 Zhaoen Yang1,2†
Zhaoen Yang1,2† Qian Gong2†
Qian Gong2† Lingling Wang2Yuying Jin2Jianping Xi2
Lingling Wang2Yuying Jin2Jianping Xi2 Zhi Li2
Zhi Li2 Wenqiang Qin1,2
Wenqiang Qin1,2 Zuoren Yang2Lili Lu2Quanjia Chen1Fuguang Li1,2*
Zuoren Yang2Lili Lu2Quanjia Chen1Fuguang Li1,2*- 1Xinjiang Research Base, State Key Laboratory of Cotton Biology, Xinjiang Agricultural University, Urumqi, China
- 2Institute of Cotton Research, Chinese Academy of Agricultural Sciences, Anyang, China
Members of the YABBY gene family, a small plant-specific family of genes, have been proposed to function in specifying abaxial cell fate. Although to date little has been learned about cotton YABBY genes, completion of the cotton genome enables a comprehensive genome-wide analysis of YABBY genes in cotton. Here, a total of 12, 12, and 23 YABBY genes were identified in Gossypium arboreum (2n = 26, A2), G. raimondii (2n = 26, D5), and G. hirsutum (2n = 4x = 52, [AD]t), respectively. Sequence analysis showed that the N-terminal zinc-finger and C-terminal YABBY domains in YABBY proteins are highly conserved among cotton, Arabidopsis, and rice. Eighty-five genes from eight sequenced species naturally clustered into five groups, and the YAB2-like group could be divided into three sub-groups, indicating that YABBYs are highly conserved among the examined species. Orthologs from the At and Dt sub-genomes (where “t” indicates tetraploid) showed good collinearity, indicating that YABBY loci are highly conserved between these two sub-genomes. Whole-genome duplication was the primary cause of upland cotton YABBY gene expansion, segmental duplication played important roles in YABBY gene expansion within the At and Dt sub-genomes, and the YAB5-like group was mainly generated by segmental duplication. The long-terminal repeat retroelements Copia and Gypsy were identified as major transposable elements accompanying the appearance of duplicated YABBY genes, suggesting that transposable element expansion might be involved in gene duplication. Selection pressure analyses using PAML revealed that relaxed purifying selection might be the main impetus during evolution of YABBY genes in the examined species. Furthermore, exon/intron pattern and motif analyses indicated that genes within the same group were significantly conserved between Arabidopsis and cotton. In addition, the expression patterns in different tissues suggest that YABBY proteins may play roles in ovule development because YABBYs are highly expressed in ovules. The expression pattern of YABBY genes showed that approximately half of the YABBYs were down-regulated under different stress treatments. Collectively, our results represent a comprehensive genome-wide study of the YABBY gene family, which should be helpful in further detailed studies on the gene function and evolution of YABBY genes in cotton.
Introduction
The small YABBY gene family is specific to seed plants (Floyd and Bowman, 2007), the members of which encode a class of transcription factors containing two conserved domains: a zinc-finger domain in the N-terminal region, and a YABBY domain (helix-loop-helix motif) in the C-terminal region (Bowman and Eshed, 2000; Kanaya et al., 2002). They have been identified in all seed plants examined to date, and fall within five different classes: CRABS CLAW (CRC), FILAMENTOUS FLOWER (FIL)/YABBY3 (YAB3), INNER NO OUTER (INO), YABBY2 (YAB2), and YABBY5 (YAB5) (Bowman, 2000b; Yamada et al., 2001). YABBY genes play roles in diverse processes such as lamina outgrowth, cell polarity maintenance, and leaf margin establishment (Chen et al., 1999; Siegfried et al., 1999; Watanabe and Okada, 2003; Finet et al., 2016). In Arabidopsis thaliana, the following six YABBY genes have been identified: FILAMENTOUS FLOWER (FIL), CRABS CLAW (CRC), INNER NO OUTER (INO), YAB2, YAB3, and YAB5 (Bowman and Smyth, 1999; Lee et al., 2005).
Arabidopsis FIL is required for normal flower development and is thought to act redundantly with YAB2 and YAB3 because of their overlapping expression patterns and sequence homologies (Chen et al., 1999; Kumaran et al., 1999; Siegfried et al., 1999). In rice, the TOB1-related YABBY genes play roles in flower development similar to their FIL orthologs in Arabidopsis (Tanaka et al., 2017). The Arabidopsis CRC gene is essential for nectary specification and carpel polarity (Alvarez and Smith, 1999), and its ortholog in Pisum sativum shows conserved functions in carpel morphogenesis (Fourquin et al., 2014). In rice and maize, homologs in the CRC clade share conserved functions in leaf development by modulating midrib development and regulating plant architecture (Nagasawa et al., 2003; Ohmori et al., 2011; Strable et al., 2017). Arabidopsis INO plays an important role in development of the outer integument (Villanueva et al., 1999). The soybean GmYABBY10 gene functions as a negative regulator in plant resistance to drought (Zhao et al., 2017).
Among the eudicots, CRC genes are expressed in abaxial carpels and nectaries (Bowman and Smyth, 1999; Lee et al., 2005), whereas INO genes are transcribed in the abaxial outer integument layers (Villanueva et al., 1999). The outer integument and carpels are considered to be typical synapomorphies for angiosperms, and are derived from sporophylls (Yamada et al., 2011). Therefore, the YABBY genes are considered to be significant candidates for participation in evolutionary stem-to-leaf transformation (Floyd and Bowman, 2010; Sarojam et al., 2010). However, the “vegetative YABBY genes” FIL-like, YAB2-like, and YAB5-like are expressed in the cotyledons, leaves, and floral organs (Golz, 2004; Bartholmes et al., 2012).
The expression patterns of vegetative YABBY genes are associated with several processes, including exclusion from apical meristems and activation of the initiation of lateral organ primordia, and often show asymmetric restriction to the abaxial domain of eudicot primordia (Siegfried et al., 1999; Golz, 2004). Evidence from core eudicots indicates that vegetative YABBY genes act in several aspects of leaf development, and some vegetative YABBY genes also regulate shoot apical meristem development and phyllotaxy (Kumaran et al., 2002; Eshed et al., 2004; Toriba et al., 2007; Sarojam et al., 2010). Thus, the evolutionary history of YABBY genes coincides with the origin of leaves in seed plants (Floyd and Bowman, 2007), and it is necessary to understand the relationship between YABBY genes in different species or within the same species.
Cotton is one of the most important economic crops and is the world's most important source of raw materials for textile fibers and edible oils (Yang et al., 2014; Gong et al., 2017b). Although RNA-seq data for cotton YABBY genes have been presented in a few studies (Jiao et al., 2013; Zhang et al., 2016), further experimental analyses are needed to clarify their biological function. Therefore, it is necessary to understand the biological consequences of YABBY gene family activity in cotton plants. In this regard, completion of the genome sequences of Gossypium raimondii, Gossypium arboreum, and Gossypium hirsutum and genome-wide analysis of the YABBY gene family have provided a valuable database resource (Wang et al., 2012; Li et al., 2014, 2015; Zhang et al., 2015). Although some YABBY genes have been reported in monocotyledonous and dicotyledonous plants, the relationships between these genes are poorly understood. In this study, we isolated YABBY sequences from eight different species and analyzed the relationship between monocotyledonous and dicotyledonous plants. Furthermore, we focused on cotton, using various methods to analyze YABBY genes, including the identification of gene families, phylogenetic tree analysis, and analyses of segmental duplication, gene structures, chromosome location, and expression patterns.
Materials and Methods
Sequence Identification
The genome sequences of the cotton species Gossypium arboreum (BJI, version 1.0), G. raimondii (JGI, version 2.0), and G. hirsutum (NAU, version 1.1) were downloaded from COTTONGEN (www.cottongen.org) (Yu et al., 2014). The genome sequences of rice (version 7.0), sorghum (version 2.1), maize (version 1.1), cacao (version 1.1), and A. thaliana were retrieved from JGI (https://phytozome.jgi.doe.gov/pz/portal.html). The amino acid sequences of YABBYs from A. thaliana were acquired from The Arabidopsis Information Resource, version 10 (TAIR 10) (http://www.arabidopsis.org). We used YABBY protein sequences from Arabidopsis as query sequences to search the G. arboreum protein database for candidate sequences, employing the blastp program. We then used Interproscan 63.0 (Jones et al., 2014) to identify the YABBY domain (PF04690) in the candidate sequences. YABBYs in rice, sorghum, maize, cacao, G. hirsutum, and G. raimondii were analyzed as described above for G. arboreum. The blastn program was also used to identify potential un-annotated genes in the cotton genome.
Conserved Sequence and Phylogenetic Analysis
We used Clustal X 2.0 for multiple-sequence alignments. For conserved domain-sequence analysis, sequence logo analysis of the zinc-finger and YABBY domains from rice, Arabidopsis, and upland cotton genes was performed using the online tool WEBLOGO (Crooks et al., 2004) and multiple sequence alignment results were used as the input file. For phylogenetic analysis, the ClustalW program (build-in MEGA 7.0) (Kumar et al., 2016) was used to align the full-length YABBY sequences from rice, sorghum, maize, cacao, Arabidopsis, G. arboreum, G. raimondii, and G. hirsutum, and a neighbor-joining (NJ) tree was constructed using the bootstrap method with 1000 replicates. Substitution was evaluated with the Poisson model using the default parameters. To validate the NJ tree, the minimum-evolution method was also used. The bootstrap method was used with 1,000 replicates.
Chromosomal Location and Collinearity Analysis
The exon loci of YABBYs were extracted from the annotation gff3 file using a Perl script. MapChart was used to display gene locations on the chromosome (Voorrips, 2002). For collinearity analysis, the entire YABBY protein sequences of upland cotton were aligned with each other using the Basic Local Alignment Search Tool (BLAST) with a cut-off e-value of 1 × 10−5. The blastp results were analyzed using MCSCAN software to produce collinearity blocks across the whole genome. Collinear pairs within the YABBY family of proteins were extracted to draw a collinearity map using CIRCOS software (Krzywinski et al., 2009).
Selective Pressure Analysis
Amino acid sequences from homologous pairs were aligned using Clustal X 2.0. The alignment results were converted to a codon alignment in PAML style using the PAL2NAL program (Suyama et al., 2006) (http://www.bork.embl.de/pal2nal/). The built-in PAML package of the CODEML program (Yang, 2007) was used to estimate the selective pressure. A branch model and site model were used in the current study. For branch model analysis, two-ratio model 2 and model 0 were used to determine the omega value of each branch. The likelihood ratio test (LRT) was used to assess which was the more suitable of the two models. For site model analysis, the amino acid sequences from each branch were extracted to construct an NJ tree and the corresponding sequences were converted to a codon alignment as an input file for the CONDEML program, as described previously (Cao et al., 2017).
Annotation and Analysis of Transposable Elements (TEs)
We used de novo prediction and a homolog search method based on Repbase (Jurka, 2000) to determine the repeat content of the genomic DNA. Three software programs, namely PILER-DF, RepeatModeler, and LTR_FINDER (Edgar and Myers, 2005; Xu and Wang, 2007), were used to predict TEs in the genome for de novo analysis. Furthermore, a known TE library was used to identify TEs at the DNA level with the RepeatMasker program (Chen, 2004), using Repbase TE (Edition-20170127). To analyze the function of TEs in the expansion of the YABBY family, we identified TEs located 10,000 and 2,000 bps upstream and downstream of the YABBY genes and performed statistical analysis of the different types of TEs present.
Gene Structure Analysis
The full-length Arabidopsis and G. hirsutum protein sequences were aligned with ClustalW, and MEGA 7.0 (Kumar et al., 2016) was used to construct an NJ tree using the method and parameters described above. The exon positions were acquired from the gff3 file using a Perl script, and the gene structure was displayed using the online tool GSDS 2.0 (Hu et al., 2014).
Transcriptome Data Analysis and Gene Expression Heatmap
The raw RNA-Seq data were downloaded from the NCBI Sequence Read Archive (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA248163/). TopHat (version: 2.0.13) was used for mapping reads, cufflinks (version: 2.2.1) were used to analyze gene expression levels, and fragments per kilobase million values were used to normalize gene expression levels (Trapnell et al., 2013). The expression levels of YABBYs were determined by extracting their respective data from the total expression matrix, using Genesis software (version: 1.7.7) (Sturn et al., 2002).
Real-Time PCR
Upland cotton seeds (CCRI24 variety) were obtained from the Institute of Cotton Research of the Chinese Academy of Agricultural Science. Seed germination was performed on a wet filter paper at 28°C for 3 days, after which the seeds were transferred to liquid culture medium (Yang et al., 2014). At the three-leaf stage, the seedlings were treated with 20% PEG6000. The roots were sampled at 0, 1, 3, and 6 h post-treatment, immediately frozen in liquid nitrogen, and stored at −80°C. In addition, the true leaf, stem, and root under normal conditions were sampled, immediately frozen in liquid nitrogen, and stored at −80°C. Total RNA was purified from the samples using the RNAprep Pure Plant Kit (TIANGEN, Beijing, China), following the manufacturer's recommended protocol. The first strand of cDNA was synthesized using the PrimeScript® RT Reagent Kit (Takara, Dalian, China). Real-time PCR amplifications were performed in an ABI 7900HT instrument (Thermo Fisher, USA) using SYBR Premix Ex Taq™ II (Takara, Dalina, China). Upland cotton histone 3 mRNA (GenBank accession number AF024716) was used as an internal control. The dissociation curves of each reaction were assessed, and the cycle threshold (CT) 2−ΔΔCT method was used to calculate the expression level of each target gene as previously reported (Gong et al., 2017a).
Results
Gene Identification and Homeobox Domain Retrieval
The sequences of six Arabidopsis YABBY genes were obtained from TAIR (http://www.arabidopsis.org) and used as queries for searching the rice, sorghum, maize, cacao, G. arboreum, G. raimondii, and G. hirsutum databases, using the blastp program with a cut-off e-value of 1 × 10−5. Thereafter, potential candidates were extracted from the database. InterProscan 63.0 (http://www.ebi.ac.uk/interpro/) was used to search for the YABBY protein (PF04690) among the obtained sequences, and 8, 8, 11, 7, 12, 12, and 23 genes were confirmed as YABBY family members in rice, sorghum, maize, cacao, G. arboreum, G. raimondii, and G. hirsutum, respectively (Supplementary Table 1). The number of YABBY genes in diploid cotton G. raimondii was the same as that in maize, but exceeded that in rice, sorghum and cacao. The number of YABBY genes in upland cotton was almost twice that in the genomes of the two diploid cottons, G. arboreum (AA) and G. raimondii (DD), in accordance with the hypothesis that upland cotton was derived from hybridization with a progenitor having a diploid genome, after which the relevant chromosomes doubled. G. raimondii has a smaller genome size and simpler genome structure compared with that of G. arboreum (AA) and G. hirsutum, which made genome sequencing and high-quality assembly much easier to accomplish with G. raimondii than with G. arboreum (AA) and G. hirsutum. As shown in Supplementary Table 2, the D genome (G. raimondii), Dt-subgenome (G. hirsutum), and A genome (G. arboreum) contain 12 YABBY genes and the At-genome has 11 genes. Ga, Gr, Gh, and At represent G. arboreum, G. raimondii, G. hirsutum, and A. thaliana, respectively, and these terms are used as prefixes before the names of the YABBY genes. On the basis of gene number and chromosome locus, we designated YABBY genes from G. raimondii as YABBY1 to YABBY12, and genes from the A-genome, At-subgenome, and Dt-subgenome were named after their orthologs in G. raimondii. We found that GhYABBY7_Dt was not available in current cotton databases (Supplementary Table 2), and therefore we used the protein sequence of its ortholog GhYABBY7_At as a query to search the upland cotton genomic sequence (Supplementary Table 3). We also extracted the genomic sequence of GhYABBY7_At to align with the upland cotton genome. As shown in Supplementary Table 3, a region (from 35585961 to 35588822 bp) on D09 is the potential locus for GhYABBY7_Dt. We extracted the genomic sequence around this region to align with the GhYABBY7_At genomic sequence and found gaps within this region that disrupted the GhYABBY7_Dt sequence (Supplementary Image 1). This region accordingly no longer retains an encoding function, and we consider that the incomplete genomic sequence may explain the absence of GhYABBY7_Dt annotation. The orthologs from G. raimondii, At-subgenome, and Dt-subgenome are almost all in homologous chromosomes, indicating that they have been conserved during evolution. GhYABBY11_Dt has 199 amino acid residues, making it larger than the orthologs GhYABB11_At (183 AA), GrYABBY11 (183 AA), and GaYABBY11 (183 AA). Further inspection of the gene structure revealed that GhYABBY11_Dt has six exons, whereas the other three orthologs have seven exons. Thus, we extracted the genomic sequence of GhYABBY11_Dt, re-annotated the gene using AUGUSTUS (Stanke et al., 2004), and found the gene has multiple splicing isoforms (Supplementary Table 4), one of which encodes 183 amino acids (similar to GhYABBY11_At and GrYABBY11). We consequently used the novel predicted isoform for subsequent study.
Conserved Amino Residues within YABBY Proteins
The YABBY gene family is a small family of transcription factors characterized by two conserved domains, a C2C2 zinc-finger-like domain (located toward the amino terminus) and a YABBY domain (located toward the carboxyl end of the protein), as shown in Figure 1A (Bowman, 2000b). Compared with the conserved domains, we found that domains I, II, and III were more variable in length (Figure 1A). To analyze the homologous domain sequences and the degree of conservation of each residue in the zinc-finger and YABBY domains, multiple sequence alignments were performed to generate sequence logos of both domains in Arabidopsis, rice, and G. hirsutum. As shown in Figure 1B, we found that both the zinc-finger and YABBY domains are significantly conserved among the three species. Some amino acid residues in the zinc-finger domain were found to be highly conserved, such as L, V, and P in sheet 2; P in coil 3; V, T, and R in sheet 3; and C, G, H, N, and L in coil 4. Most amino acids in the YABBY domain were significantly conserved (Figure 1C).
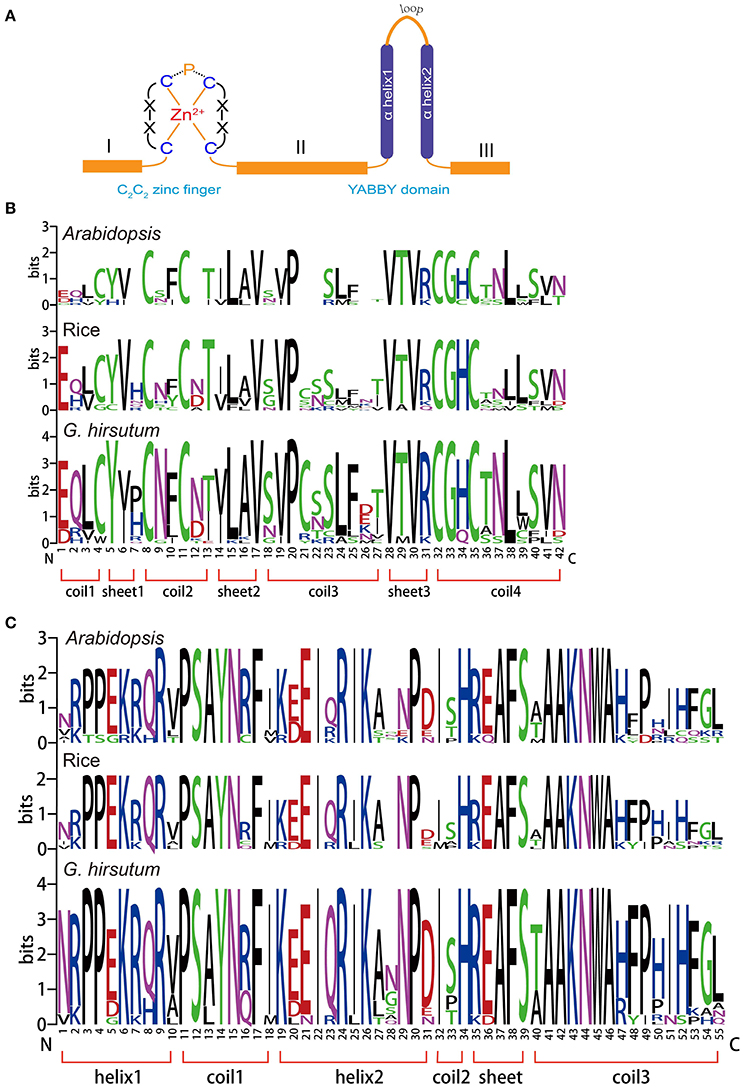
Figure 1. Conserved domains in the YABBY gene family. (A) Members of the YABBY gene family are characterized by two highly conserved domains: a C2C2 zinc-finger domain toward the N terminal and a “YABBY” domain toward the C terminal. (B) Sequence logos showing the highly conserved zinc-finger domain. (C) Sequence logos showing the highly conserved YABBY domain.
Phylogenetic Analysis of YABBY Genes among Typical Dicots and Monocots
To study the phylogenetic relationship of YABBY genes among cotton plants (G. hirsutum, G. arboreum, and G. raimondii) and other species, a comprehensive neighbor-joining (NJ) tree was constructed using MEGA 7.0. The phylogenetic tree showed that the YABBY genes can be divided into five groups, named the CRC-like, INO-like, YAB2-like, YAB5-like, and FIL-like groups, as previously reported (Bartholmes et al., 2012) (Figure 2). To evaluate the accuracy of the NJ tree, a minimum-evolution method was also used to construct a phylogenetic tree (Supplementary Image 2). We found that the topology of the two trees was almost the same, indicating that our tree was suitable for further study. The genes in the YAB2-like group can be divided into three sub-groups in the tree. The FIL-like group had the most members (26), followed by YAB5-like (22), YAB2-like (20), CRC-like (10), and INO-like (9). We found that the INO-like group contained one gene at most from each species, indicating that the group has not undergone expansion and that group members may perform similar biological functions. Similarly, with the exception of maize, the CRC-like group had only one member from each species. It is noteworthy that the FIL-like and YAB2-like groups have undergone expansion, although the expansion rates were uneven. In the YAB2-like group, the gene number from monocots has increased at least three times, whereas that in the dicots Arabidopsis and cacao has not. In contrast, the expansion rate was almost the same between dicots and monocots in the FIL-like group. With the exception of the YAB5-like group, all other groups comprised both dicot and monocot species (Figure 2), suggesting that genes from monocots in the YAB5-like group may have been lost or that this group evolved in dicots after monocots and dicots diverged.
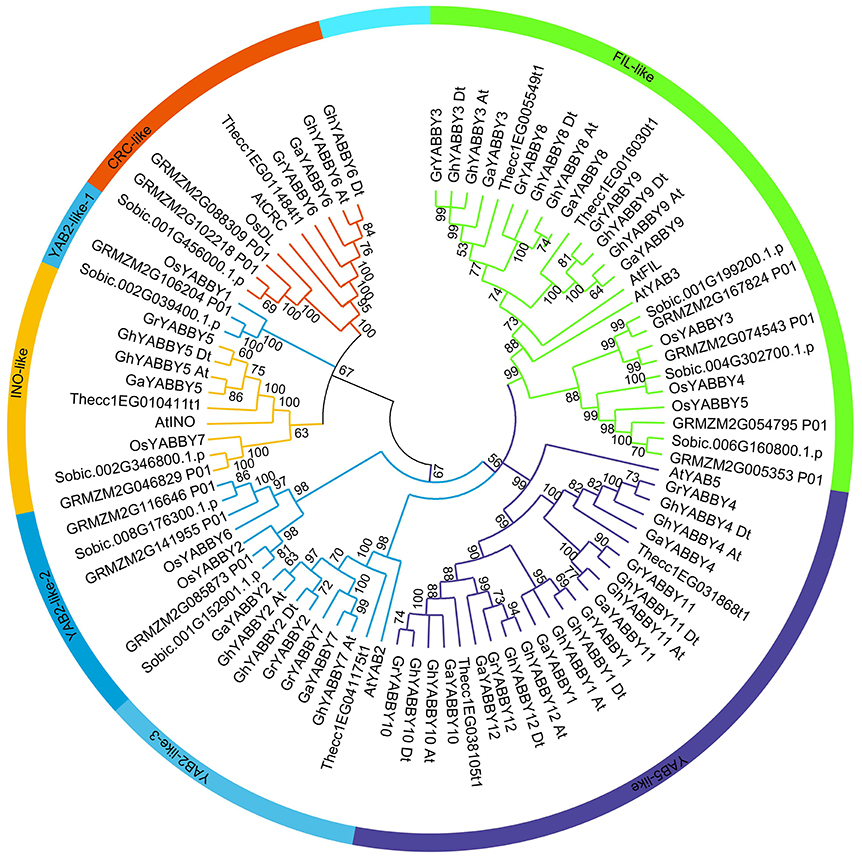
Figure 2. Phylogenetic tree of YABBY genes indicating that YABBY genes can be clustered into five groups. The NJ tree was constructed using MEGA software, version 7.0. The outer circle is marked in orange, yellow, light blue, purple, and green, which represent the CRC-like, INO-like, YAB2-like, YAB5-like, and FIL-like groups, respectively. The prefixes At, Os, GRMZM, Sobic, Thecc, Ga, Gr, and Gh, represent Arabidopsis thaliana, Oryza sativa, Zea mays, Sorghum bicolor, Theobroma cacao, Gossypium arboreum, G. raimondii, and G. hirsutum, respectively. “At” and “Dt” indicate the A- and D- sub-genomes in upland cotton, respectively. Bootstrapping was used to test the tree, and only values >50 are displayed near the nodes.
We found that cacao YABBY genes displayed a close relationship with those from cotton (Figure 2). The number of YABBY genes in G. raimondii was approximately twice that in cacao, with one cacao gene corresponding to one to three orthologs in cotton. For example, in the CRC-like group, Thecc1EG11484t1 had one ortholog in both G. arboreum and G. raimondii, whereas in the FIL-like group, Thecc1EG005549t1 had two orthologs in both G. arboreum and G. raimondii. Furthermore, in the YAB5-like group, Thecc1EG038105t1 had three orthologs in both G. arboreum and G. raimondii.
As shown in Figure 2 and Supplementary Image 2, most of the orthologs from the A genome and At sub-genome were inclined to cluster together, as was the case with orthologs from the D genome and Dt sub-genome, suggesting that the orthologs from At-A or Dt-D share a close relationship.
Gene Expansion and Synteny Analysis
G. hirsutum is a typical allotetraploid, which is an ideal material for studying genome polyploidy and its effects (Paterson et al., 2012). To better understand the collinearity relationship of orthologs between the At and Dt genomes, we performed synteny analysis after aligning the gene loci on the chromosomes. The collinearity analysis showed that most of the YABBY loci were significantly conserved between the At and Dt sub-genomes (Figure 3). Twenty-three YABBY genes were distributed unevenly among 16 chromosomes (Figure 4); for example, no YABBY genes were found in chromosomes A02, A08, A13, D03, D04, D08, D09, and D13. We found that nine pairs of orthologs were located on the homologous chromosomes and showed good collinearity. GhYBBBY4_Dt was not mapped to a chromosome because of incomplete sequencing. The orthologous pairs GhYABBY6_At-GhYABBY6_Dt and GhYABBY11_At-GhYABBY11_Dt were located on non-homologous chromosomes, which might have been caused by chromosome translocation. To validate this possibility, we inspected their orthologs in diploid cotton and found that GhYABBY6_At and GaYABBY6 were on homologous chromosomes, as were GhYABBY6_Dt and GrYABBY6, which suggested that chromosomal translocation within the GhYABBY6 locus occurred before upland cotton speciation. We found that GhYABBY11_Dt and GrYABBY11 are located on homologous chromosomes, whereas GaYBBY11 located on the scaffold.
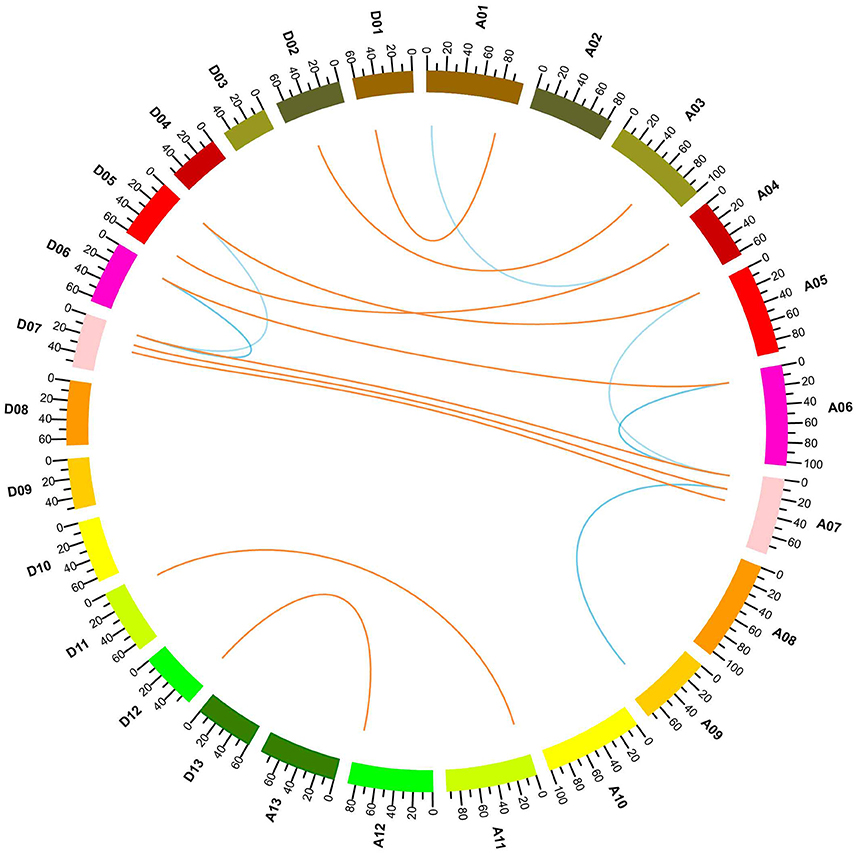
Figure 3. Collinearity analyses within upland cotton. The ends of the orange lines are oriented toward the orthologous genes from the At and Dt sub-genomes. The ends of the blue lines point toward paralog pairs derived from segmental duplication.
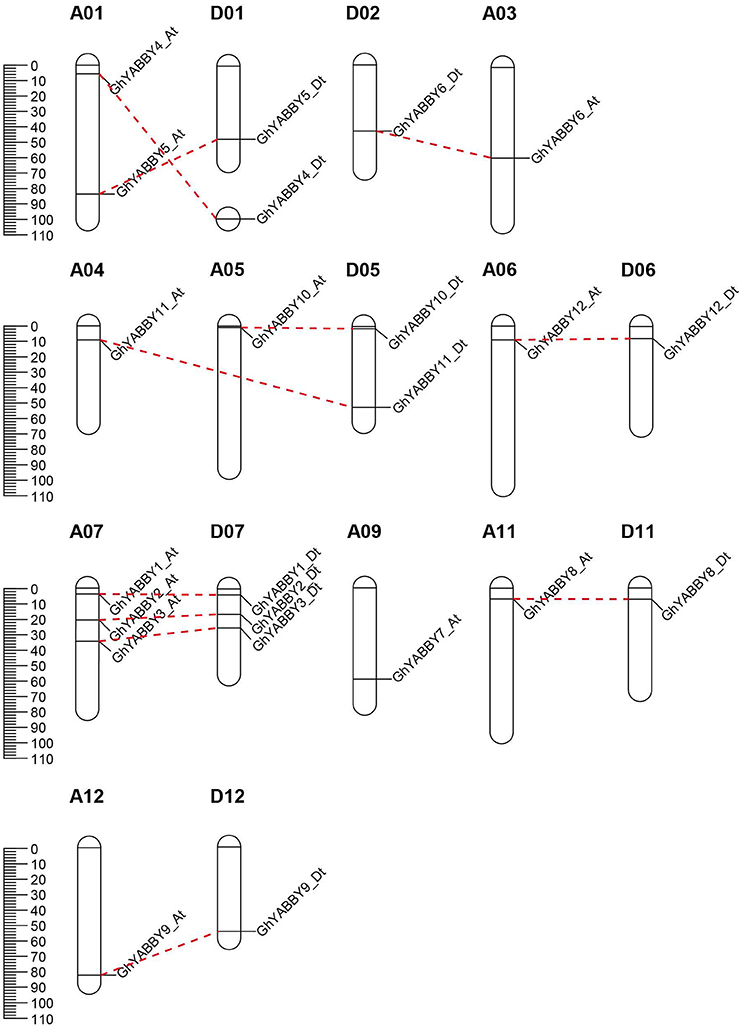
Figure 4. Chromosomal locations of YABBY genes. The red dotted lines link the orthologs located on the At and Dt sub-genomes.
MCSCAN was used to determine the duplicate gene types. Five types of gene duplication may occur, namely, singleton, dispersed, proximal, tandem, and segmental duplication. Over half of the YABBY genes were identified as singletons, and the remaining 10 out of 23 genes were found to have undergone segmental duplication. Among the duplicated genes, GhYABB1_At and GhYABBY1_Dt each formed two pairs of collinear genes, suggesting that they were active during evolution. No tandem, proximal duplications (in the nearby chromosomal region, i.e., not in the adjacent region), or dispersed genes were found among the YABBYs (Figure 3 and Supplementary Table 5).
Duplication was the major impetus underlying YABBY gene expansion during evolution. After duplication, the YABBY genes might have undergone functional divergence, and some might have lost their original functions, acquired novel functions, or maintained partition of original functions (Prince and Pickett, 2002; Vandepoele et al., 2003). During the long history of evolution, genes have been exposed to different selective pressures, including positive selection, negative selection, and purifying selection. To better understand the selection pressure among different branches of the phylogenetic tree, the branch model test was used to estimate ω. As shown in Table 1, the p-values of the likelihood ratio test (LRT) were smaller than 0.05 in FIL-like and YAB5-like branches, indicating that the one-ratio model should be rejected. Therefore, the selective pressures associated with the above mentioned two branches were different from those associated with other branches. The mean ω values for FIL-like and YAB5-like were 0.00038 and 0.01608, respectively, which are smaller than the background values, suggesting that FIL-like and YAB5-like have been under relaxed purifying selection. Moreover, the mean ω values for the other five branches were also smaller than 1.0, suggesting that they have also been under purifying selection. In addition to the branch model, we also used a site model to examine each clade. As shown in Supplementary Table 6, the ratios of dN/dS (ω) for all the branches under the M0 model were smaller than 1.0, indicating that purifying selection is the major impetus for YABBY evolution. The p-values of LRTs for comparison between the M7 model and the M8 model in FIL-like and YAB5-like clades were smaller than 0.05, indicating that some amino acid sites in these two clades may have undergone positive selection.
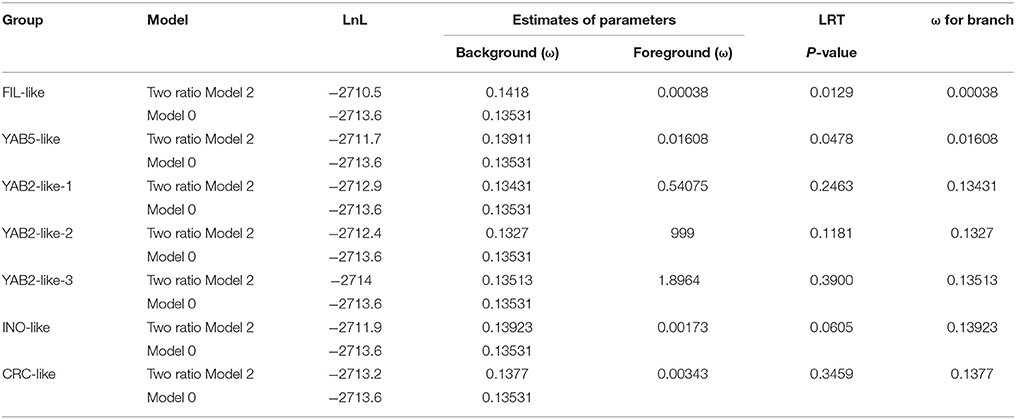
Table 1. Analysis of patterns of natural selection using PAML.
TEs are widely dispersed throughout the whole genome, and the presence of TEs is commonly associated with gene duplication (Oliver et al., 2013). TEs are not only associated with gene expansion but are also associated with the regulation of gene expression in response to stress (Bennetzen and Wang, 2014). To evaluate the relationship between TEs and the YABBY gene family, de novo prediction and homology search methods were used to identify TEs in the whole genome, as previously reported (Yang et al., 2017), and we further investigated TEs close to the YABBY genes. As shown in Table 2, analysis of the 2,000 bp region around the YABBY gene locus revealed no DNA transposons, although six retroelements were found, namely, four Copia, one Gypsy, and one Helitron element (Table 2 and Supplementary Table 7). Because only a small number of TEs were detected, we scanned regions up to 10 Kb upstream and downstream around each gene locus, and accordingly succeeded in identifying 48 TEs (one DNA transposon and 47 retroelements). The single DNA transposon was from the MULE-MuDR family, whereas the retroelements mainly contained long-terminal repeats (LTRs), including 29 Copia and 17 Gypsy retroelements (Table 2 and Supplementary Table 8). We further inspected the TE distribution and found one Gypsy TE located downstream of GhYABBY4_At and two Copia TEs located downstream of GhYABBY12_At within a 2,000 bp region. Within the 10,000 bp region, we found TEs located in the vicinity of 13 gene loci, among which 33 were located around genes showing segmental duplication. For example, four Copia TEs were located downstream of GhYABBY12_Dt, 11 Copia TEs were located upstream or downstream of GhYABBY12_At, five Gypsy TEs were located upstream or downstream of GhYABBY1_At, and two Gypsy TEs were located upstream of GhYABBY1_Dt. As reported previously, the occurrence of most TEs was correlated with the presence of duplicated genes in the WOX family (Yang et al., 2017). Consistently, we found that this general rule applied to the YABBY gene family, indicating that retroelements (particularly LTRs) may have played significant roles in YABBY family expansion. In contrast to TEs, simple repeat sequences are more abundant and variable, and are distributed widely in the upstream, downstream, and intragenic regions of YABBY genes.
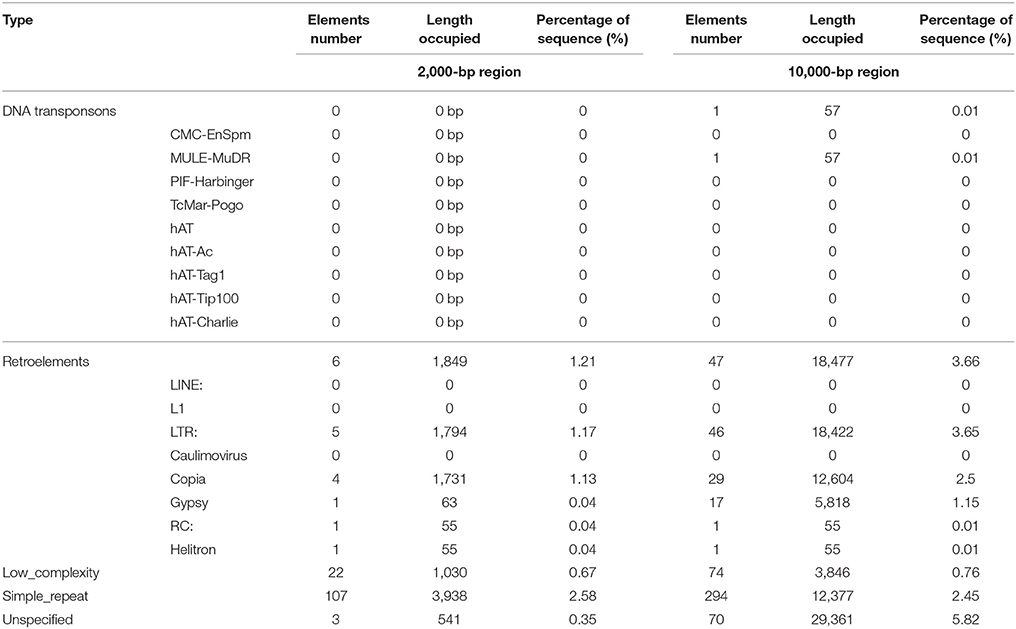
Table 2. Transporsable elements around the YABBY gene loci.
Gene Structure and Motif Analysis
To further study the phylogenetic relationship among YABBYs from upland cotton and Arabidopsis, we compared their gene structures and motifs. As shown in Figure 5A, the YABBYs were divided into the five groups described in Figure 2. Because the number of genes used for constructing the phylogenetic trees shown in Figures 2, 5A were different, the resulting topologies of these trees also differed. However, the gene members and topologies within groups were almost the same. As is evident from Figure 5, YABBYs in the YAB5-like group might have undergone expansion because one AtYAB5 gene corresponds to 10 homologous genes from upland cotton (Figure 5A), and this hypothesis was confirmed through synteny analysis with the At and Dt sub-genomes from upland cotton (Figure 3). As shown in Figure 5B, YABBYs are multiple-exon genes (five to seven exons), and the genes close to each other in the phylogenetic tree showed highly similar exon patterns. For example, all YABBY genes in the FIL-like and INO-like groups have seven exons, most YABBY genes in the YAB2-like group have six exons, and most in the YAB5-like group have seven exons. Comparing exons in orthologs from the same position, we found that the exon sequences were more conserved than the intron sequences. For example, the first intron sequence of GhYABBY11_Dt was longer than that of GhYABBY11_At, and the length of the sixth intron of GhYABB10_Dt was shorter than that of GhYABB10_At. We found that the length of the sixth exons of GhYABBY6_At/Dt were not equal, which may have been caused by a premature stop codon. Indeed, further analysis of the genomic sequences of GhYABBY6_At/Dt revealed a 4 bp indel in the sixth exon of GhYABBY6_Dt, resulting in a frameshift mutation and a premature stop codon (Supplementary Image 3).
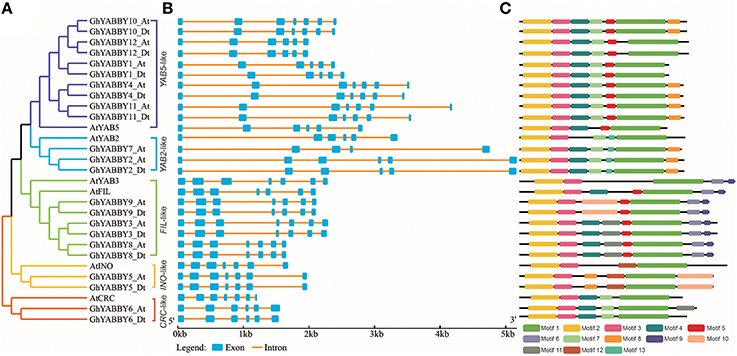
Figure 5. Comparison of the gene structures between Arabidopsis thaliana and Gossypium hirsutum. (A) NJ tree analysis of A. thaliana and G. hirsutum. Orange, yellow, light blue, purple, and green represent the CRC-like, INO-like, YAB2-like, YAB5-like, and FIL-like groups, respectively. (B) The number, length, and position of exons and introns within YABBY genes. Boxes indicate the exons and black lines indicate the introns. (C) Distribution of the predicted motifs in the YABBY genes.
MEME was used to discover the possible motifs within YABBY genes to better understand their phylogenetic relationships. A total of 13 motifs were identified in the YABBY genes, the widths of which ranged from 8 to 57 amino acids. Motif 1 is located in the YABBY domain, and motifs 2 and 3 are located in the C2C2 zinc-finger-like domain (Supplementary Table 9 and Figure 5C). As shown in Figure 5C, in addition to the common motifs 1, 2, and 3 harbored by all the YABBY genes analyzed, some private motifs were only found in genes within the same group. For example, all genes within the YAB5-like group were characterized by motifs 4 (QS[LH]S[GW][QH][DS][IF][QF][AT]P[NQ][YN][AT]L[ES][ED]YRSD) and 5 ([NE][ETN][IAT][TP][EKQ][EP][RP]VVNR), whereas genes in the FIL-like group contained motifs 5 ([NE][ETN][IAT][TP][EKQ][EP][RP]VVNR) and 6 ([RH]QQEGE[ED][MAV][VL][MV]KDGFF).
Gene Expression Patterns in Different Tissues under Multiple Stresses
Gene expression is the major step toward a gene realizing its biological function. Thus, we investigated YABBY gene expression profiles in different tissues, during seed germination and seed (fiber) development, and under biological stresses. RNA-Seq data were acquired and analyzed as described in our previous report (Yang et al., 2017). As shown in Figure 6A, YABBY genes exhibited different expression patterns in differing vegetative tissues (roots, stems, and leaves), reproductive tissues (torus, petal, stamen, pistil, and calycle), stages of seed development (−3, −1, 0, 1, 3, 5, 10, 20, 25, and 35 days post-anthesis [DPA] ovule), and stages of fiber development (5, 10, 20, and 25 DPA), suggesting that YABBYs have multiple biological functions. With the exception of GhYABBY2_At/Dt, the expression of other genes was not detectable in the stem. The YABBY genes have a relative low expression level in leaves. All the genes were transcribed in roots and calycles. Most genes were highly expressed in the late stage of ovule development (20–35 DPA) and 25 DPA fibers (Figure 6A), indicating that YABBYs might play important roles in seed development and secondary wall development, suggesting that these are potential candidate genes for further study. However, GhYABB5_At/Dt were highly expressed during the early stages of ovule development (−3 DPA and−1 DPA). GhYABBY6_At/Dt have a relatively low expression level during ovule development, but were preferentially expressed in the pistil (carpel). YABBYs might be associated with seed germination, considering that almost all these genes were highly expressed in cotyledons at different sampling stages (Figure 6B). When comparing the expression profiles of the orthologs between the At and Dt sub-genomes, the expression patterns and levels of the orthologous pairs were similar, suggesting that their biological functions might be conserved. When comparing the expression patterns of homologous pairs, we found that GhYABB1_At/Dt were expressed at all the stage of ovule development, showing a down-regulated pattern, whereas GhYABBY10_At/Dt and GhYABBY12_At/Dt were transcribed in the late stage of ovule development, suggesting that functional divergence might have occurred during evolution after duplication.
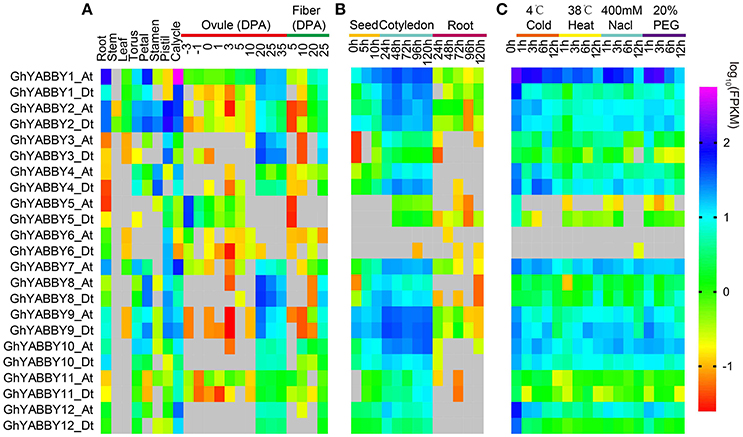
Figure 6. Gene expression profiles of YABBY genes in different tissues (A), during seed germination (B), and under different stresses (C). True leaves were harvested at 0, 1, 3, 6, and 12 h after treatment.
Cotton is the most important cash crop for fiber production; however, it faces multiple stresses during growth and development. Therefore, we performed a comprehensive study of the expression pattern of YABBYs in this study. When cotton was exposed to cold treatment, most genes displayed a down-regulated expression pattern, i.e., the expression levels of GhYABBY1_Dt, GhYABBY2_At/Dt, GhYABBY4_At, GhYABBY9_At/Dt, GhYABBY10_At/Dt, and GhYABBY12_At/Dt were down-regulated and when exposed to cold treatment. We found that only GhYABBY3_At/Dt and GhYABBY8_At/Dt from the same group were up-regulated under cold stress. When cotton was exposed to high temperatures, approximately half of the YABBY genes showed a down-regulated pattern, including GhYABBY1_Dt, GhYABBY2_At/Dt, and GhYABBY4_At/Dt. Similar to the patterns under low and high temperature treatments, approximately half of the YABBY genes displayed down-regulated patterns under PEG and NaCl treatments, indicating that YABBYs might be negative regulators under exposure to stress.
Evaluation of YABBY Gene Expression by Quantitative PCR (qPCR)
To validate and further characterize the YABBY gene expression profiles from the RNA-Seq data, qPCR was performed using root samples treated with PEG6000. Root samples were used because of their relatively higher gene expression levels in most samples. Given that orthologs from the At and Dt sub-genomes have highly similar mRNA sequences, it is difficult to distinguish such orthologous pairs by qPCR. Therefore, primers were designed to amplify the orthologous pairs together (Supplementary Table 10). Figure 6C shows that GhYABBY6_At/Dt were not detectable in response to PEG treatment, and we found that GhYABBY6_At/Dt were transcribed at a relatively low expression level since the cycle threshold (Ct) values were larger than 30 for all samples, whereas the Ct values for the internal control gene (histone 3) were in the normal range (17–18) for the same samples. As shown in Figure 7, most genes were down-regulated following PEG6000 exposure, which is similar to the expression pattern shown in Figure 6C, indicating that our RNA-Seq data were relatively accurate. The expression levels of GhYABBY2, GhYABBY3, GhYABBY8, GhYABBY9, and GhYABBY10 were down-regulated under PEG6000 treatment, suggesting that they may function as negative regulators under drought stress and are potential candidate genes for further study.
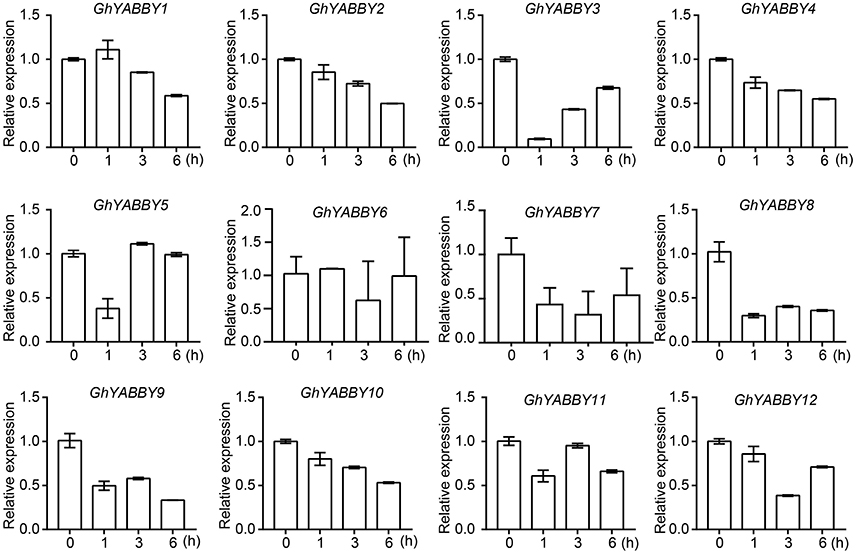
Figure 7. Expression levels of YABBY genes under PEG6000 treatment. Error bars represent the standard deviation of three independent experiments.
Discussion
YABBY genes are seed plant-specific transcription factors that are associated with leaf, shoot, and flower development. They have been well studied in A. thaliana, tomato, and rice (Siegfried et al., 1999; Toriba et al., 2007; Bartholmes et al., 2012; Huang et al., 2013). However, to date, few studies have been performed on YABBY genes in cotton. In the current study, we performed comprehensive identification and analysis of YABBY genes in cotton, with the aim of gaining a better understanding of their functional roles in further studies.
Duplication of YABBY Genes in Cotton
Upland cotton is a typical allotetraploid and is an ideal plant for studying polyploid formation in evolution (Paterson et al., 2012). Previous findings have suggested that diploid species resembling G. arboreum (A2) and G. raimondii (D5) hybridized naturally, followed by chromosome doubling, and natural and human selection, with the eventual formation of upland cotton. In our study, we found that the number of YABBY genes in upland cotton is close to the total number in G. arboreum (A2) and G. raimondii (D5), suggesting that whole genome duplication was the major impetus underlying the expansion of YABBY genes in upland cotton. Polyploidization has previously been identified in more than 90% of flowering plants, and has played important roles in the adaptation of plants to the environment (Ramsey et al., 1998). Polyploidization has resulted in the enhancement of gene dosages and has led to the strengthening of gene expression, which might have enhanced the yield, fiber quality, biomass, and stress tolerance of the allotetraploids G. hirsutum and G. barbadense compared with G. raimondii, and G. arboreum.
As described in a previous study (Fraser et al., 2005), major chromosome events such as segmental duplication and translocation have enabled plants to rapidly adapt to new environments. In the current study, we found that the YABBYs from cotton were well divided into five groups, as observed previously (Bartholmes et al., 2012), and no additional groups were identified in cotton. Among the five groups, the YAB5-like group tended to retain recently duplicated paralogs in cotton (Figure 2). Five duplicated gene pairs were found in the YAB5-like group and they displayed different expression patterns, indicating that their biological functions might have diverged after duplication and that these genes might have helped upland cotton adapt to new environments. We also found that WOX genes in upland cotton have undergone segmental duplication and that the duplicated genes displayed different expression patterns (Yang et al., 2017), suggesting that divergence is a common evolutionary process after duplication. It appears that the YAB5-like group is dicot-specific because all members within this group are dicots. Whether monocots were lost from this group or dicots acquired a specific genetic requirement necessitates further study. Homologs in the YAB5-like group appear to have been active during evolution because some duplication events were found in this group. Although the function of AtYAB5 has yet to be determined, the functions of homologs in cotton from the same group may have diverged because their expression patterns were different, although elucidating their functions requires further study.
Tandem duplication has commonly been found within plant genomes and is a basic contributor to gene family expansion. Unequal chromosome crossover events, chromosomal anomalies, transposon insertions, and other reverse transcriptase-mediated processes can result in tandem duplication, and the new genes would co-exist with the homolog(s) already present (Flagel and Wendel, 2009). One example of tandem duplication is seen in the NBS-LRR gene family, which functions as the major class of disease resistance genes in flowering plants. Among the 166 NBS-LRR genes, 40 clusters have previously been identified (Leister, 2004). Functional redundancy has been considered an important impetus for plant environmental adaption. Neofunctionalization, duplication, degeneration, complementation, subfunctionalization, escape from adaptive conflict, and dosage-balance models have been proposed to help understand gene-duplication events (Flagel and Wendel, 2009).
In addition to gene expansion, gene loss has also occurred during the evolution of upland cotton. The occurrence of gene loss has been accompanied by “genome shock,” such as the rapid arrangement of genomic sequences during hybridization and following chromosome doubling during polyploidization (Paterson et al., 2004).
Transposable elements (TEs) are mobile genetic elements that are located at varying positions in genomic DNA using element-encoded enzymatic machinery (Arkhipova, 2017). Mobile element activity together with recombination are the major processes promoting genome divergence, which plays a key role in the evolution of species. TEs are divided into two classes according to whether TE mobility requires an RNA intermediate (Class I, or retrotransposons) or not (Class II, or DNA transposons) (Finnegan, 1989). Retrotransposons are the most common TEs in genomic sequences, and can be divided into long terminal repeat retrotransposons (LTR) and non-LTR type. In our study, we analyzed the TEs around YABBY gene loci and found that most of the identified TEs were retrotransposons, which is consistent with the pattern in Helianthus species (Mascagni et al., 2017). In upland cotton, TEs account for at least 64.8% of the assembled genome, and Gypsy and Copia were the most abundant types in the assembled genome. Although we only analyzed TEs around the YABBY loci, the local TEs display a pattern similar to that detected in genome-wide analysis, which indicated that the identified TEs were mainly of the Gypsy and Copia types. Zhang et al. considered that TEs occurred in the progenitor genomes and were retained after allopolyploid formation (Zhang et al., 2015). In our study, we found the same types of TEs around GhYABBY1_At/Dt and GhYABBY12_At/Dt, suggesting that these TEs are ancient TEs that occurred in the progenitor genomes of upland cotton.
Conservation of Cotton YABBYs during Evolution
YABBY genes are seed plant-specific transcription regulators, characterized by an N-terminal zinc-finger domain and a C-terminal YABBY domain (Figure 1A). Previous studies have shown that the C-X2-C-X9-P-X11-C-X2-C motif is a core element forming the C2C2-type zinc-finger with Zn2+, and this structure has also been found in cotton (Leister, 2004; Toriba et al., 2007), indicating that C2C2-type zinc-fingers are conserved among different species. The putative helix-loop-helix “YABBY” domain has been reported to share sequence similarity with the first two helices of the high mobility group (HMG) box (Leister, 2004), and we found that cotton YABBYs also contain helices typical of HMG boxes, suggesting that YABBYs are conserved among seed plants. Although there is variation in the length of sequences in the internal region between the zinc-finger and YABBY domains, C-terminal region, and N-terminal region, no amino acid insertions/deletions were identified in the aforementioned two domains, and amino acids identities in the YABBY domains were higher than those in the zinc-finger domains, which is consistent with previous findings (Siegfried et al., 1999).
The YABBY genes from Arabidopsis and rice can be divided into five groups: the INO-like, YAB2-like, YAB5-like, FIL-like, and CRC-like groups (Finet et al., 2016). Twenty-three genes from upland cotton were well divided into these five groups (Figures 2, 4). Genes within the same groups displayed similar characteristics. Firstly, the gene sequences showed higher similarities among genes from the same group. Secondly, apart from the conserved domain regions, the other regions also showed similarities, as evidenced by the finding that different motifs from genes within the same group were more similar (Figure 3C), although they were different within the whole family. Thirdly, exon numbers within the same group were conserved. Our results indicated that genes within the same clade diverged from a common ancestor and may have similar biological functions, suggesting interesting topics for further study. Cacao and cotton plants, which belong to the Malvaceae family, share a common ancestor, and were shown to have a close relationship in the phylogenetic tree (Figure 2). However, the gene numbers in diploid cotton are considerably larger than those in cacao. Previous data have shown that diploid cotton and cacao plants underwent a common ancient duplication event, and that diploid cotton experienced a recent independent duplication event (Li et al., 2014). Therefore, the theoretical number of diploid cotton genes should be twice that of cacao, and a single ortholog in cacao should correspond to two in cotton. However, not all the orthologs in cacao have two orthologs in cotton. We speculate that some cotton genes have been lost or mutated to pseudogenes after recent duplication events during evolution, which has also been found in the HD-ZIP and MIKC MADS-box gene families (Chen et al., 2017; Ren et al., 2017).
Diverse Expression Patterns of YABBYs
Cotton is the most important cash crop for fiber and seed oil production, providing raw materials for the textile industry. Cotton fiber cells originate from the epidermis of the outer integument of ovules. In flowering plants, seeds are the most important reproductive organ formed from ovules. Angiosperm ovules have one or two integuments covering the nucellus and female gametophyte, and the integuments develop into a seed coat after fertilization. Numerous studies have focused on ovule ontogeny in Arabidopsis (Robinson-Beers et al., 1992; Schneitz, 1999), and many genes associated with ovule development have been identified (Skinner et al., 2004), among which genes involved in cell polarity have been demonstrated to play key roles in ovule development (Villanueva et al., 1999; Eshed et al., 2001; McAbee et al., 2006).
Kelley et al. showed that abaxial determinants are necessary for the initiation and maintenance of integument growth and that the KANADI and YABBY gene families are the two major putative polarity determinants involved in integument development (Kelley et al., 2009). Members of the KANADI gene family control cell polarity and play important roles in integument development. ABERRANT TESTA SHAPE (ATS, also known as KAN4), a member of the KANADI gene family, is essential for laminar extension during inner integument development and for maintaining integument separation (McAbee et al., 2006). Other KANADI members (KAN1 and KAN2) were found to be involved in outer integument development and are necessary regulators for laminar extension of the outer integument. YABBY genes, a small plant-specific gene family, were previously certified to play roles in abaxial cell fate specification (Bowman, 2000b). INO, a member of the YABBY gene family, together with KAN1 and KAN2, were found to play significant roles in polar differentiation of the outer integument (Simon et al., 2012). In a previous study, YABBY2 and YABBY3 were shown to be expressed in a polar manner in all lateral organ primordia and were proposed as key regulators responsible for the specification of abaxial cell fate in Arabidopsis (Sawa et al., 1999; Siegfried et al., 1999). CRABS CLAW (CRC) expression is restricted to nectaries and carpels. Although loss of CRC function does not result in any change in carpel polarity, a double mutant of CRC and either PICKLE or KANADI was found to exhibit abaxial abnormity (Bowman, 2000a). In addition, CRC has been reported to play derived roles in leaf vascular development, and an ancestral role of CRC orthologs has been implicated in carpel development in the Poaceae family (Fourquin et al., 2014). In the current study, we found that most YABBY genes were expressed at relatively high levels in 25–35 DPA ovules and in early-stage seed germination, which indicates that the functions of YABBYs are conserved between cotton and Arabidopsis, and play important roles in ovule development. In contrast, only GhYBBY5_At/Dt were specifically highly expressed during an early stage of ovule development, which indicates that GhYABB5_At/Dt play important roles in early-stage ovule development. Although YABBYs have undergone expansion in cotton, GhYABBY5 was the only ortholog of INO in cotton, and therefore GhYABBY5_At/Dt may share a similar function with INO in outer integument differentiation during the early stage of ovule development and are candidate genes for studying cotton ovule development. GhYABBY6_At/Dt are preferentially transcribed in the pistil (carpel), and their expression patterns are similar to those in Arabidopsis and Poaceae taxa (Bowman, 2000a; Fourquin et al., 2014), indicating that CRC and its orthologs shared conserved functions during evolution after speciation.
The establishment of polarity is a critical process in the development of nearly all lateral organs, and is also required for lamina expansion. Previous data showed that a juxtaposition of abaxial and abaxial domains is essential for lamina development (Waites and Hudson, 1995). YABBYs and KANADIs are the two major gene families required for promoting abaxial cell fate and have been proposed to play important roles in asymmetric leaf development and blade expansion in Arabidopsis (Eshed et al., 2004). In the current study, only five gene pairs were detectable in leaves with a relatively high expression level by RNA-seq, among which GhYABBY2_At/Dt and GhYABBY7_At were close to each other in the phylogenetic tree and are orthologs of AtYAB2, which is a gene that is essential for determining lamina outgrowth (Siegfried et al., 1999). Therefore, GhYABBY2_At/Dt and GhYABBY7_At may participate in lamina development. Compared with other tissues, YABBY genes were almost undetectable in stems, suggesting that YABBYs play functional roles in restricted regions.
YABBYs are associated with the growth and development of lateral organs under both normal growth and stressed conditions. Peng et al. found that YABBY genes were upregulated under cold stress and proposed that transcription factors of the YABBY family and other families, by interacting with Auxin and other hormones, were essential for regulating growth and development of paper mulberry under cold stress (Peng et al., 2015). In our study, only GhYABBY3_At/Dt and GhYABBY8_At/Dt were up-regulated under cold treatment, indicating that these genes may be key regulators of lateral organ development under cold stress. Moumeni et al. found that drought-responsive genes were characterized by the presence of a GCAC[AG][ACGT][AT]TCCC[AG]A[ACGT]G[CT] motif in the promoter region, which is common to AP1, AP2, HD-ZIP, and YABBY, indicating that these transcription factors may play roles in drought response (Moumeni et al., 2015). OsYABBY1 in rice might be associated with leaf rolling during drought stress (Dai et al., 2007). In our study, we found that most YABBY genes were down-regulated under drought and salinity stress, indicating that they might negatively regulate drought or salinity responses. Under heat stress, both up- and down-regulated YABBY genes were detected in our study, indicating that they may serve diverse functions under heat stress. Although the RNA-Seq data revealed the expression pattern of YABBYs under different stress conditions, further studies are needed to unravel their functions.
Author Contributions
FL and ZhY: Conceived and designed the study; YJ, JX, LW, ZL, WQ, and ZuY: Performed the experiments; QG: Prepared the figures; LL: Analyzed and interpreted the data; ZhY, QG, and FL: prepared the manuscript; QC: participated in the design of the experiments, wrote part of the manuscript, and performed a critical review for intellectual content. All authors have read, edited, and approved the current version of the manuscript.
Funding
This work was supported by grants from National Key R&D Program of China (2016YFD0100500).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer DH and handling Editor declared their shared affiliation.
Acknowledgments
We would like to thank the reviewers for their comments and suggestion.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2018.00033/full#supplementary-material
References
Alvarez, J., and Smith, D. R. (1999). CRABS CLAW and SPATULA, two Arabidopsis genes that control carpel development in parallel with AGAMOUS. Development 126, 2377–2386.
Arkhipova, I. R. (2017). Using bioinformatic and phylogenetic approaches to classify transposable elements and understand their complex evolutionary histories. Mob. DNA 8:19. doi: 10.1186/s13100-017-0103-2
Bartholmes, C., Hidalgo, O., and Gleissberg, S. (2012). Evolution of the YABBY gene family with emphasis on the basal eudicot Eschscholzia californica (Papaveraceae). Plant Biol. 14, 11–23. doi: 10.1111/j.1438-8677.2011.00486.x
Bennetzen, J. L., and Wang, H. (2014). The contributions of transposable elements to the structure, function, and evolution of plant genomes. Annu. Rev. Plant Biol. 65, 505–530. doi: 10.1146/annurev-arplant-050213-035811
Bowman, J. L. (2000a). Axial patterning in leaves and other lateral organs. Curr. Opin. Genet. Dev. 10, 399–404. doi: 10.1016/S0959-437X(00)00103-9
Bowman, J. L. (2000b). The YABBY gene family and abaxial cell fate. Curr. Opin. Plant Biol. 3, 17–22. doi: 10.1016/S1369-5266(99)00035-7
Bowman, J. L., and Eshed, Y. (2000). Formation and maintenance of the shoot apical meristem. Trends Plant Sci. 5, 110–115. doi: 10.1016/S1360-1385(00)01569-7
Bowman, J. L., and Smyth, D. R. (1999). CRABS CLAW, a gene that regulates carpel and nectary development in Arabidopsis, encodes a novel protein with zinc finger and helix-loop-helix domains. Development 126, 2378–2396.
Cao, Y., Han, Y., Meng, D., Li, G., Li, D., Abdullah, M., et al. (2017). Genome-wide analysis suggests the relaxed purifying selection affect the evolution of WOX genes in Pyrus bretschneideri, Prunus persica, Prunus mume, and Fragaria vesca. Front. Genet. 8:78. doi: 10.3389/fgene.2017.00078
Chen, E., Zhang, X., Yang, Z., Wang, X., Zhang, C., Wu, Z., et al. (2017). Genome-wide analysis of the HD-ZIP IV transcription factor family in Gossypium arboreum and GaHDG11 involved in osmotic tolerance in transgenic Arabidopsis. Mol. Genet. Genomics 292, 593–609. doi: 10.1007/s00438-017-1293-5
Chen, N. (2004). Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 4, 4.10.11–14.10.14. doi: 10.1002/0471250953.bi0410s05
Chen, Q., Atkinson, A., Otsuga, D., Christensen, T., Reynolds, L., and Drews, G. N. (1999). The Arabidopsis filamentous flower gene is required for flower formation. Development 126, 2715–2726.
Crooks, G. E., Hon, G., Chandonia, J.-M., and Brenner, S. E. (2004). WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190. doi: 10.1101/gr.849004
Dai, M., Zhao, Y., Ma, Q., Hu, Y., Hedden, P., Zhang, Q., et al. (2007). The rice YABBY1 gene is involved in the feedback regulation of gibberellin metabolism. Plant Physiol. 144, 121–133. doi: 10.1104/pp.107.096586
Edgar, R. C., and Myers, E. W. (2005). PILER: identification and classification of genomic repeats. Bioinformatics 21, i152–i158. doi: 10.1093/bioinformatics/bti1003
Eshed, Y., Baum, S. F., Perea, J. V., and Bowman, J. L. (2001). Establishment of polarity in lateral organs of plants. Curr. Biol. 11, 1251–1260. doi: 10.1016/S0960-9822(01)00392-X
Eshed, Y., Izhaki, A., Baum, S. F., Floyd, S. K., and Bowman, J. L. (2004). Asymmetric leaf development and blade expansion in Arabidopsis are mediated by KANADI and YABBY activities. Development 131, 2997–3006. doi: 10.1242/dev.01186
Finet, C., Floyd, S. K., Conway, S. J., Zhong, B., Scutt, C. P., and Bowman, J. L. (2016). Evolution of the YABBY gene family in seed plants. Evol. Dev. 18, 116–126. doi: 10.1111/ede.12173
Finnegan, D. J. (1989). Eukaryotic transposable elements and genome evolution. Trends Genet. 5, 103–107. doi: 10.1016/0168-9525(89)90039-5
Flagel, L. E., and Wendel, J. F. (2009). Gene duplication and evolutionary novelty in plants. New Phytol. 183, 557–564. doi: 10.1111/j.1469-8137.2009.02923.x
Floyd, S. K., and Bowman, J. L. (2007). The ancestral developmental tool kit of land plants. Int. J. Plant Sci. 1, 1–35. doi: 10.1086/509079
Floyd, S. K., and Bowman, J. L. (2010). Gene expression patterns in seed plant shoot meristems and leaves: homoplasy or homology? J. Plant Res. 123, 43–55. doi: 10.1007/s10265-009-0256-2
Fourquin, C., Primo, A., Martínez-Fernández, I., Huet-Trujillo, E., and Ferrándiz, C. (2014). The CRC orthologue from Pisum sativum shows conserved functions in carpel morphogenesis and vascular development. Ann. Bot. 114, 1535–1544. doi: 10.1093/aob/mcu129
Fraser, J. A., Huang, J. C., Pukkila-Worley, R., Alspaugh, J. A., Mitchell, T. G., and Heitman, J. (2005). Chromosomal translocation and segmental duplication in Cryptococcus neoformans. Eukaryotic Cell 4, 401–406. doi: 10.1128/EC.4.2.401-406.2005
Golz, J. F. (2004). GRAMINIFOLIA promotes growth and polarity of Antirrhinum leaves. Development 131, 3661–3670. doi: 10.1242/dev.01221
Gong, Q., Yang, Z., Chen, E., Sun, G., He, S., Butt, H. I., et al. (2017a). A phi-class glutathione S-transferase gene for Verticillium wilt resistance in Gossypium arboreum identified in a genome-wide association study. Plant Cell Physiol. doi: 10.1093/pcp/pcx180. [Epub ahead of print]
Gong, Q., Yang, Z., Wang, X., Butt, H. I., Chen, E., He, S., et al. (2017b). Salicylic acid-related cotton (Gossypium arboreum) ribosomal protein GaRPL18 contributes to resistance to Verticillium dahliae. BMC Plant Biol. 17:59. doi: 10.1186/s12870-017-1007-5
Hu, B., Jin, J., Guo, A. Y., Zhang, H., Luo, J., and Gao, G. (2014). GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics 31, 1296–1297. doi: 10.1093/bioinformatics/btu817
Huang, Z., Van Houten, J., Gonzalez, G., Xiao, H., and Van Der Knaap, E. (2013). Genome-wide identification, phylogeny and expression analysis of SUN, OFP and YABBY gene family in tomato. Mol. Genet. Genomics 288, 111–129. doi: 10.1007/s00438-013-0733-0
Jiao, X., Zhao, X., Zhou, X. R., Green, A., Fan, Y., Wang, L., et al. (2013). Comparative transcriptomic analysis of developing cotton cotyledons and embryo axis. PLoS ONE 8:e71756. doi: 10.1371/journal.pone.0071756
Jones, P., Binns, D., Chang, H. Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Jurka, J. (2000). Repbase update: a database and an electronic journal of repetitive elements. Trends Genet. 16, 418–420. doi: 10.1016/S0168-9525(00)02093-X
Kanaya, E., Nakajima, N., and Okada, K. (2002). Non-sequence-specific DNA binding by the filamentous flower protein from Arabidopsis thaliana is reduced by EDTA. J. Biol. Chem. 277, 11957–11964. doi: 10.1074/jbc.M108889200
Kelley, D. R., Skinner, D. J., and Gasser, C. S. (2009). Roles of polarity determinants in ovule development. Plant J. 57, 1054–1064. doi: 10.1111/j.1365-313X.2008.03752.x
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Kumaran, M. K., Bowman, J. L., and Sundaresan, V. (2002). YABBY polarity genes mediate the repression of KNOX homeobox genes in Arabidopsis. Plant Cell 14, 2761–2770. doi: 10.1105/tpc.004911
Kumaran, M. K., Ye, D., Yang, W.-C., Griffith, M. E., Chaudhury, A. M., and Sundaresan, V. (1999). Molecular cloning of abnormal floral organs: a gene required for flower development in Arabidopsis. Sex. Plant Reprod. 12, 118–122. doi: 10.1007/s004970050180
Lee, J. Y., Baum, S. F., Oh, S. H., Jiang, C. Z., Chen, J. C., and Bowman, J. L. (2005). Recruitment of CRABS CLAW to promote nectary development within the eudicot clade. Development 132, 5021–5032. doi: 10.1242/dev.02067
Leister, D. (2004). Tandem and segmental gene duplication and recombination in the evolution of plant disease resistance gene. Trends Genet. 20, 116–122. doi: 10.1016/j.tig.2004.01.007
Li, F., Fan, G., Lu, C., Xiao, G., Zou, C., Kohel, R. J., et al. (2015). Genome sequence of cultivated upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat. Biotechnol. 33, 524–530. doi: 10.1038/nbt.3208
Li, F., Fan, G., Wang, K., Sun, F., Yuan, Y., Song, G., et al. (2014). Genome sequence of the cultivated cotton Gossypium arboreum. Nat. Genet. 46, 567–572. doi: 10.1038/ng.2987
Mascagni, F., Giordani, T., Ceccarelli, M., Cavallini, A., and Natali, L. (2017). Genome-wide analysis of LTR-retrotransposon diversity and its impact on the evolution of the genus Helianthus (L.). BMC Genomics 18:634. doi: 10.1186/s12864-017-4050-6
McAbee, J. M., Hill, T. A., Skinner, D. J., Izhaki, A., Hauser, B. A., Meister, R. J., et al. (2006). Aberrant testa shape encodes a kanadi family member, linking polarity determination to separation and growth of Arabidopsis ovule integuments. Plant J. 46, 522–531. doi: 10.1111/j.1365-313X.2006.02717.x
Moumeni, A., Satoh, K., Venuprasad, R., Serraj, R., Kumar, A., Leung, H., et al. (2015). Transcriptional profiling of the leaves of near-isogenic rice lines with contrasting drought tolerance at the reproductive stage in response to water deficit. BMC Genomics 16:1110. doi: 10.1186/s12864-015-2335-1
Nagasawa, N., Miyoshi, M., Sano, Y., Satoh, H., Hirano, H., Sakai, H., et al. (2003). Superwoman1 and drooping leaf genes control floral organ identity in rice. Development 130, 705–718. doi: 10.1242/dev.00294
Ohmori, Y., Toriba, T., Nakamura, H., Ichikawa, H., and Hirano, H. Y. (2011). Temporal and spatial regulation of drooping leaf gene expression that promotes midrib formation in rice. Plant J. 65, 77–86. doi: 10.1111/j.1365-313X.2010.04404.x
Oliver, K. R., McComb, J. A., and Greene, W. K. (2013). Transposable elements: powerful contributors to angiosperm evolution and diversity. Genome Biol. Evol. 5, 1886–1901. doi: 10.1093/gbe/evt141
Paterson, A. H., Bowers, J. E., and Chapman, B. A. (2004). Ancient polyploidization predating divergence of the cereals, and its consequences for comparative genomics. Proc. Natl. Acad. Sci. U.S.A. 101, 9903–9908. doi: 10.1073/pnas.0307901101
Paterson, A. H., Wendel, J. F., Gundlach, H., Guo, H., Jenkins, J., Jin, D., et al. (2012). Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature 492, 423–427. doi: 10.1038/nature11798
Peng, X., Wu, Q., Teng, L., Tang, F., Pi, Z., and Shen, S. (2015). Transcriptional regulation of the paper mulberry under cold stress as revealed by a comprehensive analysis of transcription factors. BMC Plant Biol. 15:108. doi: 10.1186/s12870-015-0489-2
Prince, V. E., and Pickett, F. B. (2002). Splitting pairs: the diverging fates of duplicated genes. Nat. Rev. Genet. 3, 827–837. doi: 10.1038/nrg928
Ramsey, J., Schemske, D. W., Soltis, P. S., and Soltis, D. E. (1998). Pathways, mechanisms, and rates of polyploid formation in flowering plants. Annu. Rev. Ecol. Syst. 60, 467–501. doi: 10.1146/annurev.ecolsys.29.1.467
Ren, Z., Yu, D., Yang, Z., Li, C., Qanmber, G., Li, Y., et al. (2017). Genome-wide identification of the MIKC-type MADS-box gene family in Gossypium hirsutum L. unravels their roles in flowering. Front. Plant Sci. 8:384. doi: 10.3389/fpls.2017.00384
Robinson-Beers, K., Pruitt, R. E., and Gasser, C. S. (1992). Ovule development in wild-type Arabidopsis and two female-sterile mutants. Plant Cell 4, 1237–1249. doi: 10.1105/tpc.4.10.1237
Sarojam, R., Sappl, P. G., Goldshmidt, A., Efroni, I., Floyd, S. K., Eshed, Y., et al. (2010). Differentiating Arabidopsis shoots from leaves by combined YABBY activities. Plant Cell 22, 2113–2130. doi: 10.1105/tpc.110.075853
Sawa, S., Watanabe, K., Goto, K., Liu, Y. G., Shibata, D., Kanaya, E., et al. (1999). Filamentous flower, a meristem and organ identity gene of Arabidopsis, encodes a protein with a zinc-finger and HMG-related domains. Genes Dev. 13, 1079–1088. doi: 10.1101/gad.13.9.1079
Schneitz, K. (1999). The molecular and genetic control of ovule development. Curr. Opin. Plant Biol. 2, 13–17. doi: 10.1016/S1369-5266(99)80003-X
Siegfried, K. R., Eshed, Y., Baum, S. F., Otsuga, D., Drews, G. N., and Bowman, J. L. (1999). Members of the YABBY gene family specify abaxial cell fate in Arabidopsis. Development 126, 4117–4128.
Simon, M. K., Williams, L. A., Brady-Passerini, K., Brown, R. H., and Gasser, C. S. (2012). Positive- and negative-acting regulatory elements contribute to the tissue-specific expression of inner no outer, a YABBY-type transcription factor gene in Arabidopsis. BMC Plant Biol. 12:214. doi: 10.1186/1471-2229-12-214
Skinner, D. J., Hill, T. A., and Gasser, C. S. (2004). Regulation of ovule development. Plant Cell 16(Suppl.), S32–S45. doi: 10.1105/tpc.015933
Stanke, M., Steinkamp, R., Waack, S., and Morgenstern, B. (2004). AUGUSTUS: a web server for gene finding in eukaryotes. Nucl. Acids Res. 32, W309–W312. doi: 10.1093/nar/gkh379
Strable, J., Wallace, J. G., Unger-Wallace, E., Briggs, S., Bradbury, P. J., Buckler, E. S., et al. (2017). Maize YABBY genes drooping leaf1 and drooping leaf2 regulate plant architecture. Plant Cell 29, 1622–1641. doi: 10.1105/tpc.16.00477
Sturn, A., Quackenbush, J., and Trajanoski, Z. (2002). Genesis: cluster analysis of microarray data. Bioinformatics 18, 207–208. doi: 10.1093/bioinformatics/18.1.207
Suyama, M., Torrents, D., and Bork, P. (2006). PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucl. Acids Res. 34, W609–W612. doi: 10.1093/nar/gkl315
Tanaka, W., Toriba, T., and Hirano, H. Y. (2017). Three TOB1-related YABBY genes are required to maintain proper function of the spikelet and branch meristems in rice. New Phytol. 215, 825–839. doi: 10.1111/nph.14617
Toriba, T., Harada, K., Takamura, A., Nakamura, H., Ichikawa, H., Suzaki, T., et al. (2007). Molecular characterization the YABBY gene family in Oryza sativa and expression analysis of OsYABBY1. Mol. Genet. Genomics 277, 457–468. doi: 10.1007/s00438-006-0202-0
Trapnell, C., Roberts, A., Goff1, O., Pertea, G., Kim, D., Kelley, D. R., et al. (2013). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578. doi: 10.1038/nprot.2012.016
Vandepoele, K., Simillion, C., and Van de Peer, Y. (2003). Evidence that rice and other cereals are ancient aneuploids. Plant Cell 15, 2192–2202. doi: 10.1105/tpc.014019
Villanueva, J. M., Broadhvest, J., Hauser, B. A., Meister, R. J., Schneitz, K., and Gasser, C. S. (1999). Inner no outer regulates abaxial–adaxial patterning in Arabidopsis ovules. Genes Dev. 13, 3160–3169. doi: 10.1101/gad.13.23.3160
Voorrips, R. E. (2002). MapChart: software for the graphical presentation of linkage maps and QTLs. J. Hered. 93, 77–78. doi: 10.1093/jhered/93.1.77
Waites, R., and Hudson, A. (1995). Phantastica: a gene required for dorsoventrality of leaves in Antirrhinum majus. Development 121, 2143–2154.
Wang, K., Wang, Z., Li, F., Ye, W., Wang, J., Song, G., et al. (2012). The draft genome of a diploid cotton Gossypium raimondii. Nat. Genet. 44, 1098–1103. doi: 10.1038/ng.2371
Watanabe, K., and Okada, K. (2003). Two discrete cis elements control the abaxial side-specific expression of the filamentous flower gene in Arabidopsis. Plant Cell 15, 2592–2602. doi: 10.1105/tpc.015214
Xu, Z., and Wang, H. (2007). LTR_finder: an efficient tool for the prediction of full-length LTR retrotransposons. Nucl. Acids Res. 35, W265–W268. doi: 10.1093/nar/gkm286
Yamada, T., Tobe, H., Imaichi, R., and Kato, M. (2001). Developmental morphology of the ovules of Amborella trichopoda (Amborellaceae) and Chloranthus serratus (Chloranthaceae). Bot. J. Linn. Soc. 137, 277–290. doi: 10.1111/j.1095-8339.2001.tb01123.x
Yamada, T., Yokota, S., Hirayama, Y., Imaichi, R., Kato, M., and Gasser, C. S. (2011). Ancestral expression patterns and evolutionary diversification of YABBY genes in angiosperms. Plant J. 67, 26–36. doi: 10.1111/j.1365-313X.2011.04570.x
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Yang, Z., Gong, Q., Qin, W., Cheng, Y., Lu, L., Ge, X., et al. (2017). Genome-wide analysis of WOX genes in upland cotton and their expression pattern under different stresses. BMC Plant Biol. 17:113. doi: 10.1186/s12870-017-1065-8
Yang, Z., Zhang, C., Yang, X., Liu, K., Wu, Z., Zhang, X., et al. (2014). PAG1, a cotton brassinosteroid catabolism gene, modulates fiber elongation. New Phytol. 203, 437–448. doi: 10.1111/nph.12824
Yu, J., Jung, S., Cheng, C. H., Ficklin, S. P., Lee, T., Zheng, P., et al. (2014). CottonGen: a genomics, genetics and breeding database for cotton research. Nucl. Acids Res. 42, D1229–D1236. doi: 10.1093/nar/gkt1064
Zhang, F., Zhu, G., Du, L., Shang, X., Cheng, C., Yang, B., et al. (2016). Genetic regulation of salt stress tolerance revealed by RNA-Seq in cotton diploid wild species, Gossypium davidsonii. Sci. Rep. 6:20582. doi: 10.1038/srep20582
Zhang, T., Hu, Y., Jiang, W., Fang, L., Guan, X., Chen, J., et al. (2015). Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 33, 531–537. doi: 10.1038/nbt.3207
Keywords: polarity, Gossypium hirsutum, abiotic stress, ovule, transcription factor, segmental duplication
Citation: Yang Z, Gong Q, Wang L, Jin Y, Xi J, Li Z, Qin W, Yang Z, Lu L, Chen Q and Li F (2018) Genome-Wide Study of YABBY Genes in Upland Cotton and Their Expression Patterns under Different Stresses. Front. Genet. 9:33. doi: 10.3389/fgene.2018.00033
Received: 26 August 2017; Accepted: 25 January 2018;
Published: 07 February 2018.
Edited by:
Edward Hollox, University of Leicester, United KingdomReviewed by:
Dieter Hackenberg, University of Leicester, United KingdomLuciana Werneck Zuccherato, Universidade Federal de Minas Gerais, Brazil
Copyright © 2018 Yang, Gong, Wang, Jin, Xi, Li, Qin, Yang, Lu, Chen and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fuguang Li, YXlsaWZ1Z0AxNjMuY29t
†These authors have contributed equally to this work.