 Donal M. O'Sullivan
Donal M. O'Sullivan Deepti Angra
Deepti Angra
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet. , 22 August 2016
Sec. Plant Genetics and Genomics
Volume 7 - 2016 | https://doi.org/10.3389/fgene.2016.00150
This article is part of the Research Topic Genomic tools in legume breeding programs: status and perspectives View all 20 articles
Vicia faba L, is a globally important grain legume whose main centers of diversity are the Fertile Crescent and Mediterranean basin. Because of its small number (six) of exceptionally large and easily observed chromosomes it became a model species for plant cytogenetics the 70s and 80s. It is somewhat ironic therefore, that the emergence of more genomically tractable model plant species such as Arabidopsis and Medicago coincided with a marked decline in genome research on the formerly favored plant cytogenetic model. Thus, as ever higher density molecular marker coverage and dense genetic and even complete genome sequence maps of key crop and model species emerged through the 1990s and early 2000s, genetic and genome knowledge of Vicia faba lagged far behind other grain legumes such as soybean, common bean and pea. However, cheap sequencing technologies have stimulated the production of deep transcriptome coverage from several tissue types and numerous distinct cultivars in recent years. This has permitted the reconstruction of the faba bean meta-transcriptome and has fueled development of extensive sets of Simple Sequence Repeat and Single Nucleotide Polymorphism (SNP) markers. Genetics of faba bean stretches back to the 1930s, but it was not until 1993 that DNA markers were used to construct genetic maps. A series of Random Amplified Polymorphic DNA-based genetic studies mainly targeted at quantitative loci underlying resistance to a series of biotic and abiotic stresses were conducted during the 1990's and early 2000s. More recently, SNP-based genetic maps have permitted chromosome intervals of interest to be aligned to collinear segments of sequenced legume genomes such as the model legume Medicago truncatula, which in turn opens up the possibility for hypotheses on gene content, order and function to be translated from model to crop. Some examples of where knowledge of gene content and function have already been productively exploited are discussed. The bottleneck in associating genes and their functions has therefore moved from locating gene candidates to validating their function and the last part of this review covers mutagenesis and genetic transformation, two complementary routes to validating gene function and unlocking novel trait variation for the improvement of this important grain legume.
Vicia faba L.(Vf) or faba bean is a grain legume of great importance in world agriculture due to its high yield potential compared to alternative grain legumes, its ability to fix nitrogen through symbiosis with Rhizobium leguminosarum in its root nodules, but most crucially for its role as a staple dietary protein source in North African and Middle Eastern cultures. The species is thought to have been domesticated in the Eastern Mediterranean region, perhaps somewhere between Afghanistan and the Eastern Mediterranean (van de Wouw et al., 2001), but no extant wild relative has yet been found although new species closely related to Vf have been found in recent decades (Maxted, 1993). Nonetheless, there is great variability within the domesticated genepool, with the major center of diversity centered around the Mediterranean basin and secondary centers of diversity in the Nile Valley, South America, and Central and Eastern Asia (Duc et al., 2010), providing much untapped potential to breeders. This diversity of forms is exemplified by the botanical classification of “major” (large-seeded or “broad” bean), “equine” (mid-sized or “horse” bean), and “minor” (small, rounded seed) types. Vf is one of the earliest domesticated Old World agricultural crops, with credible archaeobotanical evidence linking it to the pre-pottery Neolithic period around 10,000 BP in a site in North-West Syria (Tanno and Willcox, 2006). However, again in keeping with its great diversity and adaptability, it has a long history of utilization outside the center of origin in diverse agroecological settings from the Boreal and Atlantic Maritime climates to arid and sub-tropical regions. The most current available statistics for dry beans show it ranking 7th in terms of global production of grain legumes with production in 2013 of 1.17 million tons (FAOSTAT, 2013)1, though this probably grossly underestimates the nutritional and societal importance of the species as a majority of faba bean worldwide is cultivated and consumed locally by subsistence farmers. Furthermore, FAOSTAT figures relate to dry beans and there are no reliable global data for large-seeded broad bean types picked green and consumed fresh. China is the leading producer, followed currently by Ethiopia, Australia, France, Egypt, Morocco, Sudan, and the UK, though rank order amongst the latter seven countries has historically been somewhat fluid. Despite the strong cultural attachment of Mediterranean basin and Middle Eastern populations to consumption of faba bean, for a variety of reasons addressed below, many of these countries have become net importers.
Faba bean is generally speaking the most productive of grain legumes in environments where rainfall is not limiting or in irrigated conditions, and can indeed be a highly profitable crop, especially if the economic benefits of biologically fixed nitrogen and enhanced weed and disease control in subsequent crops are considered (Preissel et al., 2015). However, in common with many legumes, its yields are relatively unstable, which is thought to be one important reason underlying the low inclusion rate of leguminous crops in European agriculture in particular (Cernay et al., 2015). This inherent yield instability is thought to be due, in part, to its apparently profligate flowering habit whereby many flowers spread over many nodes are produced containing far more ovules than the capacity of the plant to fertilize and fill those potential seed sites, resulting in a high but variable rate of flower and fertilized ovule abortion. Optimal yield production in faba bean is also dependent on symbiosis with Rhizobium leguminosarum biovar viciae to produce nitrogen-fixing root nodules as well as on the pollination services of wild bee populations to ensure both optimal seed set and outcrossing rates. Pollinator insufficiency has been shown recently in the UK to explain up to 64% yield loss (Nayak et al., 2015) while a large N-fixation response to rhizobial inoculation in the field has been shown even in the presence of high natural rhizobial populations (Denton et al., 2013). On top of these variable symbiotic or mutualistic interactions are layered a host of abiotic and biotic stresses, combinations of which will exact further environment-dependent losses. The many specific pests and pathogens of faba bean include fungi (Ascochyta fabae—ascochyta blight, Botrytis fabae—chocolate spot, Uromyces fabae—bean rust, Peronospora viciae f.sp. faba—downy mildew), insects (Aphis fabae—black bean aphid, Bruchus rufimanus—the bruchid seed beetle), nematodes (Ditylenchus gigas—stem nematode), and parasitic plants (Orobanche crenata and Orobanche foetida—broomrape) to name just a few of the most prominent. Detailing the fascinating coevolutionary struggles between these diverse organisms and their common host is beyond the scope of this review and interested readers can find further details in one of many recent reviews of biotic stresses on faba bean (Pérez-de-Luque et al., 2010; Sillero et al., 2010; Stoddard et al., 2010). Chief amongst abiotic stresses are heat and drought, which play all the more important a role in yield determination because of the sensitivity of flower fertility to even mild or transient levels of stress. Fertility of developed flowers has recently been shown to be highly sensitive to heat stress, though total yield losses are mitigated by the shift of yield production to higher nodes formed after a stress event has passed (Bishop et al., 2016b). And a further study showed that bee pollination can significantly mitigate damage to fertility caused by heat stress (Bishop et al., 2016a). Cold/frost tolerance is an important trait for winter beans (Sallam et al., 2015), and tolerance to salinity soils is pertinent in semi-arid regions (Tavakkoli et al., 2012). Again, abiotic stress resistance is a wide topic which has been adequately covered in recent reviews (Khan et al., 2010; Link et al., 2010; Patrick and Stoddard, 2010).
Meeting the protein demand of a growing global population represents a challenge not only from the yield perspective but also from the quality perspective. As yield potential continues to increase through the efforts of breeders, it is important for the protein density of the crop to be maintained, if not improved, from typical values of around 30% dry weight. The well-established relative deficiency of faba bean in essential sulfur-containing amino acids, cysteine and methionine, can of course be balanced in food, and feed processing, but amelioration of the balance of essential amino acids would be a valuable breeding target which has not yet been addressed (Multari et al., 2015). Finally, in order for the nutritive value of faba protein to be maximized and nitrogen- and phosphorous-rich wastes in the food chain minimized, serious research is needed to identify, and remove anti-nutritional factors that inhibit the normal digestion of starch and protein in the gut (amylase and protease inhibitors), that cause oxidative stress (e.g., vicine) or that sequester key nutrients (e.g., phytic acid). Much research on faba bean quality has focussed on two key anti-nutritional factors: tannins and vicine/convicine. Vicine and convicine are particularly intriguing as the metabolized derivatives of these pyrimidine glucosides cause a serious and potentially fatal condition known as favism in genetically predisposed humans and are found only in a handful of Vicia spp., the most notable of which is faba bean (Ray and Georges, 2010). Seed coat tannins and vicine-convicine are naturally high in a large majority of current varieties (e.g., Khamassi et al., 2013) and have been shown to lower protein digestibility and energy content in a variety of animal feeding studies. The nutritional quality of faba bean and status of research on anti-nutritional factors have been thoroughly reviewed by Crepon et al. (2010).
Against this backdrop of high yield potential and high utility in increasing the sustainability of nitrogen cycling in agro-ecosystems and security of protein supply in the food chain, it is both timely and appropriate to review how genetic and genomics are already contributing to the accelerated breeding of high yielding, climate-resilient, and nutritious faba beans and how we can expect these technologies to be exploited in the future.
The first major contribution to systematic faba bean transcriptome knowledge was the release of approximately 5000 Expressed Sequence Tags (EST) from developing embryos of a broad bean variety “Windsor” described by Ray and Georges (2010). This study provided a useful snapshot of the functional classification and relative expression level of the more abundant transcripts from the embryo transcriptome in the early to middle stages of its development, even though restricted to one genotype and one tissue. Kaur et al. (2012) and Yang et al. (2012) undertook 454 sequencing of the transcriptome specifically to underpin SSR discovery and this significantly increased the volume of transcriptome data available although in both cases from mixed genotypes. Subsequently, studies began to encompass multiple, separate inbred genotypes (Ocaña et al., 2015; Webb et al., 2016) as well as multiple tissues and genotypes (Ray et al., 2015). The deepest transcriptome coverage yet produced has come from Illumina sequencing of a library of mixed tissues enriched with embryo transfer cells from variety “Fiord” (Arun-Chinnappa and McCurdy, 2015; Zhang et al., 2015). At the time of writing, transcriptomes from nine specifically identified single genotypes and a selection of tissues including whole seedling, root, shoot, leaf, seed coat, and embryo were available (summarized in Table 1), which offers for the first time to the faba bean research community the possibility to conduct crude electronic Northern analyses and to mine genotypic variants from multiple genetic backgrounds.
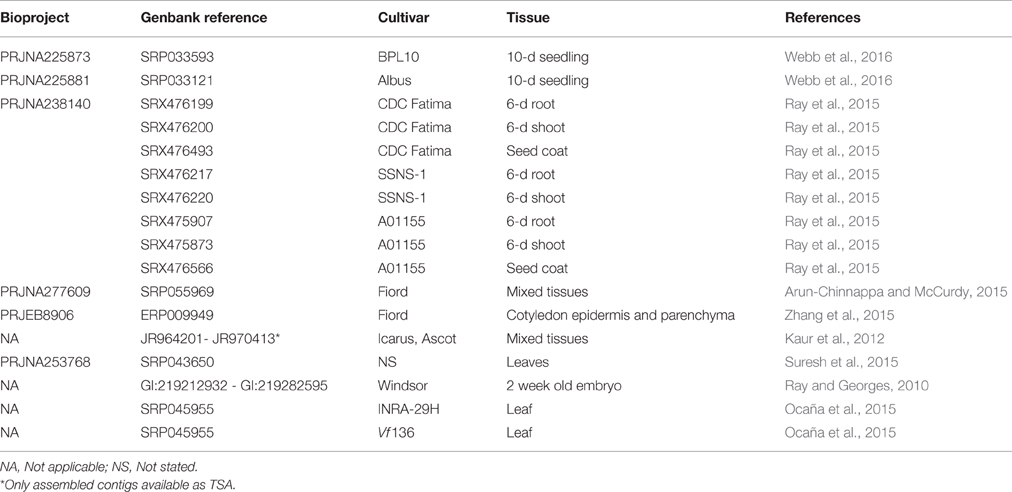
Table 1. Key Vf transcriptome datasets.
The extreme paucity of Vf sequence of any description in public databases, lamented by previous reviewers (e.g., Gnanasambandam et al., 2012), meant that very small numbers of SNPs had been discovered prior to 2014. The first genetic linkage map of faba bean to explicitly target gene-based polymorphisms was reported by Ellwood et al. (2008), who adopted a strategy of cross-species amplification of conserved orthologs in order to identify polymorphic intron-targeted markers, which were implemented at first either as Cleaved Amplified Polymorphic Sequence (CAPS) or Single Nucleotide Primer Extension (SNuPE) assays. Later, many of these polymorphic intron sequences were converted from CAPS/SNuPE to Kompetitive Allele Specific PCR (KASP) format (Cottage et al., 2012), which made the exploitation of this first suite of SNPs more accessible. The advent of RNA-Seq datasets fueled the next wave of SNP development. Kaur et al. (2014b) designed an iSelect assay based on 768 SNPs, of which 551 were placed on a genetic map generated from a RIL population of the cross Icarus × Ascot. More recently, Webb et al. (2016) reported design of individual KASP assays for 845 SNPs mined from alignment of assembled transcriptomes of “Albus” and “BPL10” inbred lines; of these, 653 were successfully mapped. The burgeoning transcriptome datasets described in the previous section permit ever greater numbers of SNPs to be called, for e.g., Ray et al. (2015) reported 5300 unique variants where alternate alleles could be found in one more of the discovery genotypes SSNS-1, A01155, or CDC Fatima and Ocaña et al. (2015) reported 39,060 SNP and 3669 InDel polymorphisms in their analysis of transcriptomes of INRA-29H and Vf 136, though these larger SNP sets remain to be validated as working SNP assays.
The alignment of genotype-specific transcriptome datasets to well annotated references is relatively straightforward and can be accomplished using user-friendly cloud-based tools e.g., BWA-MEM for alignment (Li, 2013) and Varscan for SNP calling (Koboldt et al., 2012) implemented in the Galaxy workflow environment (Afgan et al., 2010). Figure 1 shows a typical alignment of contigs from several distinct Vf genotypes to a gene predicted by synteny to lie within the published VC interval (Khazaei et al., 2015). Over the 1534 bp contig, which covers the full coding region predicted in Medicago together with some 5′ and 3′-UTR sequence, there are 16 varietal Vf SNPs.
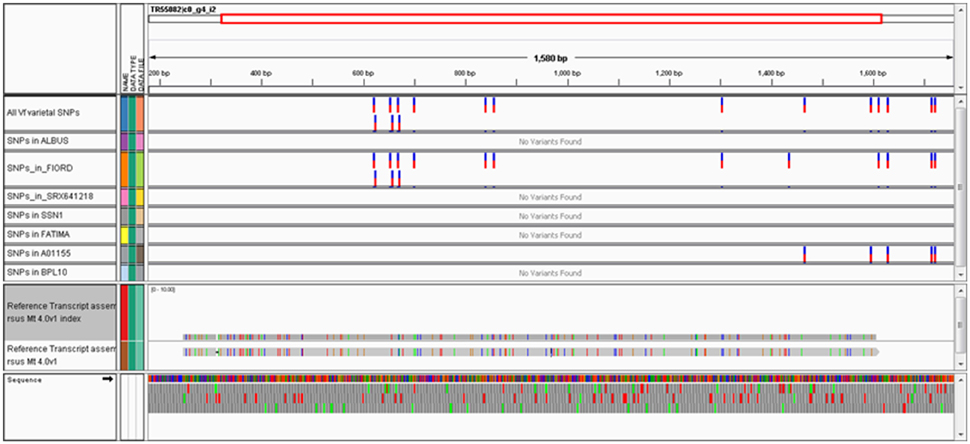
Figure 1. Integrative Genomics Viewer (IGV) snapshot of SNPs in the Vf ortholog of Medtr2g009080 mined from public trancriptome datasets. The 11 horizontal tracks shown in this view from top to bottom are: the meta-transcriptome contig TR55082¦c0_g4_i2 is shown as a schematic, all intravarietal SNP positions; positions where the minor (non-consensus) allele is found in each of the following genotypes: “Albus,” “Fiord,” “SRX641218,” “SSN1,” “Fatima,” “A01155,” and “BPL10,” alignment of the reference Vf contig TR55082¦c0_g4_i2 to the Mt4.0 genome (coding region of Medtr2g009080 in this instance), and in the bottom panel is shown a 3-frame translation of the Vf sequence. In this example, there are a total of 16 intravarietal SNPs distributed across the gene—11 unique to “Fiord,” two unique to “A001155” and three minor alleles in common to “Fiord” and “A01155,” with the remaining genotypes carrying the unchanged common allele identical to the consensus contig sequence.
The other category of molecular marker which has received much attention from the faba bean research community was Expressed Sequence Tag—Simple Sequence Repeats (EST-SSRs). As inherently highly discriminatory, co-dominant markers embedded in genic locations which can be readily associated with orthologous positions in related species, EST-SSRs are an attractive category of marker and offer the advantage that repeats can be mined from a single genetic background with relatively low sequence coverage, so were in the first wave of marker sets to be developed from transcriptome data. Initially modest numbers of EST-SSRs reported by Gong et al. (2011) and Ma et al. (2011) were followed by greater numbers of candidate and validated EST-SSRs (Kaur et al., 2012; El-Rodeny et al., 2014). Although highly portable and informative, EST-SSRs, as with all types of SSR marker, can be difficult to score in a fully automated fashion, and are inherently less amenable than SNPs and INDELs to conversion to high throughput parallel assay formats.
In contrast to EST-SSRs, genomic SSRs anchored in non-coding genomic sequence may be difficult to validate as locus-specific assays due to the prevalence of complex, repetitive DNA sequences outside coding regions, which constrain the design of locus-specific primers. Furthermore, most genomic SSRs which fall outside conserved coding regions cannot be used to make syntenic bridges to better characterized relatives. Nonetheless, genomic SSRs, mined from SSR-enriched genomic DNA libraries, have been successfully developed by Zeid et al. (2009) and Yang et al. (2012). Table 2 summarizes key molecular marker sets which have been developed to date for faba bean.
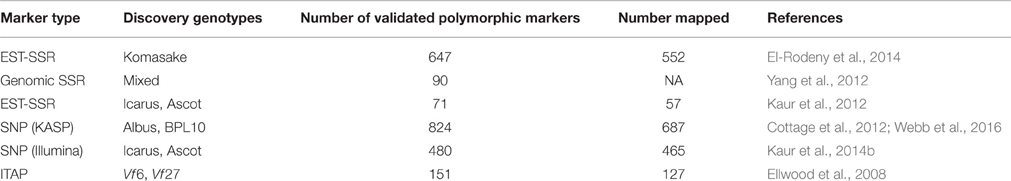
Table 2. Key molecular marker sets.
Much progress has been made in the power and possibilities of genetic analysis in faba bean since the pioneering work of Adela Erith, working at the University of Reading in the 1920s, on inheritance of color, size, and form of seeds and other traits (Erith, 1930). Erith's seminal paper showed Mendelian inheritance of genes controlling readily observable (but nonetheless important) characters including hilum color, seed, and flower color and height. However, many years were to pass before the genes underlying these or any other traits have been identified. Between then and now, Vf genetics has passed through distinct phases which will be described below in chronological order.
The period 1930–1993 was the era of molecular marker-free genetic analysis. During this period and beyond, useful foundations were laid showing simple inheritance patterns for a variety of heritable traits. As mentioned already, (Erith, 1930) found single dominant or semi-dominant genes explaining flower, seed, and hilum color as well as plant height, though there was no well-developed locus nomenclature in this early work. Faba bean received much attention in the 1960s to the early 1990s as a model for cytogenetics due to its uncommonly large haploid genome size of 13 Gb (Soltis et al., 2003) and modest haploid chromosomes number (n = 6), which made for large, readily observable chromosomes. For a time, the study of chromosome breakage response to a range of physical and chemical agents became quite fashionable (e.g., Menke et al., 2000; Sobita and Bhagirath, 2005; Milan and Upadhyay, 2007; Rybaczek et al., 2008) and Vf was one of the early crop species whose chromosomes were found to be amenable to flow sorting (Kovarova et al., 2007). The more direct relevance of cytogenetics to modern molecular genetics was the identification of asynaptic mutants (Sjödin, 1970), on which a series of trisomic stocks were founded. Genetic analyses of crosses involving trisomic parents allowed genetic markers to be assigned to physical chromosomes (Patto et al., 1999). The definitive work on pre-molecular faba bean genetics was the monumental work of Sjödin (1971) to collect, induce, classify and cross mutants and spontaneous variants in a wide variety of characters of interest. Although such comprehensive surveys of genetic variation have not since been carried out, induced and spontaneous mutants affecting a variety of traits have been described in this period. For instance, a gene controlling symbiosis, sym-1, was described by Duc and Picard (1986). Duc et al. (1999) reported a recessive allele of the i1 locus conferring a green cotyledon; this has a similar phenotype as the green cotyledon allele of the Sc locus, mapped by Khazaei et al. (2014) to chromosome IV.
Work on genetics was not confined to visible morphological phenotypes either. Ramsay (1997) showed Mendelian inheritance of a major effect seed dormany gene, denoted doz, detected by scoring the timing of seed germination semi-quantitatively in a set of segregating Recombinant Inbred Lines (RILs). Unfortunately, few of the populations generated and characterized in these early genetic studies have been maintained to the present day, though the trait sources (or at least sources carrying analogous phenotypic states) have for the most part. However, quantitative traits, especially those under oligogenic control, were always going to require a molecular mapping approach so that medium or even small size effects could be placed within a sufficiently dense phenotype-independent marker framework.
A combination of isozymes and RAPD markers formed the basis of the first Vf molecular marker map (Torres et al., 1993), consisting of 66 markers arranged in 11 linkage groups, in which, interestingly, the first indications of synteny between faba bean and other Fabaceae were noted.
From this point on, Vf genetic studies followed a standard pattern. Typically biparental RIL or F2 populations generally consisting of no more than 200 progeny lines were genotyped with non-sequence based markers, chiefly RAPD. The highlights of this phase of faba bean genetics were a series of RAPD-based QTL studies targeting resistance to pathogens and parasites—for instance, to ascochyta (Avila et al., 2004; Diaz-Ruiz et al., 2009), rust (Avila et al., 2003), and Orobanche (Diaz-Ruiz et al., 2010; Gutierrez et al., 2013). This early period of integration of molecular marker technology in genetic mapping studies has been reviewed by Torres et al. (2010) and Gnanasambandam et al. (2012).
In parallel with the acceleration in growth of faba bean sequence and marker datasets, there has been a correspondingly encouraging increase in the density and utility of gene-based genetic maps (Table 3). The first gene-based genetic map of Vf, composed in the main of 127 co-dominant, portable, Intron-Targeted Amplified Polymorphism (ITAP) markers, was that of Ellwood et al. (2008). A linkage map comprising 128 EST-SSR markers was produced by Ma et al. (2013) and a 552- locus map comprising loci generated from 235 faba bean-derived EST-SSRs and 162 markers derived by cross-amplification from red and white clover was reported recently by El-Rodeny et al. (2014). The consensus map of Satovic et al. (2013) was the first where a majority of markers mapped to just six chromosomally assigned linkage groups, though a minority of these were gene-based, transportable, co-dominant markers. More recently, 551 SNPs and 71 SSRs were combined in Kaur et al. (2014b), though not all of the 12 linkage groups reported could be definitively assigned to one of the six physical chromosomes. The densest SNP coverage available in a fully physically anchored consensus linkage map to date is that reported by Webb et al. (2016). This combined 34 SNP markers discovered in Vf 6/Vf 27 backgrounds by Ellwood et al. (2008) and converted to KASP format by Cottage et al. (2012) with 653 new “Albus” × “BPL10” SNPs into a single 687-locus consensus map with all markers mapping to just six linkage groups each of which could be assigned to a physical chromosome.
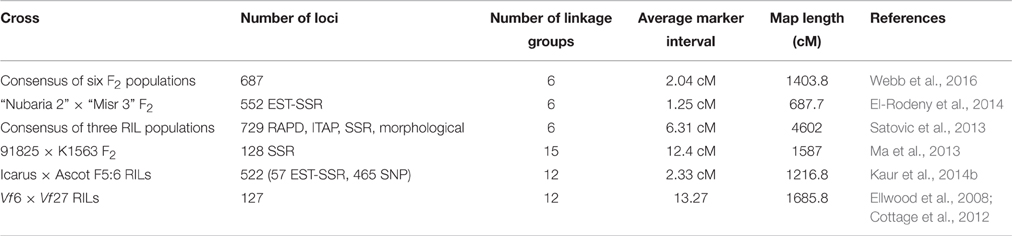
Table 3. Key genetic maps.
The great benefit of the progress in mapping ever greater numbers of sequence-based markers is that the by now well-established conservation of gene order amongst related legume genomes could be used to anchor genetic maps from unsequenced legumes (in this instance faba bean) to the Medicago truncatula (Mt) genome. The first sequence-based genetic map of faba bean which allowed the global pattern of Vf -Mt synteny to be observed was that of Ellwood et al. (2008). Although the number of markers (127) was modest, a clear picture of extensive macrosynteny emerged. El-Rodeny et al. (2014) and Kaur et al. (2014b) elaborated this picture with a greater marker density, but the clearest and most complete picture of the extent of macrosynteny between Vf and Mt comes from the Webb et al. (2016) study. All 653 newly discovered and mapped SNP markers in this latter study were selected following highly conservative filtering for single copy Vf sequences with a clear best reciprocal BLAST hit relationship with a single copy gene in M. truncatula. Reflecting these stringent marker design criteria, the name of the each new (Webb et al., 2016) Vf marker carries explicit reference to its presumed Mt ortholog. Combined with the fact that each linkage group in the Webb et al. (2016) consensus map corresponds to a full physical Vf chromosomes meant that for the first time all six Vf chromosomes could be aligned to the Mt sequence without spurious interruptions to macrosynteny pattern caused by lack of marker coverage.
The hallmark of “pre-synteny” genetic studies in all species, Vf being no exception, was that the reporting of a given gene or QTL in proximity of a certain molecular marker was quite frequently the end of the story as there was no method to target markers to a region of interest without screening vast numbers of anonymous markers such as AFLPs or RAPDs.
The year 2012 marked a new departure for faba bean with sequence-based and synteny-anchored marker maps being applied to flowering time QTL (Cruz-Izquierdo et al., 2012). Likewise, detection of ascochyta resistance QTL by Kaur et al. (2014b) permitted localization of QTL in regions with clear colinearity to fully sequenced model genomes. Khazaei et al. (2014) studying stomatal traits and Khazaei et al. (2015) mapping vicine-convicine content were able to do likewise. In theory now, a given QTL would be associated with an interval whose gene content and even order could be predicted by exploiting synteny with Medicago and marker development efficiently targeted to this predicted gene content.
As outlined in the previous sections, much of this progress has stemmed from the exploitation of sequencing and cost-effective genotyping technologies to achieve reasonably dense coverage of all six Vf chromosomes with sequence-based molecular markers that allow the Vf gene-based genetic map to be confidently aligned in large part with fully sequenced reference genomes such as Medicago (Young et al., 2011) or soybean (Schmutz et al., 2010).
The exploitation of macrosynteny between crop and model genomes has a number of benefits. Firstly, once a trait has been mapped to a genetic interval which aligns well to a segment of a model sequenced genome, knowledge of gene function in the model species can be translated back to target crop species. In an instance illustrated in Figure 2, ZT1 (controlling flower pigmentation as well as seed coat tannins) was mapped to the Vf _Mt3g092810_001–Vf _Mt3g094760_001 interval, with clear synteny to a portion of Mt3. Perusal of the annotation information for the syntenic interval in Medicago revealed a logical biological candidate in the form of the Transparent Testa Glabra 1 (TTG1) WD40 transcription factor (Medtr3g092840), which had previously been shown in Mt to determine flower color, and in the follow-up of this hypothesis, a deletion in the recessive allele sequence was found to plausibly explain the zt (unpigmented) phenotype (Webb et al., 2016). This co-called translational genomics approach leverages prior investment in model species biology and fast-tracks causative allele identification. Secondly, quite independently of translation of biological information, molecular marker targeting to specific regions of interest is possible using the syntenic framework. This is relevant in situations where the target interval is large or initial candidate functional information is absent and is illustrated in Figure 2 by the case of SNP mining targeting the VC locus. The published interval in which this gene maps is collinear with part of Mt2 (Khazaei et al., 2015) and SNPs discovered in sequences orthologous to Medicago genes in the syntenic interval can be used selectively and cost-effectively in further high-resolution mapping of the locus. Thirdly, this syntenic framework can in principle be used to reverse map genes in Vf. Here, we use the example of the Vf TERMINAL FLOWER 1 (VfTFL1) gene. Avila et al. (2007) examined the translational hypothesis that an ortholog of the TFL1 gene controls determinacy of flowering in Vf as it does in Arabidopsis, soybean and numerous other legume and non-legume species by showing correlation of the determinate type with a diagnostic non-synonymous substitution in a conserved residue of the coding region across a diverse panel of determinate and indeterminate types. They did not, however, genetically map VfTFL1. Once again, clear macro-colinearity between the Vf region corresponding to the Mt region harboring MtTFL1 suggests a working hypothesis (which remains to be proven) that the published VfTFL1 sequence should map to the long arm of Vf chromosome 1.
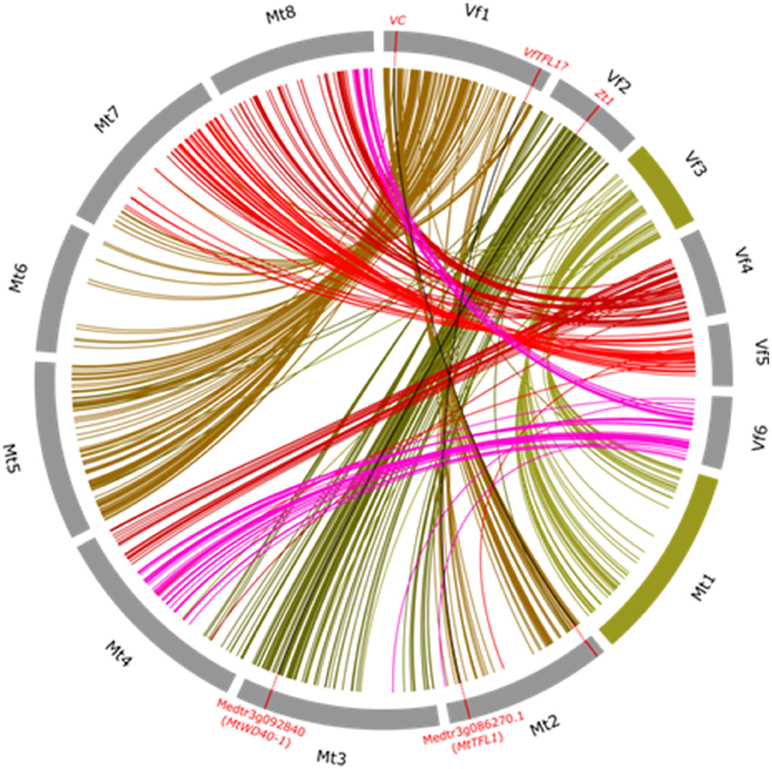
Figure 2. Wheel representation of synteny between pseudomolecule sequence of Medicago truncatula chromosomes Mt1-Mt8 and the consensus Vf linkage map (Vf1–Vf6). Syntenic relationships were as described in Webb et al. (2016) and re-drawn for this figure using “Circos” software (Krzywinski et al., 2009). Link lines to Medicago orthologs for markers on each Vf chromosome are shown in a different color. The virtually uninterrupted collinearity between the whole of Vf 3 and the whole of Mt1 is highlighted by representing these orthologous chromosomes in green. To illustrate how useful this syntenic framework is for translational genomics as well as for drawing together knowledge of the genome, three exemplar loci where there is a proven or presumed orthologous relationship are shown as red ticks on the orthologous chromosomes and linked by black lines. These are: the mapped ZT1 locus and its Medicago ortholog MtWD40-1, the mapped genetic interval corresponding to the mapped VC (vicine-convicine) locus and its corresponding Medicago syntenic interval on Mt2, and the actual position of the Medicago Terminal Flower Locus 1 (TFL1) with a link drawn to the region of Vf 1 where it is presumed the Vf ortholog (VfTFL1) is likely to be located.
Another arena in which genomics is playing a crucial role is in the description and rational exploitation of diverse, but often poorly characterized, genetic resources. Despite the lack of a known wild progenitor species, the domesticated Vf genepool, as alluded to in the introduction, is extremely diverse. This diversity is manifest in the plethora of morphological forms and adaptations to diverse agroecological settings. However, full exploitation of this diversity for faba bean improvement necessitates efficient methods for quantifying and mapping genomic diversity. In parallel with the development of SSAP, AFLP, SSR, and SNP marker types, a succession of studies have used increasingly powerful molecular marker sets to quantify and map genomic diversity.
One of the first surveys of genomic diversity in diverse Vf germplasm was carried out by Sanz et al. (2007) using retrotransposon-based Sequence-Specific Amplified Polymorphism (SSAP) markers. This study found relatively little fine genetic structure and the first molecular evidence that “major,” “minor,” and “equine” lines do not form distinct clades, but rather are completely dispersed across the Vf phylogeny. Due to the low numbers of Vf lines (n = 20), it was not possible to draw any conclusions on geographic partitioning of diversity, though it was noted that samples from the better-represented countries in the study were well dispersed across the phylogenetic tree. These themes were recapitulated by Zeid et al. (2003) using AFLP markers to genotype 79 inbred lines from Europe, North Africa and Asia. Again, “major” and “minor” botanical types were clearly shown not to be genetically distinct, though here for the first time was a suggestion that the (n = 8) lines of Asian origin formed a genetically differentiated group. The AFLP-based study of Zong et al. (2009) took this further with a study of large numbers of Chinese landraces (n = 204) from the Chinese Academy of Agricultural Sciences (CAAS) germplasm collection, mainly winter types. They found the mainland Chinese winter germplasm to be completely distinct from other Asian, African and European diverse lines included for comparison, in line with the long history of cultivation of winter-type faba bean in relatively isolated mountainous regions of China. Kwon et al. (2010), using Targeted Region Amplified Polymorphism (TRAP) markers, also observed Chinese landraces drawn from the United States Department of Agriculture (USDA) germplasm collection (n = 107) to cluster completely separately from 30 comparator lines from Asia and Europe. However, the largest study of faba bean genomic diversity to date has been reported by Wang et al. (2012a), whose large sample (n = 802) confirmed not only the genetic distinctness of Chinese germplasm from African, European, and other Asian germplasm, but for the first time, convincingly showed differentiation amongst Chinese provinces and between winter and spring ecotypes. SNP genotyping platforms have been more recently used on small samples of diversity (Cottage et al., 2012; Kaur et al., 2014a) and the prospect of wider deployment of SNP markers on larger panels of diverse material promises the possible foundations for future Vf genome-wide association studies.
The potential of mutagenesis as a tool for breeding was amply demonstrated by Sjödin (1971), reviewing not only observations made in the extensive mutagenesis programme run by Swedish breeders Svalov-Weibull in the “Primus” genetic background, but also a series of spontaneous mutants reported by a host of previous researchers. The only other mutageneis programme reported in the literature was 23 years later, when the isolation of five nodulation mutants in a screen of 20,000 M2 EMS-mutagenized lines in the cv. “Ascott” background was reported by Duc (1995).
Interest in mutagenesis has undergone something of a resurgence in recent years. For example, the control of the devastating parasitic weed, Orobanche crenata, referred to in an earlier section, could be effectively controlled, together with a range of other troublesome broad-leaved weeds, if faba bean varieties with target-site mutations that render them insensitive to particular actives were developed (Gressel, 2009). Examples are the Ser653 and Ala205 mutations in the AcetoLactate Synthase target of imidazolinone and amidosulfuron families of herbicide actives, which do not occur in nature, but have been documented to occur at low frequency under strong selection pressure in the field, and there are examples in numerous crop species of targeted isolation of induced mutants (reviewed in Tan et al., 2005). At the time of writing, a number of imazapyr resistance mutations have been identified by Mao et al. (2014) and are undergoing further characterization. As a further example of the ongoing mutagenesis programmes, Figure 3 shows a selection of phenotypes observed in ongoing M2 X-ray and EMS populations grown during 2015 at the University of Reading.

Figure 3. Mutant phenotypes observed in X-ray and EMS M2 lines grown at the University of Reading in 2015. (A) disease lesion mimic, (B) determinate flowering, (B) curled wing petal, (C) disease lesion mimic, (D) reduced flower pigmentation.
Whilst the primary driver for contemporary mutagenesis programmes remains the identification of novel induced variants for direct incorporation in active breeding programmes, a broader motivation can be ascribed to some of the recent activity. Until efficient genetic transformation systems have been adopted and made available as a service, reverse genetics in the form of TILLing (Till et al., 2003) may be one of the most amenable techniques for validation of candidate genes identified via synteny-based approaches, especially if combined with powerful re-sequencing techniques to identify the causative mutations in a single step (Wang et al., 2012b).
Genetic modification represents both a research tool, permitting testing of hypotheses on gene function by over-, mis-expression or knockdown/knockout studies and an outlet for genetic research in generation of targeted phenotypic modifications based on knowledge of gene function. Stable germline transformation of Vf using in vitro regeneration of Agrobacterium-infiltrated (non-meristematic) internode stem segments was first reported by Böttinger et al. (2001). Adopting a somewhat different strategy, Hanafy et al. (2005) infiltrated excised (meristematic) embryo axes with Agrobacterium and successfully recovered stable transgenic lines. Both methods, however, reported low primary transformation efficiencies and relied on micro-grafting of putative transgenic shoot material onto non-transgenic roots, a slow and highly manual process. Hanafy et al. (2013) later reported abiotic stress resistance phenotypes of Vf transgenic lines overexpressing potato PR10a using their previous methods. This remains to our knowledge the sole successful demonstration to date of the feasibility of a biotechnological approach to Vf improvement. In the absence of a robust and efficient transformation method, some attention has been devoted to the task of decreasing generation time using tissue-culture based embryo rescue, with some success (Mobini et al., 2015). However, the search for an efficient transformation method has been recently taken up by other groups (e.g., Abdelwahd et al., 2014).
The prospects afforded by new insights into the phenotypic effects of allelic variation and the more refined biotechnological possibilities afforded by rapidly maturing genome editing technologies (Gaj et al., 2013) could potentially stimulate renewed interest in genetic transformation. An example of a game-changing product which could readily be generated using even a medium efficiency transformation system would be herbicide resistance obtained by directed mutagenesis of endogenous herbicide target genes e.g., introduction of heterologous glyphosate resistance of bacterial origin.
Vicia faba genetics and genomics is now in a much healthier state than it was just a few years ago. We can take heart from the accelerating progress in gene identification and in production of outputs relevant to contemporary methods of breeding. We have seen the transformative effects of transcriptome re-sequencing in opening the doors to high density gene-based marker discovery and mapping. In the near future, we expect that a high throughput SNP chip incorporating many 10's of thousands of genome-wide SNPs discovered from a wide variety of genetic backgrounds will be produced and made available for community use, as well as giving rise to an ultra-high density SNP map in which perhaps as much as half of all Vf genes are genetically mapped. In parallel, we should see progress in the application of genotype-by-sequencing methods, both for the unbiased assessment of genetic relationships, breeding applications such as genomic selection and in trait mapping approaches based on bulked segregant analysis. Faba bean's modest but well-linked community of researchers would do well however, to set its sights on a series of even more ambitious targets to enable the community as a whole to elevate its work onto a higher plane of achievement and impact. For example, given the many highly cost effective sequencing and assembly technologies now available, a comprehensive genomic scaffold and haplotype map is surely now within reach. The history of modern agricultural genomics shows the transformative effects of a well-annotated reference genome. Communities of crop researchers who have organized themselves and published strategic roadmaps requiring centralized investment in a professional genome assembly and annotation have captured significant R&D investment and transformed the profile and fate of their communities (rice, wheat, soybean, Phaseolus, cowpea). Similarly, on functional genomics platforms: whilst there are no doubt training benefits to having numerous small mutagenesis programmes dotted around the globe, it could be argued that the global community needs one well-funded programme scoped to guarantee saturation mutagenesis, to catalog the mutations obtained in a public sequence database and to distribute mutant seed to research groups freely on request, an unlikely outcome from nationally funded programmes. Likewise, transformation and genome editing technologies could advantageously be developed and provided as an efficient service from a centralized laboratory.
DO drafted the manuscript and provided original Figures 1, 3. DA provided information for the section on Genomics and the original Figure 2. DO and DA edited, reviewed and approved the final version of the manuscript.
Work on genetic resources of faba bean in DOS laboratory is part-funded by the Pulse Crop Genetic Improvement Network (CH0103) project funded by the UK Department for Environment, Food and Rural Affairs (Defra).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. ^FAOSTAT (2013). Available online at: http://faostat3.fao.org/home/E (accessed on 10 December 2015).
Abdelwahd, R., Udupa, S. M., Gaboun, F., Diria, G., Mentag, R., Ibriz, M., et al. (2014). Agrobacterium-mediated transformation of cotyledonary node of Vicia faba L. Rom. Agric. Res. [Epub ahead of print].
Afgan, E., Baker, D., Coraor, N., Chapman, B., Nekrutenko, A., and Taylor, J. (2010). Galaxy CloudMan: delivering cloud compute clusters. BMC Bioinformatics 11(Suppl. 12):S4. doi: 10.1186/1471-2105-11-S12-S4
Arun-Chinnappa, K. S., and McCurdy, D. W. (2015). De novo assembly of a genome-wide transcriptome map of Vicia faba (L.) for transfer cell research. Front. Plant Sci. 6:217. doi: 10.3389/fpls.2015.00217
Avila, C. M., Atienza, S. G., Moreno, M. T., and Torres, A. M. (2007). Development of a new diagnostic marker for growth habit selection in faba bean (Vicia faba L.) breeding. Theor. Appl. Genet. 115, 1075–1082. doi: 10.1007/s00122-007-0633-y
Avila, C. M., Satovic, Z., Sillero, J. C., Rubiales, D., Moreno, M. T., and Torres, A. M. (2004). Isolate and organ-specific QTLs for ascochyta blight resistance in faba bean (Vicia faba L). Theor. Appl. Genet. 108, 1071–1078. doi: 10.1007/s00122-003-1514-7
Avila, C. M., Sillero, J. C., Rubiales, D., Moreno, M. T., and Torres, A. M. (2003). Identification of RAPD markers linked to the Uvf-1 gene conferring hypersensitive resistance against rust (Uromyces viciae-fabae) in Vicia faba L. Theor. Appl. Genet. 107, 353–358. doi: 10.1007/s00122-003-1254-8
Bishop, J., Jones, H. E., Lukac, M., and Potts, S. G. (2016a). Insect pollination reduces yield loss following heat stress in faba bean (Vicia faba L.). Agric. Ecosyst. Environ. 220, 89–96. doi: 10.1016/j.agee.2015.12.007
Bishop, J., Potts, S. G., and Jones, H. E. (2016b). Susceptibility of faba bean (Vicia faba L.) to heat stress during floral development and anthesis. J. Agron. Crop Sci. doi: 10.1111/jac.12172. [Epub ahead of print].
Böttinger, P., Steinmetz, A., Schieder, O., and Pickardt, T. (2001). Agrobacterium-mediated transformation of Vicia faba. Mol. Breed. 8, 243–254. doi: 10.1023/A:1013711210433
Cernay, C., Ben-Ari, T., Pelzer, E., Meynard, J.-M., and Makowski, D. (2015). Estimating variability in grain legume yields across Europe and the Americas. Sci. Rep. 5:11171. doi: 10.1038/srep11171
Cottage, A., Gostkiewicz, K., Thomas, J. E., Borrows, R., Torres, A. M., and O'Sullivan, D. M. (2012). Heterozygosity and diversity analysis using mapped single nucelotide polymorphisms in a faba bean inbreeding programme. Mol. Breed. 30, 1799–1809. doi: 10.1007/s11032-012-9745-4
Crepon, K., Marget, P., Peyronnet, C., Carrouee, B., Arese, P., and Duc, G. (2010). Nutritional value of faba bean (Vicia faba L.) seeds for feed and food. Field Crop Res. 115, 329–339. doi: 10.1016/j.fcr.2009.09.016
Cruz-Izquierdo, S., Avila, C. M., Satovic, Z., Palomino, C., Gutierrez, N., Ellwood, S. R., et al. (2012). Comparative genomics to bridge Vicia faba with model and closely-related legume species: stability of QTLs for flowering and yield-related traits. Theor. Appl. Genet. 125, 1767–1782. doi: 10.1007/s00122-012-1952-1
Denton, M. D., Pearce, D. J., and Peoples, M. B. (2013). Nitrogen contributions from faba bean (Vicia faba L.) reliant on soil rhizobia or inoculation. Plant Soil 365, 363–374. doi: 10.1007/s11104-012-1393-2
Diaz-Ruiz, R., Satovic, Z., Avila, C. M., Alfaro, C. M., Gutierrez, M. V., Torres, A. M., et al. (2009). Confirmation of QTLs controlling Ascochyta fabae resistance in different generations of faba bean (Vicia faba L.). Crop Pasture Sci. 60, 353–361. doi: 10.1071/CP08190
Diaz-Ruiz, R., Torres, A. M., Satovic, Z., Gutierrez, M. V., Cubero, J. I., and Roman, B. (2010). Validation of QTLs for Orobanche crenata resistance in faba bean (Vicia faba L.) across environments and generations. Theor. Appl. Genet. 120, 909–919. doi: 10.1007/s00122-009-1220-1
Duc, G. (1995). Mutagenesis of faba bean (Vicia-Faba L) and the identification of 5 different genes-controlling no nodulation, ineffective nodulation or supernodulation. Euphytica 83, 147–152. doi: 10.1007/BF01678042
Duc, G., Bao, S. Y., Baum, M., Redden, B., Sadiki, M., Suso, M. J., et al. (2010). Diversity maintenance and use of Vicia faba L. genetic resources. Field Crop Res. 115, 270–278. doi: 10.1016/j.fcr.2008.10.003
Duc, G., Moussy, F., Zong, X., and Ding, G. (1999). Single gene mutation for green cotyledons as a marker for the embryonic genotype in faba bean, Vicia faba. Plant Breed. 118, 577–578. doi: 10.1046/j.1439-0523.1999.00422.x
Duc, G., and Picard, J. (1986). Note on the presence of the Sym-1 gene in Vicia-faba hampering its symbiosis with Rhizobium-leguminosarum. Euphytica 35, 61–64. doi: 10.1007/BF00028541
Ellwood, S. R., Phan, H. T. T., Jordan, M., Hane, J., Torres, A. M., Avila, C. M., et al. (2008). Construction of a comparative genetic map in faba bean (Vicia faba L.); conservation of genome structure with Lens culinaris. BMC Genomics 9:380. doi: 10.1186/1471-2164-9-380
El-Rodeny, W., Kimura, M., Hirakawa, H., Sabah, A., Shirasawa, K., Sato, S., et al. (2014). Development of EST-SSR markers and construction of a linkage map in faba bean (Vicia faba). Breed. Sci. 64, 252–263. doi: 10.1270/jsbbs.64.252
Erith, A. G. (1930). The inheritance of colour, size and form of seeds, and of flower colour in Vicia Faba L. Genetica 12, 477–510. doi: 10.1007/BF01486760
Gaj, T., Gersbach, C. A., and Barbas Iii, C. F. (2013). ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 31, 397–405. doi: 10.1016/j.tibtech.2013.04.004
Gnanasambandam, A., Paull, J., Torres, A., Kaur, S., Leonforte, T., Li, H., et al. (2012). Impact of molecular technologies on faba bean (Vicia faba L.) breeding strategies. Agronomy 2, 132–166. doi: 10.3390/agronomy2030132
Gong, Y. M., Xu, S. C., Mao, W. H., Li, Z. Y., Hu, Q. Z., Zhang, G. W., et al. (2011). Genetic diversity analysis of faba bean (Vicia faba L.) based on EST-SSR MARKERS. Agric. Sci. China 10, 838–844. doi: 10.1016/S1671-2927(11)60069-2
Gressel, J. (2009). Crops with target-site herbicide resistance for Orobanche and Striga control. Pest Manag. Sci. 65, 560–565. doi: 10.1002/ps.1738
Gutierrez, N., Palomino, C., Satovic, Z., Ruiz-Rodriguez, M. D., Vitale, S., Gutierrez, M. V., et al. (2013). QTLs for Orobanche spp. resistance in faba bean: identification and validation across different environments. Mol. Breed. 32, 909–922. doi: 10.1007/s11032-013-9920-2
Hanafy, M., Pickardt, T., Kiesecker, H., and Jacobsen, H-J. (2005). Agrobacterium-mediated transformation of faba bean (Vicia faba L.) using embryo axes. Euphytica 142, 227–236. doi: 10.1007/s10681-005-1690-4
Hanafy, M. S., El-Banna, A., Schumacher, H. M., Jacobsen, H. J., and Hassan, F. S. (2013). Enhanced tolerance to drought and salt stresses in transgenic faba bean (Vicia faba L.) plants by heterologous expression of the PR10a gene from potato. Plant Cell Rep. 32, 663–674. doi: 10.1007/s00299-013-1401-x
Kaur, S., Cogan, N. O. I., Forster, J. W., and Paull, J. G. (2014a). Assessment of genetic diversity in Faba bean based on single nucleotide polymorphism. Diversity 6, 88–101. doi: 10.3390/d6010088
Kaur, S., Kimber, R. B. E., Cogan, N. O. I., Materne, M., Forster, J. W., and Paull, J. G. (2014b). SNP discovery and high-density genetic mapping in faba bean (Vicia faba L.) permits identification of QTLs for ascochyta blight resistance. Plant Sci. 217, 47–55. doi: 10.1016/j.plantsci.2013.11.014
Kaur, S., Pembleton, L. W., Cogan, N. O. I., Savin, K. W., Leonforte, T., Paull, J., et al. (2012). Transcriptome sequencing of field pea and faba bean for discovery and validation of SSR genetic markers. BMC Genomics 13:104. doi: 10.1186/1471-2164-13-104
Khamassi, K., Ben Jeddi, F., Hobbs, D., Irigoyen, J., Stoddard, F., O'Sullivan, D. M., et al. (2013). A baseline study of vicine-convicine levels in faba bean (Vicia faba L.) germplasm. Plant Genet. Resour. 11, 250–257. doi: 10.1017/S1479262113000105
Khan, H. R., Paull, J. G., Siddique, K. H. M., and Stoddard, F. L. (2010). Faba bean breeding for drought-affected environments: a physiological and agronomic perspective. Field Crop Res. 115, 279–286. doi: 10.1016/j.fcr.2009.09.003
Khazaei, H., O'Sullivan, D. M., Jones, H., Pitts, N., Sillanpaa, M. J., Parssinen, P., et al. (2015). Flanking SNP markers for vicine-convicine concentration in faba bean (Vicia faba L.). Mol. Breed. 35:38. doi: 10.1007/s11032-015-0214-8
Khazaei, H., O'Sullivan, D. M., Sillanpää, M. J., and Stoddard, F. L. (2014). Use of synteny to identify candidate genes underlying QTL controlling stomatal traits in faba bean (Vicia faba L.). Theor. Appl. Genet. 127, 2371–2385. doi: 10.1007/s00122-014-2383-y
Koboldt, D. C., Zhang, Q., Larson, D. E., Shen, D., McLellan, M. D., Lin, L., et al. (2012). VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576. doi: 10.1101/gr.129684.111
Kovarova, P., Navratilova, A., Macas, J., and Dolezel, J. (2007). Chromosome analysis and sorting in Vicia sativa using flow cytometry. Biol. Plant. 51, 43–48. doi: 10.1007/s10535-007-0009-9
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Kwon, S. J., Hu, J. G., and Coyne, C. J. (2010). Genetic diversity and relationship among faba bean (Vicia faba L.) germplasm entries as revealed by TRAP markers. Plant Genet. Resour. 8, 204–213. doi: 10.1017/S1479262110000201
Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:13033997.
Link, W., Balko, C., and Stoddard, F. L. (2010). Winter hardiness in faba bean: physiology and breeding. Field Crop Res. 115, 287–296. doi: 10.1016/j.fcr.2008.08.004
Ma, Y., Bao, S. Y., Yang, T., Hu, J. G., Guan, J. P., He, Y. H., et al. (2013). Genetic linkage map of Chinese native variety faba bean (Vicia faba L.) based on simple sequence repeat markers. Plant Breed. 132, 397–400. doi: 10.1111/pbr.12074
Ma, Y., Yang, T., Guan, J., Wang, S., Wang, H., Sun, X., et al. (2011). Development and characterization of 21 Est-derived microsatellite markers in Vicia Faba (Fava Bean). Am. J. Bot. 98, E22–E24. doi: 10.3732/ajb.1000407
Mao, D., Paull, J., Oldach, K. H., Preston, C., Yang, S. Y., Davies, P., et al. (2014). “The development of multiple herbicide tolerances in faba bean (Vicia faba L.) through induced mutation,” in 15th Autralasian Plant Breeding Conference (Melbourne, VIC).
Menke, M., Meister, A., and Schubert, I. (2000). N-methyl-N-nitrosourea-induced DNA damage detected by the comet assay in Vicia faba nuclei during all interphase stages is not restricted to chromatid aberration hot spots. Mutagenesis 15, 503–506. doi: 10.1093/mutage/15.6.503
Milan, P. R., and Upadhyay, S. (2007). Impact of food additives on mitotic chromosomes of Vicia faba L. Caryologia 60, 309–314. doi: 10.1080/00087114.2007.10797952
Mobini, S. H., Lulsdorf, M., Warkentin, T. D., and Vandenberg, A. (2015). Plant growth regulators improve in vitro flowering and rapid generation advancement in lentil and faba bean. In Vitro Cell. Dev. Biol. Plant 51, 71–79. doi: 10.1007/s11627-014-9647-8
Multari, S., Stewart, D., and Russell, W. R. (2015). Potential of fava bean as future protein supply to partially replace meat intake in the human diet. Compr. Rev. Food Sci. Food Saf. 14, 511–522. doi: 10.1111/1541-4337.12146
Nayak, G. K., Roberts, S. P. M., Garratt, M., Breeze, T. D., Tscheulin, T., Harrison-Cripps, J., et al. (2015). Interactive effect of floral abundance and semi-natural habitats on pollinators in field beans (Vicia faba). Agric. Ecosyst. Environ 199, 58–66. doi: 10.1016/j.agee.2014.08.016
Ocaña, S., Seoane, P., Bautista, R., Palomino, C., Claros, G. M., Torres, A. M., et al. (2015). Large-scale transcriptome analysis in faba bean (Vicia faba L.) under Ascochyta fabae infection. PLoS ONE 10:e0135143. doi: 10.1371/journal.pone.0135143.
Patrick, J. W., and Stoddard, F. L. (2010). Physiology of flowering and grain filling in faba bean. Field Crop Res. 115, 234–242. doi: 10.1016/j.fcr.2009.06.005
Patto, M. C. V., Torres, A. M., Koblizkova, A., Macas, J., and Cubero, J. I. (1999). Development of a genetic composite map of Vicia faba using F-2 populations derived from trisomic plants. Theor. Appl. Genet. 98, 736–743. doi: 10.1007/s001220051129
Pérez-de-Luque, A., Eizenberg, H., Grenz, J. H., Sillero, J. C., Ávila, C., Sauerborn, J., et al. (2010). Broomrape management in faba bean. Field Crop Res. 115, 319–328. doi: 10.1016/j.fcr.2009.02.013
Preissel, S., Reckling, M., Schläfke, N., and Zander, P. (2015). Magnitude and farm-economic value of grain legume pre-crop benefits in Europe: a review. Field Crop Res. 175, 64–79. doi: 10.1016/j.fcr.2015.01.012
Ramsay, G. (1997). Inheritance and linkage of a gene for testa-imposed seed dormancy in faba bean (Vicia faba L.). Plant Breed. 116, 287–289. doi: 10.1111/j.1439-0523.1997.tb00998.x
Ray, H., Bock, C., and Georges, F. (2015). Faba Bean: Transcriptome Analysis from etiolated seedling and developing seed coat of key cultivars for synthesis of proanthocyanidins, phytate, raffinose family oligosaccharides, vicine, and convicine. Plant Genome 8:1. doi: 10.3835/plantgenome2014.07.0028
Ray, H., and Georges, F. (2010). A genomic approach to nutritional, pharmacological and genetic issues of faba bean (Vicia faba): prospects for genetic modifications. GM Crops 1, 99–106. doi: 10.4161/gmcr.1.2.11891
Rybaczek, D., Zabka, A., Pastucha, A., and Maszewski, J. (2008). Various chemical agents can induce premature chromosome condensation in Vicia faba. Acta Physiol. Plant 30, 663–672. doi: 10.1007/s11738-008-0163-5
Sallam, A., Martsch, R., and Moursi, Y. (2015). Genetic variation in morpho-physiological traits associated with frost tolerance in faba bean (Vicia faba L.). Euphytica 205, 395–408. doi: 10.1007/s10681-015-1395-2
Sanz, A. M., Gonzalez, S. G., Syed, N. H., Suso, M. J., Saldaña, C. C., and Flavell, A. J. (2007). Genetic diversity analysis in Vicia species using retrotransposon-based SSAP markers. Mol. Genet. Genomics 278, 433–441. doi: 10.1007/s00438-007-0261-x
Satovic, Z., Avila, C. M., Cruz-Izquierdo, S., Díaz-Ruíz, R., García-Ruíz, G. M., Palomino, C., et al. (2013). A reference consensus genetic map for molecular markers and economically important traits in faba bean (Vicia faba L.). BMC Genomics 14:932. doi: 10.1186/1471-2164-14-932
Schmutz, J., Cannon, S. B., Schlueter, J., Ma, J., Mitros, T., Nelson, W., et al. (2010). Genome sequence of the palaeopolyploid soybean. Nature 463, 178–183. doi: 10.1038/nature08670
Sillero, J. C., Villegas-Fernández, A. M., Thomas, J., Rojas-Molina, M. M., Emeran, A. A., Fernández-Aparicio, M., et al. (2010). Faba bean breeding for disease resistance. Field Crop Res. 115, 297–307. doi: 10.1016/j.fcr.2009.09.012
Sjödin, J. A. N. (1970). Induced asynaptic mutants in Vicia faba L. Hereditas 66, 215–232. doi: 10.1111/j.1601-5223.1970.tb02347.x
Sjödin, J. A. N. (1971). Induced morphological variation in Vicia faba L. Hereditas 67, 155–179. doi: 10.1111/j.1601-5223.1971.tb02371.x
Sobita, K., and Bhagirath, T. (2005). Effects of some medicinal plant extracts on Vicia faba root tip chromosomes. Caryologia 58, 255–261. doi: 10.1080/00087114.2005.10589460
Soltis, D. E., Soltis, P. S., Bennett, M. D., and Leitch, I. J. (2003). Evolution of genome size in the angiosperms. Am. J. Bot. 90, 1596–1603. doi: 10.3732/ajb.90.11.1596
Stoddard, F. L., Nicholas, A. H., Rubiales, D., Thomas, J., and Villegas-Fernández, A. M. (2010). Integrated pest management in faba bean. Field Crop Res. 115, 308–318. doi: 10.1016/j.fcr.2009.07.002
Suresh, S., Kim, T.-S., Raveendar, S., Cho, J.-H., Yi, J.-Y., Lee, M. C., et al. (2015). Transcriptome characterization and large-scale identification of SSR/SNP markers in symbiotic nitrogen fixation crop faba bean (Vicia faba L.). Turk. J. Agric. For. 39, 459–469. doi: 10.3906/tar-1409-3
Tan, S., Evans, R. R., Dahmer, M. L., Singh, B. K., and Shaner, D. L. (2005). Imidazolinone-tolerant crops: history, current status and future. Pest Manag. Sci. 61, 246–257. doi: 10.1002/ps.993
Tanno, K., and Willcox, G. (2006). The origins of cultivation of Cicer arietinum L. and Vicia faba L.: early finds from Tell el-Kerkh, north-west Syria, late 10th millennium BP. Veg. Hist. Archaeobot. 15, 197–204. doi: 10.1007/s00334-005-0027-5
Tavakkoli, E., Paull, J., Rengasamy, P., and McDonald, G. K. (2012). Comparing genotypic variation in faba bean (Vicia faba L.) in response to salinity in hydroponic and field experiments. Field Crop Res. 127, 99–108. doi: 10.1016/j.fcr.2011.10.016
Till, B. J., Reynolds, S. H., Greene, E. A., Codomo, C. A., Enns, L. C., Johnson, J. E., et al. (2003). Large-scale discovery of induced point mutations with high-throughput TILLING. Genome Res. 13, 524–530. doi: 10.1101/gr.977903
Torres, A. M., Avila, C. M., Gutierrez, N., Palomino, C., Moreno, M. T., and Cubero, J. I. (2010). Marker-assisted selection in faba bean (Vicia faba L.). Field Crop Res. 115, 243–252. doi: 10.1016/j.fcr.2008.12.002
Torres, A. M., Weeden, N. F., and Martín, A. (1993). Linkage among isozyme, RFLP and RAPD markers in Vicia faba. Theor. Appl. Genet. 85, 937–945. doi: 10.1007/BF00215032
van de Wouw, M., Enneking, D., Robertson, L. D., and Maxted, N. (2001). “Vetches,” in Plant Genetics Resources of Legumes in the Mediterranean, eds N. Maxted and S. J. Bennett (Dordrecht: Springer), 134–158.
Wang, H. F., Zong, X. X., Guan, J. P., Yang, T., Sun, X. L., Ma, Y., et al. (2012a). Genetic diversity and relationship of global faba bean (Vicia faba L.) germplasm revealed by ISSR markers. Theor. Appl. Genet. 124, 789–797. doi: 10.1007/s00122-011-1750-1
Wang, T. L., Uauy, C., Robson, F., and Till, B. (2012b). TILLING in extremis. Plant Biotechnol. J. 10, 761–772. doi: 10.1111/j.1467-7652.2012.00708.x
Webb, A., Cottage, A., Wood, T., Khamassi, K., Hobbs, D., Gostkiewicz, K., et al. (2016). A SNP-based consensus genetic map for synteny-based trait targeting in faba bean (Vicia faba L.). Plant Biotechnol. J. 14, 177–185. doi: 10.1111/pbi.12371
Yang, T., Bao, S. Y., Ford, R., Jia, T. J., Guan, J. P., He, Y. H., et al. (2012). High-throughput novel microsatellite marker of faba bean via next generation sequencing. BMC Genomics 13:602. doi: 10.1186/1471-2164-13-602
Young, N. D., Debellé, F., Oldroyd, G. E., Geurts, R., Cannon, S. B., Udvardi, M. K., et al. (2011). The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature 480, 520–524. doi: 10.1038/nature10625
Zeid, M., Mitchell, S., Link, W., Carter, M., Nawar, A., Fulton, T., et al. (2009). Simple sequence repeats (SSRs) in faba bean: new loci from Orobanche-resistant cultivar ‘Giza 402’. Plant Breed. 128, 149–155. doi: 10.1111/j.1439-0523.2008.01584.x
Zeid, M., Schön, C. C., and Link, W. (2003). Genetic diversity in recent elite faba bean lines using AFLP markers. Theor. Appl. Genet. 107, 1304–1314. doi: 10.1007/s00122-003-1350-9
Zhang, H. M., Wheeler, S., Xia, X., Radchuk, R., Weber, H., Offler, C. E., et al. (2015). Differential transcriptional networks associated with key phases of ingrowth wall construction in trans-differentiating epidermal transfer cells of Vicia faba cotyledons. BMC Plant Biol. 15:103. doi: 10.1186/s12870-015-0486-5
Keywords: Vicia faba, transcriptome, single nucleotide polymorphism, genetic linkage map, synteny, mutagenesis, transformation
Citation: O'Sullivan DM and Angra D (2016) Advances in Faba Bean Genetics and Genomics. Front. Genet. 7:150. doi: 10.3389/fgene.2016.00150
Received: 24 November 2015; Accepted: 03 August 2016;
Published: 22 August 2016.
Edited by:
Janila Pasupuleti, International Crops Research Institute for the Semi-Arid Tropics, IndiaReviewed by:
Steven B. Cannon, USDA - Agricultural Research Service, USACopyright © 2016 O'Sullivan and Angra. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Donal M. O'Sullivan, ZC5tLm9zdWxsaXZhbkByZWFkaW5nLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.