 Xiujuan Du1†
Xiujuan Du1† Xueren Gao2†
Xueren Gao2† Xin Liu3Lixiao Shen3Kai Wang4Yanjie Fan2Yu Sun2Xiaomei Luo2Huili Liu2Lili Wang2Yu Wang2Zhuwen Gong2Jianguo Wang2
Xin Liu3Lixiao Shen3Kai Wang4Yanjie Fan2Yu Sun2Xiaomei Luo2Huili Liu2Lili Wang2Yu Wang2Zhuwen Gong2Jianguo Wang2 Yongguo Yu2*
Yongguo Yu2* Fei Li1*
Fei Li1*- 1Developmental and Behavioral Pediatric Department – Child Primary Care Department, Brain and Behavioral Research Unit of Shanghai Institute for Pediatric Research and MOE Shanghai Key Laboratory for Children’s Environmental Health, Xinhua Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 2Department of Pediatric Endocrinology and Genetics, Shanghai Institute for Pediatric Research, Xinhua Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 3Developmental and Behavioral Pediatric Department – Child Primary Care Department, Xinhua Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 4Shanghai Children’s Medical Center, Shanghai Jiao Tong University School of Medicine, Shanghai, China
Autism spectrum disorder (ASD) is a group of clinically and genetically heterogeneous neurodevelopmental disorders. Recent tremendous advances in the whole exome sequencing (WES) enable rapid identification of variants associated with ASD including single nucleotide variations (SNVs) and indels. To further explore genetic etiology of ASD in Chinese children with negative findings of copy number variants (CNVs), we applied WES in 80 simplex families with a single affected offspring with ASD or suspected ASD, and validated variations predicted to be damaging by Sanger sequencing. The results showed that an overall diagnostic yield of 8.8% (9.2% in the group of ASD and 6.7% in the group of suspected ASD) was observed in our cohort. Among patients with diagnosed ASD, developmental delay or intellectual disability (DD/ID) was the most common comorbidity with a diagnostic yield of 13.3%, followed by seizures (50.0%) and craniofacial anomalies (40.0%). All of identified de novo SNVs and indels among patients with ASD were loss of function (LOF) variations and were slightly more frequent among female (male vs. female: 7.3% vs. 8.5%). A total of seven presumed causative genes (CHD8, AFF2, ADNP, POGZ, SHANK3, IL1RAPL1, and PTEN) were identified in this study. In conclusion, WES is an efficient diagnostic tool for diagnosed ASD especially those with negative findings of CNVs and other neurological disorders in clinical practice, enabling early identification of disease related genes and contributing to precision and personalized medicine.
Introduction
Autism spectrum disorder (ASD) is a group of highly heterogeneous neurodevelopmental disorders affecting 1 in 59 children aged 8 years, with boys four times more likely to be affected than girls (Baio et al., 2018). It’s characterized by impaired reciprocal social interaction and communication, as well as restricted repetitive interests and behaviors (The Lancet, 2010). The symptoms could develop gradually from early childhood, affecting daily functioning and persisting throughout one’s life (Stefanatos, 2008). Given the variety of phenotypes and severity, it’s believed that genetic factors play a key role in the pathogenesis of ASD, in combination with developmental environmental factors (Hofvander et al., 2009; Mattila et al., 2010; Anagnostou et al., 2014).
The clinical and genetic heterogeneity of ASD has proved to be challenging to the diagnostic workup of affected patients. Routine testing for Fragile X syndrome, karyotyping and chromosomal microarray (CMA) have been established as the first-tier tests for patients with ASD for several years, accounting only for about 1–2%, 5%, and 5–10% cases, respectively (Shen et al., 2010; Betancur, 2011; State and Levitt, 2011; Devlin and Scherer, 2012). Certain loci were identified to confer risk for ASD, and 16p11.2, 15q11-q13, and 22q11.2 were the most frequent (Marshall et al., 2008; Weiss et al., 2008; Fernandez et al., 2010; Hogart et al., 2010; Hiroi et al., 2013). In addition, several genes identified by copy number variants (CNVs) screening and target sequencing for candidate genes were related to ASD susceptibility, such as PTCHD1, NRXN1, NLGN3, SHANK3, SHANK1 and so on (Jamain et al., 2003; Moessner et al., 2007; Kim et al., 2008; Noor et al., 2010; Sato et al., 2012; Dabell et al., 2013). Recent rapidly improved accuracy and decreased cost of whole-exome sequencing (WES) enabled the application among proband-parent trios of ASD in clinical practice, opening the way to the discovery of single nucleotide variations (SNVs) and indels (Sanders et al., 2012; O’Roak et al., 2014). By using WES, ∼ 20.0% patients with sporadic ASD could be identified and this rate even reached to ∼90% because of the highly inbred nature of the Saudi population, making it useful in complementing CMA designed to detect CNVs, and better characterizing the genetic architecture for ASD in simplex families (O’Roak et al., 2011; Yu et al., 2013; Tammimies et al., 2015; Al-Mubarak et al., 2017). However, thus far, there remains a gap in our knowledge of the diagnostic yield of trio-WES among Chinese children with autistic features when CMA is unable to detect risk-related variations, and its impacts on clinical practice.
It is estimated that more than 70% of individuals with ASD have comorbidities including developmental and psychiatric disorders (Hofvander et al., 2009; Kohane et al., 2012). Based on the suspicion that genetic mechanism of children suffered an abnormality of morphogenesis differed from those who did not, Miles et al. (2005) collected data of dysmorphisms among children with ASD. The patients were further divided into two subsets of patients with documented dysmorphology (complex group) and without evident disrupted morphogenesis (essential group). The findings demonstrated that an abnormal karyotype (2.3%) or a clinically recognized syndrome (1.9%) were identified and restricted to complex group. Triggered by this incentive, we classified patients into different subgroups according to clinical manifestations, to explore the utility of WES and better characterize the underlining genetic differences.
In an attempt to expand the genetic spectrum of ASD by identifying novel SNV and indels, and evaluate how well WES could make up the deficiency of CMA in China, trio-based WES was further implemented among 80 children diagnosed as ASD and suspected of having ASD with negative findings of CMA.
Materials and Methods
Patients
Data were collected from children visiting the outpatient clinic of Department of Developmental Behavioral Pediatric and Children Healthcare at Xinhua Hospital, Shanghai, China during March to December 2017. Without detection of CNVs related to ASD, a total of 80 unrelated children (aged 4 months to 13 years) with autistic features were enrolled to further complete trio-based whole exome sequencing (WES). All probands did not have neurological disorders (such as cerebral palsy and schizophrenia) or have the known chromosome/genetic disorders (such as trisomy 21 syndrome, trisomy 18 syndrome, trisomy 13 syndrome, Rett syndrome, Fragile X syndrome). Chromosome microarray analysis were applied by using Cyto Scan HD array (Affymetrix, Santa Clara, CA, United States).
Of these children, 65 (55 males and 10 females) were diagnosed as ASD using standard evaluation including Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DMS-5), the Autism Diagnostic Observation Schedule (ADOS), Childhood Autism Rating Scale (CARS) and the intellectual assessment by clinicians. By intellectual assessment, the patients with developmental quotient (DQ) < 75 assessed using Gesell development scales, and intelligence quotient (IQ) < 70 assessed using WISC-R or WPPSI (Wechsler Intelligence Scale for children) were diagnosed as developmental delay or intellectual disability (DD/ID). With related evaluation not available, the rest of children exhibiting clinician-reported autistic features were suspected of having ASD. The study was approved by the ethical committee at Xinhua hospital and conducted in accordance with the relevant guidelines and regulations. Written informed consent in accordance with the Declaration of Helsinki was obtained from the patients and parents. And we have been adhered to standard biosecurity and institutional safety procedures in this study.
DNA Samples, WES, and Bioinformatics Analysis
Peripheral blood leukocytes from 80 children and parents were obtained. Genomic DNA (gDNA) was extracted using Lab-Aid Nucleic Acid (DNA) Isolation Kit (Zeesan, China), according to the manufacturer’s instructions. The preparation of library of WES was completed using xGen Exome research panel v1.0 (Integrated DNA Technologies, Coralville, IA, United States). Sequencing was performed using paired 150-end, 150-cycle chemistry on the Illumina HiSeq 4000 (Illumina, San Diego, CA, United States), according to the manufacturer’s instructions. Burrows-Wheeler Aligner (BWA, version 0.7.10) was used for FASTQ files to mapping reads to the human reference genome (GRCh37/hg19). Base calling, QC analysis and coverage analysis was performed with Picard tools-1.124 and GATK software. Variants were then annotated using SnpEff version 4.2. Stepwise variant filtering are as follows: variants that demonstrated >1% frequency in the population variant databases including 1000 Genomes Project, Exome Variant Server (EVS) and Exome Aggregation Consortium (ExAC) or >5% frequency in our in-house database (based on 150 exome datasets), and intergenic and 3′/5′ untranslated region variants, none splice-related intronic and synonymous variants were filtered, with those located at canonical splice sites excluded.
Combined with clinical manifestation and modes of inheritance, Sanger sequencing and DNA-based paternity testing were performed to validate the putative pathogenic mutation for all family members. Sequencing products were analyzed using an ABI 3730xl DNA Analyzer (Applied Biosystems, Foster City, CA, United States). For DNA-based paternity testing, the IdentifilerTM system and the ABI 3730xL DNA Analyzer (Applied Biosystems) were used to perform multiplex polymerase chain reaction (PCR) amplification and genotyping of PCR products with capillary electrophoresis, respectively. Primer sequences used for validation have been showed in Supplementary Table S1.
MutationTaster1, SIFT2, and PolyPhen-23 were used to assess the effect of variants on protein function. Validated variants were classified as pathogenic, likely pathogenic, variants of uncertain clinical significance (VUS), likely benign and benign, based on standards and guidelines of the American College of Medical Genetics and Genomics (ACMG). Potential causative genetic variants have been deposited in database of LOVD4, with associated accession numbers ranging from #0000379121 to #0000379127.
Results
Clinical Characteristics of Patients
The clinical characteristics of children with diagnosed ASD and suspected ASD were summarized in Table 1. Among the patients with ASD, there were 23 children younger than 3 years, 30 children from 3 to 6 year of age (diagnostic rate = 16.7%) and 12 children older than 6 years (diagnostic rate = 8.3%) and the male/female ratio was 53/12. 41 out of 60 patients diagnosed as ASD with available behavioral assessments had CARS scores ≧37 (diagnostic rate = 7.3%), suggesting a severe autistic behavior.
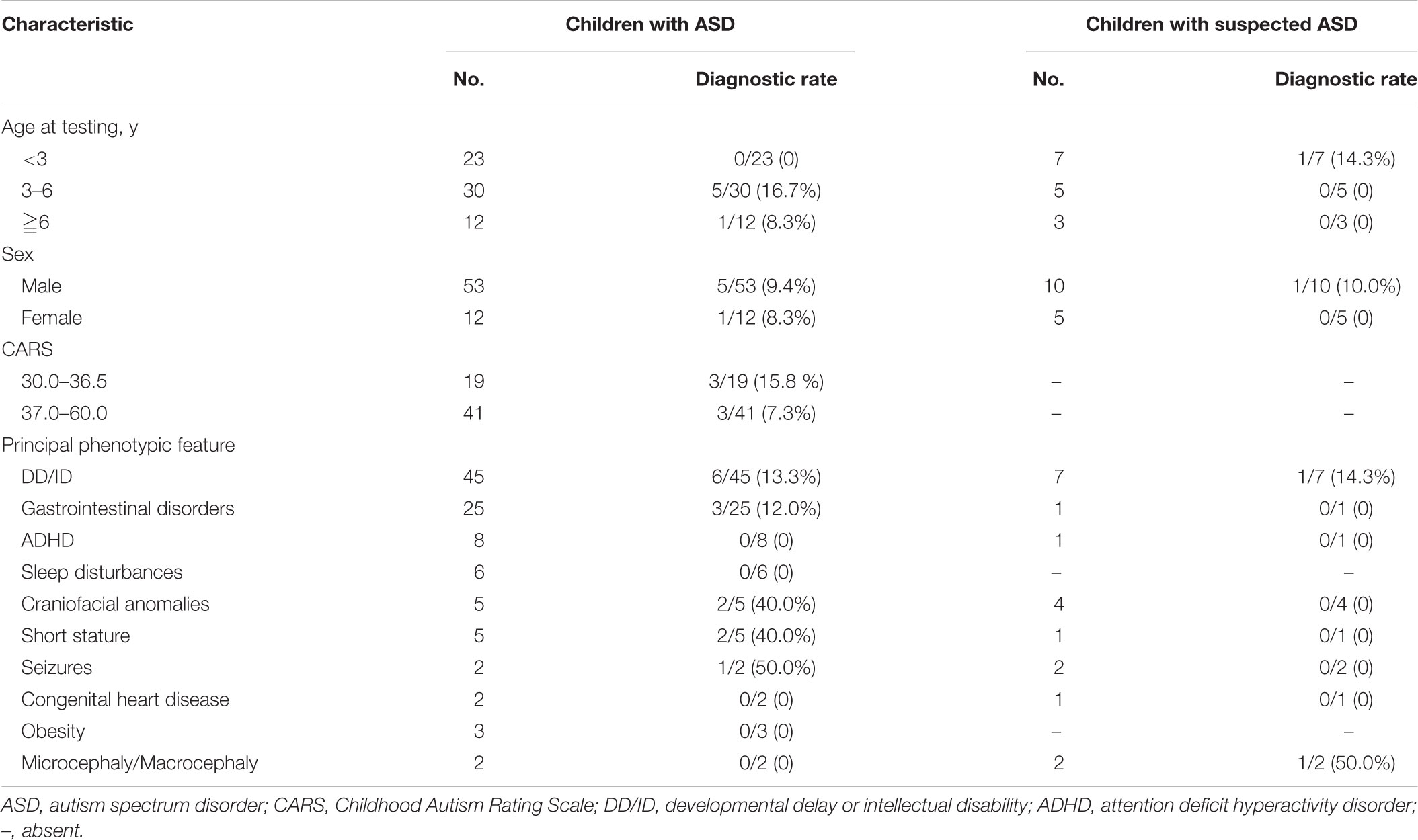
TABLE 1. Clinical characteristics of children with ASD and suspected ASD in the current study.
In terms of other clinical phenotypes, DD/ID was the most common comorbidity in both groups of patients with ASD and suspected ASD. A variety of neurological and non-neurological deficits were exhibit among patients with ASD, including DD/ID (n = 45, diagnostic rate = 13.3%), seizures (n = 2, diagnostic rate = 50.0%), craniofacial anomalies (n = 5, diagnostic rate = 40.0%), etc. (Table 1).
Molecular Genetic Findings of WES
WES was performed among 80 trios with diagnosed or suspected ASD. Quality control of sequencing showed that 97.8% of the reads were mapped to the reference genome, and 97.7% of the targeted regions were covered by ≧10X reads with enough average depth (138X) (Supplementary Table S2). And details of QC (the depth, the coverage and the target regions covered by ≧10X reads) were shown in Figure 1. Potential causative variants were subsequently confirmed by Sanger sequencing.
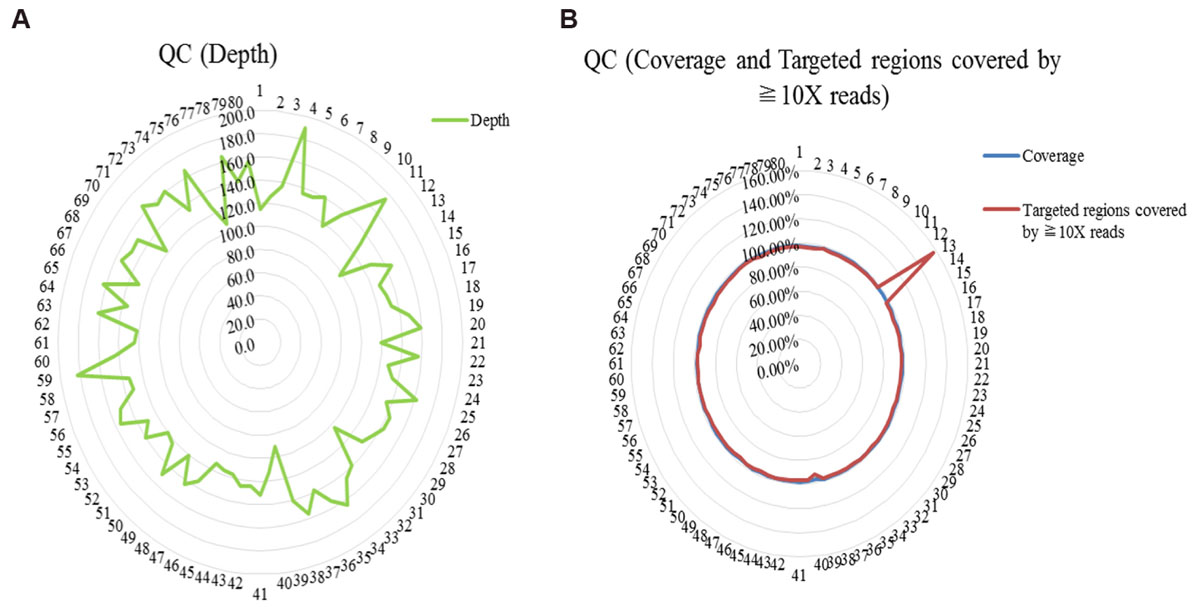
FIGURE 1. Quality control (QC) of whole exome sequencing. (A) QC of the depth; (B) QC of the coverage and the targeted regions covered by ≧10X reads.
A conclusive genetic diagnosis were obtained in seven of 80 children identified by WES, corresponding to an overall diagnostic yield of 8.8% (9.2% in the group of ASD and 6.7% in the group of suspected ASD). We detected and validated a total of seven variants and each was identified in a different gene (Table 2). Based on the distribution of the confirmed variants, frameshift were the most common (3/7), followed by the missense (2/7), stop gained (1/7) and start lost variants (1/7). Among the causative variants, the presumed mode of inheritance was autosomal dominant in 71.4% (71.4% de novo), and X-linked in 28.6% (14.3% de novo and 14.3% inherited).
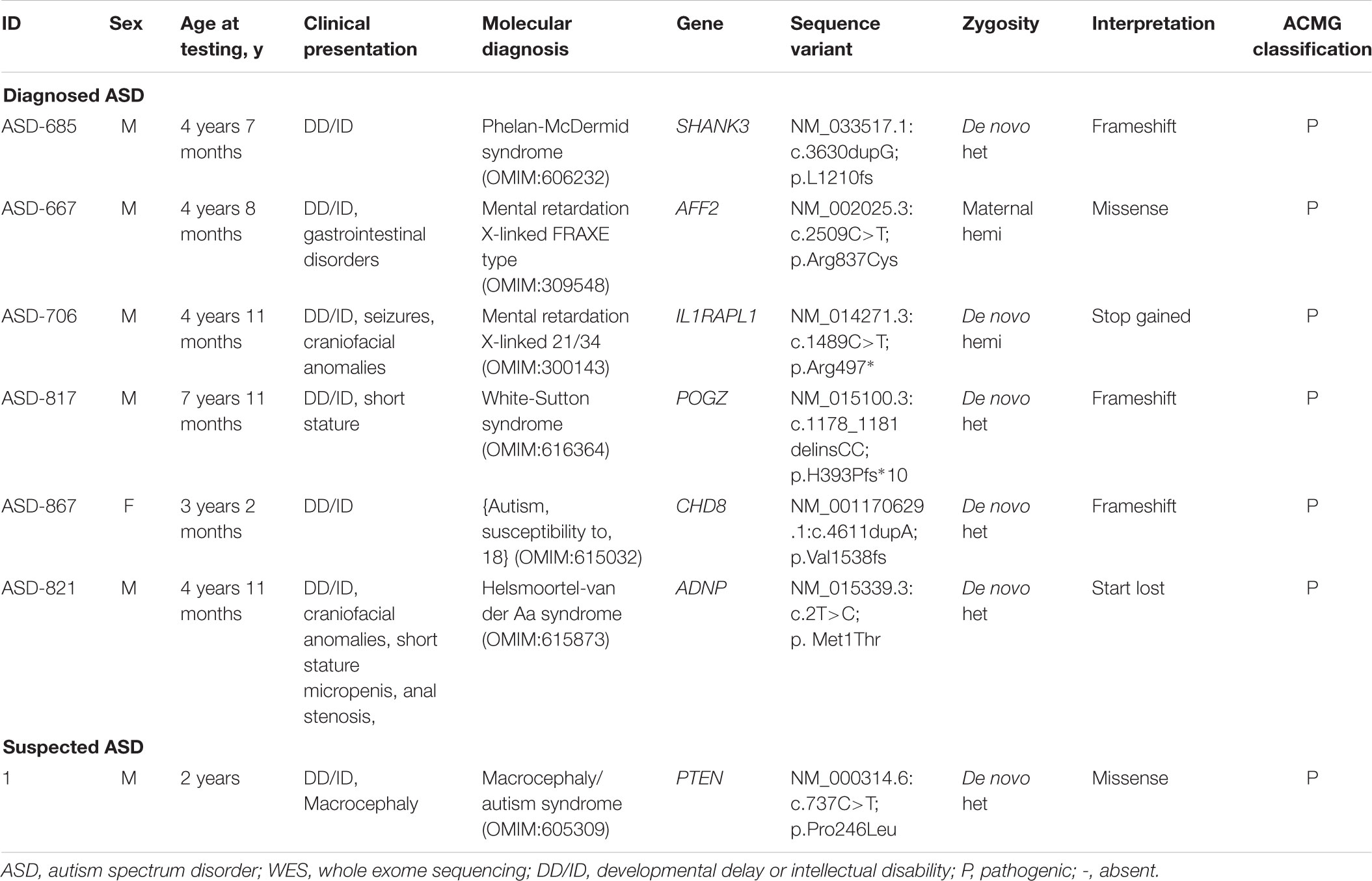
TABLE 2. Clinical and molecular findings in children with positive results of WES.
De novo mutations accounted for 85.7% (six of seven) of the overall molecular diagnoses. Among patients with diagnosed ASD, all of identified de novo SNVs and indels were loss of function (LOF) variations and were slightly more frequent among female (male vs. female: 7.3% vs. 8.5%). In addition, all of patients with diagnosed ASD revealed to carry de novo LOF variations were co-occurring with DD/ID. A de novo missense variation was identified in one patient with suspected ASD. In total, seven genes (CHD8, AFF2, ADNP, POGZ, SHANK3, IL1RAPL1, and PTEN) with presumed pathogenic variations were identified in this study.
Impact of WES on Clinical Management
The discovery of WES makes both early clinical detection and genetic counseling possible in various ways among four of seven probands with a conclusive molecular diagnosis. In addition to the following up for ASD based on the risk genes, these patients received further workup of systemic involvement in this cohort. For example, developmental and behavioral evaluation were conducted in the patients with variants of AFF2 and IL1RAPL1, respectively; seizures, short stature, abnormalities of skeleton system, eye, ear, brain and gastrointestinal tract screening, and developmental and behavioral evaluation were implemented in the patient with a de novo SNV of POGZ; hormone deficiency, short stature, obesity, hypotonia, seizures and feeding problems screening were evaluated in the patient with a de novo SNV of ADNP. And correspondingly, medication was changed. Growth hormone was applied in patients with ADNP and POGZ based on the diagnosis of short stature. Brain protein hydrolysate was discontinued in patients with seizures. Two couples with future pregnancy were informed the importance of prenatal testing and preimplantation genetic diagnosis.
Discussion
With the advent of decreasing cost combined with superior efficiency of WES, studies focusing on the contribution of de novo and/or inherited mutations become affordable as well as avoid the potential ‘diagnostic odyssey’ (Bamshad et al., 2011; O’Roak et al., 2011; Tan et al., 2017). In this study, with negative findings of ASD-related CNVs by using CMA, we further confirmed utility of trio-WES for diagnosis among children with ASD or suspected ASD in clinical practice. All de novo and inherited variants with predicted damaging effect were validated by Sanger sequencing in both patients and parents. Genetic etiology was identified in seven of 80 trios with an overall detection rate of 8.8%. Within the diagnosed ASD group, six of 65 (9.2%) patients received molecular diagnoses, which was similar to the results observed among sporadic ASD (8.4%), as well as those focusing on either de novo or inherited variations, ranging from 6.3% to 13.8% (Neale et al., 2012; Sanders et al., 2012; De Rubeis et al., 2014; Dong et al., 2014; Iossifov et al., 2014; Tammimies et al., 2015). An interesting finding emerging from this study implied the importance of completed ASD-related assessments in enabling a higher diagnostic yield among patients with suspected ASD. Our data showed that compared with children with suspected ASD (6.7%), the diagnostic yield was higher among patients with diagnosed ASD (9.2%). Moreover, diagnostic rate seemed high among ASD patients suffering from other neurodevelopmental disorders including DD/ID, suggesting that patients with these comorbidities may benefit more from WES. These findings were supported by the work of Tammimies et al. who recommended WES as a first-tier test for ASD, especially when comorbid with physical and congenital anomalies (Tammimies et al., 2015). To some extent, the relatively higher yield might result from the patients diagnosed and managed in the outpatient. Those who were diagnosed as ASD especially co-occurring with other neurodevelopmental disorders, are more likely undergone etiological testing including WES, given a high suspicion of genetic etiology.
The past few years have witnessed increasing studies of ASD trios published, highlighting the role of de novo variants and improving the identification of candidate risk genes for ASD (O’Roak et al., 2011; Iossifov et al., 2014). Given that de novo variation is less frequent and potentially more deleterious, we evaluated its diagnostic rates and effects to determine risk genes. Among children with diagnosed ASD, de novo variations were observed in 83.3% of the patients. Moreover, de novo LOF mutations contribute to 87.5% cases with ASD. Our findings that de novo variations of LOF predominant is contrary to previous population-based studies is intriguing (Iossifov et al., 2012; O’Roak et al., 2012; Sanders et al., 2012). One possible explanation is that ASD patients with other neurodevelopment disorders are prior to be tested by WES in outpatient, and may limit generalizability to the broader ASD population. Another key finding demonstrated here, was that in spite of a predominant male to female ratio (about 4:1), de novo LOF mutations were slightly more enriched in females with ASD. And this finding was consistent with the previous results (Levy et al., 2011; Iossifov et al., 2012). Genetic studies suggest that the strong male bias in liability might be attributed to a female protective effect, in which a higher load of mutations were tolerated by female (Gilman et al., 2011; Levy et al., 2011). An increasing body of evidence indicates that affected female with ASD are more susceptible to de novo SNVs and indels of LOF (Neale et al., 2012; Sanders et al., 2012; De Rubeis et al., 2014; Dong et al., 2014; Iossifov et al., 2014; Jacquemont et al., 2014). In addition to variations mentioned before, large CNVs encompassing more genes and probably more damaging, are especially abundant in affected females (Levy et al., 2011; Sanders et al., 2011; Jacquemont et al., 2014). These findings suggest to us that other underlying factors that have not yet been identified may contributes much more in males than in females.
All of genes with presumed causative mutations identified here were previously reported in ASD (Herman et al., 2007; Piton et al., 2008; Nishiyama et al., 2009; Bernier et al., 2014; Colak et al., 2014; Helsmoortel et al., 2014; Stessman et al., 2016; Yi et al., 2016). Six genes (CHD8, AFF2, ADNP, POGZ, SHANK3, and IL1RAPL1) were identified among patients with diagnosed ASD with DD/ID (Piton et al., 2008; Nishiyama et al., 2009; Bernier et al., 2014; Colak et al., 2014; Helsmoortel et al., 2014; Stessman et al., 2016; Yi et al., 2016). In spite of de novo LOF variations detected in SHANK3 and CHD8, patient ASD-685 and ASD-867 presented ASD and DD/ID without other disorders at the age of testing. SHANK3 was a gene encoding a scaffolding protein that is enriched in postsynaptic densities of excitatory synapses (Yi et al., 2016). And CHD8, allelic variants of which are associated with ASD, encoding the protein chromodomain helicase DNA binding protein 8 (Nishiyama et al., 2009), which is a chromatin regulator enzyme that is essential during fetal development (Ronan et al., 2013). At present, the mechanism of the higher incidence in males remains inconclusive, and hormones, sex-specific brain differences or variation on the sex chromosomes were speculated to play a role in. We identified two variants (one missense and one de novo LOF) in two X-chromosome genes (AFF2 and IL1RAPL1). AFF2 whose function is to encode a putative transcriptional activator that is a member of the AF4∖FMR2 gene family, was previously associated with ASD and mental retardation, X-linked, FRAXE type (Colak et al., 2014). And in patient ASD-667 with a missense mutation in AFF2 displayed gastrointestinal disorders in addition to DD/ID. Patient ASD-706 with a de novo LOF variation in IL1RAPL1 showed ASD co-occurring with DD/ID, seizures and craniofacial anomalies. This gene is highly expressed in post-natal brain structures, which functions in the hippocampal memory system, thus suggesting a key role in the physiological processes underlying memory and learning abilities (Gambino et al., 2007). A de novo LOF variation in POGZ was identified in patient ASD-817 with DD/ID and short stature. Interestingly, previous reports showed patients with variation in ADNP often displayed Helsmoortel-van der Aa syndrome (Helsmoortel et al., 2014). However, without obesity and short stature at age of diagnosis, patients ASD-821 harboring a de novo LOF mutation (start lost) in ADNP presented novel phenotype of micropenis and anal stenosis. After genetic counseling, this patient were screened by biochemical tests related to hormone deficiency, short stature and suggested to be followed up in Department of Pediatric Endocrinology/Genetics. There remains one gene (PTEN) with a de novo missense variation was detected in a child with suspected ASD. This child presented typically macrocephaly and DD/ID. PTEN identified as a tumor suppressor is mutated in a large number of cancers at high frequency (Bonneau and Longy, 2000). These results implied that a continuum of neurological and non-neurological disorders that present in varied patterns might result from candidate risk genes by interacting with other factors.
To our knowledge, this work represents the first comprehensive analysis in Chinese children with diagnosed and suspected ASD by trio-based WES. Similar to other studies by WES, one of potential limitations is that true causative variants may be omitted as a result of stringent criteria to filter false-positives. Besides, WES has limited ability to detect genomic imbalances and could not evaluate variations located on non-coding sequences. Notwithstanding the small sample size, our study in part contributes to dataset of phenotype and genetic etiology of ASD in Chinese children. Moreover, we confirmed the utility of WES in patients without positive results of CNVs, improving the detection rate in a way. Accordingly, many challenges remain, it’s hopeful for a brighter future of individuals with ASD and their families benefiting from the advantages of WES.
Conclusion
In conclusion, WES offers the advantage of early screening of the underlying ASD-related genes when related CNVs were not identified by CMA, providing genetic diagnoses across diverse clinical subgroups and contributing to precision and personalized medicine.
Data Availability Statement
The potential causative variants in this study can be found in the LOVD database (https://databases.lovd.nl/shared/variants?search_owned_by_=%3D%22Fei%20Li%22), with associated accession numbers ranging from #0000379121 to #0000379127.
Author Contributions
FL and YY conceived and designed the study. XG performed the experiments. XD and XG drafted the manuscript. XL and XD collected the samples from ASD families. FL and LS made diagnoses, and interpreted the clinical data. XG, YF, YS, XL, HL, LW, YW, ZG, and JW analyzed the exome sequencing data. XL and KW were responsible for obtaining study ethics and collected clinical data. The authors jointly discussed the experimental results throughout the duration of the study. All authors reviewed and approved the final manuscript.
Funding
The work was financially supported by funding from Shanghai Municipal Commission of Health and Family Planning (Grant Nos. 2017ZZ02026, 2018BR33, 2017EKHWYX-02, GDEK201709, and 201740192), Shanghai Shenkang Hospital Development Center (Grant No. 16CR2025B), Shanghai Municipal Education Commission (Grant No. 20152234), National Natural Science Foundation of China (Grant Nos. 81571031, 81761128035, and 81670812), Shanghai Committee of Science and Technology (Grant Nos. 17XD1403200 and 18DZ2313505), Xinhua Hospital of Shanghai Jiao Tong University School of Medicine (2018YJRC03, Talent introduction-014, Top talent-201603), Jiao Tong University Cross Biomedical Engineering (Grant No. YG2017MS72), Shanghai Shen Kang Hospital Development Center new frontier technology joint project (Grant No. SHDC12017109), Youth Research Project of the Shanghai Municipal Health and Family Planning Commission (Grant No. 20184Y0348).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors gratefully thank all the patients and their families.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2018.00594/full#supplementary-material
TABLE S1 | Primer sequences used for validation.
TABLE S2 | Quality control of sequencing depth, coverage and the targeted regions covered by ≧10X reads (138X).
Footnotes
- ^http://www.mutationtaster.org
- ^http://sift.jcvi.org
- ^http://genetics.bwh.harvard.edu/pph2/
- ^https://databases.lovd.nl/shared/variants?search_owned_by_=%3D%22Fei%20Li%22
References
Al-Mubarak, B., Abouelhoda, M., Omar, A., AlDhalaan, H., Aldosari, M., Nester, M., et al. (2017). Whole exome sequencing reveals inherited and de novo variants in autism spectrum disorder: a trio study from Saudi families. Sci. Rep. 7:5679. doi: 10.1038/s41598-017-06033-1
Anagnostou, E., Zwaigenbaum, L., Szatmari, P., Fombonne, E., Fernandez, B. A., Woodbury-Smith, M., et al. (2014). Autism spectrum disorder: advances in evidence-based practice. CMAJ 186, 509–519. doi: 10.1503/cmaj.121756
Baio, J., Wiggins, L., Christensen, D. L., Maenner, M. J., Daniels, J., Warren, Z., et al. (2018). Prevalence of autism spectrum disorder among children aged 8 years - autism and developmental disabilities monitoring network, 11 sites, United States, 2014. MMWR Surveill. Summ. 67, 1–23. doi: 10.15585/mmwr.ss6706a1
Bamshad, M. J., Ng, S. B., Bigham, A. W., Tabor, H. K., Emond, M. J., Nickerson, D. A., et al. (2011). Exome sequencing as a tool for mendelian disease gene discovery. Nat. Rev. Genet. 12, 745–755. doi: 10.1038/nrg3031
Bernier, R., Golzio, C., Xiong, B., Stessman, H. A., Coe, B. P., Penn, O., et al. (2014). Disruptive CHD8 mutations define a subtype of autism early in development. Cell 158, 263–276. doi: 10.1016/j.cell.2014.06.017
Betancur, C. (2011). Etiological heterogeneity in autism spectrum disorders: more than 100 genetic and genomic disorders and still counting. Brain Res. 1380, 42–77. doi: 10.1016/j.brainres.2010.11.078
Bonneau, D., and Longy, M. (2000). Mutations of the human PTEN gene. Hum. Mutat. 16, 109–122. doi: 10.1002/1098-1004(200008)16:2<109::AID-HUMU3>3.0.CO;2-0
Colak, D., Zaninovic, N., Cohen, M. S., Rosenwaks, Z., Yang, W. Y., Gerhardt, J., et al. (2014). Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science 343, 1002–1005. doi: 10.1126/science.1245831
Dabell, M. P., Rosenfeld, J. A., Bader, P., Escobar, L. F., El-Khechen, D., Vallee, S. E., et al. (2013). Investigation of NRXN1 deletions: clinical and molecular characterization. Am. J. Med. Genet. A 161A, 717–731. doi: 10.1002/ajmg.a.35780
De Rubeis, S., He, X., Goldberg, A. P., Poultney, C. S., Samocha, K., Cicek, A. E., et al. (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215. doi: 10.1038/nature13772
Devlin, B., and Scherer, S. W. (2012). Genetic architecture in autism spectrum disorder. Curr. Opin. Genet. Dev. 22, 229–237. doi: 10.1016/j.gde.2012.03.002
Dong, S., Walker, M. F., Carriero, N. J., DiCola, M., Willsey, A. J., Ye, A. Y., et al. (2014). De novo insertions and deletions of predominantly paternal origin are associated with autism spectrum disorder. Cell Rep. 9, 16–23. doi: 10.1016/j.celrep.2014.08.068
Fernandez, B. A., Roberts, W., Chung, B., Weksberg, R., Meyn, S., Szatmari, P., et al. (2010). Phenotypic spectrum associated with de novo and inherited deletions and duplications at 16p11.2 in individuals ascertained for diagnosis of autism spectrum disorder. J. Med. Genet. 47, 195–203. doi: 10.1136/jmg.2009.069369
Gambino, F., Pavlowsky, A., Begle, A., Dupont, J. L., Bahi, N., Courjaret, R., et al. (2007). IL1-receptor accessory protein-like 1 (IL1RAPL1), a protein involved in cognitive functions, regulates N-type Ca2+-channel and neurite elongation. Proc. Natl. Acad. Sci. U.S.A. 104, 9063–9068. doi: 10.1073/pnas.0701133104
Gilman, S. R., Iossifov, I., Levy, D., Ronemus, M., Wigler, M., and Vitkup, D. (2011). Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron 70, 898–907. doi: 10.1016/j.neuron.2011.05.021
Helsmoortel, C., Vulto-van Silfhout, A. T., Coe, B. P., Vandeweyer, G., Rooms, L., van den Ende, J., et al. (2014). A SWI/SNF-related autism syndrome caused by de novo mutations in ADNP. Nat. Genet. 46, 380–384. doi: 10.1038/ng.2899
Herman, G. E., Butter, E., Enrile, B., Pastore, M., Prior, T. W., and Sommer, A. (2007). Increasing knowledge of PTEN germline mutations: two additional patients with autism and macrocephaly. Am. J. Med. Genet. A 143A, 589–593. doi: 10.1002/ajmg.a.31619
Hiroi, N., Takahashi, T., Hishimoto, A., Izumi, T., Boku, S., and Hiramoto, T. (2013). Copy number variation at 22q11.2: from rare variants to common mechanisms of developmental neuropsychiatric disorders. Mol. Psychiatry 18, 1153–1165. doi: 10.1038/mp.2013.92
Hofvander, B., Delorme, R., Chaste, P., Nyden, A., Wentz, E., Stahlberg, O., et al. (2009). Psychiatric and psychosocial problems in adults with normal-intelligence autism spectrum disorders. BMC Psychiatry 9:35. doi: 10.1186/1471-244X-9-35
Hogart, A., Wu, D., LaSalle, J. M., and Schanen, N. C. (2010). The comorbidity of autism with the genomic disorders of chromosome 15q11.2-q13. Neurobiol. Dis. 38, 181–191. doi: 10.1016/j.nbd.2008.08.011
Iossifov, I., O’Roak, B. J., Sanders, S. J., Ronemus, M., Krumm, N., Levy, D., et al. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221. doi: 10.1038/nature13908
Iossifov, I., Ronemus, M., Levy, D., Wang, Z., Hakker, I., Rosenbaum, J., et al. (2012). De novo gene disruptions in children on the autistic spectrum. Neuron 74, 285–299. doi: 10.1016/j.neuron.2012.04.009
Jacquemont, S., Coe, B. P., Hersch, M., Duyzend, M. H., Krumm, N., Bergmann, S., et al. (2014). A higher mutational burden in females supports a ”female protective model” in neurodevelopmental disorders. Am. J. Hum. Genet. 94, 415–425. doi: 10.1016/j.ajhg.2014.02.001
Jamain, S., Quach, H., Betancur, C., Rastam, M., Colineaux, C., Gillberg, I. C., et al. (2003). Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 34, 27–29. doi: 10.1038/ng1136
Kim, H. G., Kishikawa, S., Higgins, A. W., Seong, I. S., Donovan, D. J., Shen, Y., et al. (2008). Disruption of neurexin 1 associated with autism spectrum disorder. Am. J. Hum. Genet. 82, 199–207. doi: 10.1016/j.ajhg.2007.09.011
Kohane, I. S., McMurry, A., Weber, G., MacFadden, D., Rappaport, L., Kunkel, L., et al. (2012). The co-morbidity burden of children and young adults with autism spectrum disorders. PLoS One 7:e33224. doi: 10.1371/journal.pone.0033224
Levy, D., Ronemus, M., Yamrom, B., Lee, Y. H., Leotta, A., Kendall, J., et al. (2011). Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron 70, 886–897. doi: 10.1016/j.neuron.2011.05.015
Marshall, C. R., Noor, A., Vincent, J. B., Lionel, A. C., Feuk, L., Skaug, J., et al. (2008). Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 82, 477–488. doi: 10.1016/j.ajhg.2007.12.009
Mattila, M. L., Hurtig, T., Haapsamo, H., Jussila, K., Kuusikko-Gauffin, S., Kielinen, M., et al. (2010). Comorbid psychiatric disorders associated with Asperger syndrome/high-functioning autism: a community- and clinic-based study. J. Autism Dev. Disord. 40, 1080–1093. doi: 10.1007/s10803-010-0958-2
Miles, J. H., Takahashi, T. N., Bagby, S., Sahota, P. K., Vaslow, D. F., Wang, C. H., et al. (2005). Essential versus complex autism: definition of fundamental prognostic subtypes. Am. J. Med. Genet. A 135, 171–180. doi: 10.1002/ajmg.a.30590
Moessner, R., Marshall, C. R., Sutcliffe, J. S., Skaug, J., Pinto, D., Vincent, J., et al. (2007). Contribution of SHANK3 mutations to autism spectrum disorder. Am. J. Hum. Genet. 81, 1289–1297. doi: 10.1086/522590
Neale, B. M., Kou, Y., Liu, L., Ma’ayan, A., Samocha, K. E., Sabo, A., et al. (2012). Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242–245. doi: 10.1038/nature11011
Nishiyama, M., Oshikawa, K., Tsukada, Y., Nakagawa, T., Iemura, S., Natsume, T., et al. (2009). CHD8 suppresses p53-mediated apoptosis through histone H1 recruitment during early embryogenesis. Nat. Cell Biol. 11, 172–182. doi: 10.1038/ncb1831
Noor, A., Whibley, A., Marshall, C. R., Gianakopoulos, P. J., Piton, A., Carson, A. R., et al. (2010). Disruption at the PTCHD1 Locus on Xp22.11 in Autism spectrum disorder and intellectual disability. Sci. Transl. Med. 2:49ra68. doi: 10.1126/scitranslmed.3001267
O’Roak, B. J., Deriziotis, P., Lee, C., Vives, L., Schwartz, J. J., Girirajan, S., et al. (2011). Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet. 43, 585–589. doi: 10.1038/ng.835
O’Roak, B. J., Stessman, H. A., Boyle, E. A., Witherspoon, K. T., Martin, B., Lee, C., et al. (2014). Recurrent de novo mutations implicate novel genes underlying simplex autism risk. Nat. Commun. 5:5595. doi: 10.1038/ncomms6595
O’Roak, B. J., Vives, L., Girirajan, S., Karakoc, E., Krumm, N., Coe, B. P., et al. (2012). Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250. doi: 10.1038/nature10989
Piton, A., Michaud, J. L., Peng, H., Aradhya, S., Gauthier, J., Mottron, L., et al. (2008). Mutations in the calcium-related gene IL1RAPL1 are associated with autism. Hum. Mol. Genet. 17, 3965–3974. doi: 10.1093/hmg/ddn300
Ronan, J. L., Wu, W., and Crabtree, G. R. (2013). From neural development to cognition: unexpected roles for chromatin. Nat. Rev. Genet. 14, 347–359. doi: 10.1038/nrg3413
Sanders, S. J., Ercan-Sencicek, A. G., Hus, V., Luo, R., Murtha, M. T., Moreno-De-Luca, D., et al. (2011). Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 70, 863–885. doi: 10.1016/j.neuron.2011.05.002
Sanders, S. J., Murtha, M. T., Gupta, A. R., Murdoch, J. D., Raubeson, M. J., Willsey, A. J., et al. (2012). De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237–241. doi: 10.1038/nature10945
Sato, D., Lionel, A. C., Leblond, C. S., Prasad, A., Pinto, D., Walker, S., et al. (2012). SHANK1 deletions in males with autism spectrum disorder. Am. J. Hum. Genet. 90, 879–887. doi: 10.1016/j.ajhg.2012.03.017
Shen, Y., Dies, K. A., Holm, I. A., Bridgemohan, C., Sobeih, M. M., Caronna, E. B., et al. (2010). Clinical genetic testing for patients with autism spectrum disorders. Pediatrics 125, e727–e735. doi: 10.1542/peds.2009-1684
State, M. W., and Levitt, P. (2011). The conundrums of understanding genetic risks for autism spectrum disorders. Nat. Neurosci. 14, 1499–1506. doi: 10.1038/nn.2924
Stefanatos, G. A. (2008). Regression in autistic spectrum disorders. Neuropsychol. Rev. 18, 305–319. doi: 10.1007/s11065-008-9073-y
Stessman, H. A. F., Willemsen, M. H., Fenckova, M., Penn, O., Hoischen, A., Xiong, B., et al. (2016). Disruption of POGZ is associated with intellectual disability and autism spectrum disorders. Am. J. Hum. Genet. 98, 541–552. doi: 10.1016/j.ajhg.2016.02.004
Tammimies, K., Marshall, C. R., Walker, S., Kaur, G., Thiruvahindrapuram, B., Lionel, A. C., et al. (2015). Molecular diagnostic yield of chromosomal microarray analysis and whole-exome sequencing in children with autism spectrum disorder. JAMA 314, 895–903. doi: 10.1001/jama.2015.10078
Tan, T. Y., Dillon, O. J., Stark, Z., Schofield, D., Alam, K., Shrestha, R., et al. (2017). Diagnostic impact and cost-effectiveness of whole-exome sequencing for ambulant children with suspected monogenic conditions. JAMA Pediatr. 171, 855–862. doi: 10.1001/jamapediatrics.2017.1755
Weiss, L. A., Shen, Y., Korn, J. M., Arking, D. E., Miller, D. T., Fossdal, R., et al. (2008). Association between microdeletion and microduplication at 16p11.2 and autism. N. Engl. J. Med. 358, 667–675. doi: 10.1056/NEJMoa075974
Yi, F., Danko, T., Botelho, S. C., Patzke, C., Pak, C., Wernig, M., et al. (2016). Autism-associated SHANK3 haploinsufficiency causes Ih channelopathy in human neurons. Science 352:aaf2669. doi: 10.1126/science.aaf2669
Keywords: autism spectrum disorder, whole exome sequencing, diagnostic yield, comorbidity, genetic etiology
Citation: Du X, Gao X, Liu X, Shen L, Wang K, Fan Y, Sun Y, Luo X, Liu H, Wang L, Wang Y, Gong Z, Wang J, Yu Y and Li F (2018) Genetic Diagnostic Evaluation of Trio-Based Whole Exome Sequencing Among Children With Diagnosed or Suspected Autism Spectrum Disorder. Front. Genet. 9:594. doi: 10.3389/fgene.2018.00594
Received: 01 July 2018; Accepted: 15 November 2018;
Published: 30 November 2018.
Edited by:
Wenbo Zhang, The University of Texas Medical Branch at Galveston, United StatesReviewed by:
Jing Dong, Baylor College of Medicine, United StatesSrinivas Ayyadevara, Central Arkansas Veterans Healthcare System Eugene J. Towbin Healthcare Center, United States
Copyright © 2018 Du, Gao, Liu, Shen, Wang, Fan, Sun, Luo, Liu, Wang, Wang, Gong, Wang, Yu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongguo Yu, eXV5b25nZ3VvQHNoc211LmVkdS5jbg== Fei Li, ZmVpbGlAc2hzbXUuZWR1LmNu
†These authors have contributed equally to this work