 Gonzalo Cordova
Gonzalo Cordova Elisa Negroni1
Elisa Negroni1 Claudio Cabello-Verrugio
Claudio Cabello-Verrugio Vincent Mouly
Vincent Mouly Capucine Trollet
Capucine Trollet- 1Sorbonne Université, Institut National de la Santé et de la Recherche Médicale, Association Institut de Myologie, Centre de Recherche en Myologie, UMRS974, Paris, France
- 2Laboratorio de Patologías Musculares, Fragilidad y Envejecimiento, Departamento de Ciencias Biológicas, Facultad de Ciencias Biológicas, Universidad Andres Bello, Santiago, Chile
- 3Millennium Institute on Immunology and Immunotherapy, Santiago, Chile
Duchene Muscular Dystrophy (DMD) is the most frequent muscular dystrophy and one of the most severe due to the absence of the dystrophin protein. Typical pathological features include muscle weakness, muscle wasting, degeneration, and inflammation. At advanced stages DMD muscles present exacerbated extracellular matrix and fat accumulation. Recent progress in therapeutic approaches has allowed new strategies to be investigated, including pharmacological, gene-based and cell-based therapies. Gene and cell-based therapies are still limited by poor targeting and low efficiency in fibrotic dystrophic muscle, therefore it is increasingly evident that future treatments will have to include “combined therapies” to reach maximal efficiency. The scope of this mini-review is to provide an overview of the current literature on such combined therapies for DMD. By “combined therapies” we mean those that include both a therapy to correct the genetic defect and an additional one to address one of the secondary pathological features of the disease. In this mini-review, we will not provide a comprehensive view of the literature on therapies for DMD, since many such reviews already exist, but we will focus on the characteristics, efficiency, and potential of such combined therapeutic strategies that have been described so far for DMD.
Introduction
Duchenne Muscular Dystrophy (DMD), the muscular dystrophy, is an X-linked recessive disease that affects one in 3,500 live male births (Bushby et al., 2010). DMD patients present progressive muscular weakness, in addition to orthopedic, respiratory, and cardiac complications that lead to their death around the third or fourth decade of life (McNally, 2007; Bushby et al., 2010). At the molecular level, DMD is caused by mutations in the dystrophin gene leading to the absence of the protein (Koenig et al., 1987; Kunkel et al., 1987). The dystrophin gene is one of the largest genes in the human genome with more than 2 million base pairs in Xp21.2-p21.1. The size of the dystrophin coding sequence (11 kbp) is huge with 79 exons encoding a 427 kDa protein (Guiraud et al., 2015). Most of the DMD patients carry out-of-frame and non-sense mutations leading to reduction of the transcript level and truncation of translation (Monaco et al., 1988; Roberts et al., 1994). Dystrophin is a cytoskeletal protein critical for the stability and function of myofibers in muscle: dystrophin establishes a mechanical link between the extracellular matrix and the cytoskeletal actin in muscle fibers through the dystrophin-associated protein complex (DAPC) (Ervasti and Sonnemann, 2008). Dystrophin deficiency leads to the rupture of the muscle fiber membrane during contraction (Allen and Whitehead, 2011) and causes impaired intracellular signaling (Constantin, 2014). At the cellular level, the muscles of DMD patients show evidence of necrosis, degeneration and regeneration, myofiber atrophy, fatty accumulation, fibrosis, and inflammation (Spencer and Tidball, 2001; Alvarez et al., 2002; Desguerre et al., 2009a,b; Serrano and Muñoz-Cá-noves, 2010; Zhou and Lu, 2010; Villalta et al., 2011). Different approaches (gene-based, cell-based, nano-particles, and pharmacological) have been developed to restore a functional dystrophin to DMD muscles (Negroni et al., 2016; Chamberlain and Chamberlain, 2017; Nance et al., 2017). These strategies are promising and several clinical trials are on-going or have been conducted on DMD patients: between 1995 and 2018, 127 clinical trials are found on clinicaltrials.gov, with 57% pharmacological approaches, 28% gene-based (22% antisense oligonucleotide based exon skipping, 6% AAV gene addition), and 3% cell-based approaches. To maximize the efficiency of gene- and cell-based approaches, future therapies will have to take into account the state of the muscle tissue and the secondary modifications associated to the genetic defect. For example, the integrity of the sarcolemma of muscle fibers, essential for efficient and long term gene therapy, is severely compromised in DMD (McElhanon and Bhattacharya, 2018), which leads to the concomitant loss of the therapeutic agent (Le Hir et al., 2013). In addition, in dystrophic muscle the continuous breakdown of muscle fibers causes inflammation and fibrosis (Serrano and Muñoz-Cánoves, 2017). Such a hostile environment will be detrimental for the efficacy of cell-based therapies and exacerbated extracellular matrix will affect the accessibility of all therapeutic agents to the muscle fibers (gene, cell, or pharmacological).
The aim of this review is to highlight pre-clinical studies in DMD that have tested two therapeutic strategies in combination (Figure 1). We will only focus on studies that have used “combined therapies”: one to correct the genetic defect, and a second to improve the status of the recipient muscle. To improve the dystrophic environment of the recipient tissue, strategies are being developed to eliminate the barriers that limit the access of the therapeutic vector to the fibers and to limit degeneration—even temporarily—to allow dystrophin to reach therapeutic expression levels. “Muscle conditioning” treatments ameliorating the status of the targeted muscle are being developed. Such improvements per se are beneficial to the muscle and will also improve the efficacy of gene or cell-based therapy. The final goal of any combined therapy should be to improve the efficacy of the single target therapies. Since an extensive literature on the different single target strategies developed for DMD already exist, we will not describe these here, nor shall we discuss animal models used for the same reason.
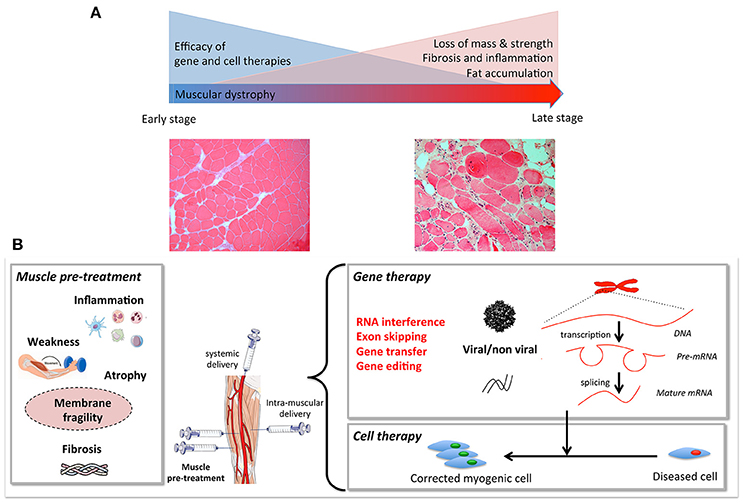
Figure 1. (A) Muscular dystrophies fibers, including loss of mass, weakness, fat, and extracellular matrix accumulation. Gene and cell based therapies will have to overcome the progressive degeneration of muscle fibers. When these histological changes become prominent, combined strategies are needed. (B) Muscle pre- or co-treatment may target inflammation, atrophy, membrane fragility, muscle weakness, and/or atrophy to pre-condition the tissue to increase efficiency of gene and cell therapy.
Improving Dystrophin Expression Using Combined Therapy
Exon skipping approaches have already shown promising results in animal models. This therapy is based on the use of antisense oligonucleotides (AON), that will interfere with the normal splicing process removing the mutation-carrying exons, allowing the production of a truncated but still functional dystrophin (Nakamura, 2017). Indeed the dystrophin structure with its central rod-domain made of 24 spectrin-like repeats, can tolerate large internal deletions while maintaining most of its function. In an elegant study, Peccate et al. recently demonstrated that a pre-treatment of the skeletal muscle of mdx mice (the most common mouse model for DMD; Bulfield et al., 1984) with peptide-phosphorodiamidate morpholino (PPMO) antisense oligonucleotides targeting dystrophin was beneficial for a subsequent AAV-based exon-skipping therapy (Peccate et al., 2016). This pre-treatment allowed temporary restoration of dystrophin at the sarcolemma, improving membrane integrity to reduce the loss of vector genome after AAV injection and improve the efficiency of gene therapy. This study emphasizes the strong potential of combined approaches to improve the benefit of AAV-based therapies since without pre-treatment the viral vector would be lost when the muscle fibers degenerate. For DMD, such pre-treatment would allow the use of lower and thus safer vector doses for a higher level of dystrophin expression in the long term. Such pre-treatment aiming at improving muscle fiber integrity could benefit also to other muscular dystrophies with degenerative features. The efficiency of exon-skipping can also be targeted. Using high-throughput screening, Kendall et al. identified Dantrolene—currently used to treat malignant hyperthermia—as a “skipping enhancer” (Kendall et al., 2012). This drug delivered to mdx mice by intraperitoneal injections enhanced antisense oligonucleotide (AON)-mediated DMD exon skipping. The use of such an enhancer will improve AON treatment by increasing the therapeutic value of AON, reducing the dose needed, and thus lowering the costs and potential toxicity. Finally, nanotechnologies have also been used to deliver therapeutic agents, such as antisense nucleotides (for a review see Falzarano et al., 2014). Such tools might in the future be used in combined therapeutic strategies.
Stimulation of Muscle Growth and Regeneration
If muscle wasting has already progressed, dystrophin expression in the surviving fibers will not be sufficient to restore function. Maintaining and stimulating higher levels of muscle regeneration could potentially have a beneficial effect in dystrophic muscles. The first attempt of a combined therapy stimulating muscle growth came from Abmayr et al. (2005), who used the co-expression of Insulin-like Growth factor-1 (IGF-1)—a known inducer of muscle hypertrophy, strength and regeneration (Philippou and Barton, 2014)—together with the expression of a functional microdystrophin (μDys) in mdx mice. Muscles treated with this combined therapy, showed increased muscle mass and specific force compared to untreated or to muscles treated with μDys alone. A similar approach was used by Rodino-Klapac et al. (2013) by combining follistatin—an inhibitor of myostatin (Sharma et al., 2015)—to increase muscle mass and strength, and μDys. They showed a potent synergistic effect of the combined therapy on muscle force and architecture. This was also demonstrated with an AON triggering exon skipping of dystrophin and another one targeting myostatin to improve muscle weakness (Kemaladewi et al., 2011; Lu-Nguyen et al., 2017) with promising results. Similar approaches to interfere with the myostatin pathways have also been used in combined therapies: RNA interference for the Activin Receptor type IIb (AcvRIIB) (Dumonceaux et al., 2010)—the receptor for Myostatin—or a soluble version of AcvRIIB (Hoogaars et al., 2012) have been used in combination with AAV-U7 based exon skipping resulting in a beneficial effect increasing both muscle mass and strength.
Controlling Fibrosis, Inflammation, and Atrophy
Fibrosis, inflammation and muscle atrophy are among the most important complications associated with muscular dystrophy, and they can severely compromise the efficiency of gene or cell therapy by limiting access to the dystrophic muscle. Fibrosis can be defined as the increased expression and accumulation of Extracellular Matrix (ECM) proteins, such as fibronectin and collagen, which contributes to muscle dysfunction (Serrano and Muñoz-Cánoves, 2017). Transforming Growth Factor type β (TGF-β) is a potent pro-fibrotic cytokine that contributes to the pathogenesis of several fibrotic disorders, including muscular dystrophies (Bernasconi et al., 1999). Interestingly, it has been found that TGF-β induces the expression of Connective Tissue Growth Factor (CTGF/CCN2) in fibroblasts (Igarashi et al., 1993) and the pro-fibrotic effects of TGF-β may be CTGF-dependent (Grotendorst, 1997; Leask and Abraham, 2004; Leask et al., 2004). The expression of microRNA-29—a family of microRNAs whose downregulation is associated with fibrosis—not only decreased TGF-β1 and ECM proteins expression but also completely restored muscle strength in dystrophic muscle when combined with μDys treatment (Heller et al., 2017). Similarly, reducing CTGF expression genetically or blocking CTGF with neutralizing antibodies, decreased fibrosis, and increased muscle strength and the efficiency of cell therapy (Morales et al., 2013b). Combining different cell types in cell therapy has also been shown to improve the fibrotic environment in dystrophic mice. Gargioli et al. showed that a pre-treatment using modified tendon fibroblasts, expressing angiogenic factors such as placenta growth factor and an antifibrotic treatment using MMP-9, improved microcirculation, reduced collagen and fat tissue deposition, decreased leukocyte infiltration, increased fiber numbers and improved cell-therapy in aged α-Sarcoglycan null mice, a model of Limb-girdle muscular dystrophy (Gargioli et al., 2008).
Inflammation is part of the normal regeneration process where macrophages play a fundamental role in both inflammation and regeneration by the sequential expression of cytokines and inflammatory molecules (Juban and Chazaud, 2017). It is now well-established that the inflammatory response in damaged muscle positively influences normal muscle repair, while its exacerbation in dystrophic muscle promotes the formation of fibrotic tissue during disease progression (Tidball, 2005). For these reasons, targeting inflammation may improve therapies for DMD (Miyatake et al., 2016). Several different combined approaches have been used to decrease inflammation and improve therapeutic outcome: anti-inflammatory prednisolone combined with AON exon skipping treatment has been shown to increase dystrophin expression (Verhaart et al., 2012). A study by Cabrera et al. (2014) showed that andrographolide—an inhibitor of NF-κB (pro-inflammatory pathway implicated in atrophy and fibrosis; Li et al., 2008)—reduces the expression of fibrotic factors and ECM proteins, while increasing muscle strength and cell therapy efficacy. Another study showed that treatment with HCT 1026—a non-steroidal anti-inflammatory drug capable of releasing nitric oxide (NO)—increased the efficiency of cell therapy in mdx mice and in a mouse model of limb girdle muscular dystrophy (Brunelli et al., 2007). NO deficiency in DMD, due to the disappearance of nNOS linked to the dystrophin complex, is also an important issue since it is a potent regulator of skeletal muscle physiology and regeneration, and could also be targeted in combined therapies (Timpani et al., 2017).
Muscular atrophy is a common feature of DMD and many pathological processes discussed in this mini-review contribute to muscle wasting (Shin et al., 2013). One of the pathways involved in the regulation of muscle mass is the Renin-Angiotensin System (RAS) (Cabello-Verrugio et al., 2012a, 2015). Several of its components are upregulated in dystrophic muscles (Sun et al., 2009) where they can also trigger a fibrotic response. Pharmacological modulation of RAS can be used to decrease atrophy (Burks et al., 2011), decrease fibrosis (Cabello-Verrugio et al., 2012b; Morales et al., 2013a; Acuna et al., 2014) and ameliorate cardiac complications related to MD (Allen et al., 2013; Sabharwal et al., 2014). Several studies have used combined therapies using Losartan, an inhibitor or the AT-1 receptor. Losartan treatment has been shown to increase the efficiency of myoblast cell therapy (Fakhfakh et al., 2012) and Adipose-Derived Stem Cell therapy (Lee et al., 2015). However, a study by Lee et al. (2014) showed that although combined therapy with Losartan and exon skipping was beneficial in terms of muscle regeneration, the efficiency of exon skipping was lower in Losartan treated mice due to decreased in vivo morpholino penetration. In this study, Losartan was added prior to morpholino treatment, it would be interesting to see what happens when losartan treatment is started after the exon skipping treatment. Moreover, as Losartan seems to increase sarcolemma stability, it could be interesting to see the effect of viral gene therapy after Losartan treatment.
In Vitro Modification of Myogenic Cells
Among “combined therapies” those combining gene- and cell- therapies should be mentioned, even though none of the two strategies improve the status of the recipient muscle. Most combined gene- and cell-based studies developed so far consist in genetic modification of adult stem cells harvested from patients or dystrophic models to produce a functional dystrophin protein. In order to accommodate DNA packaging limitations in a range of viral vectors, synthetic mini- and micro-dystrophin versions have been engineered (Athanasopoulos et al., 2004) and tested in cell transplantation studies using lentiviral vectors. Several transduced types of myogenic cells, e.g., mouse, canine, primate, and human muscle precursors (Ikemoto et al., 2007; Quenneville et al., 2007b; Pichavant et al., 2010), murine side population (SP) cells (Bachrach et al., 2004), canine (Sampaolesi et al., 2006), and human (Dellavalle et al., 2007) mesoangioblasts/pericytes, have been tested in dystrophic models. The group of J. Tremblay demonstrated that the use of electroporation combined with the introduction of a phiC31 integrase led to the stable expression of full-length dystrophin in murine and human MPCs, even if this technique is less efficient than viral vector transduction (Quenneville et al., 2007a). Kazuki et al. have also validated the use of a human artificial chromosome (HAC) to restore full-length dystrophin in mouse and human iPS cells (Kazuki et al., 2010), while genomic integration of the full-length human dystrophin has been achieved in iPS cells (Farruggio et al., 2017) and mesangioblasts (Loperfido et al., 2016). Also, a full-length dystrophin was efficiently expressed in dog mesoangioblasts using piggyBac transposons (Loperfido et al., 2016). The same technique was used to modify mouse mesoangioblasts prior to transplantation in mdx mice, showing a good level of dystrophin expression, increased number of satellite cells, reduction in fibrosis, and increased muscle function (Iyer et al., 2018). Exon skipping has also been tested in combination with cell therapy approaches using targeted antisense sequences vectorised in U7 snRNA constructs in skin fibroblasts (Chaouch et al., 2009) or CD133+ cells of DMD patients (Benchaouir et al., 2007).
More recently, direct targeting of a morbid allele has been challenged using nucleases in vitro and in vivo: meganucleases, Zinc-finger nucleases, TALENs, and CRISPR have all been used for genome editing to correct DMD cells carrying deletions and out-of-frame mutations in dystrophin gene (Ousterout et al., 2013, 2015b; Popplewell et al., 2013; Young et al., 2016; Gee et al., 2017; Pini et al., 2017; Reinig et al., 2017; Wang et al., 2017; Zhu et al., 2017). A recent study also described a multiplexed strategy using a lentiviral vector capable of editing multiple sequences at a time, allowing the correction of up to 62% of mutations causing DMD (Ousterout et al., 2015a). While none of these approaches have yet been used to our knowledge in combined therapies, they could also profit from such strategies.
Concluding Remarks
Combined Therapies for DMD
There is now an increased interest in developing combined therapies for DMD (Table 1). A combined therapy is most often designed to treat the secondary consequences of the muscular dystrophy that decrease the efficiency of single therapies, e.g., inflammation, fibrosis, or degeneration. If efficient, this therapy should have two effects: (1) improve the muscle phenotype per se and, (2) improve the combined therapy by pre- or co-conditioning the muscle that is receiving the treatment. In other words, combined therapies should have a synergistic effect. It is essential to combine the positive outcomes of distinct therapies that target these different features to enhance therapeutic efficiency. Many combinatory studies could be tested, just to name one for example, it would be interesting to know if SMT-C1100—an utrophin upregulation drug (Tinsley et al., 2011), already tested in clinical trials with promising results and no side-effects (Ricotti et al., 2016)—shows a synergistic effect when combined with other therapies, like cell therapy.

Table 1. Overview of synergistic therapies tested in muscular dystrophies.
Of course, the advances in combined therapies should not stop the efforts that have been conducted to ameliorate single target therapies for DMD since they will eventually also benefit to these combined therapies. Future technical advances in distinct approaches will help to improve combined therapeutic assays that will eventually lead to the effective treatment or even a cure for DMD.
Early Detection and Treatment vs. Symptomatic Patients
It is important to make this distinction. The early detection of dystrophin deficiency and a precise genetic diagnosis (Aartsma-Rus et al., 2016) will allow the treatment to be started before the onset of fibrosis, chronic damage, and inflammation in the muscle. A genetic correction might then be enough to avoid the progression of the disease. With this in mind, a screening test for the presence of a fully functional dystrophin for all male newborns could potentially result in an invaluable social and monetary benefit for the families and the health care system (Landfeldt et al., 2016; Ryder et al., 2017). However, the diagnosis for sporadic mutations is usually done when the patients start to show their first symptoms in early childhood and, at this moment, muscles already show extensive damage, inflammation and fibrosis. In this case, secondary effects of the gene deficiency should be addressed in combined therapies to enhance the correction of the genetic defect.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Centre National pour la Recherche Scientifique, the Association Française contre les Myopathies [Research Program 17110], the University Paris VI Pierre et Marie Curie, the Institut National de la Santé et de la Recherche Médicale, the ECOS-CONICYT program (action #C16S02), Fondecyt 1161646, Millennium Institute on Immunology and Immunotherapy [P09-016- F].
References
Aartsma-Rus, A., Ginjaar, I. B., and Bushby, K. (2016). The importance of genetic diagnosis for Duchenne muscular dystrophy. J. Med. Genet. 53, 145–151. doi: 10.1136/jmedgenet-2015-103387
Abmayr, S., Gregorevic, P., Allen, J. M., and Chamberlain, J. S. (2005). Phenotypic improvement of dystrophic muscles by rAAV/microdystrophin vectors is augmented by Igf1 codelivery. Mol. Ther. 12, 441–450. doi: 10.1016/j.ymthe.2005.04.001
Acuña, M. J., Pessina, P., Olguin, H., Cabrera, D., Vio, C. P., Bader, M., et al. (2014). Restoration of muscle strength in dystrophic muscle by angiotensin-1-7 through inhibition of TGF-beta signalling. Hum. Mol. Genet. 23, 1237–1249. doi: 10.1093/hmg/ddt514
Allen, D. G., and Whitehead, N. P. (2011). Duchenne muscular dystrophy–what causes the increased membrane permeability in skeletal muscle? Int. J. Biochem. Cell Biol. 43, 290–294. doi: 10.1016/j.biocel.2010.11.005
Allen, H. D., Flanigan, K. M., Thrush, P. T., Dvorchik, I., Yin, H., Canter, C., et al. (2013). A randomized, double-blind trial of lisinopril and losartan for the treatment of cardiomyopathy in Duchenne muscular dystrophy. PLoS Curr. 5. doi: 10.1371/currents.md.2cc69a1dae4be7dfe2bcb420024ea865
Alvarez, K., Fadic, R., and Brandan, E. (2002). Augmented synthesis and differential localization of heparan sulfate proteoglycans in Duchenne muscular dystrophy. J. Cell. Biochem. 85, 703–713. doi: 10.1002/jcb.10184
Athanasopoulos, T., Graham, I. R., Foster, H., and Dickson, G. (2004). Recombinant adeno-associated viral (rAAV) vectors as therapeutic tools for Duchenne muscular dystrophy (DMD). Gene Ther. 11 (Suppl. 1), S109–S121. doi: 10.1038/sj.gt.3302379
Bachrach, E., Li, S., Perez, A. L., Schienda, J., Liadaki, K., Volinski, J., et al. (2004). Systemic delivery of human microdystrophin to regenerating mouse dystrophic muscle by muscle progenitor cells. Proc. Natl. Acad. Sci. U.S.A. 101, 3581–3586. doi: 10.1073/pnas.0400373101
Benchaouir, R., Meregalli, M., Farini, A., D'Antona, G., Belicchi, M., Goyenvalle, A., et al. (2007). Restoration of human dystrophin following transplantation of exon-skipping-engineered DMD patient stem cells into dystrophic mice. Cell Stem Cell 1, 646–657. doi: 10.1016/j.stem.2007.09.016
Bernasconi, P., Di Blasi, C., Mora, M., Morandi, L., Galbiati, S., Confalonieri, P., et al. (1999). Transforming growth factor-beta1 and fibrosis in congenital muscular dystrophies. Neuromuscul. Disord. 9, 28–33. doi: 10.1016/S0960-8966(98)00093-5
Brunelli, S., Sciorati, C., D'Antona, G., Innocenzi, A., Covarello, D., Galvez, B. G., et al. (2007). Nitric oxide release combined with nonsteroidal antiinflammatory activity prevents muscular dystrophy pathology and enhances stem cell therapy. Proc. Natl. Acad. Sci. U.S.A. 104, 264–269. doi: 10.1073/pnas.0608277104
Bulfield, G., Siller, W. G., Wight, P. A., and Moore, K. J. (1984). X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl. Acad. Sci. U.S.A. 81, 1189–1192. doi: 10.1073/pnas.81.4.1189
Burks, T. N., Andres-Mateos, E., Marx, R., Mejias, R., Van Erp, C., Simmers, J. L., et al. (2011). Losartan restores skeletal muscle remodeling and protects against disuse atrophy in sarcopenia. Sci. Transl. Med. 3:82ra37. doi: 10.1126/scitranslmed.3002227
Bushby, K., Finkel, R., Birnkrant, D. J., Case, L. E., Clemens, P. R., Cripe, L., et al. (2010). Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 9, 77–93. doi: 10.1016/S1474-4422(09)70271-6
Cabello-Verrugio, C., Córdova, G., and Salas, J. D. (2012a). Angiotensin II: role in skeletal muscle atrophy. Curr. Protein Pept. Sci. 13, 560–569. doi: 10.2174/138920312803582933
Cabello-Verrugio, C., Morales, M. G., Cabrera, D., Vio, C. P., and Brandan, E. (2012b). Angiotensin II receptor type 1 blockade decreases CTGF/CCN2-mediated damage and fibrosis in normal and dystrophic skeletal muscles. J. Cell. Mol. Med. 16, 752–764. doi: 10.1111/j.1582-4934.2011.01354.x
Cabello-Verrugio, C., Morales, M. G., Rivera, J. C., Cabrera, D., and Simon, F. (2015). Renin-angiotensin system: an old player with novel functions in skeletal muscle. Med. Res. Rev. 35, 437–463. doi: 10.1002/med.21343
Cabrera, D., Gutiérrez, J., Cabello-Verrugio, C., Morales, M. G., Mezzano, S., Fadic, R., et al. (2014). Andrographolide attenuates skeletal muscle dystrophy in mdx mice and increases efficiency of cell therapy by reducing fibrosis. Skelet. Muscle 4:6. doi: 10.1186/2044-5040-4-6
Chamberlain, J. R., and Chamberlain, J. S. (2017). Progress toward Gene Therapy for Duchenne Muscular Dystrophy. Mol. Ther. 25, 1125–1131. doi: 10.1016/j.ymthe.2017.02.019
Chaouch, S., Mouly, V., Goyenvalle, A., Vulin, A., Mamchaoui, K., Negroni, E., et al. (2009). Immortalized skin fibroblasts expressing conditional MyoD as a renewable and reliable source of converted human muscle cells to assess therapeutic strategies for muscular dystrophies: validation of an exon-skipping approach to restore dystrophin in Duchenne muscular dystrophy cells. Hum. Gene Ther. 20, 784–790. doi: 10.1089/hum.2008.163
Constantin, B. (2014). Dystrophin complex functions as a scaffold for signalling proteins. Biochim. Biophys. Acta 1838, 635–642. doi: 10.1016/j.bbamem.2013.08.023
Dellavalle, A., Sampaolesi, M., Tonlorenzi, R., Tagliafico, E., Sacchetti, B., Perani, L., et al. (2007). Pericytes of human skeletal muscle are myogenic precursors distinct from satellite cells. Nat. Cell Biol. 9, 255–267. doi: 10.1038/ncb1542
Desguerre, I., Christov, C., Mayer, M., Zeller, R., Becane, H. M., Bastuji-Garin, S., et al. (2009a). Clinical heterogeneity of Duchenne muscular dystrophy (DMD): definition of sub-phenotypes and predictive criteria by long-term follow-up. PLoS ONE 4:e4347. doi: 10.1371/journal.pone.0004347
Desguerre, I., Mayer, M., Leturcq, F., Barbet, J. P., Gherardi, R. K., and Christov, C. (2009b). Endomysial fibrosis in Duchenne muscular dystrophy: a marker of poor outcome associated with macrophage alternative activation. J. Neuropathol. Exp. Neurol. 68, 762–773. doi: 10.1097/NEN.0b013e3181aa31c2
Dumonceaux, J., Marie, S., Beley, C., Trollet, C., Vignaud, A., Ferry, A., et al. (2010). Combination of myostatin pathway interference and dystrophin rescue enhances tetanic and specific force in dystrophic mdx mice. Mol. Ther. 18, 881–887. doi: 10.1038/mt.2009.322
Ervasti, J. M., and Sonnemann, K. J. (2008). Biology of the striated muscle dystrophin-glycoprotein complex. Int. Rev. Cytol. 265, 191–225. doi: 10.1016/S0074-7696(07)65005-0
Fakhfakh, R., Lamarre, Y., Skuk, D., and Tremblay, J. P. (2012). Losartan enhances the success of myoblast transplantation. Cell Transplant. 21, 139–152. doi: 10.3727/096368911X576045
Falzarano, M. S., Passarelli, C., and Ferlini, A. (2014). Nanoparticle delivery of antisense oligonucleotides and their application in the exon skipping strategy for Duchenne muscular dystrophy. Nucleic. Acid Ther. 24, 87–100. doi: 10.1089/nat.2013.0450
Farruggio, A. P., Bhakta, M. S., du Bois, H., Ma, J., and P Calos, M. (2017). Genomic integration of the full-length dystrophin coding sequence in Duchenne muscular dystrophy induced pluripotent stem cells. Biotechnol. J. 12. doi: 10.1002/biot.201600477
Gargioli, C., Coletta, M., De Grandis, F., Cannata, S. M., and Cossu, G. (2008). PlGF-MMP-9-expressing cells restore microcirculation and efficacy of cell therapy in aged dystrophic muscle. Nat. Med. 14, 973–978. doi: 10.1038/nm.1852
Gee, P., Xu, H., and Hotta, A. (2017). Cellular reprogramming, genome editing, and alternative CRISPR Cas9 technologies for precise gene therapy of Duchenne muscular dystrophy. Stem Cells Int. 2017:8765154. doi: 10.1155/2017/8765154
Grotendorst, G. R. (1997). Connective tissue growth factor: a mediator of TGF-beta action on fibroblasts. Cytokine Growth Factor Rev. 8, 171–179. doi: 10.1016/S1359-6101(97)00010-5
Guiraud, S., Aartsma-Rus, A., Vieira, N. M., Davies, K. E., van Ommen, G. J., and Kunkel, L. M. (2015). The pathogenesis and therapy of muscular dystrophies. Annu. Rev. Genomics Hum. Genet. 16, 281–308. doi: 10.1146/annurev-genom-090314-025003
Heller, K. N., Mendell, J. T., Mendell, J. R., and Rodino-Klapac, L. R. (2017). MicroRNA-29 overexpression by adeno-associated virus suppresses fibrosis and restores muscle function in combination with micro-dystrophin. JCI Insight 2:93309. doi: 10.1172/jci.insight.93309
Hoogaars, W. M., Mouisel, E., Pasternack, A., Hulmi, J. J., Relizani, K., Schuelke, M., et al. (2012). Combined effect of AAV-U7-induced dystrophin exon skipping and soluble activin Type IIB receptor in mdx mice. Hum. Gene Ther. 23, 1269–1279. doi: 10.1089/hum.2012.056
Igarashi, A., Okochi, H., Bradham, D. M., and Grotendorst, G. R. (1993). Regulation of connective tissue growth factor gene expression in human skin fibroblasts and during wound repair. Mol. Biol. Cell 4, 637–645. doi: 10.1091/mbc.4.6.637
Ikemoto, M., Fukada, S., Uezumi, A., Masuda, S., Miyoshi, H., Yamamoto, H., et al. (2007). Autologous transplantation of SM/C-2.6(+) satellite cells transduced with micro-dystrophin CS1 cDNA by lentiviral vector into mdx mice. Mol. Ther. 15, 2178–2185. doi: 10.1038/sj.mt.6300295
Iyer, P. S., Mavoungou, L. O., Ronzoni, F., Zemla, J., Schmid-Siegert, E., Antonini, S., et al. (2018). Autologous cell therapy approach for Duchenne muscular dystrophy using piggybac transposons and mesoangioblasts. Mol. Ther. 26, 1093–1108. doi: 10.1016/j.ymthe.2018.01.021
Juban, G., and Chazaud, B. (2017). Metabolic regulation of macrophages during tissue repair: insights from skeletal muscle regeneration. FEBS Lett. 591, 3007–3021. doi: 10.1002/1873-3468.12703
Kazuki, Y., Hiratsuka, M., Takiguchi, M., Osaki, M., Kajitani, N., Hoshiya, H., et al. (2010). Complete genetic correction of ips cells from Duchenne muscular dystrophy. Mol. Ther. 18, 386–393. doi: 10.1038/mt.2009.274
Kemaladewi, D. U., Hoogaars, W. M., van Heiningen, S. H., Terlouw, S., de Gorter, D. J., den Dunnen, J. T., et al. (2011). Dual exon skipping in myostatin and dystrophin for Duchenne muscular dystrophy. BMC Med. Genomics 4:36. doi: 10.1186/1755-8794-4-36
Kendall, G. C., Mokhonova, E. I., Moran, M., Sejbuk, N. E., Wang, D. W., Silva, O., et al. (2012). Dantrolene enhances antisense-mediated exon skipping in human and mouse models of Duchenne muscular dystrophy. Sci. Transl. Med. 4:164ra160. doi: 10.1126/scitranslmed.3005054
Koenig, M., Hoffman, E. P., Bertelson, C. J., Monaco, A. P., Feener, C., and Kunkel, L. M. (1987). Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 50, 509–517. doi: 10.1016/0092-8674(87)90504-6
Kunkel, L. M., Monaco, A. P., Hoffman, E., Koenig, M., Feener, C., and Bertelson, C. (1987). Molecular studies of progressive muscular dystrophy (Duchenne). Enzyme 38, 72–75. doi: 10.1159/000469192
Landfeldt, E., Lindgren, P., Bell, C. F., Guglieri, M., Straub, V., Lochmüller, H., et al. (2016). Quantifying the burden of caregiving in Duchenne muscular dystrophy. J. Neurol. 263, 906–915. doi: 10.1007/s00415-016-8080-9
Leask, A., and Abraham, D. J. (2004). TGF-beta signaling and the fibrotic response. FASEB J. 18, 816–827. doi: 10.1096/fj.03-1273rev
Leask, A., Denton, C. P., and Abraham, D. J. (2004). Insights into the molecular mechanism of chronic fibrosis: the role of connective tissue growth factor in scleroderma. J. Invest. Dermatol. 122, 1–6. doi: 10.1046/j.0022-202X.2003.22133.x
Lee, E. J., Kim, A. Y., Lee, E. M., Lee, M. M., Min, C. W., Kang, K. K., et al. (2014). Therapeutic effects of exon skipping and losartan on skeletal muscle of mdx mice. Pathol. Int. 64, 388–396. doi: 10.1111/pin.12190
Lee, E. M., Kim, A. Y., Lee, E. J., Park, J. K., Lee, M. M., Hwang, M., et al. (2015). Therapeutic effects of mouse adipose-derived stem cells and losartan in the skeletal muscle of injured mdx mice. Cell Transplant. 24, 939–953. doi: 10.3727/096368914X678599
Le Hir, M., Goyenvalle, A., Peccate, C., Précigout, G., Davies, K. E., Voit, T., et al. (2013). AAV genome loss from dystrophic mouse muscles during AAV-U7 snRNA-mediated exon-skipping therapy. Mol. Ther. 21, 1551–1558. doi: 10.1038/mt.2013.121
Li, H., Malhotra, S., and Kumar, A. (2008). Nuclear factor-kappa B signaling in skeletal muscle atrophy. J. Mol. Med. 86, 1113–1126. doi: 10.1007/s00109-008-0373-8
Loperfido, M., Jarmin, S., Dastidar, S., Di Matteo, M., Perini, I., Moore, M., et al. (2016). piggyBac transposons expressing full-length human dystrophin enable genetic correction of dystrophic mesoangioblasts. Nucleic Acids Res. 44, 744–760. doi: 10.1093/nar/gkv1464
Lu-Nguyen, N., Malerba, A., Popplewell, L., Schnell, F., Hanson, G., and Dickson, G. (2017). Systemic antisense therapeutics for dystrophin and myostatin exon splice modulation improve muscle pathology of adult mdx mice. Mol. Ther. Nucleic Acids 6, 15–28. doi: 10.1016/j.omtn.2016.11.009
McElhanon, K. E., and Bhattacharya, S. (2018). Altered membrane integrity in the progression of muscle diseases. Life Sci. 192, 166–172. doi: 10.1016/j.lfs.2017.11.035
McNally, E. M. (2007). New approaches in the therapy of cardiomyopathy in muscular dystrophy. Annu. Rev. Med. 58, 75–88. doi: 10.1146/annurev.med.58.011706.144703
Miyatake, S., Shimizu-Motohashi, Y., Takeda, S., and Aoki, Y. (2016). Anti-inflammatory drugs for Duchenne muscular dystrophy: focus on skeletal muscle-releasing factors. Drug Des. Dev. Ther. 10, 2745–2758. doi: 10.2147/DDDT.S110163
Monaco, A. P., Bertelson, C. J., Liechti-Gallati, S., Moser, H., and Kunkel, L. M. (1988). An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 2, 90–95. doi: 10.1016/0888-7543(88)90113-9
Morales, M. G., Cabrera, D., Céspedes, C., Vio, C. P., Vazquez, Y., Brandan, E., et al. (2013a). Inhibition of the angiotensin-converting enzyme decreases skeletal muscle fibrosis in dystrophic mice by a diminution in the expression and activity of connective tissue growth factor (CTGF/CCN-2). Cell Tissue Res. 353, 173–187. doi: 10.1007/s00441-013-1642-6
Morales, M. G., Gutierrez, J., Cabello-Verrugio, C., Cabrera, D., Lipson, K. E., Goldschmeding, R., et al. (2013b). Reducing CTGF/CCN2 slows down mdx muscle dystrophy and improves cell therapy. Hum. Mol. Genet. 22, 4938–4951. doi: 10.1093/hmg/ddt352
Nakamura, A. (2017). Moving towards successful exon-skipping therapy for Duchenne muscular dystrophy. J. Hum. Genet. 62, 871–876. doi: 10.1038/jhg.2017.57
Nance, M. E., Hakim, C. H., Yang, N. N., and Duan, D. (2017). Nanotherapy for Duchenne muscular dystrophy. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 10:e1472. doi: 10.1002/wnan.1472
Negroni, E., Bigot, A., Butler-Browne, G. S., Trollet, C., and Mouly, V. (2016). Cellular Therapies for Muscular Dystrophies: frustrations and clinical successes. Hum. Gene Ther. 27, 117–126. doi: 10.1089/hum.2015.139
Ousterout, D. G., Kabadi, A. M., Thakore, P. I., Majoros, W. H., Reddy, T. E., and Gersbach, C. A. (2015a). Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat. Commun. 6:6244. doi: 10.1038/ncomms7244
Ousterout, D. G., Kabadi, A. M., Thakore, P. I., Perez-Pinera, P., Brown, M. T., Majoros, W. H., et al. (2015b). Correction of dystrophin expression in cells from Duchenne muscular dystrophy patients through genomic excision of exon 51 by zinc finger nucleases. Mol. Ther. 23, 523–532. doi: 10.1038/mt.2014.234
Ousterout, D. G., Perez-Pinera, P., Thakore, P. I., Kabadi, A. M., Brown, M. T., Qin, X., et al. (2013). Reading frame correction by targeted genome editing restores dystrophin expression in cells from Duchenne muscular dystrophy patients. Mol. Ther. 21, 1718–1726. doi: 10.1038/mt.2013.111
Peccate, C., Mollard, A., Le Hir, M., Julien, L., McClorey, G., Jarmin, S., et al. (2016). Antisense pre-treatment increases gene therapy efficacy in dystrophic muscles. Hum. Mol. Genet. 25, 3555–3563. doi: 10.1093/hmg/ddw201
Philippou, A., and Barton, E. R. (2014). Optimizing IGF-I for skeletal muscle therapeutics. Growth Horm. IGF Res. 24, 157–163. doi: 10.1016/j.ghir.2014.06.003
Pichavant, C., Chapdelaine, P., Cerri, D. G., Dominique, J. C., Quenneville, S. P., Skuk, D., et al. (2010). Expression of dog microdystrophin in mouse and dog muscles by gene therapy. Mol. Ther. 18, 1002–1009. doi: 10.1038/mt.2010.23
Pini, V., Morgan, J. E., Muntoni, F., and O'Neill, H. C. (2017). Genome editing and muscle stem cells as a therapeutic tool for muscular dystrophies. Curr Stem Cell Rep. 3, 137–148. doi: 10.1007/s40778-017-0076-6
Popplewell, L., Koo, T., Leclerc, X., Duclert, A., Mamchaoui, K., Gouble, A., et al. (2013). Gene correction of a Duchenne muscular dystrophy mutation by meganuclease-enhanced exon knock-in. Hum. Gene Ther. 24, 692–701. doi: 10.1089/hum.2013.081
Quenneville, S. P., Chapdelaine, P., Rousseau, J., and Tremblay, J. P. (2007a). Dystrophin expression in host muscle following transplantation of muscle precursor cells modified with the phiC31 integrase. Gene Ther. 14, 514–522. doi: 10.1038/sj.gt.3302887
Quenneville, S. P., Chapdelaine, P., Skuk, D., Paradis, M., Goulet, M., Rousseau, J., et al. (2007b). Autologous transplantation of muscle precursor cells modified with a lentivirus for muscular dystrophy: human cells and primate models. Mol. Ther. 15, 431–438. doi: 10.1038/sj.mt.6300047
Reinig, A. M., Mirzaei, S., and Berlau, D. J. (2017). Advances in the treatment of Duchenne muscular dystrophy: new and emerging Pharmacotherapies. Pharmacotherapy 37, 492–499. doi: 10.1002/phar.1909
Ricotti, V., Spinty, S., Roper, H., Hughes, I., Tejura, B., Robinson, N., et al. (2016). Safety, tolerability, and pharmacokinetics of SMT C1100, a 2-Arylbenzoxazole utrophin modulator, following single- and multiple-dose administration to pediatric patients with Duchenne muscular dystrophy. PLoS ONE 11:e0152840. doi: 10.1371/journal.pone.0152840
Roberts, R. G., Gardner, R. J., and Bobrow, M. (1994). Searching for the 1 in 2,400,000: a review of dystrophin gene point mutations. Hum. Mutat. 4, 1–11. doi: 10.1002/humu.1380040102
Rodino-Klapac, L. R., Janssen, P. M., Shontz, K. M., Canan, B., Montgomery, C. L., Griffin, D., et al. (2013). Micro-dystrophin and follistatin co-delivery restores muscle function in aged DMD model. Hum. Mol. Genet. 22, 4929–4937. doi: 10.1093/hmg/ddt342
Ryder, S., Leadley, R. M., Armstrong, N., Westwood, M., de Kock, S., Butt, T., et al. (2017). The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: an evidence review. Orphanet J. Rare Dis. 12:79. doi: 10.1186/s13023-017-0631-3
Sabharwal, R., Cicha, M. Z., Sinisterra, R. D., De Sousa, F. B., Santos, R. A., and Chapleau, M. W. (2014). Chronic oral administration of Ang-(1-7) improves skeletal muscle, autonomic and locomotor phenotypes in muscular dystrophy. Clin. Sci. 127, 101–109. doi: 10.1042/CS20130602
Sampaolesi, M., Blot, S., D'Antona, G., Granger, N., Tonlorenzi, R., Innocenzi, A., et al. (2006). Mesoangioblast stem cells ameliorate muscle function in dystrophic dogs. Nature 444, 574–579. doi: 10.1038/nature05282
Serrano, A. L., and Muñoz-Cá-noves, P. (2010). Regulation and dysregulation of fibrosis in skeletal muscle. Exp. Cell Res. 316, 3050–3058. doi: 10.1016/j.yexcr.2010.05.035
Serrano, A. L., and Muñoz-Cánoves, P. (2017). Fibrosis development in early-onset muscular dystrophies: mechanisms and translational implications. Semin. Cell Dev. Biol. 64, 181–190. doi: 10.1016/j.semcdb.2016.09.013
Sharma, M., McFarlane, C., Kambadur, R., Kukreti, H., Bonala, S., and Srinivasan, S. (2015). Myostatin: expanding horizons. IUBMB Life 67, 589–600. doi: 10.1002/iub.1392
Shin, J., Tajrishi, M. M., Ogura, Y., and Kumar, A. (2013). Wasting mechanisms in muscular dystrophy. Int. J. Biochem. Cell Biol. 45, 2266–2279. doi: 10.1016/j.biocel.2013.05.001
Spencer, M. J., and Tidball, J. G. (2001). Do immune cells promote the pathology of dystrophin-deficient myopathies? Neuromuscul. Disord. 11, 556–564. doi: 10.1016/S0960-8966(01)00198-5
Sun, G., Haginoya, K., Dai, H., Chiba, Y., Uematsu, M., Hino-Fukuyo, N., et al. (2009). Intramuscular renin-angiotensin system is activated in human muscular dystrophy. J. Neurol. Sci. 280, 40–48. doi: 10.1016/j.jns.2009.01.020
Tidball, J. G. (2005). Inflammatory processes in muscle injury and repair. Am. J. Physiol. Regul. Integr. Comp. Physiol. 288, R345–353. doi: 10.1152/ajpregu.00454.2004
Timpani, C. A., Hayes, A., and Rybalka, E. (2017). Therapeutic strategies to address neuronal nitric oxide synthase deficiency and the loss of nitric oxide bioavailability in Duchenne muscular dystrophy. Orphanet J. Rare Dis. 12:100. doi: 10.1186/s13023-017-0652-y
Tinsley, J. M., Fairclough, R. J., Storer, R., Wilkes, F. J., Potter, A. C., Squire, S. E., et al. (2011). Daily treatment with SMTC1100, a novel small molecule utrophin upregulator, dramatically reduces the dystrophic symptoms in the mdx mouse. PLoS ONE 6:e19189. doi: 10.1371/journal.pone.0019189
Verhaart, I. E., Heemskerk, H., Karnaoukh, T. G., Kolfschoten, I. G., Vroon, A., van Ommen, G. J., et al. (2012). Prednisolone treatment does not interfere with 2'-O-methyl phosphorothioate antisense-mediated exon skipping in Duchenne muscular dystrophy. Hum. Gene Ther. 23, 262–273. doi: 10.1089/hum.2011.127
Villalta, S. A., Rinaldi, C., Deng, B., Liu, G., Fedor, B., and Tidball, J. G. (2011). Interleukin-10 reduces the pathology of mdx muscular dystrophy by deactivating M1 macrophages and modulating macrophage phenotype. Hum. Mol. Genet. 20, 790–805. doi: 10.1093/hmg/ddq523
Wang, J. Z., Wu, P., Shi, Z. M., Xu, Y. L., and Liu, Z. J. (2017). The AAV-mediated and RNA-guided CRISPR/Cas9 system for gene therapy of DMD and BMD. Brain Dev. 39, 547–556. doi: 10.1016/j.braindev.2017.03.024
Young, C. S., Hicks, M. R., Ermolova, N. V., Nakano, H., Jan, M., Younesi, S., et al. (2016). A Single CRISPR-Cas9 deletion strategy that targets the majority of DMD patients restores dystrophin function in hiPSC-derived muscle cells. Cell Stem Cell 18, 533–540. doi: 10.1016/j.stem.2016.01.021
Zhou, L., and Lu, H. (2010). Targeting fibrosis in Duchenne muscular dystrophy. J. Neuropathol. Exp. Neurol. 69, 771–776. doi: 10.1097/NEN.0b013e3181e9a34b
Keywords: gene therapy, cell therapy, muscle, Duchenne muscular dystrophy, dystrophin, fibrosis, inflammation, atrophy
Citation: Cordova G, Negroni E, Cabello-Verrugio C, Mouly V and Trollet C (2018) Combined Therapies for Duchenne Muscular Dystrophy to Optimize Treatment Efficacy. Front. Genet. 9:114. doi: 10.3389/fgene.2018.00114
Received: 31 October 2017; Accepted: 22 March 2018;
Published: 10 April 2018.
Edited by:
Linda Popplewell, Royal Holloway, University of London, United KingdomReviewed by:
Muhammad Jawad Hassan, National University of Sciences and Technology, PakistanVirginia Arechavala-Gomeza, BioCruces Health research Institute, Spain
Copyright © 2018 Cordova, Negroni, Cabello-Verrugio, Mouly and Trollet. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Capucine Trollet, Y2FwdWNpbmUudHJvbGxldEB1cG1jLmZy