 Thais F. Bartelli
Thais F. Bartelli Danielle C. F. Bruno
Danielle C. F. Bruno Marcelo R. S. Briones
Marcelo R. S. Briones- 1Laboratory of Evolutionary Genomics and Biocomplexity, Department of Microbiology, Immunology and Parasitology, Federal University of São Paulo, São Paulo, Brazil
- 2Laboratory of Medical Genomics, A. C. Camargo Cancer Center, São Paulo, Brazil
- 3Department of Health Informatics, Federal University of São Paulo, São Paulo, Brazil
The commensal yeast Candida albicans is an opportunistic pathogen. In order to successfully colonize or infect the human body, the fungus must adapt to the host’s environmental conditions, such as low oxygen tension (hypoxia), temperature (37∘C), and the different carbon sources available. Previous studies demonstrated the adaptive importance of C. albicans genetic variability for its pathogenicity, although the contributions of epigenetic and the influence of environmental factors are not fully understood. Mitochondria play important roles in fungal energetic metabolism, regulation of nuclear epigenetic mechanisms and pathogenicity. However, the specific impact of inter-strain mitochondrial genome variability and mitochondrial epigenetics in pathogenicity is unclear. Here, we draw attention to this relevant organelle and its potential role in C. albicans pathogenicity and provide preliminary evidence, for the first time, for methylation of the yeast mitochondrial genome. Our results indicate that environmental conditions, such as continuous exposure for 12 weeks to hypoxia and 37∘C, decrease the mitochondrial genome methylation in strains SC5314 and L757. However, the methylation decrease is quantitatively different in specific genome positions when strains SC5314 and L757 are compared. We hypothesize that this phenomenon can be promising for future research to understand how physical factors of the host affect the C. albicans mitochondrial genome and its possible impact on adaptation and pathogenicity.
The yeast Candida albicans is responsible for severe mucosal and bloodstream opportunistic infections, with nearly 400,000 nosocomial cases worldwide and high mortality rates (46–75%) (Brown et al., 2012). In normal conditions this fungus is a harmless human commensal, successfully colonizing diverse niches, such as skin, urogenital and gastrointestinal tracts (Brock, 2009; Huffnagle and Noverr, 2013; Underhill and Iliev, 2014). The metabolic plasticity of C. albicans allows its survival and response to multiple environmental stimuli simultaneously, such as temperature, oxygen, and nutrient availability (Brown et al., 2014). The remarkable genetic variability among C. albicans isolates suggests that this opportunistic pathogen undergoes adaptation and microevolution when colonizing the human body (Morschhäuser et al., 2000; Forche et al., 2009; Wartenberg et al., 2014). Although intraspecific variation in the nuclear DNA is well-established, the evolutionary rate of C. albicans mitochondrial DNA (mtDNA) and its effect on its fitness and virulence is not clear.
Besides genetic diversity, several studies address the distribution and significance of C. albicans epigenetic plasticity as an adaptive mechanism (Zordan et al., 2006; Lopes da Rosa and Kaufman, 2012; Tscherner et al., 2015; Freire-Benéitez et al., 2016). Epigenetic factors, such as DNA methylation, can regulate the cellular response in absence of sequence changes in the DNA, and these factors can be modified and reversed by internal and external cellular stimuli, such as pollutants, oxidative stress, temperature, and nutrients (Byun and Baccarelli, 2014). Currently the only data available on C. albicans epigenetic regulation focus on its nuclear genome, mainly on modulation of morphology and other virulence factors, such as white-opaque switching (Zordan et al., 2006; Mishra et al., 2011; Zhang et al., 2013; Kim et al., 2015; Tscherner et al., 2015; Freire-Benéitez et al., 2016). Mapping of C. albicans nuclear genome hypermethylated sites identified genes involved in morphogenesis and hyphal growth (16.7%), white-opaque switching (3.3%), iron use (6.7%), drugs resistance and signaling (12%), stress response (7.3%), and genes involved in regulatory activities such as chromatin organization (3.3%), cycle or cell division (7.3%), biogenesis and protein transport (12.7%), DNA/RNA processing (5.3%), pathogenesis or virulence (2%), and carbohydrate metabolism (1.3%). It was also observed that, in this species, methylation occurs at both CpG and CH sites (H = Adenine, Cytosine, or Thymine), mainly in the gene bodies, instead of the promoters (Mishra et al., 2011).
In mammalian mitochondria the DNA methyltransferase enzymes DNMT1 (Shock et al., 2011) and DNMT3A (Chestnut et al., 2011) are responsible for DNA methylation and the enzymes TET1 and TET2 (10–11 translocation) (Dzitoyeva et al., 2012; Bellizzi et al., 2013) catalyze demethylation by converting 5methylcytosines (5mC) into 5-hydroxymethylcytosines (5hmC), which are passively eliminated during DNA replication or actively reversed in cytosines by iterative oxidation followed by base excision repair (Kohli and Zhang, 2013). In addition to being involved in energy production, mitochondria produce several epigenetic-related metabolites, such as NAD+, ATP, alpha-ketoglutarate and acetyl coenzyme A, which are necessary substrates for nuclear transcriptional and epigenetic processes, such as chromatin remodeling, histone modification, and nucleosome positioning (Shaughnessy et al., 2014). In the human mitochondrial genome, the methylation pattern is uniform and constant throughout the molecule, although there are differentially methylated sites between different tissues, different samples collected at different time points and at different gene start sites, suggesting the existence of a regulatory mechanism of the mtDNA methylation that needs to be further elucidated (Ghosh et al., 2014). The induction of reactive oxygen species in human cells, for example, leads to a decrease in the mtDNA methylation, probably due to the greater compactness of the mtDNA in order to protect it, since the nucleoid protein TFAM has greater DNA binding affinity with damaged DNA (Rebelo et al., 2009). Although recent studies detected histones in the mitochondria (Choi et al., 2011), the general consensus is that mitochondrial histone complexes do not exist and that the methylation of mtDNA also plays a role in its stability and functioning (Byun and Baccarelli, 2014).
Data on mtDNA methylation are focused on human samples, given the increasing evidence of mtDNA methylation role in human diseases and its potential use as a biomarker for harmful environmental and nutritional factors (Iacobazzi et al., 2013). In addition, although an increasing amount of data indicate that mitochondria are important in fungal virulence and survival (Bambach et al., 2009; Qu et al., 2012; Thomas et al., 2013), little is known about C. albicans mtDNA variability and its effect in pathogenicity. There is no currently available data on C. albicans mtDNA methylation and how host conditions may influence it.
To test the hypothesis that methylated cytosines are present in C. albicans mtDNA, we performed Whole-Genome Bisulfite Sequencing (WGBS) using DNA isolated from the reference strain SC5314. Yeast cells were grown on YPD plates (1% w/v yeast extract; 2% w/v peptone, 2% w/v dextrose, 2% w/v agar) and a single colony was used for overnight growth on YPD broth at 28°C, 150 rpm. Total yeast DNA was extracted from samples as described previously (Wach et al., 1994). DNA was treated with sodium bisulfite (Zymo EZ DNA Methylation Gold kit) prior to the addition of the adapters during a preparation of the libraries, according to the Illumina TruSeq Nano kit manufacturer’s instructions. Paired-end 2 bp × 300 bp runs were performed by the MiSeq Illumina method. FastQC v.0.11.4 software (Andrews, 2010) was used to evaluate sequencing quality and trimming was performed with Trim Galore 0.4.0v. Read alignment was carried out with Bismark Bisulfite Mapper 0.13.0v (Krueger and Andrews, 2011) and duplicated mapped reads removed. Alignment was performed using the reference sequence publicly available for the SC5314 strain (Assembly 22: A22-s05-m04-r02, from candidagenome.org). Interestingly, a previous analysis showed that the mtDNA of the strain L757 had only 1% sequence variability (approximately 300 nucleotides) in comparison to the SC5314 mtDNA sequence (Bartelli et al., 2013). The percent methylation was calculated with the bismark_methylation_extractor tool implemented in the Bismark program (bedGraph files) and the R package MethylKit v. 0.9.5 (Akalin et al., 2012) for the different CpG, CHH and CHG contexts. The sites analyzed were filtered and only those with Phred score > 20 and minimum 10 reads coverage were considered. Histograms with coverage distribution were checked and no duplication bias was detected. Differentially methylated cytosine calculations (Wang et al., 2011) and Pearson correlation coefficients were calculated as implemented in MethylKit v. 0.9.5 (Akalin et al., 2012). For this analysis, only sites with satisfactory quality and coverage on both samples were considered. MtDNA mappings were visualized in the Integrative Genomics Viewer v.2.3.57 (Robinson et al., 2011).
The complete mtDNA of this pathogen is 40 kb long, and at least 85% of the C. albicans SC5314 mtDNA sequence (34,233 bp) with an average coverage of 162x was obtained after bisulfite treatment, sequencing and assembly. Fragments of mtDNA not assembled correspond to the two repetitive and inverted regions present at the two ends of C. albicans mtDNA, with approximately 7 kb each (Figure 1A, top row). Methylation levels were analyzed in three different contexts, CpG, CHH and CHG, on both strands. This sample exhibited a global hypermethylation pattern, with a mean methylation per cytosine of 99%. In addition, the methylation distribution shows that approximately 97, 96, and 94% of the mtDNA cytosines have the methylation rate equal to or greater than 95% in the CpG, CHH, and CHG contexts, respectively. Another strain, L757, a more recent C. albicans pathogenic isolated from a candidemia patient (Padovan et al., 2009), was analyzed in parallel after growth under the same conditions and similar results were identified (Figure 1B, top row).
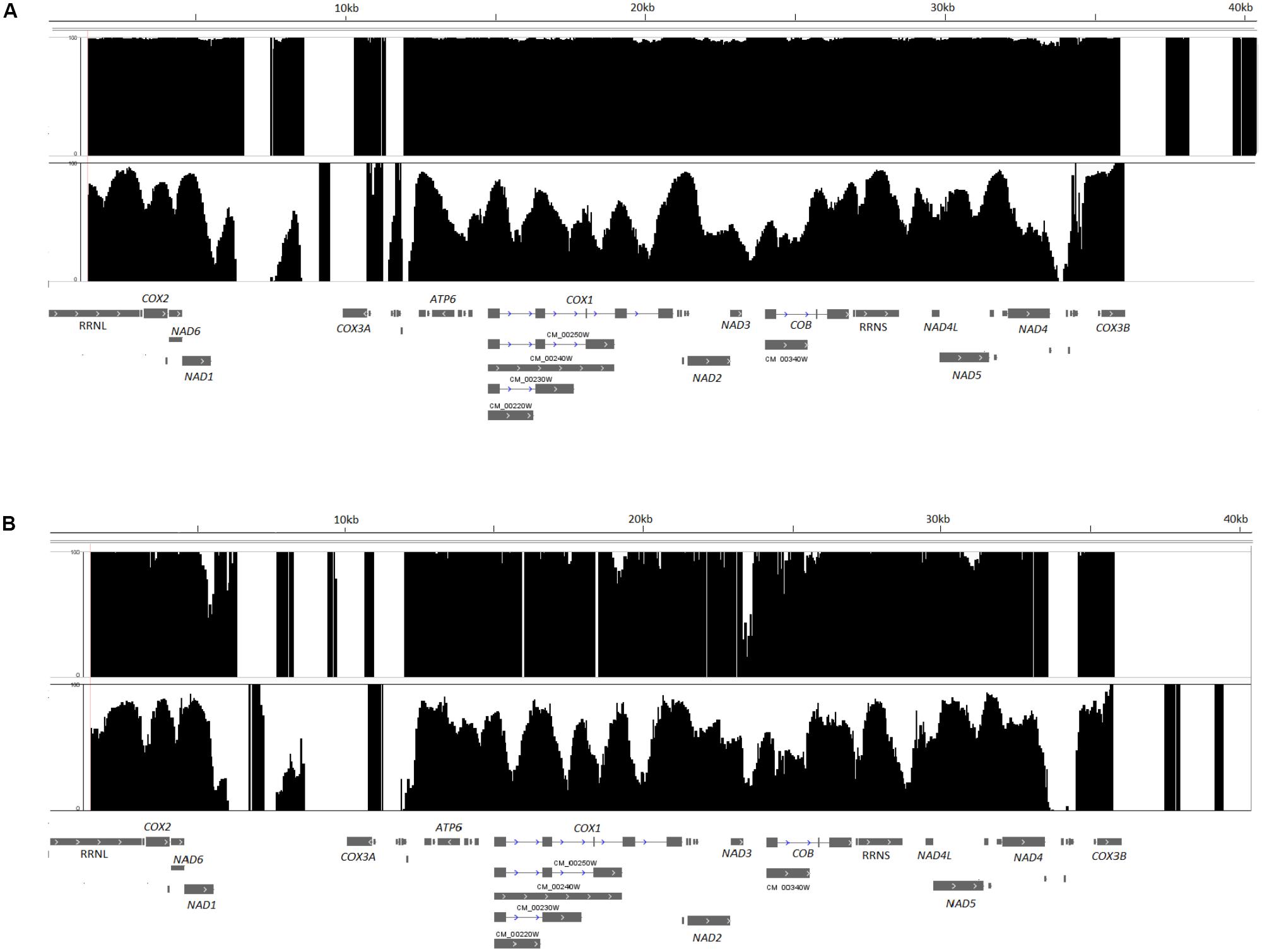
FIGURE 1. Methylation maps of Candida albicans mtDNA, strains SC5314 (A) and L757 (B). The figure depicts the percentage of methylation per cytosine on CpG, CHH, and CHG contexts for SC5314 (A) or L757 (B) (top row) showing hypermethylation and SC5314-GTH12 or L757-GTH12 (bottom row) with variable methylation distribution and hypomethylated cytosines. The two large portions of the mtDNA without percentage of methylation in the figure correspond to the two repetitive inverted portions of C. albicans mitochondrial genome (5,540–12,382 and 33,578–40,420 bp).
To test the hypothesis that growth conditions affect the methylation profile of C. albicans mtDNA, strains SC5314 and L757 were analyzed after 12 weeks of continuous growth under 5–15% oxygen level (hypoxia), YPG broth (1% w/v yeast extract; 2% w/v peptone, 2% w/v glycerol) at 37°C, named SC5314-GTH12 or L757-GTH12 (Glycerol, Thirty-seven °C, Hypoxia, 12 weeks). The oxygen tension in the atmosphere is approximately 21% (O2 pressure of 159 mmHg at sea level), known as normoxia, and healthy tissues in the human body range from 2.5 to 9% O2 (Grahl et al., 2012). This lower oxygen tension (<21%), known as hypoxia, varies depending on the anatomical site or tissue inflammation (Grahl et al., 2012). When subjected to temperatures above 30°C C. albicans activates its heat shock response, leading to the transcription of several genes, especially chaperones, by the heat shock factor 1 (Hsf1) (Nicholls et al., 2009). While the response to heat shock and hypoxia can be related and dependent, several studies show that changes in the carbon source strongly influence C. albicans stress resistance, and therefore, it is essential to consider the pathogen response to combinatorial stressors rather than to individual stress factors commonly studied separately in vitro (Brown et al., 2014). The combinatorial stresses are relevant when representing many host niches, such as a mucosal invasion (oxidative stresses plus water balance) or a kidney infection (cell adaptation to high salt concentrations plus endogenous reactive oxygen species) (Brown et al., 2014).
We obtained at least 82% (33,151 bp) of the sequence of the mtDNA of strain SC5314-GTH12, with 119x average coverage. This sample showed reduced levels of methylation, with higher heterogeneity and greater variability in the methylation levels of the mtDNA with a mean methylation of 60%, including non-methylated sites (0%) that were not seen in the SC5314 mitochondrial methylome previously analyzed (Figure 1A, bottom row). The methylation distribution showed that only 14, 9.1, and 12.2% of the cytosines analyzed had values equal to or greater than 90% methylation in CpG, CHH, and CHG, respectively, values that are much lower than the ones observed for strain SC5314. Again, similar results were identified for the other C. albicans strain (L757-GTH12) (Figure 1B, bottom row) when grown under this same condition.
The variability in cytosine methylation percentages, with values ranging from 0 to 100% at each site, is related to the heterogeneity of the sample, which is composed of a mixture of C. albicans cells and/or the occurrence of several copies of the mtDNA per cell, which may have heterogeneous methylation profiles. Also, for some genes, within a predominantly methylated population, the occurrence of unmethylated copies between them is common. This indicates that methylases may have a limiting rate, which results in incomplete methylation or that the transition from active to inactive transcription occurs through a passive dilution of methylated copies during replication (Mishra et al., 2011).
To identify differentially methylated cytosines between samples SC5314 and SC5314-GTH12, bases with q-value < 0.01 and % methylation difference > 30% were considered (Wang et al., 2011; Akalin et al., 2012). When comparing SC5314 and SC5314-GTH12, we identified 5,520 differentially methylated cytosines, 530 in the CpG, 4,355 in the CHH and 635 in the CHG context, all of them hypomethylated in SC5314-GTH12 as compared to SC5314. Only 1,949 cytosines (35.3%) were located in coding regions. Among the genes with the highest number of differentially methylated cytosines between SC5314-GTH12 and SC5314, were RRNL, COX1, COB and NAD4, including possible endonucleases involved with COX1 and COB splicing (Table 1). When analyzing differentially methylated cytosines between strain SC5314 and L757, we could detect genes with cytosines hypo and/or hypermethylated between SC5314 versus L757 and SC5314-GTH12 versus L757-GTH12 (Table 1), indicating that mtDNA methylation is likely strain specific and associated with the different adaptation patterns of C. albicans clinical isolates in the human body.
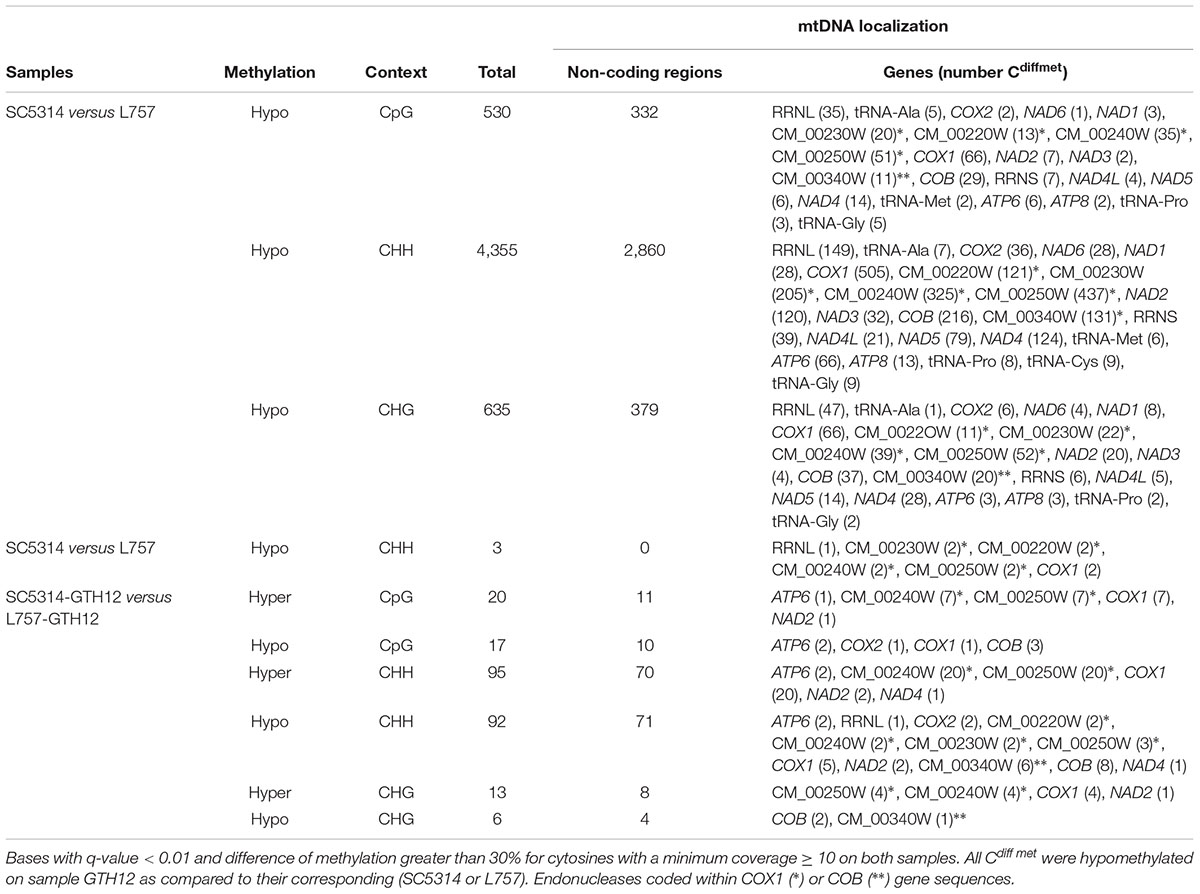
TABLE 1. Differentially methylated cytosines (Cdiffmet) between samples SC5314, SC5314-GTH12, L757, and L757-GTH12.
We observed that the C. albicans methylated mtDNA cytosines were mainly distributed inside introns, exons, and intergenic regions (Figure 1). C. albicans mtDNA transcription is polycistronic, and there are only eight promoters involved in the regulation of eight transcription units (TUs) (Kolondra et al., 2015). Among them, only TU7 promoter (CTCCTTATA), controlling the expression of NAD4L, NAD5 and three tRNAs genes, located between positions 29,532 and 29,540 bp, has cytosines in its sequence. Therefore, these data indicate that the methylation of mtDNA gene promoters is not a mechanism as relevant as the gene body methylation for C. albicans may be. For strain SC5314, cytosine methylation in this TU7 promoter was close to 98%, while the values dropped to 67% for sample SC5314-GTH12.
Using the Pearson correlation coefficient, we identified that the mtDNA methylation of cytosines between samples SC5314 and SC5314-GTH12 is not strongly correlated, with values ≤0.35 (0.35 CpG, 0.25 CHH, and 0.28 CHG), suggesting an environmental influence on the overall level of methylation of C. albicans mtDNA. Global methylation profiles were also analyzed by the Spearman correlation coefficient, with similar results (Bartelli, 2016).
In our samples, methylation occurred in the CpG and non-CpG (CHH and CHG) contexts, uniformly throughout the molecule, in both gene bodies and non-coding regions. In C. albicans, other authors (Mishra et al., 2011) observed that the methylation of nuclear genome cytosines is also distributed throughout the gene bodies, and the presence of methylation plays a direct role in the inhibition of transcription. In the human nuclear genome the methylation in the gene body is a very frequent phenomenon and CpG methylation in nuclear gene promoters may not play such a large role in the gene regulation, with histone acetylation or methylation being more relevant in this context (Maunakea et al., 2010). Increased methylation along the gene sequence is associated with increased transcription (Flanagan and Wild, 2007; Cokus et al., 2008; Rauch et al., 2009; Maunakea et al., 2010). In our samples, we observed a decrease in the overall methylation profile of the SC5314-GTH12 sample, which may be associated with the transcriptional profile of C. albicans in adaptation to hypoxia, which is characterized by increased expression of genes associated with glycolysis and decreased expression of genes involved in the mitochondrial tricarboxylic cycle (Krebs cycle) and oxidative phosphorylation, that are oxygen dependent (Setiadi et al., 2006; Synnott et al., 2010).
The mtDNA methylation pattern and its consequences to the cell are still poorly understood and may be associated with cellular and mitochondrial responses to environmental stressors (Shaughnessy et al., 2014). Although there are few studies, mtDNA methylation is likely to influence gene expression, biogenesis and mitochondrial functions (Iacobazzi et al., 2013). In humans, the mtDNA methyltransferase 1 (mtDNMT1) is associated with mtDNA CpG, especially in D-loop, where the origin of replication and promoters are located, and also in rRNA and protein-coding gene sequences (Shock et al., 2011). Non-methylated sites in the mtDNA are caused by proteins that hinder DNMT access and consequently, cytosine methylation (Rebelo et al., 2009). In humans, DNMTs have differential access to different mtDNA sites, based on the level of proteins in their nucleoids. The ratio between the concentrations of TFAM, the major constituent protein of human nucleoids, and mtDNA may be one of the major regulators of mitochondrial activity (e.g., replication and gene expression) and may vary according to the metabolic demand of the cell. Methylation levels may be influenced by factors known to alter nucleoid structure, such as a decreased TFAM/mtDNA ratio, which leads to a less compacted mtDNA that is more accessible to DNMT, and therefore, have higher methylation rates (Rebelo et al., 2009). Therefore, unlike the common effect expected in nuclear DNA, higher levels of methylation in mtDNA may be associated with its lower compaction and, consequently, increased gene activation. In S. cerevisiae, Abf2 level, which is homologous to TFAM, is known to be variable in yeast cells according to its metabolic necessity. The Abf2/mtDNA ratio is reduced under conditions that favor aerobic respiration, while their levels increase under conditions unfavorable to respiration (Xiong and Laird, 1997), suggesting an increase in mtDNA compaction under conditions unfavorable to respiration that would lead to lower methylation rates by hindering the access of DNMTs.
In our experiments we also observed that several sites remained with high methylation levels in SC5314-GTH12 or exhibited differential methylation between different strains (SC5314 and L757) (Figure 1 and Table 1). This probably occurs because the distribution of DNA binding proteins, such as the human TFAM, is not constant throughout mtDNA, and some regions have a more discrete decrease in methylation even with increased concentration of this protein in the cell (Rebelo et al., 2009). In C. albicans, nucleoid proteins, such as Gcf1 which is the most important described so far, participate in several functions, such as mtDNA replication (Visacka et al., 2009) and may have a role in the regulation of methylation of specific sites in the mtDNA to which they are associated and of the level of compactness they may induce to the molecule. When analyzing the Pearson correlation coefficients between global methylation profiles of samples SC5314 and SC5314-GTH12, we observed medium to low values of correlation and occurrence of differentially methylated sites between different culture conditions and strains. These results indicate that in addition to environmental conditions affecting the mtDNA methylation patterns, this response could be lineage-specific and related to adaptation and differential virulence of these strains, as previously described by our group (Padovan et al., 2009).
Accession Numbers
Raw sequencing data are available from the NIH SRA (BioProject PRJNA395439), under BioSample Accession Numbers SAMN07406734 (SC5314), SAMN07406735 (SC5314-GTH12), SAMN07406736 (L757), and SAMN07406737 (L757-GTH12).
Author Contributions
TB: planned and performed the experiments, analyzed the data, discussed the results and implications, performed the sequencing and bioinformatics analysis, and wrote the manuscript. DB: discussed the results and implications and edited the manuscript. MB: planned and supervised the experiments, data analysis, obtained funding, and edited the manuscript.
Funding
TB received a fellowship from Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP), Brazil (2013/08457-0). This work was supported by grants to MB from FAPESP (2013/07838-0) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Brazil (303905/2013-1).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Akalin, A., Kormaksson, M., Li, S., Garrett-Bakelman, F. E., Figueroa, M. E., Melnick, A., et al. (2012). methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 13:R87. doi: 10.1186/gb-2012-13-10-r87
Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data. Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc
Bambach, A., Fernandes, M. P., Ghosh, A., Kruppa, M., Alex, D., Li, D., et al. (2009). Goa1p of Candida albicans localizes to the mitochondria during stress and is required for mitochondrial function and virulence. Eukaryot. Cell 8, 1706–1720. doi: 10.1128/EC.00066-09
Bartelli, T. F. (2016). Mutational and Epigenetic Dynamics of Candida albicans Genome During in vitro Evolution under Hypoxia and Heat Shock. Ph.D. thesis, Universidade Federal de São Paulo, Sao Paulo.
Bartelli, T. F., Ferreira, R. C., Colombo, A. L., and Briones, M. R. (2013). Intraspecific comparative genomics of Candida albicans mitochondria reveals non-coding regions under neutral evolution. Infect. Genet. Evol. 14, 302–312. doi: 10.1016/j.meegid.2012.12.012
Bellizzi, D., D’Aquila, P., Scafone, T., Giordano, M., Riso, V., Riccio, A., et al. (2013). The control region of mitochondrial DNA shows an unusual CpG and non-CpG methylation pattern. DNA Res. 20, 537–547. doi: 10.1093/dnares/dst029
Brock, M. (2009). Fungal metabolism in host niches. Curr. Opin. Microbiol. 12, 371–376. doi: 10.1016/j.mib.2009.05.004
Brown, G. D., Denning, D. W., Gow, N. A., Levitz, S. M., Netea, M. G., and White, T. C. (2012). Hidden killers: human fungal infections. Sci. Transl. Med. 4:165rv13. doi: 10.1126/scitranslmed.3004404
Brown, A. J. P., Budge, S., Kaloriti, D., Tillmann, A., Jacobsen, M. D., Yin, Z., et al. (2014). Stress adaptation in a pathogenic fungus. J. Exp. Biol. 217, 144–155. doi: 10.1242/jeb.088930
Byun, H.-M., and Baccarelli, A. A. (2014). Environmental exposure and mitochondrial epigenetics: study design and analytical challenges. Hum. Genet. 133, 247–257. doi: 10.1007/s00439-013-1417-x
Chestnut, B. A., Chang, Q., Price, A., Lesuisse, C., Wong, M., and Martin, L. J. (2011). Epigenetic regulation of motor neuron cell death through DNA methylation. J. Neurosci. 31, 16619–16636. doi: 10.1523/JNEUROSCI.1639-11.2011
Choi, Y.-S., Hoon Jeong, J., Min, H.-K., Jung, H.-J., Hwang, D., Lee, S.-W., et al. (2011). Shot-gun proteomic analysis of mitochondrial D-loop DNA binding proteins: identification of mitochondrial histones. Mol. Biosyst. 7, 1523–1536. doi: 10.1039/c0mb00277a
Cokus, S. J., Feng, S., Zhang, X., Chen, Z., Merriman, B., Haudenschild, C. D., et al. (2008). Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 452, 215–219. doi: 10.1038/nature06745
Dzitoyeva, S., Chen, H., and Manev, H. (2012). Effect of aging on 5-hydroxymethylcytosine in brain mitochondria. Neurobiol. Aging 33, 2881–2891. doi: 10.1016/j.neurobiolaging.2012.02.006
Flanagan, J. M., and Wild, L. (2007). An epigenetic role for noncoding RNAs and intragenic DNA methylation. Genome Biol. 8:307. doi: 10.1186/gb-2007-8-6-307
Forche, A., Magee, P. T., Selmecki, A., Berman, J., and May, G. (2009). Evolution in Candida albicans populations during a single passage through a mouse host. Genetics 182, 799–811. doi: 10.1534/genetics.109.103325
Freire-Benéitez, V., Price, R. J., Tarrant, D., Berman, J., and Buscaino, A. (2016). Candida albicans repetitive elements display epigenetic diversity and plasticity. Sci. Rep. 6:22989. doi: 10.1038/srep22989
Ghosh, S., Sengupta, S., and Scaria, V. (2014). Comparative analysis of human mitochondrial methylomes shows distinct patterns of epigenetic regulation in mitochondria. Mitochondrion 18, 58–62. doi: 10.1016/j.mito.2014.07.007
Grahl, N., Shepardson, K. M., Chung, D., and Cramer, R. A. (2012). Hypoxia and fungal pathogenesis: to air or not to air? Eukaryot. Cell 11, 560–570. doi: 10.1128/EC.00031-12
Huffnagle, G. B., and Noverr, M. C. (2013). The emerging world of the fungal microbiome. Trends Microbiol. 21, 334–341. doi: 10.1016/j.tim.2013.04.002
Iacobazzi, V., Castegna, A., Infantino, V., and Andria, G. (2013). Mitochondrial DNA methylation as a next-generation biomarker and diagnostic tool. Mol. Genet. Metab. 110, 25–34. doi: 10.1016/j.ymgme.2013.07.012
Kim, J., Lee, J.-E., and Lee, J.-S. (2015). Histone deacetylase-mediated morphological transition in Candida albicans. J. Microbiol. 53, 805–811. doi: 10.1007/s12275-015-5488-3
Kohli, R. M., and Zhang, Y. (2013). TET enzymes, TDG and the dynamics of DNA demethylation. Nature 502, 472–479. doi: 10.1038/nature12750
Kolondra, A., Labedzka-Dmoch, K., Wenda, J. M., Drzewicka, K., and Golik, P. (2015). The transcriptome of Candida albicans mitochondria and the evolution of organellar transcription units in yeasts. BMC Genomics 16:827. doi: 10.1186/s12864-015-2078-z
Krueger, F., and Andrews, S. R. (2011). Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27, 1571–1572. doi: 10.1093/bioinformatics/btr167
Lopes da Rosa, J., and Kaufman, P. D. (2012). Chromatin-Mediated Candida albicans Virulence. Biochim. Biophys. Acta 1819, 349–355. doi: 10.1016/j.bbagrm.2011.08.007
Maunakea, A. K., Nagarajan, R. P., Bilenky, M., Ballinger, T. J., D’Souza, C., Fouse, S. D., et al. (2010). Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 466, 253–257. doi: 10.1038/nature09165
Mishra, P. K., Baum, M., and Carbon, J. (2011). DNA methylation regulates phenotype-dependent transcriptional activity in Candida albicans. Proc. Natl. Acad. Sci. U.S.A. 108, 11965–11970. doi: 10.1073/pnas.1109631108
Morschhäuser, J., Köhler, G., Ziebuhr, W., Blum-Oehler, G., Dobrindt, U., and Hacker, J. (2000). Evolution of microbial pathogens. Philos. Trans. R. Soc. B Biol. Sci. 355, 695–704. doi: 10.1098/rstb.2000.0609
Nicholls, S., Leach, M. D., Priest, C. L., and Brown, A. J. P. (2009). Role of the heat shock transcription factor, Hsf1, in a major fungal pathogen that is obligately associated with warm-blooded animals. Mol. Microbiol. 74, 844–861. doi: 10.1111/j.1365-2958.2009.06883.x
Padovan, A. C. B., Chaves, G. M., Colombo, A. L., and Briones, M. R. S. (2009). A novel allele of HWP1, isolated from a clinical strain of Candida albicans with defective hyphal growth and biofilm formation, has deletions of Gln/Pro and Ser/Thr repeats involved in cellular adhesion. Med. Mycol. 47, 824–835. doi: 10.3109/13693780802669574
Qu, Y., Jelicic, B., Pettolino, F., Perry, A., Lo, T. L., Hewitt, V. L., et al. (2012). Mitochondrial sorting and assembly machinery subunit Sam37 in Candida albicans: insight into the roles of mitochondria in fitness, cell wall integrity, and virulence. Eukaryot. Cell 11, 532–544. doi: 10.1128/EC.05292-11
Rauch, T. A., Wu, X., Zhong, X., Riggs, A. D., and Pfeifer, G. P. (2009). A human B cell methylome at 100-base pair resolution. Proc. Natl. Acad. Sci. U.S.A. 106, 671–678. doi: 10.1073/pnas.0812399106
Rebelo, A. P., Williams, S. L., and Moraes, C. T. (2009). In vivo methylation of mtDNA reveals the dynamics of protein-mtDNA interactions. Nucleic Acids Res. 37, 6701–6715. doi: 10.1093/nar/gkp727
Robinson, J. T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., et al. (2011). Integrative genomics viewer. Nat. Biotechnol. 29, 24–26. doi: 10.1038/nbt.1754
Setiadi, E. R., Doedt, T., Cottier, F., Noffz, C., and Ernst, J. F. (2006). Transcriptional response of Candida albicans to hypoxia: linkage of oxygen sensing and Efg1p-regulatory networks. J. Mol. Biol. 361, 399–411. doi: 10.1016/j.jmb.2006.06.040
Shaughnessy, D. T., McAllister, K., Worth, L., Haugen, A. C., Meyer, J. N., Domann, F. E., et al. (2014). Mitochondria, energetics, epigenetics, and cellular responses to stress. Environ. Health Perspect. 122, 1271–1278. doi: 10.1289/ehp.1408418
Shock, L. S., Thakkar, P. V., Peterson, E. J., Moran, R. G., and Taylor, S. M. (2011). DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc. Natl. Acad. Sci. U.S.A. 108, 3630–3635. doi: 10.1073/pnas.1012311108
Synnott, J. M., Guida, A., Mulhern-Haughey, S., Higgins, D. G., and Butler, G. (2010). Regulation of the hypoxic response in Candida albicans. Eukaryot. Cell 9, 1734–1746. doi: 10.1128/EC.00159-10
Thomas, E., Roman, E., Claypool, S., Manzoor, N., Pla, J., and Panwar, S. L. (2013). Mitochondria influence CDR1 efflux pump activity, Hog1-mediated oxidative stress pathway, iron homeostasis, and ergosterol levels in Candida albicans. Antimicrob. Agents Chemother. 57, 5580–5599. doi: 10.1128/AAC.00889-13
Tscherner, M., Zwolanek, F., Jenull, S., Sedlazeck, F. J., Petryshyn, A., Frohner, I. E., et al. (2015). The Candida albicans histone acetyltransferase hat1 regulates stress resistance and virulence via distinct chromatin assembly pathways. PLoS Pathog. 11:e1005218. doi: 10.1371/journal.ppat.1005218
Underhill, D. M., and Iliev, I. D. (2014). The mycobiota: interactions between commensal fungi and the host immune system. Nat. Rev. Immunol. 14, 405–416. doi: 10.1038/nri3684
Visacka, K., Gerhold, J. M., Petrovicova, J., Kinsky, S., Jõers, P., Nosek, J., et al. (2009). Novel subfamily of mitochondrial HMG box-containing proteins: functional analysis of Gcf1p from Candida albicans. Microbiology 155, 1226–1240. doi: 10.1099/mic.0.025759-0
Wach, A., Pick, H., and Philippsen, P. (1994). “Procedures for isolating yeast DNA for different purposes,” in Molecular Genetics of Yeast, ed. J. R. Johnston (Oxford: IRL Press).
Wang, H.-Q., Tuominen, L. K., and Tsai, C.-J. (2011). SLIM: a sliding linear model for estimating the proportion of true null hypotheses in datasets with dependence structures. Bioinformatics 27, 225–231. doi: 10.1093/bioinformatics/btq650
Wartenberg, A., Linde, J., Martin, R., Schreiner, M., Horn, F., Jacobsen, I. D., et al. (2014). Microevolution of Candida albicans in macrophages restores filamentation in a nonfilamentous mutant. PLoS Genet. 10:e1004824. doi: 10.1371/journal.pgen.1004824
Xiong, Z., and Laird, P. W. (1997). COBRA: a sensitive and quantitative DNA methylation assay. Nucleic Acids Res. 25, 2532–2534. doi: 10.1093/nar/25.12.2532
Zhang, A., Liu, Z., and Myers, L. C. (2013). Differential regulation of white-opaque switching by individual subunits of Candida albicans mediator. Eukaryot. Cell 12, 1293–1304. doi: 10.1128/EC.00137-13
Keywords: Candida albicans, mitochondrial genome methylation, mtDNA, hypoxia, heat shock
Citation: Bartelli TF, Bruno DCF and Briones MRS (2018) Evidence for Mitochondrial Genome Methylation in the Yeast Candida albicans: A Potential Novel Epigenetic Mechanism Affecting Adaptation and Pathogenicity? Front. Genet. 9:166. doi: 10.3389/fgene.2018.00166
Received: 27 November 2017; Accepted: 26 April 2018;
Published: 29 May 2018.
Edited by:
Trygve Tollefsbol, University of Alabama at Birmingham, United StatesReviewed by:
Luis G. Brieba, Centro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional, MexicoTomas J. Ekstrom, Karolinska Institute (KI), Sweden
Copyright © 2018 Bartelli, Bruno and Briones. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thais F. Bartelli, dGhhaXMuYmFydGVsbGlAaG90bWFpbC5jb20=