 Anne K. Ludwig
Anne K. Ludwig Peng Zhang
Peng Zhang M. C. Cardoso
M. C. Cardoso
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet. , 21 June 2016
Sec. Epigenomics and Epigenetics
Volume 7 - 2016 | https://doi.org/10.3389/fgene.2016.00115
This article is part of the Research Topic How Does Nuclear Structure Control Genome Function Throughout the Life-Span and Then Fail in Disease? View all 7 articles
Cytosine base modifications in mammals underwent a recent expansion with the addition of several naturally occurring further modifications of methylcytosine in the last years. This expansion was accompanied by the identification of the respective enzymes and proteins reading and translating the different modifications into chromatin higher order organization as well as genome activity and stability, leading to the hypothesis of a cytosine code. Here, we summarize the current state-of-the-art on DNA modifications, the enzyme families setting the cytosine modifications and the protein families reading and translating the different modifications with emphasis on the mouse protein homologs. Throughout this review, we focus on functional and mechanistic studies performed on mammalian cells, corresponding mouse models and associated human diseases.
In mammals, the modified cytosine was initially described by Hotchkiss (1948) and was further extensively studied since the 1970s (Razin and Cedar, 1977). Recently, evidence for methylation of adenine has been also reported in mammals (Koziol et al., 2016). Here, we will focus on cytosine modifications in mammals.
DNA cytosine methylation is catalyzed by DNA methyltransferases (Dnmts) that transfer a methyl group from S-adenosyl methionine to the fifth carbon of a cytosine residue to form 5-methylcytosine (5mC). The majority of 5mC bases are present in CpG dinucleotides, however, non-CpG methylation was also observed especially in mouse embryonic stem cells (mESCs) and brain tissue (Guo et al., 2014). DNA methylation plays a major role in gene expression, cellular differentiation, genomic imprinting, X-inactivation, inactivation of transposable elements, and embryogenesis.
Cytosine methylation patterns are mainly established by de novo methyltransferases Dnmt3a, Dnmt3b and their regulatory unit Dnmt3l during early embryonic and germ cell development. Once the patterns are established, they are maintained throughout cell generations by Dnmt1 (Bestor et al., 1988; Li et al., 1992). Unlike Dnmt1 and Dnmt3a/3b, Dnmt2 is a RNA methyltransferase rather than a DNA methyltransferase (Okano et al., 1998; Yoder and Bestor, 1998; Goll et al., 2006). A summary of the mouse Dnmt protein family and their domains is shown in Figure 1 and a summary of the respective knockout mice phenotypes is shown in Table 1.
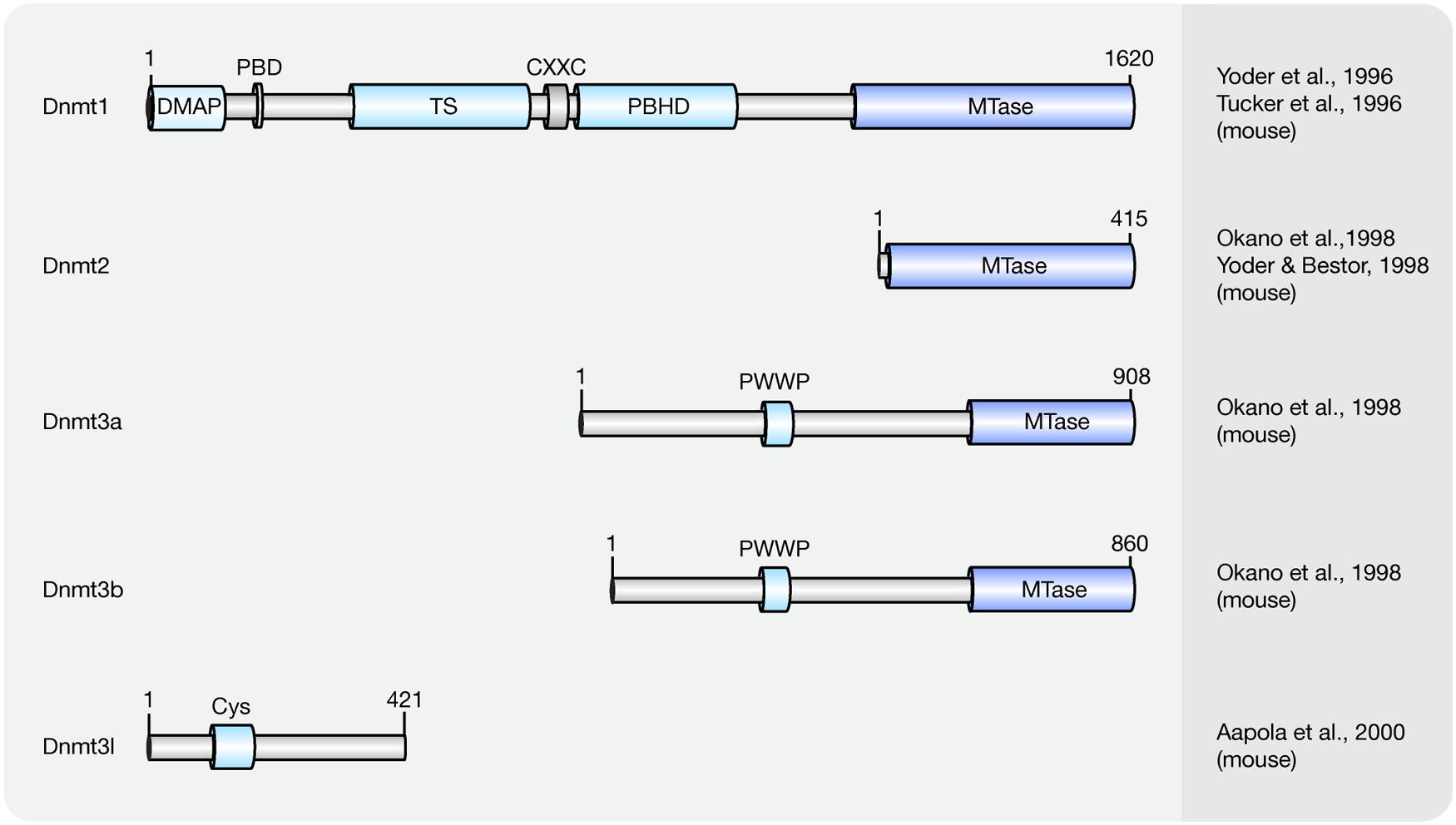
FIGURE 1. Schematic representation of the Dnmt protein family. Shown are domain structures of mouse Dnmt proteins and the initial references. Numbers represent amino acid positions. DMAP, Dnmt1-associated protein binding domain; PBD, proliferating cell nuclear antigen (PCNA)-binding domain; TS, targeting sequence; CXXC, CXXC zinc finger domain; PBHD, polybromo-1 protein homologous domain; MTase, methyltransferase; PWWP, proline-tryptophan-tryptophan-proline motif; Cys, cysteine-rich domain.

TABLE 1. Phenotypes of initial Dnmt knockout mouse models.
Overexpression of Dnmt3a and Dnmt3b is capable of methylating both native and synthetic DNA with no preference for hemimethylated DNA (Okano et al., 1999). The domain structure for de novo methyltransferases Dnmt3a and Dnmt3b is similar, including a DNA binding domain PWWP domain (Qiu et al., 2002) and a C-terminal catalytic domain (Okano et al., 1999; Figure 1). However, several studies showed that the distribution and expression of Dnmt3a and Dnmt3b varies among cell types.
Dnmt3a is expressed relatively ubiquitously and two isoforms of Dnmt3a have been identified. One binds to euchromatic and the other to heterochromatic regions (Okano et al., 1998; Chen et al., 2002). Dnmt3a knockout mice developed to term and appeared to be normal at birth but most of the homozygous mutant mice became runted and died at about 4 weeks of age (Table 1).
Dnmt3b is highly expressed in embryonic implantation stages, as well as in stem cells and progenitor cells and is the major methyltransferase in early embryogenesis (Watanabe et al., 2002, 2004). Several isoforms were identified and among all isoforms only Dnmt3b1 and Dnmt3b2 possess DNA methyltransferase activity (Aoki et al., 2001). No viable Dnmt3b knockout mice were recovered at birth, further highlighting its functions in early embryogenesis (Table 1). The major substrates of Dnmt3a/3b are CpGs, but non-CpG methylation activity of Dnmt3a/3b was also detected (Aoki et al., 2001).
Although Dnmt3l does not possess DNA methylation activity (Bourc’his et al., 2001), it strongly interacts with Dnmt3a/3b and enhances their methylation activity (Aapola et al., 2000; Suetake et al., 2004; Hu et al., 2008). However, high expression levels of Dnmt3l are found only in germ cells and early stage embryos but not in somatic cells (Watanabe et al., 2004) indicating that the methylation activity enhancement is cell type and developmental stage dependent. Disruption of Dnmt3l caused azoospermia in homozygous males and heterozygous progeny of homozygous female died before midgestation (Table 1).
Dnmt1 has a preference for hemi-methylated DNA substrates (Song et al., 2011) and is the enzyme responsible for the maintenance of DNA methylation after DNA replication (Leonhardt et al., 1992). Homozygous knockout Dnmt1 mice were runted, delayed in development and did not survive past midgestation (Table 1). The major isoform of Dnmt1 in mice contains 1620 amino acids and includes an N-terminal regulatory domain and a C-terminal catalytic domain (Tucker et al., 1996; Yoder et al., 1996). However, one isoform lacking the most N-terminal 118 amino acids was shown to accumulate in mouse oocytes (Mertineit et al., 1998).
The Dnmt1-associated protein (DMAP) binding domain is located at the beginning of the N-terminus of Dnmt1 and it recruits DMAP1 to further maintain the heterochromatin state (Rountree et al., 2000). With the contribution of Uhrf1 [ubiquitin-like with plant homeodomain (PHD) and ring finger domains 1], Dnmt1 methylates hemi-methylated DNA generated upon DNA replication by a mechanism encompassing base flipping (Song et al., 2011, 2012).
In most mouse cells, Dnmt1 localizes to the cell nucleus. In fact, Dnmt1 contains several functional nuclear localization sequences within its N-terminal regulatory domain (Cardoso and Leonhardt, 1999). In early embryos (Cardoso and Leonhardt, 1999) and in post-mitotic neurons (Inano et al., 2000) though, it is retained in the cytoplasm. Although highly expressed in mouse embryos, the exclusion of Dnmt1 from nuclei might inhibit DNA methylation conservation after DNA replication (Grohmann et al., 2005), implying that localization of Dnmt1 also regulates its methylation activity. Within the cell nucleus, the distribution of Dnmt1 is cell cycle dependent (Leonhardt et al., 1992). In G1-phase, it is diffusely distributed throughout the nucleoplasm. In early S-phase, its proliferating cell nuclear antigen (PCNA)-binding domain (PBD) targets Dnmt1 to replication sites and in late S-phase, the targeting sequence (TS) further enhances Dnmt1 binding to replicating pericentromeric heterochromatin (Schermelleh et al., 2007; Schneider et al., 2013). In G2-phase, Dnmt1 is de novo loaded onto pericentromeric heterochromatin via a replication independent mechanism (Easwaran et al., 2004). Besides its PBD and TS domains, the polybromo-1 protein homologous domain (PBHD) is also involved in targeting Dnmt1 to replication foci (Liu et al., 1998). Between the TS and PBHD domains, a CXXC domain can be found in Dnmt1. The CXXC domain of Dnmt1 occludes access of Dnmt1 catalytic site to non-methylated CpGs and allows Dnmt1 to bind and specifically methylate hemi-methylated CpGs (Song et al., 2011).
The stable covalent C–C bond formed between the methyl group and the cytosine is difficult to be directly removed and, therefore, 5mC is thought to be a long-lived epigenetic mark. After DNA replication, Dnmt1 association with the replication machinery ensures the maintenance of the methylation pattern onto the newly synthesized strand. Failure to do so, e.g., by retention in the cytoplasm as mentioned above, leads to gradual passive loss of DNA methylation over cell generations. DNA replication independent (active) loss of global DNA methylation was also observed in some biological processes such as reprogramming of the paternal genome after fertilization (Mayer et al., 2000) and development of primordial germ cells (PGC; Hajkova et al., 2002). The active loss of DNA methylation allows rapid reprogramming of the genome in a short time. Similar observations were made in post-mitotic neurons indicating that active loss of DNA methylation also occurs in somatic cells and might have important roles in the regulation of gene expression (Martinowich et al., 2003).
For several decades, scientists have been interested in identifying pathways or proteins involved in the active loss of DNA methylation. Lacking the evidence to show that C–C bonds can be directly broken in mammals, multi-step processes have been proposed to be involved in the active removal of DNA methylation marks. In 1972, several additional modifications of cytosines were described in rat, mouse, and frog brain tissue including 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), 5-carboxylcytosine (5caC), and 5-hydroxymethyluracil (5hmU; Penn et al., 1972; Figure 2). However, these modifications were considered to be oxidative damage products of DNA (de Rojas-Walker et al., 1995; Tardy-Planechaud et al., 1997). Three decades later, 5hmC was re-discovered in mouse brain tissue (Kriaucionis and Heintz, 2009) and embryonic stem cells (ESCs; Tahiliani et al., 2009). Furthermore, a family of proteins (ten-eleven translocation, TET) was identified that oxidize 5mC to 5hmC both in humans (Tahiliani et al., 2009) and mice (Ito et al., 2010). TET1 was first described in 2003 as a fusion partner of the mixed lineage leukemia (MLL) gene in acute myeloid leukemia (AML; Lorsbach et al., 2003) and 6 years later it was re-discovered as an oxygenase, which can convert 5mC to 5hmC (Tahiliani et al., 2009). Further studies showed that Tet proteins also convert 5hmC to 5fC and 5caC (Ito et al., 2011; Pfaffeneder et al., 2011).
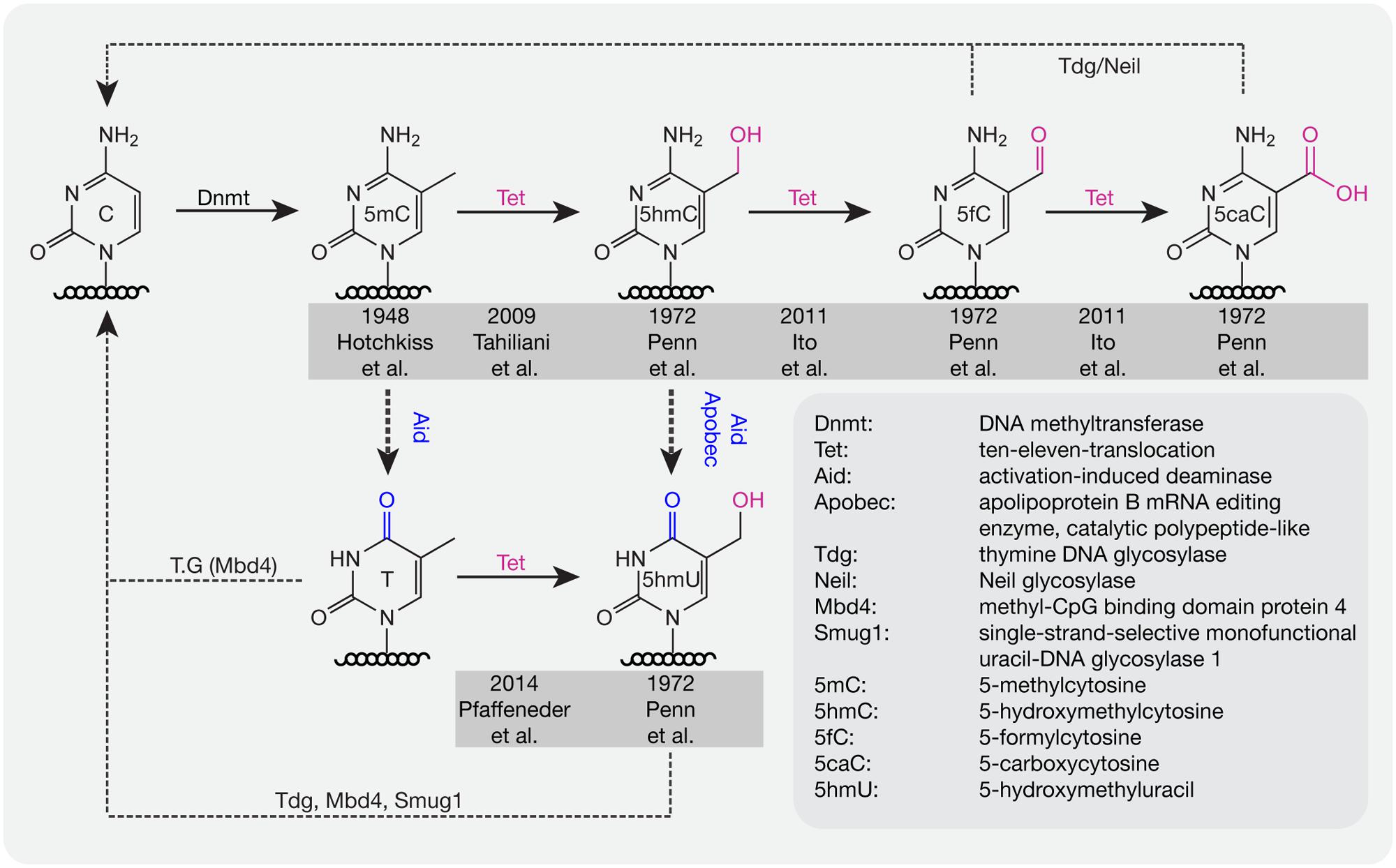
FIGURE 2. DNA base modifications with respective enzymes. Dnmts catalyze the addition of a methyl group to cytosine bases. Tet proteins oxidize methylated cytosines to 5hmC, 5fC, and 5caC in an iterative manner. 5mC and 5hmC can be further deaminated by Aid/Apobec to T and 5hmU. T, 5hmU, 5fC, and 5caC can be removed by the indicated glycosylases. Initial references are indicated.
Deaminases such as Aid and Apobec can recognize 5mC and 5hmC and further convert 5mC to thymine (T) and 5hmC to 5hmU. Although the deaminase activity is quite low, it is still a possible pathway for DNA demethylation (Guo et al., 2011). In addition, Tets were also shown to oxidize T to 5hmU in mESCs (Pfaffeneder et al., 2014), which additionally leads to loss of DNA methylation. The oxidation products like 5fC, 5caC, and 5hmU can be recognized and excised by the glycosylases Tdg (Maiti and Drohat, 2011) and Neil (Muller et al., 2014) to create an abasic site on DNA, which is further repaired by enzymes of the base excision repair (BER) pathway. In addition to Tdg, 5hmU can also be recognized by other glycosylases like Mbd4 (Hashimoto et al., 2012b) and Smug1 (Kemmerich et al., 2012). Accordingly, a combination of oxidation, deamination and BER might contribute to the active removal of DNA methylation. In mouse zygotes, the decrease of 5mC and increase of 5hmC suggests that 5hmC might be an intermediate of DNA methylation removal. However, recent studies showed that loss of 5mC mainly happens before S-phase, whereas gain of 5hmC occurred after DNA replication (Amouroux et al., 2016), indicating that besides the conversion of 5mC to 5hmC, other pathways might contribute to methylation removal before DNA replication in mouse zygotes.
Until now three members of the Tet protein family named Tet1 (mouse homolog of human TET1), Tet2 (mouse homolog of human TET2), and Tet3 (mouse homolog of human TET3) have been identified in mice and humans. All three Tets share a conserved C-terminal catalytic domain including a cysteine-rich and a double-stranded β-helix (DSBH) domain, which belong to the cupin-like dioxygenase superfamily; and exhibit iterative iron- and oxoglutarate-dependent oxidation activity (Figure 3).
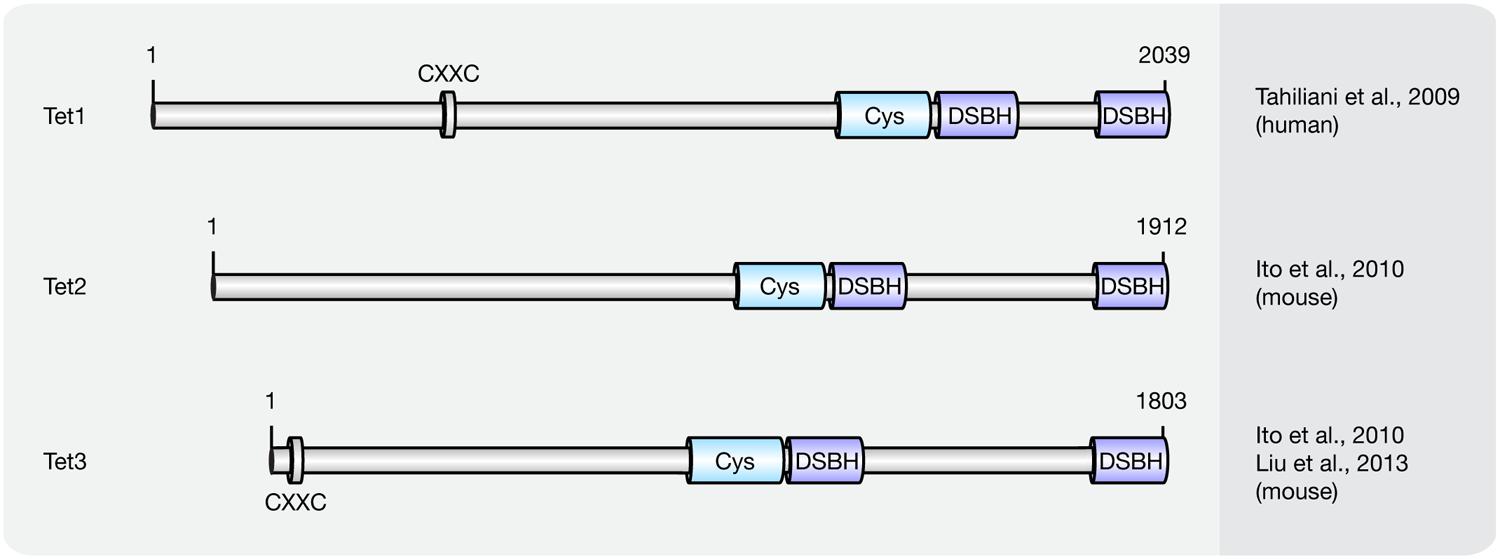
FIGURE 3. Schematic representation of the Tet protein family. Shown are domain structures of mouse Tet proteins and the initial references. Numbers represent amino acid positions. CXXC, CXXC zinc finger domain; Cys, cysteine-rich domain; DSBH, double-stranded β-helix.
During mouse embryo development, Tet3 is highly expressed in oocytes and zygotes. Female mice depleted of Tet3 in the germ line showed severely reduced fecundity and their heterozygous mutant offspring lacking maternal Tet3 suffer an increased incidence of developmental failure. Since Tet1, Tet2 as well as Tet1 and Tet2 double knockout mice are viable, this suggests that Tet1 and Tet2 are not essential for mouse development (Table 2).

TABLE 2. Phenotypes of initial Tet knockout mouse models.
Tet-mediated 5mC to 5hmC conversion is though involved in reprogramming the paternal genome (Gu et al., 2011; Iqbal et al., 2011; Wossidlo et al., 2011; Zhang et al., 2012) and also in reprogramming donor cell DNA during somatic cell nuclear transfer (Gu et al., 2011). In addition, HIV-1 Vpr binding protein (VprBP)-mediated monoubiquitylation promotes Tet binding to chromatin and enhances 5hmC formation (Nakagawa et al., 2015) in mouse embryos. This process is involved in female germ cell development and genome reprogramming in zygotes (Yu et al., 2013).
During PGC reprogramming, Tet1 and Tet2 are highly expressed (Hackett et al., 2013). However, genome-wide DNA methylation removal is unaffected by the absence of Tet1 and Tet2 and, thus, 5hmC, indicating that the first comprehensive 5mC loss does not involve 5hmC formation. Instead Tet1 and Tet2 have a locus specific role in shaping the PGC epigenome during subsequent development (Vincent et al., 2013). Further studies showed that Tet1 has a critical role in the erasure of genomic imprinting (Yamaguchi et al., 2013) and it controls meiosis by regulating meiotic gene expression (Yamaguchi et al., 2012).
In mESCs, both Tet1 and Tet2, as well as their oxidation product 5hmC are highly abundant (Ito et al., 2010). While Tet2 preferentially acts on gene bodies, Tet1 preferentially acts on promoters and transcription start sites (TSS; Huang et al., 2014). Tet1 and Tet2 double knockout ESCs remained pluripotent, but were depleted of 5hmC and caused developmental defects in chimeric embryos (Dawlaty et al., 2014). During somatic reprogramming, Tet2 is required for 5hmC formation at the Nanog locus (Doege et al., 2012). Further studies showed that the recruitment of Tet1 by Nanog facilitates the expression of a subset of reprogramming target genes, such as Oct4 (Costa et al., 2013). Accordingly, Tet1 can replace Oct4 during somatic cell reprogramming in conjunction with Sox2, Klf4, and c-Myc (Gao et al., 2013). The data above indicate that Tet-mediated 5hmC formation is not only important for ESCs differentiation but also for somatic reprogramming.
In mouse brain, 5hmC is a constituent of nuclear DNA (Kriaucionis and Heintz, 2009). Tet1 plays an important role in regulating neural progenitor cell (NPC) proliferation in adult mouse brain (Zhang et al., 2013) and is critical for neuronal activity-regulated gene expression and memory extinction (Table 2; Rudenko et al., 2013).
Similar to Dnmt1, Tet proteins use a base flipping mechanism to oxidize 5mC, which includes binding of DNA by a Watson–Crick polar hydrogen and van der Waals interactions, flipping out 5mC (Hu et al., 2013; Hashimoto et al., 2014) and oxidation of 5mC to 5hmC (Hashimoto et al., 2015; Hu et al., 2015). Although Tet proteins successively oxidize 5mC to 5caC, recent experimental data showed that, in comparison with 5hmC and 5fC, 5mC is the preferential substrate for Tet2 (Hu et al., 2015). This preference was further confirmed by computer simulations (Lu et al., 2016). In cultured cells, the majority of genomic 5hmC nucleotides are stable (Bachman et al., 2014), indicating that 5hmC is not only involved in loss of DNA methylation, but represents an additional stable epigenetic mark. The global content of 5hmC varies in mouse tissues, does not correlate with 5mC content and rapidly decreases as the cells adapt to cell culture conditions (Nestor et al., 2012). The cell-, tissue-, and developmental stage-specific distribution of 5hmC indicates that the conversion of 5mC to 5hmC is highly regulated.
Although the N-terminal domain (NTD) of Tet proteins was shown to be dispensable for their catalytic activity, it was shown to possess regulatory functions. A CXXC domain, which usually binds specifically to unmethylated CpGs can be found in the N-terminus of Tet1 and Tet3 (Liu et al., 2013). While the CXXC domain of Tet1 cannot bind to DNA in vitro (Frauer et al., 2011b), it binds to unmodified C, 5mC- or 5hmC-modified CpGs in vivo (Zhang et al., 2010; Xu Y. et al., 2011). Moreover, binding of the CXXC domain to DNA was shown to control DNA methylation levels by preventing unwanted DNA methyltransferase activity in ESCs (Xu Y. et al., 2011) or aberrant methylation spreading into CpG islands (CGIs) in differentiated cells (Jin et al., 2014). The CXXC domain of Xenopus Tet3 recognizes non-methylated cytosines in either CpG or non-CpG context, and it is critical for specific Tet3 targeting (Xu et al., 2012). Although Tet2 proteins do not have a CXXC domain, recent studies showed that the ancestral CXXC domain of Tet2 is encoded by a distinct gene named Idax. Unlike the CXXC domain of Tet1 and Tet3, the CXXC domain of Idax binds unmethylated CpGs. Through direct protein–protein interactions of Tet2 and Idax, Tet2 is recruited to DNA. Furthermore, Tet2 is degraded by caspase activation, which is triggered by the CXXC of Idax (Ko et al., 2013).
Two parts of the DSBH domain are connected by a potential regulatory spacer region. Although the spacer region was shown to be dispensable for 5mC catalytic activity (Hu et al., 2013), post-translational modifications (PTMs), such as phosphorylation and O-GlcNAcylation were observed in the spacer region (Bauer et al., 2015) indicating that it might exhibit regulatory functions. O-GlcNAc transferase (Ogt) directly interacts with Tet proteins and consequently Tet proteins are GlcNAcylated. The GlcNAcylation does not affect the hydroxylation activity of Tet2 and Tet3, rather Tet2 and Tet3 were shown to promote Ogt activity (Deplus et al., 2013) by enhancing the localization of Ogt to chromatin (Chen et al., 2013; Ito et al., 2014). However, it was shown that Ogt drives Tet3 out of the nucleus further affecting its activity on DNA (Zhang et al., 2014). In mESCs, Ogt is recruited to unmethylated CpG promoters in a Tet1-dependent manner (Vella et al., 2013). In addition to PTMs, mutations within the spacer region of Tet2 were observed in myelodysplastic syndrome (MDS), thus further highlighting the importance of this region (Ko et al., 2010).
In vivo, besides PTMs, Tet activity is regulated by protein–protein interactions, such as with Sin3a. In mESCs, the interaction between Sin3a and Tet1 allows Sin3a to repress a subset of Tet1 target genes (Williams et al., 2011). In mouse zygotes, Tet3-mediated 5mC to 5hmC conversion is involved in reprogramming of the paternal but not the maternal genome although they share the same cytoplasm (Mayer et al., 2000). The resistance of the maternal genome to reprogramming is achieved by a protein named developmental pluripotency associated 3 (Dppa3, or PGC7). Dppa3 binds to histone H3K9me2 (Nakamura et al., 2012) and interacts with Tet3 further blocking the activity of Tet3 (Bian and Yu, 2014). Dazl, an RNA-binding protein known to play a key role in germ cell development, was shown to enhance Tet1-mediated 5mC to 5hmC conversion by enhancing Tet1 protein translation (Welling et al., 2015). In addition, growth arrest and DNA damage inducible protein 45 (Gadd45) interacts with Tet1 and Tdg and promotes loss of DNA methylation by enhancing 5fC/5caC removal (Kienhöfer et al., 2015; Li et al., 2015).
Finally, Tet-mediated 5mC to 5hmC conversion was shown to be regulated by Tet cofactors. 2-Ketoglutarate (2-KG), one of the cofactors for Tet oxidation is produced by isocitrate dehydrogenase 1/2 (Idh1/2) in vivo. However, mutated Idh1/2 produce 2-hydroxyglutarate, a competitive inhibitor of 2-KG, which can further inhibit 5mC to 5hmC conversion (Konstandin et al., 2011). Vitamin C is a potential cofactor for Tet-mediated oxidation and was shown to enhance Tet activity, which leads to increased global 5hmC in ESCs (Blaschke et al., 2013). ATP was also shown to be involved in regulating Tet activity. In vitro, the reaction of Tet-mediated 5mC to 5caC can be enhanced by addition of ATP (He et al., 2011).
Dnmt1 recognizes hemi-mC DNA and methylates the nascent DNA strand after replication during the S-phase of the cell cycle. However, in vitro studies showed a 60-fold decreased binding ability of Dnmt1 to hemi-hmC DNA compared to hemi-mC DNA (Hashimoto et al., 2012a), indicating that hemi-hmC DNA might not be a substrate for Dnmt1. Previous studies showed that Np95 can recognize 5hmC and bind to hemi-hmC DNA (Frauer et al., 2011a), indicating that Np95 might target Dnmt1 to hemi-hmC containing replication forks to maintain hmC after DNA replication. In addition, Dnmt3a and Dnmt3b recognize hemi-hmC DNA (Hashimoto et al., 2012a) and are necessary for methylation maintenance at repeat genomic elements (Chen et al., 2003) suggesting Dnmt3a/3b might play a role in maintaining 5hmC after DNA replication in repeat elements.
In vivo, the majority of 5hmC is present in CpG dinucleotides. However, 5hmC has also been observed in non-CpG context, especially in gene bodies (Pastor et al., 2011; Xu Y. et al., 2011). One important role of CpG methylation in gene promoter regions is the repression of gene expression by directly or indirectly preventing interactions between promoter and transcription factors. Hydroxymethylated CpGs might affect binding of transcription factors and/or 5mC readers to DNA.
In mammals, the methylome is specifically read by a variety of proteins known as methyl-CpG binding proteins (MBPs), which based on structural features are further classified into three main families: the methyl-CpG binding domain (MBD) protein family (Lewis et al., 1992; Cross et al., 1997; Hendrich and Bird, 1998; Hendrich and Tweedie, 2003; Laget et al., 2010; Baymaz et al., 2014), the Kaiso protein family (Daniel and Reynolds, 1999; Filion et al., 2006), and the SET and RING (really interesting new gene) finger associated (SRA) domain protein family (Hopfner et al., 2000; Mori et al., 2002). While initially identified as 5mC binding proteins, recent studies indicate that a distinct and dynamic set of MBPs binds the Tet oxidation product 5hmC during differentiation (Figure 4; Frauer et al., 2011a; Mellen et al., 2012; Spruijt et al., 2013). Through further interactions with multiple protein partners, MBPs provide a link between cytosine derivatives and functional chromatin states in a temporally and spatially regulated fashion.

FIGURE 4. 5-(Hydroxy)methylcytosine readers. Shown are MBPs that read 5mC and 5hmC as indicated and the initial references.
Presently, the MBD protein family consists of eleven members (Mecp2, Mbd1–6, SETDB1, SETDB2, TIP5/BAZ2A, and BAZ2B; Lewis et al., 1992; Cross et al., 1997; Hendrich and Bird, 1998; Hendrich and Tweedie, 2003; Laget et al., 2010; Baymaz et al., 2014). All of them share a common protein motif, the 70–85 amino acids long MBD, which enables some, but not all family members, to selectively bind to single methylated CpG dinucleotides. With the exception of Mbd2 and Mbd3, MBD proteins bear little resemblance outside their MBD (Hendrich and Bird, 1998). Instead, MBD proteins comprise several distinct domains that confer unique DNA binding, as well as other functional features. Since this review covers DNA (hydroxy)methylation-dependent processes, we will thereafter focus on MBD family members (Figure 5) capable of binding to (hydroxy)methylated CpG dinucleotides, i.e., methyl-CpG binding protein 2 (Mecp2) and methyl-CpG binding domain proteins 1–4 (Mbd1–4).
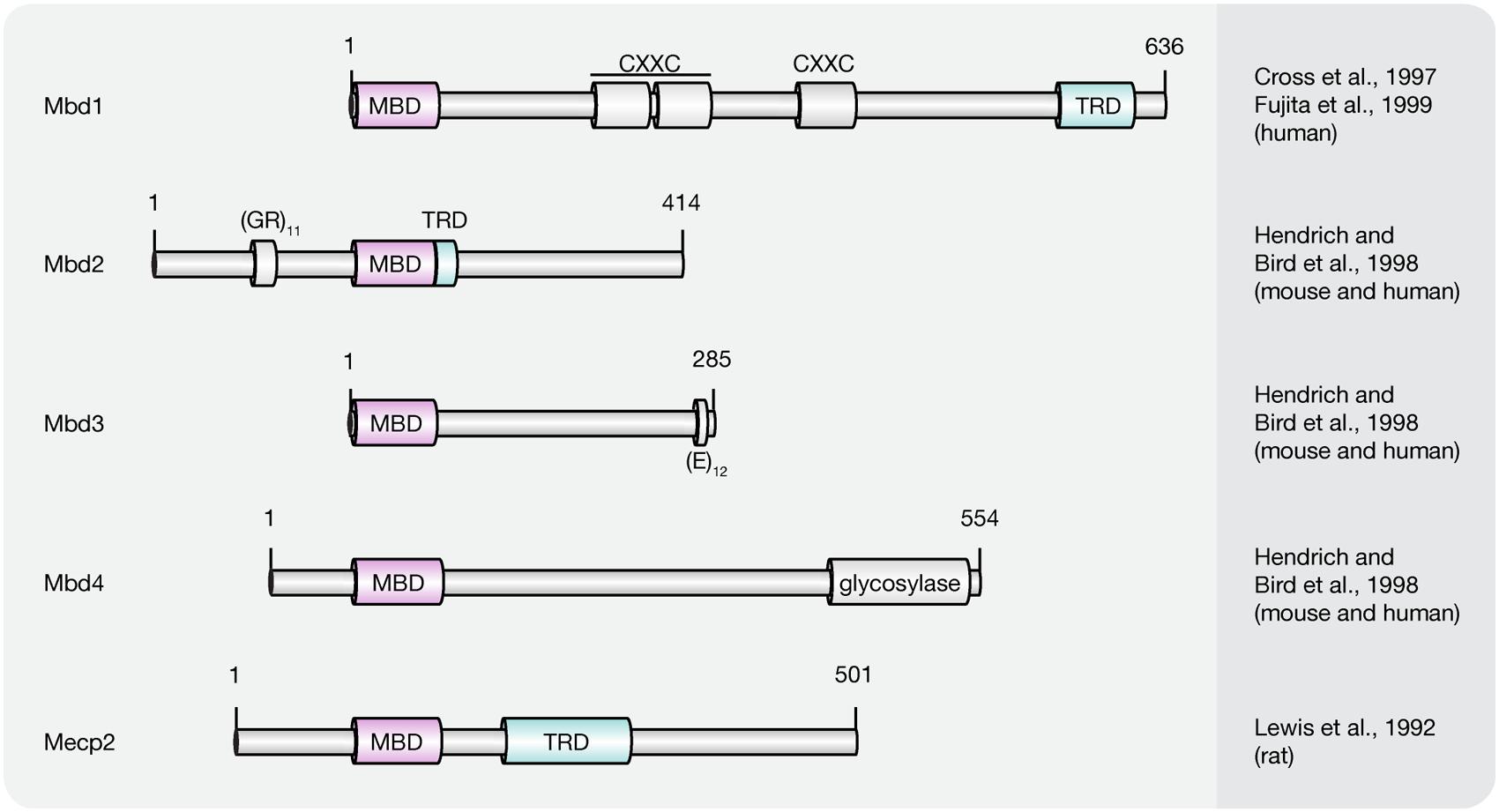
FIGURE 5. Schematic representation of the MBD protein family. Shown are domain structures of mouse MBD proteins and the initial references. Numbers represent amino acid positions. MBD, methyl-CpG binding domain; CXXC, CXXC zinc finger domain; TRD, transcriptional repression domain; GR, glycine/arginine; E, glutamic acid.
The first protein described to selectively recognize and bind single, symmetrically methylated CpG dinucleotides was Mecp2 (Lewis et al., 1992). It is abundantly expressed in the central nervous system with the highest protein levels in post-mitotic neurons (Akbarian et al., 2001; Traynor et al., 2002; Jung et al., 2003). Of the two alternatively spliced isoforms (Mecp2 e1 and e2), which differ in their N-terminus, Mecp2 e2 was first identified and is, therefore, best characterized (Kriaucionis and Bird, 2004; Mnatzakanian et al., 2004). Although both isoforms distribute differently in developing and post-natal mouse brains, no functional differences have been identified so far (Dragich et al., 2007).
Both Mecp2 variants include two functionally characterized domains, the MBD and the transcriptional repression domain (TRD). While the MBD proved sufficient to direct specific binding to methylated cytosines (Nan et al., 1993), the TRD was originally identified as the region required for transcriptional repression in vitro and in vivo (Lewis et al., 1992; Nan et al., 1997; Jones et al., 1998; Kaludov and Wolffe, 2000). Circular dichroism and protease digestion analysis revealed that outside these functional domains the full-length protein is largely devoid of secondary structure (Adams et al., 2007). With almost 60% unstructured regions, Mecp2 is reckoned among the intrinsically disordered proteins, which often undergo a disorder-to-order transition upon binding to other macromolecules (Adams et al., 2007). Indeed, recent studies demonstrate that Mecp2 gains secondary structure and acquires substantial thermal stabilization upon binding to DNA (Ghosh et al., 2010). Unlike its name implies DNA binding is, however, not solely mediated via its 5mC specific MBD. Instead, as indicated by the release of Mecp2 upon salt extraction, regions outside the MBD contribute to the overall binding energy through electrostatic interactions (Meehan et al., 1992). As shown by electrophoretic mobility shift assays (EMSAs), these sequence-unspecific DNA binding motifs include the TRD and, based on their relative location to the MBD and TRD, the so-called intervening domain, as well as the C-terminal domain alpha (Ghosh et al., 2010). The NTD of Mecp2 in contrast, contributes indirectly to the overall binding affinity by enhancing the methylation specificity of the MBD through conformational coupling (Ghosh et al., 2010). An analog synergistic increase in DNA binding efficiency was observed through interdomain interactions between the TRD and the C-terminal part of the protein (Ghosh et al., 2010). Similar to the NTD, the C-terminal domain beta (CTD beta) does not directly interact with DNA (Ghosh et al., 2010). Nevertheless, the overall chromatin binding efficiency was lost upon its deletion (Nikitina et al., 2007b). Consistent with this, the CTD beta induced moderate and reproducible shifts with nucleosomal arrays, but not with naked DNA (Ghosh et al., 2010), suggesting that the most C-terminal 192 residues of Mecp2 harbor a chromatin interaction surface (Nikitina et al., 2007b). Indeed, Mecp2 has been shown to interact with histone H3 and, similar to the linker histone H1, binds to nucleosomes close to the linker DNA entry–exit site (Nikitina et al., 2007b). As a result, the entering and exiting linker DNA segments are brought in close proximity to form a stem-like motif (Nikitina et al., 2007a), which bears strong resemblance to structures induced by H1 (Hamiche et al., 1996; Bednar et al., 1998). The modes of chromatin compaction, however, differ significantly from each other. While histone H1 arranges nucleosomes and linker DNA into regular zigzag-shaped chromatin fibers (Woodcock, 2006), Mecp2 forms highly compacted globular structures in vitro due to its multiple DNA and chromatin binding domains (Georgel et al., 2003). Accordingly, Mecp2 was shown to induce clustering of pericentric heterochromatin in a dose-dependent manner in vivo to establish a locally repressive chromatin environment (Brero et al., 2005; Agarwal et al., 2011). More recently, Szulwach et al. (2011) provided evidence that binding of Mecp2 to methylated CpG dinucleotides may protect 5mC against Tet-mediated oxidation thereby preventing reactivation of silenced genes. The underlying mechanism, however, has so far not been described.
An additional level of regulation is achieved through various protein–protein interactions. While direct homo- and hetero-interactions of Mecp2 and Mbd2 were shown to cross-link chromatin fibers (Becker et al., 2013), physical associations of Mecp2 with the transcriptional co-repressor Sin3a and histone deacetylase 2 (HDAC2) via its TRD contribute to the global heterochromatin architecture through histone hypoacetylation (Jones et al., 1998; Nan et al., 1998). Consequently, Mecp2 deficiency was demonstrated to result in global changes in neuronal chromatin architecture, elevated histone acetylation levels, and increased transcriptional noise in a DNA methylation-dependent manner (Skene et al., 2010; Cohen et al., 2011). A number of other repressive protein partners of Mecp2 have been identified including the co-repressors c-Ski (Kokura et al., 2001), CoREST (Lunyak et al., 2002), and NCoR/SMRT (Stancheva et al., 2003), as well as DNA methyltransferase Dnmt1 (Kimura and Shiota, 2003) and H3K9 methyltransferase (Fuks et al., 2003).
Both, binding of Mecp2 to DNA, as well as interactions with protein partners are affected by PTMs. Neuronal activity induced phosphorylation and dephosphorylation of Mecp2 was shown to modulate its association with promoters of specific genes, as well as with interaction partners (reviewed in Li and Chang, 2014). More recently, poly(ADP-ribosyl)ation of Mecp2 in mouse brain tissue was reported, which anticorrelated with its chromatin binding affinity and clustering ability (Becker et al., 2016). Furthermore, ubiquitylation (Gonzales et al., 2012), SUMOylation (Cheng et al., 2014), acetylation (Zocchi and Sassone-Corsi, 2012), and methylation (Jung et al., 2008) were shown to substantially contribute to the functional versatility of Mecp2.
Another unanticipated level of functional complexity was demonstrated by recent work of Spruijt et al. (2013) who identified Mecp2 as reader of 5hmC in mESC by quantitative mass-spectrometry-based proteomics. Moreover, independent studies of Mellen et al. (2012), revealed Mecp2 as the major 5hmC-binding protein in mouse brain, which moreover turned out to bind both, 5hmC- and 5mC-containing substrates with similar affinity.
Finally, chip-chip analysis using antibodies against MECP2 in a human neuronal cell line demonstrated that around two-third of strongly MECP2 bound promoters were transcriptionally active (Yasui et al., 2007). Subsequent analysis of gene expression patterns in Mecp2 knockout and overexpressing mice concurred that Mecp2 functions as an activator as well as a repressor of transcription (Chahrour et al., 2008).
Hence, the traditional view of Mecp2 as a 5mC-dependent transcriptional silencer may be incomplete and its biology appears far more complicated than previously assumed.
Both, male and female mice lacking Mecp2 (Table 3) developed an uncoordinated gait and reduced spontaneous movement between 3 and 8 weeks of age and most died between 6 and 12 weeks (Chen et al., 2001; Guy et al., 2001). Furthermore, most animals developed hind limb clasping, irregular breathing, misaligned jaws and uneven wearing of teeth. Mutant brains were reduced in weight, however, no structural abnormalities or signs of neurodegeneration were detected, suggesting that stability of brain function, not brain development per se, is impaired in the absence of Mecp2. Consistent with this hypothesis, re-expression of the Mecp2 gene in Mecp2lox-Stop/y mice proved sufficient to reverse the neurological symptoms of Rett syndrome (RTT), indicating that Mecp2-deficient neurons develop normally and are not irreversibly damaged (Guy et al., 2007). Further microarray analyses revealed that knockout of Mecp2 implicates only minor changes in gene expression (Tudor et al., 2002). Subsequent studies demonstrating increased expression restricted to non-coding RNA in brain of Mecp2-deficient mice (Muotri et al., 2010; Skene et al., 2010), indicated that Mecp2 may not act as a gene-specific transcriptional repressor, but might instead dampen transcriptional noise genome-wide in a DNA methylation-dependent manner (Skene et al., 2010). Accordingly, expression of repetitive elements (Muotri et al., 2010; Skene et al., 2010) as well as retrotransposition of LINE1 was increased in brain of Mecp2-deficient mice (Muotri et al., 2010).

TABLE 3. Phenotype of initial MBP-deficient mouse models.
Mbd1, initially termed PCM1, is expressed in somatic cells and represents the largest member of the MBD family (Cross et al., 1997; Hendrich and Bird, 1998). Similar to Mecp2, Mbd1 contains a MBD and a TRD, which have analog functions to that of Mecp2 (Ng et al., 2000). In addition, depending on the isoform, Mbd1 contains two or three CXXC zinc finger motifs (Fujita et al., 1999; Jorgensen et al., 2004). The most C-terminal one, referred to as CXXC3, is homolog to zinc fingers found in Dnmt1, CpG binding protein CGBP, histone H3K4 methyltransferase MLL and histone H3K36 deacetylases of the Jumonji family JHDM1A and JHDM1B (Jorgensen et al., 2004; Lee and Skalnik, 2005; Tsukada et al., 2006). While CXXC3 was shown to bind unmethylated CpG dinucleotides in vitro (Birke et al., 2002; Lee and Skalnik, 2002, 2005; Jorgensen et al., 2004), the remaining zinc finger motifs of Mbd1 lack a conserved glutamine residue and the characteristic KFFG motif necessary for binding to DNA (Jorgensen et al., 2004). Accordingly, Mbd1 isoforms containing the first two CXXC domains preferentially bind methylated DNA via their MBD, whereas isoforms comprising a complete set of zinc fingers have the ability to bind both, methylated and unmethylated substrates (Jorgensen et al., 2004; Baubec et al., 2013).
As a transcriptional repressor, Mbd1 was thus shown to inhibit transcription from both, methylated and unmethylated promoters in reporter gene assays (Fujita et al., 1999; Jorgensen et al., 2004). While methylation-dependent silencing is mediated by the MBD and TRD, suppression of non-methylated reporter constructs required the presence of the CXXC3 domain (Jorgensen et al., 2004). Although, a precise association between Mbd1 and HDACs has not been described, transcriptional repression was partially sensitive to trichostatin A (TSA), an HDAC inhibitor (Ng et al., 2000). In most assays, however, Mbd1 behaved as an HDAC-independent repressor (Ng et al., 2000).
Instead, MBD1 has been found associated with histone H3K9 methyltransferases SETDB1 (Sarraf and Stancheva, 2004) and Suv39h1 (Fujita et al., 2003). Association to SETDB1 mediates transcriptional repression throughout the cell cycle (Sarraf and Stancheva, 2004). During S-phase, however, MBD1 was shown to recruit SETDB1 to the large subunit of chromatin assembly factor CAF-1 to form an S-phase specific complex that mediates methylation of H3K9 in a post-replicative manner (Sarraf and Stancheva, 2004). Accordingly, H3K9 methylation is lost in the absence of MBD1 and results in activation of specific genes, such as p53BP2 (Sarraf and Stancheva, 2004).
MBD1-mediated transcriptional repression and hetero chromatin maintenance was shown to be regulated by SUMOylation (Lyst et al., 2006; Uchimura et al., 2006). In human cells, two E3 SUMO-ligases (PIAS1 and PIAS3) were shown to SUMOylate MBD1 (Lyst et al., 2006). While SUMO1-conjugation blocks the MBD1 and SETDB1 interaction, modification with SUMO2/3 recruits SETDB1 thereby stimulating its repressive function (Uchimura et al., 2006).
Although mice lacking Mbd1 (Table 3) developed normally and appeared healthy throughout life, they were impaired in spatial learning, had decreased neurogenesis and reduced long-term potentiation in the dentate gyrus of the hippocampus (Zhao et al., 2003). Moreover, Mbd1-deficient neural stem cells differentiated less and had decreased genomic stability (Zhao et al., 2003).
Mbd2 and Mbd3 are the only known members of the MBD protein family with significant sequence similarity beyond the MBD (Hendrich and Bird, 1998) and, thus, are believed to have arisen from an ancient duplication during evolution of the vertebrate lineage (Hendrich and Tweedie, 2003). Consistent with this, a homolog Mbd2/3 like protein was identified in invertebrates, including Drosophila (Lyko et al., 2000; Marhold et al., 2004). Despite the high degree of sequence similarity, Mbd3 lacks the amino-terminal extension of Mbd2, which contains a repeat consisting of glycine and arginine residues (Hendrich and Bird, 1998). While both, Mbd2 and Mbd3 contain a C-terminal coiled coil (CC) domain that mediates protein–protein interactions, Mbd3 was shown to comprise an additional glutamic acid repeat at its extreme COOH-terminus (Hendrich and Bird, 1998; Gnanapragasam et al., 2011; Becker et al., 2013).
Mbd2 contains two in-frame start codons, which give rise to Mbd2a and the truncated version Mbd2b, which lacks the first 140 amino acids (Hendrich and Bird, 1998). In vivo, however, only Mbd2a, but not Mbd2b, has been detected (Ng et al., 1999). Inclusion of an alternative third exon gives rise to an additional isoform of Mbd2, named Mbd2c, which lacks the C-terminal TRD and CC domain due to an early stop codon (Hendrich and Bird, 1998).
Tethering of Mbd2a near a promoter via a GAL4 DNA binding domain was shown to mediate transcriptional repression that is sensitive to TSA (Ng et al., 1999). Similarly, Mbd2b enhanced transcriptional repression of methylated reporter constructs in co-transfection assays (Boeke et al., 2000). Different from other MBD family members, the sequence required for TRD partially overlapped with the MBD (Boeke et al., 2000), indicating a strong interrelation of methylation binding and transcriptional silencing. In line with this, the TRD directly interacts with the transcriptional repressor Sin3A (Boeke et al., 2000). Moreover, Mbd2 co-purified with a large protein complex known as NuRD (nucleosome remodeling and histone deacetylation), which includes chromatin remodeling ATPase Mi-2, as well as HDAC1 and HDAC2 (Ng et al., 1999; Wade et al., 1999; Zhang et al., 1999; Mahajan et al., 2005; Le Guezennec et al., 2006). EMSAs indicated that Mbd2a directs the NuRD complex, which is implicated in transcriptional silencing, to methylated DNA (Zhang et al., 1999). Finally, immunoprecipitation analysis showed that Mbd2 associates with HDAC1 in mammalian cells and is the long sought methyl-CpG binding component of the 400–800 kDa MeCP1 complex (Meehan et al., 1989; Ng et al., 1999).
Mbd2 was shown to bind 5mC in a manner similar to the isolated MBD of Mecp2 (Hendrich and Bird, 1998; Wade et al., 1999). Binding of oxidative 5mC derivatives, however, has not been observed (Hashimoto et al., 2012a; Mellen et al., 2012; Spruijt et al., 2013).
Mbd2b has also been reported to have DNA demethylase activity (Bhattacharya et al., 1999), but this finding has been questioned (Ng et al., 1999; Wade et al., 1999).
Mbd2-deficient mice (Table 3) are viable and fertile, but exhibit a maternal nurturing defect resulting in reduced litter size and weight of pups (Hendrich et al., 2001).
The smallest member of the MBD family, coding for a protein of approximately 30 kDa is Mbd3 (Hendrich and Bird, 1998). It appears in a rich diversity of splice variants and is expressed in ESCs as well as somatic tissues (Hendrich and Bird, 1998; Roloff et al., 2003).
DNA binding properties of Mbd3 seem to vary with species. While mammalian Mbd3 is unable to interact with methylated DNA, its amphibian counterpart binds methylated CpG dinucleotides in vitro and in vivo (Hendrich and Bird, 1998; Wade et al., 1999; Saito and Ishikawa, 2002). Sequence comparison of 5mC binding competent MBD domains revealed two highly conserved residues, which are altered in mammalian Mbd3: a largely solvent exposed tyrosine, as well as an amino-terminal lysine or arginine residue (Ohki et al., 1999; Wakefield et al., 1999; Saito and Ishikawa, 2002).
Despite its inability to recognize 5mC, three different Mbd3 isoforms (Mbd3a–c) that vary in their amino termini were detected within the NuRD repression complex in embryonic stem cells (Zhang et al., 1999; Kaji et al., 2006). ESCs lacking Mbd3-NuRD displayed a severe defect in differentiation that lead to persistent self-renewal even in the absence of leukemia inhibitory factor (Kaji et al., 2006). More recently, depletion of Mbd3 in somatic cells was shown to enhance the reprogramming efficiency of the four Yamanaka factors (Oct4, Sox2, Klf4, and Myc; Luo et al., 2013; Rais et al., 2013). Accordingly, Mbd3 was proposed to play a key role in lineage commitment and pluripotency (Yildirim et al., 2011; Reynolds et al., 2012; Whyte et al., 2012). Contradictory studies using neural and epiblast-derived stem cells, however, indicate a role for Mbd3 in facilitating induction of pluripotency and argue that its function may be context specific (dos Santos et al., 2014).
Binding sites of Mbd3 have been mapped genome-wide in mouse and human cells (Yildirim et al., 2011; Baubec et al., 2013; Gunther et al., 2013; Shimbo et al., 2013). While Yildirim et al. (2011) identified Mbd3 bound to TSS of CpG-rich, hydroxymethylation marked promoters, Baubec et al. (2013) found Mbd3 bound to enhancers independent of CpG density and (hydroxy)methylation status. Further data questioning the interaction of Mbd3 with hydroxymethylated DNA was provided by Spruijt et al. (2013), who did not detect Mbd3 among hydroxymethylation-specific readers.
Although both, Mbd2 and Mbd3 associate with the NuRD complex, the two MBD containing complexes appear to have no functional overlap since knockout of Mbd3 in mice is embryonic lethal, whereas Mbd2-deficient mice are viable and fertile (Hendrich et al., 2001; Table 3).
Mbd4, also referred to as MED1 (Bellacosa et al., 1999), is the only known member of the MBD protein family not associated with HDAC activity (Hendrich and Bird, 1998). Instead, several lines of evidence suggest that Mbd4 plays a role in DNA repair (Bader et al., 1999; Bellacosa et al., 1999; Hendrich et al., 1999; Riccio et al., 1999; Petronzelli et al., 2000; Millar et al., 2002). In addition to its MBD, Mbd4 contains a C-terminal catalytic domain that is highly homologous to bacterial DNA damage specific endonucleases that exhibit glycosylase activity during BER (Michaels et al., 1990; Hendrich and Bird, 1998). Accordingly, Mbd4 was shown to remove thymine or uracil from mismatched CpG sites through glycosidic bond cleavage. As genomic G/T mismatches are the expected product of 5mCpG deamination, Mbd4 has been designated a methylation specific DNA repair enzyme (Hendrich et al., 1999; Petronzelli et al., 2000; Hashimoto et al., 2012b). Furthermore, Mbd4 has been implicated in DNA demethylation as it was shown in vitro to excise 5hmU, the deamination product of 5hmC (Hashimoto et al., 2012b).
Knockout and rescue experiments in embryonic stem cells, however, demonstrated that oxidation-dependent reactivation of methylated reporter genes is mediated by the action of thymine DNA glycosylase (TDG), but not by Mbd4 (Muller et al., 2014). Accordingly, deamination of 5hmC to 5hmU and subsequent excision by Mbd4 does not play a major role in ESCs (Hashimoto et al., 2012b). A contribution of Mbd4 to Tet-initiated DNA demethylation in NPCs, however, cannot be excluded, since Mbd4 was shown to bind to 5hmC at this developmental stage (Spruijt et al., 2013).
Mice lacking Mbd4 are viable and fertile (Millar et al., 2002). However, compared to wild-type mice, Mbd4 knockout lead to a 3.3-fold higher number of C:G to T:A transitions at CpG sites (Millar et al., 2002). Moreover, Mbd4-/- mice that were made heterozygous for the Min allele of the adenomatous polyposis coli gene (ApcMin), which pre-disposes mice to develop spontaneous intestinal neoplasia (Su et al., 1992), showed markedly reduced survival compared to Mbd4+/+ controls. Accordingly, Mbd4 plays an important role in the repair of 5mC deamination at mCpGs. The relatively mild phenotype of Mbd4 knockout mice (Table 3), however, suggests that its absence might be compensated for by other glycosylases, such as TDG.
Members of the Kaiso-like protein family (Figure 6) present a second class of proteins capable of binding specifically to methylated DNA (Filion et al., 2006). In contrast to members of the MBD protein family, Kaiso, Zbtb4, and Zbtb38 contain a conserved BTB/POZ (Bric-a-brac, tramtrack, broad complex/poxvirus and zinc finger) domain involved in protein–protein interactions and three Kruppel-like C2H2 zinc finger motifs, of which two were found essential for binding to methylated DNA (Filion et al., 2006). Similar to MBD proteins, members of the Kaiso family function as HDAC-dependent transcriptional repressors (Sasai et al., 2005). Several lines of evidence, however, including their variable binding modes, protein partners and expression patterns, suggest that Kaiso-like proteins have different biological functions (Daniel and Reynolds, 1999; Kiefer et al., 2005; Park et al., 2005; Filion et al., 2006).
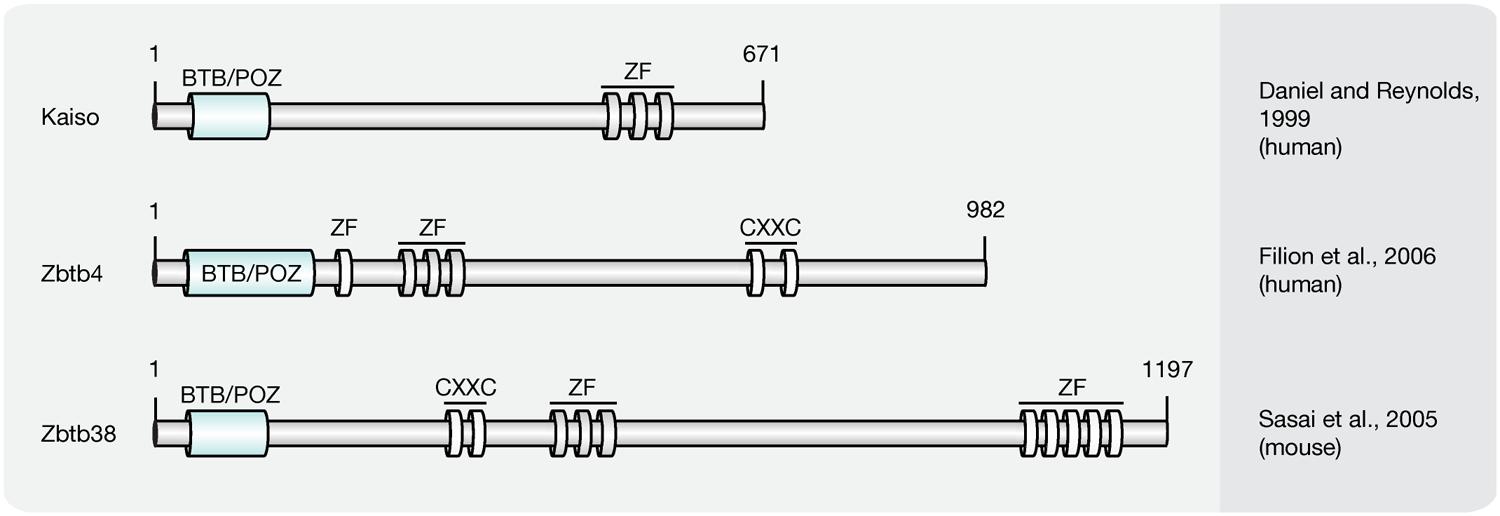
FIGURE 6. Schematic representation of the Kaiso-like protein family. Shown are domain structures of mouse Kaiso-like proteins and the initial references. Numbers represent amino acid positions. BTB/POZ, broad complex, tramtrack and bric a brac/poxvirus and zinc finger domain; ZF, zinc finger; CXXC, CXXC zinc finger domain.
While Kaiso was shown to require at least two methylated CpG dinucleotides, a single mCpG proved sufficient for efficient binding of the Zbtb4 and Zbtb38 proteins (Prokhortchouk et al., 2001; Filion et al., 2006). Besides its ability to bind methylated DNA, in vitro synthesized Kaiso was shown to interact specifically with an unmethylated consensus sequence, the Kaiso binding site (KBS, TCCTGCNA), which can be found at promoters of Wnt target genes (Daniel and Reynolds, 1999; Park et al., 2005). Accordingly, the xWnt11 gene, a target of non-canonical Wnt signaling, was shown to be regulated by Kaiso in Xenopus (Daniel and Reynolds, 1999; Prokhortchouk et al., 2001). Moreover, Kaiso-mediated repression of non-canonical and canonical Wnt targets was repressed by interactions with p120-catenin (Kim et al., 2004), as it competes with DNA for the access to the Kaiso zinc finger domains (Daniel et al., 2002). The ability to bind unmethylated KBS sequences is shared by Zbtb4. Zbtb38, however, was shown to interact with the E-box motif (CACCTG) of the rat tyrosine hydroxylase gene promoter (Kiefer et al., 2005), but failed to bind a labeled KBS probe (Filion et al., 2006). More recently, Kaiso was found to bind 5hmC in NPCs and Zbtb4 was pulled down with hydroxymethylated DNA from brain tissue (Spruijt et al., 2013). The 5hmC binding domains, as well as the biological function, however, remain to be determined.
Kaiso-like proteins contain a BTB/POZ domain, which facilitates interaction with different sets of co-repressors and mediate transcriptional repression.
Kaiso was shown to recruit the NCoR complex to promoters of target genes to introduce histone hypoacetylation, as well as H3K9 methylation (Yoon et al., 2003). Moreover, Kaiso was identified as component of an alternative MeCP1 complex in NIH3T3 cells (Prokhortchouk et al., 2001). Zbtb38 was found to interact with the co-repressors CtBPs (C-terminal binding proteins), which include HDAC, methyltransferase, and demethylase activities (Sasai et al., 2005; Zocchi and Sassone-Corsi, 2012). Zbtb4 was shown to associate with the Sin3A/HDAC complex to repress expression of p21CIP1 in response to stimuli that activate p53 (Weber et al., 2008).
Kaiso-like proteins exhibit diverging expression patterns. While Kaiso is ubiquitously expressed, Zenon, the rat homolog of ZBTB38, is primarily transcribed in brain and neuroendocrine tissues (Kiefer et al., 2005). For Zbtb4, in contrast, high expression levels were identified in brain, lung, kidney, muscle, and heart (Filion et al., 2006).
Kaiso-null mice (Table 3) are viable and fertile, with no detectable changes in gene expression profiles or developmental abnormalities. However, when crossed with tumor-susceptible Apc(Min/+) mice, Kaiso-deficient animals showed resistance to intestinal cancer (Prokhortchouk et al., 2006).
Recent studies implicate that yet another protein fold, the SRA domain could read DNA (hydroxy)methylation marks in vitro and in vivo (Unoki et al., 2004; Johnson et al., 2007; Woo et al., 2007; Frauer et al., 2011a; Spruijt et al., 2013). In mammals, two SRA domain-containing proteins (Figure 7), Np95 (mouse homolog of human ICBP90, gene name UHRF1) and Np97 (mouse homolog of human NIRF, gene name UHRF2), have been characterized (Unoki et al., 2004; Woo et al., 2007; Zhang et al., 2011). While Np95 was first discovered during the generation process of antibodies against murine thymic lymphoma (Fujimori et al., 1998), NIRF was identified through screenings for PCNP (PEST containing nuclear protein) interaction partners (Mori et al., 2002).
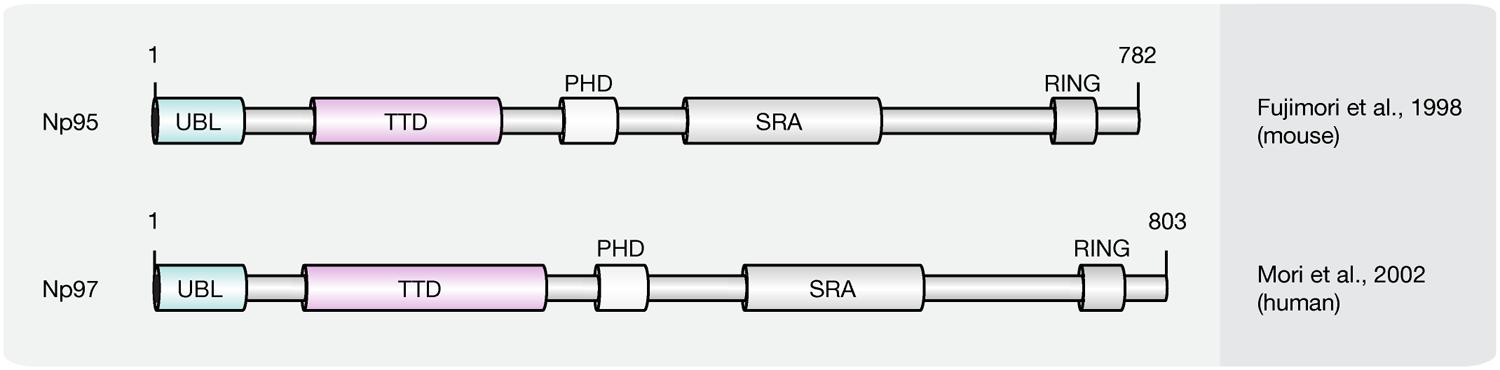
FIGURE 7. Schematic representation of the SRA domain protein family. Shown are domain structures of mouse SRA domain proteins and the initial references. Numbers represent amino acid positions. UBL, ubiquitin-like domain; TTD, tandem tudor domain; PHD, plant homeodomain; SRA, SET, and RING finger associated domain; RING, really interesting new gene.
Besides the eponymous SRA domain, ICBP90 contains at least four additional functional motifs (Hashimoto et al., 2009): an N-terminal ubiquitin-like domain (Ubl, or NIRF_N); a tandem Tudor domain (TTD) that binds histone H3 tails di/tri-methylated at lysine 9 (H3K9me2/3; Karagianni et al., 2008; Papait et al., 2008; Rottach et al., 2010); a PHD, which binds (un)modified histones; and a C-terminal RING, which exhibits ubiquitin E3 ligase activity.
ICBP90 and Np95 play a critical role in epigenetic inheritance and maintenance of DNA methylation (Bostick et al., 2007; Sharif et al., 2007). Accordingly, ICBP90/Np95 was shown to colocalize with PCNA during S phase and to interact with Dnmt3a, Dnmt3b and several histone-modifying enzymes like HDAC1, as well as histone methyltransferase G9a (Achour et al., 2009; Kim et al., 2009; Meilinger et al., 2009). Moreover, besides its ability to bind and flip out hemi-methylated DNA, the SRA domain of ICBP90 was shown to target Dnmt1 to replicating pericentric heterochromatin for maintenance methylation (Bostick et al., 2007; Arita et al., 2008; Avvakumov et al., 2008; Hashimoto et al., 2008; Papait et al., 2008). In addition, ICBP90 was shown to bind histone H3K9me2/3 via its TTD, thus connecting repressive histone marks with DNA methylation (Rottach et al., 2010; Nady et al., 2011; Rothbart et al., 2012). The PHD of ICBP90, on the other hand, was found associated with the N-terminal tail of histone H3 (Papait et al., 2007; Hu et al., 2011; Rajakumara et al., 2011; Wang et al., 2011; Arita et al., 2012; Cheng et al., 2013). More recently, the SRA domain of Np95 was demonstrated to bind 5hmC and 5mC containing DNA substrates with similar affinity in vitro (Frauer et al., 2011a). Consistent with this, Np95 was identified as 5hmC reader in mESCs and NPCs. In mouse brain tissue, however, association with 5hmC remained undetected likely due to its low expression levels. Although the structure of NIRF, the second member of the SRA domain protein family, is closely related to ICBP90, both proteins possess significantly different expression patterns. While ICBP90 is mainly expressed in proliferating cells (Fujimori et al., 1998), NIRF protein levels increase during differentiation (Pichler et al., 2011). NIRF binds hemi-methylated DNA and H3K9me2/3 containing heterochromatin marks in a cooperative manner, whereby localization and in vivo binding dynamics of NIRF, were shown to require an intact TTD and depend on H3K9me3 but not on DNA methylation (Pichler et al., 2011). While Np95 was shown to bind 5hmC in mESCs and NPCs, the interaction of Np97 and 5hmC was specific for NPCs. Furthermore, Np97 exhibited higher binding affinity for 5hmC than for 5mC in NPCs (Spruijt et al., 2013). Finally, Np97 was proposed to promote repetitive oxidation of 5mC by Tet proteins, since the levels of the oxidative cytosine derivatives 5hmC, 5fC and 5caC were increased upon coexpression of Np97 and Tet1 in HEK293T cells (Spruijt et al., 2013). Consequently, Spruijt et al. (2013), hypothesized that flipping of the modified base, as previously described for Np95, may enhance the accessibility of Tet enzymes to the hydroxymethylated base, whereby further oxidation is promoted.
Furthermore, ectopic Np97 was unable to rescue DNA methylation defects observed in Np95-/- ESCs. Neither DNA methylation levels, nor pericentric heterochromatin localization of Dnmt1 in S-phase could be restored upon overexpression of Np97 arguing for functional differences between both proteins (Pichler et al., 2011). NIRF was found to interact with cell cycle proteins including cyclins, cyclin-dependent kinases (CDKs), retinoblastoma protein (pRB), p53, PCNA, HDAC1, DNMTs, and G9a (Mori et al., 2012). It was shown to ubiquitinate cyclins D1 and E1, and to induce G1 arrest. Accordingly, NIRF was proposed to link the cell cycle regulatory network with the epigenetic landscape (Mori et al., 2012).
While knockout of Np95 leads to developmental arrest shortly after gastrulation and early gestational lethality (Sharif et al., 2007), the phenotype of Np97 null mice has not been analyzed (Li et al., 2013; Table 3).
Mutations in proteins involved in writing, reading, and modifying the epigenetic landscape have been implicated in various severe human disorders. Due to their high sequence (Table 4) and functional similarity (Kumar et al., 1994; Hendrich and Bird, 1998; Mori et al., 2002; Filion et al., 2006; Bostick et al., 2007; Ito et al., 2010; Qin et al., 2011), we, hereafter, summarize the state-of-the-art regarding the role of the human orthologs of the aforementioned mouse Dnmts, Tets, and MBPs in human diseases.
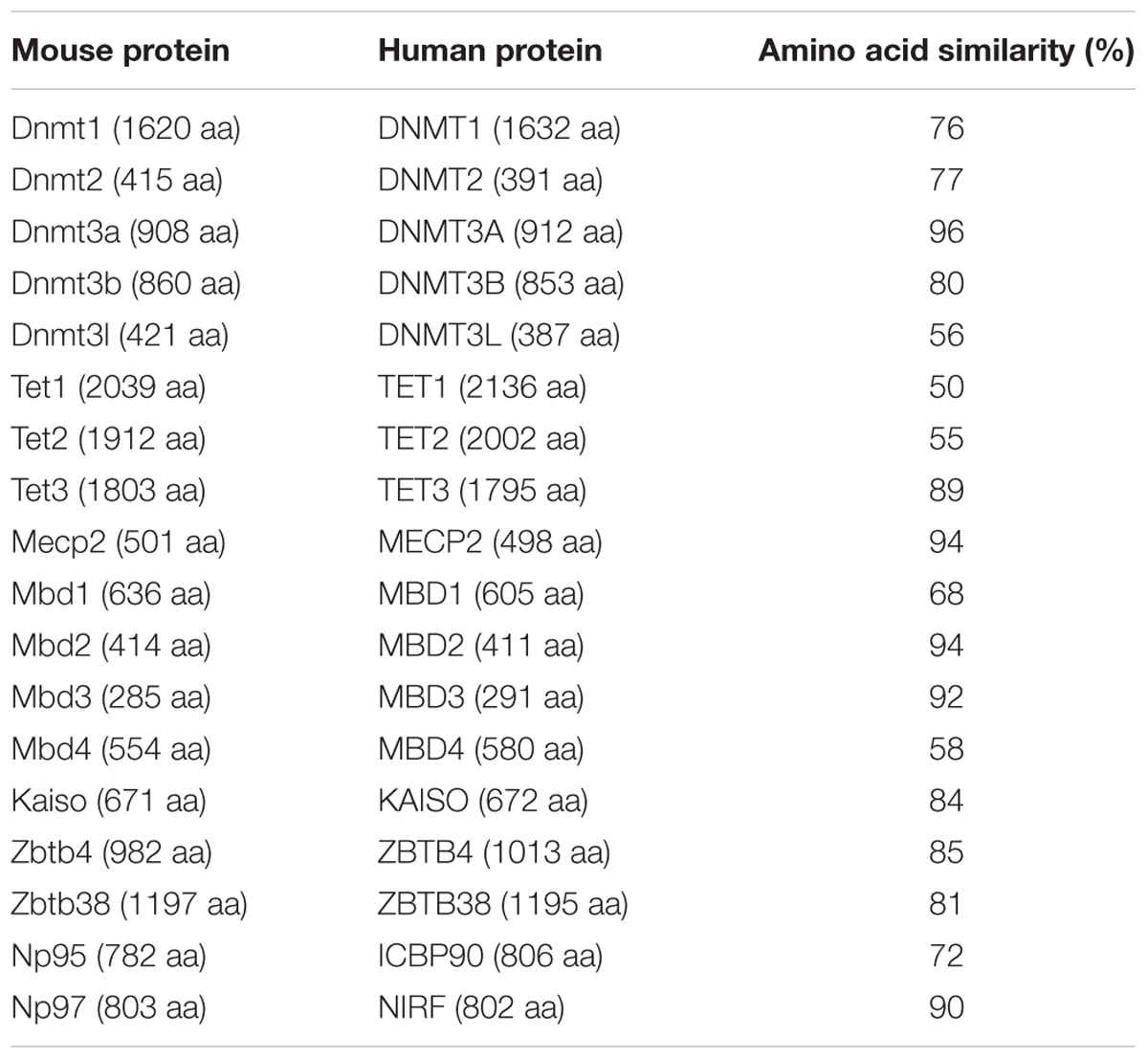
TABLE 4. Comparison of human proteins and their mouse orthologs.
Since Dnmt1 knockout is embryonic lethal in mice, it is unlikely to expect a human disease linked to a DNMT1 catalytic domain mutation. But mutations in the regulatory domain of DNMT1 were found (Table 5). Mutations in the TS domain of DNMT1 cause neurodegeneration like hereditary sensory autonomic neuropathy with dementia and hearing loss (HSAN1E; Klein et al., 2011) and autosomal dominant cerebellar ataxia, deafness and narcolepsy (ADCA-DN; Winkelmann et al., 2012). Mutations of Y495C, Y495H, D490E-P491Y (Klein et al., 2011, 2013) in exon 20 cause HSAN1E. Those mutations caused premature degradation of mutant proteins, reduced methyltransferase activity and impaired heterochromatin binding during G2 phase leading to global hypomethylation and site-specific hypermethylation (Klein et al., 2011). ADCA-DN is a polymorphic disorder first described in 1995 in a Swedish pedigree. Unlike mutations in HSAN1E located in exon 20, mutations in ADCA-DN including A570V, G605A, and V606F were found in exon 21 of the DNMT1 gene.
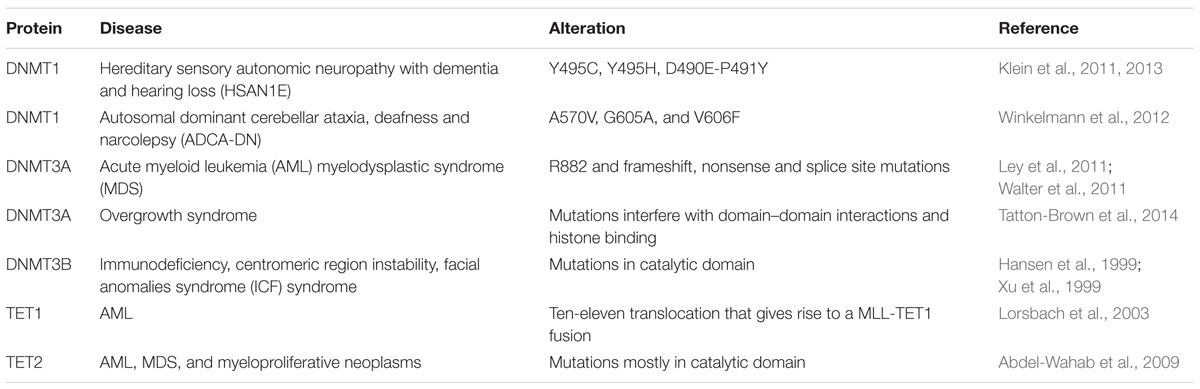
TABLE 5. Summary of disease-related DNMT and TET mutations.
Mutations in DNMT3A were found in de novo AML and are associated with poor survival (Table 5; Ley et al., 2011). The most frequent mutation occurred in amino acid R882, however, frameshift, nonsense and splice site mutations were also reported (Ley et al., 2011). Mutations of DNMT3A are not only observed in AML patients, but also in MDS. Similar to mutations leading to AML, amino acid R882 located in the methyltransferase domain of DNMT3A is the most common mutation site (Walter et al., 2011). Unlike in AML and MDS, most mutations in overgrowth syndrome do not directly affect the catalytic activity of DNMT3A, but interfere with domain–domain interactions and histone binding, which further affect the activity of DNMT3A (Tatton-Brown et al., 2014).
ICF syndrome (immunodeficiency, chromosomal instability, and facial anomalies), a human genetic disorder is caused by DNMT3B mutations (Table 5; Hansen et al., 1999; Xu et al., 1999). Several mutations were identified and most mutations are located in the catalytic domain of DNMT3B and directly affect the activity of DNMT3B (Xu et al., 1999). However, mutations, which do not directly affect its catalytic activity were also observed in ICF syndrome. Two mutations, A766P and R840Q displayed similar methylation activity than the wild-type enzyme but lost the ability to interact with DNMT3L, which further leads to loss of activity in vivo (Xie et al., 2006). Direct or indirect loss of DNMT3B activity consequently decreased satellite DNA methylation in ICF syndrome patients, indicating that DNMT3B is involved in maintaining genome stability.
5mC, the product of DNMTs is related to tumorigenesis. It was shown that the genome of cancer cells is globally hypomethylated relative to their normal counterparts. Usually, hypomethylation leads to gene activation. In cancer cells, the activation of genes is caused by hypomethylation of nearby CGIs, which are silenced in somatic tissues by DNA methylation (Strichman-almashanu et al., 2002). Satellite sequences and repetitive sequences such as LINE1, SINE, IAP, and Alu elements are silenced mainly by DNA methylation in normal cells. However, in tumor cells, hypomethylation of L1 promoter was detected and the activation of L1 might promote chromosomal rearrangements and genome instability (Suter et al., 2004). Although the cancer genome is hypomethylated, several studies showed that Dnmts are upregulated in cancer cells (Ahluwalia et al., 2001; Lin et al., 2007; Roll et al., 2008), suggesting that demethylation enzymes might be additionally involved in loss of DNA methylation in cancer.
MLL gene is located in 11q23 and is the most frequent cytogenetic finding in AML. In AML, MLL is translocated to chromosome 10 as a fusion with the TET1 gene. The MLL-TET1 fusion protein contains the AT hooks, subnuclear localization domains, and the CXXC domain of MLL and the C-terminus of TET1 (Table 5; Lorsbach et al., 2003). The function of MLL-TET1 fusion protein is still unknown, but it was showed that TET1 is involved in MLL-rearranged leukemia. TET1 is a direct target of the MLL-fusion protein and is significantly upregulated in MLL-rearranged leukemia, leading to a global increase 5hmC, thus playing an oncogenic role (Huang et al., 2013).
In myeloproliferative neoplasms, mutations of TET2 but not TET1 and TET3 were observed (Table 5; Abdel-Wahab et al., 2009). Mutations of TET2 were also observed in AML with varied frequency and most of them occurred in the catalytic domain of TET2. In AML, TET2 mutations correlate with genomic 5hmC level (Konstandin et al., 2011). TET2 is one of the most frequently mutated genes in MDS. Mutations of TET2 were detected in most of the bone marrow cells in MDS and these mutations contribute to the malignant transformation of bone marrow cells (Langemeijer et al., 2009), which consequently displayed uniformly low levels of 5hmC in genomic DNA compared to bone marrow samples from healthy controls (Ko et al., 2010).
Besides the hematopoietic malignancies, 5hmC levels are also changed in solid tumors. 5hmC level were profoundly reduced in glioma, colon cancer, breast cancer, and melanoma compared to normal tissues (Haffner et al., 2011; Jin et al., 2011; Li and Liu, 2011; Xu W. et al., 2011; Kraus et al., 2012).
Unlike in cancer, in the hippocampus/parahippocampal gyrus (HPG) of preclinical and later-stage Alzheimer’s disease patients, significantly increased levels of TET1, 5mC, and 5hmC were observed. In contrast, levels of 5fC and 5caC were significantly decreased in the HPG of these patients (Bradley-Whitman and Lovell, 2013). This indicates that DNA methylation might play an important role in memory-related disease.
As readers and translators of epigenetic information, alterations in MBP sequences affect the precisely coordinated link between DNA methylation, histone modification and higher order chromatin structure.
Mutations in the X-linked MECP2 gene give rise to RTT (Table 6), a late onset (6–18 months post-birth) debilitating neurological disease that affects 1 in 10,000–15,000 female live births (Hagberg et al., 1983; Amir et al., 1999). After a period of normal development (6–18 months), RTT patients usually lose speech and acquired motor skills (Hagberg et al., 1983). They are afflicted with seizures, autism, loss of motor coordination, abnormal breathing and develop stereotypical, repetitive hand movements (Hagberg et al., 1983). After the initial regression, however, conditions often stabilize and allow viability until adulthood (Rett, 1966; Hagberg et al., 1983).
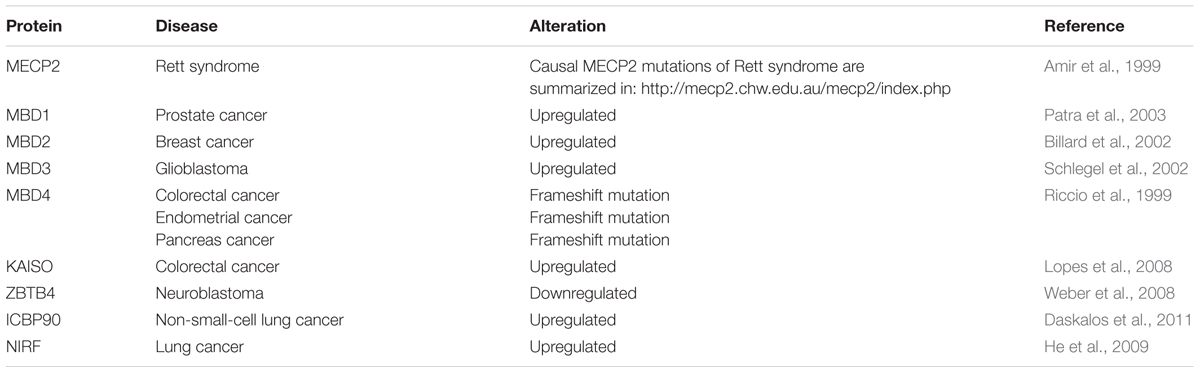
TABLE 6. Summary of disease-related MBP alterations.
Although the first patients were described in 1966 by Andreas Rett (Rett, 1966), more than 30 years passed before mutations within the MECP2 gene located in Xq28 were identified as the cause of the neurological disorder (Amir et al., 1999). The most frequent mutations observed in patients suffering from RTT are missense mutations that cluster within the MBD (aa 78–162), as well as nonsense mutations primarily found within the TRD (aa 207–310; Christodoulou et al., 2003). In Xenopus, missense mutations R106W, R133C, F155S, and T158M were shown to reduce the binding ability of Mecp2 to methylated DNA (Ballestar et al., 2000). Studies in mouse cells showed that the majority of MBD-related missense mutations affected the heterochromatin binding and/or clustering ability of Mecp2 (Agarwal et al., 2011). By artificially targeting chromatin binding deficient Rett mutants (R111G, R133L, and F155S) to constitutive heterochromatic regions, however, Casas-Delucchi et al. (2012) revealed that some of these mutations exclusively affect the chromatin binding but not linking ability. Mutations within the TRD have been shown to influence protein–protein interactions. In knock-in mice bearing the common RTT mutation R306C, neuronal activity fails to induce T308 phosphorylation, a PTM required to suppress the interaction of Mecp2 with the co-repressor complex NCoR. Accordingly, R306C mutations result in persistent association of both proteins leading to decreased induction of a subset of activity-related genes (Ebert et al., 2013; Lyst et al., 2013). In addition to missense and nonsense mutations, reading frame shifts and C-term deletions were shown to give rise to RTT. Mice bearing a truncating mutation similar to those found in RTT patients showed normally localized Mecp2 proteins (Shahbazian et al., 2002). Histone H3, however, was hyperacetylated indicating abnormal chromatin architecture and misregulated gene expression (Shahbazian et al., 2002). Moreover, Muotri et al. (2010) identified increased susceptibility for L1 transposition and genome instability in RTT patients with truncating mutations.
In addition to RTT, Mecp2 was implicated in other neurological diseases, including Hirschsprung’s disease, autism spectrum disorder, schizophrenia, Prader-Willi, and Angelman syndromes (Carney et al., 2003; Shibayama et al., 2004; Nagarajan et al., 2006; Loat et al., 2008; Ramocki et al., 2009; Zhou et al., 2013).
More recently MBP have been associated with several types of human cancers (Table 6). While Mecp2 was overexpressed in estrogen receptor positive human breast cancer (Muller et al., 2003), MBD1 mRNA and protein levels were increased in prostate cancer (Patra et al., 2003). Accordingly, Patra et al. (2003) proposed MBD1 as the major cause of hypermethylated chromatin regions in prostate cancer through the recruitment of HDAC1/2 and subsequent histone deacetylation. MBD2 mRNA level were shown to be significantly elevated in benign tumors of the breast and correlated with tumor size of invasive ductal carcinomas, the most common type of breast cancer (Billard et al., 2002). Accordingly, upregulation of MBD2 was proposed to be associated with breast cell proliferation (Billard et al., 2002). Increased expression of MBD3 and MBD4 were associated with malignant glioma of the brain, and the grade of malignancy correlated with MBD3/4 expression level (Schlegel et al., 2002). Furthermore, frameshift mutations of MBD4 have been identified in colorectal, endometrial and pancreatic cancer with microsatellite instability (Riccio et al., 1999). MBD4 mutations consisted of 1- to 2-bp deletions or 1-bp insertions that caused frameshifts and premature stop codons. The resultant truncated MBD4 proteins were predicted to be non-functional, as they lack the C-terminal catalytic domain, whereby genomic instability was proposed to steadily increase (Riccio et al., 1999). As a regulator of target genes of the canonical and non-canonical Wnt pathway, Kaiso was shown to mediate silencing of tumor suppressor genes CDKN2A and HIC1 in Wnt-driven human colon cancer cell lines (Lopes et al., 2008). Kaiso depletion induced expression of tumor suppressor genes without altering DNA methylation levels (Lopes et al., 2008). As a result, colon cancer cells became susceptible to cell cycle arrest and cell death induced by chemotherapy (Lopes et al., 2008). Accordingly, Lopes et al. (2008) suggested Kaiso as a methylation-dependent oncogene that represses hypermethylated tumor suppressor genes. ZBTB4 expression was shown to be downregulated in advanced stages of human neuroblastoma and multiple human solid tumors (Weber et al., 2008). As a repressor of the P21CIP1 gene, an inhibitor of the Cdk2 kinase, ZBTB4 usually blocks cell cycle arrest in response to p53 activation (Weber et al., 2008). Consequently, loss of ZBTB4 inhibits apoptosis and favors long-term survival of affected cells (Weber et al., 2008). In tumors, where many promoter-associated CGIs are hypermethylated, maintenance of methylation plays a major role. Accordingly, elevated levels of ICBP90 were shown to control cell cycle through maintenance of promoter methylation at CDK2A and RASSF1 in non-small-cell lung cancer (Daskalos et al., 2011). Finally, decreased expression of let-7a miRNA in lung cancer was shown to result in elevated NIRF and reduced P21CIP1 protein level, thereby most likely contributing to lung carcinogenesis (He et al., 2009).
In summary, alterations in 5mC writers, readers, and modifiers that affect their level, PTMs, ability to bind and/or modify DNA and protein interactions are each and all potential mechanisms contributing to altered chromatin composition and structure as well as genome activity and stability (Figure 8) and contribute to an overwhelming variety of human diseases. Despite intensive research, genotype–phenotype connections have been generally difficult to establish and subsequent studies are urgently needed to elucidate potential strategies for diagnostic and therapeutic applications.
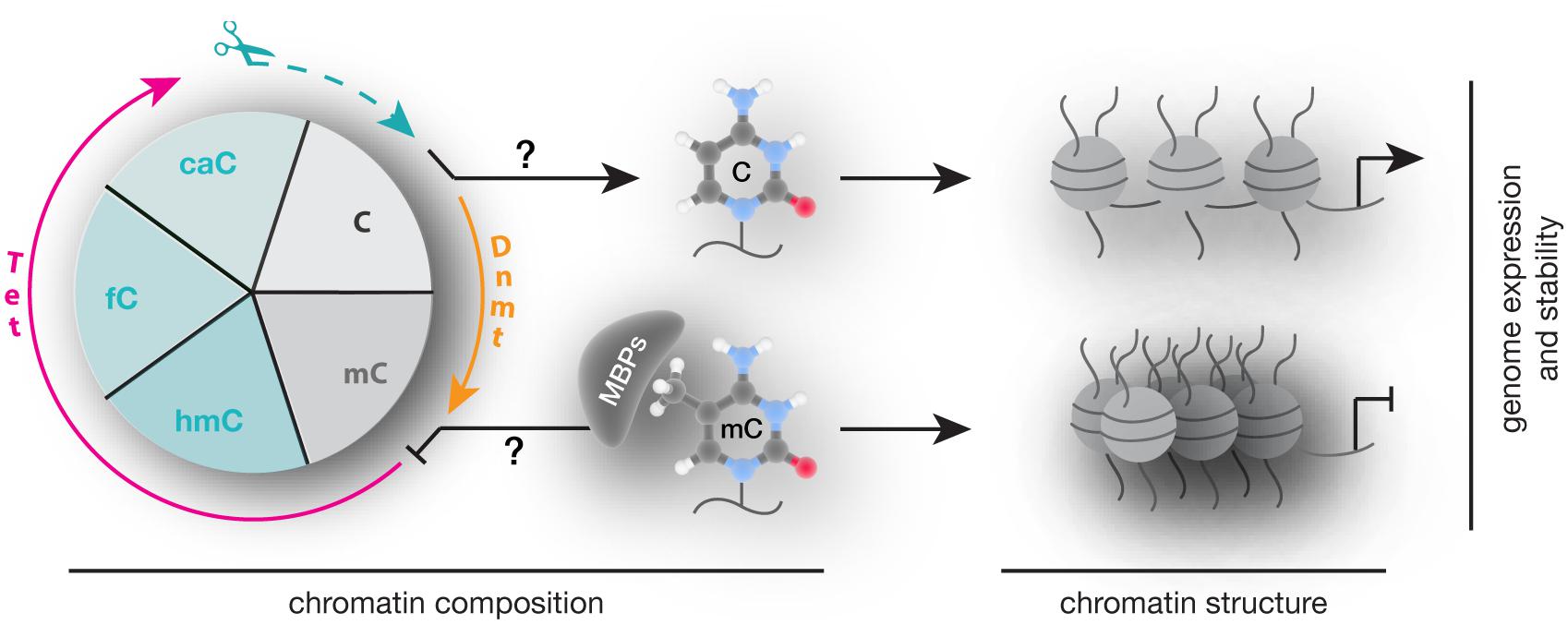
FIGURE 8. Writing, reading, and translating DNA modifications. Graphical summary of how DNA modification writers, readers, and translators can impact on chromatin composition, structure (nucleosomes are represented as balls, DNA as line) as well as genome expression (arrow represents active promoters) and stability.
All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
Our research has been supported by grants of the German Research Foundation (DFG), the Volkswagen Foundation, and the German Ministry for Education and Research (BMBF). PZ was funded by a fellowship of the Chinese Scholarship Council.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We apologize to the colleagues whose work was not cited due to space constraints. We thank all the past and present members of our laboratory for their many contributions along the years. Last but not least, we thank our collaborators over the years, which have made our work so much more enjoyable.
Aapola, U., Kawasaki, K., Scott, H. S., Ollila, J., Vihinen, M., Heino, M., et al. (2000). Isolation and initial characterization of a novel zinc finger gene, DNMT3L, on 21q22.3, related to the cytosine-5-methyltransferase 3 gene family. Genomics 65, 293–298. doi: 10.1006/geno.2000.6168
Abdel-Wahab, O., Mullally, A., Hedvat, C., Garcia-Manero, G., Patel, J., Wadleigh, M., et al. (2009). Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood 114, 144–147. doi: 10.1182/blood-2009-03-210039
Achour, M., Fuhrmann, G., Alhosin, M., Ronde, P., Chataigneau, T., Mousli, M., et al. (2009). UHRF1 recruits the histone acetyltransferase Tip60 and controls its expression and activity. Biochem. Biophys. Res. Commun. 390, 523–528. doi: 10.1016/j.bbrc.2009.09.131
Adams, V. H., McBryant, S. J., Wade, P. A., Woodcock, C. L., and Hansen, J. C. (2007). Intrinsic disorder and autonomous domain function in the multifunctional nuclear protein, MeCP2. J. Biol. Chem. 282, 15057–15064. doi: 10.1074/jbc.M700855200
Agarwal, N., Becker, A., Jost, K. L., Haase, S., Thakur, B. K., Brero, A., et al. (2011). MeCP2 Rett mutations affect large scale chromatin organization. Hum. Mol. Genet. 20, 4187–4195. doi: 10.1093/hmg/ddr346
Ahluwalia, A., Hurteau, J. A., Bigsby, R. M., and Nephew, K. P. (2001). DNA methylation in ovarian cancer. II. Expression of DNA methyltransferases in ovarian cancer cell lines and normal ovarian epithelial cells. Gynecol. Oncol. 82, 299–304. doi: 10.1006/gyno.2001.6284
Akbarian, S., Chen, R. Z., Gribnau, J., Rasmussen, T. P., Fong, H., Jaenisch, R., et al. (2001). Expression pattern of the Rett syndrome gene MeCP2 in primate prefrontal cortex. Neurobiol. Dis. 8, 784–791. doi: 10.1006/nbdi.2001.0420
Amir, R. E., Van den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188. doi: 10.1038/13810
Amouroux, R., Nashun, B., Shirane, K., Nakagawa, S., Hill, P. W. S., D’Souza, Z., et al. (2016). De novo DNA methylation drives 5hmC accumulation in mouse zygotes. Nat. Cell Biol. 18, 225–233. doi: 10.1038/ncb3296
Aoki, A., Suetake, I., Miyagawa, J., Fujio, T., Chijiwa, T., Sasaki, H., et al. (2001). Enzymatic properties of de novo-type mouse DNA (cytosine-5) methyltransferases. Nucleic Acids Res. 29, 3506–3512. doi: 10.1093/nar/29.17.3506
Arita, K., Ariyoshi, M., Tochio, H., Nakamura, Y., and Shirakawa, M. (2008). Recognition of hemi-methylated DNA by the SRA protein UHRF1 by a base-flipping mechanism. Nature 455, 818–821. doi: 10.1038/nature07249
Arita, K., Isogai, S., Oda, T., Unoki, M., Sugita, K., Sekiyama, N., et al. (2012). Recognition of modification status on a histone H3 tail by linked histone reader modules of the epigenetic regulator UHRF1. Proc. Natl. Acad. Sci. U.S.A. 109, 12950–12955. doi: 10.1073/pnas.1203701109
Avvakumov, G. V., Walker, J. R., Xue, S., Li, Y., Duan, S., Bronner, C., et al. (2008). Structural basis for recognition of hemi-methylated DNA by the SRA domain of human UHRF1. Nature 455, 822–825. doi: 10.1038/nature07273
Bachman, M., Uribe-Lewis, S., Yang, X., Williams, M., Murrell, A., and Balasubramanian, S. (2014). 5-Hydroxymethylcytosine is a predominantly stable DNA modification. Nat. Chem. 6, 1049–1055. doi: 10.1038/nchem.2064
Bader, S., Walker, M., Hendrich, B., Bird, A., Bird, C., Hooper, M., et al. (1999). Somatic frameshift mutations in the MBD4 gene of sporadic colon cancers with mismatch repair deficiency. Oncogene 18, 8044–8047. doi: 10.1038/sj.onc.1203229
Ballestar, E., Yusufzai, T. M., and Wolffe, A. P. (2000). Effects of Rett syndrome mutations of the methyl-CpG binding domain of the transcriptional repressor MeCP2 on selectivity for association with methylated DNA. Biochemistry 39, 7100–7106. doi: 10.1021/bi0001271
Baubec, T., Ivanek, R., Lienert, F., and Schubeler, D. (2013). Methylation-dependent and -independent genomic targeting principles of the MBD protein family. Cell 153, 480–492. doi: 10.1016/j.cell.2013.03.011
Bauer, C., Göbel, K., Nagaraj, N., Colantuoni, C., Wang, M., Müller, U., et al. (2015). Phosphorylation of TET proteins is regulated via O-GlcNAcylation by the O-Linked N-Acetylglucosamine transferase (OGT). J. Biol. Chem. 290, 4801–4812. doi: 10.1074/jbc.M114.605881
Baymaz, H. I., Fournier, A., Laget, S., Ji, Z., Jansen, P. W., Smits, A. H., et al. (2014). MBD5 and MBD6 interact with the human PR-DUB complex through their methyl-CpG-binding domain. Proteomics 14, 2179–2189. doi: 10.1002/pmic.201400013
Becker, A., Allmann, L., Hofstatter, M., Casa, V., Weber, P., Lehmkuhl, A., et al. (2013). Direct homo- and hetero-interactions of MeCP2 and MBD2. PLoS ONE 8:e53730. doi: 10.1371/journal.pone.0053730
Becker, A., Zhang, P., Allmann, L., Meilinger, D., Bertulat, B., Eck, D., et al. (2016). Poly(ADP-ribosyl)ation of Methyl CpG Binding Domain Protein 2 regulates chromatin structure. J. Biol. Chem. 291, 4873–4881. doi: 10.1074/jbc.M115.698357
Bednar, J., Horowitz, R. A., Grigoryev, S. A., Carruthers, L. M., Hansen, J. C., Koster, A. J., et al. (1998). Nucleosomes, linker DNA, and linker histone form a unique structural motif that directs the higher-order folding and compaction of chromatin. Proc. Natl. Acad. Sci. U.S.A. 95, 14173–14178. doi: 10.1073/pnas.95.24.14173
Bellacosa, A., Cicchillitti, L., Schepis, F., Riccio, A., Yeung, A. T., Matsumoto, Y., et al. (1999). MED1, a novel human methyl-CpG-binding endonuclease, interacts with DNA mismatch repair protein MLH1. Proc. Natl. Acad. Sci. U.S.A. 96, 3969–3974. doi: 10.1073/pnas.96.7.3969
Bestor, T., Laudano, A., Mattaliano, R., and Ingram, V. (1988). Cloning and sequencing of a cDNA encoding DNA methyltransferase of mouse cells. J. Mol. Biol. 203, 971–983. doi: 10.1016/0022-2836(88)90122-2
Bhattacharya, S. K., Ramchandani, S., Cervoni, N., and Szyf, M. (1999). A mammalian protein with specific demethylase activity for mCpG DNA. Nature 397, 579–583. doi: 10.1038/17533
Bian, C., and Yu, X. (2014). PGC7 suppresses TET3 for protecting DNA methylation. Nucleic Acids Res. 42, 2893–2905. doi: 10.1093/nar/gkt1261
Billard, L. M., Magdinier, F., Lenoir, G. M., Frappart, L., and Dante, R. (2002). MeCP2 and MBD2 expression during normal and pathological growth of the human mammary gland. Oncogene 21, 2704–2712. doi: 10.1038/sj/onc/1205357
Birke, M., Schreiner, S., Garcia-Cuellar, M. P., Mahr, K., Titgemeyer, F., and Slany, R. K. (2002). The MT domain of the proto-oncoprotein MLL binds to CpG-containing DNA and discriminates against methylation. Nucleic Acids Res. 30, 958–965. doi: 10.1093/nar/30.4.958
Blaschke, K., Ebata, K. T., Karimi, M. M., Zepeda-Martinez, J. A., Goyal, P., Mahapatra, S., et al. (2013). Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature 500, 222–226. doi: 10.1038/nature12362
Boeke, J., Ammerpohl, O., Kegel, S., Moehren, U., and Renkawitz, R. (2000). The minimal repression domain of MBD2b overlaps with the methyl-CpG-binding domain and binds directly to Sin3A. J. Biol. Chem. 275, 34963–34967. doi: 10.1074/jbc.M005929200
Bostick, M., Kim, J. K., Esteve, P. O., Clark, A., Pradhan, S., and Jacobsen, S. E. (2007). UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 317, 1760–1764. doi: 10.1126/science.1147939
Bourc’his, D., Xu, G. L., Lin, C. S., Bollman, B., and Bestor, T. H. (2001). Dnmt3L and the establishment of maternal genomic imprints. Science (New York) 294, 2536–2539. doi: 10.1126/science.1065848
Bradley-Whitman, M. A., and Lovell, M. A. (2013). Epigenetic changes in the progression of Alzheimer’s disease. Mech. Ageing Dev. 134, 486–495. doi: 10.1016/j.mad.2013.08.005
Brero, A., Easwaran, H. P., Nowak, D., Grunewald, I., Cremer, T., Leonhardt, H., et al. (2005). Methyl CpG-binding proteins induce large-scale chromatin reorganization during terminal differentiation. J. Cell Biol. 169, 733–743. doi: 10.1083/jcb.20002062
Cardoso, M. C., and Leonhardt, H. (1999). DNA methyltransferase is actively retained in the cytoplasm during early development. J. Cell Biol. 147, 25–32. doi: 10.1083/jcb.147.1.25
Carney, R. M., Wolpert, C. M., Ravan, S. A., Shahbazian, M., Ashley-Koch, A., Cuccaro, M. L., et al. (2003). Identification of MeCP2 mutations in a series of females with autistic disorder. Pediatr. Neurol. 28, 205–211. doi: 10.1016/S0887-8994(02)00624-0
Casas-Delucchi, C. S., Becker, A., Bolius, J. J., and Cardoso, M. C. (2012). Targeted manipulation of heterochromatin rescues MeCP2 Rett mutants and re-establishes higher order chromatin organization. Nucleic Acids Res. 40, e176. doi: 10.1093/nar/gks784
Chahrour, M., Jung, S. Y., Shaw, C., Zhou, X., Wong, S. T., Qin, J., et al. (2008). MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320, 1224–1229. doi: 10.1126/science.1153252
Chen, Q., Chen, Y., Bian, C., Fujiki, R., and Yu, X. (2013). TET2 promotes histone O-GlcNAcylation during gene transcription. Nature 493, 561–564. doi: 10.1038/nature11742
Chen, R. Z., Akbarian, S., Tudor, M., and Jaenisch, R. (2001). Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat. Genet. 27, 327–331. doi: 10.1038/85906
Chen, T., Ueda, Y., Dodge, J. E., Wang, Z., and Li, E. (2003). Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol. Cell. Biol. 23, 5594–5605. doi: 10.1128/MCB.23.16.5594
Chen, T., Ueda, Y., Xie, S., and Li, E. (2002). A novel Dnmt3a isoform produced from an alternative promoter localizes to euchromatin and its expression correlates with active de novo methylation. J. Biol. Chem. 277, 38746–38754. doi: 10.1074/jbc.M205312200
Cheng, J., Huang, M., Zhu, Y., Xin, Y. J., Zhao, Y. K., Huang, J., et al. (2014). SUMOylation of MeCP2 is essential for transcriptional repression and hippocampal synapse development. J. Neurochem. 128, 798–806. doi: 10.1111/jnc.12523
Cheng, J., Yang, Y., Fang, J., Xiao, J., Zhu, T., Chen, F., et al. (2013). Structural insight into coordinated recognition of trimethylated histone H3 lysine 9 (H3K9me3) by the plant homeodomain (PHD) and tandem tudor domain (TTD) of UHRF1 (ubiquitin-like, containing PHD and RING finger domains, 1) protein. J. Biol. Chem. 288, 1329–1339. doi: 10.1074/jbc.M112.415398
Christodoulou, J., Grimm, A., Maher, T., and Bennetts, B. (2003). RettBASE: the IRSA MECP2 variation database-a new mutation database in evolution. Hum. Mutat. 21, 466–472. doi: 10.1002/humu.10194
Cohen, S., Gabel, H. W., Hemberg, M., Hutchinson, A. N., Sadacca, L. A., Ebert, D. H., et al. (2011). Genome-wide activity-dependent MeCP2 phosphorylation regulates nervous system development and function. Neuron 72, 72–85. doi: 10.1016/j.neuron.2011.08.022
Costa, Y., Ding, J., Theunissen, T. W., Faiola, F., Hore, T. A., Shliaha, P. V., et al. (2013). NANOG-dependent function of TET1 and TET2 in establishment of pluripotency. Nature 495, 370–374. doi: 10.1038/nature11925
Cross, S. H., Meehan, R. R., Nan, X., and Bird, A. (1997). A component of the transcriptional repressor MeCP1 shares a motif with DNA methyltransferase and HRX proteins. Nat. Genet. 16, 256–259. doi: 10.1038/ng0797-256
Daniel, J. M., and Reynolds, A. B. (1999). The catenin p120(ctn) interacts with Kaiso, a novel BTB/POZ domain zinc finger transcription factor. Mol. Cell. Biol. 19, 3614–3623. doi: 10.1128/MCB.19.5.3614
Daniel, J. M., Spring, C. M., Crawford, H. C., Reynolds, A. B., and Baig, A. (2002). The p120(ctn)-binding partner Kaiso is a bi-modal DNA-binding protein that recognizes both a sequence-specific consensus and methylated CpG dinucleotides. Nucleic Acids Res. 30, 2911–2919. doi: 10.1093/nar/gkf398
Daskalos, A., Oleksiewicz, U., Filia, A., Nikolaidis, G., Xinarianos, G., Gosney, J. R., et al. (2011). UHRF1-mediated tumor suppressor gene inactivation in nonsmall cell lung cancer. Cancer 117, 1027–1037. doi: 10.1002/cncr.25531
Dawlaty, M. M., Breiling, A., Le, T., Raddatz, G., Inmaculada, M., Cheng, A. W., et al. (2014). Combined deficiency of Tet1 and Tet2 causes epigenetic abnormalities but is compatible with postnatal development. Dev. Cell 24, 310–323. doi: 10.1016/j.devcel.2012.12.015.Combined
Dawlaty, M. M., Ganz, K., Powell, B. E., Hu, Y. C., Markoulaki, S., Cheng, A. W., et al. (2011). Tet1 is dispensable for maintaining pluripotency and its loss is compatible with embryonic and postnatal development. Cell Stem Cell 9, 166–175. doi: 10.1016/j.stem.2011.07.010
de Rojas-Walker, T., Tamir, S., Ji, H., Wishnok, J. S., and Tannenbaum, S. R. (1995). Nitric oxide induces oxidative damage in addition to deamination in macrophage DNA. Chem. Res. Toxicol. 8, 473–477. doi: 10.1021/tx00045a020
Deplus, R., Delatte, B., Schwinn, M. K., Defrance, M., Mendez, J., Murphy, N., et al. (2013). TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J. 32, 645–655. doi: 10.1038/emboj.2012.357
Doege, C. A., Inoue, K., Yamashita, T., Rhee, D. B., Travis, S., Fujita, R., et al. (2012). Early-stage epigenetic modification during somatic cell reprogramming by Parp1 and Tet2. Nature 488, 652–655. doi: 10.1038/nature11333
dos Santos, R. L., Tosti, L., Radzisheuskaya, A., Caballero, I. M., Kaji, K., Hendrich, B., et al. (2014). MBD3/NuRD facilitates induction of pluripotency in a context-dependent manner. Cell Stem Cell 15, 102–110. doi: 10.1016/j.stem.2014.04.019
Dragich, J. M., Kim, Y. H., Arnold, A. P., and Schanen, N. C. (2007). Differential distribution of the MeCP2 splice variants in the postnatal mouse brain. J. Comp. Neurol. 501, 526–542. doi: 10.1002/cne.21264
Easwaran, H. P., Schermelleh, L., Leonhardt, H., and Cardoso, M. C. (2004). Replication-independent chromatin loading of Dnmt1 during G2 and M phases. EMBO Rep. 5, 1181–1186. doi: 10.1038/sj.embor.7400295
Ebert, D. H., Gabel, H. W., Robinson, N. D., Kastan, N. R., Hu, L. S., Cohen, S., et al. (2013). Activity-dependent phosphorylation of MeCP2 threonine 308 regulates interaction with NCoR. Nature 499, 341–345. doi: 10.1038/nature12348
Filion, G. J., Zhenilo, S., Salozhin, S., Yamada, D., Prokhortchouk, E., and Defossez, P. A. (2006). A family of human zinc finger proteins that bind methylated DNA and repress transcription. Mol. Cell. Biol. 26, 169–181. doi: 10.1128/MCB.26.1.169-181.2006
Frauer, C., Hoffmann, T., Bultmann, S., Casa, V., Cardoso, M. C., Antes, I., et al. (2011a). Recognition of 5-hydroxymethylcytosine by the Uhrf1 SRA domain. PLoS ONE 6:e21306. doi: 10.1371/journal.pone.0021306
Frauer, C., Rottach, A., Meilinger, D., Bultmann, S., Fellinger, K., Hasenöder, S., et al. (2011b). Different binding properties and function of CXXC zinc finger domains in Dnmt1 and Tet1. PLoS ONE 6:e16627. doi: 10.1371/journal.pone.0016627
Fujimori, A., Matsuda, Y., Takemoto, Y., Hashimoto, Y., Kubo, E., Araki, R., et al. (1998). Cloning and mapping of Np95 gene which encodes a novel nuclear protein associated with cell proliferation. Mamm. Genome 9, 1032–1035. doi: 10.1007/s003359900920
Fujita, N., Takebayashi, S., Okumura, K., Kudo, S., Chiba, T., Saya, H., et al. (1999). Methylation-mediated transcriptional silencing in euchromatin by methyl-CpG binding protein MBD1 isoforms. Mol. Cell. Biol. 19, 6415–6426. doi: 10.1128/MCB.19.9.6415
Fujita, N., Watanabe, S., Ichimura, T., Tsuruzoe, S., Shinkai, Y., Tachibana, M., et al. (2003). Methyl-CpG binding domain 1 (MBD1) interacts with the Suv39h1-HP1 heterochromatic complex for DNA methylation-based transcriptional repression. J. Biol. Chem. 278, 24132–24138. doi: 10.1074/jbc.M302283200
Fuks, F., Hurd, P. J., Wolf, D., Nan, X., Bird, A. P., and Kouzarides, T. (2003). The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J. Biol. Chem. 278, 4035–4040. doi: 10.1074/jbc.M210256200
Gao, Y., Chen, J., Li, K., Wu, T., Huang, B., Liu, W., et al. (2013). Replacement of Oct4 by Tet1 during iPSC induction reveals an important role of DNA methylation and hydroxymethylation in reprogramming. Cell Stem Cell 12, 453–469. doi: 10.1016/j.stem.2013.02.005
Georgel, P. T., Horowitz-Scherer, R. A., Adkins, N., Woodcock, C. L., Wade, P. A., and Hansen, J. C. (2003). Chromatin compaction by human MeCP2. Assembly of novel secondary chromatin structures in the absence of DNA methylation. J. Biol. Chem. 278, 32181–32188. doi: 10.1074/jbc.M305308200
Ghosh, R. P., Nikitina, T., Horowitz-Scherer, R. A., Gierasch, L. M., Uversky, V. N., Hite, K., et al. (2010). Unique physical properties and interactions of the domains of methylated DNA binding protein 2. Biochemistry 49, 4395–4410. doi: 10.1021/bi9019753
Gnanapragasam, M. N., Scarsdale, J. N., Amaya, M. L., Webb, H. D., Desai, M. A., Walavalkar, N. M., et al. (2011). p66Alpha-MBD2 coiled-coil interaction and recruitment of Mi-2 are critical for globin gene silencing by the MBD2-NuRD complex. Proc. Natl. Acad. Sci. U.S.A. 108, 7487–7492. doi: 10.1073/pnas.1015341108
Goll, M. G., Kirpekar, F., Maggert, K. A., Yoder, J. A., Hsieh, C.-L., Zhang, X., et al. (2006). Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 311, 395–398. doi: 10.1126/science.1120976
Gonzales, M. L., Adams, S., Dunaway, K. W., and LaSalle, J. M. (2012). Phosphorylation of distinct sites in MeCP2 modifies cofactor associations and the dynamics of transcriptional regulation. Mol. Cell. Biol. 32, 2894–2903. doi: 10.1128/MCB.06728-11
Grohmann, M., Spada, F., Schermelleh, L., Alenina, N., Bader, M., Cardoso, M. C., et al. (2005). Restricted mobility of Dnmt1 in preimplantation embryos: implications for epigenetic reprogramming. BMC Dev. Biol. 5:18. doi: 10.1186/1471-213X-5-18
Gu, T. P., Guo, F., Yang, H., Wu, H. P., Xu, G. F., Liu, W., et al. (2011). The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 477, 606–610. doi: 10.1038/nature10443
Gunther, K., Rust, M., Leers, J., Boettger, T., Scharfe, M., Jarek, M., et al. (2013). Differential roles for MBD2 and MBD3 at methylated CpG islands, active promoters and binding to exon sequences. Nucleic Acids Res. 41, 3010–3021. doi: 10.1093/nar/gkt035
Guo, J. U., Su, Y., Shin, J. H., Shin, J., Li, H., Xie, B., et al. (2014). Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat. Neurosci. 17, 215–222. doi: 10.1038/nn.3607
Guo, J. U., Su, Y., Zhong, C., Ming, G. L., and Song, H. (2011). Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 145, 423–434. doi: 10.1016/j.cell.2011.03.022
Guy, J., Gan, J., Selfridge, J., Cobb, S., and Bird, A. (2007). Reversal of neurological defects in a mouse model of Rett syndrome. Science 315, 1143–1147. doi: 10.1126/science.1138389
Guy, J., Hendrich, B., Holmes, M., Martin, J. E., and Bird, A. (2001). A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 27, 322–326. doi: 10.1038/85899
Hackett, J. A., Sengupta, R., Zylicz, J. J., Murakami, K., Lee, C., Down, T. A., et al. (2013). Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science 339, 448–452. doi: 10.1126/science.1229277
Haffner, M. C., Chaux, A., Meeker, A. K., Esopi, D. M., Gerber, J., Pellakuru, L. G., et al. (2011). Global 5-hydroxymethylcytosine content is significantly reduced in tissue stem/progenitor cell compartments and in human cancers. Oncotarget 2, 627–637. doi: 10.18632/oncotarget.316
Hagberg, B., Aicardi, J., Dias, K., and Ramos, O. (1983). A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: rett’s syndrome: report of 35 cases. Ann. Neurol. 14, 471–479. doi: 10.1002/ana.410140412
Hajkova, P., Erhardt, S., Lane, N., Haaf, T., El-Maarri, O., Reik, W., et al. (2002). Epigenetic reprogramming in mouse primordial germ cells. Mech. Dev. 117, 15–23. doi: 10.1016/S0925-4773(02)00181-8
Hamiche, A., Schultz, P., Ramakrishnan, V., Oudet, P., and Prunell, A. (1996). Linker histone-dependent DNA structure in linear mononucleosomes. J. Mol. Biol. 257, 30–42. doi: 10.1006/jmbi.1996.0144
Hansen, R. S., Wijmenga, C., Luo, P., Stanek, A. M., Canfield, T. K., Weemaes, C. M., et al. (1999). The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc. Natl. Acad. Sci. U.S.A. 96, 14412–14417. doi: 10.1073/pnas.96.25.14412
Hashimoto, H., Horton, J. R., Zhang, X., Bostick, M., Jacobsen, S. E., and Cheng, X. (2008). The SRA domain of UHRF1 flips 5-methylcytosine out of the DNA helix. Nature 455, 826–829. doi: 10.1038/nature07280
Hashimoto, H., Horton, J. R., Zhang, X., and Cheng, X. (2009). UHRF1, a modular multi-domain protein, regulates replication-coupled crosstalk between DNA methylation and histone modifications. Epigenetics 4, 8–14. doi: 10.4161/epi.4.1.7370
Hashimoto, H., Liu, Y., Upadhyay, A. K., Chang, Y., Howerton, S. B., Vertino, P. M., et al. (2012a). Recognition and potential mechanisms for replication and erasure of cytosine hydroxymethylation. Nucleic Acids Res. 40, 4841–4849. doi: 10.1093/nar/gks155
Hashimoto, H., Pais, J. E., Dai, N., Corrêa, I. R., Zhang, X., Zheng, Y., et al. (2015). Structure of Naegleria Tet-like dioxygenase (NgTet1) in complexes with a reaction intermediate 5-hydroxymethylcytosine DNA. Nucleic Acids Res. 43, 10713–10721. doi: 10.1093/nar/gkv870
Hashimoto, H., Pais, J. E., Zhang, X., Saleh, L., Fu, Z. Q., Dai, N., et al. (2014). Structure of a Naegleria Tet-like dioxygenase in complex with 5-methylcytosine DNA. Nature 506, 391–395. doi: 10.1038/nature12905
Hashimoto, H., Zhang, X., and Cheng, X. (2012b). Excision of thymine and 5-hydroxymethyluracil by the MBD4 DNA glycosylase domain: structural basis and implications for active DNA demethylation. Nucleic Acids Res. 40, 8276–8284. doi: 10.1093/nar/gks628
He, X., Duan, C., Chen, J., Ou-Yang, X., Zhang, Z., Li, C., et al. (2009). Let-7a elevates p21(WAF1) levels by targeting of NIRF and suppresses the growth of A549 lung cancer cells. FEBS Lett. 583, 3501–3507. doi: 10.1016/j.febslet.2009.10.007
He, Y. F., Li, B. Z., Li, Z., Liu, P., Wang, Y., Tang, Q., et al. (2011). Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333, 1303–1307. doi: 10.1126/science.1210944
Hendrich, B., and Bird, A. (1998). Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol. Cell. Biol. 18, 6538–6547. doi: 10.1128/MCB.18.11.6538
Hendrich, B., Guy, J., Ramsahoye, B., Wilson, V. A., and Bird, A. (2001). Closely related proteins MBD2 and MBD3 play distinctive but interacting roles in mouse development. Genes Dev. 15, 710–723. doi: 10.1101/gad.194101
Hendrich, B., Hardeland, U., Ng, H. H., Jiricny, J., and Bird, A. (1999). The thymine glycosylase MBD4 can bind to the product of deamination at methylated CpG sites. Nature 401, 301–304. doi: 10.1038/45843
Hendrich, B., and Tweedie, S. (2003). The methyl-CpG binding domain and the evolving role of DNA methylation in animals. Trends Genet. 19, 269–277. doi: 10.1016/S0168-9525(03)00080-5
Hopfner, R., Mousli, M., Jeltsch, J. M., Voulgaris, A., Lutz, Y., Marin, C., et al. (2000). ICBP90, a novel human CCAAT binding protein, involved in the regulation of topoisomerase IIalpha expression. Cancer Res. 60, 121–128.
Hotchkiss, D. (1948). THE QUANTITATIVE SEPARATION OF PURINES, and NUCLEOSIDES BY paper chromatography. J. Biol. Chem. 175, 315–332.
Hu, L., Li, Z., Cheng, J., Rao, Q., Gong, W., Liu, M., et al. (2013). Crystal structure of TET2-DNA complex: insight into TET-mediated 5mC oxidation. Cell 155, 1545–1555. doi: 10.1016/j.cell.2013.11.020
Hu, L., Li, Z., Wang, P., Lin, Y., and Xu, Y. (2011). Crystal structure of PHD domain of UHRF1 and insights into recognition of unmodified histone H3 arginine residue 2. Cell Res. 21, 1374–1378. doi: 10.1038/cr.2011.124
Hu, L., Lu, J., Cheng, J., Rao, Q., Li, Z., Hou, H., et al. (2015). Structural insight into substrate preference for TET-mediated oxidation. Nature 527, 118–122. doi: 10.1038/nature15713
Hu, Y. G., Hirasawa, R., Hu, J. L., Hata, K., Li, C. L., Jin, Y., et al. (2008). Regulation of DNA methylation activity through Dnmt3L promoter methylation by Dnmt3 enzymes in embryonic development. Hum. Mol. Genet. 17, 2654–2664. doi: 10.1093/hmg/ddn165
Huang, H., Jiang, X., Li, Z., Li, Y., Song, C.-X., He, C., et al. (2013). TET1 plays an essential oncogenic role in MLL-rearranged leukemia. Proc. Natl. Acad. Sci. U.S.A. 110, 11994–11999. doi: 10.1073/pnas.1310656110
Huang, Y., Chavez, L., Chang, X., Wang, X., Pastor, W. A., Kang, J., et al. (2014). Distinct roles of the methylcytosine oxidases Tet1 and Tet2 in mouse embryonic stem cells. Proc. Natl. Acad. Sci. U.S.A. 111, 1361–1366. doi: 10.1073/pnas.1322921111
Inano, K., Suetake, I., Ueda, T., Miyake, Y., Nakamura, M., Okada, M., et al. (2000). Maintenance-type DNA methyltransferase is highly expressed in post-mitotic neurons and localized in the cytoplasmic compartment. J. Biochem. 128, 315–321. doi: 10.1093/oxfordjournals.jbchem.a022755
Iqbal, K., Jin, S. G., Pfeifer, G. P., and Szabo, P. E. (2011). Reprogramming of the paternal genome upon fertilization involves genome-wide oxidation of 5-methylcytosine. Proc. Natl. Acad. Sci. U.S.A. 108, 3642–3647. doi: 10.1073/pnas.1014033108
Ito, R., Katsura, S., Shimada, H., Tsuchiya, H., Hada, M., Okumura, T., et al. (2014). TET3-OGT interaction increases the stability and the presence of OGT in chromatin. Genes Cells 19, 52–65. doi: 10.1111/gtc.12107
Ito, S., D’Alessio, A. C., Taranova, O. V., Hong, K., Sowers, L. C., and Zhang, Y. (2010). Role of tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 466, 1129–1133. doi: 10.1038/nature09303
Ito, S., Shen, L., Dai, Q., Wu, S. C., Collins, L. B., Swenberg, J. A., et al. (2011). Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333, 1300–1303. doi: 10.1126/science.1210597
Jin, C., Lu, Y., Jelinek, J., Liang, S., Estecio, M. R. H., Barton, M. C., et al. (2014). TET1 is a maintenance DNA demethylase that prevents methylation spreading in differentiated cells. Nucleic Acids Res. 42, 6956–6971. doi: 10.1093/nar/gku372
Jin, S. G., Wu, X., Li, A. X., and Pfeifer, G. P. (2011). Genomic mapping of 5-hydroxymethylcytosine in the human brain. Nucleic Acids Res. 39, 5015–5024. doi: 10.1093/nar/gkr120
Johnson, L. M., Bostick, M., Zhang, X., Kraft, E., Henderson, I., Callis, J., et al. (2007). The SRA methyl-cytosine-binding domain links DNA and histone methylation. Curr. Biol. 17, 379–384. doi: 10.1016/j.cub.2007.01.009
Jones, P. L., Veenstra, G. J., Wade, P. A., Vermaak, D., Kass, S. U., Landsberger, N., et al. (1998). Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 19, 187–191. doi: 10.1038/561
Jorgensen, H. F., Ben-Porath, I., and Bird, A. P. (2004). Mbd1 is recruited to both methylated and nonmethylated CpGs via distinct DNA binding domains. Mol. Cell. Biol. 24, 3387–3395. doi: 10.1128/MCB.24.8.3387-3395.2004
Jung, B. P., Jugloff, D. G., Zhang, G., Logan, R., Brown, S., and Eubanks, J. H. (2003). The expression of methyl CpG binding factor MeCP2 correlates with cellular differentiation in the developing rat brain and in cultured cells. J. Neurobiol. 55, 86–96. doi: 10.1002/neu.10201
Jung, S. Y., Li, Y., Wang, Y., Chen, Y., Zhao, Y., and Qin, J. (2008). Complications in the assignment of 14 and 28 Da mass shift detected by mass spectrometry as in vivo methylation from endogenous proteins. Anal. Chem. 80, 1721–1729. doi: 10.1021/ac7021025
Kaji, K., Caballero, I. M., MacLeod, R., Nichols, J., Wilson, V. A., and Hendrich, B. (2006). The NuRD component Mbd3 is required for pluripotency of embryonic stem cells. Nat. Cell Biol. 8, 285–292. doi: 10.1038/ncb1372
Kaludov, N. K., and Wolffe, A. P. (2000). MeCP2 driven transcriptional repression in vitro: selectivity for methylated DNA, action at a distance and contacts with the basal transcription machinery. Nucleic Acids Res. 28, 1921–1928. doi: 10.1093/nar/28.9.1921
Karagianni, P., Amazit, L., Qin, J., and Wong, J. (2008). ICBP90, a novel methyl K9 H3 binding protein linking protein ubiquitination with heterochromatin formation. Mol. Cell. Biol. 28, 705–717. doi: 10.1128/MCB.01598-07
Kemmerich, K., Dingler, F. A., Rada, C., and Neuberger, M. S. (2012). Germline ablation of SMUG1 DNA glycosylase causes loss of 5-hydroxymethyluracil- and UNG-backup uracil-excision activities and increases cancer predisposition of Ung-/-Msh2-/- mice. Nucleic Acids Res. 40, 6016–6025. doi: 10.1093/nar/gks259
Kiefer, H., Chatail-Hermitte, F., Ravassard, P., Bayard, E., Brunet, I., and Mallet, J. (2005). ZENON, a novel POZ Kruppel-like DNA binding protein associated with differentiation and/or survival of late postmitotic neurons. Mol. Cell. Biol. 25, 1713–1729. doi: 10.1128/MCB.25.5.1713-1729.2005
Kienhöfer, S., Musheev, M. U., Stapf, U., Helm, M., Schomacher, L., Niehrs, C., et al. (2015). GADD45a physically and functionally interacts with TET1. Differentiation 90, 59–68. doi: 10.1016/j.diff.2015.10.003
Kim, J. K., Esteve, P. O., Jacobsen, S. E., and Pradhan, S. (2009). UHRF1 binds G9a and participates in p21 transcriptional regulation in mammalian cells. Nucleic Acids Res. 37, 493–505. doi: 10.1093/nar/gkn961
Kim, S. W., Park, J. I., Spring, C. M., Sater, A. K., Ji, H., Otchere, A. A., et al. (2004). Non-canonical Wnt signals are modulated by the Kaiso transcriptional repressor and p120-catenin. Nat. Cell Biol. 6, 1212–1220. doi: 10.1038/ncb1191
Kimura, H., and Shiota, K. (2003). Methyl-CpG-binding protein, MeCP2, is a target molecule for maintenance DNA methyltransferase, Dnmt1. J. Biol. Chem. 278, 4806–4812. doi: 10.1074/jbc.M209923200
Klein, C. J., Bird, T., Ertekin-Taner, N., Lincoln, S., Hjorth, R., Wu, Y., et al. (2013). DNMT1 mutation hot spot causes varied phenotypes of HSAN1 with dementia and hearing loss. Neurology 80, 824–828. doi: 10.1212/WNL.0b013e31828
Klein, C. J., Botuyan, M. V., Wu, Y., Ward, C. J., Nicholson, G. A., Hammans, S., et al. (2011). Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat. Genet. 43, 595–600. doi: 10.1038/ng.830
Ko, M., An, J., Bandukwala, H. S., Chavez, L., Aijö, T., Pastor, W. A., et al. (2013). Modulation of TET2 expression and 5-methylcytosine oxidation by the CXXC domain protein IDAX. Nature 497, 122–126. doi: 10.1038/nature12052
Ko, M., Huang, Y., Jankowska, A. M., Pape, U. J., Tahiliani, M., Bandukwala, H. S., et al. (2010). Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature 468, 839–843. doi: 10.1038/nature09586
Kokura, K., Kaul, S. C., Wadhwa, R., Nomura, T., Khan, M. M., Shinagawa, T., et al. (2001). The Ski protein family is required for MeCP2-mediated transcriptional repression. J. Biol. Chem. 276, 34115–34121. doi: 10.1074/jbc.M105747200
Konstandin, N., Bultmann, S., Szwagierczak, A., Dufour, A., Ksienzyk, B., Schneider, F., et al. (2011). Genomic 5-hydroxymethylcytosine levels correlate with TET2 mutations and a distinct global gene expression pattern in secondary acute myeloid leukemia. Leukemia 25, 1649–1652. doi: 10.1038/leu.2011.134leu2011134
Koziol, M. J., Bradshaw, C. R., Allen, G. E., Costa, A. S., Frezza, C., and Gurdon, J. B. (2016). Identification of methylated deoxyadenosines in vertebrates reveals diversity in DNA modifications. Nat. Struct. Mol. Biol. 23, 24–30. doi: 10.1038/nsmb.3145
Kraus, T. F. J., Globisch, D., Wagner, M., Eigenbrod, S., Widmann, D., Münzel, M., et al. (2012). Low values of 5-hydroxymethylcytosine (5hmC), the “sixth base,” are associated with anaplasia in human brain tumors. Int. J. Cancer 131, 1577–1590. doi: 10.1002/ijc.27429
Kriaucionis, S., and Bird, A. (2004). The major form of MeCP2 has a novel N-terminus generated by alternative splicing. Nucleic Acids Res. 32, 1818–1823. doi: 10.1093/nar/gkh349
Kriaucionis, S., and Heintz, N. (2009). The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 324, 929–930. doi: 10.1126/science.1169786
Kumar, S., Cheng, X., Klimasauskas, S., Mi, S., Posfai, J., Roberts, R. J., et al. (1994). The DNA (cytosine-5) methyltransferases. Nucleic Acids Res. 22, 1–10. doi: 10.1093/nar/22.1.1
Laget, S., Joulie, M., Le Masson, F., Sasai, N., Christians, E., Pradhan, S., et al. (2010). The human proteins MBD5 and MBD6 associate with heterochromatin but they do not bind methylated DNA. PLoS ONE 5:e11982. doi: 10.1371/journal.pone.0011982
Ley, T. J., Ding, L., Walter, M. J., McLellan, M. D., Lamprecht, T., Larson, D. E., et al. (2011). DNMT3A Mutations in Acute Myeloid Leukemia. N. Engl. J. Med. 363, 2424–2433. doi: 10.1056/NEJMoa1005143
Langemeijer, S., Kuiper, R., Berends, M., Knops, R., Aslanyan, M., Massop, M., et al. (2009). Acquired mutations in TET2 are common in myelodysplastic syndromes. Leuk. Res. 33, 838–842. doi: 10.1016/S0145-2126(09)70132-8
Le Guezennec, X., Vermeulen, M., Brinkman, A. B., Hoeijmakers, W. A., Cohen, A., Lasonder, E., et al. (2006). MBD2/NuRD and MBD3/NuRD, two distinct complexes with different biochemical and functional properties. Mol. Cell. Biol. 26, 843–851. doi: 10.1128/MCB.26.3.843-851.2006
Lee, J. H., and Skalnik, D. G. (2002). CpG-binding protein is a nuclear matrix- and euchromatin-associated protein localized to nuclear speckles containing human trithorax. Identification of nuclear matrix targeting signals. J. Biol. Chem. 277, 42259–42267. doi: 10.1074/jbc.M205054200
Lee, J. H., and Skalnik, D. G. (2005). CpG-binding protein (CXXC finger protein 1) is a component of the mammalian Set1 histone H3-Lys4 methyltransferase complex, the analogue of the yeast Set1/COMPASS complex. J. Biol. Chem. 280, 41725–41731. doi: 10.1074/jbc.M508312200
Leonhardt, H., Page, A. W., Weier, H. U., and Bestor, T. H. (1992). A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei. Cell 71, 865–873. doi: 10.1016/0092-8674(92)90561-P
Lewis, J. D., Meehan, R. R., Henzel, W. J., Maurer-Fogy, I., Jeppesen, P., Klein, F., et al. (1992). Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell 69, 905–914. doi: 10.1016/0092-8674(92)90610-O
Li, D., Qiu, Z., Shao, Y., Chen, Y., Guan, Y., Liu, M., et al. (2013). Heritable gene targeting in the mouse and rat using a CRISPR-Cas system. Nat. Biotechnol. 31, 681–683. doi: 10.1038/nbt.2661
Li, E., Bestor, T. H., and Jaenisch, R. (1992). Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 69, 915–926. doi: 10.1016/0092-8674(92)90611-F
Li, H., and Chang, Q. (2014). Regulation and function of stimulus-induced phosphorylation of MeCP2. Front. Biol. (Beijing) 9:367–375. doi: 10.1007/s11515-014-1330-2
Li, W., and Liu, M. (2011). Distribution of 5-hydroxymethylcytosine in different human tissues. J. Nucleic Acids 2011, 870726. doi: 10.4061/2011/870726
Li, Z., Cai, X., Cai, C. L., Wang, J., Zhang, W., Petersen, B. E., et al. (2011). Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood 118, 4509–4518. doi: 10.1182/blood-2010-12-325241
Li, Z., Gu, T. P., Weber, A. R., Shen, J. Z., Li, B. Z., Xie, Z. G., et al. (2015). Gadd45a promotes DNA demethylation through TDG. Nucleic Acids Res. 43, 3986–3997. doi: 10.1093/nar/gkv283
Lin, R. K., Hsu, H. S., Chang, J. W., Chen, C. Y., Chen, J. T., and Wang, Y. C. (2007). Alteration of DNA methyltransferases contributes to 5’CpG methylation and poor prognosis in lung cancer. Lung Cancer 55, 205–213. doi: 10.1016/j.lungcan.2006.10.022
Liu, N., Wang, M., Deng, W., Schmidt, C. S., Qin, W., Leonhardt, H., et al. (2013). Intrinsic and extrinsic connections of Tet3 dioxygenase with CXXC zinc finger modules. PLoS ONE 8:e62755. doi: 10.1371/journal.pone.0062755
Liu, Y., Oakeley, E. J., Sun, L., and Jost, J. P. (1998). Multiple domains are involved in the targeting of the mouse DNA methyltransferase to the DNA replication foci. Nucleic Acids Res. 26, 1038–1045. doi: 10.1093/nar/26.4.1038
Loat, C. S., Curran, S., Lewis, C. M., Duvall, J., Geschwind, D., Bolton, P., et al. (2008). Methyl-CpG-binding protein 2 polymorphisms and vulnerability to autism. Genes Brain Behav. 7, 754–760. doi: 10.1111/j.1601-183X.2008.00414.x
Lopes, E. C., Valls, E., Figueroa, M. E., Mazur, A., Meng, F. G., Chiosis, G., et al. (2008). Kaiso contributes to DNA methylation-dependent silencing of tumor suppressor genes in colon cancer cell lines. Cancer Res. 68, 7258–7263. doi: 10.1158/0008-5472.CAN-08-0344
Lorsbach, R. B., Moore, J., Mathew, S., Raimondi, S. C., Mukatira, S. T., and Downing, J. R. (2003). TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t(10;11)(q22;q23). Leukemia 17, 637–641. doi: 10.1038/sj.leu.2402834
Lu, J., Hu, L., Cheng, J., Dong, F., Wang, C., Yu, K., et al. (2016). A computational investigation on the substrate preference of TET2. Phys. Chem. Chem. Phys 18, 4728–4738. doi: 10.1039/C5CP07266B
Lunyak, V. V., Burgess, R., Prefontaine, G. G., Nelson, C., Sze, S. H., Chenoweth, J., et al. (2002). Corepressor-dependent silencing of chromosomal regions encoding neuronal genes. Science 298, 1747–1752. doi: 10.1126/science.1076469
Luo, M., Ling, T., Xie, W., Sun, H., Zhou, Y., Zhu, Q., et al. (2013). NuRD blocks reprogramming of mouse somatic cells into pluripotent stem cells. Stem Cells 31, 1278–1286. doi: 10.1002/stem.1374
Lyko, F., Ramsahoye, B. H., and Jaenisch, R. (2000). DNA methylation in Drosophila melanogaster. Nature 408, 538–540. doi: 10.1038/35046205
Lyst, M. J., Ekiert, R., Ebert, D. H., Merusi, C., Nowak, J., Selfridge, J., et al. (2013). Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nat. Neurosci. 16, 898–902. doi: 10.1038/nn.3434
Lyst, M. J., Nan, X., and Stancheva, I. (2006). Regulation of MBD1-mediated transcriptional 1478 repression by SUMO and PIAS proteins. EMBO J. 25, 5317-5328. doi: 10.1038/sj.emboj.7601404
Mahajan, M. C., Narlikar, G. J., Boyapaty, G., Kingston, R. E., and Weissman, S. M. (2005). Heterogeneous nuclear ribonucleoprotein C1/C2, MeCP1, and SWI/SNF form a chromatin remodeling complex at the beta-globin locus control region. Proc. Natl. Acad. Sci. U.S.A. 102, 15012–15017. doi: 10.1073/pnas.0507596102
Maiti, A., and Drohat, A. C. (2011). Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J. Biol. Chem. 286, 35334–35338. doi: 10.1074/jbc.C111.284620
Marhold, J., Kramer, K., Kremmer, E., and Lyko, F. (2004). The Drosophila MBD2/3 protein mediates interactions between the MI-2 chromatin complex and CpT/A-methylated DNA. Development 131, 6033–6039. doi: 10.1242/dev.01531
Martinowich, K., Hattori, D., Wu, H., Fouse, S., He, F., Hu, Y., et al. (2003). DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 302, 890–893. doi: 10.1126/science.1090842
Mayer, W., Niveleau, A., Walter, J., Fundele, R., and Haaf, T. (2000). Demethylation of the zygotic paternal genome. Nature 403, 501–502. doi: 10.1038/35000654
Meehan, R. R., Lewis, J. D., and Bird, A. P. (1992). Characterization of MeCP2, a vertebrate DNA binding protein with affinity for methylated DNA. Nucleic Acids Res. 20, 5085–5092. doi: 10.1093/nar/20.19.5085
Meehan, R. R., Lewis, J. D., McKay, S., Kleiner, E. L., and Bird, A. P. (1989). Identification of a mammalian protein that binds specifically to DNA containing methylated CpGs. Cell 58, 499–507. doi: 10.1016/0092-8674(89)90430-3
Meilinger, D., Fellinger, K., Bultmann, S., Rothbauer, U., Bonapace, I. M., Klinkert, W. E., et al. (2009). Np95 interacts with de novo DNA methyltransferases, Dnmt3a and Dnmt3b, and mediates epigenetic silencing of the viral CMV promoter in embryonic stem cells. EMBO Rep. 10, 1259–1264. doi: 10.1038/embor.2009.201
Mellen, M., Ayata, P., Dewell, S., Kriaucionis, S., and Heintz, N. (2012). MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell 151, 1417–1430. doi: 10.1016/j.cell.2012.11.022
Mertineit, C., Yoder, J. A., Taketo, T., Laird, D. W., Trasler, J. M., and Bestor, T. H. (1998). Sex-specific exons control DNA methyltransferase in mammalian germ cells. Development 125, 889–897.
Michaels, M. L., Pham, L., Nghiem, Y., Cruz, C., and Miller, J. H. (1990). MutY, an adenine glycosylase active on G-A mispairs, has homology to endonuclease III. Nucleic Acids Res. 18, 3841–3845. doi: 10.1093/nar/18.13.3841
Millar, C. B., Guy, J., Sansom, O. J., Selfridge, J., MacDougall, E., Hendrich, B., et al. (2002). Enhanced CpG mutability and tumorigenesis in MBD4-deficient mice. Science 297, 403–405. doi: 10.1126/science.1073354
Mnatzakanian, G. N., Lohi, H., Munteanu, I., Alfred, S. E., Yamada, T., MacLeod, P. J., et al. (2004). A previously unidentified MECP2 open reading frame defines a new protein isoform relevant to Rett syndrome. Nat. Genet. 36, 339–341. doi: 10.1038/ng1327
Mori, T., Ikeda, D. D., Yamaguchi, Y., Unoki, M., and Project, N. (2012). NIRF/UHRF2 occupies a central position in the cell cycle network and allows coupling with the epigenetic landscape. FEBS Lett. 586, 1570–1583. doi: 10.1016/j.febslet.2012.04.038
Mori, T., Li, Y., Hata, H., Ono, K., and Kochi, H. (2002). NIRF, a novel RING finger protein, is involved in cell-cycle regulation. Biochem. Biophys. Res. Commun. 296, 530–536. doi: 10.1016/S0006-291X(02)00890-2
Muller, H. M., Fiegl, H., Goebel, G., Hubalek, M. M., Widschwendter, A., Muller-Holzner, E., et al. (2003). MeCP2 and MBD2 expression in human neoplastic and non-neoplastic breast tissue and its association with oestrogen receptor status. Br. J. Cancer 89, 1934–1939. doi: 10.1038/sj.bjc.6601392
Muller, U., Bauer, C., Siegl, M., Rottach, A., and Leonhardt, H. (2014). TET-mediated oxidation of methylcytosine causes TDG or NEIL glycosylase dependent gene reactivation. Nucleic Acids Res. 42, 8592–8604. doi: 10.1093/nar/gku552
Muotri, A. R., Marchetto, M. C., Coufal, N. G., Oefner, R., Yeo, G., Nakashima, K., et al. (2010). L1 retrotransposition in neurons is modulated by MeCP2. Nature 468, 443–446. doi: 10.1038/nature09544
Nady, N., Lemak, A., Walker, J. R., Avvakumov, G. V., Kareta, M. S., Achour, M., et al. (2011). Recognition of multivalent histone states associated with heterochromatin by UHRF1 protein. J. Biol. Chem. 286, 24300–24311. doi: 10.1074/jbc.M111.234104
Nagarajan, R. P., Hogart, A. R., Gwye, Y., Martin, M. R., and LaSalle, J. M. (2006). Reduced MeCP2 expression is frequent in autism frontal cortex and correlates with aberrant MECP2 promoter methylation. Epigenetics 1, e1–e11. doi: 10.4161/epi.1.4.3514
Nakagawa, T., Lv, L., Nakagawa, M., Yu, Y., Yu, C., D’Alessio, A. C., et al. (2015). CRL4VprBP E3 ligase promotes monoubiquitylation and chromatin binding of TET dioxygenases. Mol. Cell. 57, 247–260. doi: 10.1016/j.molcel.2014.12.002
Nakamura, T., Liu, Y. J., Nakashima, H., Umehara, H., Inoue, K., Matoba, S., et al. (2012). PGC7 binds histone H3K9me2 to protect against conversion of 5mC to 5hmC in early embryos. Nature 486, 415–419. doi: 10.1038/nature11093
Nan, X., Campoy, F. J., and Bird, A. (1997). MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell 88, 471–481. doi: 10.1016/S0092-8674(00)81887-5
Nan, X., Meehan, R. R., and Bird, A. (1993). Dissection of the methyl-CpG binding domain from the chromosomal protein MeCP2. Nucleic Acids Res. 21, 4886–4892. doi: 10.1093/nar/21.21.4886
Nan, X., Ng, H. H., Johnson, C. A., Laherty, C. D., Turner, B. M., Eisenman, R. N., et al. (1998). Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 393, 386–389. doi: 10.1038/30764
Nestor, C. E., Ottaviano, R., Reddington, J., Sproul, D., Reinhardt, D., Dunican, D., et al. (2012). Tissue type is a major modifier of the 5-hydroxymethylcytosine content of human genes. Genome Res. 22, 467–477. doi: 10.1101/gr.126417.111
Ng, H. H., Jeppesen, P., and Bird, A. (2000). Active repression of methylated genes by the chromosomal protein MBD1. Mol. Cell. Biol. 20, 1394–1406. doi: 10.1128/MCB.20.4.1394-1406.2000
Ng, H. H., Zhang, Y., Hendrich, B., Johnson, C. A., Turner, B. M., Erdjument-Bromage, H., et al. (1999). MBD2 is a transcriptional repressor belonging to the MeCP1 histone deacetylase complex. Nat. Genet. 23, 58–61. doi: 10.1038/12659
Nikitina, T., Ghosh, R. P., Horowitz-Scherer, R. A., Hansen, J. C., Grigoryev, S. A., and Woodcock, C. L. (2007a). MeCP2-chromatin interactions include the formation of chromatosome-like structures and are altered in mutations causing Rett syndrome. J. Biol. Chem. 282, 28237–28245. doi: 10.1074/jbc.M704304200
Nikitina, T., Shi, X., Ghosh, R. P., Horowitz-Scherer, R. A., Hansen, J. C., and Woodcock, C. L. (2007b). Multiple modes of interaction between the methylated DNA binding protein MeCP2 and chromatin. Mol. Cell. Biol. 27, 864–877. doi: 10.1128/MCB.01593-06
Ohki, I., Shimotake, N., Fujita, N., Nakao, M., and Shirakawa, M. (1999). Solution structure of the methyl-CpG-binding domain of the methylation-dependent transcriptional repressor MBD1. EMBO J. 18, 6653–6661. doi: 10.1093/emboj/18.23.6653
Okano, M., Bell, D. W., Haber, D. A., and Li, E. (1999). DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–257. doi: 10.1016/S0092-8674(00)81656-6
Okano, M., Xie, S., and Li, E. (1998). Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nature Am. Inc. 19, 219–220. doi: 10.1038/890
Papait, R., Pistore, C., Grazini, U., Babbio, F., Cogliati, S., Pecoraro, D., et al. (2008). The PHD domain of Np95 (mUHRF1) is involved in large-scale reorganization of pericentromeric heterochromatin. Mol. Biol. Cell 19, 3554–3563. doi: 10.1091/mbc.E07-10-1059
Papait, R., Pistore, C., Negri, D., Pecoraro, D., Cantarini, L., and Bonapace, I. M. (2007). Np95 is implicated in pericentromeric heterochromatin replication and in major satellite silencing. Mol. Biol. Cell 18, 1098–1106. doi: 10.1091/mbc.E06-09-0874
Park, J. I., Kim, S. W., Lyons, J. P., Ji, H., Nguyen, T. T., Cho, K., et al. (2005). Kaiso/p120-catenin and TCF/beta-catenin complexes coordinately regulate canonical Wnt gene targets. Dev. Cell 8, 843–854. doi: 10.1016/j.devcel.2005.04.010
Pastor, W. A., Pape, U. J., Huang, Y., Henderson, H. R., Lister, R., Ko, M., et al. (2011). Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature 473, 394–397. doi: 10.1038/nature10102
Patra, S. K., Patra, A., Zhao, H., Carroll, P., and Dahiya, R. (2003). Methyl-CpG-DNA binding proteins in human prostate cancer: expression of CXXC sequence containing MBD1 and repression of MBD2 and MeCP2. Biochem. Biophys. Res. Commun. 302, 759–766. doi: 10.1016/S0006-291X(03)00253-5
Penn, N. W., Suwalski, R., O’Riley, C., Bojanowski, K., and Yura, R. (1972). The presence of 5-hydroxymethylcytosine in animal deoxyribonucleic acid. Biochem. J. 126, 781–790. doi: 10.1042/bj1260781
Petronzelli, F., Riccio, A., Markham, G. D., Seeholzer, S. H., Stoerker, J., Genuardi, M., et al. (2000). Biphasic kinetics of the human DNA repair protein MED1 (MBD4), a mismatch-specific DNA N-glycosylase. J. Biol. Chem. 275, 32422–32429. doi: 10.1074/jbc.M004535200
Pfaffeneder, T., Hackner, B., Truss, M., Münzel, M., Müller, M., Deiml, C. A., et al. (2011). The discovery of 5-formylcytosine in embryonic stem cell DNA. Angew. Chem. Int. Edn. 50, 7008–7012. doi: 10.1002/anie.201103899
Pfaffeneder, T., Spada, F., Wagner, M., Brandmayr, C., Laube, S. K., Eisen, D., et al. (2014). Tet oxidizes thymine to 5-hydroxymethyluracil in mouse embryonic stem cell DNA. Nat. Chem. Biol. 10, 574–581. doi: 10.1038/nchembio.1532
Pichler, G., Wolf, P., Schmidt, C. S., Meilinger, D., Schneider, K., Frauer, C., et al. (2011). Cooperative DNA and histone binding by Uhrf2 links the two major repressive epigenetic pathways. J. Cell. Biochem. 112, 2585–2593. doi: 10.1002/jcb.23185
Prokhortchouk, A., Hendrich, B., Jorgensen, H., Ruzov, A., Wilm, M., Georgiev, G., et al. (2001). The p120 catenin partner Kaiso is a DNA methylation-dependent transcriptional repressor. Genes Dev. 15, 1613–1618. doi: 10.1101/gad.198501
Prokhortchouk, A., Sansom, O., Selfridge, J., Caballero, I. M., Salozhin, S., Aithozhina, D., et al. (2006). Kaiso-deficient mice show resistance to intestinal cancer. Mol. Cell. Biol. 26, 199–208. doi: 10.1128/MCB.26.1.199-208.2006
Qin, W., Leonhardt, H., and Pichler, G. (2011). Regulation of DNA methyltransferase 1 by interactions and modifications. Nucleus 2, 392–402. doi: 10.4161/nucl.2.5.17928
Qiu, C., Sawada, K., Zhang, X., and Cheng, X. (2002). The PWWP domain of mammalian DNA methyltransferase Dnmt3b defines a new family of DNA-binding folds. Nat. Struct. Biol. 9, 217–224. doi: 10.1038/nsb759
Rais, Y., Zviran, A., Geula, S., Gafni, O., Chomsky, E., Viukov, S., et al. (2013). Deterministic direct reprogramming of somatic cells to pluripotency. Nature 502, 65–70. doi: 10.1038/nature12587
Rajakumara, E., Wang, Z., Ma, H., Hu, L., Chen, H., Lin, Y., et al. (2011). PHD finger recognition of unmodified histone H3R2 links UHRF1 to regulation of euchromatic gene expression. Mol. Cell. 43, 275–284. doi: 10.1016/j.molcel.2011.07.006
Ramocki, M. B., Peters, S. U., Tavyev, Y. J., Zhang, F., Carvalho, C. M., Schaaf, C. P., et al. (2009). Autism and other neuropsychiatric symptoms are prevalent in individuals with MeCP2 duplication syndrome. Ann. Neurol. 66, 771–782. doi: 10.1002/ana.21715
Razin, A., and Cedar, H. (1977). Distribution of 5-methylcytosine in chromatin. Proc. Natl. Acad. Sci. U.S.A. 74, 2725–2728. doi: 10.1073/pnas.74.7.2725
Rett, A. (1966). [On an until now unknown disease of a congenital metabolic disorder]. Krankenschwester 19, 121–122.
Reynolds, N., Latos, P., Hynes-Allen, A., Loos, R., Leaford, D., O’Shaughnessy, A., et al. (2012). NuRD suppresses pluripotency gene expression to promote transcriptional heterogeneity and lineage commitment. Cell Stem Cell 10, 583–594. doi: 10.1016/j.stem.2012.02.020
Riccio, A., Aaltonen, L. A., Godwin, A. K., Loukola, A., Percesepe, A., Salovaara, R., et al. (1999). The DNA repair gene MBD4 (MED1) is mutated in human carcinomas with microsatellite instability. Nat. Genet. 23, 266–268. doi: 10.1038/15443
Roll, J. D., Rivenbark, A. G., Jones, W. D., and Coleman, W. B. (2008). DNMT3b overexpression contributes to a hypermethylator phenotype in human breast cancer cell lines. Mol. Cancer 7, 15. doi: 10.1186/1476-4598-7-15
Roloff, T. C., Ropers, H. H., and Nuber, U. A. (2003). Comparative study of methyl-CpG-binding domain proteins. BMC Genomics 4:1. doi: 10.1186/1471-2164-4-1
Rothbart, S. B., Krajewski, K., Nady, N., Tempel, W., Xue, S., Badeaux, A. I., et al. (2012). Association of UHRF1 with methylated H3K9 directs the maintenance of DNA methylation. Nat. Struct. Mol. Biol. 19, 1155–1160. doi: 10.1038/nsmb.2391
Rottach, A., Frauer, C., Pichler, G., Bonapace, I. M., Spada, F., and Leonhardt, H. (2010). The multi-domain protein Np95 connects DNA methylation and histone modification. Nucleic Acids Res. 38, 1796–1804. doi: 10.1093/nar/gkp1152
Rountree, M. R., Bachman, K. E., and Baylin, S. B. (2000). DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat. Genet. 25, 269–277. doi: 10.1038/77023
Rudenko, A., Dawlaty, M. M., Seo, J., Cheng, A. W., Meng, J., Le, T., et al. (2013). Tet1 is critical for neuronal activity-regulated gene expression and memory extinction. Neuron 79, 1109–1122. doi: 10.1016/j.neuron.2013.08.003
Saito, M., and Ishikawa, F. (2002). The mCpG-binding domain of human MBD3 does not bind to mCpG but interacts with NuRD/Mi2 components HDAC1 and MTA2. J. Biol. Chem. 277, 35434–35439. doi: 10.1074/jbc.M203455200
Sarraf, S. A., and Stancheva, I. (2004). Methyl-CpG binding protein MBD1 couples histone H3 methylation at lysine 9 by SETDB1 to DNA replication and chromatin assembly. Mol. Cell. 15, 595–605. doi: 10.1016/j.molcel.2004.06.043
Sasai, N., Matsuda, E., Sarashina, E., Ishida, Y., and Kawaichi, M. (2005). Identification of a novel BTB-zinc finger transcriptional repressor, CIBZ, that interacts with CtBP corepressor. Genes Cells 10, 871–885. doi: 10.1111/j.1365-2443.2005.00885.x
Schermelleh, L., Haemmer, A., Spada, F., Rosing, N., Meilinger, D., Rothbauer, U., et al. (2007). Dynamics of Dnmt1 interaction with the replication machinery and its role in postreplicative maintenance of DNA methylation. Nucleic Acids Res. 35, 4301–4312. doi: 10.1093/nar/gkm432
Schlegel, J., Guneysu, S., and Mennel, H. D. (2002). Expression of the genes of methyl-binding domain proteins in human gliomas. Oncol. Rep. 9, 393–395.
Schneider, K., Fuchs, C., Dobay, A., Rottach, A., Qin, W., Wolf, P., et al. (2013). Dissection of cell cycle-dependent dynamics of Dnmt1 by FRAP and diffusion-coupled modeling. Nucleic Acids Res. 41, 4860–4876. doi: 10.1093/nar/gkt191
Shahbazian, M., Young, J., Yuva-Paylor, L., Spencer, C., Antalffy, B., Noebels, J., et al. (2002). Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron 35, 243–254. doi: 10.1016/S0896-6273(02)00768-7
Sharif, J., Muto, M., Takebayashi, S., Suetake, I., Iwamatsu, A., Endo, T. A., et al. (2007). The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature 450, 908–912. doi: 10.1038/nature06397
Shibayama, A., Cook, E. H. Jr., Feng, J., Glanzmann, C., Yan, J., Craddock, N., et al. (2004). MECP2 structural and 3’-UTR variants in schizophrenia, autism and other psychiatric diseases: a possible association with autism. Am. J. Med. Genet. B Neuropsychiatr. Genet. 128B, 50–53. doi: 10.1002/ajmg.b.30016
Shimbo, T., Du, Y., Grimm, S. A., Dhasarathy, A., Mav, D., Shah, R. R., et al. (2013). MBD3 localizes at promoters, gene bodies and enhancers of active genes. PLoS Genet. 9:e1004028. doi: 10.1371/journal.pgen.1004028
Skene, P. J., Illingworth, R. S., Webb, S., Kerr, A. R., James, K. D., Turner, D. J., et al. (2010). Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol. Cell. 37, 457–468. doi: 10.1016/j.molcel.2010.01.030
Song, J., Rechkoblit, O., Bestor, T. H., and Patel, D. J. (2011). Structure of DNMT1-DNA complex reveals a role for autoinhibition in maintenance DNA methylation. Science 331, 1036–1040. doi: 10.1126/science.1195380
Song, J., Teplova, M., Ishibe-Murakami, S., and Patel, D. J. (2012). Structure-based mechanistic insights into DNMT1-mediated maintenance DNA methylation. Science 335, 709–712. doi: 10.1126/science.1214453
Spruijt, C. G., Gnerlich, F., Smits, A. H., Pfaffeneder, T., Jansen, P. W., Bauer, C., et al. (2013). Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell 152, 1146–1159. doi: 10.1016/j.cell.2013.02.004
Stancheva, I., Collins, A. L., Van den Veyver, I. B., Zoghbi, H., and Meehan, R. R. (2003). A mutant form of MeCP2 protein associated with human Rett syndrome cannot be displaced from methylated DNA by notch in Xenopus embryos. Mol. Cell. 12, 425–435. doi: 10.1016/S1097-2765(03)00276-4
Strichman-almashanu, L. Z., Lee, R. S., Onyango, P. O., Perlman, E., Flam, F., Frieman, M. B., et al. (2002). A genome-wide screen for normally methylated human cpg islands that can identify novel imprinted genes a genome-wide screen for normally methylated human cpg islands that can identify novel imprinted genes. Genome Res 12, 543–554. doi: 10.1101/gr.224102
Su, L. K., Kinzler, K. W., Vogelstein, B., Preisinger, A. C., Moser, A. R., Luongo, C., et al. (1992). Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science 256, 668–670. doi: 10.1126/science.256.5060.1114-c
Suetake, I., Shinozaki, F., Miyagawa, J., Takeshima, H., and Tajima, S. (2004). DNMT3L stimulates the DNA methylation activity of Dnmt3a and Dnmt3b through a direct interaction. J. Biol. Chem. 279, 27816–27823. doi: 10.1074/jbc.M400181200
Suter, C. M., Martin, D. I., and Ward, R. L. (2004). Hypomethylation of L1 retrotransposons in colorectal cancer and adjacent normal tissue. Int. J. Colorectal. Dis. 19, 95–101. doi: 10.1007/s00384-003-0539-3
Szulwach, K. E., Li, X., Li, Y., Song, C. X., Wu, H., Dai, Q., et al. (2011). 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat. Neurosci. 14, 1607–1616. doi: 10.1038/nn.2959
Tahiliani, M., Koh, K. P., Shen, Y., Pastor, W. A., Bandukwala, H., Brudno, Y., et al. (2009). Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935. doi: 10.1126/science.1170116
Tardy-Planechaud, S., Fujimoto, J., Lin, S. S., and Sowers, L. C. (1997). Solid phase synthesis and restriction endonuclease cleavage of oligodeoxynucleotides containing 5-(hydroxymethyl)-cytosine. Nucleic Acids Res. 25, 553–559. doi: 10.1093/nar/25.3.553
Tatton-Brown, K., Seal, S., Ruark, E., Harmer, J., Ramsay, E., Del Vecchio Duarte, S., et al. (2014). Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nat. Genet. 46, 385–388. doi: 10.1038/ng.2917
Traynor, J., Agarwal, P., Lazzeroni, L., and Francke, U. (2002). Gene expression patterns vary in clonal cell cultures from Rett syndrome females with eight different MECP2 mutations. BMC Med. Genet. 3:12. doi: 10.1186/1471-2350-3-12
Tsukada, Y., Fang, J., Erdjument-Bromage, H., Warren, M. E., Borchers, C. H., Tempst, P., et al. (2006). Histone demethylation by a family of JmjC domain-containing proteins. Nature 439, 811–816. doi: 10.1038/nature04433
Tucker, K. L., Talbot, D., Lee, M. A., Leonhardt, H., and Jaenisch, R. (1996). Complementation of methylation deficiency in embryonic stem cells by a DNA methyltransferase minigene. Proc. Natl. Acad. Sci. U.S.A. 93, 12920–12925. doi: 10.1073/pnas.93.23.12920
Tudor, M., Akbarian, S., Chen, R. Z., and Jaenisch, R. (2002). Transcriptional profiling of a mouse model for Rett syndrome reveals subtle transcriptional changes in the brain. Proc. Natl. Acad. Sci. U.S.A. 99, 15536–15541. doi: 10.1073/pnas.242566899
Uchimura, Y., Ichimura, T., Uwada, J., Tachibana, T., Sugahara, S., Nakao, M., et al. (2006). Involvement of SUMO modification in MBD1- and MCAF1-mediated heterochromatin formation. J. Biol. Chem. 281, 23180–23190. doi: 10.1074/jbc.M602280200
Unoki, M., Nishidate, T., and Nakamura, Y. (2004). ICBP90, an E2F-1 target, recruits HDAC1 and binds to methyl-CpG through its SRA domain. Oncogene 23, 7601–7610. doi: 10.1038/sj.onc.1208053
Vella, P., Scelfo, A., Jammula, S., Chiacchiera, F., Williams, K., Cuomo, A., et al. (2013). Tet proteins connect the O-Linked N-acetylglucosamine transferase Ogt to chromatin in embryonic stem cells. Mol. Cell 49, 645–656. doi: 10.1016/j.molcel.2012.12.019
Vincent, J. J., Huang, Y., Chen, P.-Y., Feng, S., Calvopiña, J. H., Nee, K., et al. (2013). Stage-specific roles for Tet1 and Tet2 in DNA demethylation in primordial germ cells. Cell Stem Cell 12, 470–478. doi: 10.1016/j.stem.2013.01.016
Wade, P. A., Gegonne, A., Jones, P. L., Ballestar, E., Aubry, F., and Wolffe, A. P. (1999). Mi-2 complex couples DNA methylation to chromatin remodelling and histone deacetylation. Nat. Genet. 23, 62–66. doi: 10.1038/12664
Wakefield, R. I., Smith, B. O., Nan, X., Free, A., Soteriou, A., Uhrin, D., et al. (1999). The solution structure of the domain from MeCP2 that binds to methylated DNA. J. Mol. Biol. 291, 1055–1065. doi: 10.1006/jmbi.1999.3023
Walter, M. J., Ding, L., Shen, D., Shao, J., Grillot, M., McLellan, M., et al. (2011). Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia 25, 1153–1158. doi: 10.1038/leu.2011.44
Wang, C., Shen, J., Yang, Z., Chen, P., Zhao, B., Hu, W., et al. (2011). Structural basis for site-specific reading of unmodified R2 of histone H3 tail by UHRF1 PHD finger. Cell Res. 21, 1379–1382. doi: 10.1038/cr.2011.123
Watanabe, D., Suetake, I., Tada, T., and Tajima, S. (2002). Stage- and cell-specific expression of Dnmt3a and Dnmt3b during embryogenesis. Mech. Dev. 118, 187–190. doi: 10.1016/S0925-4773(02)00242-3
Watanabe, D., Suetake, I., Tajima, S., and Hanaoka, K. (2004). Expression of Dnmt3b in mouse hematopoietic progenitor cells and spermatogonia at specific stages. Gene Expr. Patterns 5, 43–49. doi: 10.1016/j.modgep.2004.06.008
Weber, A., Marquardt, J., Elzi, D., Forster, N., Starke, S., Glaum, A., et al. (2008). Zbtb4 represses transcription of P21CIP1 and controls the cellular response to p53 activation. EMBO J. 27, 1563–1574. doi: 10.1038/emboj.2008.85
Welling, M., Chen, H.-H., Muñoz, J., Musheev, M. U., Kester, L., Junker, J. P., et al. (2015). DAZL regulates Tet 1 translation in murine embryonic stem cells. EMBO Rep. 16, 791–802. doi: 10.15252/embr.201540538
Whyte, W. A., Bilodeau, S., Orlando, D. A., Hoke, H. A., Frampton, G. M., Foster, C. T., et al. (2012). Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature 482, 221–225. doi: 10.1038/nature10805
Williams, K., Christensen, J., Pedersen, M. T., Johansen, J. V., Cloos, P. A., Rappsilber, J., et al. (2011). TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature 473, 343–348. doi: 10.1038/nature10066
Winkelmann, J., Lin, L., Schormair, B., Kornum, B. R., Faraco, J., Plazzi, G., et al. (2012). Mutations in DNMT1 cause autosomal dominant cerebellar ataxia, deafness and narcolepsy. Hum. Mol. Genet. 21, 2205–2210. doi: 10.1093/hmg/dds035
Woo, H. R., Pontes, O., Pikaard, C. S., and Richards, E. J. (2007). VIM1, a methylcytosine-binding protein required for centromeric heterochromatinization. Genes Dev. 21, 267–277. doi: 10.1101/gad.1512007
Woodcock, C. L. (2006). Chromatin architecture. Curr. Opin. Struct. Biol. 16, 213–220. doi: 10.1016/j.sbi.2006.02.005
Wossidlo, M., Nakamura, T., Lepikhov, K., Marques, C. J., Zakhartchenko, V., Boiani, M., et al. (2011). 5-Hydroxymethylcytosine in the mammalian zygote is linked with epigenetic reprogramming. Nat. Commun. 2:241. doi: 10.1038/ncomms1240
Xie, Z. H., Huang, Y. N., Chen, Z. X., Riggs, A. D., Ding, J. P., Gowher, H., et al. (2006). Mutations in DNA methyltransferase DNMT3B in ICF syndrome affect its regulation by DNMT3L. Hum. Mol. Genet. 15, 1375–1385. doi: 10.1093/hmg/ddl059
Xu, G. L., Bestor, T. H., Bourc’His, D., Hsieh, C. L., Tommerup, N., Bugge, M., et al. (1999). Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 402, 187–191. doi: 10.1038/46052
Xu, W., Yang, H., Liu, Y., Yang, Y., Wang, P., Kim, S. H., et al. (2011). Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 19, 17–30. doi: 10.1016/j.ccr.2010.12.014
Xu, Y., Wu, F., Tan, L., Kong, L., Xiong, L., Deng, J., et al. (2011). Genome-wide regulation of 5hmC, 5mC, and gene expression by Tet1 hydroxylase in mouse embryonic stem cells. Mol. Cell 42, 451–464. doi: 10.1016/j.molcel.2011.04.005
Xu, Y., Xu, C., Kato, A., Tempel, W., Abreu, J. G., Bian, C., et al. (2012). Tet3 CXXC domain and dioxygenase activity cooperatively regulate key genes for xenopus eye and neural development. Cell 151, 1200–1213. doi: 10.1016/j.cell.2012.11.014
Yamaguchi, S., Hong, K., Liu, R., Shen, L., Inoue, A., Diep, D., et al. (2012). Tet1 controls meiosis by regulating meiotic gene expression. Nature 492, 443–447. doi: 10.1038/nature11709
Yamaguchi, S., Shen, L., Liu, Y., Sendler, D., and Zhang, Y. (2013). Role of Tet1 in erasure of genomic imprinting. Nature 504, 460–464. doi: 10.1038/nature12805
Yasui, D. H., Peddada, S., Bieda, M. C., Vallero, R. O., Hogart, A., Nagarajan, R. P., et al. (2007). Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. Proc. Natl. Acad. Sci. U.S.A. 104, 19416–19421. doi: 10.1073/pnas.0707442104
Yildirim, O., Li, R., Hung, J. H., Chen, P. B., Dong, X., Ee, L. S., et al. (2011). Mbd3/NURD complex regulates expression of 5-hydroxymethylcytosine marked genes in embryonic stem cells. Cell 147, 1498–1510. doi: 10.1016/j.cell.2011.11.054
Yoder, J. A., and Bestor, T. H. (1998). A candidate mammalian DNA methyltransferase related to pmt1p of fission yeast. Hum. Mol. Genet. 7, 279–284. doi: 10.1093/hmg/7.2.279
Yoder, J. A., Yen, R. W., Vertino, P. M., Bestor, T. H., and Baylin, S. B. (1996). New 5’ regions of the murine and human genes for DNA (cytosine-5)-methyltransferase. J. Biol. Chem. 271, 31092–31097. doi: 10.1074/jbc.271.49.31092
Yoon, H. G., Chan, D. W., Reynolds, A. B., Qin, J., and Wong, J. (2003). N-CoR mediates DNA methylation-dependent repression through a methyl CpG binding protein Kaiso. Mol. Cell 12, 723–734. doi: 10.1016/j.molcel.2003.08.008
Yu, C., Zhang, Y. L., Pan, W. W., Li, X. M., Wang, Z. W., Ge, Z. J., et al. (2013). CRL4 complex regulates mammalian oocyte survival and reprogramming by activation of TET proteins. Science 342, 1518–1521. doi: 10.1126/science.1244587
Zhang, H., Zhang, X., Clark, E., Mulcahey, M., Huang, S., and Shi, Y. G. (2010). TET1 is a DNA-binding protein that modulates DNA methylation and gene transcription via hydroxylation of 5-methylcytosine. Cell Res. 20, 1390–1393. doi: 10.1038/cr.2010.156
Zhang, J., Gao, Q., Li, P., Liu, X., Jia, Y., Wu, W., et al. (2011). S phase-dependent interaction with DNMT1 dictates the role of UHRF1 but not UHRF2 in DNA methylation maintenance. Cell Res. 21, 1723–1739. doi: 10.1038/cr.2011.176
Zhang, P., Su, L., Wang, Z., Zhang, S., Guan, J., Chen, Y., et al. (2012). The involvement of 5-hydroxymethylcytosine in Active DNA demethylation in Mice. Biol. Reprod. 86, 104. doi: 10.1095/biolreprod.111.096073
Zhang, Q., Liu, X., Gao, W., Li, P., Hou, J., Li, J., et al. (2014). Differential regulation of the ten-eleven translocation (TET) family of dioxygenases by O-linked beta-N-acetylglucosamine transferase (OGT). J. Biol. Chem. 289, 5986–5996. doi: 10.1074/jbc.M113.524140
Zhang, R. R., Cui, Q. Y., Murai, K., Lim, Y. C., Smith, Z. D., Jin, S., et al. (2013). Tet1 regulates adult hippocampal neurogenesis and cognition. Cell Stem Cell 13, 237–245. doi: 10.1016/j.stem.2013.05.006
Zhang, Y., Ng, H. H., Erdjument-Bromage, H., Tempst, P., Bird, A., and Reinberg, D. (1999). Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev. 13, 1924–1935.
Zhao, X., Ueba, T., Christie, B. R., Barkho, B., McConnell, M. J., Nakashima, K., et al. (2003). Mice lacking methyl-CpG binding protein 1 have deficits in adult neurogenesis and hippocampal function. Proc. Natl. Acad. Sci. U.S.A. 100, 6777–6782. doi: 10.1073/pnas.1131928100
Zhou, Z., Qin, J., Tang, J., Li, B., Geng, Q., Jiang, W., et al. (2013). Down-regulation of MeCP2 in Hirschsprung’s disease. J. Pediatr. Surg. 48, 2099–2105. doi: 10.1016/j.jpedsurg.2013.07.011
Keywords: cytosine modifications, Dnmt, epigenetics, hydroxymethylcytosine, MBD, methylcytosine, mouse models, Tet
Citation: Ludwig AK, Zhang P and Cardoso MC (2016) Modifiers and Readers of DNA Modifications and Their Impact on Genome Structure, Expression, and Stability in Disease. Front. Genet. 7:115. doi: 10.3389/fgene.2016.00115
Received: 31 March 2016; Accepted: 06 June 2016;
Published: 21 June 2016.
Edited by:
Joanna Mary Bridger, Brunel University London, UKReviewed by:
Abhijit Shukla, Harvard Medical School, USACopyright © 2016 Ludwig, Zhang and Cardoso. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: M. C. Cardoso, Y2FyZG9zb0BiaW8udHUtZGFybXN0YWR0LmRl
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.