 Aristides G. Eliopoulos
Aristides G. Eliopoulos Sophia Havaki
Sophia Havaki Vassilis G. Gorgoulis
Vassilis G. Gorgoulis
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet. , 21 November 2016
Sec. Systems Biology Archive
Volume 7 - 2016 | https://doi.org/10.3389/fgene.2016.00204
This article is part of the Research Topic DNA Damage and Inflammation under stress View all 6 articles
Autophagy and the DNA damage response (DDR) are biological processes essential for cellular and organismal homeostasis. Herein, we summarize and discuss emerging evidence linking DDR to autophagy. We highlight published data suggesting that autophagy is activated by DNA damage and is required for several functional outcomes of DDR signaling, including repair of DNA lesions, senescence, cell death, and cytokine secretion. Uncovering the mechanisms by which autophagy and DDR are intertwined provides novel insight into the pathobiology of conditions associated with accumulation of DNA damage, including cancer and aging, and novel concepts for the development of improved therapeutic strategies against these pathologies.
The term ‘DNA damage response’ (DDR) refers to a network of intracellular pathways that sense and resolve damaged DNA. If unrepaired, DNA lesions may result in cell death (Roos et al., 2016) but can also be a major source of genomic instability particularly when cell death pathways have been deactivated (Halazonetis et al., 2008). DDR signaling has been extensively reviewed elsewhere (Halazonetis et al., 2008; Jackson and Bartek, 2009; Marechal and Zou, 2013; Ribezzo et al., 2016); below we provide a summary of some of its components and their functions which are most relevant to this review.
DNA damage response utilizes proteins involved in sensing, signaling, and repair of DNA damage. While the early activation events that follow DNA breaks are well elucidated, the primary signal which triggers DDR remains incompletely understood. It has, however, been proposed that when a DNA lesion occurs, it is accompanied by relaxation of chromatin through a series of post-translational histone modifications that include poly-(ADP-ribosylation) which is catalyzed by poly (ADP-ribose) polymerases (PARPs), phosphorylation and acetylation (Lukas et al., 2011). These chromatin responses “freeze” transcription and replication around the site of DNA lesion to facilitate subsequent repair. They also provide access to DDR sensors such as the Mre11–Rad50–Nbs1 (MRN) complex which binds double strand breaks (DSBs) and recruits ATM kinase (Figure 1) and the replication protein A (RPA) complex which responds predominantly to single strand DNA lesions and recruits ATR kinase. The binding of these kinases to damaged DNA triggers the recruitment of additional proteins, many of which become phosphorylated and activated to further transduce signals that orchestrate DNA replication, cell cycle control, transcription, repair of damage, and/or survival versus death.
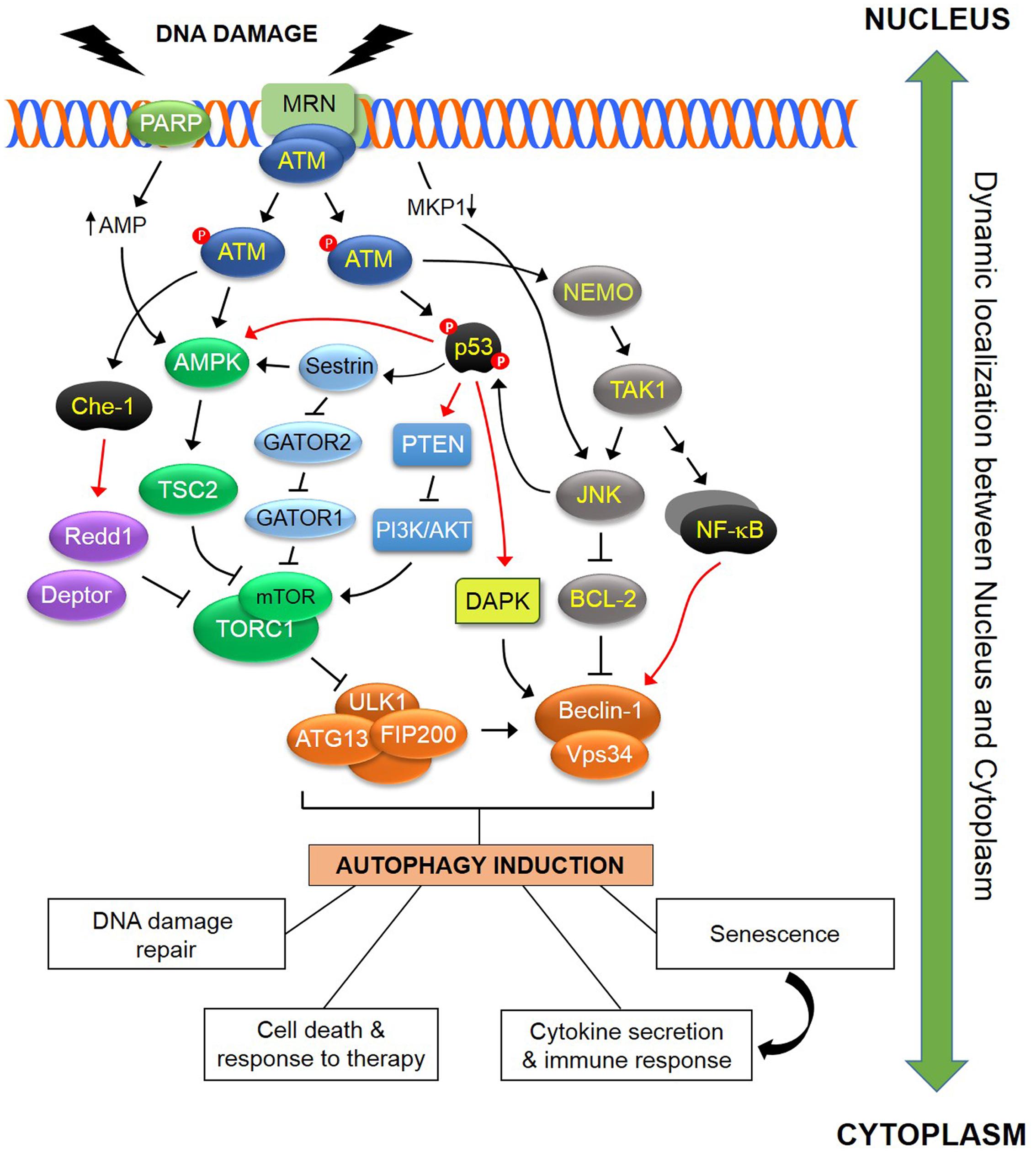
FIGURE 1. Schematic representation of some of the known pathways linking DDR to autophagy. In response to DNA damage, ATM is autophosphorylated within a MRN multiprotein complex that binds DSBs. Activated ATM initiates a pathway that results in activation of AMPK and its target TSC2 which functions as inhibitor of TORC1. As ULK1-dependent autophagosome formation is negatively regulated by TORC1, this ATM pathway induces autophagy. In addition, ATM directly phosphorylates and stabilizes p53 which transcriptionally regulates various regulators of the autophagic pathway including AMPK, DAPK and PTEN. Sestrins, which are regulated by p53, influence TORC1 activity in an AMPK-dependent and AMPK-independent manner, the latter through a GATOR2-GATOR1-RAGB/A signaling pathway (see text for details). The ATM-mediated phosphorylation and activation of the RNA polymerase II-binding protein Che-1, leads to increased transcription of two mTOR inhibitors, Redd1 and Deptor. ATM contributes to the activation of a NEMO-dependent TAK1-ATM-NEMO-NFκB pathway that transcriptionally modulates Beclin-1. TAK1 may also mediate activation of JNK which in turn phosphorylates Beclin-1 and releases it from the inhibitory effect of BCL-2 or BCL-XL. DNA damage also blocks transcription of the JNK phosphatase MKP-1, releasing its inhibitory effect over JNK which in turn induces autophagy. PARP1 activation in response to DNA damage leads to reduction in both NAD+ and ATP pools, with the latter causing activation of AMPK and induction of autophagy. The DDR-autophagy axis has major implications for DNA damage repair, senescence, cell survival versus death, cytokine secretion and modulation of the immune response (see text for details). Black arrows (↑) and perpendicular lines (⊥) denote activation and suppression, respectively. Red arrows denote transcriptional regulation.
Thus, ATM bound to DSBs in conjunction with the MRN complex, undergoes autophosphorylation and activation. In turn, ATM activates various downstream effector proteins (Marechal and Zou, 2013), including Chk2 and Chk1 involved in cell cycle control, the tumor suppressor p53 which regulates cell survival versus death, HDAC1 and HDAC2 which are responsible for chromatin remodeling (Kim et al., 1999), the BASC complex containing DNA damage repair proteins (Wang et al., 2000), the senescence regulator ARF (Velimezi et al., 2013) and transcription factors such as FOXO3 which regulates genes involved in DNA repair (Tran et al., 2002). ATR shares at least some of these ATM functions, for example capacity to phosphorylate p53 and Chk1 in response to irradiation (Liu et al., 2000; Zhao and Piwnica-Worms, 2001).
In addition to the aforementioned phosphorylation targets, ATM forms a nuclear complex with PARP1 and NEMO (also known as IKKγ). Within this complex, NEMO is subjected to a series of post-translational modifications including phosphorylation, SUMOylation and ubiquitination that lead to its nuclear export (Mabb et al., 2006; Wu et al., 2006). Once in the cytoplasm, NEMO orchestrates the formation of a high molecular weight multiprotein complex which includes the kinases TAK1, IKKα, and IKKβ which are responsible for a signaling cascade that leads to translocation of cytoplasmic RelA NF-κB to the nucleus. Thus, ATM links DNA damage to NF-κB activation (Figure 1).
TAK1 also engages the JNK pathway which activates various transcription factors, including AP1. NF-κB and AP1 transcribe genes involved in cytokine synthesis and, depending on the duration of activation, cell survival versus death. Moreover, ATM utilizes JNK to coordinate optimal p53 activation. Thus, on one hand, ATM directly phosphorylates p53 at Ser15 which reduces its affinity for the ubiquitin ligase HDM2 leading to p53 stabilization. However, ATM also impacts on p53 indirectly through activation of JNK which phosphorylates p53 at Thr81 enhancing its transcriptional activity (Buschmann et al., 2001) (Figure 1). DNA damage has been reported to activate JNK also through the transcriptional repression of the JNK phosphatase MKP1 caused by transcription-blocking DNA lesions (Hamdi et al., 2005). Further studies are needed to establish the relative contribution of the ATM-NEMO-TAK1 versus MKP1 pathways to JNK activation which is likely to depend on the extent of DNA damage.
A major end-point of DDR is the activation of the DNA damage repair system. Depending on the type of damage and the phase of the cell cycle, different repair mechanisms are utilized to restore DNA integrity (Jackson and Bartek, 2009; Ribezzo et al., 2016). For example, PARP1 is involved in the repair of single strand DNA breaks by recruiting enzymes necessary for base excision repair such as XRCC1, polymerase β and DNA ligase III to the sites of damage (de Murcia et al., 1997). DSBs are mainly repaired by HR, an error-proof process operating predominantly in the S and G2 phases of the cell cycle, and by NHEJ which is operational in all phases of the cell cycle but is error-prone because of the lack of appropriate undamaged DNA template (Lieber et al., 2003). In general, ATM/ATR-mediated DDR signaling regulates repair by: (a) inducing the transcription of DNA-repair genes, (b) modulating DNA-repair protein activity through post-translational modifications such as phosphorylation, acetylation, ubiquitination or SUMOylation, and (c) by recruiting repair factors to the DNA lesion. If the damage cannot be resolved, chronic DDR signaling triggers cell death or senescence (Roos et al., 2016).
In addition to nuclear DNA, mtDNA is subject to damage following exposure to radiation or chemotherapy. Moreover, a by-product of oxidative production of ATP is the generation of reactive oxygen species (ROS) which may damage both mitochondrial and nuclear DNA (Sedelnikova et al., 2010; Cline, 2012). MtDNA lacks histones which makes it even more susceptible to injury than nuclear DNA. To counteract mtDNA damage, mitochondria possess quality control systems that include antioxidant enzymes and a dedicated repair system which appears to be less elaborate and effective than the nuclear DNA damage repair machinery (LeDoux et al., 1992; Cline, 2012).
Autophagy is a cellular ‘self-eating’ degradation process in which proteins or whole organelles are degraded in lysosomes and recycled to meet the anabolic and bioenergetic needs of the cell. As such, it plays a pivotal role in tissue homeostasis and various human pathologies, including cancer, neurodegeneration, autoimmunity and aging, have been associated with deregulated autophagy (Mizushima et al., 2008).
Depending on the mechanism of delivery of the cargo to the lysosomes, three main types of autophagy have been so-far recognized: micro-autophagy that involves the direct delivery of cargo to lysosomes through lysosomal membrane invaginations; CMA which is typified by the lysosomal import of proteins through their interaction with specialized chaperones; and macro-autophagy which is the most widely studied mechanism of autophagy. In macro-autophagy (hereafter termed autophagy), the cargo is sequestered in double membrane vesicles known as autophagosomes, which are progressively formed by the finely interconnected activities of around 15 autophagy-related (ATG) proteins (Rubinsztein et al., 2012; Wirth et al., 2013). Autophagosomes can engulf cytoplasmic material, protein aggregates, organelles including mitochondria (mitophagy), peroxisomes (pexophagy) and lipid droplets (lipophagy), as well as ribosomes (ribophagy) and parts of the nucleus (nucleophagy).
Autophagosome biogenesis entails three steps: initiation, nucleation, and elongation. Initiation requires the activation of a complex containing ULK1, ATG13, the FAK-interacting protein FIP200 and ATG101. Following activation of this ULK complex, additional proteins are recruited to form an initial double membrane structure called the phagophore. This step requires Vps34 class III PI3K that operates within a progressively formed large macromolecular “nucleation” complex involving Beclin-1, ATG14, and Vps15. Subsequent vesicle elongation allows engulfment and sequestration of organelles or bulk cytosolic material. The elongation step is driven by the covalent conjugation of ATG12 to ATG5, supported by ATG7 and ATG10, and is responsible for the end product of a second reaction, the conjugation of phosphatidylethanolamine (PE) to the microtubule-associated protein light chain 3 (LC3). PE-conjugated (“lipidated”) LC3, known as LC3-II, decorates mature autophagosomes which are targeted to lysosomes together with their cargo. The complete process of autophagy from nucleation to degradation is referred to as autophagic flux and reflects the actual ability of autophagosomes to degrade intracellular components. Autophagic flux can be monitored by determining the degradation of specific autophagic substrates such as SQSTM1 (also known as p62) which interacts with LC3-II (Ichimura et al., 2008). SQSTM1/p62 has attracted significant attention also because of its role in facilitating the degradation of Lys63-ubiquitinated, p62-sequestered proteins by autophagy (Pankiv et al., 2007) and its ability to interact with several key components of the NF-κB pathway (Moscat and Diaz-Meco, 2012).
Phagophore formation is typically triggered by the interaction of ATG family proteins with two major regulatory complexes, TORC1 and AMPK. Under nutrient-rich conditions, high TORC1 activity prevents ULK1 activation by phosphorylating ULK1 at Ser757 and disrupting the interaction between ULK1 and its activating kinase AMPK (Figure 1) (Alexander et al., 2010a,b; Kim et al., 2011). Under starvation conditions, AMPK is activated and induces autophagy by phosphorylating ULK1 at Ser317 and Ser777 and by activating TSC2 thereby inhibiting TORC1 (Ganley et al., 2009; Laplante and Sabatini, 2012). Recent studies identified an additional, AMPK-independent mechanism of TORC1 inhibition that entails Sestrins, the RAG GTPases and a multiprotein complex called GATOR (GTPase-activating protein activity toward RAGs) composed of two complexes, GATOR1 and GATOR2. GATOR1 plays an essential role in switching off TORC1 upon amino acid depletion by functioning as a GAP for the RAGA/B heterodimer, incapacitating it from interacting with TORC1. GATOR2 is required for lysosomal recruitment of TORC1 by amino acids and is a negative regulator for GATOR1 by inhibiting its GAP activity. The interaction of Sestrins with GATOR2 under nutrient starvation liberates GATOR1 from GATOR2-mediated inhibition (Figure 1). Released GATOR1 subsequently binds to and inactivates RAGA/B, resulting in TORC1 suppression (Kim et al., 2015). Among various amino acids, cytoplasmic leucine was found to bind to Sestrins with the highest affinity and to disrupt their interaction with GATOR2 leading to TORC1 activation (Wolfson et al., 2016). Arginine, on the other hand, does not utilize Sestrins but a complex called CASTOR to release GATOR1 from GATOR2-mediated inhibition (Chantranupong et al., 2016). Therefore, different amino acid sensors are involved in TORC1 regulation and, by inference, on autophagy activation.
However, it should be noted that in addition to the aforementioned “canonical” pathway of autophagy, alternative mechanisms of autophagosome formation that do not require the hierarchical participation of all ATG proteins have been described and represent an area of intense investigations (Codogno et al., 2012). For example, unlike amino acid starvation, autophagy induced by deprivation of glucose or inhibition of glucose metabolism does not require ULK1 (Cheong et al., 2011). The topoisomerase inhibitor and clinically relevant chemotherapeutic agent etoposide induces non-canonical autophagosome formation that depends on ULK1 and Beclin but not ATG5, ATG7 or LC3-II (Nishida et al., 2009). Moreover, basal autophagy under nutrient rich conditions operates in an ATG5-independent manner and lipidation of LC3 depends on ATG3 following its conjugation to ATG12 (Murrow et al., 2015).
There is, therefore, a remarkable plasticity in autophagy-related pathways which is likely to be influenced by the type of autophagic stimulus and the expression levels of autophagy regulators. For example, the anti-apoptotic proteins BCL-2 and BCL-XL which are over-expressed in certain lymphomas and carcinomas (Eliopoulos et al., 1995), prevent the induction of autophagy by binding to and inhibiting Beclin-1 (Pattingre et al., 2005). PTEN antagonizes the effects of the PI3K/AKT pathway on TORC1 and thus, positively regulates autophagy (Arico et al., 2001). Conversely, inactivation of PTEN which occurs with high frequency in certain tumor types, may suppress autophagic responses through the concomitant constitutive activation of the PI3K/AKT pathway. The nuclear protein HMGB1 can induce autophagy in a cell-intrinsic manner following its translocation to the cytoplasm and interaction with Beclin-1 to facilitate autophagosome formation (Tang et al., 2010b). However, HMGB1 is also released extracellularly during ‘immunogenic’ cell death (Kepp et al., 2014), and functions in conjunction with other DAMP molecules to induce autophagy in neighboring tumor cells (Tang et al., 2010a). MicroRNA expression may also influence the autophagic process by targeting various autophagy pathway components (Zhai et al., 2013).
Accumulating evidence suggests that autophagy can be activated by DNA damage (Robert et al., 2011; Orlotti et al., 2012; Eapen and Haber, 2013) through various, albeit not exclusive routes, summarized in Figure 1.
As described above, ATM is a major sensor of DSBs induced by genotoxic stress. ATM links DDR to the induction of autophagy by activating AMPK which in turn phosphorylates TSC2 and removes the inhibitory effect of TORC1 on autophagy (Alexander et al., 2010a,b). The aforementioned ATM-AMPK-TSC2-mediated suppression of TORC1 operates in response to oxidative and nitrosative stress (Alexander et al., 2010a; Tripathi et al., 2013), both of which induce DNA damage and may involve mobilization of ATM to the cytoplasm (Alexander et al., 2010a). AMPK can also activate ULK1 to promote autophagosome formation (Kim et al., 2011).
PARP1, a NAD+ dependent chromatin-associated enzyme involved in base-excision repair of small adducts such as those induced by alkylating agents and ROS, is another DDR protein involved in autophagy regulation (de Murcia et al., 1997). DNA damage-induced PARP1 activation is associated with a reduction in both the NAD+ and the ATP pool. The latter is paralleled by elevated AMP levels that are sensed by AMPK leading to its activation and induction of autophagy (Rodriguez-Vargas et al., 2012). DNA damage may induce autophagy also through JNK. JNK phosphorylates BCL-2 leading to its displacement from the Beclin-1 complex that primes autophagosomal membrane formation (Wei et al., 2008).
Whereas the rapid induction of autophagy is mediated by post-translational modifications such as phosphorylation, ubiquitination, acetylation and lipidation, the regulation of autophagy may also depend on the execution of particular transcriptional and post-transcriptional programs. Indeed, the β1 and β2 subunits of AMPK are transcriptionally regulated by p53 (Feng et al., 2007) and are indirectly activated by p53 through Sestrin1 and Sestrin2 (Budanov and Karin, 2008). The recent discovery of the Sestrin2-GATOR-RAG pathway regulating TORC1 raises the possibility that the effects of p53 on TORC1 may be influenced by leucine availability. Additionally, p53 up-regulates PTEN expression leading to TORC1 inactivation (Stambolic et al., 2001; Feng et al., 2007). Moreover, DAPK, a transcriptional target of p53, triggers autophagy by phosphorylating Beclin-1 on Thr119 thereby releasing it from BCL-2 and BCL-XL, and by phosphorylating protein kinase D (PKD), both of which result in Vps34 class III PI3K complex activation and autophagy initiation (Zalckvar et al., 2009a,b; Eisenberg-Lerner and Kimchi, 2012). Another relevant transcriptional target of p53 is the gene encoding DRAM, a lysosomal protein that facilitates the end-stage of the autophagic process (Crighton et al., 2006). However, it should be noted that unlike its nuclear counterpart, cytoplasmic p53 has been associated with activation of mTOR and repression of autophagy but the underlying mechanism remains unclear at present (Tasdemir et al., 2008).
P53 is not the only DDR pathway protein that may transcriptionally regulate autophagy components (Pietrocola et al., 2013). For example, tumor protein p63 isoform ΔNp63α which is phosphorylated by ATM in response to genotoxic stress, transactivates various autophagy regulators including ULK1, ATG3, ATG5, Beclin-1, ATG7, and ATG10 (Huang et al., 2012). The same study showed that phosphorylated ΔNp63α also modulates the expression levels of ATG5, Beclin-1, ATG10, ATG12, ATG16L1, and UVRAG indirectly through the up-regulation of miR-181a, miR-519a, miR-374a, and miR-630, underscoring the contribution of post-transcriptional mechanisms to DNA damage-induced autophagy (Zhai et al., 2013). ATM also mediates the phosphorylation and activation of Che-1, a RNA polymerase II-binding protein which acts to increase the transcription of two mTOR inhibitor genes, Redd1 and Deptor (Desantis et al., 2015). Interestingly, Che-1 expression correlates with the progression of multiple myeloma, a malignancy characterized by high autophagy responses (Desantis et al., 2015). Another example of DDR-mediated transcriptional regulation of autophagy is provided by NF-κB which is activated by ATM-emanating signals and reported to transcriptionally upregulate Beclin-1 (Copetti et al., 2009).
As mentioned above, mtDNA is also subject to damage by irradiation, chemotherapy or ROS. If the extent of this damage exceeds the capacity of the mitochondrial quality control mechanisms, a form of autophagy is activated termed ‘mitophagy’ that leads to the lysosomal degradation of the damaged mitochondria. A critical regulator of this pathway is PINK1 which acts as sensor for mitochondrial damage. PINK1 is physiologically imported to the inner mitochondrial membrane; however, in response to stress-induced loss of mitochondrial membrane potential, PINK1 fails to be imported and is instead retained at the outer mitochondrial membrane where it phosphorylates a number of substrates. One of its targets is the ubiquitin ligase Parkin which catalyzes Lys63 and Lys48-linked ubiquitination of outer mitochondrial membrane proteins. Lys48-linked ubiquitination of target proteins results in their proteasomal degradation whereas Lys63-linked ubiquitination leads to recruitment of autophagy adaptors such as optineurin (OPT), nuclear dot protein 52 (NDP52) and SQSTM1/p62, which serve as a bridge for the assembly of the ULK1 complex and autophagosome formation (Youle and Narendra, 2011). The entire mitochondrion eventually becomes engulfed in the autophagosome which then fuses with a lysosome. This autophagy pathway ensures a healthy mitochondrial pool which is essential for normal energy metabolism and cellular homeostasis.
An interesting connection between nuclear DDR signaling and induction of mitophagy has been reported (Fang et al., 2014, 2016). It has been shown that nuclear DNA damage repair defects in xeroderma pigmentosum group A (XPA), ataxia-telangiectasia (AT), or Cockayne syndrome (CS) patients lead to defective mitophagy and the accumulation of dysfunctional mitochondria producing damaging levels of ROS (Fang et al., 2014). In this pathway, systemic DNA damage causes hyperactivation of PARP1 accompanied by NAD+ depletion and functional impairment of other NAD+-dependent enzymes such as Sirtuins (SIRT). Among them, SIRT1 is of particular interest as beyond its role as deacetylase of proteins involved in DNA repair pathways (Luna et al., 2013; Fang et al., 2016), it also deacetylates PGC1α which is a master transcriptional regulator of several mitochondrial biogenesis genes. Fang et al. (2016) found that reduced SIRT1 activity in XPA disease models resulted in diminished expression of the mitochondrial uncoupling protein 2 (UCP2), a transcriptional target of PGC1α responsible for mitochondrial hyperpolarization and increased import, cleavage and removal of PINK1. The in vivo relevance of these findings is highlighted by the observation that XPA, CS and AT patients as well as nematode (Caenorhabditis elegans) and rodent models of XPA possess dysfunctional mitochondria that contribute to the neurological and other pathologies manifested in these diseases (Scheibye-Knudsen et al., 2012; Valentin-Vega et al., 2012; Fang et al., 2014). Agents that recover NAD+ levels may thus represent a novel approach for therapeutic intervention in XPA, CS, and AT disease.
Overall, there is robust evidence that autophagy is activated by DDR pathways at multiple levels, raising the important question of whether autophagy impinges on functional outcomes of DDR.
One of the most fascinating functional outcomes of the DDR-autophagy axis is the regulation of DNA damage repair with major implications in genomic stability, aging and aging-related pathologies including cancer. Indeed, there is now significant evidence to suggest that autophagy is required for the function of ‘error-proof’ HR and NER (Liu et al., 2015; Park et al., 2015; Hewitt et al., 2016; Qiang et al., 2016; Wang et al., 2016). Conversely, autophagy-deficient cells rely mostly on the error-prone NHEJ repair process, which may explain the genomic instability observed in experimental mouse models with defective autophagy (Karantza-Wadsworth et al., 2007; Mathew et al., 2007b) and the observation that in human breast, ovarian and prostate cancers, beclin-1 is monoallelically deleted (Aita et al., 1999).
SQSTM1/p62 emerges as an important mediator of the effects of autophagy on DNA damage repair. This role has been attributed to a nuclear pool of p62 that accumulates upon autophagy blockade and binds RNF168, inhibiting its E3 ubiquitin ligase activity toward histone H2A. The ensued reduction in chromatin ubiquitination hinders the recruitment of DNA repair proteins such as BRCA1, RAD51, and RAP80 to sites of DSBs and impacts on their ability to repair radiation-induced DNA damage (Wang et al., 2016). RAD51 is also regulated by SQSTM1/p62 through filamin A which physiologically responds to DNA damage by recruiting RAD51 to DSBs. Autophagy impairment increases the interaction of p62 with filamin A, causing proteasomal degradation of both filamin A and RAD51 (Hewitt et al., 2016). Therefore, nuclear p62 that accrues from defective autophagy compromises DNA damage repair and genomic integrity. In line with these findings, nuclear levels and co-localization of p62 with DNA damage foci have been reported to increase with aging, underscoring the potential contribution of this pathway to aging and age-related diseases (Hewitt et al., 2016).
Autophagy is also responsible for the degradation of another chromatin component, HP1α. HP1α maintains a condensed chromatin configuration that hinders the formation of RAD51 nucleoprotein filaments at DSBs. The successful completion of HR repair requires access of RAD51 to DSBs which is achieved by RAD6-mediated ubiquitination and autophagy-mediated degradation of HP1α (Chen et al., 2015).
Chk1, a regulator of DNA damage repair by HR, is another recently described target of autophagy. One study showed that loss of (macro)autophagy by ablation of ATG7 leads to Chk1 ubiquitination and proteasomal degradation which in turn impairs DNA damage repair by HR (but not NHEJ) and genomic integrity (Liu et al., 2015). Another study identified Chk1 as target of CMA. CMA is upregulated in response to DNA damage inflicted by irradiation or chemotherapy, leading to lysosomal degradation of Ser345-phosphorylated Chk1 (Park et al., 2015). In contrast, Ser317-phosphorylated Chk1 which is the preferred substrate of the proteasome-dependent degradation of Chk1 remains unaffected by CMA inhibition, raising the possibility that different autophagy pathways (e.g., CMA vs. macro-autophagy) may target distinct Chk1 pools. Intriguingly, nuclear accumulation of Chk1 that ensues from defective CMA leads to destabilization of the MRN complex involved in the initial processing of DSBs prior to DNA repair by HR (Park et al., 2015). Thus, loss of CMA may facilitate genomic instability through excessive nuclear accumulation of Chk1 and deregulation of the MRN complex.
There is also evidence that autophagy positively regulates NER, the primary mechanism of repair of UV-induced lesions, by modulating the levels of NER-specific damage recognition proteins XPC, UVRAG, and DDB1/DDB2 (Qiang et al., 2016; Yang et al., 2016). UVRAG is of particular relevance as it binds the Beclin-1/Vps34 complex and increases the catalytic activity of Vps34, therefore acting as an inducer of autophagy (Liang et al., 2006). UVRAG is upregulated by radiation (Yin et al., 2011), localizes to photolesions and associates with DDB1 to promote the assembly and activity of the DDB2–DDB1–Cul4A–Roc1 ubiquitin ligase complex, leading to XPC recruitment and NER (Yang et al., 2016). Conversely, autophagy deficiency has been reported to impair the recruitment of DDB1/2 to UVB-induced DNA damage sites (Qiang et al., 2016). Impaired autophagy also leads to both transcriptional suppression of XPC and reduction in UVB-induced XPC ubiquitination, a process critical for DNA damage recognition. The in vivo relevance of these observations is underscored by pharmacological studies in mice showing that inhibition of autophagy by the chemical spautin-1 promotes, whereas stimulation of autophagy by rapamycin reduces UVB-induced tumorigenesis (Qiang et al., 2016).
Notwithstanding the impact of autophagic pathways on the turnover of DNA damage repair proteins, autophagy may have a generic role in DNA repair by regulating the supply of ATP, NAD+, and dNTPs that are necessary for this process. For example, repair of DSB requires ATP-dependent chromatin remodeling (Bao and Shen, 2007) and the unwinding of DNA by helicases during NER is an ATP-dependent process (de Laat et al., 1999). NAD+ is necessary for the function of PARP1 which is involved in base-excision repair (de Murcia et al., 1997). Moreover, a dNTP pool that is required for DNA replication and repair is maintained by autophagy-mediated degradation of ribonucleotide reductase subunits (Kumar et al., 2011). Likewise, autophagy removes nuclear membrane-enclosed chromosome fragments containing damaged DNA called micronuclei (Rello-Varona et al., 2012), a process that may also contribute to maintenance of genomic integrity.
Overall, the reported findings support a prominent role for autophagy in coordinating the execution of DNA damage repair and warrant further studies into how different repair mechanisms are controlled by distinct modes of autophagy and, conversely, how different autophagic pathways interact to finely balance distinct DNA repair systems.
Activation of autophagy contributes to DNA damage-induced senescence. A study by Kang et al. (2015) has recently unveiled a novel role for selective autophagy in linking DDR to the secretion of cytokines, chemokines, growth factors and proteases that collectively formulate the so-called SASP. The regulation of SASP has attracted significant attention as factors secreted by senescent cells establish an inflammatory environment that may foster the initiation and progression of several pathologies, including aging and aging-related diseases. The work by Kang et al. (2015) has implicated the transcription factor GATA4, which is physiologically targeted for degradation by autophagy through interaction with the autophagy adaptor p62, in SASP regulation. Specifically, upon irradiation or oncogene-induced senescence, GATA4 dissociates from p62 and becomes stabilized, causing induction of TRAF3IP2 and interleukin-1α (IL-1α) expression. In turn, TRAF3IP2 and IL-1α activate NF-κB which regulates major SASP components. Interestingly, activation of this GATA4 pathway depends on the DDR kinases ATM and ATR but is independent of p53 and p16INK4a. Thus, selective autophagy for GATA4 functions as an anti-senescence mechanism. Conversely, stimuli that induce senescence activate ATM and ATR to block p62-dependent autophagic degradation of GATA4, resulting in NF-κB activation and SASP induction. The in vivo relevance of these findings is highlighted by the fact that GATA4 accumulates in tissues of aged mice and humans as well as in various tissues of irradiated mice and may well contribute to the low level inflammation that typifies aging and aging-related pathologies including cancer (Kang et al., 2015). Interestingly, mice lacking p62 also exhibit an accelerated aging phenotype (Kwon et al., 2012).
Autophagy may also regulate SASP post-transcriptionally through spatial coupling to mTOR (Narita et al., 2011). In cells undergoing H-RasV 12-induced senescence, TORC1-positive lysosomes are recruited at the vicinity of the nucleus, resulting in de-inhibition of ULK1 at more distal locations where autophagosomes can form. As autophagosomes mature, they fuse with TORC1-positive lysosomes, giving rise to a novel membrane compartment called ‘TOR-Autophagy Spatial Coupling Compartment’ or TASCC. The flow of amino acids and other metabolites from autophagolysosomes activates TORC1 and also provides basic building blocks for the synthesis of SASP components. Indeed, blocking the localization of TORC1 to the TASCC by knocking-down the RAG GTPases, results in a reduction in the synthesis and secretion of two major SASP components, IL6 and IL8 (Narita et al., 2011).
In addition to SASP, autophagy may regulate senescence in a cell-autonomous manner through degradation of nuclear lamina components. Dou et al showed that in response to oncogenic stress or chemotherapy-induced DNA damage, Lamin B1 is exported from the nucleus together with chromatin domains, interacts with LC3 and undergoes autophagic degradation (Dou et al., 2015). Interestingly, oncogenic stress fails to induce Lamin B1 degradation and senescence when autophagy is impaired. Moreover, Lamin B1 is not processed by autophagy during starvation. Collectively, these data suggest that the activation of autophagy is required but is not sufficient for the establishment of oncogene-induced senescence and underscore the diversity of autophagic responses to different signals. As Lamin B1 anchors proteins that participate in NER (Butin-Israeli et al., 2013) it would be of interest to examine putative links between nucleophagy and DNA damage repair and their impact on senescence.
A key regulator of cellular senescence is ARF (Liontos et al., 2012). Activated oncogenes, oxidative stress and heat shock can induce the expression of both nucleolar and mitochondria-localized ARF (smARF) in a positive or a negative manner (Sideridou et al., 2011; Liontos et al., 2012; Evangelou et al., 2013; Velimezi et al., 2013; Kotsinas et al., 2014; Sherr et al., 2016). When overexpressed, smARF interacts with BCL-XL and decreases its interaction with Beclin-1. As a result, cell lines overexpressing smARF are more sensitive to the induction of autophagy and senescence caused by starvation or hydrogen peroxide (Pimkina et al., 2009). In contrast, expression of nucleolar ARF does not confer a similar effect (Reef and Kimchi, 2008). These observations indicate that smARF may link autophagy to senescence but whether this applies also in response to DNA damage remains to be determined. It would also be of interest to identify targets of smARF-mediated autophagy. In this regard, a recent study has shown that smARF overexpression depolarizes mitochondria and promotes mitophagy in a Parkin/PINK1-dependent manner (Grenier et al., 2014); the relevance of this finding to DNA damage-induced senescence requires additional studies.
Very recently, a revolutionary hybrid histo/immuno-chemical method of a biotin-linked Sudan Black-B analog has been established to assess the senescent status in in vitro and in vivo biological settings (Evangelou et al., 2016). This method may constitute an invaluable research tool for the study of the interplay between the molecular pathways implicated in autophagy and DNA damage-induced senescence.
Resistance to chemotherapy represents a major clinical problem for the management of cancer patients. The effects of autophagy on cell survival versus death have attracted particular attention in the context of malignancy as they may affect the outcome of DNA-targeted drug treatments.
As discussed above, autophagy intersects with DDR in the regulation of DNA damage repair pathways which are thought to play protective roles in genotoxic cancer therapy. Indeed, the autophagy inhibitor chloroquine has been shown to impair DNA damage repair and to increase the cytotoxic effect of the chemotherapeutic agent carboplatin in breast cancer stem cells (Liang et al., 2016). Accumulating evidence indicates that autophagy is exploited by tumor cells to resist radiation or chemotherapy-induced cell death and that genetic or pharmacological inhibition of autophagy sensitizes malignant cells to genotoxic therapy both in vitro and in experimental mouse models (Amaravadi et al., 2007; Pan et al., 2011; Chittaranjan et al., 2014; Wang and Wu, 2014; Filippi-Chiela et al., 2015; Liang et al., 2016; Piya et al., 2016). In line with this is the presence of autophagic vacuoles in Saos2 cells (Figure 2), a p53 null human osteosarcoma cell line, which after prolonged expression of p21WAF1/Cip1 exhibit enhanced aggressiveness and chemoresistance by deregulating the replication licensing machinery causing replication stress and fuelling genomic instability (Galanos et al., 2016).
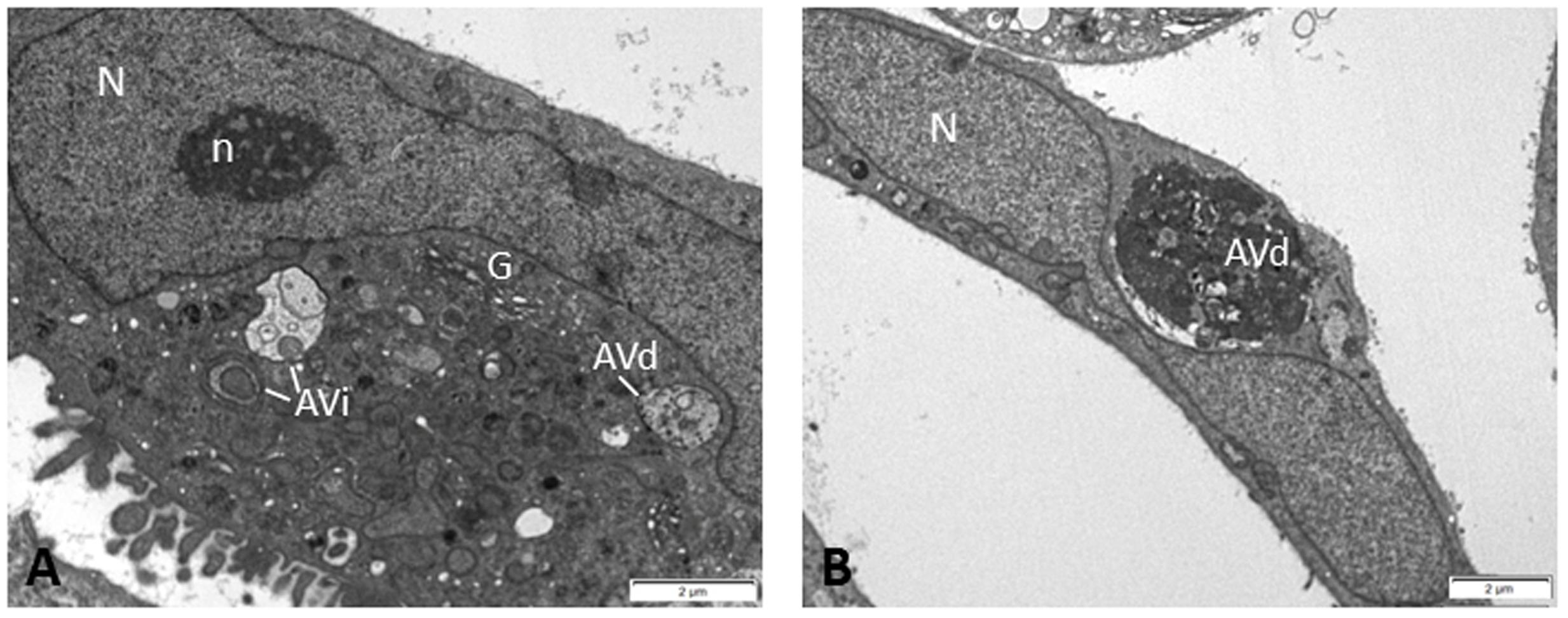
FIGURE 2. Electron micrographs of Saos2 p21WAF1/Cip1 Tet-ON human osteosarcoma cells after prolonged p21 overexpression (25 days) evading senescence and exhibiting aggressiveness and chemoresistance (Galanos et al., 2016). In the cytoplasm of these cells, several autophagic vacuoles were observed at different stages of autophagic process, i.e., initial autophagic vacuoles (AVi) (A) and degradative autophagic vacuoles (AVd) (A,B), indicating that autophagy may support the chemoresistant features of these cells. Percentage quantitative analysis showed increased number of autophagic vacuoles in Saos2 p21WAF1/Cip1 Tet-ON cells after prolonged p21 overexpression (30.1 ± 4.3) compared to control cells (without p21 induction) (3.3 ± 0.7). N, nucleus; n, nucleolus; G, Golgi apparatus.
These studies have provided the rationale for the clinical application of autophagy inhibitors in combination with established anti-cancer therapies (Huang et al., 2016). Fortuitously, the autophagy inhibitor hydroxychloroquine (a chloroquine derivative) has been used for many years in the management of patients with systemic lupus erythematosus, rheumatoid arthritis and malaria and therefore information about dosage, safety and side-effects has been available. The first phase I clinical trials incorporating hydroxychloroquine to chemo- or radio-therapeutic regimens aim at assessing the tolerability of the combination treatments, as well as dose and schedule optimization and are expected to guide subsequent clinical studies of efficacy (Poklepovic and Gewirtz, 2014).
Whereas as described above, autophagy mediates survival and drug resistance at low DNA damage levels, excessive lesions that cannot be repaired may lead to persistent, unrestraint autophagy which in turn induces a form of cell death termed ‘autophagic cell death’ (ACD). ACD is morphologically distinct from apoptosis and necrosis and is characterized by the sequestration of cytoplasmic materials in autophagosomes (Mathew et al., 2007a; Kroemer et al., 2009). Therefore, depending on the extent of DNA damage, autophagy may play a cytoprotective or cytotoxic role in the determination of cellular fate. The status of the apoptotic versus autophagic machinery components is likely to contribute to this outcome.
There is significant evidence supporting that DDR and immune response networks functionally interact (Pateras et al., 2015) and that autophagy participates in the regulation of inflammatory pathways (Netea-Maier et al., 2016).
Beyond SASP, autophagy may link DDR to the generation of local and systemic immune responses through the regulation of the so-called ‘immunogenic cell death,’ a form of cell death accompanied by the emission of immunostimulatory DAMPs. These include cell surface-exposed calreticulin and ATP and HMGB1 that are released by dying tumor cells following their exposure to certain genotoxic agents such as ionizing radiation (IR), doxorubicin and oxaliplatin (Kepp et al., 2014). A study by Guido Kroemer’s group has shown that autophagy is critically required for the release of ATP by irradiated tumor cells (Michaud et al., 2011). In turn, ATP stimulates the P2X7 receptor on antigen-presenting cells (APCs) leading to activation of the inflammasome, a large multiprotein signaling complex that regulates the processing of inactive pro-IL-1β to mature, secretable IL-1β (Michaud et al., 2011).
Intriguingly, autophagy also operates in APCs in a cell-intrinsic manner to control secretion of IL-1β and other cytokines. Thus, basal autophagy inhibits IL-1β secretion by degrading both inflammasome proteins (Shi et al., 2012) and pro-IL-1β (Harris et al., 2011). In contrast, under conditions of dual inflammasome and autophagy activation, IL-1β secretion increases through a non-canonical secretory pathway that depends on autophagy components (Dupont et al., 2011). Whereas these studies have not addressed the impact of autophagy on IL-1β biogenesis and secretion in response to genotoxic stress, it is likely that autophagy has a broader role in influencing the extent and duration of inflammation. This may be of particular relevance to and warrants further investigations in aging which is typified in humans by chronic, low-level inflammation, termed ‘inflammaging’ (Franceschi and Campisi, 2014), accumulated genomic damage (Ribezzo et al., 2016) and reduced autophagic activity (Martinez-Lopez et al., 2015).
The DDR has been conserved during evolution from bacteria to mammals, serving pivotal cellular functions that include repair of DNA lesions and maintenance of DNA integrity. As prokaryotic cells do not possess lysosomes, autophagy must have originated at a later phase of evolution than the DDR. Indeed, despite some differences in the biochemical pathways involved, autophagy-specific machineries have been identified in all eukaryotes examined, including yeast, plants (Arabidopsis thaliana), amoebozoa (Dictyostelium discoideum), and metazoa (Caenorhabditis elegans, Drosophila melanogaster, Mus musculus, and Homo sapiens) but not in prokaryotes (Hughes and Rusten, 2007). It has been proposed that the original function of autophagy may have been the adaptation to conditions of starvation through the recycling of intracellular components, and/or an early form of innate immune system allowing the destruction of intracellular bacterial pathogens (Hughes and Rusten, 2007).
However, emerging evidence links autophagy also to the DDR. Herein we have reviewed this evidence and highlighted in particular that: (1) autophagy is activated upon exposure to diverse DNA damaging factors including radiation, chemicals, ROS and oncogenes; (2) the DDR-autophagy link operates in a variety of eukaryotic cell types representing different evolutionary and developmental stages; (3) autophagy is required for several fundamental processes associated with the cellular response to DNA damage such as repair of DNA lesions, senescence, cell death, and cytokine secretion; (4) reduced autophagic flux characterizes human pathologies that are associated with accumulation of DNA damage, including cancer and aging.
Together, these observations raise the possibility that autophagy may have evolved as a quality control system that responds to a wide range of stress conditions including DNA damage, a major cellular stress factor. Unlike the more broadly conserved ubiquitin-proteasome system which regulates the turnover of short-lived proteins in both prokaryotes and eukaryotes, autophagy can target organelles or bulk cytoplasmic and nuclear material for lysosomal degradation. This is a property relevant to DDR in eukaryotic cells that enables them, for example, to deplete mitochondria bearing irreparable DNA damage and to control the number of mitochondria under stress conditions. Notably, the autophagic degradation of dysfunctional mitochondria (mitophagy) operates across the Eukaryota domain, including yeast, nematodes, flies, and mammals (Hirota et al., 2012), contributes to lifespan extension in model organisms (Palikaras et al., 2015) and protects against aging-related pathologies such as cancer and neurodegenerative diseases (Mathew et al., 2007a; Menzies et al., 2015; Fang et al., 2016). Further studies are needed to address the impact of DDR signaling on autophagy-mediated degradation of organelles other than mitochondria and their role in aging and aging-related disease pathogenesis. For example, defects in autophagy-mediated lipid droplet degradation (lipophagy) have been associated with metabolic disease and liver steatosis (Madrigal-Matute and Cuervo, 2016) but whether DDR utilizes this degradation pathway remains largely unexplored. Intriguingly, systemic DNA damage ensued by NER deficiency in mouse adipocytes leads to destruction of white adipose tissue depots (Karakasilioti et al., 2013). Whilst the contribution of autophagic degradation of lipid droplets to this phenotype remains elusive, the aforementioned observation does raise the possibility of putative links between DDR and lipophagy.
The exciting findings that continue to emerge in this field warrant further studies into the complex and often contrasting roles of autophagy in the onset, progression and therapy of various human diseases. This need is underscored by its dual role in malignancy: whereas autophagy prevents genomic instability, a hallmark of cancer, it may promote survival of tumor cells under stress conditions, including those induced by anticancer therapy (Mathew et al., 2007a). As the pathological basis of the vast majority of human diseases is typified by the involvement of multiple cell types and inter-organ communication, further studies are needed to address how tissue-specific deregulation of the DDR-autophagy axis may lead to systemic effects underpinning disease pathogenesis. Along these lines, a recently published study revealed a mechanism by which autophagy in the CNS and periphery coordinate lipophagy in the liver and adipose tissue (Martinez-Lopez et al., 2016). Likewise, the pursuit of autophagic pathways and their functional outcomes will no doubt continue to provide an invaluable side entrance into the complex but fascinating biology of DDR.
AE, SH, and VG contributed to the preparation, drafting and final review of the manuscript. SH provided the data depicted in Figure 2.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Konstantina Georgila of the Eliopoulos’ laboratory for critical reading of the manuscript and for contributing to the preparation of Figure 1.
AMPK, AMP-kinase; AP1, activator protein 1; APC, antigen-presenting cells; ATM, ataxia-telangiectasia mutated; ATR, ATM- and Rad3-Related; BASC, BRCA1-associated genome surveillance complex; CMA, chaperone-mediated autophagy; Chk1/Chk2, Checkpoint kinases 1/2; DAMP, damage-associated molecular pattern; DAPK, death-associated protein kinase; DDB, damage specific DNA binding proteins 1 and 2; DNA-PK, DNA-dependent protein kinase; DRAM, damage-regulated autophagy modulator; FOXO3, Forkhead box O3; GATA4, GATA Binding Protein 4; HDAC, histone deacetylases; HMGB1, High Mobility Group Box 1; HP1α, heterochromatin protein 1α; HR, homologous recombination; IKKγ, inhibitor of nuclear factor kappa-B kinase subunit gamma; JNK, cJun N-terminal kinase; LC3, microtubule-associated protein light chain 3; MRN, Mre11-Rad50-Nbs1 complex; mtDNA, mitochondrial DNA; NEMO, NF-κB essential modulator; NER, nucleotide excision repair; NHEJ, non-homologous end joining; PI3K, phosphatidylinositol 3-kinase; PINK1, PTEN-induced putative kinase 1; PTEN, Phosphatase and Tensin homolog; RAG, Ras-related GTPases; SASP, senescence-associated secretory phenotype; SQSTM1, sequestosome 1; TAK1, transforming growth factor beta-activated kinase 1; TASCC, TOR-Autophagy Spatial Coupling Compartment; TORC1, mechanistic target of rapamycin complex 1; TRAF3IP2, TNF receptor–associated factor interacting protein 2; TSC2, Tuberous Sclerosis Complex 2; ULK1, unc-51-like autophagy-activating kinase 1; UVRAG, UV radiation resistance-associated gene; XPA, xeroderma pigmentosum group A; XPC, xeroderma pigmentosum, complementation group C.
Aita, V. M., Liang, X. H., Murty, V. V., Pincus, D. L., Yu, W., Cayanis, E., et al. (1999). Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics 59, 59–65. doi: 10.1006/geno.1999.5851
Alexander, A., Cai, S. L., Kim, J., Nanez, A., Sahin, M., MacLean, K. H., et al. (2010a). ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc. Natl. Acad. Sci. U.S.A. 107, 4153–4158. doi: 10.1073/pnas.0911852107
Alexander, A., Kim, J., and Walker, C. L. (2010b). ATM engages the TSC2/mTORC1 signaling node to regulate autophagy. Autophagy 6, 672–673. doi: 10.4161/auto.6.5.12509
Amaravadi, R. K., Yu, D., Lum, J. J., Bui, T., Christophorou, M. A., Evan, G., et al. (2007). Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J. Clin. Invest. 117, 326–336. doi: 10.1172/JCI28833
Arico, S., Petiot, A., Bauvy, C., Dubbelhuis, P. F., Meijer, A. J., Codogno, P., et al. (2001). The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J. Biol. Chem. 276, 35243–35246. doi: 10.1074/jbc.C100319200
Bao, Y., and Shen, X. (2007). Chromatin remodeling in DNA double-strand break repair. Curr. Opin. Genet. Dev. 17, 126–131. doi: 10.1016/j.gde.2007.02.010
Budanov, A. V., and Karin, M. (2008). p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 134, 451–460. doi: 10.1016/j.cell.2008.06.028
Buschmann, T., Potapova, O., Bar-Shira, A., Ivanov, V. N., Fuchs, S. Y., Henderson, S., et al. (2001). Jun NH2-terminal kinase phosphorylation of p53 on Thr-81 is important for p53 stabilization and transcriptional activities in response to stress. Mol. Cell Biol. 21, 2743–2754. doi: 10.1128/MCB.21.8.2743-2754.2001
Butin-Israeli, V., Adam, S. A., and Goldman, R. D. (2013). Regulation of nucleotide excision repair by nuclear lamin b1. PLoS ONE 8:e69169. doi: 10.1371/journal.pone.0069169
Chantranupong, L., Scaria, S. M., Saxton, R. A., Gygi, M. P., Shen, K., Wyant, G. A., et al. (2016). The CASTOR proteins are arginine sensors for the mTORC1 pathway. Cell 165, 153–164. doi: 10.1016/j.cell.2016.02.035
Chen, S., Wang, C., Sun, L., Wang, D. L., Chen, L., Huang, Z., et al. (2015). RAD6 promotes homologous recombination repair by activating the autophagy-mediated degradation of heterochromatin protein HP1. Mol. Cell Biol. 35, 406–416. doi: 10.1128/MCB.00121-15
Cheong, H., Lindsten, T., Wu, J., Lu, C., and Thompson, C. B. (2011). Ammonia-induced autophagy is independent of ULK1/ULK2 kinases. Proc. Natl. Acad. Sci. U.S.A. 108, 11121–11126. doi: 10.1073/pnas.1108004108
Chittaranjan, S., Bortnik, S., Dragowska, W. H., Xu, J., Abeysundara, N., Leung, A., et al. (2014). Autophagy inhibition augments the anticancer effects of epirubicin treatment in anthracycline-sensitive and -resistant triple-negative breast cancer. Clin. Cancer Res. 20, 3159–3173. doi: 10.1158/1078-0432.CCR-13-2060
Cline, S. D. (2012). Mitochondrial DNA damage and its consequences for mitochondrial gene expression. Biochim. Biophys. Acta 1819, 979–991. doi: 10.1016/j.bbagrm.2012.06.002
Codogno, P., Mehrpour, M., and Proikas-Cezanne, T. (2012). Canonical and non-canonical autophagy: variations on a common theme of self-eating? Nat. Rev. Mol. Cell. Biol. 13, 7–12.
Copetti, T., Bertoli, C., Dalla, E., Demarchi, F., and Schneider, C. (2009). p65/RelA modulates BECN1 transcription and autophagy. Mol. Cell. Biol. 29, 2594–2608. doi: 10.1128/MCB.01396-08
Crighton, D., Wilkinson, S., O’Prey, J., Syed, N., Smith, P., Harrison, P. R., et al. (2006). DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 126, 121–134. doi: 10.1016/j.cell.2006.05.034
de Laat, W. L., Jaspers, N. G., and Hoeijmakers, J. H. (1999). Molecular mechanism of nucleotide excision repair. Genes Dev. 13, 768–785. doi: 10.1101/gad.13.7.768
de Murcia, J. M., Niedergang, C., Trucco, C., Ricoul, M., Dutrillaux, B., Mark, M., et al. (1997). Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc. Natl. Acad. Sci. U.S.A. 94, 7303–7307. doi: 10.1073/pnas.94.14.7303
Desantis, A., Bruno, T., Catena, V., De Nicola, F., Goeman, F., Iezzi, S., et al. (2015). Che-1-induced inhibition of mTOR pathway enables stress-induced autophagy. EMBO J. 34, 1214–1230. doi: 10.15252/embj.201489920
Dou, Z., Xu, C., Donahue, G., Shimi, T., Pan, J. A., Zhu, J., et al. (2015). Autophagy mediates degradation of nuclear lamina. Nature 527, 105–109. doi: 10.1038/527S105a
Dupont, N., Jiang, S., Pilli, M., Ornatowski, W., Bhattacharya, D., and Deretic, V. (2011). Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. EMBO J. 30, 4701–4711. doi: 10.1038/emboj.2011.398
Eapen, V. V., and Haber, J. E. (2013). DNA damage signaling triggers the cytoplasm-to-vacuole pathway of autophagy to regulate cell cycle progression. Autophagy 9, 440–441. doi: 10.4161/auto.23280
Eisenberg-Lerner, A., and Kimchi, A. (2012). PKD is a kinase of Vps34 that mediates ROS-induced autophagy downstream of DAPk. Cell Death Differ. 19, 788–797. doi: 10.1038/cdd.2011.149
Eliopoulos, A. G., Kerr, D. J., Herod, J., Hodgkins, L., Krajewski, S., Reed, J. C., et al. (1995). The control of apoptosis and drug resistance in ovarian cancer: influence of p53 and Bcl-2. Oncogene 11, 1217–1228.
Evangelou, K., Bartkova, J., Kotsinas, A., Pateras, I. S., Liontos, M., Velimezi, G., et al. (2013). The DNA damage checkpoint precedes activation of ARF in response to escalating oncogenic stress during tumorigenesis. Cell Death Differ. 20, 1485–1497. doi: 10.1038/cdd.2013.76
Evangelou, K., Lougiakis, N., Rizou, S. V., Kotsinas, A., Kletsas, D., Muñoz-Espín, D., et al. (2016). Robust, universal biomarker assay to detect senescent cells in biological specimens. Aging Cell (in press).
Fang, E. F., Scheibye-Knudsen, M., Brace, L. E., Kassahun, H., SenGupta, T., Nilsen, H., et al. (2014). Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell 157, 882–896. doi: 10.1016/j.cell.2014.03.026
Fang, E. F., Scheibye-Knudsen, M., Chua, K. F., Mattson, M. P., Croteau, D. L., and Bohr, V. A. (2016). Nuclear DNA damage signalling to mitochondria in ageing. Nat. Rev. Mol. Cell Biol. 17, 308–321. doi: 10.1038/nrm.2016.14
Feng, Z., Hu, W., de Stanchina, E., Teresky, A. K., Jin, S., Lowe, S., et al. (2007). The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 67, 3043–3053. doi: 10.1158/0008-5472.CAN-06-4149
Filippi-Chiela, E. C., Bueno e Silva, M. M., Thomé, M. P., and Lenz, G. (2015). Single-cell analysis challenges the connection between autophagy and senescence induced by DNA damage. Autophagy 11, 1099–1113. doi: 10.1080/15548627.2015.1009795
Franceschi, C., and Campisi, J. (2014). Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci. 69(Suppl. 1), S4–S9. doi: 10.1093/gerona/glu057
Galanos, P., Vougas, K., Walter, D., Polyzos, A., Maya-Mendoza, A., Haagensen, E. J., et al. (2016). Chronic p53-independent p21 expression causes genomic instability by deregulating replication licensing. Nat. Cell Biol. 18, 777–789.
Ganley, I. G., Lam du, H., Wang, J., Ding, X., Chen, S., and Jiang, X. (2009). ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 284, 12297–12305. doi: 10.1074/jbc.M900573200
Grenier, K., Kontogiannea, M., and Fon, E. A. (2014). Short mitochondrial ARF triggers Parkin/PINK1-dependent mitophagy. J. Biol. Chem. 289, 29519–29530. doi: 10.1074/jbc.M114.607150
Halazonetis, T. D., Gorgoulis, V. G., and Bartek, J. (2008). An oncogene-induced DNA damage model for cancer development. Science 319, 1352–1355. doi: 10.1126/science.1140735
Hamdi, M., Kool, J., Cornelissen-Steijger, P., Carlotti, F., Popeijus, H. E., van der Burgt, C., et al. (2005). DNA damage in transcribed genes induces apoptosis via the JNK pathway and the JNK-phosphatase MKP-1. Oncogene 24, 7135–7144. doi: 10.1038/sj.onc.1208875
Harris, J., Hartman, M., Roche, C., Zeng, S. G., O’Shea, A., Sharp, F. A., et al. (2011). Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J. Biol. Chem. 286, 9587–9597. doi: 10.1074/jbc.M110.196808
Hewitt, G., Carroll, B., Sarallah, R., Correia-Melo, C., Ogrodnik, M., Nelson, G., et al. (2016). SQSTM1/p62 mediates crosstalk between autophagy and the UPS in DNA repair. Autophagy 12, 1917–1930. doi: 10.1080/15548627.2016.1210368
Hirota, Y., Kang, D., and Kanki, T. (2012). The physiological role of mitophagy: new insights into phosphorylation events. Int. J. Cell Biol. 2012:354914. doi: 10.1155/2012/354914
Huang, Y., Guerrero-Preston, R., and Ratovitski, E. A. (2012). Phospho-DeltaNp63alpha-dependent regulation of autophagic signaling through transcription and micro-RNA modulation. Cell Cycle 11, 1247–1259. doi: 10.4161/cc.11.1.18430
Huang, Z., Zhou, L., Chen Z., Nice, E. C., and Huang, C. (2016). Stress management by autophagy: Implications for chemoresistance. Int. J. Cancer 139, 23–32. doi: 10.1002/ijc.30066
Hughes, T., and Rusten, T. E. (2007). Origin and evolution of self-consumption: autophagy. Adv. Exp. Med. Biol. 607, 111–118. doi: 10.1007/978-0-387-74021-8_9
Ichimura, Y., Kumanomidou, T., Sou, Y. S., Mizushima, T., Ezaki, J., Ueno, T., et al. (2008). Structural basis for sorting mechanism of p62 in selective autophagy. J. Biol. Chem. 283, 22847–22857. doi: 10.1074/jbc.M802182200
Jackson, S. P., and Bartek, J. (2009). The DNA-damage response in human biology and disease. Nature 461, 1071–1078. doi: 10.1038/nature08467
Kang, C., Xu, Q., Martin, T. D., Li, M. Z., Demaria, M., Aron, L., et al. (2015). The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 349:aaa5612. doi: 10.1126/science.aaa5612
Karakasilioti, I., Kamileri, I., Chatzinikolaou, G., Kosteas, T., Vergadi, E., Robinson, A. R., et al. (2013). DNA damage triggers a chronic autoinflammatory response, leading to fat depletion in NER progeria. Cell Metab. 18, 403–415. doi: 10.1016/j.cmet.2013.08.011
Karantza-Wadsworth, V., Patel, S., Kravchuk, O., Chen, G., Mathew, R., Jin, S., et al. (2007). Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 21, 1621–1635. doi: 10.1101/gad.1565707
Kepp, O., Senovilla, L., Vitale, I., Vacchelli, E., Adjemian, S., Agostinis, P., et al. (2014). Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 3:e955691. doi: 10.4161/21624011.2014.955691
Kim, G. D., Choi, Y. H., Dimtchev, A., Jeong, S. J., Dritschilo, A., and Jung, M. (1999). Sensing of ionizing radiation-induced DNA damage by ATM through interaction with histone deacetylase. J. Biol. Chem. 274, 31127–31130. doi: 10.1074/jbc.274.3.1233
Kim, J., Kundu, M., Viollet, B., and Guan, K. L. (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141. doi: 10.1038/ncb2152
Kim, J. S., Ro, S. H., Kim, M., Park, H. W., Semple, I. A., Park, H., et al. (2015). Sestrin2 inhibits mTORC1 through modulation of GATOR complexes. Sci. Rep. 5:9502. doi: 10.1038/srep10309
Kotsinas, A., Papanagnou, P., Evangelou, K., Trigas, G. C., Kostourou, V., Townsend, P., et al. (2014). ARF: a versatile DNA damage response ally at the crossroads of development and tumorigenesis. Front. Genet. 5:236. doi: 10.3389/fgene.2014.00236
Kroemer, G., Galluzzi, L., Vandenabeele, P., Abrams, J., Alnemri, E. S., Baehrecke, E. H., et al. (2009). Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 16, 3–11. doi: 10.1038/cdd.2008.150
Kumar, D., Abdulovic, A. L., Viberg, J., Nilsson, A. K., Kunkel, T. A., and Chabes, A. (2011). Mechanisms of mutagenesis in vivo due to imbalanced dNTP pools. Nucleic Acids Res. 39, 1360–1371. doi: 10.1093/nar/gkq1198
Kwon, J., Han, E., Bui, C. B., Shin, W., Lee, J., Lee, S., et al. (2012). Assurance of mitochondrial integrity and mammalian longevity by the p62-Keap1-Nrf2-Nqo1 cascade. EMBO Rep. 13, 150–156. doi: 10.1038/embor.2011.246
Laplante, M., and Sabatini, D. M. (2012). mTOR signaling in growth control and disease. Cell 149, 274–293. doi: 10.1016/j.cell.2012.03.017
LeDoux, S. P., Wilson, G. L., Beecham, E. J., Stevnsner, T., Wassermann, K., and Bohr, V. A. (1992). Repair of mitochondrial DNA after various types of DNA damage in Chinese hamster ovary cells. Carcinogenesis 13, 1967–1973. doi: 10.1093/carcin/13.11.1967
Liang, C., Feng, P., Ku, B., Dotan, I., Canaani, D., Oh, B. H., et al. (2006). Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat. Cell Biol. 8, 688–699. doi: 10.1038/ncb1426
Liang, D. H., Choi, D. S., Ensor, J. E., Kaipparettu, B. A., Bass, B. L., and Chang, J. C. (2016). The autophagy inhibitor chloroquine targets cancer stem cells in triple negative breast cancer by inducing mitochondrial damage and impairing DNA break repair. Cancer Lett. 376, 249–258. doi: 10.1016/j.canlet.2016.04.002
Lieber, M. R., Ma, Y., Pannicke, U., and Schwarz, K. (2003). Mechanism and regulation of human non-homologous DNA end-joining. Nat. Rev. Mol. Cell Biol. 4, 712–720. doi: 10.1038/nrm1202
Liontos, M. I., Pateras, S., Evangelou, K., and Gorgoulis, V. G. (2012). The tumor suppressor gene ARF as a sensor of oxidative stress. Curr. Mol. Med. 12, 704–715. doi: 10.2174/156652412800792633
Liu, E. Y., Xu, N., O’Prey, J., Lao, L. Y., Joshi, S., Long, J. S., et al. (2015). Loss of autophagy causes a synthetic lethal deficiency in DNA repair. Proc. Natl. Acad. Sci. U.S.A. 112, 773–778. doi: 10.1073/pnas.1504817112
Liu, Q., Guntuku, S., Cui, X. S., Matsuoka, S., Cortez, D., Tamai, K., et al. (2000). Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 14, 1448–1459. doi: 10.1101/gad.840500
Lukas, J., Lukas, C., and Bartek, J. (2011). More than just a focus: the chromatin response to DNA damage and its role in genome integrity maintenance. Nat. Cell. Biol. 13, 1161–1169. doi: 10.1038/ncb2344
Luna, A., Aladjem, M. I., and Kohn, K. W. (2013). SIRT1/PARP1 crosstalk: connecting DNA damage and metabolism. Genome Integr. 4:6. doi: 10.1186/2041-9414-4-6
Mabb, A. M., Wuerzberger-Davis, S. M., and Miyamoto, S. (2006). PIASy mediates NEMO sumoylation and NF-kappaB activation in response to genotoxic stress. Nat. Cell Biol. 8, 986–993. doi: 10.1038/ncb1458
Madrigal-Matute, J., and Cuervo, A. M. (2016). Regulation of liver metabolism by autophagy. Gastroenterology 150, 328–339. doi: 10.1016/S0016-5085(16)31153-2
Marechal, A., and Zou, L. (2013). DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 5:a012716. doi: 10.1101/cshperspect.a012716
Martinez-Lopez, N., Athonvarangkul, D., and Singh, R. (2015). Autophagy and aging. Adv. Exp. Med. Biol. 847, 73–87. doi: 10.1007/978-1-4939-2404-2_3
Martinez-Lopez, N., Garcia-Macia, M., Sahu, S., Athonvarangkul, D., Liebling, E., Merlo, P., et al. (2016). Autophagy in the CNS and periphery coordinate lipophagy and lipolysis in the brown adipose tissue and liver. Cell Metab. 23, 113–127. doi: 10.1016/j.cmet.2015.10.008
Mathew, R., Karantza-Wadsworth, V., and White, E. (2007a). Role of autophagy in cancer. Nat. Rev. Cancer 7, 961–967. doi: 10.1038/nrc2254
Mathew, R., Kongara, S., Beaudoin, B., Karp, C. M., Bray, K., Degenhardt, K., et al. (2007b). Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 21, 1367–1381. doi: 10.1101/gad.1545107
Menzies, F. M., Fleming, A., and Rubinsztein, D. C. (2015). Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci. 16, 345–357. doi: 10.1038/nrn3961
Michaud, M., Martins, I., Sukkurwala, A. Q., Adjemian, S., Ma, Y., Pellegatti, P., et al. (2011). Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 334, 1573–1577. doi: 10.1126/science.1208347
Mizushima, N., Levine, B., Cuervo, A. M., and Klionsky, D. J. (2008). Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075. doi: 10.1038/nature06639
Moscat, J., and Diaz-Meco, M. T. (2012). p62: a versatile multitasker takes on cancer. Trends Biochem. Sci. 37, 230–236. doi: 10.1016/j.tibs.2012.02.008
Murrow, L., Malhotra, R., and Debnath, J. (2015). ATG12-ATG3 interacts with Alix to promote basal autophagic flux and late endosome function. Nat. Cell Biol. 17, 300–310. doi: 10.1038/ncb3112
Narita, M., Young, A. R., Arakawa, S., Samarajiwa, S. A., Nakashima, T., Yoshida, S., et al. (2011). Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science 332, 966–970. doi: 10.1126/science.1205407
Netea-Maier, R. T., Plantinga, T. S., van de Veerdonk, F. L., Smit, J. W., and Netea, M. G. (2016). Modulation of inflammation by autophagy: consequences for human disease. Autophagy 12, 245–260. doi: 10.1080/15548627.2015.1071759
Nishida, Y., Arakawa, S., Fujitani, K., Yamaguchi, H., Mizuta, T., Kanaseki, T., et al. (2009). Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 461, 654–658. doi: 10.1038/nature08455
Orlotti, N. I., Cimino-Reale, G., Borghini, E., Pennati, M., Sissi, C., Perrone, F., et al. (2012). Autophagy acts as a safeguard mechanism against G-quadruplex ligand-mediated DNA damage. Autophagy 8, 1185–1196. doi: 10.4161/auto.20519
Palikaras, K., Lionaki, E., and Tavernarakis, N. (2015). Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 521, 525–528. doi: 10.1038/nature14300
Pan, Y., Gao, Y., Chen, L., Gao, G., Dong, H., Yang, Y., et al. (2011). Targeting autophagy augments in vitro and in vivo antimyeloma activity of DNA-damaging chemotherapy. Clin. Cancer Res. 17, 3248–3258. doi: 10.1158/1078-0432.CCR-10-0890
Pankiv, S., Clausen, T. H., Lamark, T., Brech, A., Bruun, J. A., Outzen, H., et al. (2007). p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282, 24131–24145. doi: 10.1074/jbc.M702824200
Park, C., Suh, Y., and Cuervo, A. M. (2015). Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage. Nat. Commun. 6:6823. doi: 10.1038/ncomms7552
Pateras, I. S., Havaki, S., Nikitopoulou, X., Vougas, K., Townsend, P. A., Panayiotidis, M. I., et al. (2015). The DNA damage response and immune signaling alliance: Is it good or bad? Nature decides when and where. Pharmacol. Ther. 154, 36–56. doi: 10.1016/j.pharmthera.2015.06.011
Pattingre, S., Tassa, A., Qu, X., Garuti, R., Liang, X. H., Mizushima, N., et al. (2005). Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122, 927–939. doi: 10.1016/j.cell.2005.07.002
Pietrocola, F., Izzo, V., Niso-Santano, M., Vacchelli, E., Galluzzi, L., Maiuri, M. C., et al. (2013). Regulation of autophagy by stress-responsive transcription factors. Semin. Cancer Biol. 23, 310–322. doi: 10.1016/j.semcancer.2013.05.008
Pimkina, J., Humbey, O., Zilfou, J. T., Jarnik, M., and Murphy, M. E. (2009). ARF induces autophagy by virtue of interaction with Bcl-xl. J. Biol. Chem. 284, 2803–2810. doi: 10.1074/jbc.M804705200
Piya, S., Kornblau, S. M., Ruvolo, V. R., Mu, H., Ruvolo, P. P., McQueen, T., et al. (2016). Atg7 suppression enhances chemotherapeutic agent sensitivity and overcomes stroma-mediated chemoresistance in acute myeloid leukemia. Blood 128, 1260–1269. doi: 10.1182/blood-2016-01-692244
Poklepovic, A., and Gewirtz, D. A. (2014). Outcome of early clinical trials of the combination of hydroxychloroquine with chemotherapy in cancer. Autophagy 10, 1478–1480. doi: 10.4161/auto.29428
Qiang, L., Zhao, B., Shah, P., Sample, A., Yang, S., and He, Y. Y. (2016). Autophagy positively regulates DNA damage recognition by nucleotide excision repair. Autophagy 12, 357–368. doi: 10.1080/15548627.2015.1110667
Reef, S., and Kimchi, A. (2008). Nucleolar p19ARF, unlike mitochondrial smARF, is incapable of inducing p53-independent autophagy. Autophagy 4, 866–869. doi: 10.4161/auto.6691
Rello-Varona, S., Lissa, D., Shen, S., Niso-Santano, M., Senovilla, L., Mariño, G., et al. (2012). Autophagic removal of micronuclei. Cell Cycle 11, 170–176. doi: 10.4161/cc.11.1.18564
Ribezzo, F., Shiloh, Y., and Schumacher, B. (2016). Systemic DNA damage responses in aging and diseases. Semin. Cancer Biol. 3, 26–35. doi: 10.1016/j.semcancer.2015.12.005
Robert, T., Vanoli, F., Chiolo, I., Shubassi, G., Bernstein, K. A., Rothstein, R., et al. (2011). HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature 471, 74–79. doi: 10.1038/nature09803
Rodriguez-Vargas, J. M., Ruiz-Magana, M. J., Ruiz-Ruiz, C., Majuelos-Melguizo, J., Peralta-Leal, A., Rodríguez, M. I., et al. (2012). ROS-induced DNA damage and PARP-1 are required for optimal induction of starvation-induced autophagy. Cell Res. 22, 1181–1198. doi: 10.1038/cr.2012.70
Roos, W. P., Thomas, A. D., and Kaina, B. (2016). DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 16, 20–33. doi: 10.1038/nrc.2015.2
Rubinsztein, D. C., Shpilka, T., and Elazar, Z. (2012). Mechanisms of autophagosome biogenesis. Curr. Biol. 22, R29–R34. doi: 10.1016/j.cub.2011.11.034
Scheibye-Knudsen, M., Ramamoorthy, M., Sykora, P., Maynard, S., Lin, P. C., Minor, R. K., et al. (2012). Cockayne syndrome group B protein prevents the accumulation of damaged mitochondria by promoting mitochondrial autophagy. J. Exp. Med. 209, 855–869. doi: 10.1084/jem.20111721
Sedelnikova, O. A., Redon, C. E., Dickey, J. S., Nakamura, A. J., Georgakilas, A. G., and Bonner, W. M. (2010). Role of oxidatively induced DNA lesions in human pathogenesis. Mutat. Res. 704, 152–159. doi: 10.1016/j.mrrev.2009.12.005
Sherr, C. J., Beach, D., and Shapiro, G. I. (2016). Targeting CDK4 and CDK6: from discovery TO Therapy. Cancer Discov. 6, 353–367. doi: 10.1158/2159-8290.CD-15-0894
Shi, C. S., Shenderov, K., Huang, N. N., Kabat, J., Abu-Asab, M., Fitzgerald, K. A., et al. (2012). Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 13, 255–263. doi: 10.1038/ni.2215
Sideridou, M., Zakopoulou, R., Evangelou, K., Liontos, M., Kotsinas, A., Rampakakis, E., et al. (2011). Cdc6 expression represses E-cadherin transcription and activates adjacent replication origins. J. Cell Biol. 195, 1123–1140. doi: 10.1083/jcb.201108121
Stambolic, V., MacPherson, D., Sas, D., Lin, Y., Snow, B., Jang, Y., et al. (2001). Regulation of PTEN transcription by p53. Mol. Cell 8, 317–325. doi: 10.1016/S1097-2765(01)00323-9
Tang, D., Kang, R., Cheh, C. W., Livesey, K. M., Liang, X., Schapiro, N. E., et al. (2010a). HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene 29, 5299–5310. doi: 10.1038/onc.2010.261
Tang, D., Loze, M. T., Zeh, H. J., and Kang, R. (2010b). The redox protein HMGB1 regulates cell death and survival in cancer treatment. Autophagy 6, 1181–1183. doi: 10.4161/auto.6.8.13367
Tasdemir, E., Maiuri, M. C., Galluzzi, L., Vitale, I., Djavaheri-Mergny, M., D’Amelio, M., et al. (2008). Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 10, 676–687. doi: 10.1038/ncb1730
Tran, H., Brunet, A., Grenier, J. M., Datta, S. R., Fornace, A. J. Jr., DiStefano, P. S., et al. (2002). DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science 296, 530–534. doi: 10.1126/science.1068712
Tripathi, D. N., Chowdhury, R., Trudel, L. J., Tee, A. R., Slack, R. S., Walker, C. L., et al. (2013). Reactive nitrogen species regulate autophagy through ATM-AMPK-TSC2-mediated suppression of mTORC1. Proc. Natl. Acad. Sci. U.S.A. 110, E2950–E2957. doi: 10.1073/pnas.1307736110
Valentin-Vega, Y. A., Maclean, K. H., Tait-Mulder, J., Milasta, S., Steeves, M., Dorsey, F. C., et al. (2012). Mitochondrial dysfunction in ataxia-telangiectasia. Blood 119, 1490–1500. doi: 10.1182/blood-2011-08-373639
Velimezi, G., Liontos, M., Vougas, K., Roumeliotis, T., Bartkova, J., Sideridou, M., et al. (2013). Functional interplay between the DNA-damage-response kinase ATM and ARF tumour suppressor protein in human cancer. Nat. Cell Biol. 15, 967–977. doi: 10.1038/ncb2795
Wang, J., and Wu, G. S. (2014). Role of autophagy in cisplatin resistance in ovarian cancer cells. J. Biol. Chem. 289, 17163–17173. doi: 10.1074/jbc.M113.527671
Wang, Y., Cortez, D., Yazdi, P., Neff, N., Elledge, S. J., Qin, J., et al. (2000). BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev. 14, 927–939. doi: 10.1101/gad.827000
Wang, Y., Zhang, N., Zhang, L., Li, R., Fu, W., Ma, K., et al. (2016). Autophagy regulates chromatin ubiquitination in DNA damage response through elimination of SQSTM1/p62. Mol. Cell 63, 34–48. doi: 10.1016/j.molcel.2016.08.021
Wei, Y., Sinha, S., and Levine B. (2008). Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy 4, 949–951. doi: 10.4161/auto.6788
Wirth, M., Joachim, J., and Tooze, S. A. (2013). Autophagosome formation–the role of ULK1 and Beclin1-PI3KC3 complexes in setting the stage. Semin. Cancer Biol. 23, 301–309. doi: 10.1016/j.semcancer.2013.05.007
Wolfson, R. L., Chantranupong, L., Saxton, R. A., Shen, K., Scaria, S. M., Cantor, J. R., et al. (2016). Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 351, 43–48. doi: 10.1126/science.aad3526
Wu, Z. H., Shi, Y., Tibbetts, R. S., and Miyamoto, S. (2006). Molecular linkage between the kinase ATM and NF-kappaB signaling in response to genotoxic stimuli. Science 311, 1141–1146. doi: 10.1126/science.1121513
Yang, Y., He, S., Wang, Q., Li, F., Kwak, M. J., Chen, S., et al. (2016). Autophagic UVRAG promotes UV-induced photolesion repair by activation of the CRL4(DDB2) E3 ligase. Mol. Cell 62, 507–519. doi: 10.1016/j.molcel.2016.04.014
Yin, X., Cao, L., Kang, R., Yang, M., Wang, Z., Peng, Y., et al. (2011). UV irradiation resistance-associated gene suppresses apoptosis by interfering with BAX activation. EMBO Rep. 12, 727–734. doi: 10.1038/embor.2011.79
Youle, R. J., and Narendra, D. P. (2011). Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 12, 9–14. doi: 10.1038/nrm3028
Zalckvar, E., Berissi, H., Eisenstein, M., and Kimchi, A. (2009a). Phosphorylation of Beclin 1 by DAP-kinase promotes autophagy by weakening its interactions with Bcl-2 and Bcl-XL. Autophagy 5, 720–722. doi: 10.4161/auto.5.5.8625
Zalckvar, E., Berissi, H., Mizrachy, L., Idelchuk, Y., Koren, I., Eisenstein, M., et al. (2009b). DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep. 10, 285–292. doi: 10.1038/embor.2008.246
Zhai, H., Fesler, A., and Ju, J. (2013). MicroRNA: a third dimension in autophagy. Cell Cycle 12, 246–250. doi: 10.4161/cc.23273
Keywords: DNA damage, autophagy, mitophagy, evolution, senescence, repair, inflammation, cell death
Citation: Eliopoulos AG, Havaki S and Gorgoulis VG (2016) DNA Damage Response and Autophagy: A Meaningful Partnership. Front. Genet. 7:204. doi: 10.3389/fgene.2016.00204
Received: 14 October 2016; Accepted: 02 November 2016;
Published: 21 November 2016.
Edited by:
Pierre De Meyts, de Duve Institute, BelgiumReviewed by:
Alma Balestrazzi, University of Pavia, ItalyCopyright © 2016 Eliopoulos, Havaki and Gorgoulis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aristides G. Eliopoulos, ZWxpb3BhZ0BtZWQudW9jLmdy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.