 Monnat Pongpanich
Monnat Pongpanich Maturada Patchsung
Maturada Patchsung Apiwat Mutirangura
Apiwat Mutirangura
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 25 October 2018
Sec. Genetics of Aging
Volume 9 - 2018 | https://doi.org/10.3389/fgene.2018.00501
This article is part of the Research TopicBiology of Aging: Impactful Interventions to Extend Health-spanView all 19 articles
Reduction of physiologic replication-independent endogenous DNA double strand breaks (Phy-RIND-EDSBs) in chronological aging yeast increases pathologic RIND-EDSBs (Path-RIND-EDSBs). Path-RIND-EDSBs can occur spontaneously in non-dividing cells without any inductive agents, and they must be repaired immediately otherwise their accumulation can lead to senescence. If yeasts have DSB repair defect, retention of Path-RIND-EDSBs can be found. Previously, we found that Path-RIND-EDSBs are not only produced but also retained in chronological aging yeast. Here, we evaluated if chronological aging yeasts have a DSB repair defect. We found a significant accumulation of Path-RIND-EDSBs around the same level in aging cells and caffeine treated cells and at a much higher level in the DSB repair mutant cells. Especially in the mutant, some unknown sequence was found inserted at the breaks. In addition, % difference of cell viability between HO induced and non-induced cells was significantly greater in aging cells. Our results suggested that RIND-EDSBs repair efficiency declines, but is not absent, in chronological aging yeast which might promote senescence phenotype. When a repair protein is deficient, an alternative pathway might be employed or an end modification process might occur as inserted sequences at the breaks were observed. Restoring repair defects might slow down the deterioration of cells from chronological aging.
We previously proved that pathologic endogenous DNA double-strand breaks (EDSBs) can occur spontaneously, even without exposure to radiation or DNA damaging agents, and were excessively detectable when non-dividing cells had functional double-strand breaks (DSB) repair defects. In other words, they are produced independently of replication, hence we named them pathologic replication-independent EDSBs (Path-RIND-EDSBs) (Thongsroy et al., 2018). They are lethal and must be repaired just as other DNA lesions (Hoeijmakers, 2009). They are associated with a decrease in the cell’s viability (Thongsroy et al., 2013). In addition, the findings that γ-H2AX foci were observed in mice brains supported the existence of Path-RIND-EDSBs in non-dividing cells (Barral et al., 2014; Hare et al., 2018). When non-dividing cells age, they are produced in large numbers, possibly due to a reduction of Physiologic RIND-EDSBs (Phy-RIND-EDSBs) another type of RIND-EDSBs. Phy-RIND-EDSBs, in contrast to Path-RIND-EDSBs, were shown to possess a functional role as epigenetic markers as they affected genome integrity and were generated by non-random mechanism (Pornthanakasem et al., 2008; Thongsroy et al., 2018). Previous studies showed evidence that a reduction of Phy-RIND-EDSBs decreased cell viability and promoted genome instability (Thongsroy et al., 2018). In addition, they are preferentially located in hypermethylated genome regions, were retained in facultative heterochromatin in human cells, preferentially occurred immediately after certain 4-bp sequence and were prevented from occurring after certain 4-bp in yeast (Kongruttanachok et al., 2010; Pongpanich et al., 2014).
RIND-EDSBs can be divided into three groups: odds ratio (OR) > 1 breaks, OR ≤ 1 breaks and modified ends with insertion at the breaks (MIB) (Thongsroy et al., 2018). OR > 1 breaks are breaks that were found more frequently in wild type; therefore, they represent Phy-RIND-EDSBs. OR ≤ 1 breaks are breaks that were unlikely to be found in wild type. MIB are breaks that had a certain sequence inserted. In yeast, JKM179 treated with caffeine (a repair inhibitor), OR ≤ 1 breaks and MIB increased (Thongsroy et al., 2018). Therefore, OR ≤ 1 breaks and MIB represent Path-RIND-EDSBs.
In our previous work, we found that in a condition where cells possessed a low level of Phy-RIND-EDSBs and were treated with chemical to inhibit repair (caffeine), Path-RIND-EDSBs and viability reduction was significantly greater than cells with only low level of Phy-RIND-EDSBs (Thongsroy et al., 2018). We have shown that aging cells have a low level of Phy-RIND-EDSBs which cause a high level of Path-RIND-EDSBs. Therefore, if aging cells are decreased in DSB repair function, production and retention of Path-RIND-EDSBs can promote DNA damage response leading to cellular senescence.
The general notion has been that DNA repair machinery is becoming more compromised and error-prone with age (Gorbunova et al., 2007; Li and Vijg, 2012). Major DNA repair pathways are base excision repair (BER), nucleotide excision repair (NER), mismatch repair (MMR), and double-strand break repair (DSBR). The repair pathway used depends on the type of lesion (Maynard et al., 2015). BER repairs DNA lesions such as oxidation products generated by ROS; NER removes bulky lesions; MMR removes mismatches in DNA and DSBR works on double-strand break (Gredilla, 2011). Numerous studies have found a significant decline of all four major repair pathways activities: BER (Intano et al., 2002, 2003; Zhang et al., 2010; Løhr et al., 2015; Xu et al., 2015), NER (Goukassian et al., 2000; Boyle et al., 2005; Yamada et al., 2006), MMR (Yehuda et al., 2001; Neri et al., 2005) and DSBR (Seluanov et al., 2004; Ju et al., 2006; Vyjayanti and Rao, 2006; Mao et al., 2012; Vaidya et al., 2014; Li et al., 2016; Delabaere et al., 2017) with aging. Increasing evidence suggests that deficiencies in BER, NER, and DSBR indeed induce aging-associated phenotypes (Borgesius et al., 2011; Pan et al., 2016; Guedj et al., 2017). Werner syndrome patients with an inherited defect in BER show features of premature aging (Pan et al., 2016). Reduction of the activity of enzymes involved in mitochondrial BER has been shown to promote aging (Pan et al., 2016). Defects in NER cause three distinct premature aging disorders in human: xeroderma pigmentosum, Cockayne’s syndrome and trichothiodystrophy (Coppede and Migliore, 2010). Trichothiodystrophy (TTD) mice, defective in NER, exhibit phenotypes associated with advanced aging, including osteoporosis, osteosclerosis, gray hair, cachexia, and reduced life-span (de Boer et al., 2002). Xpdm/m mice, which have a partial defect in both GG-NER and TC-NER, exhibited prominent premature aging features and about 20% reduction in lifespan (Dollé et al., 2006). Mice with a defect in Ku80 (Ku86), a NHEJ gene, exhibit an early onset of aging characteristics (Hasty et al., 2003).
Many studies have found a significant decline of double-strand break repair (DSBR) activities with aging (Seluanov et al., 2004; Ju et al., 2006; Vyjayanti and Rao, 2006; Mao et al., 2012; Vaidya et al., 2014; Li et al., 2016; Delabaere et al., 2017). Two major mechanisms of DSBR are homologous recombination (HR) and non-homologous end joining (NHEJ). HR repair has been shown to decline with increasing replicative age and can be restored by supplementation with SIRT6 (Mao et al., 2012). Rad51 does not change in expression level with age (Li et al., 2016); however, age-related HR defects are possibly caused by malfunctions of steps after Rad51 recruitment (Delabaere et al., 2017). NHEJ efficiency declines with age (Seluanov et al., 2004). End joining activity became less efficient (Vyjayanti and Rao, 2006) and its fidelity declined with age as it was associated with extended deletions and insertions in old cells (Seluanov et al., 2004; Vaidya et al., 2014). In addition, the frequency of microhomology-mediated end joining (MMEJ) increased with age, suggesting that as NHEJ declined in function it was compensated by MMEJ (Vaidya et al., 2014). It has been shown that declining DSB repair is associated with decreased expression of Ku70 and Mre11 (Ju et al., 2006). Another study showed that restoration of XRCC4 and Lig4 significantly promotes the NHEJ fidelity and efficiency in aged fibroblasts (Li et al., 2016). Although there are many studies on aged-related changes in DSB repair, DSB repair defect in chronological aging cell has not been extensively studied.
In this work, we aim to find evidence that DSB repair defect occurs in chronological aging yeast and in turn causes retention of Path-RIND-EDSBs. To study age-related changes in RIND-EDSBs repair, we perform sequencing at RIND-EDSBs in cells treated with caffeine, repair deficient mutant yeast and chronologically aging yeast.
Yeast strains used in this study were BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) as wild type, mec1Δ (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 mec1Δ sml1Δ), mre11Δ (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 mre11Δ::KanMX), nej1Δ (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 nej1Δ::KanMX), rad51Δ (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 rad51Δ::KanMX), tel1Δ (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 tel1Δ::KanMX), yku70Δ (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 yku70Δ::KanMX), yku80Δ (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 yku80Δ::KanMX), nhp6aΔ (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 nh6aΔ::KanMX) and JKM179 (MATα Δho hmlΔ::ADE1 hmrΔ::ADE1 lys5 leu2 ura3 trp1ade3::GALHO) from the same sources as previously described (Thongsroy et al., 2013; Pongpanich et al., 2014). Asynchronous yeast cultures were grown in YPD (Sigma, United States) until an optical density (OD) at 600 nm reached 0.4–0.6 (cell density ∼0.53–0.87 × 107 cells/ml) then yeast cells were arrested at G0 phase by culturing in YP medium containing 2% raffinose (Sigma, United States) for 2 days. Cells in the G0 phase that were small and without buds were confirmed by their morphology under a microscope. All cells of all yeast strains consisted of G0 phase cells (100% unbudded stationary-phase cells). Cell viability in all experiments was monitored by following these steps. First, cell density of yeast cultures was determined by measuring OD600. This value was used to calculate the volume required for making 1 ml of 106 cells/ml dilute solution. Then ten-fold serial dilutions was performed to produce 1 ml of 103 cells/ml solution. An aliquot of 100 μl of 103 cells/ml solution (approximately 100 cells) was plated on a YPD plate and counting the number of visible colony forming units (CFUs). To inhibit the ATM/ATR-dependent DNA repair pathways, yeast cells were treated with 10 mM caffeine (Sigma, United States) for 24 h (Moser et al., 2000).
For the two chronological aging experiments (experiment 1: wild type and wild type day 50; experiment 2: wild type, yku70Δ, nej1Δ and nhp6aΔ), yeast cells were grown in YP + 2% glucose at 30°C 250 rpm overnight and then switched to YP + 2% raffinose for 2 days before being washed and resuspended in sterile deionized water. The cell pellets were collect at the first day in water (day 1) for all strains, the next 10 days (day 10) for wild type, yku70Δ, nej1Δ and nhp6aΔ and the next 50 days (day 50) for wild type day 50. % cell viability was calculated using the following formula (Figure 3)
For the HO induction experiment, JKM179 was used (Kim et al., 2007). Expression of the HO endonuclease was induced by the addition of galactose to the media for 3 h to produce a DSB in the yeast genome. After induction, cells were placed in media containing 2% glucose to repress HO expression and washed with sterile deionized water. Approximately 1 × 108 cells/ml were resuspended in sterile deionized water (0 hr) and maintained at 30°C in a rotary shaker at 250 rpm until the end of the experiment. After HO induction, cell viability was measured by plating an aliquot of the culture on a YPD plate and counting the number of visible colony forming units (CFUs). Cell viability was monitored every 10 days until 30 days. % cell viability was calculated using the following formula (Figure 1A)
% difference of cell viability between induced and non-induced cells (control) was calculated from (Figure 1B)
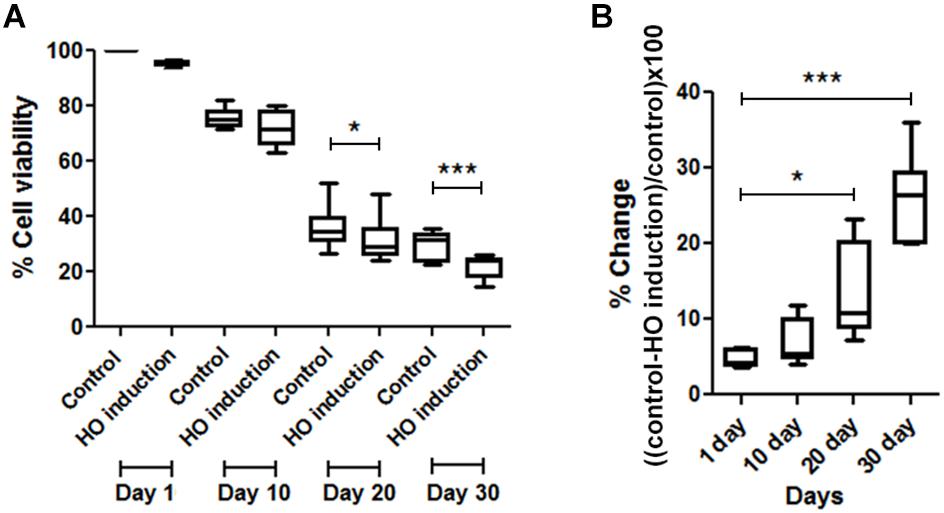
FIGURE 1. Comparisons of cell viability between induced and non-induced cells every 10 days. (A) Percent viability in JKM179 with and without induction every 10 days. There was a significant difference of % cell viability between control and induced cells at day 20 (∗P < 0.05) and day 30 (∗∗∗P < 0.001) (B) % difference of cell viability between JKM179 with and without induction HO induction every 10 days. There was a significant increase between day 1 and day 20 (∗P < 0.05) and day 1 and day 30 (∗∗∗P < 0.001) of % difference of cell viability between induced and non-induced cells.
High Molecular weight DNA preparation was performed as previously described (Pongpanich et al., 2014). In brief, yeast cells were treated with 1 mg/ml lyticase for yeast cell wall degradation, mixed with 1% low melting point agarose and embedded in plug molds. The yeast cell plugs were digested in a cell membrane digesting buffer, treated with T4 DNA polymerase to polish the cohesive end EDSBs. Then we performed the first linker ligation, digested the first linker ligated DNA with RsaI and ligated again with the second linkers.
Library preparation and EDSB sequencing were performed as previously described (Pongpanich et al., 2014). In brief, to detect RIND-EDSBs, the first linker was ligated to one of the EDSBs end. Note that, because the size of the first linker-ligated DNA was long, we added RsaI, which does not cut within the linker, to digest the first linker-ligated DNA. After digestion, the second linker is ligated. Therefore, the intact sequence would contain the first linker, the DNA sequence and the second linker. HMW DNA with two-linker ligation was then subjected to PCR and sequenced on an Ion Torrent sequencer (Ion TorrentTM Personal Genome Machine® (PGM), Life Technologies, United States).
There are two linkers: the first is ligated to EDSBs and the second is ligated to RsaI digested DNA. The first linker is composed of two parts: 5′ end have the same sequence as primer for amplification and 3′ end is a unique sequence. For each sample, we extracted the sequence following the intact 3′ end of first linker sequence by first aligned reads and their reverse complement against the first linker sequence to determine the strand. The aligned position was used to determine the position that sequence was extracted. As there was a heterogeneity in the PCR product, we retained only reads with intact 3′ end of the first linker sequence. Only reads with intact 3′ end of first linker sequence contained RIND-EDSBs, which occur between the last base of linker sequence and its next base. Then we aligned the retained reads against the Saccharomyces cerevisiae strain B4741 reference genome, downloaded from the Saccharomyces Genome Database (SGD) using Nucleotide-Nucleotide BLAST 2.3.0+ (Altschul et al., 1990). Thus the first base of aligned reads corresponded to the position after breaks.
Using the BLAST results, we divided reads into two groups based on the position of the first base that got mapped. The first group contained reads that got mapped from the first base and the second group contained reads that were not mapped from the first base. For the second group, we interpret the unaligned portion as an inserted sequence at the break and called them modified ends with insertion at the breaks (MIB). The unmapped portion is extracted and shown in Supplementary Tables 1–10. Regardless of the mapped position of the first base, reads could be uniquely mapped or mapped to multiple locations (multi-mapped reads). We retained multi-mapped reads if the prior four bases of 90% of the mapped location were all the same.
We counted the number of reads that could not be mapped from the first base and quantified them as MIB. Then we extracted the 4-bp sequence immediately prior to the breaks for each read, whether those reads were mapped from the first base or not. Then we counted the reads by prior 4-bp for all possible combinations of 4-bp (i.e., AAAA, AAAC and so on, denoted by XXXX) for reads mapped from the first base and did the same thing for reads not mapped from first base in all samples.
Next, we counted the occurrence of each possible combination of 4-bp sequence in the reference genome. Two hundred and fifty-six 2 × 2 contingency tables were then constructed for wild type only, one table per one 4-bp sequence. For each table the first column represents the specific 4-bp sequence; the second column represents all other possible 4-bp sequences. The first row represents the number of reads mapped from first base in our sample; the second row the number of occurrence in the reference genome. The tables were formulated as below.

where XXXX represents each possible combination of 4-bp (i.e., AAAA, AAAC and so on) resulting in 256 tables in total.
OR was computed from these contingency tables using
Any break in wild type which had prior 4-bp sequence with OR > 1 was regarded as an OR > 1 break and any 4-bp sequence with OR ≤ 1 was regarded as an OR ≤ 1 break. A percentage of reads that had the same prior 4-bp sequences as sequences in OR > 1 breaks and OR ≤ 1 in wild type was calculated in all samples (Figure 2).
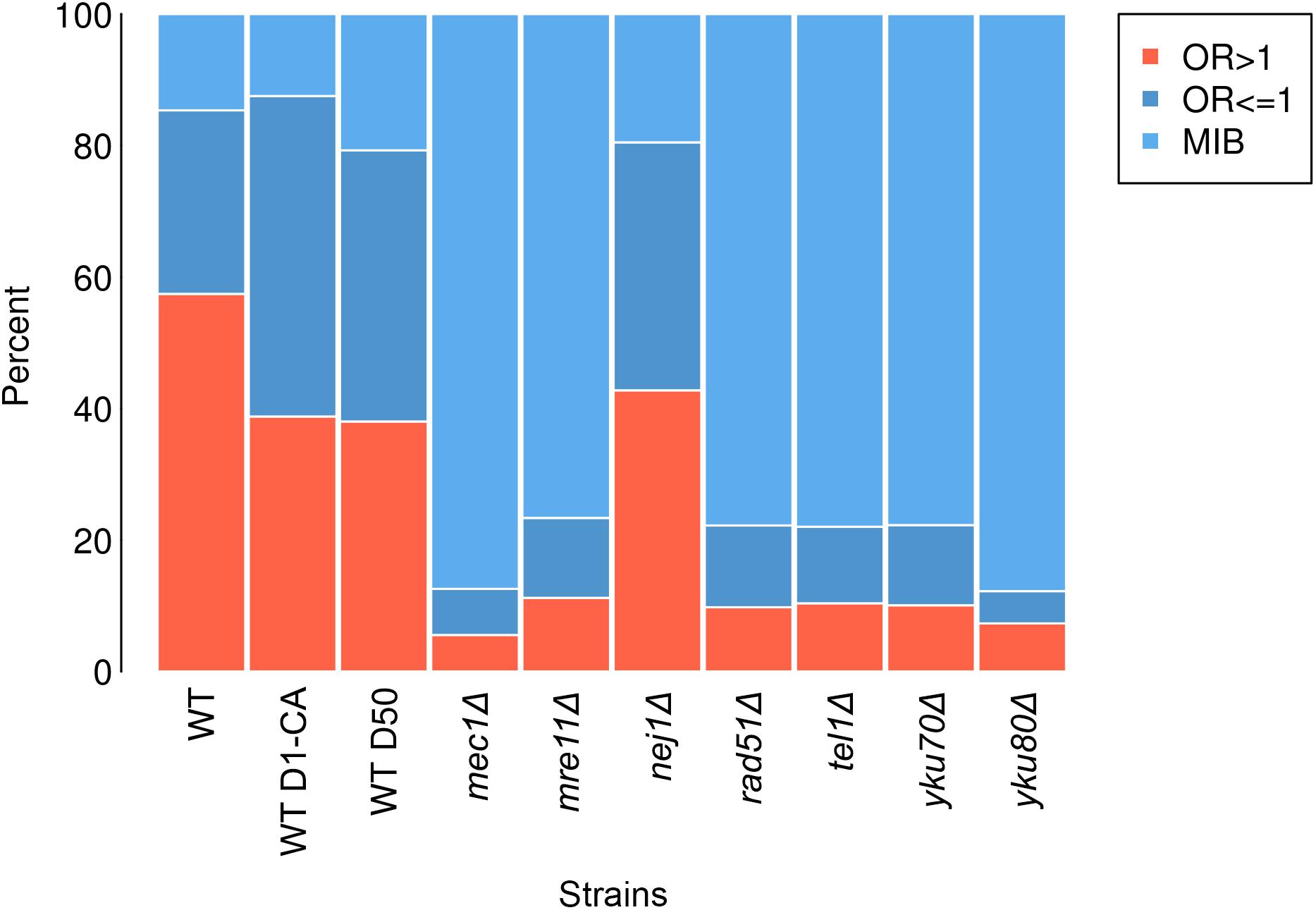
FIGURE 2. Percentage of each break type—OR > 1 breaks (salmon), OR ≤ 1 breaks (dark blue) and MIB (light blue) in wild type (WT), wild type treated with caffeine (WT D1-CA), wild type day 50 (WT D50), mec1Δ, mre11Δ, nej1Δ, rad51Δ, tel1Δ, yku70Δ, and yku80Δ.
To determine whether having repair defect increased the number of OR ≤ 1 breaks and MIB significantly, we performed Fisher’s exact tests (Table 1). We calculated nine 2 × 2 contingency tables considering the number of MIB and OR ≤ 1 breaks (first column) and the number of OR > 1 breaks (second column) in each of the samples (first row) compared to the same values in wild type (second row). The tables were formulated as below.

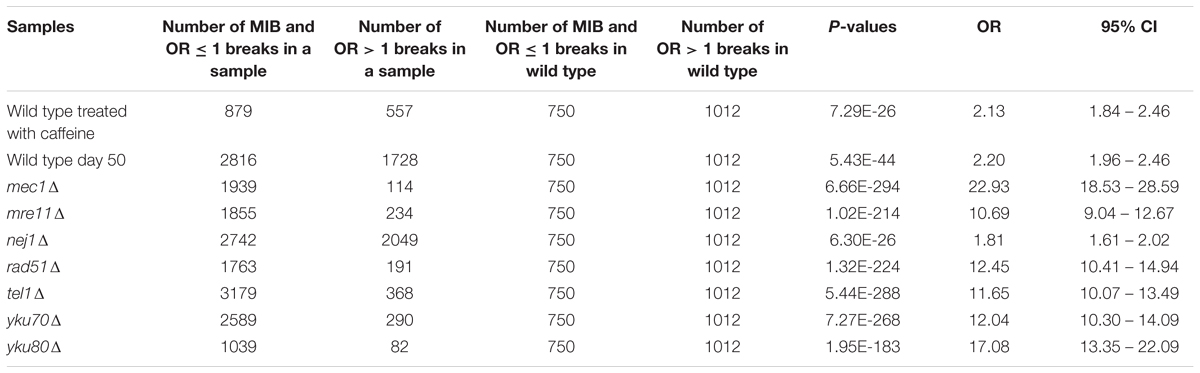
TABLE 1. Fisher’s exact test P-values for determining any association between number of MIB and OR ≤ 1 breaks and repair defect.
To determine whether MIB occurrence was associated with types of breaks, we performed Fisher’s exact tests from these contingency tables (Table 2). We calculated ten 2 × 2 contingency tables where A and B were the number of OR ≤ 1 breaks and OR > 1 breaks in reads not mapped from first base, respectively, C and D were the number of the number of OR ≤ 1 breaks and OR > 1 breaks in reads mapped from first base, respectively. Fisher’s exact test was then computed from these contingency tables.
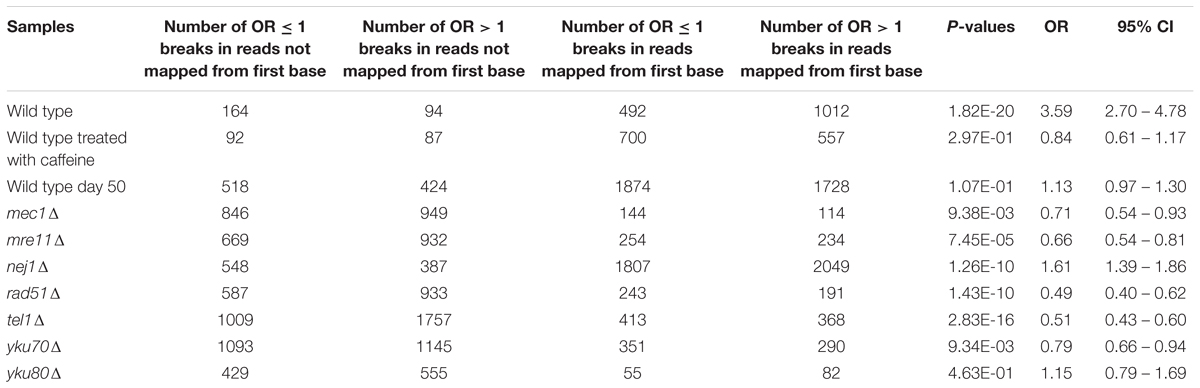
TABLE 2. Fisher’s exact test P-values for determining any association between MIB occurrence and types of breaks.
Statistical analyses were performed using Student’s t-test for data with a normal distribution or a Mann–Whiney test for data that did not have a normal distribution, to determine (1) if % cell viability was significantly different between control and induced cells at each day (Figure 1A), (2) if the % differences were significantly different between day 10 vs. day 1, day 20 vs. day 1 and day 30 vs. day 1 (Figure 1B) and (3) if % viability was significantly different between wild type and each mutant (Figure 3).
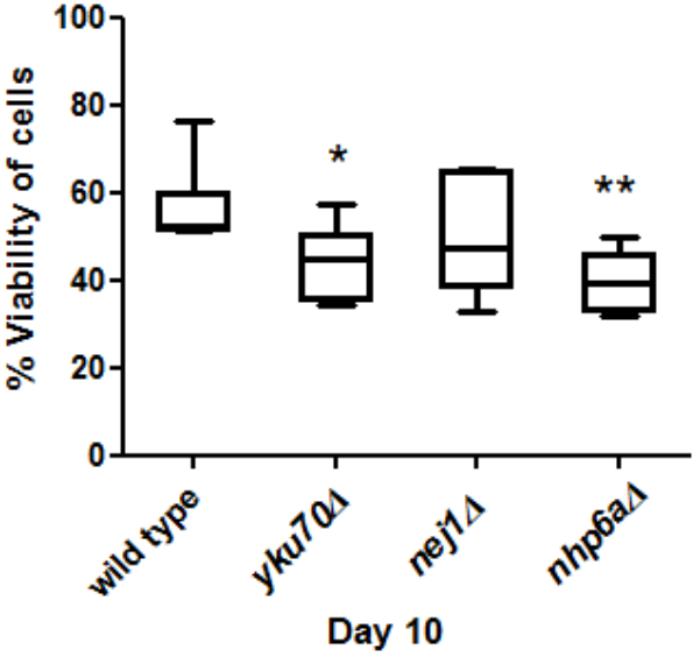
FIGURE 3. Comparisons of cell viability in yku70Δ, nej1Δ, nhp6aΔ and wild type at day 10. There was a significant decrease between yku70Δ and wild type (∗P < 0.05) and nhp6aΔ and wild type (∗∗P < 0.01).
To show that chronological aging yeast has declined DNA repair capacity, we induced a break every 10 days in JKM179 by HO induction and observed whether those cells could repair the break by measuring the viability compared to control (cells with no induction). We found a significant % difference in viability between HO-induced JKM179 cells and the control cells (JKM179 cells with no induction) at days 20 and 30 (Figure 1A) and that % differences were significantly different between day 20 vs. day 1 and day 30 vs. day 1 (p-value < 0.05 and 0.001, respectively) (Figure 1B). Hence, we observed that aging cells are more sensitive to HO induction.
To explore if Path-RIND-EDSBs increased when DNA repair is defective, we looked at the proportion of the three break types, OR > 1 breaks, OR ≤ 1 breaks and MIB (see OR > 1 Breaks, OR ≤ 1 Breaks and MIB Calculation) in wild type, wild type treated with caffeine, wild type day 50, and seven repair defect yeast strains: mec1Δ, mre11Δ, nej1Δ, rad51Δ, tel1Δ, yku70Δ, and yku80Δ. The percentage of OR ≤ 1 breaks and MIB is significantly increased in all samples when compared to wild type (Figure 2 and Table 1). Wild type treated with caffeine and wild type day 50 had about the same increment of OR ≤ 1 breaks and MIB. All mutant except nej1Δ had similar increase of MIB which was much higher than both wild type treated with caffeine and wild type day 50. Therefore, we conclude that Path-RIND-EDSBs (OR ≤ 1 breaks and MIB) are increased in DNA repair defect cells.
We examined if there was an association between MIB and types of breaks (OR > 1 breaks and OR ≤ 1 breaks) (see Statistical Analyses). Results showed that the odds of MIB occurring at OR ≤ 1 breaks is 3.59 higher than no MIB in wild type (Table 2). On the contrary, in mec1Δ, mre11Δ, rad51Δ, tel1Δ, yku70Δ, the odds of MIB occurring at OR ≤ 1 breaks is lower than no MIB (OR = 0.71, 0.66, 0.49, 0.51, and 0.78, respectively, Table 2). In cells treated with caffeine, wild type day 50 and yku80Δ, MIB did not preferentially occur in either types of breaks. Therefore, we conclude that MIB tend to occur more frequently at Path-RIND-EDSBs in normal condition but occur at Phy-RIND-EDSBs in DNA repair defect.
Viability was used as a measure of aging to test if mutant strains aged faster than wild type. We grew yku70Δ, nej1Δ, nhp6aΔ and wild type cells for 10 days and then measured their viability at day 10. We found that the viability of nhp6aΔ and yku70Δ significantly decreased (p < 0.01 and p < 0.05, respectively) compared to wild type but not nej1Δ (Figure 3).
We found that chronological aging yeast has impaired DNA repair and found the accumulation of Path-RIND-EDSBs – as OR ≤ 1 breaks and MIB – increased in cells with DSB repair defects. Wild type treated with caffeine in this study had similar results to JKM179 treated with caffeine in the study of Thongsroy et al. (2018) in terms of the increment of proportion of OR ≤ 1 breaks and MIB. In mutant yeast with impaired DNA repair, Path-RIND-EDSBs, specifically MIB, increased even greater than cells treated with caffeine (DNA repair is defect by chemical). The results of wild type day 50 was similar to cells treated with caffeine. When we performed HO induction every 10 days, we found that % difference of cell viability between induced and non-induced cells increased significantly in aging cells compared to day 1 cells. These two results provide evidence that chronological aging cells may have DSB repair defect: firstly, RIND-EDSBs level in aging occur in the same direction as cells with repair inhibition; secondly, a reduction of viability of HO-induced cells compared to control increased as cells age.
DSB repair defects in chronological aging yeast may give rise to senescence phenotype by retention of Path-RIND-EDSBs. Pathologic EDSBs is a crisis state that requires immediate repair for cells. In response to DSB, players in damage response are activated, e.g., H2AX, p21, and p53 (Kulaberoglu et al., 2016; Ji et al., 2017). p53 triggers cell cycle arrest and promotes DSB repair. If damage cannot be repaired, apoptosis and/or senescence occurs (d’Adda di Fagagna, 2008), therefore, accumulation of DSBs can trigger senescence (Bielak-Zmijewska et al., 2018). We found that aging cells had Path-RIND-EDSBs retention due to DSB repair defect. Thus, we posit that DSB repair defect leads to senescence.
We postulate that a decline in repair in aging is due to the exhaustion of substrate of DNA repair. We previously found that Phy-RIND-EDSBs levels decreased in chronological aging yeast and associated with viability (Pearson correlation coefficient = 0.94) i.e., the lower the level of Phy-RIND-EDSBs, the lower the cells viability. This in turn increased the Path-RIND-EDSBs level. We observed an accumulation of Path-RIND-EDSBs in aging yeast cells (Thongsroy et al., 2018). Therefore, aging yeast cells have to repair Path-RIND-EDSBs more than younger cells. As cells age, more DNA repair machinery is required and DNA repair substrates are depleted resulting in DNA repair defect.
Aging cells have a decreased DNA repair efficiency; however, the repair mechanisms are not completely absent. While mutant strains had a markedly increase of MIB, aging cells had proportion of each breaks type in similar to caffeine treated cells. The number of MIB increased in aging cells but not at the same level as with mutant strains. This suggested that when cells are depleted of one of the repair proteins, they use an alternate repair pathway or their repair is compensated by some mechanism resulting in numerous MIB. Our results implied that MIB might be a mechanism that compensates repair defect (Table 2). We observed that MIB tended to occur at Path-RIND-EDSBs in wild type while the opposite was seen in repair defect cells: i.e., MIB tended to occur at Phy-RIND-EDSBs. This indicates that in normal condition, MIB might be an alternative pathway for Path-RIND-EDSBs, while Phy-RIND-EDSBs might have a specific mechanism to avoid MIB since they are retained in cells and need not be repaired immediately. However, when Phy-RIND-EDSBs cannot be repaired due to repair defect, MIB might be an alternative pathway as Phy-RIND-EDSBs must be repaired too. Another possibility is MIB might be an end modification process that occurred after DSB repair defect. This end modification process might leads to cell death i.e., its role might be to tag the damage sites to be recognized by certain apoptosis pathway, similar to ubiquitin tagging in protein degradation pathway (Pickart, 2004). Another possibility is the end modification process might prevent cell death i.e., its role might be to disguise Phy-RIND-EDSBs from apoptosis pathway just as telomere ends that were protected from damage response pathway (de Lange, 2009).
Nej1 was either not involved in repairing RIND-EDSBs or it might have other proteins with a redundancy role. Our results showed that its Path-RIND-EDSBs level increased slightly: nej1Δ results were very different from other mutant strains and this was in accord with (Thongsroy et al., 2013). They observed no change in RIND-EDSBs level in nej1Δ but the level was significantly increased in mec1Δ, tel1Δ, mre11Δ, rad51Δ, yku70Δ and yku80Δ. Therefore, DSB repair complex of Path-RIND-EDSBs might be different from other pathologic DSBs. Results from Yang et al. (2015) perhaps support the possibility that Nej1 is compensatory. Nej1 together with Dnl4/Lif1 carried out DNA end joining (Pannunzio et al., 2014). Both Nej1 and Dnl4/Lif1 additively but independently recruit end processing factors, Pol4 and Rad27. They found that no recruitment of Pol4 and Rad27 was observed in lif1Δnej1Δ. However, loss of either Nej1 or Dnl4/Lif1 reduced the rate of recruitment. They also showed that flap cleavage and ligation product were detected in reactions that lack Nej1. However, when Nej1 was added, the product was enhanced, indicating a stimulatory effect of Nej1.
Our finding that yku70Δ and nhp6aΔ aged faster than wild type is in agreement with some other studies. Ju et al. (2006) showed that expression of Ku70 in the longevity group was higher than in control group suggesting that people who possess higher level of Ku70 expression have a longer life span. Li et al. (2007) found that Ku70, Ku80 mutant and double-mutant mice exhibit early aging with very low cancer levels. Giavara et al. (2005) found that nhp6a/b mutant strain had a significantly shorter lifespan than NHP6A/B. The results of nej1Δ that a significant decrease of viability at day 10 was not observed is perhaps consistent with our other finding that pathologic RIND-EDSBs level increased slightly in nej1Δ.
Our data showed an accumulation of Path-RIND-EDSBs in aging cells and cells with repair defects either chemically defected (caffeine) or genetically defected (mutant strains). In addition, a more pronounced increment of % difference of cell viability in aging cells was observed when cells were induced with HO. These support the conclusion that chronological aging yeast might have impaired DNA repair. MIB is employed to process Path-RIND-EDSBs in normal condition; however, it is used to handle Phy-RIND-EDSBs if repair is defective. Since DSB repair defect might lead to senescence phenotype, restoring repair defects is an important aim to slow down deterioration of cells from chronological aging.
The datasets analyzed for this study can be found in the Sequence Read Archive under the accession numbers SRP095646.
MoP performed all of the bioinformatics analyses and wrote the manuscript. MaP prepared the samples for sequencing and helped to draft the manuscript. AM conceived and designed the study, analyzed the data, and edited the manuscript. All the authors have read and approved the final manuscript.
This work was supported by the Thailand Research Fund (TRG5880214 and DPG5980005); Grants for Development of New Faculty Staff, Ratchadaphiseksomphot Endowment Fund, Chulalongkorn University (GDNS 59-009-23-005); Anantara Siam Bangkok Hotel and Four Seasons Bangkok Hotel Care for Cancer Fun Run in coordination with the Thai Red Cross Society; and Postdoctoral Fellowship 2018 under Ratchadapisek Somphot Fund, Chulalongkorn University. Funding for open access charge: Chulalongkorn University.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2018.00501/full#supplementary-material
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Barral, S., Beltramo, R., Salio, C., Aimar, P., Lossi, L., and Merighi, A. (2014). Phosphorylation of histone H2AX in the mouse brain from development to senescence. Int. J. Mol. Sci. 15, 1554–1573. doi: 10.3390/ijms15011554
Bielak-Zmijewska, A., Mosieniak, G., and Sikora, E. (2018). Is DNA damage indispensable for stress-induced senescence? Mech. Ageing Dev. 170, 13–21. doi: 10.1016/j.mad.2017.08.004
Borgesius, N. Z., de Waard, M. C., van der Pluijm, I., Omrani, A., Zondag, G. C., van der Horst, G. T., et al. (2011). Accelerated age-related cognitive decline and neurodegeneration, caused by deficient DNA repair. J. Neurosci. 31, 12543–12553. doi: 10.1523/JNEUROSCI.1589-11.2011
Boyle, J., Kill, I. R., and Parris, C. N. (2005). Heterogeneity of dimer excision in young and senescent human dermal fibroblasts. Aging Cell 4, 247–255. doi: 10.1111/j.1474-9726.2005.00167.x
Coppede, F., and Migliore, L. (2010). DNA repair in premature aging disorders and neurodegeneration. Curr. Aging Sci. 3, 3–19. doi: 10.2174/1874609811003010003
d’Adda di Fagagna, F. (2008). Living on a break: cellular senescence as a DNA-damage response. Nat. Rev. Cancer 8, 512–522. doi: 10.1038/nrc2440
de Boer, J., Andressoo, J. O., de Wit, J., Huijmans, J., Beems, R. B., van Steeg, H., et al. (2002). Premature aging in mice deficient in DNA repair and transcription. Science 296, 1276–1279. doi: 10.1126/science.1070174
de Lange, T. (2009). How telomeres solve the end-protection problem. Science 326, 948–952. doi: 10.1126/science.1170633
Delabaere, L., Ertl, H. A., Massey, D. J., Hofley, C. M., Sohail, F., Bienenstock, E. J., et al. (2017). Aging impairs double-strand break repair by homologous recombination in Drosophila germ cells. Aging Cell 16, 320–328. doi: 10.1111/acel.12556
Dollé, M. E. T., Busuttil, R. A., Garcia, A. M., Wijnhoven, S., van Drunen, E., Niedernhofer, L. J., et al. (2006). Increased genomic instability is not a prerequisite for shortened lifespan in DNA repair deficient mice. Mutat. Res. 596, 22–35. doi: 10.1016/j.mrfmmm.2005.11.008
Giavara, S., Kosmidou, E., Hande, M. P., Bianchi, M. E., Morgan, A., d’Adda di Fagagna, F., et al. (2005). Yeast Nhp6A/B and mammalian Hmgb1 facilitate the maintenance of genome stability. Curr. Biol. 15, 68–72. doi: 10.1016/j.cub.2004.12.065
Gorbunova, V., Seluanov, A., Mao, Z., and Hine, C. (2007). Changes in DNA repair during aging. Nucleic Acids Res. 35, 7466–7474. doi: 10.1093/nar/gkm756
Goukassian, D., Gad, F., Yaar, M., Eller, M. S., Nehal, U. S., and Gilchrest, B. A. (2000). Mechanisms and implications of the age-associated decrease in DNA repair capacity. FASEB J. 14, 1325–1334. doi: 10.1096/fasebj.14.10.1325
Gredilla, R. (2011). DNA damage and base excision repair in mitochondria and their role in aging. J. Aging Res. 2011:257093. doi: 10.4061/2011/257093
Guedj, A., Geiger-Maor, A., Benyamini, H., Nevo, Y., Elgavish, S., Galun, E., et al. (2017). Correction: early age decline in DNA repair capacity in the liver: in depth profile of differential gene expression. Aging 9:1640. doi: 10.18632/aging.101242
Hare, B. D., Thornton, T. M., Rincon, M., Golijanin, B., King, S. B., Jaworski, D. M., et al. (2018). Two weeks of variable stress increases gamma-H2AX levels in the mouse bed nucleus of the stria terminalis. Neuroscience 373, 137–144. doi: 10.1016/j.neuroscience.2018.01.024
Hasty, P., Campisi, J., Hoeijmakers, J., van Steeg, H., and Vijg, J. (2003). Aging and genome maintenance: lessons from the mouse? Science 299, 1355–1359. doi: 10.1126/science.1079161
Hoeijmakers, J. H. (2009). DNA damage, aging, and cancer. N. Engl. J. Med. 361, 1475–1485. doi: 10.1056/NEJMra0804615
Intano, G. W., Cho, E. J., McMahan, C. A., and Walter, C. A. (2003). Age-related base excision repair activity in mouse brain and liver nuclear extracts. J. Gerontol. A Biol. Sci. Med. Sci. 58, B205–B211. doi: 10.1093/gerona/58.3.B205
Intano, G. W., McMahan, C. A., McCarrey, J. R., Walter, R. B., McKenna, A. E., Matsumoto, Y., et al. (2002). Base excision repair is limited by different proteins in male germ cell nuclear extracts prepared from young and old mice. Mol. Cell. Biol. 22, 2410–2418. doi: 10.1128/MCB.22.7.2410-2418.2002
Ji, J., Zhang, Y., Redon, C. E., Reinhold, W. C., Chen, A. P., Fogli, L. K., et al. (2017). Phosphorylated fraction of H2AX as a measurement for DNA damage in cancer cells and potential applications of a novel assay. PLoS One 12:e0171582. doi: 10.1371/journal.pone.0171582
Ju, Y. J., Lee, K. H., Park, J. E., Yi, Y. S., Yun, M. Y., Ham, Y. H., et al. (2006). Decreased expression of DNA repair proteins Ku70 and Mre11 is associated with aging and may contribute to the cellular senescence. Exp. Mol. Med. 38, 686–693. doi: 10.1038/emm.2006.81
Kim, J. A., Kruhlak, M., Dotiwala, F., Nussenzweig, A., and Haber, J. E. (2007). Heterochromatin is refractory to γ-H2AX modification in yeast and mammals. J. Cell Biol. 178, 209–218. doi: 10.1083/jcb.200612031
Kongruttanachok, N., Phuangphairoj, C., Thongnak, A., Ponyeam, W., Rattanatanyong, P., Pornthanakasem, W., et al. (2010). Research replication independent DNA double-strand break retention may prevent genomic instability. Mol. Cancer 9:70. doi: 10.1186/1476-4598-9-70
Kulaberoglu, Y., Gundogdu, R., and Hergovich, A. (2016). Chapter 15 - The Role of p53/p21/p16 in DNA-Damage Signaling and DNA Repair. Boston: Academic Press, 243–256.
Li, H., Vogel, H., Holcomb, V. B., Gu, Y., and Hasty, P. (2007). Deletion of Ku70, Ku80, or both causes early aging without substantially increased cancer. Mol. Cell. Biol. 27, 8205–8214. doi: 10.1128/MCB.00785-07
Li, W., and Vijg, J. (2012). Measuring genome instability in aging – a mini-review. Gerontology 58, 129–138. doi: 10.1159/000334368
Li, Z., Zhang, W., Chen, Y., Guo, W., Zhang, J., Tang, H., et al. (2016). Impaired DNA double-strand break repair contributes to the age-associated rise of genomic instability in humans. Cell Death Differ. 23, 1765. doi: 10.1038/cdd.2016.65
Løhr, M., Jensen, A., Eriksen, L., Grønbæk, M., Loft, S., and Møller, P. (2015). Association between age and repair of oxidatively damaged DNA in human peripheral blood mononuclear cells. Mutagenesis 30, 695–700. doi: 10.1093/mutage/gev031
Mao, Z., Tian, X., Van Meter, M., Ke, Z., Gorbunova, V., and Seluanov, A. (2012). Sirtuin 6 (SIRT6) rescues the decline of homologous recombination repair during replicative senescence. Proc. Natl. Acad. Sci. U.S.A. 109, 11800–11805. doi: 10.1073/pnas.1200583109
Maynard, S., Fang, E. F., Scheibye-Knudsen, M., Croteau, D. L., and Bohr, V. A. (2015). DNA damage, DNA repair, aging, and neurodegeneration. Cold Spring Harb. Perspect. Med. 5:a025130. doi: 10.1101/cshperspect.a025130
Moser, B. A., Brondello, J. M., Baber-Furnari, B., and Russell, P. (2000). Mechanism of caffeine-induced checkpoint override in fission yeast. Mol. Cell. Biol. 20, 4288–4294. doi: 10.1128/MCB.20.12.4288-4294.2000
Neri, S., Gardini, A., Facchini, A., Olivieri, F., Franceschi, C., Ravaglia, G., et al. (2005). Mismatch repair system and aging: microsatellite instability in peripheral blood cells from differently aged participants. J. Gerontol. A Biol. Sci. Med. Sci. 60, 285–292. doi: 10.1093/gerona/60.3.285
Pan, M. R., Li, K., Lin, S. Y., and Hung, W. C. (2016). Connecting the dots: from DNA damage and repair to aging. Int. J. Mol. Sci. 17, E685. doi: 10.3390/ijms17050685
Pannunzio, N. R., Li, S. C., Watanabe, G., and Lieber, M. R. (2014). Non-homologous end joining often uses microhomology: implications for alternative end joining. DNA Repair 17, 74–80. doi: 10.1016/j.dnarep.2014.02.006
Pickart, C. M. (2004). Back to the future with ubiquitin. Cell 116, 181–190. doi: 10.1016/S0092-8674(03)01074-2
Pongpanich, M., Patchsung, M., Thongsroy, J., and Mutirangura, A. (2014). Characteristics of replication-independent endogenous double-strand breaks in Saccharomyces cerevisiae. BMC Genomics 15:750. doi: 10.1186/1471-2164-15-750
Pornthanakasem, W., Kongruttanachok, N., Phuangphairoj, C., Suyarnsestakorn, C., Sanghangthum, T., Oonsiri, S., et al. (2008). LINE-1 methylation status of endogenous DNA double-strand breaks. Nucleic Acids Res. 36, 3667–3675. doi: 10.1093/nar/gkn261
Seluanov, A., Mittelman, D., Pereira-Smith, O. M., Wilson, J. H., and Gorbunova, V. (2004). DNA end joining becomes less efficient and more error-prone during cellular senescence. Proc. Natl. Acad. Sci. U.S.A. 101, 7624–7629. doi: 10.1073/pnas.0400726101
Thongsroy, J., Matangkasombut, O., Thongnak, A., Rattanatanyong, P., Jirawatnotai, S., and Mutirangura, A. (2013). Replication-independent endogenous DNA double-strand breaks in Saccharomyces cerevisiae model. PLoS One 8:e72706. doi: 10.1371/journal.pone.0072706
Thongsroy, J., Patchsung, M., Pongpanich, M., Settayanon, S., and Mutirangura, A. (2018). Reduction in replication-independent endogenous DNA double-strand breaks promotes genomic instability during chronological aging in yeast. FASEB J. doi: 10.1096/fj.201800218RR [Epub ahead of print].
Vaidya, A., Mao, Z., Tian, X., Spencer, B., Seluanov, A., and Gorbunova, V. (2014). Knock-in reporter mice demonstrate that DNA repair by non-homologous end joining declines with age. PLoS Genet. 10:e1004511. doi: 10.1371/journal.pgen.1004511
Vyjayanti, V. N., and Rao, K. S. (2006). DNA double strand break repair in brain: reduced NHEJ activity in aging rat neurons. Neurosci. Lett. 393, 18–22. doi: 10.1016/j.neulet.2005.09.053
Xu, Z., Zhang, L., Zhang, W., Meng, D., Zhang, H., Jiang, Y., et al. (2015). SIRT6 rescues the age related decline in base excision repair in a PARP1-dependent manner. Cell Cycle 14, 269–276. doi: 10.4161/15384101.2014.980641
Yamada, M., Udono, M. U., Hori, M., Hirose, R., Sato, S., Mori, T., et al. (2006). Aged human skin removes UVB-induced pyrimidine dimers from the epidermis more slowly than younger adult skin in vivo. Arch. Dermatol. Res. 297, 294–302. doi: 10.1007/s00403-005-0618-0
Yang, H., Matsumoto, Y., Trujillo, K. M., Lees-Miller, S. P., Osley, M. A., and Tomkinson, A. E. (2015). Role of the yeast DNA repair protein Nej1 in end processing during the repair of DNA double strand breaks by non-homologous end joining. DNA Repair (Amst). 31, 1–10. doi: 10.1016/j.dnarep.2015.04.003
Yehuda, A. B., Globerson, A., Krichevsky, S., Bar On, H., Kidron, M., Friedlander, Y., et al. (2001). Ageing and the mismatch repair system. Mech. Ageing Dev. 121, 173–179. doi: 10.1016/S0047-6374(00)00208-6
Keywords: replication-independent endogenous DNA double-strand breaks, repair defect, modified ends with insertion, Saccharomyces cerevisiae, chronological aging
Citation: Pongpanich M, Patchsung M and Mutirangura A (2018) Pathologic Replication-Independent Endogenous DNA Double-Strand Breaks Repair Defect in Chronological Aging Yeast. Front. Genet. 9:501. doi: 10.3389/fgene.2018.00501
Received: 07 August 2018; Accepted: 05 October 2018;
Published: 25 October 2018.
Edited by:
Richard C. Siow, King’s College London, United KingdomReviewed by:
George A. Garinis, Foundation for Research and Technology Hellas, GreeceCopyright © 2018 Pongpanich, Patchsung and Mutirangura. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Monnat Pongpanich, bW9ubmF0LnBAY2h1bGEuYWMudGg=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.