 Vishnu Hosur
Vishnu Hosur Michelle L. Farley
Michelle L. Farley- The Jackson Laboratory, Bar Harbor, ME, United States
Tylosis with esophageal cancer syndrome (TOC) is a rare autosomal dominant proliferative skin disease caused by missense mutations in the rhomboid 5 homolog 2 (RHBDF2) gene. TOC is characterized by thickening of the skin in the palms and feet and is strongly linked with the development of esophageal squamous cell carcinoma. Murine models of human diseases have been valuable tools for investigating the underlying genetic and molecular mechanisms of a broad range of diseases. Although current mouse models do not fully recapitulate all aspects of human TOC, and the molecular mechanisms underlying TOC are still emerging, the available mouse models exhibit several key aspects of the disease, including a proliferative skin phenotype, a rapid wound healing phenotype, susceptibility to epithelial cancer, and aberrant epidermal growth factor receptor (EGFR) signaling. Furthermore, we and other investigators have used these models to generate new insights into the causes and progression of TOC, including findings suggesting a tissue-specific role of the RHBDF2-EGFR pathway, rather than a role of the immune system, in mediating TOC; and indicating that amphiregulin, an EGFR ligand, is a functional driver of the disease. This review highlights the mouse models of TOC available to researchers for use in investigating the disease mechanisms and possible therapies, and the significance of genetic modifiers of the disease identified in these models in delineating the underlying molecular mechanisms.
Introduction
Gain-of-function (GOF) mutations in the human rhomboid 5 homolog 2 (RHBDF2) gene constitutively activate epidermal growth factor receptor (EGFR) signaling to cause tylosis with esophageal cancer syndrome (TOC; OMIM: 148500) (Blaydon et al., 2012; Saarinen et al., 2012; Mokoena et al., 2018). RHBDF2 encodes the highly conserved, seven-transmembrane, rhomboid protein family member RHBDF2 (Freeman, 2014). Although the mechanisms underlying RHBDF2-mediated EGFR signaling are still emerging, gaining insights into these mechanisms will contribute substantially to the discovery of unique drug targets for treating TOC and potentially other related skin diseases.
Substantial literature suggests that RHBDF2 regulates the EGFR signaling pathway and its downstream signaling events, including rapid wound healing and epithelial tumorigenesis, through enhanced secretion of the EGFR ligand amphiregulin (AREG) (Brooke et al., 2014; Hosur et al., 2014, 2017a). AREG is synthesized as a pro-protein that requires conversion to an active protein by a disintegrin and metalloprotease 17 (ADAM17), in a process known as ectodomain shedding (Blobel, 2005). Two models offer possible explanations of the role of RHBDF2 in regulating AREG secretion. The first proposes that RHBDF2 directly interacts with ADAM17 and regulates its maturation, trafficking, and ectodomain shedding activity. According to the second model, RHBDF2 regulates AREG secretion through ADAM17, as in the first model, but importantly, it does so without altering ADAM17 sheddase activity. Further studies are needed to establish specific biological functions and properties of RHBDF2 in mediating AREG secretion in health and disease.
Currently there is no cure for TOC. To find a cure, two hurdles must be addressed immediately. First, mouse models of human TOC are critically needed to identify the relevant cell types, and to understand the pathogenesis and how to approach therapeutic treatment. Second, it is imperative to better understand the mechanisms underlying RHBDF2-mediated EGFR signaling by corroborating the significance of in vitro mechanistic findings in mouse models of human TOC.
Murine Models
Mouse models of human disease have long served a valuable role in biomedical research. In recent years, we have generated and validated mice carrying the human TOC disease mutation p.P189L (Hosur et al., 2017a; Figure 1A, top panel, Figure 1B), characterized a spontaneous mutation that results in a TOC phenotype (Hosur et al., 2014), and used these mouse models of TOC in studies in which we identified AREG as a functional driver of the disease (Hosur et al., 2014, 2017a); established an essential role for ADAM17 in mediating the disease (Hosur et al., 2018); and, most recently, demonstrated that RHBDF2-AREG-EGFR signaling, and not the immune system or surrounding microenvironment, plays a role in mediating the disease (Hosur et al., 2017b). These and other murine models of TOC (Table 1) will be useful for methodically studying the molecular pathways underlying TOC in further detail, for investigating the molecular pathways underlying related proliferative skin diseases, and for testing novel and existing therapeutics.
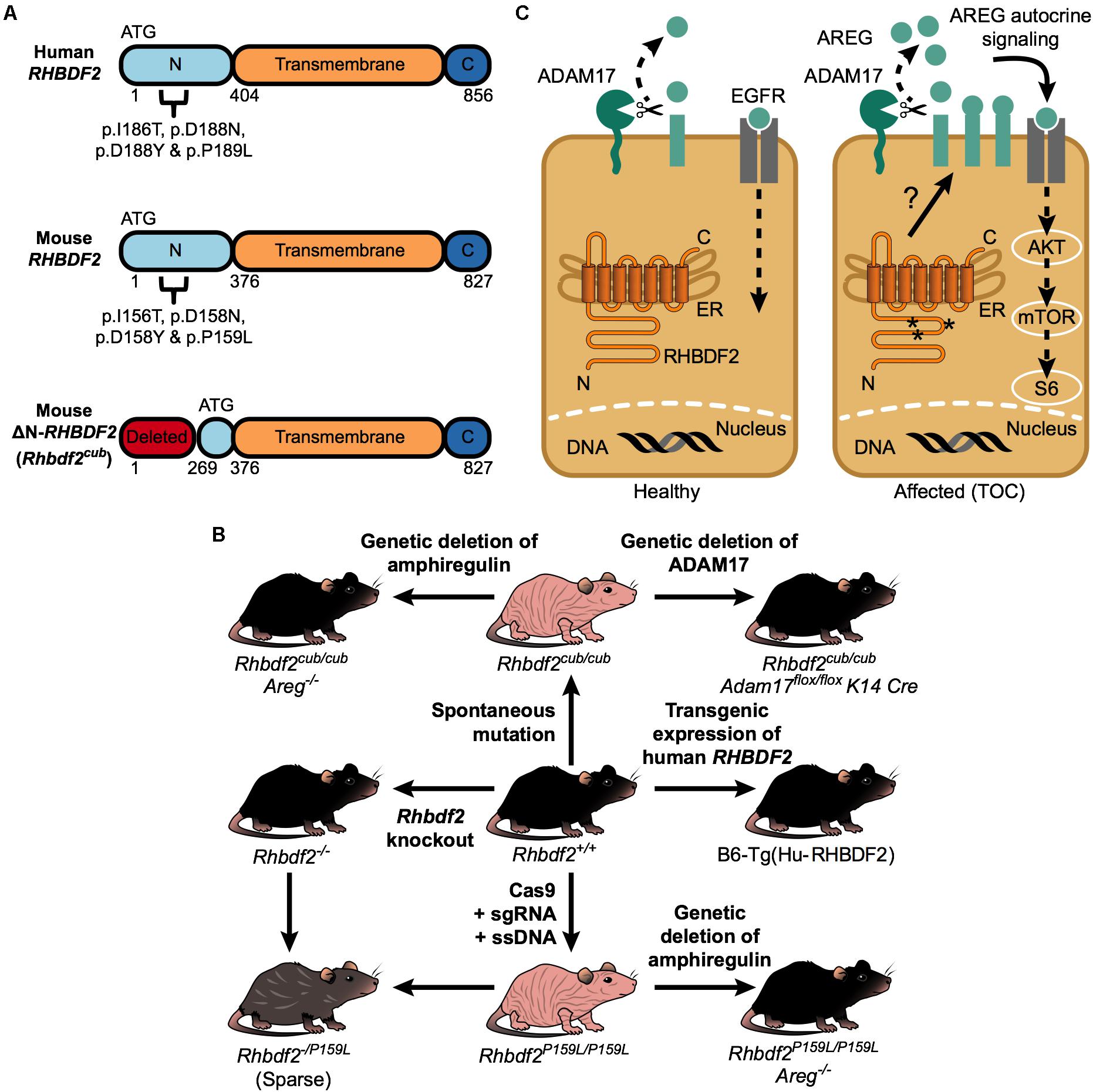
FIGURE 1. RHBDF2-regulated epidermal growth factor receptor signaling in TOC. (A) A schematic of the human RHBDF2 protein showing all four missense mutations in the cytosolic N-terminus domain that underlie human TOC (top); a schematic of the mouse RHBDF2 protein showing the analogous human TOC missense mutations (middle); a schematic of the truncated mouse RHBDF2 protein, encoded by the Rhbdf2cub gene, showing deletion of the cytosolic N-terminus domain (bottom). Notably, in the truncated protein, the cytosolic N-terminus domain still retains amino acids 269–375. (B) Diagram showing that both spontaneous (Rhbdf2cub/cub) and genetically engineered (Rhbdf2P159L/P159L) mouse models of human TOC exhibit a loss-of-hair phenotype. First, to validate that Rhbdf2cub/cub and Rhbdf2P159L/P159L are gain-of-function (GOF) mutations rather than null mutations, we generated Rhbdf2 knockout (Rhbdf2−/−) mice and observed that Rhbdf2−/− mice present with a normal hair coat, similarly to the wild-type (Rhbdf2+/+) mice, but very different from Rhbdf2cub/cub or Rhbdf2P159L/P159L mice. Second, to validate that Rhbdf2cub and Rhbdf2P159L are mutant alleles of the Rhbdf2 gene, we generated Rhbdf2cub/− and Rhbdf2P159L/− mice, which exhibit a sparse hair coat phenotype, suggesting that Rhbdf2cub and Rhbdf2P159L are mutant alleles of the Rhbdf2 gene. Third, we identified AREG and ADAM17 as genetic modifiers of the TOC phenotype; genetic deletion of Areg or Adam17 in both Rhbdf2cub/cub and Rhbdf2P159L/P159L mice restores the normal skin phenotype in these mice, suggesting that AREG and ADAM17 are essential mediators of the TOC phenotype. Lastly, we generated a B6-Tg(Hu-RHBDF2) mouse strain using a bacterial artificial chromosome (BAC) containing the human RHBDF2 gene, which, similarly to Rhbdf2+/+ and Rhbdf2−/− mice, exhibits a normal hair coat. Together, our results suggest that neither loss of RHBDF2 nor overexpression of RHBDF2, but GOF mutations, such as Rhbdf2cub and Rhbdf2P159L, induce an overt “EGFR hyperactive” phenotype to cause TOC. (C) RHBDF2 GOF mutations enhance AREG secretion through ADAM17 in a tissue-specific manner, leading to EGFR hyperactivation and in turn TOC.
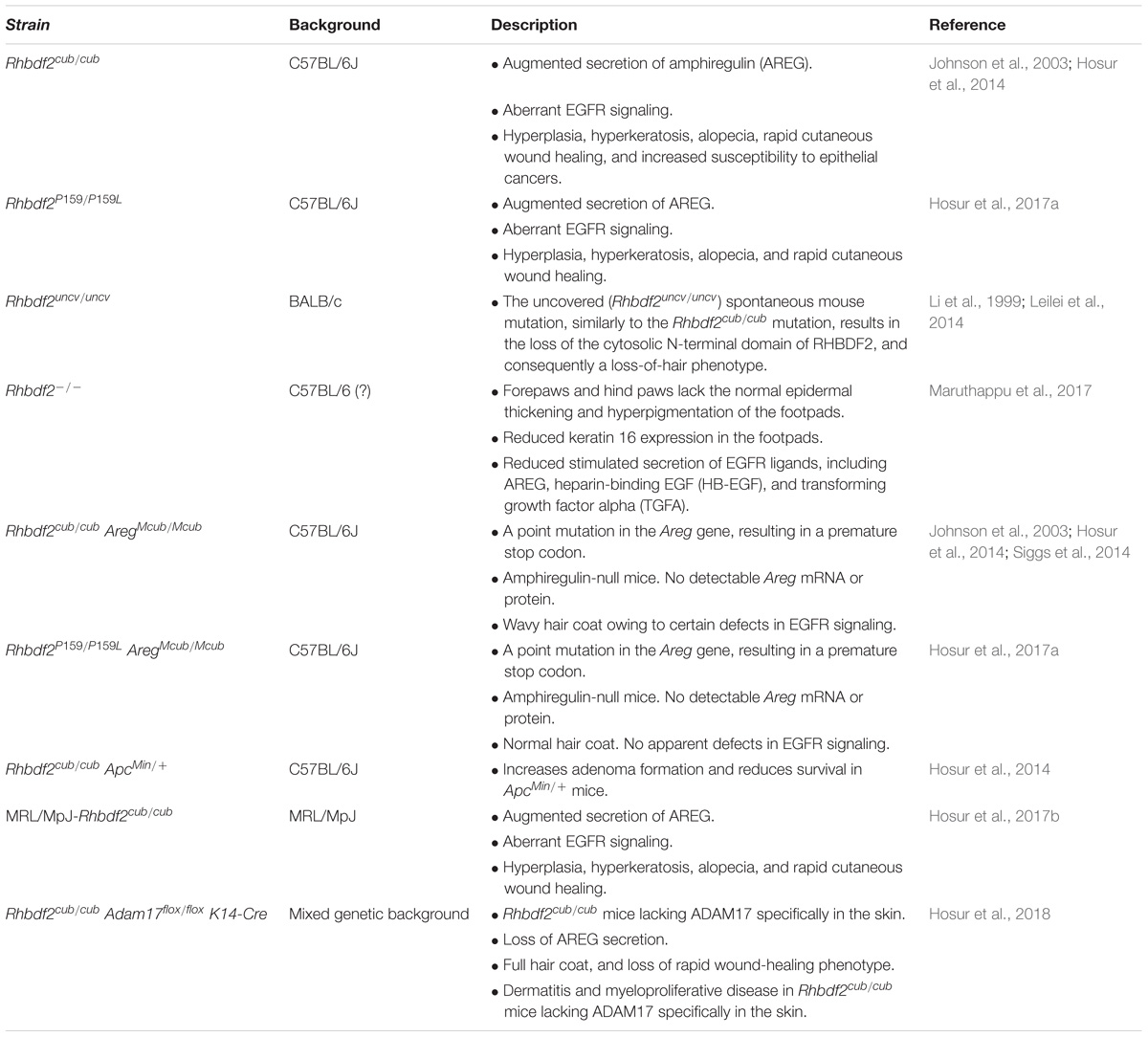
TABLE 1. Mouse models of the human skin disease tylosis with esophageal cancer syndrome.
A Spontaneous Mouse Mutation: Curly Bare (Rhbdf2cub/cub)
Spontaneous mouse mutations have been a valuable source of animal models for studying heritable human diseases, both for discovering the role of gene function and for understanding the biological pathways involved. Johnson et al. (2003) described a spontaneous recessive mouse mutation, named curly bare (cub) that maps to the distal region of Chr 11 and exhibits a hairless phenotype (Figure 1B). Hosur et al. (2014) reported that the cub mouse mutation is an unusually large (12,681 bp) deletion in the Rhbdf2 gene, resulting in the loss of exons 2–6. We also showed that even though the normal translation initiation site (ATG) in exon 3 is missing, there is translation from a downstream initiation site in exon 8, leading to a ∼63.5-kDa truncated protein rather than a full-length ∼100-kDa protein (Figure 1A; bottom panel). Histological analyses of skin sections from Rhbdf2cub/cub mice revealed epidermal hyperplasia, enlarged sebaceous glands, alopecia, and rapid cutaneous wound healing (data not shown). Additionally, embryonic fibroblasts isolated from Rhbdf2cub/cub mice exhibited a hyperactive EGFR signaling phenotype mediated by enhanced secretion of AREG (data not shown). Surprisingly, Rhbdf2 knockout (Rhbdf2−/−) mice appeared normal (Figure 1B), with no skin or rapid cutaneous wound-healing phenotype (Hosur et al., 2014; Siggs et al., 2014), suggesting that Rhbdf2cub/cub is a GOF mutation rather than a null mutation.
Next, to test the influence of mouse strain background on the Rhbdf2cub/cub phenotype, we backcrossed the Rhbdf2cub/cub mutation from the C57BL/6J background onto the MRL/MpJ background for more than 20 generations, creating a true congenic strain, MRL/MpJ-Rhbdf2cub/cub mice (Hosur et al., 2017b). We observed not only the rapid cutaneous wound-healing phenotype but also the hyperproliferative-skin phenotype in these mice, suggesting that the Rhbdf2cub/cub mutation results in a similar phenotype regardless of the mouse inbred strain background.
Tylosis is strongly associated with esophageal squamous cell carcinoma (Howel-Evans et al., 1958; Harper et al., 1970; Ellis et al., 1994; Hennies et al., 1995); however, there is no spontaneous incidence of esophageal cancer in Rhbdf2cub/cub mice. Because cigarette smoking and alcohol consumption are known risk factors for esophageal squamous cell carcinoma (Montgomery et al., 2014), we reasoned that the Rhbdf2cub mutation could increase the susceptibility to epithelial cancers and tested the role of RHBDF2 in adenoma formation in ApcMin/+ mice, a mouse model of human colon cancer. We found that a single allele of Rhbdf2cub can accelerate polyp formation and reduce the survival rates of ApcMin/+ mice (data not shown) (Hosur et al., 2014). Histological analysis revealed significantly increased tumor size and an increased number of polyps in ApcMin/+ Rhbdf2+/cub mice compared with ApcMin/+ Rhbdf2+/+ mice. Moreover, stimulated secretion of AREG was significantly increased in intestinal epithelial cells isolated from ApcMin/+ Rhbdf2+/cub mice compared with ApcMin/+ Rhbdf2+/+ mice. Together, these data suggest that GOF mutations in RHBDF2 accelerate tumor growth with an associated increase in AREG secretion, suggesting a critical role for RHBDF2 in controlling tumor growth through enhanced secretion of AREG.
A Genetically Engineered Mouse Model: Rhbdf2P159L/P159L
The capacity of CRISPR/Cas9 technology to be used to recapitulate patient variants in mouse models for various human diseases is well established (Birling et al., 2017). Using CRISPR/Cas9-mediated targeting and homology-directed repair in C57BL/6J zygotes, we recently generated mice carrying the human TOC mutation p.P189L (p.P159L in mice) in RHBDF2 (Figure 1A, top and middle panels, Figure 1B) and showed that the Rhbdf2P159L GOF mutation enhances AREG secretion to cause alopecia, rapid cutaneous wound healing, hyperplasia, and hyperkeratosis (Hosur et al., 2017a). Also, immunohistochemical analyses of skin sections from Rhbdf2+/+ and Rhbdf2P159L/P159L mice revealed a considerable increase in the activity of downstream effectors of the epidermal EGFR signaling pathway, including phospho-ERK1/2 and phospho-mTOR, in Rhbdf2P159L/P159L mice. Together, these data suggest that, analogous to the phenomena in human TOC mutations, Rhbdf2P159L/P159L is a GOF mutation in the mouse Rhbdf2 gene, and that these GOF mutations enhance secretion of AREG and lead to constitutive activation of EGFR signaling to cause TOC.
Rhbdf2 Loss-of-Function Mice: Rhbdf2−/−
Since GOF mutations in RHBDF2 cause skin hyperkeratosis and hyperplasia (Blaydon et al., 2012; Saarinen et al., 2012; Mokoena et al., 2018), Maruthappu et al. (2017) examined whether loss of RHBDF2 has any effect on skin thickness. The authors examined hematoxylin and eosin-stained skin sections of adult Rhbdf2−/− and Rhbdf2+/+ mice and reported that both the forepaws and hind paws, but not the back skin, of Rhbdf2−/− mice present with an epidermis thinner than that of Rhbdf2+/+ mice. Additionally, the authors showed that the thinner epidermis in Rhbdf2−/− mice is associated with reduced expression of cytoskeletal stress-associated protein keratin 16 (K16). Because of the reduced K16 expression associated with loss of RHBDF2 and a thinner epidermis, the authors examined K16 expression in the skin of human TOC patients using immunohistochemistry and observed a considerable increase in K16 expression. Collectively, these findings suggest that loss of RHBDF2 results in a thinner epidermis, and that, correspondingly, GOF mutations in RHBDF2, such as those observed in TOC, result in epidermal thickening through interaction with and regulation of K16.
Potential Mechanisms
Amphiregulin as a Functional Driver of the Disease
Genetic modifier genes can accelerate disease progression, slow down disease progression, or completely reverse a disease, and thus provide valuable information regarding the underlying biological pathways and the potential pharmaceutical targets. The Rhbdf2cub mouse modifier gene Modifier of cub phenotype (Mcub) was mapped to a position that coincides with a cluster of four EGFR ligand-encoding genes on chromosome 5 (Johnson et al., 2003). A single Mcub allele in combination with the Rhbdf2cub/cub genotype restores a full, wavy hair coat in Rhbdf2cub/cub mice. Mcub was later identified as a point mutation in the Areg gene that results in a premature stop codon, which in turn results in the absence of any detectable Areg mRNA or protein (Hosur et al., 2014). The rapid cutaneous wound-healing and alopecia phenotype observed in Rhbdf2cub/cub mice is lost when a single copy of the Mcub allele is present in combination with the Rhbdf2cub/cub genotype. Concordantly, in Rhbdf2P159L/P159L mice, genetic deletion of Areg restores the normal skin phenotype (Hosur et al., 2017a), suggesting that AREG mediates the hyperplasia, hyperkeratosis, alopecia, and rapid wound-healing phenotypes in Rhbdf2P159L/P159L and Rhbdf2cub/cub mice.
The Metalloprotease ADAM17 Is Essential for Shedding of Amphiregulin
The metalloprotease ADAM17 is the major ectodomain sheddase of AREG (Sunnarborg et al., 2002; Sahin et al., 2004; Sternlicht et al., 2005). However, in studies using Rhbdf2cub/cub mice, we (Hosur et al., 2014) and others (Leilei et al., 2014; Siggs et al., 2014) found that ADAM17 activity is reduced in Rhbdf2cub/cub mice, suggesting that RHBDF2 regulates secretion of AREG independently of ADAM17 activity. Thus, to examine whether RHBDF2 participates directly in shedding of EGFR ligands, similarly to ADAM17, we generated Rhbdf2cub/cub mice with ADAM17-deficient keratinocytes (Rhbdf2cub/cub Adam17flox/flox K14-Cre) and studied their phenotype (Hosur et al., 2018). We found that ADAM17 deficiency in the skin of Rhbdf2cub/cub mice restored a full hair coat and impaired the rapid wound-healing phenotype observed in Rhbdf2cub/cub mice, demonstrating that ADAM17 is the major ectodomain sheddase of AREG and that RHBDF2 does not directly participate in the shedding of AREG. In addition, Rhbdf2cub/cub mouse embryonic keratinocytes lacking ADAM17 failed to secrete AREG both under stimulated and unstimulated conditions, further demonstrating that ADAM17 is the principal sheddase of AREG and, importantly, that RHBDF2 does not directly participate in shedding of AREG (Figure 1C).
Tissue-Specific Role of the RHBDF2-AREG-ADAM17-EGFR Pathway
The immune system plays a key role in the pathogenesis of many proliferative skin diseases, including psoriasis and atopic dermatitis (Reich, 2012; Czarnowicki et al., 2014). To determine whether the hyperproliferative-skin and rapid-wound-healing phenotypes mediated by RHBDF2-AREG in TOC are tissue-specific and persist independently of the immune system, we performed bone marrow and reciprocal skin graft experiments (Hosur et al., 2017b). Bone marrow transfer from C57BL/6J(B6)-Rhbdf2cub/cub donor mice to B6 recipient mice failed to transfer the proliferative-skin and cutaneous regenerative phenotype in B6 mice, suggesting that the surrounding microenvironment or the immune system are unlikely to contribute to the TOC phenotype. Moreover, reciprocal skin grafts—from B6 mice to the dorsal skin of B6-Rhbdf2cub/cub mice and from B6-Rhbdf2cub/cub mice to the dorsal skin of B6 mice—maintained the phenotype of the donor mice. Together, these findings suggest a tissue-specific function of the RHBDF2-AREG-ADAM17-EGFR pathway in TOC.
Revisiting the Role of RHBDF2 in Regulating AREG Secretion
The mechanism by which RHBDF2 regulates AREG secretion is unclear. Substantial literature suggests that RHBDF2 regulates ectodomain shedding of AREG secretion by controlling the sheddase activity of ADAM17, the principle sheddase of AREG. This model is based primarily on the hypothesis that RHBDF2 is essential for not only the maturation and trafficking but also the activation of ADAM17 (Adrain et al., 2012; McIlwain et al., 2012; Christova et al., 2013; Li et al., 2015; Cavadas et al., 2017; Grieve et al., 2017). In further support of this model, it has been shown that lack of RHBDF2 significantly decreases ADAM17-dependent AREG secretion after stimulation with the protein kinase C stimulant PMA, whereas GOF RHBDF2 mutations such as those observed in human TOC enhance ADAM17 sheddase activity to significantly increase AREG secretion (Brooke et al., 2014). Consistent with this model, Maney et al. (2015) recently showed in vitro that GOF mutations in RHBDF2 enhance ADAM17 sheddase activity. Furthermore, it has been suggested that RHBDF1, a paralog of RHBDF2, can compensate for the loss of RHBDF2 in regulating ADAM17 maturation, trafficking, and activity (Issuree et al., 2013).
Conversely, the following evidence argues against a role for RHBDF1/RHBDF2 in regulating ADAM17 trafficking and maturation or activity to drive AREG secretion. Strong in vivo evidence includes the finding that mice overexpressing ADAM17 are viable and exhibit no overt phenotype (Yoda et al., 2013). Concordantly, overexpression of ADAM17 in vivo does not elicit any enhanced ADAM17 sheddase activity, or a significant increase in secretion of AREG or other ADAM17 substrates (Fukaya et al., 2013; Yoda et al., 2013). In support of these observations, Dang et al. (2013) demonstrated in vitro that phorbol-ester-stimulated shedding of EGF family members and ADAM17 substrates AREG, HB-EGF, and TGFα occurs independently of major changes in ADAM17 sheddase activity. Studies using Rhbdf1 knockout and/or Rhbdf1/Rhbdf2 double knockout mice suggest that it is unlikely that RHBDF1 is involved in regulating ADAM17 activity or compensating for its loss. Neither Rhbdf1 knockout mice nor Rhbdf1/Rhbdf2 double knockout mice phenocopy Adam17 knockout mice; whereas as Rhbdf1 knockout and Rhbdf1/Rhbdf2 double knockout mice exhibit postnatal and embryonic lethality (Christova et al., 2013), respectively, Adam17 knockout mice exhibit perinatal lethality with an “open eyelids at birth” phenotype and hair-follicle defects (Peschon et al., 1998). Together, these in vitro and in vivo findings strongly argue against a role for RHBDF1 and RHBDF2 in either maturation or trafficking of ADAM17, or regulation of its activity, to drive AREG secretion. We propose that RHBDF2 likely regulates the availability of AREG without directly influencing ADAM17 maturation or trafficking, or sheddase activity (Figure 1C, right panel).
Conclusion
As mice and humans share between 95 and 98% of their genes, mouse models of human skin diseases have been valuable resources for studying the normal biological processes involved in skin function, understanding the molecular mechanism causing the diseases, identifying potential targets for intervention, and testing therapeutics. While current murine models of human TOC, both spontaneous mutant and genetically engineered mice, do not recapitulate all characteristics of the human disease, these models have yielded valuable insights into disease mechanisms, including the identification of AREG as a functional driver of the disease, verification of the essential role of metalloprotease ADAM17 in shedding of AREG, demonstration of the susceptibility of mice carrying Rhbdf2 GOF mutations to epithelial cancer, and determination of the tissue-specific role of the RHBDF2-AREG-ADAM17-EGFR pathway. In addition, by further leveraging these murine models in future studies, researchers can continue to generate and test mechanistic hypotheses, and study the physiological and pathogenic molecular machinery underlying the role of the RHBDF2-regulated EGFR signaling pathway in TOC, and in other skin diseases and epithelial cancers.
Author Contributions
VH designed the experiments, involved in generation and characterization of mouse models, wrote and reviewed the manuscript. MF, BL, and LB involved in generation and characterization of mouse models. LS and MW designed, wrote, and reviewed the manuscript. All authors read and approved the final manuscript.
Funding
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health grants P30CA034196 and R21 OD023800-01 (MW).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adrain, C., Zettl, M., Christova, Y., Taylor, N., and Freeman, M. (2012). Tumor necrosis factor signaling requires iRhom2 to promote trafficking and activation of TACE. Science 335, 225–228. doi: 10.1126/science.1214400
Birling, M. C., Herault, Y., and Pavlovic, G. (2017). Modeling human disease in rodents by CRISPR/Cas9 genome editing. Mamm. Genome 28, 291–301.
Blaydon, D. C., Etheridge, S. L., Risk, J. M., Hennies, H. C., Gay, L. J., Carroll, R., et al. (2012). RHBDF2 mutations are associated with tylosis, a familial esophageal cancer syndrome. Am. J. Hum. Genet. 90, 340–346. doi: 10.1016/j.ajhg.2011.12.008
Blobel, C. P. (2005). ADAMs: key components in EGFR signalling and development. Nat. Rev. Mol. Cell Biol. 6, 32–43. doi: 10.1038/nrm1548
Brooke, M. A., Etheridge, S. L., Kaplan, N., Simpson, C., O’Toole, E. A., Ishida-Yamamoto, A., et al. (2014). iRHOM2-dependent regulation of ADAM17 in cutaneous disease and epidermal barrier function. Hum. Mol. Genet. 23, 4064–4076. doi: 10.1093/hmg/ddu120
Cavadas, M., Oikonomidi, I., Gaspar, C. J., Burbridge, E., Badenes, M., Felix, I., et al. (2017). Phosphorylation of iRhom2 controls stimulated proteolytic shedding by the metalloprotease ADAM17/TACE. Cell Rep. 21, 745–757. doi: 10.1016/j.celrep.2017.09.074
Christova, Y., Adrain, C., Bambrough, P., Ibrahim, A., and Freeman, M. (2013). Mammalian iRhoms have distinct physiological functions including an essential role in TACE regulation. EMBO Rep. 14, 884–890. doi: 10.1038/embor.2013.128
Czarnowicki, T., Krueger, J. G., and Guttman-Yassky, E. (2014). Skin barrier and immune dysregulation in atopic dermatitis: an evolving story with important clinical implications. J. Allergy Clin. Immunol. Pract. 2, 371–379.
Dang, M., Armbruster, N., Miller, M. A., Cermeno, E., Hartmann, M., Bell, G. W., et al. (2013). Regulated ADAM17-dependent EGF family ligand release by substrate-selecting signaling pathways. Proc. Natl. Acad. Sci. U.S.A. 110, 9776–9781. doi: 10.1073/pnas.1307478110
Ellis, A., Field, J. K., Field, E. A., Friedmann, P. S., Fryer, A., Howard, P., et al. (1994). Tylosis associated with carcinoma of the oesophagus and oral leukoplakia in a large Liverpool family–a review of six generations. Eur. J. Cancer B Oral Oncol. 30b, 102–112. doi: 10.1016/0964-1955(94)90061-2
Freeman, M. (2014). The rhomboid-like superfamily: molecular mechanisms and biological roles. Annu. Rev. Cell Dev. Biol. 30, 235–254. doi: 10.1146/annurev-cellbio-100913-012944
Fukaya, S., Matsui, Y., Tomaru, U., Kawakami, A., Sogo, S., Bohgaki, T., et al. (2013). Overexpression of TNF-alpha-converting enzyme in fibroblasts augments dermal fibrosis after inflammation. Lab. Invest. 93, 72–80. doi: 10.1038/labinvest.2012.153
Grieve, A. G., Xu, H., Kunzel, U., Bambrough, P., Sieber, B., and Freeman, M. (2017). Phosphorylation of iRhom2 at the plasma membrane controls mammalian TACE-dependent inflammatory and growth factor signalling. eLife 6:e23968.
Harper, P. S., Harper, R. M., and Howel-Evans, A. W. (1970). Carcinoma of the oesophagus with tylosis. Q. J. Med. 39, 317–333.
Hennies, H. C., Hagedorn, M., and Reis, A. (1995). Palmoplantar keratoderma in association with carcinoma of the esophagus maps to chromosome 17q distal to the keratin gene cluster. Genomics 29, 537–540.
Hosur, V., Farley, M. L., Burzenski, L. M., Shultz, L. D., and Wiles, M. V. (2018). ADAM17 is essential for ectodomain shedding of the EGF-receptor ligand amphiregulin. FEBS Open Bio 8, 702–710.
Hosur, V., Johnson, K. R., Burzenski, L. M., Stearns, T. M., Maser, R. S., and Shultz, L. D. (2014). Rhbdf2 mutations increase its protein stability and drive EGFR hyperactivation through enhanced secretion of amphiregulin. Proc. Natl. Acad. Sci. U.S.A. 111, E2200–E2209.
Hosur, V., Low, B. E., Shultz, L. D., and Wiles, M. V. (2017a). Genetic deletion of amphiregulin restores the normal skin phenotype in a mouse model of the human skin disease tylosis. Biol. Open 6, 1174–1179.
Hosur, V., Lyons, B. L., Burzenski, L. M., and Shultz, L. D. (2017b). Tissue-specific role of RHBDF2 in cutaneous wound healing and hyperproliferative skin disease. BMC Res. Notes 10:573. doi: 10.1186/s13104-017-2899-8
Howel-Evans, W., Mc, C. R., Clarke, C. A., and Sheppard, P. M. (1958). Carcinoma of the oesophagus with keratosis palmaris et plantaris (tylosis): a study of two families. Q. J. Med. 27, 413–429.
Issuree, P. D., Maretzky, T., McIlwain, D. R., Monette, S., Qing, X., Lang, P. A., et al. (2013). iRHOM2 is a critical pathogenic mediator of inflammatory arthritis. J. Clin. Invest. 123, 928–932. doi: 10.1172/JCI66168
Johnson, K. R., Lane, P. W., Cook, S. A., Harris, B. S., Ward-Bailey, P. F., Bronson, R. T., et al. (2003). Curly bare (cub), a new mouse mutation on chromosome 11 causing skin and hair abnormalities, and a modifier gene (mcub) on chromosome 5. Genomics 81, 6–14. doi: 10.1016/S0888-7543(02)00013-7
Leilei, Y., Bing, L., Yang, L., Shaoxia, W., Yuan, X., Dongping, W., et al. (2014). iRhom2 mutation leads to aberrant hair follicle differentiation in mice. PLoS One 9:e115114. doi: 10.1371/journal.pone.0115114
Li, S. R., Wang, D. P., Yu, X. L., Ge, B. S., Wang, C. E., Lu, Y. F., et al. (1999). Uncv (uncovered): a new mutation causing hairloss on mouse chromosome 11. Genet. Res. 73, 233–238. doi: 10.1017/S0016672399003808
Li, X., Maretzky, T., Weskamp, G., Monette, S., Qing, X., Issuree, P. D., et al. (2015). iRhoms 1 and 2 are essential upstream regulators of ADAM17-dependent EGFR signaling. Proc. Natl. Acad. Sci. U.S.A. 112, 6080–6085. doi: 10.1073/pnas.1505649112
Maney, S. K., McIlwain, D. R., Polz, R., Pandyra, A. A., Sundaram, B., Wolff, D., et al. (2015). Deletions in the cytoplasmic domain of iRhom1 and iRhom2 promote shedding of the TNF receptor by the protease ADAM17. Sci. Signal. 8:ra109. doi: 10.1126/scisignal.aac5356
Maruthappu, T., Chikh, A., Fell, B., Delaney, P. J., Brooke, M. A., Levet, C., et al. (2017). Rhomboid family member 2 regulates cytoskeletal stress-associated Keratin 16. Nat. Commun. 8:14174. doi: 10.1038/ncomms14174
McIlwain, D. R., Lang, P. A., Maretzky, T., Hamada, K., Ohishi, K., Maney, S. K., et al. (2012). iRhom2 regulation of TACE controls TNF-mediated protection against Listeria and responses to LPS. Science 335, 229–232. doi: 10.1126/science.1214448
Mokoena, T., Smit, J. G. M., Karusseit, V. O., Dorfling, C. M., and van Rensburg, E. J. (2018). Tylosis associated with squamous cell carcinoma of the oesophagus (TOC): report of an African family with a novel RHBDF2 variant. Clin. Genet. 93, 1114–1116.
Montgomery, E., Basman, F., Brennan, P., and Malekzadeh, R. (2014). “Oesophageal cancer,” in World Cancer Report, eds B. W. Stewart, and C. P. Wild, (Geneva: World Health Organization), 374–382.
Peschon, J. J., Slack, J. L., Reddy, P., Stocking, K. L., Sunnarborg, W. S., Lee, D. C., et al. (1998). An essential role for ectodomain shedding in mammalian development. Science 282, 1281–1284. doi: 10.1126/science.282.5392.1281
Reich, K. (2012). The concept of psoriasis as a systemic inflammation: implications for disease management. J. Eur. Acad. Dermatol. Venereol. 26(Suppl. 2), 3–11. doi: 10.1111/j.1468-3083.2011.04410.x
Saarinen, S., Vahteristo, P., Lehtonen, R., Aittomaki, K., Launonen, V., Kiviluoto, T., et al. (2012). Analysis of a Finnish family confirms RHBDF2 mutations as the underlying factor in tylosis with esophageal cancer. Fam. Cancer 11, 525–528. doi: 10.1007/s10689-012-9532-8
Sahin, U., Weskamp, G., Kelly, K., Zhou, H. M., Higashiyama, S., Peschon, J., et al. (2004). Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J. Cell Biol. 164, 769–779. doi: 10.1083/jcb.200307137
Siggs, O. M., Grieve, A., Xu, H., Bambrough, P., Christova, Y., and Freeman, M. (2014). Genetic interaction implicates iRhom2 in the regulation of EGF receptor signalling in mice. Biol. Open 3, 1151–1157.
Sternlicht, M. D., Sunnarborg, S. W., Kouros-Mehr, H., Yu, Y., Lee, D. C., and Werb, Z. (2005). Mammary ductal morphogenesis requires paracrine activation of stromal EGFR via ADAM17-dependent shedding of epithelial amphiregulin. Development 132, 3923–3933. doi: 10.1242/dev.01966
Sunnarborg, S. W., Hinkle, C. L., Stevenson, M., Russell, W. E., Raska, C. S., Peschon, J. J., et al. (2002). Tumor necrosis factor-alpha converting enzyme (TACE) regulates epidermal growth factor receptor ligand availability. J. Biol. Chem. 277, 12838–12845. doi: 10.1074/jbc.M112050200
Keywords: Rhbdf2, TOC, EGFR, ADAM17/TACE, AREG, CRISPR-Cas9
Citation: Hosur V, Farley ML, Low BE, Burzenski LM, Shultz LD and Wiles MV (2018) RHBDF2-Regulated Growth Factor Signaling in a Rare Human Disease, Tylosis With Esophageal Cancer: What Can We Learn From Murine Models? Front. Genet. 9:233. doi: 10.3389/fgene.2018.00233
Received: 06 April 2018; Accepted: 11 June 2018;
Published: 04 July 2018.
Edited by:
Noor Ahmad Shaik, King Abdulaziz University, Saudi ArabiaReviewed by:
Arif Mohammed, Jeddah University, Saudi ArabiaParikipandla Sridevi, Indira Gandhi National Tribal University, India
Copyright © 2018 Hosur, Farley, Low, Burzenski, Shultz and Wiles. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vishnu Hosur, dmlzaG51Lmhvc3VyQGpheC5vcmc=