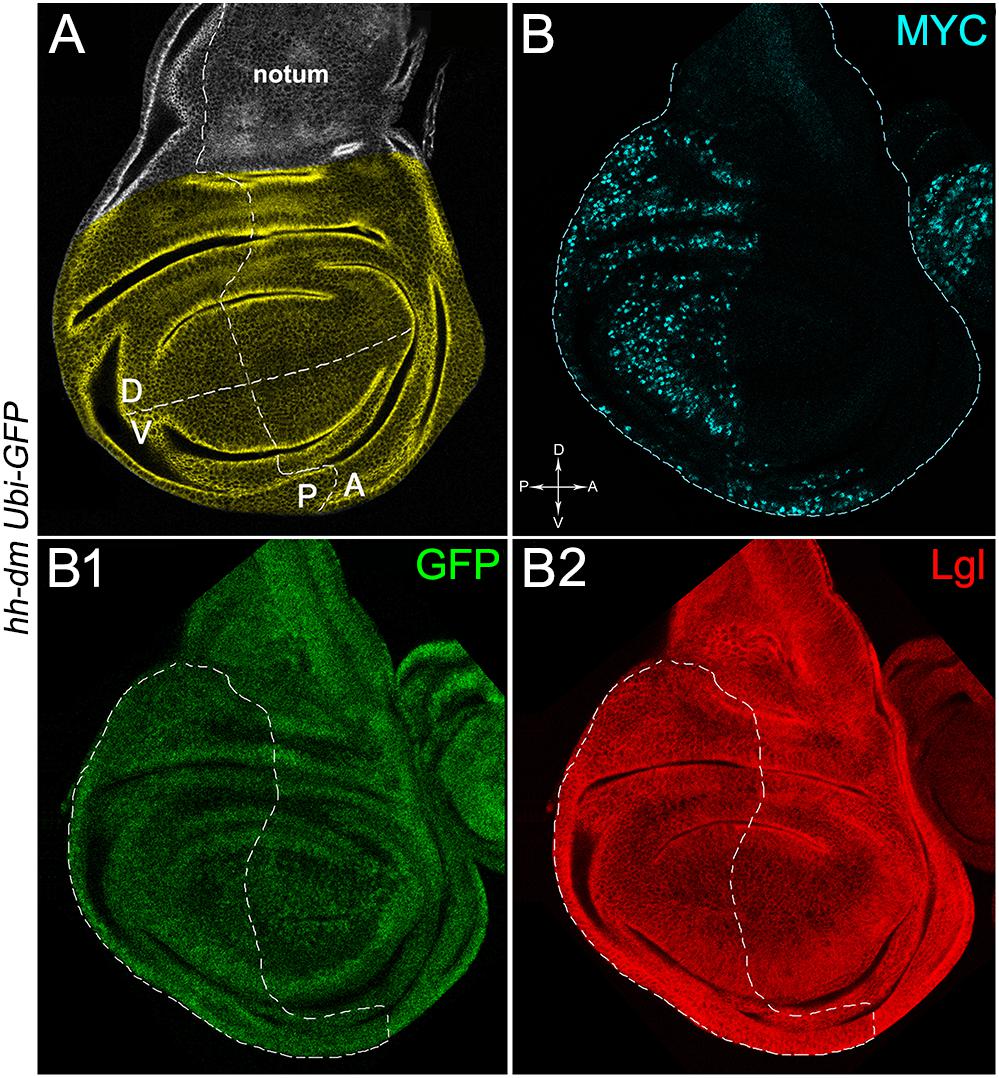
Figure 1. MYC overexpression in the posterior compartment of the wing disc does not cause morphological alterations. (A) Representation of an imaginal wing disc from a wild-type late Drosophila larva. The Posterior/Anterior (P/A) and the Dorsal/Ventral (D/V) boundaries are indicated by dotted lines. All the measures for this study have been taken in the yellowish area. (B–B2) Immunostaining for MYC (B, cyan) and Lgl (B2, red) on wing discs from late yw; Ubi-GFPnls, FRT40A/+; hh-Gal4/UAS-dm larvae. GFP is shown in B1. The basic genotype is indicated on the left of the figure panel. P compartments are outlined in B1,B2, and disc axes are indicated in B. Magnification is 400×.
Pre-cancerous fields are defined as tissue areas composed of histologically normal but genetically altered cells, shown to be more susceptible than wild-type counterparts to the onset of new mutations, promoting in time the development of multifocal tumours (Slaughter et al., 1953; Dotto, 2014). Since these areas are found to surround primary masses in several epithelial malignancies (Nonn et al., 2009; Zeki et al., 2011; Park et al., 2016), a pre-neoplastic field can be considered, borrowing Paget’s hypothesis, a soil providing “bad seeds” with the capacity to initiate malignant growth, including those that would normally fail. The wide series of aberrations underlying the process of field cancerisation can hardly be attributed to a single cellular event, but deregulation of a gene piloting a number of cell behaviours may greatly favour its formation. MYC represents an excellent candidate, because its misexpression does not account on gene mutation but is rather caused by alterations in many, if not all, signalling pathways (Nussinov et al., 2016). As an example, activated forms of RAS are frequently found in human pre-neoplastic tissues (Braakhuis et al., 2003), and it is known that activated RAS stabilises MYC protein in Drosophila (Prober and Edgar, 2002) and mammals (Sears et al., 2000). Stabilised MYC is in turn able to remodulate cell growth and proliferation, metabolism and stress response (Meyer and Penn, 2008). Moreover, a founder cell upregulating MYC could easily expand into a MYC-upregulating field through MMCC (Johnston, 2014). Therefore, MYC could play a causative role both in driving the expansion and in determining the intrinsic characteristics of a pre-cancerous field. To investigate this issue we bypassed field formation, since it is well established that Drosophila epithelial cells upregulating MYC eliminate the wild-type neighbours during development and colonise a large fraction of the tissue through MMCC (de la Cova et al., 2004; Moreno and Basler, 2004).
We then took advantage of the UAS-Gal4 binary system (Brand and Perrimon, 1993) to drive MYC overexpression (hereafter referred to as MYCOV ER) under the control of the hedgehog (hh) promoter in the posterior compartment of the wing disc, a Drosophila larval epithelial organ (Bryant, 1975). Figure 1A shows the Posterior/Anterior (P/A) and the Dorsal/Ventral (D/V) axes of the larval wing disc, while the yellowish region represents the area subjected to measurements and P vs. A comparisons, being the notum mostly composed of anterior cells (see P/A boundary in the notum region). As can be appreciated in Figure 1B, MYCOV ER is confined to the P compartment (representing the pre-cancerous field), where it does not seem to cause evident alterations in tissue morphology with respect to the A compartment (representing the wild-type field), as noted in Figures 1B1,B2, where a Ubi-GFPnls transgene and the Lgl protein mark cell nuclei and cell membranes, respectively. To demonstrate MYC’s specificity in providing cells with a complex pre-cancerisation signature, we compared the results of each experiment with those obtained following overexpression of a membrane-tethered form of PI3K (PI3KCAAX), another potent growth inducer (de la Cova et al., 2004). We first verified if overexpression of the PI3KCAAX transgene (PI3KCAAX-OV ER) caused consistent activation of the PI3K/AKT signalling pathway. As noted in Figures 2A,A1, the phosphorylated form of AKT was detected in the P compartment of the wing disc following PI3KCAAX-OV ER (GFP+ region in Figure 2A). Moreover, it did not impact MYC endogenous levels (Figure 2A2, the P/A border is outlined), being the MFI of MYC staining statistically comparable in P and A compartments (Figure 2B). Following MYCOV ER, the levels of phosphorylated AKT in the P compartment (GFP+ region, Figures 2C,C1, the P/A border is outlined) were also comparable to those observed in the A compartment (Figure 2D), confirming that, differently from what has been observed in a previous study (Levayer et al., 2015), in our genetic system and under our working conditions, the two growth inducers do not significantly cross-regulate each other, making it suitable for the successive analyses.
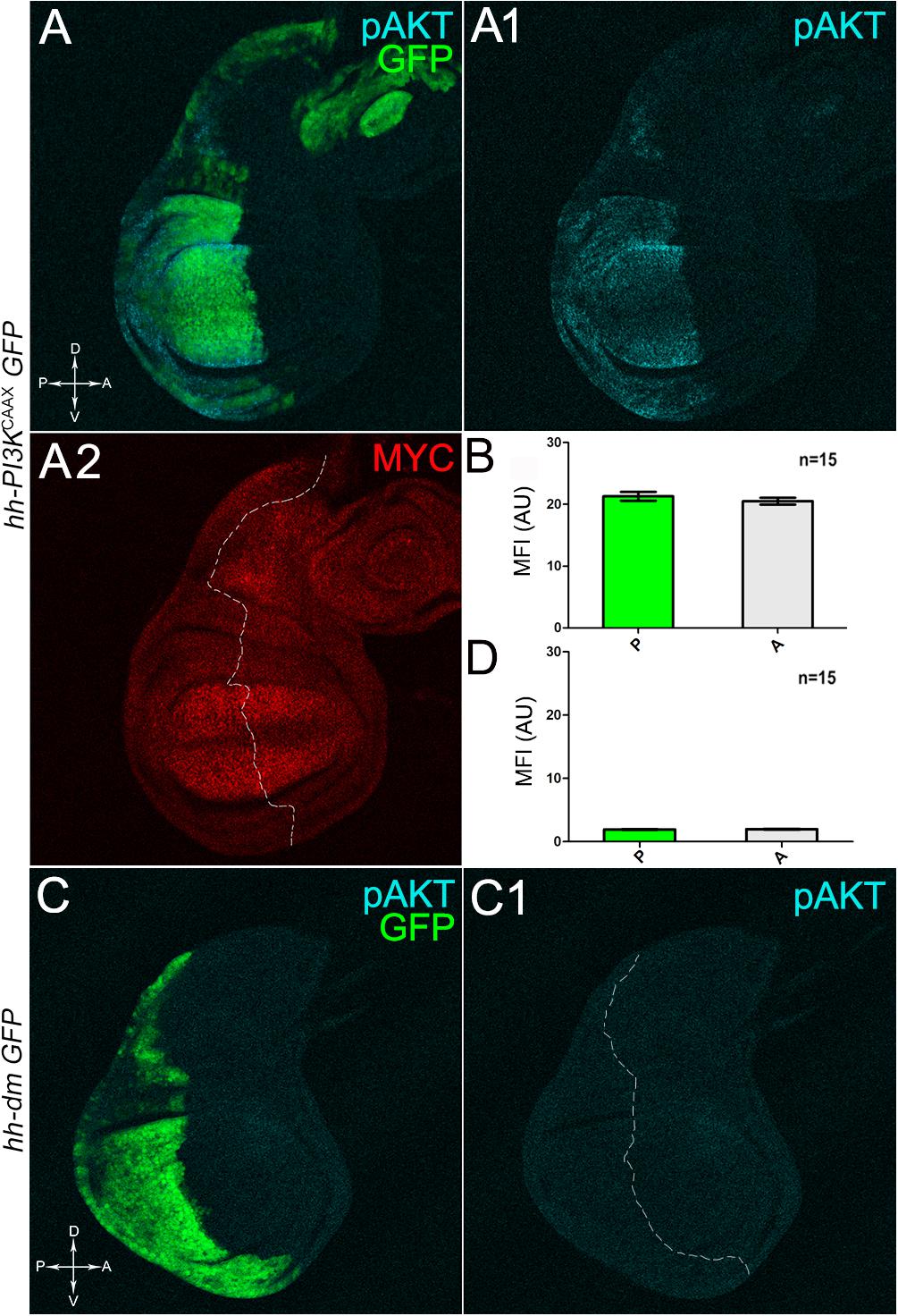
Figure 2. MYC and PI3KCAAX overexpression do not induce reciprocal activation. (A–A2) Immunostaining for pAKT (A,A1, cyan) and MYC (A2, red) on wing discs from late yw/yw, UAS-PI3KCAAX; hh-Gal4, UAS-GFP/+ larvae. (B) Graph comparing the Mean Fluorescence Intensity Arbitrary Units (MFI-AU) of MYC staining measured in the P (green bar) and A (grey bar) compartments of 15 wing discs from different larvae. (C,C1) Immunostaining for pAKT (cyan) on wing discs from late yw; hh-Gal4, UAS-GFP/UAS-dm larvae. (D) Graph comparing the Mean Fluorescence Intensity Arbitrary Units (MFI-AU) of pAKT staining measured in the P (green bar) and A (grey bar) compartments of 15 wing discs from different larvae. Basic genotypes are indicated on the left of the figure panel. P compartments are outlined in A2,C1, and disc axes are indicated in A,C. Magnification is 400×.
We started by investigating in the MYCOV ER tissue a number of markers characteristic of human pre-neoplastic fields. Since it is known that pre-malignant areas may display a higher proliferative index than normal tissues (Mohan and Jagannathan, 2014), we first checked the mitotic activity of MYCOV ER cells by immunostaining for the phosphorylated histone H3 (PH3), which is known to play a key role during mitosis both in Drosophila and mammals (Kamakaka and Biggins, 2005). A mitotic index analysis highlighted a 32% increase of PH3-positive nuclei in MYCOV ER P compartments with respect to their A counterparts (Figures 3A,A1,B), and a 20% increase in the PI3KCAAX-OV ER P vs. A compartments (Figures 3C,C1,D). This result was not unexpected, as PI3K activation plays important roles in cell growth and proliferation (Leevers et al., 1996). The mitotic index of the MYCOV ER tissue was, however, significantly higher than that observed in the PI3KCAAX-OV ER samples, as in the graph reported in Figure 3E. To assess genetic instability, another feature of pre-cancerous fields with obvious mutagenic effects (Bhattacharjee and Nandi, 2016), we used an antibody against the γ variant of the phosphorylated histone H2, which is recognised as the first modification occurring following DNA double strand breaks, resulting in the assembling of multi-protein complexes which attempt to repair DNA damage (Dronamraju and Mason, 2011). As can be seen in Figures 4A,A1, the γH2Av foci (red) in the MYCOV ER P compartment (GFP+, outlined in Figure 4A1) were about twice compared to the A compartment (Figure 4B), while they resulted comparable in the P and A compartments of the PI3KCAAX-OV ER samples (Figures 4C,C1,D). Our study continued by evaluating the presence and abundance of ROS in the presumptive pre-cancerous field. As noted in Figures 5A,A1, a strong increase in ROS generation (red) was found in the MYCOV ER P compartment of the wing disc (GFP+, outlined in Figure 5A1), quantified as about 20 arbitrary units (AU) MFI vs. the 6.5 AU found in the A compartment (compare green and grey bars in the graph Figure 5B). Contrariwise, no significant differences were found between the P (GFP+, outlined in Figure 5C1) and A compartments following PI3KCAAX-OV ER, as is appreciable in Figures 5C,C1,D. As MYCOV ER and PI3KCAAX-OV ER samples underwent parallel enzymatic reactions, we could also compare ROS levels in the respective wild-type A compartments, and found that MYCOV ER A compartment showed a twofold ROS increase with respect to the PI3KCAAX-OV ER A compartment (Figure 5E). This was an interesting finding, as ROS are diffusible ions and molecules and they may freely move away from the producing cells, thus expanding MYC’s pre-cancerisation effect to adjacent tissues by a non-autonomous mechanism. In this sense, a recent study demonstrated that, in Drosophila epithelial tumours, apoptotic caspases enhance tumour malignancy by generating ROS, which in turn recruit immune cells that signal back to the epithelium to activate cancer pathways (Perez et al., 2017). Although MYCOV ER tissues cannot be compared to overt cancers, similar cell-cell interactions may be at work that cooperate with MMCC to expand the pre-cancerous field. An analysis of apoptotic cell death carried out by immunostaining for the activated form of the effector Caspase 3 (Cas3) revealed that MYCOV ER epithelial cells were highly prone to apoptotic death (see Figures 6A,A1), with about 9 MFI AU in the P compartment vs. 1.5 in the A counterpart, as can be noted in Figure 6B. By contrast, no significant differences were noticed between P and A compartments overexpressing PI3KCAAX (Figures 6C,C1,D). Consistently with Caspase 3 activation, MYCOV ER cells downregulated the anti-apoptotic protein dIAP1 (Wang et al., 1999), as shown in Figure 7A1 (Figure 7A shows the GFP+ P compartment), with 7.4 MFI AU in the P vs. 13.5 in the A compartment (Figure 7B); dIAP1 indeed functions by inhibiting the initiator caspase DRONC (Meier et al., 2000) that, in turn, activates the effector caspases. Also in this case, PI3KCAAX-OV ER tissues did not show significant differences in dIAP1 staining between the P and A compartments (Figures 7C,C1,D). Finally, with regard to changes in the epigenetic signature of human pre-neoplastic tissues (Grady, 2005; Lee et al., 2011), we analysed the effect of MYCOV ER on the chromatin modifier Polycomb (Pc), known to shape cellular plasticity through large-scale epigenetic regulation (Klebes et al., 2005). We previously showed that Pc expression is nearly absent in Drosophila epithelial cancers (Grifoni et al., 2015). As it is known that Pc and other proteins of the Pc group (PcG) are necessary to MYC auto-repression in Drosophila (Goodliffe et al., 2005; Khan et al., 2009), Pc downregulation in overt cancers may help sustain high MYC cellular levels, so allowing it to impact many different phenotypic traits. As can be observed in Figure 7A2, Pc resulted downregulated also in our pre-cancerisation model, with 9.8 vs. 19.4 MFI AU in the P (GFP+ in Figure 7A, outlined in 7A2), and A compartments, respectively (Figure 7B). This is consistent with MYC and PcG proteins trans-regulation (Benetatos et al., 2014), and since low Pc levels result in a higher chromatin accessibility, this condition would favour additional mutational insults through inappropriate entrance of DNA cleaving enzymes (Zhang et al., 2008). Also in this case, PI3KCAAX-OV ER tissues did not display significant differences compared to the wild-type counterparts (Figures 7C2,D).
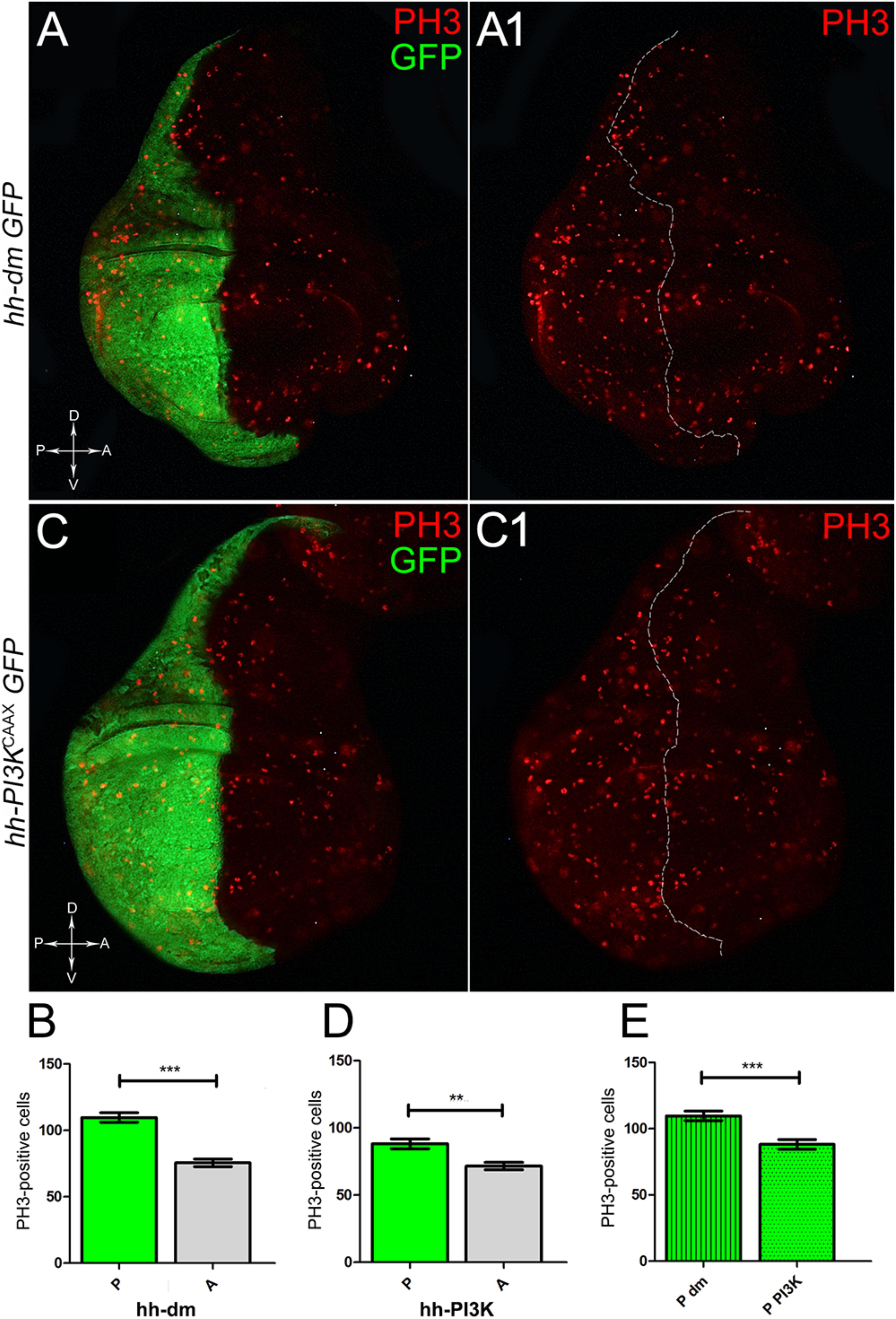
Figure 3. MYC and PI3KCAAX overexpression increases mitotic activity. (A,A1) Immunostaining for PH3 (red) on wing discs from late yw; hh-Gal4, UAS-GFP/UAS-dm larvae. (B) Graph comparing the PH3-positive nuclei counted in the P (green bar) and A (grey bar) compartments, ∗∗∗p ≤ 0.001. (C,C1) Immunostaining for PH3 (red) on wing discs from late yw/yw, UAS-PI3KCAAX; hh-Gal4, UAS-GFP/+ larvae. (D) Graph comparing the PH3-positive nuclei counted in the P (green bar) and A (grey bar) compartments, ∗∗p ≤ 0.01. (E) Graph comparing the PH3-positive nuclei counted in the P compartments of yw; hh-Gal4, UAS-GFP/UAS-dm (striped green bar) and yw/yw, UAS-PI3KCAAX; hh-Gal4, UAS-GFP/+ (dotted green bar) larvae, ∗∗∗p ≤ 0.001. Basic genotypes are indicated on the left of the figure panels and under the graphs. P compartments are outlined in A1,C1, and disc axes are indicated in A,C. Magnification is 400×.
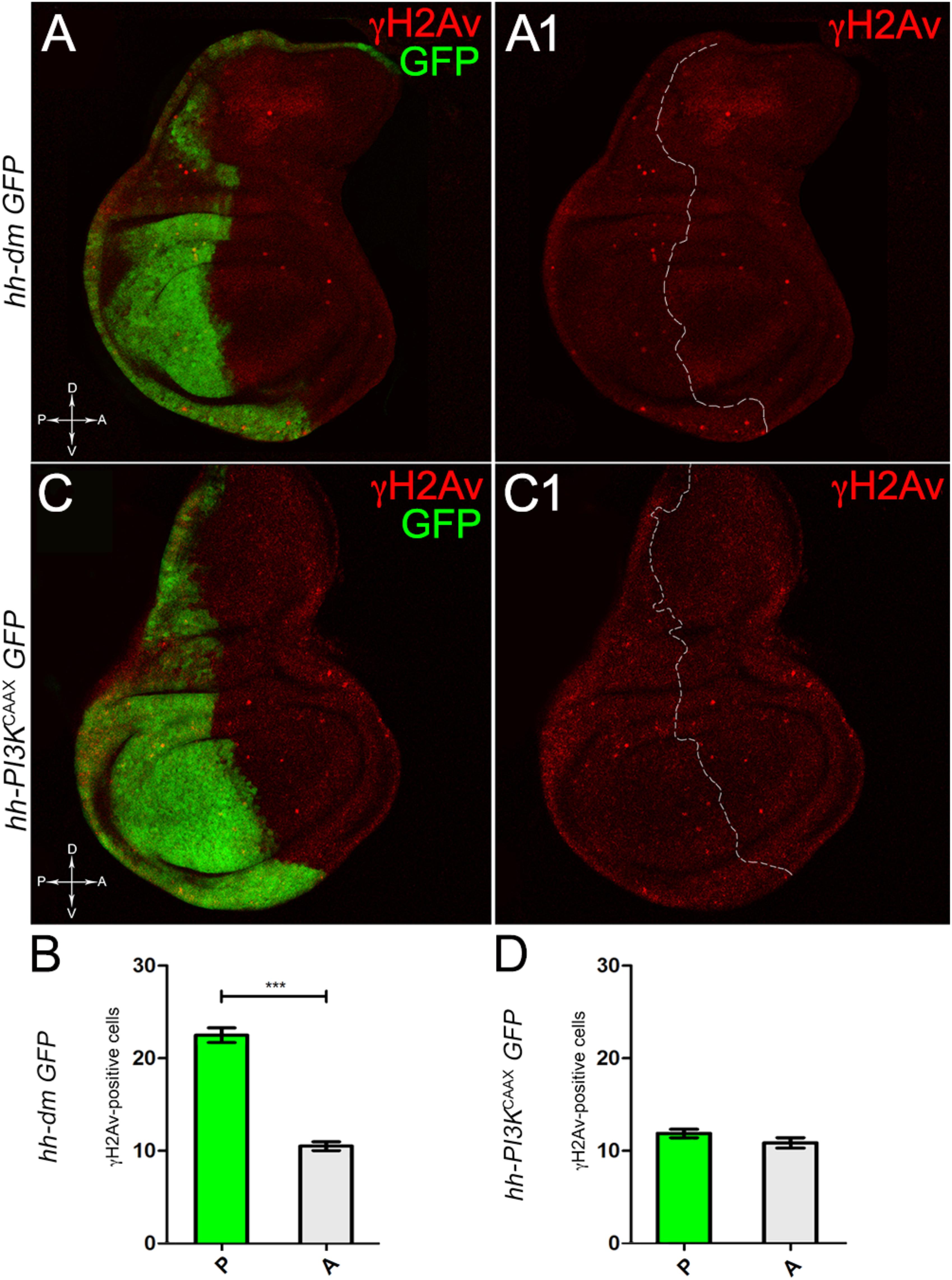
Figure 4. MYC overexpression increases genetic instability. (A,A1) Immunostaining for γH2Av (red) on wing discs from late yw; hh-Gal4, UAS-GFP/UAS-dm larvae. (B) Graph comparing the γH2Av-positive foci counted in the P (green bar) and A (grey bar) compartments, ∗∗∗p ≤ 0.001. (C,C1) Immunostaining for γH2Av (red) on wing discs from late yw/yw, UAS-PI3KCAAX; hh-Gal4, UAS-GFP/+ larvae. (D) Graph comparing the γH2Av-positive foci counted in the P (green bar) and A (grey bar) compartments. Basic genotypes are indicated on the left of both the figure panels and the graphs. P compartments are outlined in A1,C1, and disc axes are indicated in A,C. Magnification is 400×.
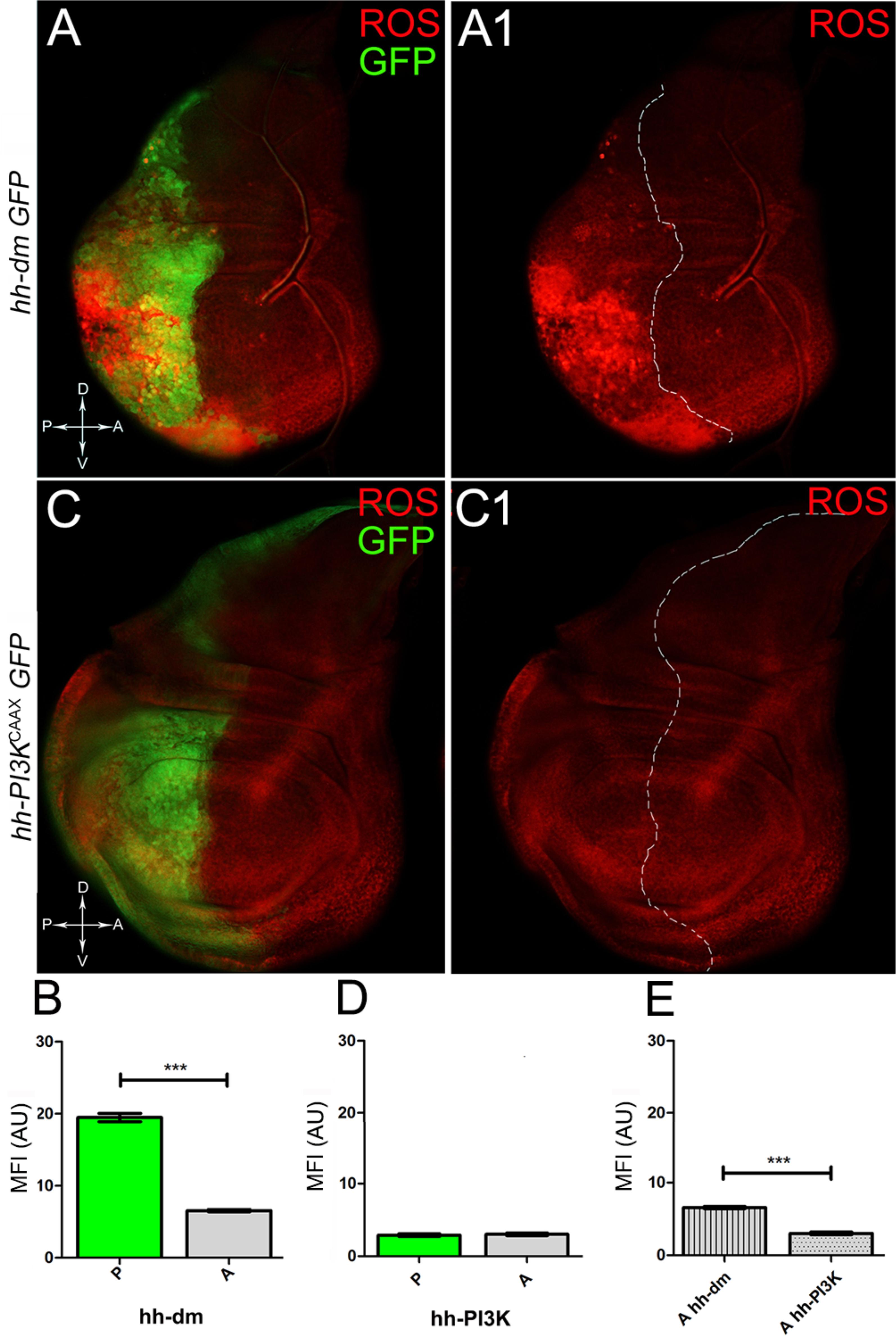
Figure 5. MYC overexpression causes intense ROS production. (A,A1) ROS production (red) in wing discs from late yw; hh-Gal4, UAS-GFP/UAS-dm larvae. (B) Graph comparing the Mean Fluorescence Intensity of ROS positivity measured in the P (green bar) and A (grey bar) compartments, ∗∗∗p ≤ 0.001. (C,C1) ROS production (red) in wing discs from late yw/yw, UAS-PI3KCAAX; hh-Gal4, UAS-GFP/+ larvae. (D) Graph comparing the Mean Fluorescence Intensity Arbitrary Units (MFI-AU) of ROS positivity measured in the P (green bar) and A (grey bar) compartments. (E) Graph comparing the Mean Fluorescence Intensity Arbitrary Units (MFI-AU) of ROS positivity measured in the A compartments of yw; hh-Gal4, UAS-GFP/UAS-dm (striped grey bar) and yw/yw, UAS-PI3KCAAX; hh-Gal4, UAS-GFP/+ (dotted grey bar) larvae, ∗∗∗p ≤ 0.001. Basic genotypes are indicated on the left of the figure panels and under the graphs. P compartments are outlined in A1,C1, and disc axes are indicated in A,C. Magnification is 400×.
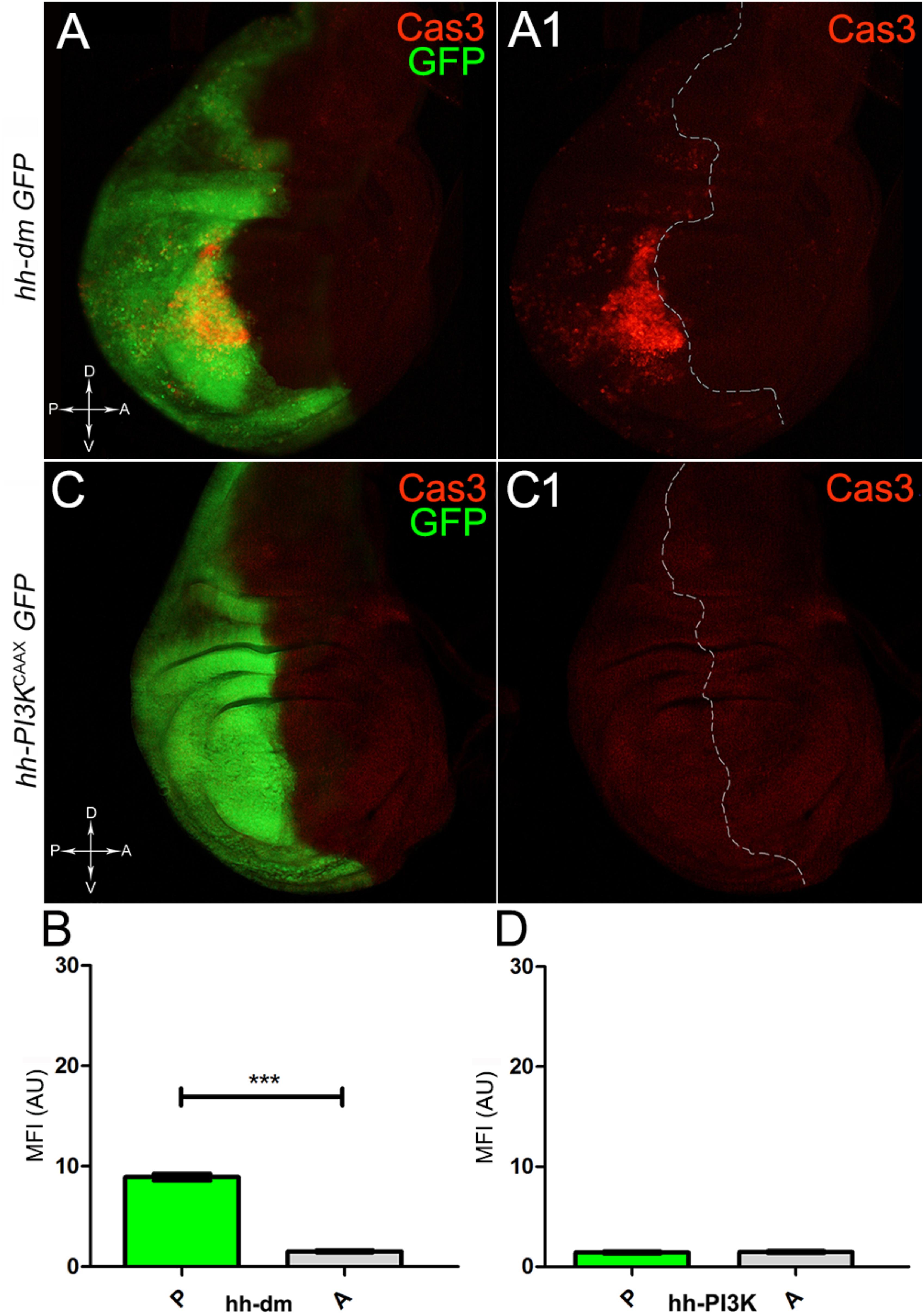
Figure 6. MYC overexpression triggers apoptotic death. (A,A1) Immunostaining for Cas3 (red) on wing discs from late yw; hh-Gal4, UAS-GFP/UAS-dm larvae. (B) Graph comparing the Mean Fluorescence Intensity Arbitrary Units (MFI-AU) of Cas3 staining measured in the P (green bar) and A (grey bar) compartments, ∗∗∗p ≤ 0.001. (C,C1) Immunostaining for Cas3 (red) on wing discs from late yw/yw, UAS-PI3KCAAX; hh-Gal4, UAS-GFP/+ larvae. (D) Graph comparing the Mean Fluorescence Intensity Arbitrary Units (MFI-AU) of Cas3 staining measured in the P (green bar) and A (grey bar) compartments. Basic genotypes are indicated on the left of the figure panels and under the graphs. P compartments are outlined in A1,C1, and disc axes are indicated in A,C. Magnification is 400×.
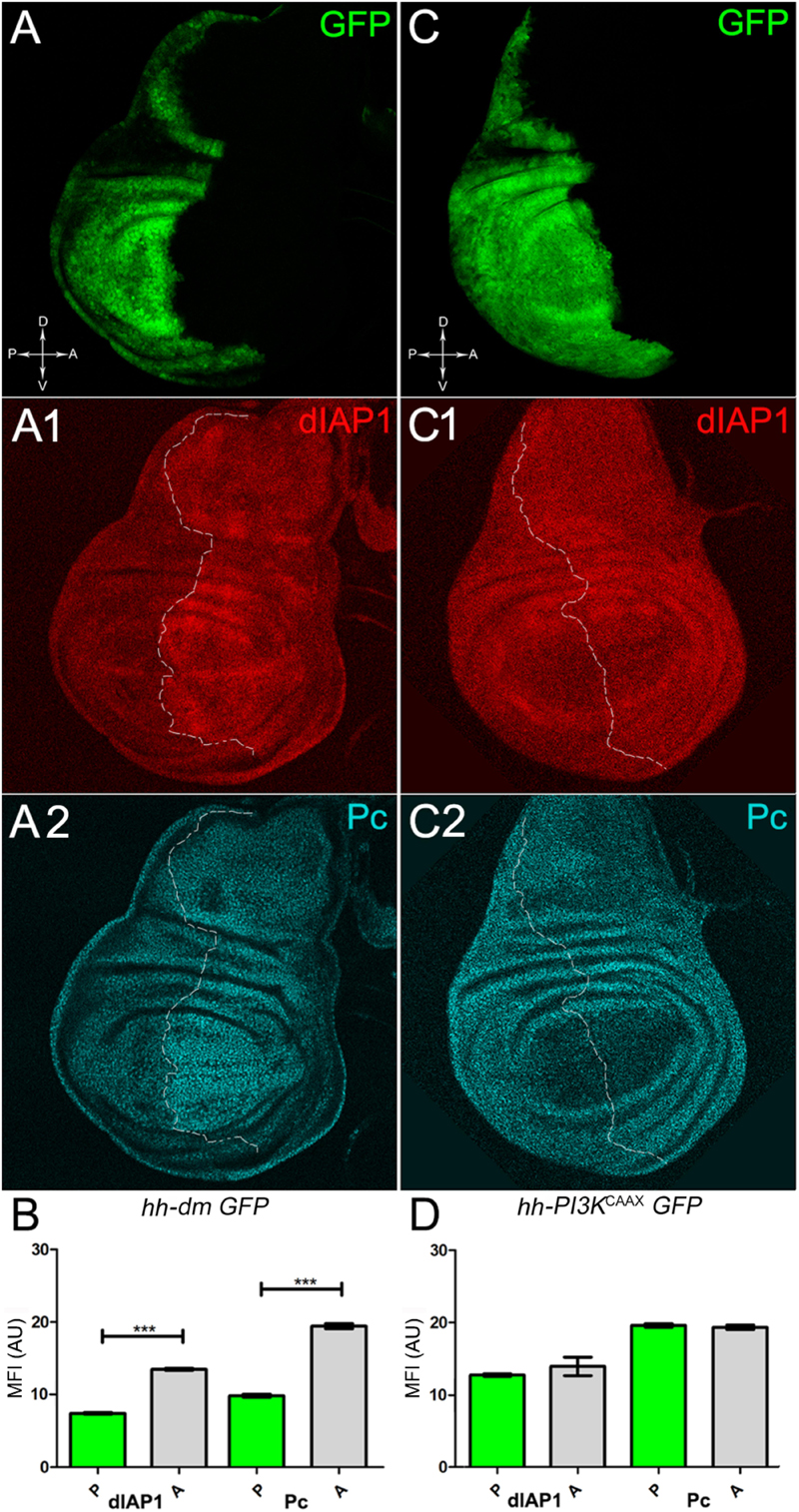
Figure 7. MYC overexpression downregulates survival and epigenetic markers. (A–A2) Immunostaining for dIAP1 (A1, red) and Pc (A2, cyan) on wing discs from late yw; hh-Gal4, UAS-GFP/UAS-dm larvae. (B) Graph comparing the Mean Fluorescence Intensity Arbitrary Units (MFI-AU) of dIAP and Pc staining measured in the P (green bars) and A (grey bars) compartments, ∗∗∗p ≤ 0.001. (C–C2) Immunostaining for dIAP1 (C1, red) and Pc (C2, cyan) on wing discs from late yw/yw, UAS-PI3KCAAX; hh-Gal4, UAS-GFP/+ larvae. (D) Graph comparing the Mean Fluorescence Intensity Arbitrary Units (MFI-AU) of dIAP and Pc staining measured in the P (green bars) and A (grey bars) compartments. Basic genotypes are indicated under the figure panels. P compartments are outlined in A1,A2,C1,C2, and disc axes are indicated in A,C. Magnification is 400×.
Altogether, these results support our hypothesis that high levels of MYC are sufficient as to induce a series of molecular changes, which are likely to turn the affected tissue into a pre-malignant field. Moreover, this ability seems to be specific to MYC, as an active form of the growth inducer PI3K failed to promote significant alterations of the markers analysed.
With the aim to translate the evidence described above into a functional demonstration of MYCOV ER’s capacity to establish a pre-malignant condition, we investigated the phenotypic consequences of the induction of second mutations in a MYCOV ER background. We used a genetic model which, through a combination of the UAS-Gal4 (Brand and Perrimon, 1993) and Flp-FRP (Xu and Rubin, 1993) binary systems, allowed us to express MYC in the P compartment and to induce second mutations of interest later in time, so reproducing the temporal sequence that is likely to occur during cancer initiation. The A compartment has been used as a control, to assess the clonal phenotype promoted by the same second mutations in a region carrying endogenous MYC expression.
As described in the Introduction, we previously showed that hyperplastic TSGs (hTSGs) exploit excess MYC to grow more rapidly, but are not able to initiate malignant transformation (Ziosi et al., 2010); we thus aimed at exploring MYCOV ER’s effect on the clonal behaviour of neoplastic TSGs (nTSGs). We first analysed the lethal giant larvae (lgl) mutation. Lgl protein regulates the apical-basal cell polarity in the epithelia (Grifoni et al., 2013); we previously demonstrated its functional conservation from Drosophila to humans (Grifoni et al., 2004), and we and others found the human orthologue HUGL-1 involved in cancers from different organs (Grifoni et al., 2004, 2007; Schimanski et al., 2005; Lu et al., 2009). In the Drosophila wing disc, lgl mutant cells are unable to grow in a wild-type background, especially in the regions where MYC levels are high, and are eliminated by MMCC (Froldi et al., 2010). In the same wild-type background, MYCOV ER in lgl mutant clones rescues them from death and transforms lgl-/- cells from losers into super-competitors (Froldi et al., 2010). But what happens to newly formed lgl, MYCOV ER cells when they are surrounded by MYCOV ER neighbours? As can be seen in Figure 8, while lgl-/- clones were smaller than wild-type twins in the A control compartment of the disc (Figures 8A,A1,C), in the 28% of the wing discs analysed the lgl-/- clones growing in the MYCOV ER P compartment appeared significantly larger than the wild-type twins (Figures 8A,A1,B). As an example, the arrow in Figure 8A1 points to a wild-type clone (double red) which appears much smaller than the lgl mutant twin (black). In addition, the arrowhead indicates an lgl mutant clone in the hinge region of the P compartment with no apparent wild-type twin clone. This suggests that the lgl mutant cells have a greater ability to exploit the excess MYC protein than the surrounding neighbours, hence the gain of a competitive advantage over the wild-type tissue. However, the average clone area occupied by the lgl-/- cells in this system was about 5000 px2, whereas it was found to be around 24000 px2 in a previous study where lgl-/-, MYCOV ER cells were induced in a wild-type background (Froldi et al., 2010), demonstrating the MYCOV ER neighbours exert a competitive pressure against the growth of the lgl mutant clones, which translates into a limited capability of lgl-/- cells to form large masses in a uniform MYCOV ER field.
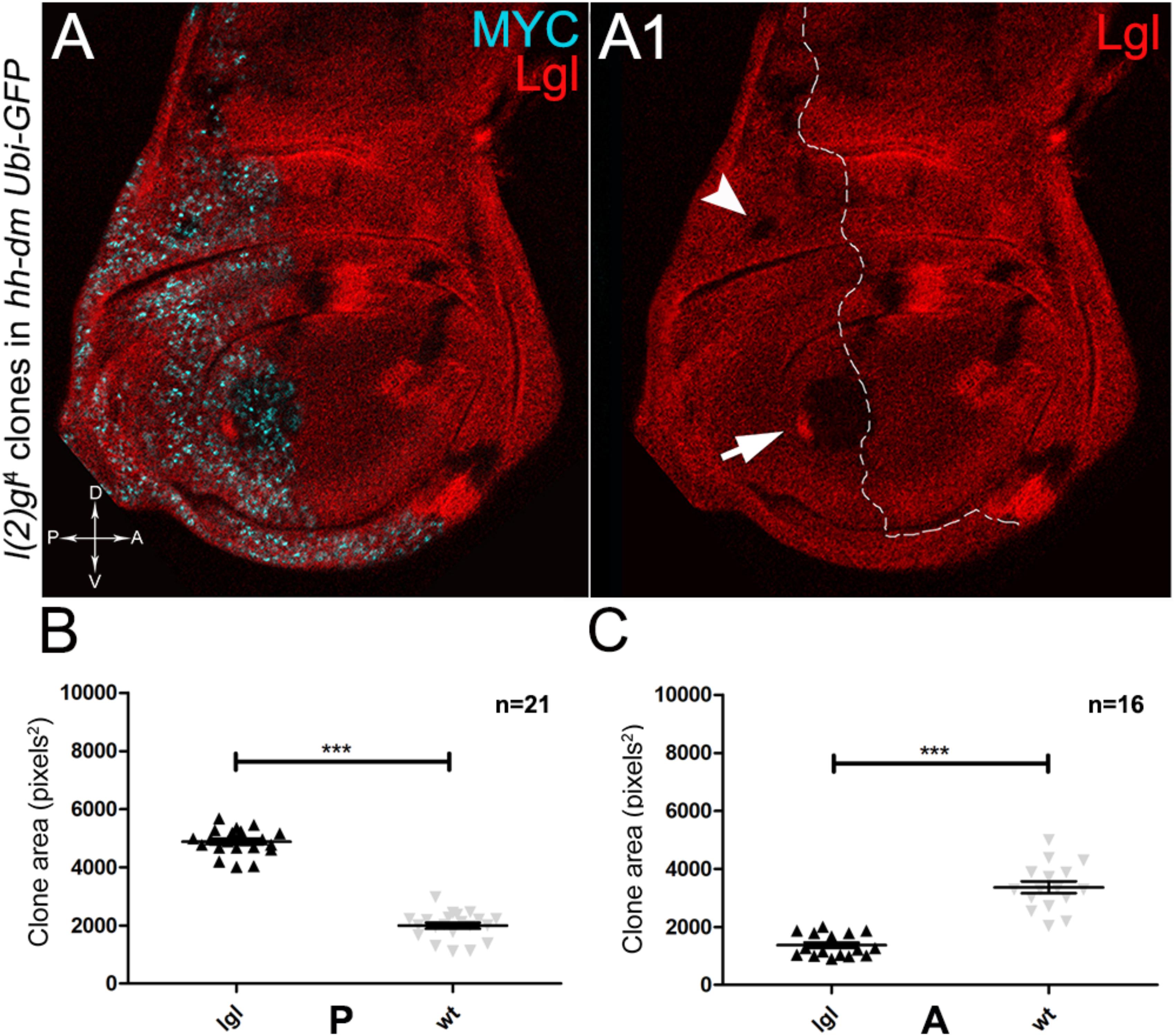
Figure 8. About one/third of lgl- clones overgrows at the expense of the surrounding wild-type tissue in a MYC-overexpressing background. (A,A1) Immunostaining for Lgl (red) and MYC (cyan) on wing discs from late w; l(2)gl4, FRT40A/Ubi-GFPnls, FRT40A; hh-Gal4/UAS-dm larvae. The arrow points to a posterior wild-type twin clone and the arrowhead indicates a scattered lgl-/- clone with no obvious wild-type twin in the posterior hinge region (A1). (B,C) Graphs comparing the average clone area of lgl-/- (black triangles, black in the images) and wild-type twins (grey triangles, double red in the images) in the P (B) and A (C) compartments, ∗∗∗p ≤ 0.001. The basic genotype is indicated on the left of the figure. The P compartment is outlined in A1, and disc axes are indicated in A. Magnification is 400×.
The remaining 72% of the wing discs analysed displayed a novel phenotype: the lgl mutant tissue grew as a multitude of spots scattered all across the MYCOV ER P compartment. Figure 9 shows two typical samples that we classified as “mild multifocal” (Figure 9A), which represented the 38% of the total samples (Figure 9C), and “severe multifocal” (Figure 9B), which represented the 34% of the total samples (Figure 9C). We classified multifocality as “mild” when the lgl mutant clones (black), though colonising a large fraction of the P compartment, did not alter its width (Figure 9A1), and “severe” when the lgl-/- cells filled the entire P compartment, which appeared dramatically enlarged (see how the P/A border moved from P to A comparing Figures 9A1,B1). This deep organ alteration suggests a locally invasive, malignant behaviour of these mutant cells that may be favoured by clone confluence during growth, as it is with other tumour models in Drosophila (Menendez et al., 2010; Ballesteros-Arias et al., 2014), with MYC protein levels that appeared to increase along with phenotype severity (compare Figures 9A,B). lgl mutant cells displayed preferential MYC accumulation, as can be appreciated in Figure 10A2, where arrowheads indicate some of the mutant cells (black, see Figure 10A1) accumulating MYC. Again, the organs displaying larger mutant spots (Figure 10B, squared area) showed an obvious increase in MYC protein levels (Figure 10B2). The most interesting aspect of this model is that it faithfully reproduced a distinctive feature of human pre-cancerous fields, i.e., multifocality (Dotto, 2014). The multifocal phenotype has never been associated with lgl mutations in Drosophila; therefore, it represents a novel trait acquired by a cell subject to a mutation in the lgl nTSG while being part of a MYCOV ER field.
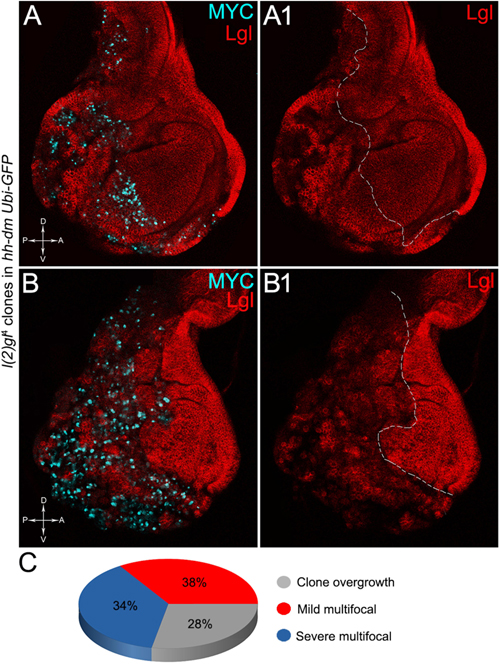
Figure 9. The majority of lgl-/- cells forms multifocal nests which colonise a large fraction of the MYC-overexpressing tissue. (A–B1) Immunostaining for Lgl (red) and MYC (cyan) on wing discs from late w; l(2)gl4, FRT40A/Ubi-GFPnls, FRT40A; hh-Gal4/UAS-dm larvae. (C) Pie chart illustrating the numerical proportions of overgrown, mild and severe multifocal lgl-/- clones found in the P compartment of w; l(2)gl4, FRT40A/Ubi-GFPnls, FRT40A; hh-Gal4/UAS-dm wing discs. The basic genotype is indicated on the left of the figure panel. P compartments are outlined in A1,B1, and disc axes are indicated in A,B. Magnification is 400×.
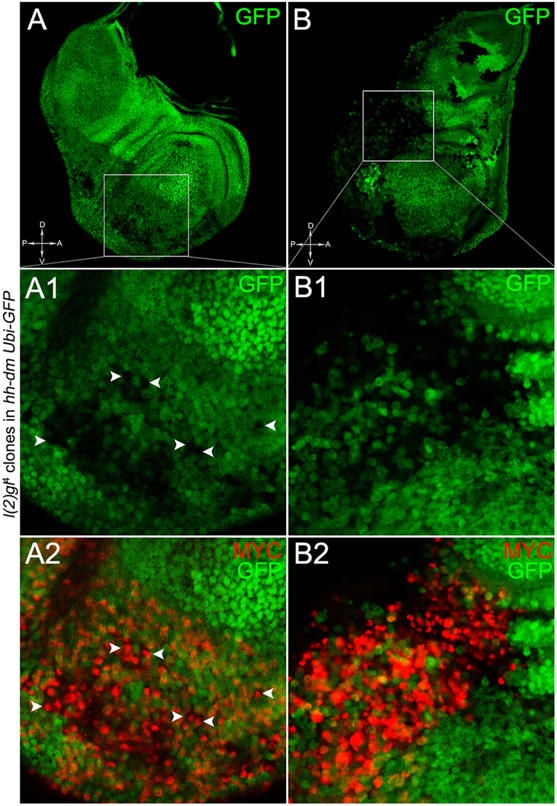
Figure 10. lgl-/- cells undergoing severe multifocal growth accumulate MYC protein. Immunostaining for MYC (red) on wing discs from late w; l(2)gl4, FRT40A/Ubi-GFPnls, FRT40A; hh-Gal4/UAS-dm larvae. In A1,A2, arrowheads indicate MYC-accumulating mutant cells. The basic genotype is indicated on the left of the figure panel. Disc axes are indicated in A,B. A1–B2 are magnified views of the squares drawn in A,B. Magnification is 400× in A,B, 1000× in A1,A2, and 1200× in B1,B2.
To verify that a MYCOV ER field represented a bona fide pre-cancerous area, and that multifocality did not result from a specific interaction between lgl and MYC, we induced a LOF mutation of a different nTSG in the MYCOV ER field. Rab5 is an evolutionarily conserved core component of the vesicle trafficking machinery (Lu and Bilder, 2005), implicated in various aspects of human tumourigenesis (Torres and Stupack, 2011; Mendoza et al., 2014). Like lgl, entire fly organs mutated for Rab5 show neoplastic growth (Lu and Bilder, 2005; Vaccari and Bilder, 2009), and Rab5 mutant cells induced in a wild-type wing disc suffer from cell competition and are eliminated from the organ (Ballesteros-Arias et al., 2014). Using the same clonal system as above, we induced Rab5 LOF clones in animals whose P compartments overexpressed MYC. As can be seen in Figure 11, the multifocal phenotype was evident also for the Rab5-/- cells (Figures 11A,B and respective magnifications A1 and B1). Also in this case, mutant cells showed MYC accumulation (Figures 11A2,B2, arrowheads in A2 indicate some mutant nests accumulating MYC). The 100% of the organs analysed showed a multifocal phenotype, subdivided in 71% mild and 29% severe. Moreover, Rab5 mutant cells showed loss of apical-basal cell polarity, a central feature of epithelial cancers (Wodarz and Nathke, 2007): in Figure 12, the magnifications in A1 and B1 show a region of the disc outer border where one can appreciate that the normal epithelium (arrowheads in A1 and B1) displays a wild-type localisation of the apical marker atypical PKC (aPKC, cyan, asterisks in B1). On the contrary, the mutant cells in the region indicated by the arrows in A1 and B1 (black in A1) show a redistribution of the polarity marker from the apical side to the entire cell cortex, together with aberrant, three-dimensional growth. In Figures 12C,C1, the impairment of aPKC expression (cyan) is evident across the entire MYCOV ER P compartment. This characteristic is consistent with Rab5 function: the endocytic trafficking is indeed essential in the maintenance of cell polarity, and mutations in genes involved in endocytosis provoke the expansion of cell’s apical domain (Shivas et al., 2010).
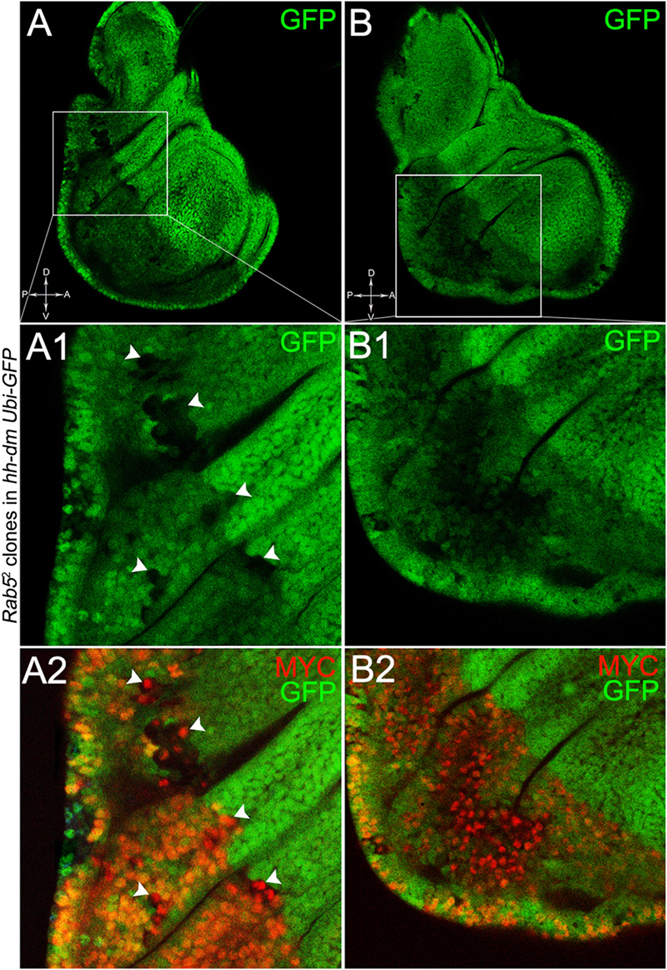
Figure 11. Rab5-/- cells show a fully penetrant, multifocal phenotype in a MYC-overexpressing background. Immunostaining for MYC (red) on wing discs from late w; Rab52, FRT40A/Ubi-GFPnls, FRT40A; hh-Gal4/UAS-dm larvae. In A1,A2, arrowheads indicate MYC-accumulating mutant cells. The basic genotype is indicated on the left of the figure panel. Disc axes are indicated in A,B. A1–B2 are magnified views of the squares drawn in A,B. Magnification is 400× in A,B, 1000× in A1,A2, and 800× in B1,B2.
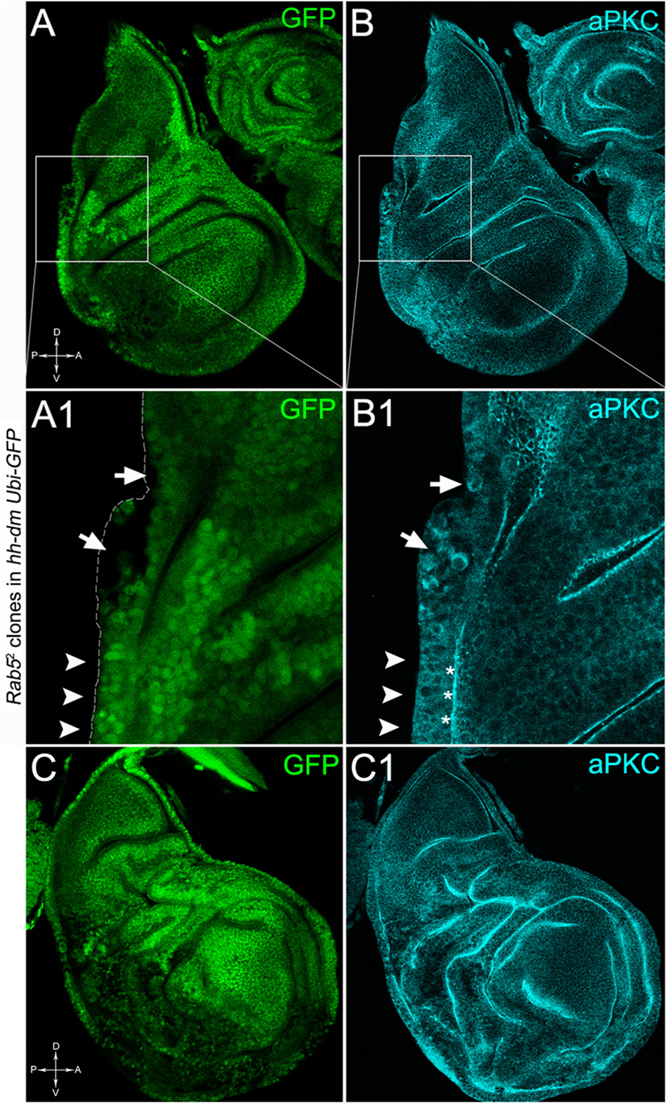
Figure 12. Rab5-/- cells show loss of cell polarity in a MYC-overexpressing background. Immunostaining for aPKC (cyan) on wing discs from late w; Rab52, FRT40A/Ubi-GFPnls, FRT40A; hh-Gal4/UAS-dm larvae. In A1,B1, arrows indicate mutant cells (black) displaying membrane redistribution of the apical marker aPKC, while the arrowheads point to a normal region of the disc border where the pseudostratified epithelium shows apical aPKC staining (asterisks). The dashed line in A1 marks the disc outer border. The basic genotype is indicated on the left of the figure panel. Disc axes are indicated in A,C. A1,B1 are magnified views of the squares drawn in A,B. Magnification is 400× in A,B,C,C1 and 1000× in A1,B1.
Altogether, this evidence indicated that MYC overexpression in an epithelial tissue is sufficient to promote multifocal malignant lesions following single-cell mutations of different nTSGs.
To assess if multifocality may be considered a trait arising from specific properties conferred by the MYC field to the mutant cells, we repeated the same experiments as above in a PI3KCAAX-OV ER territory. Using the same system as above, we first analysed lgl mutant behaviour. In Figures 13A,A1, we can observe lgl mutant clones (GFP-, indicated by the arrows) in the PI3KCAAX P compartment (marked in red by En staining in A). They are located outside the central region of the disc where, instead, we observed the presence of wild-type clones (GFP2+), indicating that mutant twins were eliminated by MMCC. Therefore, despite the over-expression of PI3KCAAX, lgl-/- clones continue to die in this area of the wing discs where MYC is normally highly expressed (see Figure 2A2). A statistical analysis of the clone area in the P and A compartments showed that lgl-/- mutant clones were significantly larger in P, with an average size of about 5000 px2, compared to A, where they displayed an average size of about 2000 px2 (Figure 13B). The most important observation was, however, the total absence of multifocal growth. We then analysed the behaviour of the Rab52 mutation in a PI3KCAAX background. In Figures 13C,C1, a wing disc is shown where small mutant clones of comparable size are present in both compartments (black, arrows in Figure 13C1). In Figure 13D, the graph indicates that the mutant clones do not show significant differences in size between the P and A compartments. Finally, as it was for lgl, no multifocal growth was observed in all the Rab5 samples analysed.
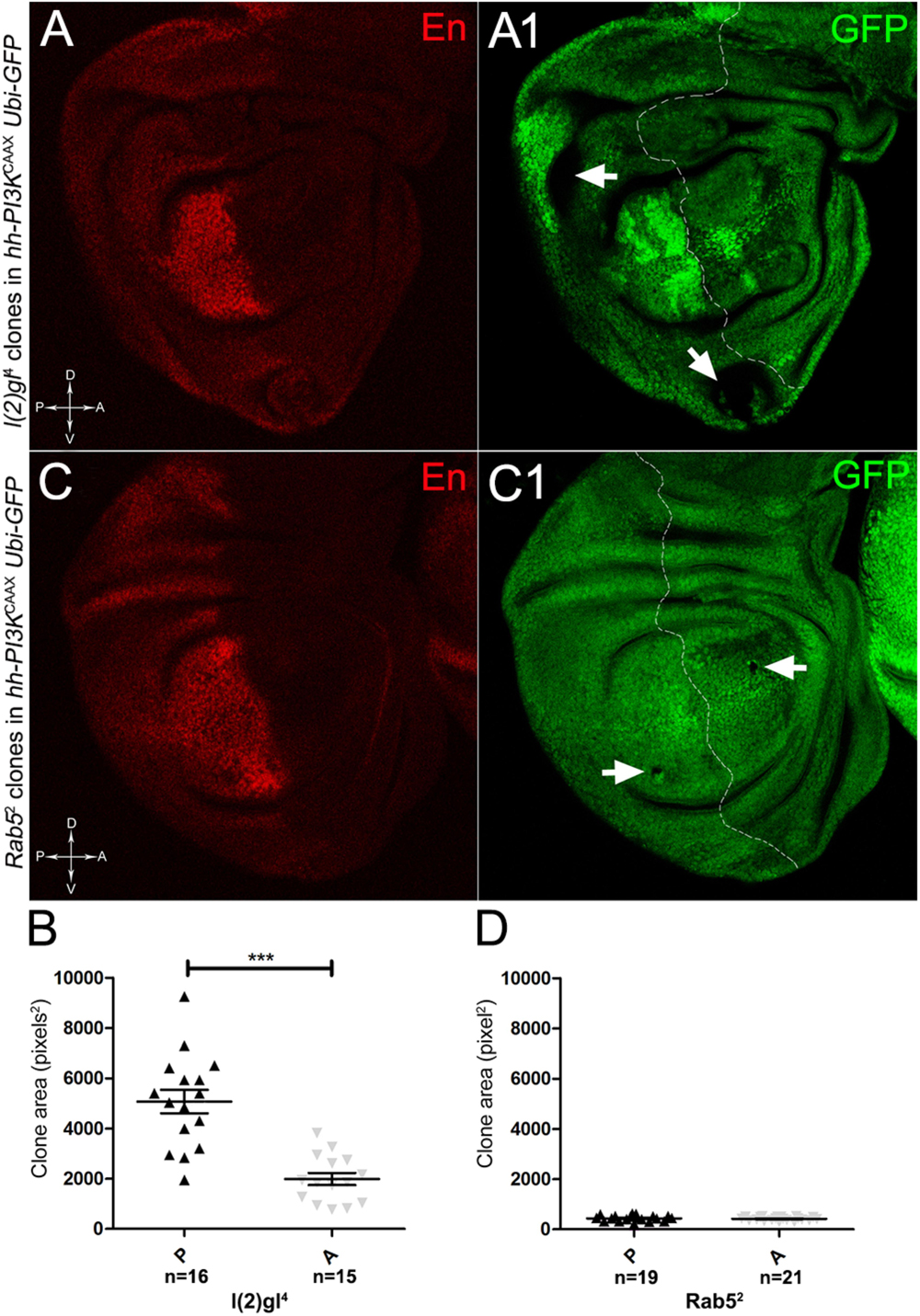
Figure 13. lgl-/- and Rab5-/- cells do not display multifocal growth in a PI3KCAAX-overexpressing background. (A,A1) Immunostaining for En (red) on wing discs from late w/yw, UAS-PI3KCAAX; l(2)gl4, FRT40A/Ubi-GFPnls; hh-Gal4/+ larvae. The arrows in A1 indicate the mutant clones (black). (B) Graph comparing the average clone area of lgl-/- clones in the P (black triangles) and A (grey triangles) compartments, ∗∗∗p ≤ 0.001. (C,C1) Immunostaining for En (red) on wing discs from late w/yw, UAS-PI3KCAAX; Rab52, FRT40A/Ubi-GFPnls; hh-Gal4/+ larvae. The arrows in C1 indicate the mutant clones (black). (D) Graph comparing the average clone area of Rab5-/- clones in the P (black triangles) and A (grey triangles) compartments. Basic genotypes are indicated on the left of the figure panel. The P compartment is outlined in A1,C1, and disc axes are indicated in A,C. Magnification is 400×.
These latter findings indicate that MYC confers on cells mutant for different nTSGs the ability to grow in multiple foci dispersed all across the modified territory. This seems to be a specific characteristic of MYC, as the growth inducer PI3K did not promote this peculiar phenotype. MYC upregulation emerges from our study as an excellent candidate to foster field cancerisation, by inducing a complex pre-cancerisation molecular signature able to provide cells hit by non-competitive mutations with the ability to initiate carcinogenesis.
Field cancerisation is studied in the effort to understand if essential events recur in tumour initiation that may help develop early therapeutic interventions. It is now recognised that many types of cancers start from cells owing some, but not all, phenotypic traits necessary for malignancy, and those traits may result from various mutagenic insults, on the basis of which the most performant cells are selected for clonal expansion (Curtius et al., 2018). This process may be driven by cell competition, which is intensively studied both in Drosophila (Merino et al., 2016) and mammals (Di Gregorio et al., 2016). In this context, we focused our attention on MMCC, a process based on steep differences in MYC levels in confronting cells, which ultimately favour the expansion of high MYC-expressing cells at the expense of the less fit neighbours (Grifoni and Bellosta, 2015). Given the broad implication of MYC protein in human cancers (Gabay et al., 2014), its myriad functions inside the cell (Dang et al., 2006) and its regulation at both the transcriptional and post-transcriptional levels by a number of signalling pathways (Nussinov et al., 2016), it seems an excellent candidate to pioneer field cancerisation (Moreno, 2008).
To address this question, we first investigated the cellular responses to MYC overexpression (MYCOV ER) in the imaginal wing disc, a Drosophila epithelial tissue widely used to model development, cell competition and cancer (Herranz et al., 2016). We found that MYCOV ER was per se sufficient to activate a series of cellular behaviours consistent with the formation of a pre-neoplastic field, such as ROS production, genetic instability, changes in apoptotic and proliferation activity and alteration of epigenetic markers. Moreover, we showed that these cellular responses were not elicited by a MYC’s generic pro-growth function, as an active form of the powerful growth inducer PI3K was not able to induce similar phenotypes, except a mild pro-proliferative effect. High MYC levels seem rather to prime field cancerisation by triggering a cascade of molecular changes that cooperate in taking cells a step closer to malignancy.
This bona fide pre-cancerous tissue was then tested for the ability to initiate tumourigenesis following mutations in neoplastic TSGs (nTSGs). We previously studied the effects of MYCOV ER on three hyperplastic TSGs (hTSGs) owing to the Hippo signalling pathway: ds, ft and ex, and found that mutant clones grew more rapidly while killing the MYCOV ER wild-type neighbours with higher efficiency, but they did not show any signs of malignancy (Ziosi et al., 2010). We and others demonstrated that most hTSGs upregulate MYC (Neto-Silva et al., 2010; Ziosi et al., 2010), hence their competitive capability, while some nTSGs downregulate MYC, hence their elimination from the tissue (Froldi et al., 2010). It is also recognised that the behaviour of both hTSGs and nTSGs depends on tissue’s MYC levels (Froldi et al., 2010; Neto-Silva et al., 2010; Ziosi et al., 2010): in a uniform background, as with our model, mutant behaviour should rather be dictated by the intrinsic features of the given mutation.
In the Drosophila wing disc, wild-type cells hit by nTSGs mutations are usually irrelevant: they are indeed eliminated rapidly or contribute to the tissue without overgrowing (Froldi et al., 2010; Ballesteros-Arias et al., 2014). The same mutations induced in a MYCOV ER field were rather capable to initiate multifocal, three-dimensional growth accompanied by loss of apical-basal cell polarity and aberrant tissue architecture. This was convincing evidence that MYC upregulation was sufficient as to establish a specific, complex pre-cancerisation signature, which predisposes the tissue to undergo malignant multifocal growth following certain second mutations.
Our findings lay the basis for future studies focused on early tumourigenesis. These studies are as essential as difficult: while understanding the very first phases of cancer is mandatory to conceive novel preventive and therapeutic interventions, investigations carried out in complex systems may lead to discouraging results. In this sense, the use of a genetically amenable animal model may greatly help dissect and dismantle the intricate networks implicated in cancer initiation.
MS and DG conceived and designed the study. MS, CG, and SP performed the experiments. DG and DdB analysed the experimental data. SDG performed the statistical analysis. AP and DG wrote the manuscript. All authors contributed to manuscript revision and read and approved the submitted version.
This study was supported by AIRC IG 17252 to AP and Research Fellowships from the University of Bologna to MS, SDG, SP, and DG.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We acknowledge the Bloomington Drosophila Stock Center (NIH P40OD018537) for the fly stocks used in this study, and the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at The University of Iowa, for monoclonal antibodies. We thank Paola Bellosta, Bruce Hay, and Dennis Strand for reagents and Silvia Strocchi for manuscript revision.
Abdalla, M., Tran-Thanh, D., Moreno, J., Iakovlev, V., Nair, R., Kanwar, N., et al. (2017). Mapping genomic and transcriptomic alterations spatially in epithelial cells adjacent to human breast carcinoma. Nat. Commun. 8:1245. doi: 10.1038/s41467-017-01357-y
Agrawal, N., Kango, M., Mishra, A., and Sinha, P. (1995). Neoplastic transformation and aberrant cell-cell interactions in genetic mosaics of lethal(2)giant larvae (lgl), a tumor suppressor gene of Drosophila. Dev. Biol. 172, 218–229. doi: 10.1006/dbio.1995.0017
Akai, N., Igaki, T., and Ohsawa, S. (2018). Wingless signaling regulates winner/loser status in Minute cell competition. Genes Cells 23, 234–240. doi: 10.1111/gtc.12568
Aldaz, S., and Escudero, L. M. (2010). Imaginal discs. Curr. Biol. 20, R429–R431. doi: 10.1016/j.cub.2010.03.010
Ballesteros-Arias, L., Saavedra, V., and Morata, G. (2014). Cell competition may function either as tumour-suppressing or as tumour-stimulating factor in Drosophila. Oncogene 33, 4377–4384. doi: 10.1038/onc.2013.407
Bascones-Martinez, A., Rodriguez-Gutierrez, C., Rodriguez-Gomez, E., Gil-Montoya, J. A., Gomez-Font, R., and Gonzalez-Moles, M. A. (2013). Evaluation of p53, caspase-3, Bcl-2, and Ki-67 markers in oral squamous cell carcinoma and premalignant epithelium in a sample from Alava Province (Spain). Med. Oral Patol. Oral Cir. Bucal 18, e846–e850. doi: 10.4317/medoral.18901
Benetatos, L., Vartholomatos, G., and Hatzimichael, E. (2014). Polycomb group proteins and MYC: the cancer connection. Cell. Mol. Life Sci. 71, 257–269. doi: 10.1007/s00018-013-1426-x
Bhattacharjee, S., and Nandi, S. (2016). Choices have consequences: the nexus between DNA repair pathways and genomic instability in cancer. Clin. Transl. Med. 5:45. doi: 10.1186/s40169-016-0128-z
Bilder, D. (2004). Epithelial polarity and proliferation control: links from the Drosophila neoplastic tumor suppressors. Genes Dev. 18, 1909–1925. doi: 10.1101/gad.1211604
Bilder, D., Li, M., and Perrimon, N. (2000). Cooperative regulation of cell polarity and growth by Drosophila tumor suppressors. Science 289, 113–116. doi: 10.1126/science.289.5476.113
Birchall, M. A., Schock, E., Harmon, B. V., and Gobe, G. (1997). Apoptosis, mitosis, PCNA and bcl-2 in normal, leukoplakic and malignant epithelia of the human oral cavity: prospective, in vivo study. Oral Oncol. 33, 419–425. doi: 10.1016/S0964-1955(97)00033-X
Bongers, V., Snow, G. B., de Vries, N., Cattan, A. R., Hall, A. G., van der Waal, I., et al. (1995). Second primary head and neck squamous cell carcinoma predicted by the glutathione S-transferase expression in healthy tissue in the direct vicinity of the first tumor. Lab. Invest. 73, 503–510.
Braakhuis, B. J., Tabor, M. P., Kummer, J. A., Leemans, C. R., and Brakenhoff, R. H. (2003). A genetic explanation of Slaughter’s concept of field cancerization: evidence and clinical implications. Cancer Res. 63, 1727–1730.
Braakhuis, B. J., Tabor, M. P., Leemans, C. R., van der Waal, I., Snow, G. B., and Brakenhoff, R. H. (2002). Second primary tumors and field cancerization in oral and oropharyngeal cancer: molecular techniques provide new insights and definitions. Head Neck 24, 198–206. doi: 10.1002/hed.10042
Brand, A. H., and Perrimon, N. (1993). Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118, 401–415.
Bryant, P. J. (1975). Pattern formation in the imaginal wing disc of Drosophila melanogaster: fate map, regeneration and duplication. J. Exp. Zool. 193, 49–77. doi: 10.1002/jez.1401930106
Buratovich, M. A., and Bryant, P. J. (1997). Enhancement of overgrowth by gene interactions in lethal(2)giant discs imaginal discs from Drosophila melanogaster. Genetics 147, 657–670.
Castven, D., Fischer, M., Becker, D., Heinrich, S., Andersen, J. B., Strand, D., et al. (2017). Adverse genomic alterations and stemness features are induced by field cancerization in the microenvironment of hepatocellular carcinomas. Oncotarget 8, 48688–48700. doi: 10.18632/oncotarget.16231
Chan, J. S., Tan, M. J., Sng, M. K., Teo, Z., Phua, T., Choo, C. C., et al. (2017). Cancer-associated fibroblasts enact field cancerization by promoting extratumoral oxidative stress. Cell Death Dis. 8:e2562. doi: 10.1038/cddis.2016.492
Chandler, C. H., Chari, S., and Dworkin, I. (2013). Does your gene need a background check? How genetic background impacts the analysis of mutations, genes, and evolution. Trends Genet. 29, 358–366. doi: 10.1016/j.tig.2013.01.009
Choi, W., Kim, J., Park, J., Lee, D. H., Hwang, D., Kim, J. H., et al. (2018). YAP/TAZ initiates gastric tumorigenesis via upregulation of MYC. Cancer Res. 78, 3306–3320. doi: 10.1158/0008-5472.CAN-17-3487
Claveria, C., Giovinazzo, G., Sierra, R., and Torres, M. (2013). Myc-driven endogenous cell competition in the early mammalian embryo. Nature 500, 39–44. doi: 10.1038/nature12389
Curtius, K., Wright, N. A., and Graham, T. A. (2017). Evolution of premalignant disease. Cold Spring Harb. Perspect. Med. 7:a026542. doi: 10.1101/cshperspect.a026542
Curtius, K., Wright, N. A., and Graham, T. A. (2018). An evolutionary perspective on field cancerization. Nat. Rev. Cancer. 18, 19–32. doi: 10.1038/nrc.2017.102
Dakubo, G. D., Jakupciak, J. P., Birch-Machin, M. A., and Parr, R. L. (2007). Clinical implications and utility of field cancerization. Cancer Cell Int. 7:2. doi: 10.1186/1475-2867-7-2
Dang, C. V., O’Donnell, K. A., Zeller, K. I., Nguyen, T., Osthus, R. C., and Li, F. (2006). The c-Myc target gene network. Semin. Cancer Biol. 16, 253–264. doi: 10.1016/j.semcancer.2006.07.014
de Bruin, E. C., McGranahan, N., Mitter, R., Salm, M., Wedge, D. C., Yates, L., et al. (2014). Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 346, 251–256. doi: 10.1126/science.1253462
de la Cova, C., Abril, M., Bellosta, P., Gallant, P., and Johnston, L. A. (2004). Drosophila myc regulates organ size by inducing cell competition. Cell 117, 107–116. doi: 10.1016/S0092-8674(04)00214-4
del Valle, Rodriguez, A., Didiano, D., and Desplan, C. (2012). Power tools for gene expression and clonal analysis in Drosophila. Nat. Methods 9, 47–55. doi: 10.1038/nmeth.1800
Di Giacomo, S., Sollazzo, M., de Biase, D., Ragazzi, M., Bellosta, P., Pession, A., et al. (2017). Human cancer cells signal their competitive fitness through MYC activity. Sci. Rep. 7:12568. doi: 10.1038/s41598-017-13002-1
Di Gregorio, A., Bowling, S., and Rodriguez, T. A. (2016). Cell competition and its role in the regulation of cell fitness from development to cancer. Dev. Cell 38, 621–634. doi: 10.1016/j.devcel.2016.08.012
Diaz-Diaz, C., Fernandez, de Manuel, L., Jimenez-Carretero, D., Montoya, M. C., Claveria, C., et al. (2017). Pluripotency surveillance by Myc-driven competitive elimination of differentiating cells. Dev. Cell 42, 585–599.e4. doi: 10.1016/j.devcel.2017.08.011
Dotto, G. P. (2014). Multifocal epithelial tumors and field cancerization: stroma as a primary determinant. J. Clin. Investig. 124, 1446–1453. doi: 10.1172/JCI72589
Driessens, G., Beck, B., Caauwe, A., Simons, B. D., and Blanpain, C. (2012). Defining the mode of tumour growth by clonal analysis. Nature 488, 527–530. doi: 10.1038/nature11344
Dronamraju, R., and Mason, J. M. (2011). MU2 and HP1a regulate the recognition of double strand breaks in Drosophila melanogaster. PLoS One 6:e25439. doi: 10.1371/journal.pone.0025439
Eichenlaub, T., Cohen, S. M., and Herranz, H. (2016). Cell competition drives the formation of metastatic tumors in a Drosophila model of epithelial tumor formation. Curr. Biol. 26, 419–427. doi: 10.1016/j.cub.2015.12.042
Ellsworth, D. L., Ellsworth, R. E., Love, B., Deyarmin, B., Lubert, S. M., Mittal, V., et al. (2004). Outer breast quadrants demonstrate increased levels of genomic instability. Ann. Surg. Oncol. 11, 861–868. doi: 10.1245/ASO.2004.03.024
Enomoto, M., and Igaki, T. (2011). Deciphering tumor-suppressor signaling in flies: genetic link between Scribble/Dlg/Lgl and the Hippo pathways. J. Genet. Genomics 38, 461–470. doi: 10.1016/j.jgg.2011.09.005
Evan, G. I., and Littlewood, T. D. (1993). The role of c-myc in cell growth. Curr. Opin. Genet. Dev. 3, 44–49. doi: 10.1016/S0959-437X(05)80339-9
Feinberg, A. P., Koldobskiy, M. A., and Gondor, A. (2016). Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 17, 284–299. doi: 10.1038/nrg.2016.13
Froldi, F., Ziosi, M., Garoia, F., Pession, A., Grzeschik, N. A., Bellosta, P., et al. (2010). The lethal giant larvae tumour suppressor mutation requires dMyc oncoprotein to promote clonal malignancy. BMC Biol. 8:33. doi: 10.1186/1741-7007-8-33
Gabay, M., Li, Y., and Felsher, D. W. (2014). MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb. Perspect. Med. 4:a014241. doi: 10.1101/cshperspect.a014241
Gagneur, J., Stegle, O., Zhu, C., Jakob, P., Tekkedil, M. M., Aiyar, R. S., et al. (2013). Genotype-environment interactions reveal causal pathways that mediate genetic effects on phenotype. PLoS Genet. 9:e1003803. doi: 10.1371/journal.pgen.1003803
Galandiuk, S., Rodriguez-Justo, M., Jeffery, R., Nicholson, A. M., Cheng, Y., Oukrif, D., et al. (2012). Field cancerization in the intestinal epithelium of patients with Crohn’s ileocolitis. Gastroenterology 142, 855–864.e8. doi: 10.1053/j.gastro.2011.12.004
Gallant, P. (2013). Myc function in Drosophila. Cold Spring Harb. Perspect. Med. 3:a014324. doi: 10.1101/cshperspect.a014324
Giaretti, W., Pentenero, M., Gandolfo, S., and Castagnola, P. (2012). Chromosomal instability, aneuploidy and routine high-resolution DNA content analysis in oral cancer risk evaluation. Future Oncol. 8, 1257–1271. doi: 10.2217/fon.12.116
Gogna, R., Shee, K., and Moreno, E. (2015). Cell competition during growth and regeneration. Annu. Rev. Genet. 49, 697–718. doi: 10.1146/annurev-genet-112414-055214
Goodliffe, J. M., Wieschaus, E., and Cole, M. D. (2005). Polycomb mediates Myc autorepression and its transcriptional control of many loci in Drosophila. Genes Dev. 19, 2941–2946. doi: 10.1101/gad.1352305
Grady, W. M. (2005). Epigenetic events in the colorectum and in colon cancer. Biochem. Soc. Trans. 33(Pt 4), 684–688. doi: 10.1042/BST0330684
Greer, C., Lee, M., Westerhof, M., Milholland, B., Spokony, R., Vijg, J., et al. (2013). Myc-dependent genome instability and lifespan in Drosophila. PLoS One 8:e74641. doi: 10.1371/journal.pone.0074641
Grewal, S. S., Li, L., Orian, A., Eisenman, R. N., and Edgar, B. A. (2005). Myc-dependent regulation of ribosomal RNA synthesis during Drosophila development. Nat. Cell Biol. 7, 295–302. doi: 10.1038/ncb1223
Grifoni, D., and Bellosta, P. (2015). Drosophila Myc: a master regulator of cellular performance. Biochim. Biophys. Acta 1849, 570–581. doi: 10.1016/j.bbagrm.2014.06.021
Grifoni, D., Froldi, F., and Pession, A. (2013). Connecting epithelial polarity, proliferation and cancer in Drosophila: the many faces of lgl loss of function. Int. J. Dev. Biol. 57, 677–687. doi: 10.1387/ijdb.130285dg
Grifoni, D., Garoia, F., Bellosta, P., Parisi, F., De Biase, D., Collina, G., et al. (2007). aPKCzeta cortical loading is associated with Lgl cytoplasmic release and tumor growth in Drosophila and human epithelia. Oncogene 26, 5960–5965. doi: 10.1038/sj.onc.1210389
Grifoni, D., Garoia, F., Schimanski, C. C., Schmitz, G., Laurenti, E., Galle, P. R., et al. (2004). The human protein Hugl-1 substitutes for Drosophila lethal giant larvae tumour suppressor function in vivo. Oncogene 23, 8688–8694. doi: 10.1038/sj.onc.1208023
Grifoni, D., Sollazzo, M., Fontana, E., Froldi, F., and Pession, A. (2015). Multiple strategies of oxygen supply in Drosophila malignancies identify tracheogenesis as a novel cancer hallmark. Sci. Rep. 5:9061. doi: 10.1038/srep09061
Grusche, F. A., Richardson, H. E., and Harvey, K. F. (2010). Upstream regulation of the hippo size control pathway. Curr. Biol. 20, R574–R582. doi: 10.1016/j.cub.2010.05.023
Gurel, B., Iwata, T., Koh, C. M., Jenkins, R. B., Lan, F., Van Dang, C., et al. (2008). Nuclear MYC protein overexpression is an early alteration in human prostate carcinogenesis. Mod. Pathol. 21, 1156–1167. doi: 10.1038/modpathol.2008.111
Haaland, C. M., Heaphy, C. M., Butler, K. S., Fischer, E. G., Griffith, J. K., and Bisoffi, M. (2009). Differential gene expression in tumor adjacent histologically normal prostatic tissue indicates field cancerization. Int. J. Oncol. 35, 537–546.
Halder, G., and Johnson, R. L. (2011). Hippo signaling: growth control and beyond. Development 138, 9–22. doi: 10.1242/dev.045500
Hariharan, I. K., and Bilder, D. (2006). Regulation of imaginal disc growth by tumor-suppressor genes in Drosophila. Annu. Rev. Genet. 40, 335–361. doi: 10.1146/annurev.genet.39.073003.100738
Herranz, H., Eichenlaub, T., and Cohen, S. M. (2016). Cancer in Drosophila: imaginal discs as a model for epithelial tumor formation. Curr. Top. Dev. Biol. 116, 181–199. doi: 10.1016/bs.ctdb.2015.11.037
Hofstad, B., Vatn, M. H., Andersen, S. N., Huitfeldt, H. S., Rognum, T., Larsen, S., et al. (1996). Growth of colorectal polyps: redetection and evaluation of unresected polyps for a period of three years. Gut 39, 449–456. doi: 10.1136/gut.39.3.449
Iwata, T., Schultz, D., Hicks, J., Hubbard, G. K., Mutton, L. N., Lotan, T. L., et al. (2010). MYC overexpression induces prostatic intraepithelial neoplasia and loss of Nkx3.1 in mouse luminal epithelial cells. PLoS One 5:e9427. doi: 10.1371/journal.pone.0009427
Jakubek, Y., Lang, W., Vattathil, S., Garcia, M., Xu, L., Huang, L., et al. (2016). Genomic landscape established by allelic imbalance in the cancerization field of a normal appearing airway. Cancer Res. 76, 3676–3683. doi: 10.1158/0008-5472.CAN-15-3064
Jaloszynski, P., Jaruga, P., Olinski, R., Biczysko, W., Szyfter, W., Nagy, E., et al. (2003). Oxidative DNA base modifications and polycyclic aromatic hydrocarbon DNA adducts in squamous cell carcinoma of larynx. Free Radic. Res. 37, 231–240. doi: 10.1080/1071576021000041014
Johnston, L. A. (2014). Socializing with MYC: cell competition in development and as a model for premalignant cancer. Cold Spring Harb. Perspect. Med. 4:a014274. doi: 10.1101/cshperspect.a014274
Kamakaka, R. T., and Biggins, S. (2005). Histone variants: deviants? Genes Dev. 19, 295–310. doi: 10.1101/gad.1272805
Kamiyama, H., Suzuki, K., Maeda, T., Koizumi, K., Miyaki, Y., Okada, S., et al. (2012). DNA demethylation in normal colon tissue predicts predisposition to multiple cancers. Oncogene 31, 5029–5037. doi: 10.1038/onc.2011.652
Kato, S., Lippman, S. M., Flaherty, K. T., and Kurzrock, R. (2016). The conundrum of genetic “Drivers” in benign conditions. J. Natl. Cancer Inst. 108:djw036. doi: 10.1093/jnci/djw036
Khan, A., Shover, W., and Goodliffe, J. M. (2009). Su(z)2 antagonizes auto-repression of Myc in Drosophila, increasing Myc levels and subsequent trans-activation. PLoS One 4:e5076. doi: 10.1371/journal.pone.0005076
Kim, J., Eltoum, I. E., Roh, M., Wang, J., and Abdulkadir, S. A. (2009). Interactions between cells with distinct mutations in c-MYC and Pten in prostate cancer. PLoS Genet. 5:e1000542. doi: 10.1371/journal.pgen.1000542
Klebes, A., Sustar, A., Kechris, K., Li, H., Schubiger, G., and Kornberg, T. B. (2005). Regulation of cellular plasticity in Drosophila imaginal disc cells by the Polycomb group, trithorax group and lama genes. Development 132, 3753–3765. doi: 10.1242/dev.01927
Kuzyk, A., and Mai, S. (2014). c-MYC-induced genomic instability. Cold Spring Harb. Perspect. Med. 4:a014373. doi: 10.1101/cshperspect.a014373
Lawrence, M. S., Stojanov, P., Mermel, C. H., Robinson, J. T., Garraway, L. A., Golub, T. R., et al. (2014). Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505, 495–501. doi: 10.1038/nature12912
Lee, Y. C., Wang, H. P., Wang, C. P., Ko, J. Y., Lee, J. M., Chiu, H. M., et al. (2011). Revisit of field cancerization in squamous cell carcinoma of upper aerodigestive tract: better risk assessment with epigenetic markers. Cancer Prev. Res. 4, 1982–1992. doi: 10.1158/1940-6207.CAPR-11-0096
Leevers, S. J., Weinkove, D., MacDougall, L. K., Hafen, E., and Waterfield, M. D. (1996). The Drosophila phosphoinositide 3-kinase Dp110 promotes cell growth. EMBO J. 15, 6584–6594. doi: 10.1002/j.1460-2075.1996.tb01049.x
Levayer, R., Hauert, B., and Moreno, E. (2015). Cell mixing induced by myc is required for competitive tissue invasion and destruction. Nature 524, 476–480. doi: 10.1038/nature14684
Levayer, R., and Moreno, E. (2013). Mechanisms of cell competition: themes and variations. J. Cell Biol. 200, 689–698. doi: 10.1083/jcb.201301051
Levayer, R., and Moreno, E. (2016). How to be in a good shape? The influence of clone morphology on cell competition. Commun. Integr. Biol. 9:e1102806. doi: 10.1080/19420889.2015.1102806
Lu, H., and Bilder, D. (2005). Endocytic control of epithelial polarity and proliferation in Drosophila. Nat. Cell Biol. 7, 1232–1239. doi: 10.1038/ncb1324
Lu, X., Feng, X., Man, X., Yang, G., Tang, L., Du, D., et al. (2009). Aberrant splicing of Hugl-1 is associated with hepatocellular carcinoma progression. Clin. Cancer Res. 15, 3287–3296. doi: 10.1158/1078-0432.CCR-08-2078
Lu, Z., Sheng, J., Zhang, Y., Deng, J., Li, Y., Lu, A., et al. (2016). Clonality analysis of multifocal papillary thyroid carcinoma by using genetic profiles. J. Pathol. 239, 72–83. doi: 10.1002/path.4696
Luo, Y., Yu, M., and Grady, W. M. (2014). Field cancerization in the colon: a role for aberrant DNA methylation? Gastroenterol. Rep. 2, 16–20. doi: 10.1093/gastro/got039
Maggrah, A., Robinson, K., Creed, J., Wittock, R., Gehman, K., Gehman, T., et al. (2013). Paired ductal carcinoma in situ and invasive breast cancer lesions in the D-loop of the mitochondrial genome indicate a cancerization field effect. Biomed Res. Int. 2013:379438. doi: 10.1155/2013/379438
Maitra, S., Kulikauskas, R. M., Gavilan, H., and Fehon, R. G. (2006). The tumor suppressors Merlin and Expanded function cooperatively to modulate receptor endocytosis and signaling. Curr. Biol. 16, 702–709. doi: 10.1016/j.cub.2006.02.063
Maley, C. C., Galipeau, P. C., Li, X., Sanchez, C. A., Paulson, T. G., and Reid, B. J. (2004). Selectively advantageous mutations and hitchhikers in neoplasms: p16 lesions are selected in Barrett’s esophagus. Cancer Res. 64, 3414–3427. doi: 10.1158/0008-5472.CAN-03-3249
Martincorena, I., Roshan, A., Gerstung, M., Ellis, P., Van Loo, P., McLaren, S., et al. (2015). Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 348, 880–886. doi: 10.1126/science.aaa6806
McDonald, S. A., Greaves, L. C., Gutierrez-Gonzalez, L., Rodriguez-Justo, M., Deheragoda, M., Leedham, S. J., et al. (2008). Mechanisms of field cancerization in the human stomach: the expansion and spread of mutated gastric stem cells. Gastroenterology 134, 500–510. doi: 10.1053/j.gastro.2007.11.035
McMahon, S. B. (2014). MYC and the control of apoptosis. Cold Spring Harb. Perspect. Med. 4:a014407. doi: 10.1101/cshperspect.a014407
Meier, P., Silke, J., Leevers, S. J., and Evan, G. I. (2000). The Drosophila caspase DRONC is regulated by DIAP1. EMBO J. 19, 598–611. doi: 10.1093/emboj/19.4.598
Mendoza, P., Diaz, J., and Torres, V. A. (2014). On the role of Rab5 in cell migration. Curr. Mol. Med. 14, 235–245. doi: 10.2174/1566524014666140128111347
Menendez, J., Perez-Garijo, A., Calleja, M., and Morata, G. (2010). A tumor-suppressing mechanism in Drosophila involving cell competition and the Hippo pathway. Proc. Natl. Acad. Sci. U.S.A. 107, 14651–14656. doi: 10.1073/pnas.1009376107
Menut, L., Vaccari, T., Dionne, H., Hill, J., Wu, G., and Bilder, D. (2007). A mosaic genetic screen for Drosophila neoplastic tumor suppressor genes based on defective pupation. Genetics 177, 1667–1677. doi: 10.1534/genetics.107.078360
Merino, M. M., Levayer, R., and Moreno, E. (2016). Survival of the fittest: essential roles of cell competition in development, aging, and cancer. Trends Cell Biol. 26, 776–788. doi: 10.1016/j.tcb.2016.05.009
Metzker, M. L. (2010). Sequencing technologies - the next generation. Nat. Rev. Genet. 11, 31–46. doi: 10.1038/nrg2626
Meyer, N., and Penn, L. Z. (2008). Reflecting on 25 years with MYC. Nat. Rev. Cancer 8, 976–990. doi: 10.1038/nrc2231
Michor, F., Iwasa, Y., and Nowak, M. A. (2004). Dynamics of cancer progression. Nat. Rev. Cancer 4, 197–205. doi: 10.1038/nrc1295
Mohan, M., and Jagannathan, N. (2014). Oral field cancerization: an update on current concepts. Oncol. Rev. 8:244. doi: 10.4081/oncol.2014.244
Montero, L., Muller, N., and Gallant, P. (2008). Induction of apoptosis by Drosophila Myc. Genesis 46, 104–111. doi: 10.1002/dvg.20373
Morata, G., and Ripoll, P. (1975). Minutes: mutants of drosophila autonomously affecting cell division rate. Dev. Biol. 42, 211–221. doi: 10.1016/0012-1606(75)90330-9
Moreno, E. (2008). Is cell competition relevant to cancer? Nat. Rev. Cancer 8, 141–147. doi: 10.1038/nrc2252
Moreno, E., and Basler, K. (2004). dMyc transforms cells into super-competitors. Cell 117, 117–129. doi: 10.1016/S0092-8674(04)00262-4
Moreno, E., Basler, K., and Morata, G. (2002). Cells compete for decapentaplegic survival factor to prevent apoptosis in Drosophila wing development. Nature 416, 755–759. doi: 10.1038/416755a
Neto-Silva, R. M., de Beco, S., and Johnston, L. A. (2010). Evidence for a growth-stabilizing regulatory feedback mechanism between Myc and Yorkie, the Drosophila homolog of Yap. Dev. Cell 19, 507–520. doi: 10.1016/j.devcel.2010.09.009
Nonn, L., Ananthanarayanan, V., and Gann, P. H. (2009). Evidence for field cancerization of the prostate. Prostate 69, 1470–1479. doi: 10.1002/pros.20983
Nussinov, R., Tsai, C. J., Jang, H., Korcsmaros, T., and Csermely, P. (2016). Oncogenic KRAS signaling and YAP1/beta-catenin: similar cell cycle control in tumor initiation. Semin. Cell Dev. Biol. 58, 79–85. doi: 10.1016/j.semcdb.2016.04.001
Oertel, M., Menthena, A., Dabeva, M. D., and Shafritz, D. A. (2006). Cell competition leads to a high level of normal liver reconstitution by transplanted fetal liver stem/progenitor cells. Gastroenterology 130, 507–520; quiz 590. doi: 10.1053/j.gastro.2005.10.049
Papadimitrakopoulou, V. A., Shin, D. M., and Hong, W. K. (1996). Molecular and cellular biomarkers for field cancerization and multistep process in head and neck tumorigenesis. Cancer Metastasis Rev. 15, 53–76. doi: 10.1007/BF00049487
Park, S. K., Song, C. S., Yang, H. J., Jung, Y. S., Choi, K. Y., Koo, D. H., et al. (2016). Field cancerization in sporadic colon cancer. Gut Liver 10, 773–780. doi: 10.5009/gnl15334
Parr, R. L., Mills, J., Harbottle, A., Creed, J. M., Crewdson, G., Reguly, B., et al. (2013). Mitochondria, prostate cancer, and biopsy sampling error. Discov. Med. 15, 213–220.
Patel, M. S., Shah, H. S., and Shrivastava, N. (2016). c-Myc dependent cell competition in human cancer cells. J. Cell. Biochem. 118, 1782–1791. doi: 10.1002/jcb.25846
Penzo-Mendez, A. I., and Stanger, B. Z. (2014). Cell competition in vertebrate organ size regulation. Wiley Interdiscip. Rev. Dev. Biol. 3, 419–427. doi: 10.1002/wdev.148
Perez, E., Lindblad, J. L., and Bergmann, A. (2017). Tumor-promoting function of apoptotic caspases by an amplification loop involving ROS, macrophages and JNK in Drosophila. eLife 6:e26747. doi: 10.7554/eLife.26747
Prober, D. A., and Edgar, B. A. (2002). Interactions between Ras1, dMyc, and dPI3K signaling in the developing Drosophila wing. Genes Dev. 16, 2286–2299. doi: 10.1101/gad.991102
Rhiner, C., Diaz, B., Portela, M., Poyatos, J. F., Fernandez-Ruiz, I., Lopez-Gay, J. M., et al. (2009). Persistent competition among stem cells and their daughters in the Drosophila ovary germline niche. Development 136, 995–1006. doi: 10.1242/dev.033340
Rhiner, C., and Moreno, E. (2009). Super competition as a possible mechanism to pioneer precancerous fields. Carcinogenesis 30, 723–728. doi: 10.1093/carcin/bgp003
Rodrigues, A. B., Zoranovic, T., Ayala-Camargo, A., Grewal, S., Reyes-Robles, T., Krasny, M., et al. (2012). Activated STAT regulates growth and induces competitive interactions independently of Myc, Yorkie, Wingless and ribosome biogenesis. Development 139, 4051–4061. doi: 10.1242/dev.076760
Rosen, C., Shezen, E., Aronovich, A., Klionsky, Y. Z., Yaakov, Y., Assayag, M., et al. (2015). Preconditioning allows engraftment of mouse and human embryonic lung cells, enabling lung repair in mice. Nat. Med. 21, 869–879. doi: 10.1038/nm.3889
Sancho, M., Di-Gregorio, A., George, N., Pozzi, S., Sanchez, J. M., Pernaute, B., et al. (2013). Competitive interactions eliminate unfit embryonic stem cells at the onset of differentiation. Dev. Cell 26, 19–30. doi: 10.1016/j.devcel.2013.06.012
Santos-Garcia, A., Abad-Hernandez, M. M., Fonseca-Sanchez, E., Cruz-Hernandez, J. J., and Bullon-Sopelana, A. (2005). Proteic expression of p53 and cellular proliferation in oral leukoplakias. Med. Oral Patol. Oral Cir. Bucal 10, 5–8; 1–5.
Schimanski, C. C., Schmitz, G., Kashyap, A., Bosserhoff, A. K., Bataille, F., Schafer, S. C., et al. (2005). Reduced expression of Hugl-1, the human homologue of Drosophila tumour suppressor gene lgl, contributes to progression of colorectal cancer. Oncogene 24, 3100–3109. doi: 10.1038/sj.onc.1208520
Sears, R., Nuckolls, F., Haura, E., Taya, Y., Tamai, K., and Nevins, J. R. (2000). Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 14, 2501–2514. doi: 10.1101/gad.836800
Shakiba, N., and Zandstra, P. W. (2017). Engineering cell fitness: lessons for regenerative medicine. Curr. Opin. Biotechnol. 47, 7–15. doi: 10.1016/j.copbio.2017.05.005
Shivas, J. M., Morrison, H. A., Bilder, D., and Skop, A. R. (2010). Polarity and endocytosis: reciprocal regulation. Trends Cell Biol. 20, 445–452. doi: 10.1016/j.tcb.2010.04.003
Slaughter, D. P., Southwick, H. W., and Smejkal, W. (1953). Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer 6, 963–968. doi: 10.1002/1097-0142(195309)6:5<963::AID-CNCR2820060515>3.0.CO;2-Q
Stine, Z. E., Walton, Z. E., Altman, B. J., Hsieh, A. L., and Dang, C. V. (2015). MYC, Metabolism, and Cancer. Cancer Discov. 5, 1024–1039. doi: 10.1158/2159-8290.CD-15-0507
Suijkerbuijk, S. J., Kolahgar, G., Kucinski, I., and Piddini, E. (2016). Cell competition drives the growth of intestinal adenomas in Drosophila. Curr. Biol. 26, 428–438. doi: 10.1016/j.cub.2015.12.043
Tamori, Y., and Deng, W. M. (2013). Tissue repair through cell competition and compensatory cellular hypertrophy in postmitotic epithelia. Dev. Cell 25, 350–363. doi: 10.1016/j.devcel.2013.04.013
Torres, V. A., and Stupack, D. G. (2011). Rab5 in the regulation of cell motility and invasion. Curr. Protein Pept. Sci. 12, 43–51. doi: 10.2174/138920311795659461
Trujillo, K. A., Heaphy, C. M., Mai, M., Vargas, K. M., Jones, A. C., Vo, P., et al. (2011). Markers of fibrosis and epithelial to mesenchymal transition demonstrate field cancerization in histologically normal tissue adjacent to breast tumors. Int. J. Cancer 129, 1310–1321. doi: 10.1002/ijc.25788
Tsuboi, A., Ohsawa, S., Umetsu, D., Sando, Y., Kuranaga, E., Igaki, T., et al. (2018). Competition for space is controlled by apoptosis-induced change of local epithelial topology. Curr. Biol. 28, 2115–2128.e5. doi: 10.1016/j.cub.2018.05.029
Tyler, D. M., Li, W., Zhuo, N., Pellock, B., and Baker, N. E. (2007). Genes affecting cell competition in Drosophila. Genetics 175, 643–657. doi: 10.1534/genetics.106.061929
Udan, R. S., Kango-Singh, M., Nolo, R., Tao, C., and Halder, G. (2003). Hippo promotes proliferation arrest and apoptosis in the Salvador/Warts pathway. Nat. Cell Biol. 5, 914–920. doi: 10.1038/ncb1050
Vaccari, T., and Bilder, D. (2009). At the crossroads of polarity, proliferation and apoptosis: the use of Drosophila to unravel the multifaceted role of endocytosis in tumor suppression. Mol. Oncol. 3, 354–365. doi: 10.1016/j.molonc.2009.05.005
Vafa, O., Wade, M., Kern, S., Beeche, M., Pandita, T. K., Hampton, G. M., et al. (2002). c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol. Cell 9, 1031–1044. doi: 10.1016/S1097-2765(02)00520-8
Vermeulen, L., Morrissey, E., van der Heijden, M., Nicholson, A. M., Sottoriva, A., Buczacki, S., et al. (2013). Defining stem cell dynamics in models of intestinal tumor initiation. Science 342, 995–998. doi: 10.1126/science.1243148
Villa Del Campo, C., Claveria, C., Sierra, R., and Torres, M. (2014). Cell competition promotes phenotypically silent cardiomyocyte replacement in the mammalian heart. Cell Rep. 8, 1741–1751. doi: 10.1016/j.celrep.2014.08.005
Villa del Campo, C., Lioux, G., Carmona, R., Sierra, R., Munoz-Chapuli, R., et al. (2016). Myc overexpression enhances of epicardial contribution to the developing heart and promotes extensive expansion of the cardiomyocyte population. Sci. Rep. 6:35366. doi: 10.1038/srep35366
Vincent, J. P., Kolahgar, G., Gagliardi, M., and Piddini, E. (2011). Steep differences in wingless signaling trigger Myc-independent competitive cell interactions. Dev. Cell 21, 366–374. doi: 10.1016/j.devcel.2011.06.021
Vogelstein, B., Papadopoulos, N., Velculescu, V. E., Zhou, S., Diaz, L. A. Jr., and Kinzler, K. W. (2013). Cancer genome landscapes. Science 339, 1546–1558. doi: 10.1126/science.1235122
Wang, S. L., Hawkins, C. J., Yoo, S. J., Muller, H. A., and Hay, B. A. (1999). The Drosophila caspase inhibitor DIAP1 is essential for cell survival and is negatively regulated by HID. Cell 98, 453–463. doi: 10.1016/S0092-8674(00)81974-1
Wodarz, A., and Nathke, I. (2007). Cell polarity in development and cancer. Nat. Cell Biol. 9, 1016–1024. doi: 10.1038/ncb433
Wodarz, D., Iwasa, Y., and Komarova, N. L. (2004). On the emergence of multifocal cancers. J. Carcinog. 3:13. doi: 10.1186/1477-3163-3-13
Xiong, D., Pan, J., Zhang, Q., Szabo, E., Miller, M. S., Lubet, R. A., et al. (2017). Bronchial airway gene expression signatures in mouse lung squamous cell carcinoma and their modulation by cancer chemopreventive agents. Oncotarget 8, 18885–18900. doi: 10.18632/oncotarget.13806
Xu, T., and Rubin, G. M. (1993). Analysis of genetic mosaics in developing and adult Drosophila tissues. Development 117, 1223–1237.
Xu, T., Wang, W., Zhang, S., Stewart, R. A., and Yu, W. (1995). Identifying tumor suppressors in genetic mosaics: the Drosophila lats gene encodes a putative protein kinase. Development 121, 1053–1063.
Zaky, A. H., Watari, J., Tanabe, H., Sato, R., Moriichi, K., Tanaka, A., et al. (2008). Clinicopathologic implications of genetic instability in intestinal-type gastric cancer and intestinal metaplasia as a precancerous lesion: proof of field cancerization in the stomach. Am. J. Clin. Pathol. 129, 613–621. doi: 10.1309/DFLELPGPNV5LK6B1
Zeki, S. S., McDonald, S. A., and Graham, T. A. (2011). Field cancerization in Barrett’s esophagus. Discov. Med. 12, 371–379.
Zhang, Y., Lin, N., Carroll, P. M., Chan, G., Guan, B., Xiao, H., et al. (2008). Epigenetic blocking of an enhancer region controls irradiation-induced proapoptotic gene expression in Drosophila embryos. Dev. Cell 14, 481–493. doi: 10.1016/j.devcel.2008.01.018
Keywords: MYC, field cancerisation, multifocality, Drosophila, TSGs, cell competition
Citation: Sollazzo M, Genchi C, Paglia S, Di Giacomo S, Pession A, de Biase D and Grifoni D (2018) High MYC Levels Favour Multifocal Carcinogenesis. Front. Genet. 9:612. doi: 10.3389/fgene.2018.00612
Received: 15 September 2018; Accepted: 20 November 2018;
Published: 11 December 2018.
Edited by:
Maria Grazia Giansanti, Consiglio Nazionale delle Ricerche (CNR), ItalyReviewed by:
Louise Cheng, Peter MacCallum Cancer Centre, AustraliaCopyright © 2018 Sollazzo, Genchi, Paglia, Di Giacomo, Pession, de Biase and Grifoni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manuela Sollazzo, bWFudWVsYS5zb2xsYXp6bzJAdW5pYm8uaXQ= Daniela Grifoni, ZGFuaWVsYS5ncmlmb25pQHVuaWJvLml0
†Present address: Manuela Sollazzo, Cellular Biochemistry Laboratory, Department of Pharmacy and Biotechnology, University of Bologna, Bologna, Italy
China Genchi, Laboratory of Membrane Trafficking, Signaling and Development, Department of Biosciences, University of Milan, Milan, Italy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.