 Sijia Huang
Sijia Huang Kumardeep Chaudhary
Kumardeep Chaudhary Lana X. Garmire
Lana X. Garmire
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Genet., 16 June 2017
Sec. Computational Genomics
Volume 8 - 2017 | https://doi.org/10.3389/fgene.2017.00084
Multi-omics data integration is one of the major challenges in the era of precision medicine. Considerable work has been done with the advent of high-throughput studies, which have enabled the data access for downstream analyses. To improve the clinical outcome prediction, a gamut of software tools has been developed. This review outlines the progress done in the field of multi-omics integration and comprehensive tools developed so far in this field. Further, we discuss the integration methods to predict patient survival at the end of the review.
A new era of personalized medicine has arrived, which proposes an individualized health care model with tailored medical target treatment and management for each patient (Chin et al., 2011). Under this regime, not only clinical profiles of patients but also their molecular profiles are personally managed to drive for advanced treatment. Cancer studies that are focused on one-dimensional omics data have only provided limited information regarding the etiology of oncogenesis and tumor progression. To overcome this, tremendous efforts have been made to obtain multi-platform based genomic data from biospecimen.
The Cancer Genome Atlas (TCGA) is by far the largest endeavor in the USA to collect and analyze the tumor specimens from over 10,000 cancer patients (Weinstein et al., 2013). Measurements of these specimens include tissue exome sequencing, copy number variation (CNV), DNA methylation, gene expression, and microRNA (miRNA) expression, as well as some physiological and clinical data such as race, tumor stage, relapse, and treatment response. However, relative to the genomic data of different levels that are available to the public, the clinical information is more limited. A scale-up of TCGA is the International Cancer Genome Consortium (ICGC), which provides the information of genomic, transcriptomic and epigenomic abnormalities, and somatic mutations over 50 different cancer types (Hudson et al., 2010). These consortia have created unprecedented opportunities to reveal underlying oncogenic molecular signatures beneath phenotypes.
However, human genomes are complex and regulated at multiple levels, which can be manifested by various genomic assays mentioned above. While each of these assays offers a peek of the complex system, these events are rather interdependent (or interactive). Thus, when combining several different omics data to discover the coherent biological signatures, it is challenging to incorporate different biological layers of information to predict phenotypic outcomes (tumor/normal, early/late stage, survival, etc.). It is herein our goal to address the pressing and challenging issues for developing novel algorithms and theoretical methods for multi-omics data integration, in the hope to extract biologically meaningful information of clinical relevance.
The outline of this review is as follows. First, we will discuss the unsupervised data integration algorithms. Among them, we will highlight matrix factorization methods, Bayesian methods, and network-based methods. Next, we will review in-depth the supervised data integration methods, including network-based models, multiple kernel learning methods, and multi-step analysis based models. Subsequently, we will elaborate semi-supervised data integration methods. Finally, we will discuss the advancement of data integration methods for the aim of prognosis prediction and the biological insights underneath the data integration methods.
Unsupervised data integration refers to the cluster of methods that draw an inference from input datasets without labeled response variables. The different approaches under the umbrella of unsupervised data integration are presented in Figure 1 and Table 1. We have categorized them below into five areas: matrix factorization methods, Bayesian methods, network-based methods and multiple kernel learning, and multi-step analysis.
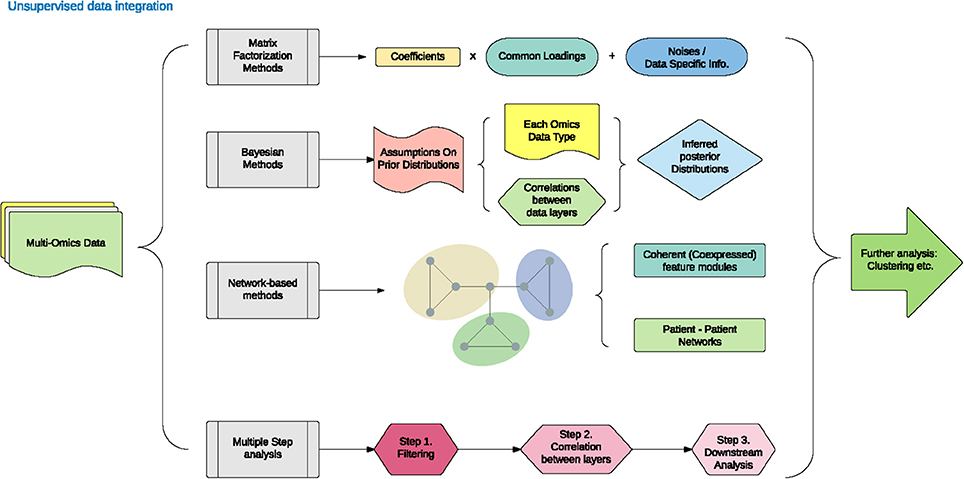
Figure 1. Unsupervised data integration methodology.
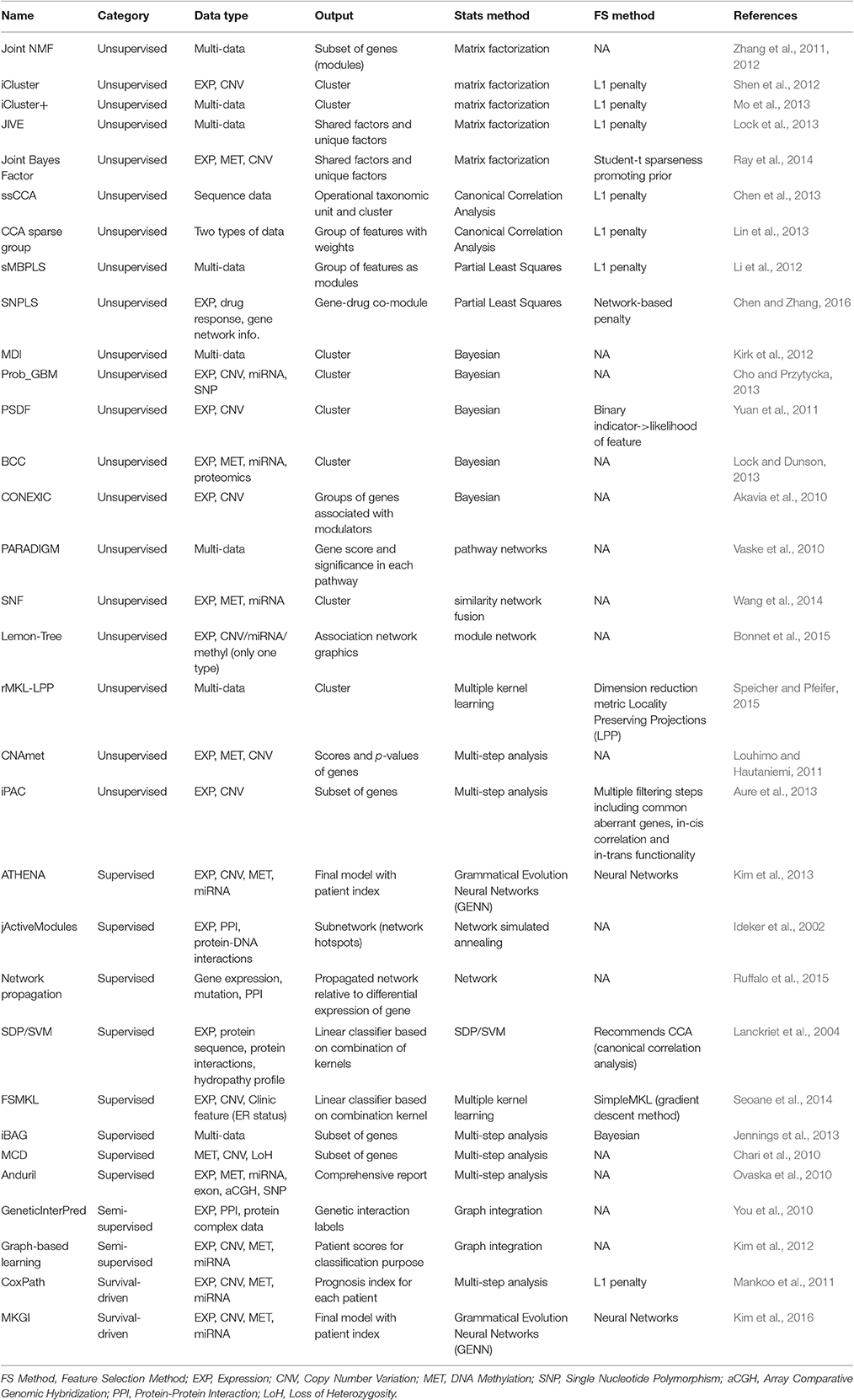
Table 1. Summary of data integration tools.
The most straightforward method for unsupervised data integration falls into the matrix factorization category, which focuses on the projection of variations among data sets onto dimension-reduced space (Lee and Seung, 2001). Zhang et al. proposed NMF framework for multi-omics data integration (Zhang et al., 2011, 2012). This method is based on decomposing a non-negative matrix into non-negative loadings and non-negative factors:
where X is the matrix of mRNA transcriptome, methylome, or other omics data that has M × N dimensions, W is the common factor for M × K dimension matrix and H is the K × N dimension coefficient matrix. Rather than simple correlation, the rationale is to project data onto common basis space, so that one can detect the coherent patterns among data, by examining the elements having significant z-scores. However, NMF is quite time-consuming and requires bulk memory space. For NMF, it is worth noting that not only it requires non-negative input matrices, but also proper normalization step for these input data sets as they have quite different distributions and variability.
Like NMF, iCluster (Shen et al., 2009, 2012) assumes a regularized joint latent variable, which is similar to W in NMF but without non-negative constraints. H is the loading factor (coefficient), the imposed sparsity with different types of penalty functions for various data types. iCluster uses E to represent the error/noise term, and the underlying decomposition equation is:
The upgraded iCluster+ expands iCluster by making the assumption of different modeling approaches for the relationships of X and W within different data platforms. It allows for diverse data types including binary, continuous, categorical, and sequential data with different modeling assumptions including logistic, normal linear, multilogit, and Poisson distributions (Mo et al., 2013). The common latent variable vector W represents the underlying driving factors that can be used for disease subtype assignment. Least absolute shrinkage and selection operator (LASSO) penalty is introduced to address the sparsity issue in H (Tibshirani, 1994). Since this approach requires high computational complexity, it is necessary to preselect the features critical for clustering results (Wang et al., 2014; Speicher and Pfeifer, 2015). Both iCluster and iCluster+ do not require non-negative input data, unlike NMF.
Another variation of NMF category is Joint and Individual Variation Explained (JIVE) method. JIVE decomposes the original data of each layer into three parts, including an approximation of joint variation across data types, approximation of specific structured variation for each data type, and residual noise. In other words, JIVE factors the original data input matrix (gene expression etc.) into two lower ranked representative portions Wc (shared factor) and Ws (data-specific factor), dependent on Hc and Hs (Lock et al., 2013). H matrix is contributed from one sub-matrix Hc common for all data types, and the other sub-matrix Hs specific to each data type.
It should be noted that there can be separate loading factors (Hc and Hs) for the shared factor and data-specific factor (Wc and Ws). The ranks of the two loading factors can be different. An application of JIVE on gene expression data and microRNA data on Glioblastoma (GBM) samples provided information to better characterize samples into different subtypes and strong clues for associations between each input layer (gene expression and microRNA). Based on PCA for factorization, JIVE suffers from outliers, thus the robustness of JIVE is a major concern. L1 penalties are also placed to reduce the dimensions in JIVE, giving non-zero loadings representing larger and significant contributions to the variation of data.
On the other hand, an alternate called Joint Bayes Factor, assumes a common factor loadings H for both shared and data-specific factor Wc and Ws (Ray et al., 2014). Like JIVE, the original data input (e.g., gene expression data matrix) is decomposed into shared common factors across data types, data-type specific factors, and residual noise. However, unlike JIVE, which introduces sparsity using L1 penalties, the Joint Bayes Factor model assumes a beta-Bernoulli process for both the common factors and data specific factors (Wc and Ws; Griffiths and Ghahramani, 2005; Thibaux and Jordan, 2007). For factor loadings (H), the model uses the student-t sparseness-promoting prior, to impose sparsity (Tipping, 2001). As a result, both shared features from each data type and unique features for individual layers can be identified for further analysis. One limitation of Joint Bayes Factor lies within the linear relationship between the latent space and the observational space, and it also assumes very close relationship for different levels of data. Joint analysis of gene expression data with CNV data through this approach identified experimentally validated key drivers, as well as important candidates for further validation for ovarian cancer.
Canonical correlation analysis (CCA), a traditional method to investigate the relationship between two sets of variables, has been modified and applied to the data integration field. In CCA, two datasets can be decomposed as:
Hx and Hy stand for loading factors for each data set. CCA aims to find the loading factors ( representing the ith column for loading factors) which maximize the correlation:
Traditional CCA doesn't account for dimension reduction techniques to compute the inverse of a covariance matrix. For the integration purpose, penalization and regularization terms are added cooperatively to create more stable and sparse solutions of loading factors. L1-penalized sCCA (sparse CCA) together with elastic net CCA were proposed to filter the number of variables to make the results more biologically interpretable (Parkhomenko et al., 2009; Witten and Tibshirani, 2009). Recent research on CCA includes consideration of grouped effects of features as structures embedded within the data sets, such as structure-constrained CCA (ssCCA) and CCA-sparse group (Chen et al., 2013; Lin et al., 2013).
Partial least squares (PLS) is focused on maximizing covariance and can potentially avoid the issue of sensitivity to outliers. It projects variables onto a new hyperplane while maximizing the variance to find the fundamental relationship between the two sets of data.
Hx and Hy stand for loading factors for each data set. The aim of PLS is to find the loading factors which maximize the covariance between Wx and Wy:
However, in some cases such as in high dimensional biological omics data, it is desired to obtain sparse solutions for better interpretations of the result. More recently, sparse solutions of PLS such as sPLS has been shown to perform equivalently with that of the CCA-elastic net (Lê Cao et al., 2009). Other implementations of PLS with different objective functions and various constraints were also reported. For example, sparse Multi-Block Partial Least Squares (sMBPLS) overcomes the limit of two data block computation through redefining the objective function as a weighted sum of latent variables in different layers (n ≥ 2; Li et al., 2012). And Sparse Network regularized Partial Least Square (SNPLS) is specialized in identification of gene expression and drug-response relationship co-modules through incorporating gene interaction network structures (Chen and Zhang, 2016). It showed significantly better performance in accuracy compared to sPLS in simulated data.
Bayesian methods have been applied to data integration for over a decade (Imoto et al., 2004; Zhao et al., 2012). The main advantage of Bayesian methods in data integration is that they can make assumptions not only on different types of data sets with various distributions but also on the correlations among data sets. We briefly overview these methods below:
It offers to model each data set using the Dirichlet-Multinomial Allocation (DMA) mixture model, thus can explore the shared information through deriving statistical dependencies (Kirk et al., 2012). In this approach, the allocation of genes from one data set has an influence on those in another set. Apart from bi-clustering (clustering two dimensions from the same data set simultaneously), MDI can cluster a single dimension (e.g., genes) across multiple data sets, under the assumption that these genes are measured in all different levels. It can be extended flexibly by allowing variable associations from different groups of genes across data types. This method excels in identifying genes having their protein products in the same complex, apart from the co-regulated genes. Finally, after learning the similarity of clusters in different data sets, MDI obtains a single-dimension cluster among all the input data sets.
Prob_GBM is another probabilistic framework to construct patient similarity network, where patients are represented by nodes and phenotypic similarities among the patients are edges (Cho and Przytycka, 2013). This method uses the genetic phenotype, which is the gene expression data of each patient, to assign corresponding disease subtype. Explanatory features (e.g., CNVs, mutations and miRNA expression) are used to explain phenotypic similarities constructed from gene expression data, among patients. Thus, each disease subtype is modeled by a distribution of these features, and each patient is characterized as the mixture of the genetic characteristic of each subtype. Finally, patients are labeled by the most likely subtype assignment. This method considers the biological relationships among several genomic layers including mutation, CNVs, and miRNA expression data, but it is limited in terms of the types of input data.
It is based on a two-level hierarchy of Dirichlet Process model, a widely used Bayesian non-parametric model for clustering (Yuan et al., 2011). It checks the concordance between expression and the CNV for each patient. Moreover, it also selects informative features and estimates the number of disease subtypes from the given data. However, this method limits the input for only two types of data (gene expression and CNV), thus reduces its flexibility within multi-platform analysis.
This method is a flexible clustering approach capable of simultaneously modeling the dependence and the heterogeneity of various data sources (Lock and Dunson, 2013). It allows for separate clustering of the objects from each data source and performs post-hoc integration of separated clusters. Consensus clustering is applied to model the source-specific structures as well as to determine the overall clustering.
It is a Bayesian network-based method to integrate CNV and gene expression data (Akavia et al., 2010). A score-guided search is applied to identify the combination of modulators (genes). A ranked list of high-scoring modulators (candidate driver genes) is produced, representing genes that are both correlated with differential gene expression modules across tumor samples and are present in significantly amplified/deleted regions. The key feature of the CONEXIC goes beyond identifying mutation drivers, as it provides the insights into the roles of drivers and associated genes.
Network-based approaches can identify modules, symbolic representations of the disease-associated mechanisms. In this regime, nodes represent genes and edges are links between two genes if there exists interaction between them. Under the unsupervised category, network-based methods are mostly applied for detecting significant genes within pathways, discovering sub-clusters, or finding co-expression network modules (Vaske et al., 2010; Wang et al., 2014; Bonnet et al., 2015).
It is a probabilistic graphical model framework to infer patient-specific genetic variations, with the incorporation of curated pathway interactions among genes (Vaske et al., 2010). PARADIGM converts each pathway in National Cancer Institute (NCI) Pathway Interaction Database (PID) into a distinct probabilistic model, represented as a factor graph with both hidden and observed states. Variables in the graph are used to describe molecules, protein-coding genes and complexes (all three assigned as physical entities) apart from gene families and abstract processes. A pathway is modeled as a directed acyclic graph where edges are defined as either positive or negative influence on the downstream nodes, and the nodes are determined by combining all input signals. The output of PARADIGM includes the integrated pathway activity (IPA) score, representing a patient specific measure for the degree of alteration for a specific pathway, through summarizing information from input data sets such as gene expression and CNVs. PARADIGM claims to provide more robust and consistent signatures for subgrouping patients through demonstration in breast cancer and glioblastoma samples. However, in PARADIGM pathways are measured independently, and interactions among pathways are not considered.
This approach aims at discovering the patient subgroup clusters. SNF integrates different data types by constructing a network of samples (rather than genomic features) for each data type, and then fusing these networks into one comprehensive network (Wang et al., 2014). It has two main steps for data integration: First, it constructs a sample-by-sample similarity matrix for each data type, acting as an individual network. Similarity matrices help to identify universal clusters and networks. It also detects different types of data that give support to each connection in the network. Then, by using the non-linear method of message passing theory (KNN and graph diffusion), SNF fuses different similarity matrices and networks, making the combined networks more coherent during each iteration. As a result, weak similarities (e.g., noises) are removed, and strong similarities are added. SNF is relative flexible without constraints for input data format and but only matched samples across different omics layers. By outputting combined similarities among patients across various layers, SNF offers deeper insight into the comprehensive biological relationship, beyond the scope of basic classification and subtyping methods.
It is another unsupervised method focused on reconstructing module networks (Bonnet et al., 2015). After finding co-expressed clusters from the expression data matrix, Lemon-Tree helps to identify consensus modules and upstream regulatory programs through ensemble methods. First, a gene expression matrix is employed to infer co-expressed gene clusters through a model-based Gibbs sampler. Consensus modules of co-expressed genes are merged through spectral edge clustering algorithm with an ensemble of the gene cluster results. On the other side, additional candidate regulator types of data such as miRNA expression, CNV and methylation data are combined with the consensus module to infer a regulatory score calculated by a decision tree structure. The above separation of module learning and regulator assignment steps provides much more flexibility allowing combination with the other methods. According to the authors, Lemon-Tree has the advantage of inferring more closely related short-path networks with more significant gene ontology-related categories, in comparison to CONEXIC. However, it limits the input data types to be only gene expression and additional one data type, as it is focused on finding co-expressed clusters.
Multi-step (or multi-stage) methods are commonly used to find relationships between the different data types first, and then between the data types and the trait or phenotypes (Ritchie et al., 2015). Kernel methods are defined by the use of kernel functions, which enables to operate in a high-dimensional feature space by simply computing the inner products among the images of all pairs of data in the feature space (Hofmann et al., 2008). Kernel-based data integration methods are usually multi-steps, thus we exemplify multi-kernel and multi-step methods together.
This approach can deal with multiple omics data integration such as gene expression, DNA methylation, and microRNA expression profiles (Speicher and Pfeifer, 2015). It is an extension of the current multiple kernel learning with dimensional reduction (MKL-DR) method, where the data are projected into a lower dimensional and integrative subspace. A regularization term is added to avoid overfitting during the optimization procedure, and it allows using several different kernel types. The Locality Preserving Projections (LPP) is applied to conserve the sum of distances for each sample's k-Nearest Neighbors. The finalized clustering is done through applying k-means on the distance summation. Compared to SNF, rMKL-LPP claims to offer comparable results with much more flexibility, as it provides different choices of dimension reduction methods and a variety of kernels per data type.
It is a state-of-the-art multi-step integration tool for CNV, DNA methylation, and gene expression data (Louhimo and Hautaniemi, 2011). The major goal of CNAmet is to identify genes that are both amplified and upregulated or both deleted and downregulated. This tool integrates CNV and DNA methylation data through their functions on gene regulation. The underlying hypothesis is that the gene upregulation is due to both amplified copy number and hypomethylation, whereas gene downregulation is the result of deleted copy number and hypermethylation. It uses three steps to detect the significant genes: weight calculation, score calculation, and significance evaluation. During the first weight calculation step, the signal-to-noise statistics is calculated to measure the copy number and methylation aberrations relative to the expression values. In the second score calculation step, the weight values are combined to infer a deterministic score, which informs the causes of the alterations in the gene expression. Finally, the permutation test is performed on the combined scores and the P-values are corrected. Identification of the genes which are synergistically regulated by methylation and CNV data leads to better characterization of these genes and better understanding of biological process underlying cancer progression.
It is a multi-step method to identify genes that are in-cis correlated through integrating gene expression and CNV data, as well as genes that are in-trans associated to the biological processes (Aure et al., 2013). The novelty of this method is the capability to adjust for confounding effects of co-occurring copy number aberrations. This analysis module combines correlation analysis, regression, gene set enrichment, and adjustment for co-occurring copy number aberrations with avoidance of confounding effects. In the in-cis correlation, it proposes a linear model where log gene expression is a linear function of log copy number and noise. In the in-trans association, it imposes a direct integration through a statistical enrichment step to get the confidence level of in-trans associations between the genes and biological processes.
Contrary to the unsupervised data integration methods, the supervised methods consider the phenotype labels of samples (disease or normal), and invoke machine training approaches to evaluate the models. Supervised data integration methods are built via information of available known labels from the training omics data. In the following section, we enlist representative network-based, multi-kernel and multi-step based methods (Figure 2 and Table 1).
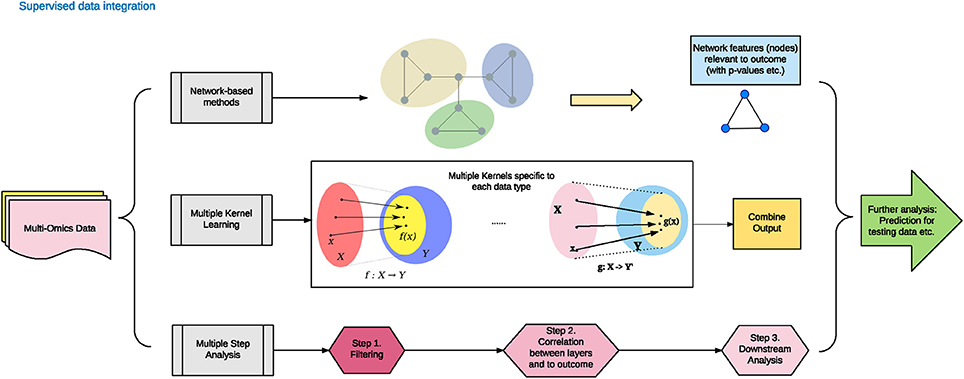
Figure 2. Supervised data integration methodology.
Analysis Tool for Heritable and Environmental Network Associations (ATHENA) is a neural network approach to integrate different omics data with a supervised model which can further be extended to do prognosis analysis (Kim et al., 2013). In ATHENA, grammatical evolution neural networks (GENN) algorithm is utilized to train individual models from different data platforms. Based on neural networks, grammatical evolution algorithm is utilized to train the model with selected features that are less noisy and significantly associated with clinical outcomes. After selecting the features, individual models are summed up to a final integrative model, which can be utilized for multiple purposes including diagnosis and prognosis. Overall, ATHENA provides a comprehensive way of visualizing genomics data's correlation with clinical features such as survival outcomes, making it stand out compared to other network-based integration methods. One limitation of ATHENA lies in lacking interaction terms among different layers, as the features are selected from individual data type first and then combined into an integrated model.
It is another network-based Cytoscape plug-in which seeks underlying network hotspots through the integration of gene expression, protein-protein interaction, and protein-DNA interaction data (Ideker et al., 2002). This method is based on the hypothesis that molecular interactions linking the genes are more likely to correlate expression profiles than randomly chosen genes in the network. This method requires an external input of significance measurements over genes for significance calculation of sub-networks. The external filtering step is a supervised feature selection for genes based on the P-values in the differential expression tests, while the integration method itself doesn't require additional outcomes as inputs. Through random sampling approach and iterative calculations, jActiveModules determines the highest-scoring sub-network circuits in a full network of molecular interactions, leading to further biologically interesting discoveries (Cline et al., 2007). Compared to other clustering methods, jActiveModules is subject to the molecular interaction network and can include genes without dramatically expression fold changes.
Another network-based integration method (Ruffalo et al., 2015) claims to identify key proteins at sample-level using propagated protein networks, based on integrated mutation and differential gene expression (DGE) data sets. Propagated mutation and DGE profiles for each gene are generated with the help of prior knowledge in PPI framework (Schaefer et al., 2012). Feature selection is then done on these propagated profiles in a supervised fashion, with top features being most relevant to outcomes, and a final set of proteins is selected based on the network proximity across the samples. The final step involves logistic regression using the selected genes. This method is useful to find the hidden repertoire of genes/proteins at pathway level with impact on tumor progression/clinical outcome, which might be overlooked by individual mutational or differential expression analysis.
It offers a pioneering kernel-based framework for data integration (Lanckriet et al., 2004). Each data set is represented by a specific kernel function that defines similarity between pairs of entities. Then the kernel functions, derived from different omics data, are combined directly using the SDP (Semidefinite Programming) techniques to reduce the integration problem to a convex optimization problem. The SDP method outperforms the classifier trained with a naïve and unweighted combination of kernels. Different kernels correspond to different transformation of the data, with an extraction of a specific type of information from each data set. The Fast Fourier Transform (FFT) kernel is specific for the membrane protein recognition, by directly incorporating information of hydrophobicity patterns. Higher-order polynomials such as radial basis kernels can be used to capture higher-order non-linear associations of a trait with genotypes. Diffusion kernels are applied to exploit unlabeled data. SDP/SVM is a prototype work for kernel-based data integration methods (published in 2004) and doesn't include a programming package.
Feature Selection Multiple Kernel Learning (FSMKL) is another method implementing the multiple kernel learning-based supervised learning (Seoane et al., 2014). This new scheme uses the statistical score for feature selection per data type per pathway. By employing additional kernels based on clinical covariates, it improves the prediction accuracy for cancer detection. Multiple kernel learning constructs classifiers with a decision function dependent on a variety of different types of input data (gene expression & CNV) using pathway-based kernels. Each type of data (omics) is encapsulated into an object called base kernel; a composite kernel is built as a linear combination of these base kernels. To further incorporate biological information into the algorithm, not only individual feature (such as genes) are independently used to construct kernels, but also specific groups of genes, which are known to have membership from a KEGG pathway, are combined to derive other base kernels. The most appropriate decision function over kernels is finalized after feature selection steps, contributing to an integrative function over base kernels. This method stands out among other kernel-based methods with the inclusion of pathway-based information to build kernels, as prior knowledge. Pathway membership is a central criterion for FSMKL to group samples into different clusters, bringing more biological knowledge compared to basic statistical priors from other methods. Combining clinical factors along with high-throughput profiles into the classifier also brings power for prediction accuracy. FSMKL claimed that this method competes with the winner methods from the DREAM challenge for breast cancer prognosis.
Integrative Bayesian analysis of genomics data (iBAG) is a flexible tool to integrate data from an arbitrary number of platforms (Jennings et al., 2013). A hierarchical model is built to incorporate the information from different genomic layers with biological sense. Basically, this multi-step analysis consists of two-stage models. The first-stage mechanistic model is a regression model which is constructed to partition gene expression data into small segments including methylation principal component, CNV principal component and unknown components other than the previous two. In the second stage of developing clinical a model, clinical data such as binary outcome and survival information is modeled as the response of joint regression from those factors in the previous regression. Normal-Gamma (NG) prior is applied to improve the effect size estimation and address sparsity. This study considers gene expression, methylation and CNV data, in specific, to identify genes having a significant impact on patient survival. The hypothesis of this research lies in the linear relationship between methylation data and CNV, together with the effect of gene expression on survival outcome. These relationships may not reflect the actual biological process underneath, thus the output prognostic genes may not be considered as causal factors. Independent functional experiments and other datasets are needed to validate the results.
This method allows to integrate CNV, DNA methylation, and allelic (loss of heterozygosity) status to find genes representing key nodes in the pathways as well as genes which exhibit prognostic significance (Chari et al., 2010). For each differentially expressed gene, the CNV, methylation and allelic statuses are examined for whether the observed expression change would match the expected change in the DNA level. This multi-step tool can be broken down into several sequential steps: First, a set of most frequent differentially expressed genes is identified for each sample with a pre-defined frequency threshold. Next, this subset of genes is further checked according to the concerted pattern of the expression change and also in at least another DNA dimension (CNV, methylation or loss of heterozygosity). Finally, genes are selected which have a role in multiple disruption mechanisms and changes in expression. As a pioneering work in data integration field, MCD offers a biologically sensible way to select genes step wisely by incorporating parallel analysis in genomic and epigenomic layers. However, it is more like a filtering step to finalize a group of genes rather than a systematic way to integrate information embedded from multiple layers.
It is a bioinformatics workflow proposed to generate integrative results from multiple platforms into a report for biologists for better comprehension (Ovaska et al., 2010). It is a flexible and intuitive analysis tool, which facilitates the integration of various data formats, bio-databases and analysis techniques to identify the genes and loci with high impact on survival. It supports data input including gene expression, miRNA expression, methylation, CNV, exome sequencing, and array CGH data. The workflow maneuvers to manage and automate the sequence of multi-platform analyses from importing the raw data to reporting and visualizing the results. The generated comprehensive website collects all the analyses results and thus facilitates the interpretation of the data. However, this framework is more of a platform to collect and process multiple types of data, rather than a package that performs data integration with sophisticated statistical or machine learning methods.
Semi-supervised integration methods, lies between supervised and unsupervised methods, takes both labeled and unlabeled samples to develop learning algorithm. Most of the semi-supervised data integration methods are graph-based, as illustrated with a few examples below (Table 1).
It is a tool to predict the genetic interactions through combining the protein-protein interaction, protein complex, and gene expression data (You et al., 2010). This method starts with building a high-coverage, high-precision weighted functional gene network by integrating gene expression, protein complex, and protein-protein interaction data. The topological properties of the protein pairs and gene expression in the function gene network are used as input for the subsequent classification step. A weight matrix is built summarizing the information among the edges in the graph, which is made symmetric. A similarity matrix is inferred from the weight matrix iteratively, using local connectivity in the gene network until convergence. Using connected weighted graph, the graph-based semi-supervised learning (SSL) method can infer the information of the unlabeled interactions in the graph. The final product is a classification matrix where all the unlabeled interactions are assigned. This method is specifically designed for prediction of genetic interaction from integrated functional gene networks. Moreover, the semi-supervised idea of inferring unlabeled data from labeled data in the connected graph of similarity matrix can be applied to clinical predictions like cancer diagnosis and prognosis.
Another pilot framework employing graph-based SSL uses the multi-level genomic data sources (including CNV, gene expression, methylation, and miRNA expression) for molecular classification of clinical outcomes (Kim et al., 2012). This method uses the genomic relationship to define the edges (relationship) between the nodes (samples), and the unlabeled samples are influenced by the propagation of their annotated neighbors. In the end, diverse graphs from different layers are combined by the linear combination of coefficients for the individual graphs. It allows the flexibility to extend to integrate multiple levels of genomic data (n > 3), while preserving the level-specific properties from the different and heterogeneous layers. In summary, this work pioneered in combining genomics, epigenomics, and transcriptomics data to predict for cancer phenotypes. However, the interaction relationships among different layers were not considered, such as the regulatory role of methylation or microRNA on gene expression.
By now we discussed a variety of integration methods in three categories: unsupervised, supervised, and semi-supervised. Unsupervised methods recruit different approaches (factorization, Bayesian, network etc.) to explore their biological profiles to assign objects into different subgroups (clusters). Supervised methods employ the biological information of labeled objects to derive patterns for different phenotypes and assign labels to unlabeled data by comparing the patterns. Semi-supervised methods are mostly building object-wise similarity networks through compiling omics data and assign labels to unknown objects through their relationship to labeled objects.
Interactions among different layers are major concerns for data integration strategies. The corresponding mapping relationship among different layers such as methylation to gene expression, microRNA to gene expression etc. should not only be considered independently but also together during the integrative process. At the initiating stage of data integration, many integrative methods are independently working on different layers (such as multi-step analysis) and then find the common subset of biological identities (e.g., genes) which are significantly differentially expressed in each layer. The more recent emerging state-of-the-art integrative tools are considering interactions while integrating different layers. SNF, for example, tries to integrate patient-wise similarities as a combined network, which both strengthens the coherent relationships from each network and reduces the noise of weak signals from the individual network. iCluster+, on the other hand, aims to discover the common latent variable (structure) from all different omics layers with different modeling assumptions. Thus, the internal relationship of different layers is considered as the driving factor that acts in a concerted manner from each omics data.
Nowadays cancer prognosis prediction is a keen point of interest for physicians, cancer patients, and healthcare-providers. Information about cancer prognosis helps all kinds of decisions regarding the patient management and therapeutic treatments etc. (Hagerty et al., 2005; Rabin et al., 2013). Prognostic biomarkers have been used for more effective selection of patient subgroups with different therapeutic strategies (Huang et al., 2014, 2016). Therefore, molecular data with increasing power to detect personalized molecular characteristics has been studied widely in the past decade (Van 't Veer et al., 2002; Kim and Ritchie, 2014). However, methods to integrate multi-omics data optimized for prognosis prediction (rather than being post-hoc evaluation) are far fewer (Table 1). We enlist some representative methods below:
It is a vector space integration methodology that can handle CNV, gene expression, DNA methylation, and miRNA expression data (Mankoo et al., 2011). First, the Spearman rank correlations among different data types are computed, and separate cut-offs are used to filter the correlated data pairs. After the filtering, L1-penalty is combined with Cox proportional hazards model for feature selection and model shrinkage simultaneously. This metric is a typical multi-step analysis method to predict survival.
This framework performs knowledge-based integration of multi-omics genomics data at pathway level (Kim et al., 2014, 2016), to predict the clinical outcome of patients. The strength of the framework lies in capturing genomic interactions by integrating pathways with the metadimensional models to achieve improved prognosis and diagnosis. In transformation phase, each genomic layer is converted to pathway-based knowledge-driven matrix. In modeling phase, an evolutionary algorithm-based method called grammatical evolution neural networks (GENN) is used to develop knowledge-driven models for predicting clinical outcome. GENN is essentially an artificial neural network (ANN) based on grammar rules, which optimizes the high-dimensional input features, network structure, and weights. Further, different genomic interaction models are integrated to develop MKGI models to predict survival, stage and grade. This method concludes that knowledge-driven (pathway-based) genomic models overall perform better than single genomic-based models where gene expression is most contributing at the pathway level.
A plethora of data is accruing with the high-end experimental set-ups in the field of pathology. Advanced technologies are coupled with the computational challenges to deliver the most relevant biological interpretation of data. In this direction, a considerable number of tools have been developed to make the most out of the multi-tier data sets. This review summarizes the diverse computational tools developed over the years, their advantages and limitations. As this field flourishes, comparisons among different methods will be critical, to aid decision-making by investigators with big data needs. Despite these accomplishments, there needs to be more accurate and efficient tools, especially when clinical outcome (e.g., survival) is to be modeled. Biological knowledge guided integrative methods will continue to be desirable, with consideration of the interactive relationship among different omics layers. Moreover, given that most studies have only a single or a few omics layers, integrating heterogeneous data from multiple cohorts, rather than coupled samples, will need rigorous investigation (Wei et al., 2016). For the purpose of precision medicine, additional benefits may be obtained by integrating omics data with other data types, such as imaging and electronic health record (EHR) data.
LG envisioned the project, SH and LG designed the workflow. SH, KC, and LG wrote the manuscript. All authors have read, revised, and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer JC and handling Editor declared their shared affiliation, and the handling Editor states that the process nevertheless met the standards of a fair and objective review.
This research was supported by grants K01ES025434 awarded by NIEHS through funds provided by the trans-NIH Big Data to Knowledge (BD2K) initiative (http://datascience.nih.gov/bd2k), P20 COBRE GM103457 awarded by NIH/NIGMS, NICHD R01 HD084633, NLM R01 LM012373 and Hawaii Community Foundation Medical Research Grant 14ADVC-64566 to LG.
Akavia, U. D., Litvin, O., Kim, J., Sanchez-Garcia, F., Kotliar, D., Causton, H. C., et al. (2010). An integrated approach to uncover drivers of cancer. Cell 143, 1005–1017. doi: 10.1016/j.cell.2010.11.013
Aure, M. R., Steinfeld, I., Baumbusch, L. O., Liestøl, K., Lipson, D., Nyberg, S., et al. (2013). Identifying in-trans process associated genes in breast cancer by integrated analysis of copy number and expression data. PLoS ONE 8:e53014. doi: 10.1371/journal.pone.0053014
Bonnet, E., Calzone, L., and Michoel, T. (2015). Integrative multi-omics module network inference with Lemon-Tree. PLoS Comput. Biol. 11:e1003983. doi: 10.1371/journal.pcbi.1003983
Chari, R., Coe, B. P., Vucic, E. A., Lockwood, W. W., and Lam, W. L. (2010). An integrative multi-dimensional genetic and epigenetic strategy to identify aberrant genes and pathways in cancer. BMC Syst. Biol. 4:67. doi: 10.1186/1752-0509-4-67
Chen, J., and Zhang, S. (2016). Integrative analysis for identifying joint modular patterns of gene-expression and drug-response data. Bioinformatics 32, 1724–1732. doi: 10.1093/bioinformatics/btw059
Chen, J., Bushman, F. D., Lewis, J. D., Wu, G. D., and Li, H. (2013). Structure-constrained sparse canonical correlation analysis with an application to microbiome data analysis. Biostatistics 14, 244–258. doi: 10.1093/biostatistics/kxs038
Chin, L., Andersen, J. N., and Futreal, P. A. (2011). Cancer genomics: from discovery science to personalized medicine. Nat. Med. 17, 297–303. doi: 10.1038/nm.2323
Cho, D.-Y., and Przytycka, T. M. (2013). Dissecting cancer heterogeneity with a probabilistic genotype-phenotype model. Nucleic Acids Res. 41, 8011–8020. doi: 10.1093/nar/gkt577
Cline, M. S., Smoot, M., Cerami, E., Kuchinsky, A., Landys, N., Workman, C., et al. (2007). Integration of biological networks and gene expression data using Cytoscape. Nat. Protoc. 2, 2366–2382. doi: 10.1038/nprot.2007.324
Griffiths, T. L., and Ghahramani, Z. (2005). “Infinite latent feature models and the Indian buffet process,” in Advances in Neural Information Processing Systems 18 (NIPS 2005) (Vancouver, BC), 475–482.
Hagerty, R. G., Butow, P. N., Ellis, P. M., Dimitry, S., and Tattersall, M. H. N. (2005). Communicating prognosis in cancer care: a systematic review of the literature. Ann. Oncol. 16, 1005–1053. doi: 10.1093/annonc/mdi211
Hofmann, T., Schölkopf, B., and Smola, A. J. (2008). Kernel methods in machine learning. Ann. Stat. 36, 1171–1220. doi: 10.1214/009053607000000677
Huang, S., Chong, N., Lewis, N. E., Jia, W., Xie, G., and Garmire, L. X. (2016). Novel personalized pathway-based metabolomics models reveal key metabolic pathways for breast cancer diagnosis. Genome Med. 8:34. doi: 10.1186/s13073-016-0289-9
Huang, S., Yee, C., Ching, T., Yu, H., and Garmire, L. X. (2014). A novel model to combine clinical and pathway-based transcriptomic information for the prognosis prediction of breast cancer. PLoS Comput. Biol. 10:e1003851. doi: 10.1371/journal.pcbi.1003851
Hudson, T. J., Anderson, W., Aretz, A., Barker, A. D., Bell, C., Bernabé, R. R., et al. (2010). International network of cancer genome projects. Nature 464, 993–998. doi: 10.1038/nature08987
Ideker, T., Ozier, O., Schwikowski, B., and Siegel, A. F. (2002). Discovering regulatory and signalling circuits in molecular interaction networks. Bioinformatics 18, S233–S240. doi: 10.1093/bioinformatics/18.suppl_1.S233
Imoto, S., Higuchi, T., Goto, T., Tashiro, K., Kuhara, S., and Miyano, S. (2004). Combining microarrays and biological knowledge for estimating gene networks via Bayesian networks. J. Bioinform. Comput. Biol. 2, 77–98. doi: 10.1142/S021972000400048X
Jennings, E. M., Morris, J. S., Carroll, R. J., Manyam, G. C., and Baladandayuthapani, V. (2013). Bayesian methods for expression-based integration of various types of genomics data. EURASIP J. Bioinforma. Syst. Biol. 2013:13. doi: 10.1186/1687-4153-2013-13
Kim, D., and Ritchie, M. D. (2014). Data integration for cancer clinical outcome prediction. J. Heal. Med. Informatics 5:e122. doi: 10.4172/2157-7420.1000e122
Kim, D., Li, R., Dudek, S. M., and Ritchie, M. D. (2013). ATHENA: Identifying interactions between different levels of genomic data associated with cancer clinical outcomes using grammatical evolution neural network. BioData Min. 6:23. doi: 10.1186/1756-0381-6-23
Kim, D., Li, R., Dudek, S. M., Frase, A. T., Pendergrass, S. A., and Ritchie, M. D. (2014). Knowledge-driven genomic interactions: an application in ovarian cancer. BioData Min. 7:20. doi: 10.1186/1756-0381-7-20
Kim, D., Li, R., Lucas, A., Verma, S. S., Dudek, S. M., and Ritchie, M. D. (2016). Using knowledge-driven genomic interactions for multi-omics data analysis: metadimensional models for predicting clinical outcomes in ovarian carcinoma. J. Am. Med. Inform. Assoc. 24, 577–587. doi: 10.1093/jamia/ocw165
Kim, D., Shin, H., Song, Y. S., and Kim, J. H. (2012). Synergistic effect of different levels of genomic data for cancer clinical outcome prediction. J. Biomed. Inform. 45, 1191–1198. doi: 10.1016/j.jbi.2012.07.008
Kirk, P., Griffin, J. E., Savage, R. S., Ghahramani, Z., and Wild, D. L. (2012). Bayesian correlated clustering to integrate multiple datasets. Bioinformatics 28, 3290–3297. doi: 10.1093/bioinformatics/bts595
Lê Cao, K.-A., Martin, P. G., Robert-Granié, C., and Besse, P. (2009). Sparse canonical methods for biological data integration: application to a cross-platform study. BMC Bioinform. 10:34. doi: 10.1186/1471-2105-10-34
Lanckriet, G. R. G., De Bie, T., Cristianini, N., Jordan, M. I., and Noble, W. S. (2004). A statistical framework for genomic data fusion. Bioinformatics 20, 2626–2635. doi: 10.1093/bioinformatics/bth,294
Lee, D. D., and Seung, H. S. (2001). “Algorithms for non-negative matrix factorization,” in Advances in Neural Information Processing Systems (Denver), 556–562.
Li, W., Zhang, S., Liu, C.-C., and Zhou, X. J. (2012). Identifying multi-layer gene regulatory modules from multi-dimensional genomic data. Bioinformatics 28, 2458–2466. doi: 10.1093/bioinformatics/bts476
Lin, D., Zhang, J., Li, J., Calhoun, V. D., Deng, H.-W., and Wang, Y.-P. (2013). Group sparse canonical correlation analysis for genomic data integration. BMC Bioinform. 14:245. doi: 10.1186/1471-2105-14-245
Lock, E. F., and Dunson, D. B. (2013). Bayesian consensus clustering. Bioinformatics 29, 2610–2616. doi: 10.1093/bioinformatics/btt425
Lock, E. F., Hoadley, K. A., Marron, J. S., and Nobel, A. B. (2013). Joint and individual variation explained (JIVE) for integrated analysis of multiple data types. Ann. Appl. Stat. 7, 523–542. doi: 10.1214/12-AOAS597
Louhimo, R., and Hautaniemi, S. (2011). CNAmet: an R package for integrating copy number, methylation and expression data. Bioinformatics 27, 887–888. doi: 10.1093/bioinformatics/btr019
Mankoo, P. K., Shen, R., Schultz, N., Levine, D. A., and Sander, C. (2011). Time to recurrence and survival in serous ovarian tumors predicted from integrated genomic profiles. PLoS ONE 6:e24709. doi: 10.1371/journal.pone.0024709
Mo, Q., Wang, S., Seshan, V. E., Olshen, A. B., Schultz, N., Sander, C., et al. (2013). Pattern discovery and cancer gene identification in integrated cancer genomic data. Proc. Natl. Acad. Sci. U.S.A. 110, 4245–4250. doi: 10.1073/pnas.1208949110
Ovaska, K., Laakso, M., Haapa-Paananen, S., Louhimo, R., Chen, P., Aittomäki, V., et al. (2010). Large-scale data integration framework provides a comprehensive view on glioblastoma multiforme. Genome Med. 2:65. doi: 10.1186/gm186
Parkhomenko, E., Tritchler, D., and Beyene, J. (2009). Sparse canonical correlation analysis with application to genomic data integration. Stat. Appl. Genet. Mol. Biol. 8, 1–34. doi: 10.2202/1544-6115.1406
Rabin, B. A., Gaglio, B., Sanders, T., Nekhlyudov, L., Dearing, J. W., Bull, S., et al. (2013). Predicting cancer prognosis using interactive online tools: a systematic review and implications for cancer care providers. Cancer Epidemiol. Biomarkers Prev. 22, 1645–1656. doi: 10.1158/1055-9965.EPI-13-0513
Ray, P., Zheng, L., Lucas, J., and Carin, L. (2014). Bayesian joint analysis of heterogeneous genomics data. Bioinformatics 30, 1370–1376. doi: 10.1093/bioinformatics/btu064
Ritchie, M. D., Holzinger, E. R., Li, R., Pendergrass, S. A., and Kim, D. (2015). Methods of integrating data to uncover genotype–phenotype interactions. Nat. Rev. Genet. 16, 85–97. doi: 10.1038/nrg3868
Ruffalo, M., Koyutürk, M., and Sharan, R. (2015). Network-based integration of disparate omic data to identify “Silent Players” in cancer. PLOS Comput. Biol. 11:e1004595. doi: 10.1371/journal.pcbi.1004595
Schaefer, M. H., Fontaine, J.-F., Vinayagam, A., Porras, P., Wanker, E. E., and Andrade-Navarro, M. A. (2012). HIPPIE: integrating protein interaction networks with experiment based quality scores. PLoS ONE 7:e31826. doi: 10.1371/journal.pone.0031826
Seoane, J. A., Day, I. N. M., Gaunt, T. R., and Campbell, C. (2014). A pathway-based data integration framework for prediction of disease progression. Bioinformatics 30, 838–845. doi: 10.1093/bioinformatics/btt610
Shen, R., Mo, Q., Schultz, N., Seshan, V. E., Olshen, A. B., Huse, J., et al. (2012). Integrative subtype discovery in glioblastoma using iCluster. PLoS ONE 7:e35236. doi: 10.1371/journal.pone.0035236
Shen, R., Olshen, A. B., and Ladanyi, M. (2009). Integrative clustering of multiple genomic data types using a joint latent variable model with application to breast and lung cancer subtype analysis. Bioinformatics 25, 2906–2912. doi: 10.1093/bioinformatics/btp543
Speicher, N. K., and Pfeifer, N. (2015). Integrating different data types by regularized unsupervised multiple kernel learning with application to cancer subtype discovery. Bioinformatics 31, i268–i275. doi: 10.1093/bioinformatics/btv244
Thibaux, R., and Jordan, M. I. (2007). “Hierarchical beta processes and the indian buffet process,” in Artificial Intelligence and Statistics (AISTATS) (San Juan), 564–571.
Tibshirani, R. (1994). Regression shrinkage and selection via the lasso. J. R. Stat. Soc. Ser. B 58, 267–288.
Tipping, M. E. (2001). Sparse Bayesian learning and the relevance vector machine. J. Mach. Learn. Res. 1, 211–244. doi: 10.1162/15324430152748236
Van 't Veer, L. J., Dai, H., van de Vijver, M. J., He, Y. D., Hart, A. A. M., Mao, M., et al. (2002). Gene expression profiling predicts clinical outcome of breast cancer. Nature 415, 530–536. doi: 10.1038/415530a
Vaske, C. J., Benz, S. C., Sanborn, J. Z., Earl, D., Szeto, C., Zhu, J., et al. (2010). Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics 26, i237–i245. doi: 10.1093/bioinformatics/btq182
Wang, B., Mezlini, A. M., Demir, F., Fiume, M., Tu, Z., Brudno, M., et al. (2014). Similarity network fusion for aggregating data types on a genomic scale. Nat. Methods 11, 333–337. doi: 10.1038/nmeth.2810
Wei, R., De Vivo, I., Huang, S., Zhu, X., Risch, H., Moore, J. H., et al. (2016). Meta-dimensional data integration identifies critical pathways for susceptibility, tumorigenesis and progression of endometrial cancer. Oncotarget 7, 55249–55263. doi: 10.18632/oncotarget.10509
Weinstein, J. N., Collisson, E. A., Mills, G. B., Shaw, K. R. M., Ozenberger, B. A., Ellrott, K., et al. (2013). The cancer genome atlas pan-cancer analysis project. Nat. Genet. 45, 1113–1120. doi: 10.1038/ng.2764
Witten, D. M., and Tibshirani, R. J. (2009). Extensions of sparse canonical correlation analysis with applications to genomic data. Stat. Appl. Genet. Mol. Biol. 8, 1–27. doi: 10.2202/1544-6115.1470
You, Z.-H., Yin, Z., Han, K., Huang, D.-S., and Zhou, X. (2010). A semi-supervised learning approach to predict synthetic genetic interactions by combining functional and topological properties of functional gene network. BMC Bioinform. 11:343. doi: 10.1186/1471-2105-11-343
Yuan, Y., Savage, R. S., and Markowetz, F. (2011). Patient-specific data fusion defines prognostic cancer subtypes. PLoS Comput. Biol. 7:e1002227. doi: 10.1371/journal.pcbi.1002227
Zhang, S., Li, Q., Liu, J., and Zhou, X. J. (2011). A novel computational framework for simultaneous integration of multiple types of genomic data to identify microRNA-gene regulatory modules. Bioinformatics 27, i401–i409. doi: 10.1093/bioinformatics/btr206
Zhang, S., Liu, C.-C., Li, W., Shen, H., Laird, P. W., and Zhou, X. J. (2012). Discovery of multi-dimensional modules by integrative analysis of cancer genomic data. Nucleic Acids Res. 40, 9379–9391. doi: 10.1093/nar/gks725
Keywords: multi-omics, integration, prognosis, prediction, precision medicine, supervised learning, unsupervised learning
Citation: Huang S, Chaudhary K and Garmire LX (2017) More Is Better: Recent Progress in Multi-Omics Data Integration Methods. Front. Genet. 8:84. doi: 10.3389/fgene.2017.00084
Received: 24 March 2017; Accepted: 01 June 2017;
Published: 16 June 2017.
Edited by:
Shihua Zhang, Academy of Mathematics and Systems Science (CAS), ChinaReviewed by:
Hyunju Lee, Gwangju Institute of Science and Technology, South KoreaCopyright © 2017 Huang, Chaudhary and Garmire. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lana X. Garmire, bGdhcm1pcmVAY2MuaGF3YWlpLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.