 Ayman S. El-Seedy
Ayman S. El-Seedy Hanaa Shafiek3
Hanaa Shafiek3- 1Department of Genetics, Alexandria University, Alexandria, Egypt
- 2EA3808, Groupe Génétique des Maladies Rares, Université de Poitiers, Poitiers, France
- 3Department of Chest Diseases, Alexandria University, Alexandria, Egypt
- 4Centre Hospitalier Universitaire de Poitiers, Poitiers, France
Cystic Fibrosis (CF) is the most common lethal rare genetic disease in the Caucasian populations. It is caused by a variety of sequence alterations in the Cystic Fibrosis Transmembrane Regulator (CFTR) gene. In Caucasian, one over 3,500 new born children suffers from the disease and one over 30 of them is at least carrier of a severe mutation in the CFTR gene. CF and CFTR-related disorders (CFTR-RDs) are two distinct clinical outcomes of the gene mutations. The CF mutation induces a severe phenotype involving different organs, whereas a CFTR-RD mutation induces less life-threatening symptoms with three main clinical entities including congenital bilateral absence of the vas deferens (CBAVD), acute recurrent or chronic pancreatitis, and disseminated bronchiectasis (Bombieri et al., 2011).
In the Arab countries, the spectrum of CF mutations, incidence and prevalence of the disease are largely unknown in the Arab populations (Wei et al., 2006). This is due to the lack of disease awareness, and diagnosis facilities that mislead the identification of CF during many decades. Additionally, epidemiological studies that realized were revealed a distinguished mutational spectrum between Arab countries if compared to White-European populations. Furthermore, Arab Mediterranean countries have a different CFTR mutational profile if compared to the Arabian Peninsula.
Egypt as Mediterranean North African country, this strategic position attracted many invaders throughout its history. Therefore, in addition to its Pharaonic origin, gene flow to its population occurred from the Ethiopian, Greco-Roman, Arab, Turkish, French and English settlers (Temtamy et al., 2010). The common heritage among the countries bordering the Mediterranean is not restricted to historical or cultural aspects. There are considerable commonalities in the gene pools of the Mediterranean Northern and Southern countries. This “genetic sharing” has resulted from considerable human movements (i.e., migration, invasion, and trade) throughout history in this area (Temtamy et al., 2010). Consequently, Egypt is not like other Arab countries and mutations in the CFTR gene have been influenced by gene flow coming from different populations. Furthermore, the high rate of consanguinity, infant and neonatal mortality in the Egyptian society will, therefore, increase CF incidence and private mutations. Indeed, another important issue is the increase number of CFTR-related disorders patients such as idiopathic bronchiectasis, congenital bilateral absence of the vas deferens, idiopathic (non-alcoholic) pancreatitis, and severe sinusitis in the Egyptian population. This observation is in agreement with data previously published (Lissens et al., 1999; Hussein et al., 2011; Fathy et al., 2016).
In Egypt, there is no available data on the nature and frequency of CF gene mutations. There are a few previous reports of CFTR gene mutations in Egyptian patients that have been published. In these papers, a CF screening strategy of CFTR mutations was realized using commercial kits or a panel defined by the American College of Medical Genetics and Genomics (ACMG) and American College of Obstetricians and Gynecologists (ACOG), (Naguib et al., 2007; El-Falaki et al., 2014; Fathy et al., 2016; Shahin et al., 2016), which is not suitable for screening of the CFTR gene mutations in the Egyptian population. This emphasizes the need for establishing a correct genetic screening for CF mutations in Egyptian patients and to determine the carrier status in their healthy relatives.
In our recent study, we have performed a complete CFTR gene screening in CF or CFTR-RD patients from Alexandria, Northern Egypt, by direct sequencing of the entire CFTR gene that identified four novel CF mutations in the CFTR gene amongst 13 other known mutations in the other populations (Figure 1A). Furthermore, this is the first comprehensive profile of CFTR gene mutations and their corresponding haplotypes in the Egyptian population (El-Seedy et al., 2016). Besides, CFTR mutations in the other Egyptian regions have not been analyzed. Thus, the identification of CFTR mutations is become increasingly important in genetic counseling and prenatal diagnosis particularly in families with multiple affected children (Wang et al., 2000).
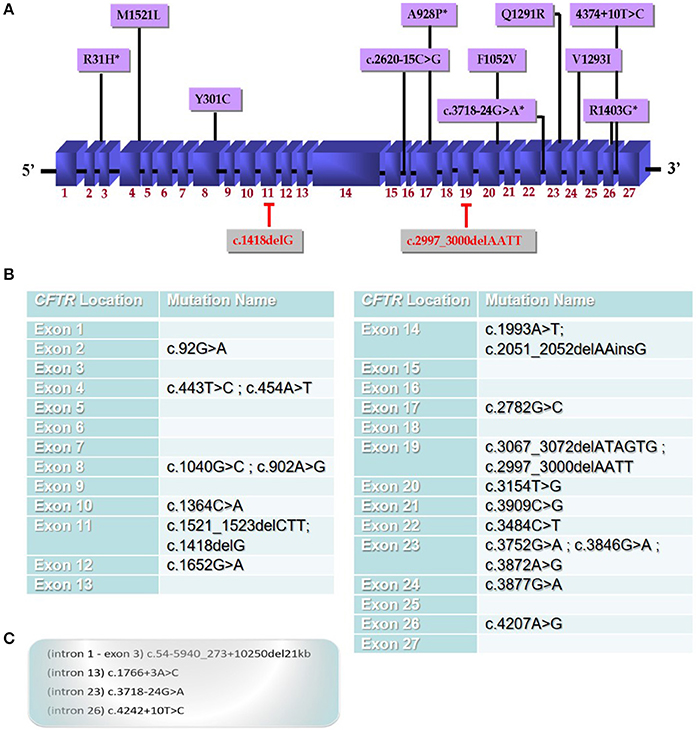
Figure 1. Mutational spectrum of CFTR gene mutations in the Egyptian population. (A) CFTR mutations identified in our recent investigation of Egyptian CF or CFTR-RD patients from Alexandria city, Northern Egypt which performed a complete CFTR gene screening by direct sequencing of the entire CFTR gene. This study is the first to identify novel mutations (indicated with a star*) in the Egyptian population amongst 13 other known mutations that appear to be a specific to Egyptian CF patients (2); (B) a panel of the most common mutations detected in the Egyptian population; (C) intronic mutations identified in the Egyptian patients that will be also present in the panel.
According to available data of CFTR gene mutations, we propose general national screening panel comprising 27 mutations reported in Egyptian patients until now. This will help to detect about 50% of CFTR gene mutations in Egypt and further extensive studies will help to improve this panel. The establishment of the first panel of the CFTR gene mutations in the Egyptian population (Figures 1B,C) will help physicians for designing an appropriate strategy for future genetic diagnosis for patients and families at risk.
In this regard, defining a population-specific mutational panel for CF in Egyptian population including: c.92G>A (exon 2), c.454A>T (exon 4), c.902A>G (exon 8), c.1418delG (exon 11), c.2620-15C>G (intron 15), c.2782G>C (exon 17), c.2997_3000delAATT (exon 19), c.3154T>G (exon 20), c.3718-24G>A (intron 23), c.3872A>G (exon 23), c.3877G>A (exon 24), c.4207A>G (exon 26), c.4242+10T>C (intron 26) mutations identified in our recent study (Figure 1A), and together with previously reported mutations in Egyptian patients (Figures 1B,C) [c.1766+3A>C (intron 13), c.1993A>T (exon 14), c.3909C>G (exon21), c.1521_1523delCTT (exon11), (Naguib et al., 2007); c.54-5940_273+10250del21kb (intron 1-exon 3), c.443T>C (exon 4), c.1040G>C (exon8), c.1364C>A (exon10), c.1652G>A (exon12), c.2051_2052delAAinsG (exon 14), c.3067_3072delATAGTG (exon19), c.3484C>T (exon 22), c.3846G>A (exon 23), (Shahin et al., 2016); c.1040G>C (exon 8), c.1364C>A (exon 10), c.3484C>T (exon 22), c.3752G>A (exon 23), c.3846G>A (exon 23), (Fathy et al., 2016)] should be highly recommended for Egyptian patients. However, full clinical manifestations of additional CF and CFTR-RDs patients could lead to clarify the pathogenicity of these mutants in this population.
The overall purpose of this paper, in our opinion, is to develop and validate a strategy for increasing the sensitivity of CFTR screening in Egyptian population. This strategy includes establishing of an Egyptian CF Registry Network (ECFRN) where there is no reliable estimates on the number of CF patients and clinical pattern as well as the most common CFTR mutations in this population. Hence, this registry is essential in order to have data about prevalence and incidence of CF in Egypt for improving clinic diagnostic outcomes and planning health policies. In addition, an extensive molecular analysis of CFTR gene using next-generation sequencing (NGS) technology combined with multiplex ligation-dependent probe amplification (MLPA) will provide a wider coverage of the CFTR locus and expand our proposed panel in CF molecular diagnosis.
In summary, our findings confirm the importance of direct gene sequencing of the entire CFTR gene in combination with molecular and functional studies (in isolation and in complex alleles) in order to perform a correct genetic counseling and to estimate the prevalence of the carriers in the general population. Moreover, collaborative studies, on the clinical and fundamental levels, are necessary to give an adequate genotype-phenotype correlation. Thus, sharing the obtained data between CFTR laboratories, clinical research centers and concerned communities in Egypt are also from a great importance to ensure a progression in mutation researches and to help clinicians in giving a suitable prenatal diagnosis, patients advising and treatments prescription when available.
Author Contributions
AE: Wrote the paper. HS, AK, and VL: Revised the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We appreciate the valuable support of French Embassy in Cairo and Dr. Louis Moreau “attaché scientific” for their kind help and encouragement the fellowship to AE. This work is also supported by Poitiers University Hospital and University of Poitiers, France.
References
Bombieri, C., Claustres, M., De Boeck, K., Derichs, N., Dodge, J., Girodon, E., et al. (2011). Recommendations for the classification of diseases as CFTR-related disorders. J. Cyst. Fibros. 10, S86–S102. doi: 10.1016/s1569-1993(11)60014-3
El-Falaki, M. M., Shahin, W. A., El-Basha, N. R., Ali, A. A., Mehaney, D. A., and El-Attar, M. M. (2014). Profile of cystic fibrosis in a single referral center in Egypt. J. Adv. Res. 5, 563–568. doi: 10.1016/j.jare.2013.07.005
El-Seedy, A., Pasquet, M. C., Shafiek, H., Morsi, T., Kitzis, A., and Ladevèze, V. (2016). Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene mutations in North Egyptian population: implications for the genetic diagnosis in Egypt. Cell. Mol. Bio. 62, 21–28. doi: 10.14715/cmb/2016.62.13.5
Fathy, M. R., Ramzy, T., Elmonem, M. A., Amer, M., Zeidan, A., and Hassan, F. A. (2016). Molecular screening of CFTR gene in Egyptian patients with congenital bilateral absence of the vas deferens: a preliminary study. Andrologia 48, 1307–1312. doi: 10.1111/and.12563
Hussein, T. M., Zakaria, N. H., and Zahran, A. M. (2011). Clinical, laboratory and genetic assessment of patients with congenital bilateral absent vas deferens. Andrologia 43, 16–22. doi: 10.1111/j.1439-0272.2009.01001.x
Lissens, W., Mahmoud, K. Z., El-Gindi, E., Abdel-Sattar, A., Seneca, S., Van Steirteghem, A., et al. (1999). Molecular analysis of the cystic fibrosis gene reveals a high frequency of the intron 8 splice variant 5T in Egyptian males with congenital bilateral absence of the vas deferens. Mol. Hum. Reprod. 5, 10–13. doi: 10.1093/molehr/5.1.10
Naguib, M. L., Schrijver, I., Gardner, P., Pique, L. M., Doss, S. S., Abu Zekry, M. A., et al. (2007). Cystic fibrosis detection in high-risk Egyptian children and CFTR mutation analysis. J. Cyst. Fibros. 6, 111–116. doi: 10.1016/j.jcf.2006.04.004
Shahin, W. A., Mehaney, D. A., and El-Falaki, M. M. (2016). Mutation spectrum of Egyptian children with cystic fibrosis. Springerplus 5, 686. doi: 10.1186/s40064-016-2338-7
Temtamy, S. A., Aglan, M. S., and Meguid, N. A. (2010). “Genetic disorders in Egypt,” in Genetic Disorders among Arab Populations, ed S. T. Ahmad, 2nd Edn. (Cairo: Springer), 219–272. doi: 10.1007/978-3-642-05080-0_8
Wang, X., Moylan, B., Leopold, D. A., Kim, J., Rubenstein, R. C., Togias, A., et al. (2000). Mutation in the gene responsible for cystic fibrosis and predisposition to chronic rhinosinusitis in the general population. JAMA 284, 1814–1819. doi: 10.1001/jama.284.14.1814
Keywords: Cystic fibrosis, CFTR mutations, genetic screening, Egyptian patients
Citation: El-Seedy AS, Shafiek H, Kitzis A and Ladevèze V (2017) CFTR Gene Mutations in the Egyptian Population: Current and Future Insights for Genetic Screening Strategy. Front. Genet. 8:37. doi: 10.3389/fgene.2017.00037
Received: 01 March 2017; Accepted: 16 March 2017;
Published: 31 March 2017.
Edited by:
Enrico Baruffini, University of Parma, ItalyReviewed by:
Loic Guillot, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceCopyright © 2017 El-Seedy, Shafiek, Kitzis and Ladevèze. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Véronique Ladevèze, dmVyb25pcXVlLmxhZGV2ZXplQHVuaXYtcG9pdGllcnMuZnI=