 Koffi N. Maglo
Koffi N. Maglo Tesfaye B. Mersha
Tesfaye B. Mersha Lisa J. Martin
Lisa J. Martin
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet. , 17 February 2016
Sec. Applied Genetic Epidemiology
Volume 7 - 2016 | https://doi.org/10.3389/fgene.2016.00022
The biological status and biomedical significance of the concept of race as applied to humans continue to be contentious issues despite the use of advanced statistical and clustering methods to determine continental ancestry. It is thus imperative for researchers to understand the limitations as well as potential uses of the concept of race in biology and biomedicine. This paper deals with the theoretical assumptions behind cluster analysis in human population genomics. Adopting an interdisciplinary approach, it demonstrates that the hypothesis that attributes the clustering of human populations to “frictional” effects of landform barriers at continental boundaries is empirically incoherent. It then contrasts the scientific status of the “cluster” and “cline” constructs in human population genomics, and shows how cluster may be instrumentally produced. It also shows how statistical values of race vindicate Darwin's argument that race is evolutionarily meaningless. Finally, the paper explains why, due to spatiotemporal parameters, evolutionary forces, and socio-cultural factors influencing population structure, continental ancestry may be pragmatically relevant to global and public health genomics. Overall, this work demonstrates that, from a biological systematic and evolutionary taxonomical perspective, human races/continental groups or clusters have no natural meaning or objective biological reality. In fact, the utility of racial categorizations in research and in clinics can be explained by spatiotemporal parameters, socio-cultural factors, and evolutionary forces affecting disease causation and treatment response.
While pervasive, the concept of race is both a problematic and highly misunderstood concept in biological and biomedical research (Smedley and Smedley, 2005). This is not new, and for centuries, the debate on whether biological human races exist has raged (Risch et al., 2002; Burchard et al., 2003; Ossorio and Duster, 2005; Caspari, 2009; Frank, 2014; Guo et al., 2014; Duster, 2015). The completion of the Human Genome Project seems to have added fuel to the ongoing debate. Indeed, when looking at continental ancestry, a relatively small number of genetic markers can separate populations into meta-populations (Paschou et al., 2008; Nelis et al., 2009). On the other hand, when examining larger number of markers, there is a tremendous diversity within groups (Hunley et al., 2009; Baye et al., 2011).
The relevance of racial classifications to biomedical research is also unclear. There are many examples in the literature where racial differences in health related phenotypes exist (e.g., obesity, asthma, and breast cancer; Ogden et al., 2012; Romero et al., 2012; Howlader et al., 2014b; Keet et al., 2015). However, the predictive ability of race in epidemiological and clinical research is generally weakened by potential confounders. Yet, the mandate by funding agencies like the National Institutes of Health on capturing racial information of research participants ensures that race cannot be ignored (Stevens, 2003; Maglo and Martin, 2012; Bliss, 2013; Maglo et al., 2014). Thus it is crucial for researchers and clinicians to understand the issues surrounding the biological status and biomedical significance of the concept of race (Maglo, 2010, 2012; Mersha and Abebe, 2015).
The purpose of this paper is to describe the scientific basis of the concept of race in biological systematic and evolutionary classification and its applications in current biomedical research by reviewing and evaluating the current literature. Specifically, we will scrutinize the issue of the biological basis by discussing the phylogenetic and evolutionary criteria for the objective existence or natural reality of biological groupings/taxa of organisms. We will apply genetic data to evaluate the evidence for the putative existence of biological human races. Lastly, we will consider the implications and utility of racial grouping to biomedical research. Taken together, this paper demonstrates that, despites technical and technological advances in clustering methods, cline remains the foundational concept in human population genomics, that continental clusters are merely instrumentally produced, and that human races/continental groups have no natural meaning or objective reality from a biological systematic and evolutionary taxonomical perspective. There are gradations (clines) in human population genetic profiles, and without understanding allelic distributions across human populations and their practical biomedical implications, the potential for reification and misinterpretation of racial disparities in epidemiological and clinical research is great.
There are two evolutionary theoretical criteria for naturally objective groupings of biological organisms. These are common ancestry and degree of similarity (Mayr and Bock, 2002; Schuh and Brower, 2009; Wiley and Lieberman, 2011; Templeton, 2013). Phylogenetic systematics and Darwinian/evolutionary taxonomy use “common descent” as a criterion for biological classification but the similarity criterion is used only in the latter. Systematics and evolutionary classification are concerned with organic diversity and evolutionary relationships. The assumptions underlying the primary use of neutral markers in human genetic diversity studies suggest that their objective biological meaning needs to be evaluated based on the above two criteria. Yet as researchers increasingly point out, the debate is “free floating” to the extent that what counts as “biological reality” of human races is elusive, ranging from “trivial” to “obscure,” and often construed in a non-Darwinian biological framework (Cavalli-Sforza, 2000; Cooper et al., 2003; Graves, 2011; Maglo, 2011).
The mounting questionable assumptions underlying biological race theories have recently led scholars to remind the research community about Dobzhansky's (1973) paper tellingly entitled “Nothing in biology makes sense except in light of evolution” (Dobzhansky, 1973; Graves, 2011). In fact, although biologists and ordinary people are interested in various forms of classifications of biological entities, not every classification reflects an evolutionary ordering of living things (Dupré, 1993; Mayr and Bock, 2002). Phylogenetic systematics, for one, posits that the various other types of biological relationships researchers are concerned with, including ecological relationships and similarity, “have maximum relevance when understood within the context of genealogical descent” (Wiley and Lieberman, 2011).
Darwinian or evolutionary classification on its part deploys the two criteria for ordering organisms, discussed above, and is also different even from biological classifications “of cell, tissue and organ types of different groups of organisms, of ecological communities, of behavioral activities and so forth” (Mayr and Bock, 2002). While the objectivity of evolutionary kinds, understood as evolutionary ordered taxa of organisms, is defined either by common descent or genetic similarity, the similarity itself is construed as deriving from homologous characteristics due to shared ancestry rather than deriving from homoplastic characteristics due to parallelism, convergence, or reversal (Mayr and Bock, 2002; Fujimura and Rajagopalan, 2011).
Accordingly, a taxon of organisms may be said to have an objective independent biological existence in Darwinian classification if either of the following two conditions obtains: (1) It constitutes a phylognetic clade by comprising all, but only all, the descendants of its originating biological common ancestor (Templeton, 1998, 2013; Schuh and Brower, 2009; Claridge, 2010; Mishler, 2010; Maglo, 2011; Wiley and Lieberman, 2011); and/or (2) It has reached a degree of genetic differentiation deemed taxonomically meaningful in system biology (Mayr and Bock, 2002; Keita et al., 2004; Graves, 2011). Thus, it follows from these evolutionary theoretical constraints that races must be evolutionary distinct human subpopulations by virtue of (1) or (2) or some combination of both in order to be a valid biological category.
In Darwinian classification (but also in phylogenetic systematics), a biological grouping of organisms that does not meet the above criteria is referred to as a wastebasket taxon. It is so called because it is evolutionary unordered and functions in science merely as a “warehouse kind” that taxonomically lumped together disparate organisms having no objectively definable evolutionary relationship. Wastebasket taxa lack natural reality (Parfrey et al., 2006; Schuh and Brower, 2009; Claridge, 2010; Mishler, 2010; Wiley and Lieberman, 2011) and granting them objective biological existence constitutes an erroneous attribution of ontological status called the fallacy of reification (Gannett, 2004, 2014; Duster, 2005; Glasgow, 2009; Maglo and Martin, 2012; Hochman, 2013).
One way to explain the reification problem in the biological classification of organisms is to consider the hierarchical population structure model which would seem to lend support to the identification of distinct clusters of human populations. For the cluster approach, one defines distinct subgroups from the genetic substructure within a population, (Figure 1A). Under the hierarchical population structure model, a population is made up of subpopulations deriving from a fragmentation history. Fragmentation may be due to various causes including habitat fragmentation which itself may be induced by natural factors (geological and climatic) or human factors (social and cultural). Population fragmentation engenders substructures and may lead to the emergence of meta-populations (Table 1). The total genetic variation in the population is then the sum of the genetic variation of the fragmented subpopulations (Relethford, 2012). But a population with a history of fragmentation may have various divisionary levels (DLs), i.e., structural layers (Maglo, 2010, 2012).

Figure 1. (A) The effect of sampling strategies (adopted from Handley et al. 2007) [“Heterogeneous sampling can reveal genetic clusters that are biologically meaningless. The gradation in color from blue to orange represents a hypothetical situation of strictly continuous variation in allele frequencies. If sampling is heterogeneous (population samples represented here by circles) then the pattern of clinal variation can be mistaken for genetically distinct clusters (black ellipses)”] (Handley et al., 2007). (B) Global genetic diversity in humans are distributed in gradients among and within continents, emphasizes intercontinental variation. The inset figure of Africa highlights finer gradations over shorter geographic spans, emphasizing the “clinality” of human genetic variation (adopted from Cavalli-Sforza et al., 1994). Figure reproduced with permission from Cell Press/Elsevier.
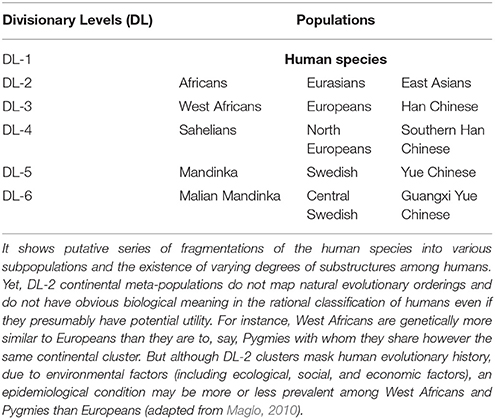
Table 1. Illustration of the concept of divisionary level (DL) with three continental groups.
For instance, continental clusters are believed to correspond to meta-populations representing the major divisions among humans at the infra-species level. So if one pragmatically takes all humans as a population (divisionary level 1; DL-1), major subgroups within DL-1 will correspond to divisionary level 2 (DL-2). Using the genetic subgroups, DL-2 is reflective of “continental groups” (e.g., Africa, Eurasia, West Asia). But each DL-2 meta-population, may be further subdivided along finer substructures according to the fragmentation of the population history (Cavalli-Sforza et al., 1994; Tishkoff and Verrelli, 2003; Maglo, 2010; Table 1). One of the major factors thought to contribute to fragmentation of populations is geomorphologic barriers constraining population dispersal. For example, the Sahara, Himalayas, and oceans, hereafter SHO, are construed as cluster enabling factors that increase genetic distance between human populations. We shall call this view “the SHO hypothesis.” According to this hypothesis, continental clusters are natural biological groupings because human populations are naturally and distinctly classified based on their genetic differences (Rosenberg et al., 2002, 2005; Rosenberg, 2011).
One vexing question raised by the steady progress in genomics and computational bioinformatics is how to conceptualize a putative correspondence between race and genomic groupings into continental metapopulations (Cavalli-Sforza, 2000; Glasgow, 2009; Maglo, 2010; Fullwiley, 2011). If we conceive the biological reality or natural character of race as group of organisms in cladistic terms as in criterion (1) above, then phylogenomics supplies the appropriate answer to that question. If continental human populations can be shown to have different recent common ancestors and that each comprises all of the descendants of the respective common ancestor, then the matter can be straightforwardly considered settled according to the cladistic concept of race. However, the problem with cladistic theories of race is that human populations show crisscrossing lineages to the extent that: “A classification that takes into account evolutionary relationships and the nested pattern of diversity would require that Sub-Saharan Africans are not a race because the most exclusive group that includes all Sub-Saharan African populations also includes every non-Sub-Saharan African population…” (Long et al., 2009; Templeton, 2013).
However, cluster analysis is a phenetic method and presupposes, from the perspective of a rational biological classification, the genetic similarity criterion of evolutionary taxonomy. Yet, unlike cluster, it is cline that best accounts for human evolutionary diversity. In fact, the cline model maps continuous genetic gradation in a dataset and indicates that there is no natural break in a population's genetic profile (Figure 1B). Although cluster and cline models are not incompatible, they may lead to competing interpretations. If the population is shown to have a clinal genetic structure but cluster arises in some situations (Ramachandran et al., 2005; Handley et al., 2007; Underhill and Kivisild, 2007), then clustering results cannot be interpreted, in biological taxonomy, as indicative of natural differentiations of biological subpopulations. In this case, cline will be the representation of the natural evolutionary ordering of the population, while cluster will be an artifact, a construct that indicates instrumental, i.e., convenient, cutoff points for various scientific purposes.
There are various ways to measure genetic differences between populations, but perhaps the most popular is the fixation index Fst. Fst is a measure of differentiation between two populations. Values range from 0 (no difference between populations) to 1 (fixed differences between populations). Stemming from Sewall Wright's guideline, Fst-values between 0 and 0.05 indicate “no to little genetic differentiation” while Fst-values between 0.05 and 0.15 represent moderate differentiation. Fst-values between 0.15 and 0.25 are considered large and Fst values above 0.25 show a very large degree of genetic differentiation (Wright, 1978; Balloux and Lugon-Moulin, 2002; Tishkoff and Kidd, 2004; Elhaik, 2012; Bhatia et al., 2013). Researchers consider Fst of 0.25 as a minimum value for genetically distinct races (Templeton, 1998; Graves, 2011).
Another way to measure population differentiation is by using statistical cluster based methods. These methods seek to group individuals together who are genetically similar. Clusters may be defined by calculating pairwise distance matrix and identified graphically. Or model based methods could be used. These models require a priori specification of model parameters including the number of clusters. Both types of approaches are utilized in current population structure software packages such as Eignstrat (genetic distance) and Structure (model based). In this paper, we will focus on results using the computer program Structure. Structure uses a Bayesian cluster analysis approach where the researcher arbitrarily determines the number of K clusters into which the data should be portioned (Bolnick, 2008). If the number of clusters is not known, Structure allows researchers to define the interval of the values of K from 1 to an arbitrary number N (1 ≤ K ≤ N) and then to compute the maximum likelihood of K-clusters in order to determine the most supported K within the defined interval. However, Evanno et al.'s (2005) ad-hoc second order statistic ΔK does not allow K = 1 (Evanno et al., 2005; Schwartz and McKelvey, 2009).
Genomic research is continually providing an improved understanding of factors affecting population differentiation. Take for example the study by Rosenberg et al. (2005). In this study, Fst was modeled as a function of geographic distance (D) and other barriers (B) (Rosenberg et al., 2002). D is a continuous measures while B = 1 if a barrier but zero otherwise. After examining pair-wise Fst between individual populations the following regression equation: Fst = 0.0032 + 0.0049D + 0.0153B was generated. This equation suggests that the Sahara, Himalayas and oceans introduce genetic discontinuities between pairs of populations on the opposite side (R2 = 0.0153). Crossing any of these barriers amounts to traveling over 3100 km on the same side of the barrier. That is, barriers so different in nature and settlement history add each nonetheless the same value to the Fst. However, geographical distance (or isolation by distance; IBD) explains the bulk of the variance (R2 = 0.690). Yet according to the SHO hypothesis, it is the abrupt tiny increase in the Fst values (R2 = 0.0153), putatively caused by spatial resisting forces, that enables computer algorithms to partition humans into continental genomic clusters (Rosenberg et al., 2005; Rosenberg, 2011). Nonetheless, using multilocus genetic markers, about 93% of the total human genetic variation was found at the individual level while an Fst of 4.3% was apportioned to “continental” regions (Rosenberg et al., 2002).
Yet even if one aims lower on the aformentioned scale of genetic differentiation, it is still clear that the Fst-value of 0.043, measuring the genetic difference between continental clusters (Rosenberg et al., 2002), unambiguously lies in the interval of no to little degree of differentiation on Wright's guideline. Continental subpopulations are also very similar and do not reach, any meaningful degree of differentiation in Darwinian classification. These results suggest that human races, understood as continental clusters, have no taxonomic meaning that warrants granting them an objective biological existence. Actually, the Fst-value is even lower, 0.036, with a partition scheme that identified 7 DL-2 meta-populations (Rosenberg et al., 2002). Nevertheless, although generally below the threshold of taxonomic meaningfulness, Fst-values, particularly pair-wise Fst, vary and are influenced by many factors (Lewontin, 1972; Barbujani et al., 1997; Jorde and Wooding, 2004; Tishkoff and Kidd, 2004; Al Sweih et al., 2010; Graves, 2011; Elhaik, 2012). For instance, data from the International HAPMAP consortium estimated differences among a limited number of selected continental populations to be between 0.11 and 0.19 (Nelis et al., 2009).
The problem is that taxonomic groupings that map actual evolutionary relationships among populations in a worldwide comparison identify series of meta-populations different from continental clustering schemes (Zhivotovsky et al., 2003). Pair-wise Fst computations confirm for example that genetic distances between sub-Saharan African “hunter and gatherers,” the first series of meta-populations (hereafter S1-Metapopulations), and sub-Saharan African “farmers” (S2-Metapopulations) are greater than between the latter and Europeans (Zhivotovsky et al., 2003; Tishkoff et al., 2009; Kalinowski, 2010). In addition, some populations in the S1-Metapopulations such as “the southern Bushmen, central forest Pygmies, and the Hadza compared with Europeans, have Fst estimates in excess of 0.23, approximately twice the average Fst between other global populations” (Henn et al., 2011, 2012). Even so, “East Africans” and the Maasai are more similar to Europeans than to the KhoeSan populations. Moreover, within the S1-Metapopulations, the Sandawe for example, are more similar to Europeans than they are to the Hadza (Henn et al., 2011, 2012). Actually, genetic patterns from all four modes of human inheritance (mtDNA, Y-chromosome, X-linked and autosome), along with protein markers, showed that continental clustering represents no natural classification of humans (Maglo, 2011; Mersha and Abebe, 2015).
Be that as it may, the emerging scientific consensus is that while isolation by distance explained a large proportion of human population pair-wise Fst-values, cluster, the computational placeholder for race, explained <2% (Rosenberg et al., 2005; Handley et al., 2007). Thus, the indistinctiveness Fst-value argument, as construed here against the idea of biological reality of human races, is not simply about crude Fst measures. It also takes into consideration the part of Fst quantitatively explained by cluster/race (Table 2). So it goes beyond Wright's qualitative guideline about the use of Fst. The argument thus has two quantitative components, the unadjusted Fst-values and the adjusted values of cluster/race covariate. It can thus be considered the quantitative equivalent of the qualitative argument of “lack of distinction” Darwin used to question the taxonomic wisdom of categorizing humans into races in natural classification since the categories cannot be objectively defined (Maglo, 2011).
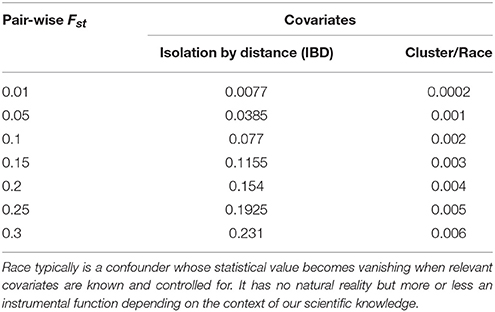
Table 2. Hypothetical pair-wise Fst values with corresponding adjusted values of IBD and cluster/race (using 77% for IBD and 2% for cluster).
The above considerations underscore the claim that “cluster” is likely a byproduct of isolation by distance and sampling procedures (Serre and Paabo, 2004; Handley et al., 2007; Schwartz and McKelvey, 2009). For instance, geographic discontinuous samples of Africans, Europeans and East Asians yield clustered representations of the datasets. But when South Asian samples are included in the analyses, clinal representations emerge (Bamshad et al., 2001; Jorde and Wooding, 2004; Tishkoff and Kidd, 2004). The sampling of geographically isolated populations has been called “island model” sampling procedure (Bamshad et al., 2004; Maglo, 2011). It produces a misleading representation of the human genetic continuum (see Figure 1A).
There are two different statistical models within Structure. One assumes uncorrelated allele frequencies and one assumes correlated allele frequencies. As early as 2004, critics pointed out that the SHO hypothesis failed to be confirmed when one combines an uncorrelated frequencies model with a sampling strategy that assumes a continuous geographic dispersal of human populations. The uncorrelated model yielded a clinal, rather than a clustered, representation of human population genetic structure (Serre and Paabo, 2004). The model used by Rosenberg et al. (2002, 2005) assumes that allele frequencies between continental clusters are correlated, due to common ancestry. Actually, the statistical model of correlated allele frequencies assumes a sharing of a recent common ancestor and admixture between human populations. However, the admixture assumption is contrary to the criteria for biologic natural reality of phylogentic classification. Furthermore, both the correlated and the uncorrelated models of Structure converge in showing empirically that human populations are not monophyletic groups (Serre and Paabo, 2004; Rosenberg et al., 2005).
An examination of the assumptions behind the correlated and uncorrelated allele frequencies models will yield insight into the controversy about the biological meaning of continental clusters. The “historical” context of the dispute is that the uncorrelated model was implemented in the computer program Structure earlier while the correlated allele frequencies used by Rosenberg and his colleagues was a later revised model (Pritchard et al., 2000; Falush et al., 2003). From an epistemic perspective, the novelty in the revised model was to distinguish between two conditions described as “harsh prior” and “permissive prior.” It is the harsh prior model that assumes that allele frequencies are statistically dependent and hence very similar. So the allele frequency distribution for one cluster provides information about the frequency distributions of the other clusters. The permissive prior makes no such assumptions and population movement is not subjected to the “unrealistic” condition that all subpopulations simultaneously drifted away from a common ancestral population (Falush et al., 2003).
Now, while the harsh prior of statistically dependent frequencies model is described as more efficient at detecting finer population substructures and best suited for “subtle admixture problems,” the authors of the revised Structure model also stated that “if the values of Fk are being used to make evolutionary inferences, a permissive prior is more appropriate” (Falush et al., 2003). Additionally, they warned about a crude attribution of biological meaning to variance partitions, including K-clusters with the highest probability. As they put it: “(1) it is computationally difficult to obtain accurate estimates of Pr(X/K), and our method merely provides an ad-hoc approximation, and (2) the biological interpretation of K may not be straightforward” (Pritchard et al., 2007). The question then is about how to interpret the Fst values (Fk in the program Structure F model) in clustering analysis of human genetic variation.
Furthermore, it has been shown that the rate of individuals having membership in multiple clusters increases with the inclusion of admixed populations in studies. This does not however negate the computational possibility of clustering admixed individuals. But under this scenario, many individuals will typically have mixed membership in different clusters (Pritchard et al., 2007; Bryc et al., 2010; Maglo, 2011; Jin et al., 2012). As mentioned above, the correlated allele model was specifically designed to resolve “subtle admixture problems.” Curiously, some researchers perform cluster analysis on admixed populations by bypassing this model (Tang et al., 2005), raising questions about their findings (Graves, 2011). Yet the user guide of Structure states that “Admixture is a common feature of real data, and you probably won't find it if you use the no-admixture model” (Pritchard et al., 2000; Elhaik, 2012).
The admixture-based argument holds indeed that, in a global partition of human genetic variation, the number of individuals with membership in multiple clusters will increase with the inclusion of more admixed populations, causing continental clusters to dissolve into a cline (Bamshad et al., 2003, 2004; Maglo, 2011). Admixture does not however occur only in a demographic melting pot situation. Neighboring-mating likely plays a role in the dissolution of cluster into cline in the “island model” cases discussed above. Neighboring-mating engenders local genetic autocorrelation which is shown to impact strongly the determination and reliability of clusters (Schwartz and McKelvey, 2009).
Spatial genetic autocorrelation results from proximal mating between individuals at the periphery of a geographically dispersed population and neighbors from the surrounding populations. The authors of Structure acknowledged the limitations of the program in this respect by stating that in the case of datasets structured by IBD, “allele frequencies vary gradually across the region. The underlying structure model is not well suited to data from this kind of scenario. When this occurs, the inferred value of K, and the corresponding allele frequencies in each group can be rather arbitrary. Depending on the sampling scheme, most individuals may have mixed membership in multiple groups” (Falush et al., 2003; Schwartz and McKelvey, 2009). In a word, computational success does not by itself alone entail the natural reality of clustered entities in evolutionary classification (Maglo and Martin, 2012).
A study on short tandem repeats (STRs)/microsatellite markers from the Diversity Panel dataset demonstrated that continental clusters masks evolutionary relationships among human populations. Estimates of divergence time (TD) showed that S1-Metapopulations (see above) were the first series of meta-populations to split from the common ancestral human population. S2-Metapopulations, the second series of African meta-populations, and “non-Africans” split from each other at a later time. Principal Component Analysis showed S1-Metapopulations at the edge of the sub-Saharan African cluster. The authors consequently wrote: “Each of the large population groups (sub-Saharan African farmers, Eurasia, and East Asia) can be considered as a metapopulation consisting of populations with some genetic exchange between them and with a common ancestry. This is suggested by the value of the statistic S4, which is substantially greater for the pooled regional groups than for single populations within those regions…” (Zhivotovsky et al., 2003). But the pooled statistical value (0.89) of three S1-Metapopulations' samples was very close to their individual values (0.85). This suggests, under the hierarchical structure model, that they do not come from one single meta-population. In fact, S1-Metapopulations provide crucial information about human origin and evolutionary history (Zhivotovsky et al., 2003; Henn et al., 2012; Veeramah et al., 2012).
This study thus showed not only that there are many sub-Saharan African DL-2 meta-populations at a global level (Figure 2B) but also that S2-Metapopulations are evolutionary closer than their neighbor S1-Metapopulations, to “non-Africans” (Figure 2A). Nevertheless, this study illustrates also how continental clusters may be instrumentally produced with real data. In fact, when the study goals were altered, the researchers were able to reduce the evolutionary tree by considering sub-Saharan African populations pragmatically “as a single group” (Zhivotovsky et al., 2003). The reduced tree then helped generate a continentally clustered picture of the dataset, a picture that masks the fragmentation history and the evolutionary relationship among human populations (Figure 3).
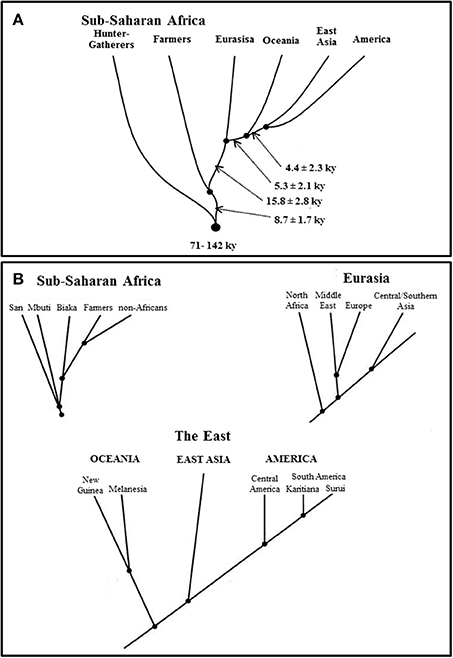
Figure 2. Human metapopulations based on their divergence time estimates and evolutionary relationships (adopted from Zhivotovsky et al., 2003, Figure 5). Zhivotovsky et al. (2003)'s Figures 5A,B clearly indicate that human metapopulations with evolutionary implications do not correspond to continental clusters considered as the placeholders for race in human population genomics. [“Population tree based on TD estimates of divergence time. (A). Divergence among major groups. The time estimates are based on 374 STRs (three outlying STRs with tetranucleotide repeats were omitted). Arrows indicate the time (lower bounds, in ky) between adjacent nodes, assuming a generation length of 25 years. (B). Schemes of divergence within the major groups, based on the 374 STRs. Time estimates within each continental group were omitted, because they may be biased owing to possible differential gene flows from other groups”] (Zhivotovsky et al., 2003). Figure reproduced with permission from Cell Press/Elsevier.
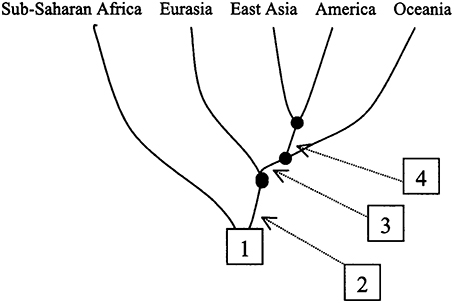
Figure 3. Conveniently defined metapopulations that mask human evolutionary history and relationships (adopted from Zhivotovsky et al., 2003, Figure 6). This tree is simply instrumentally produced to meet the demand of our interest in continental groups/human races and may function under some circumstances as useful problem-solving tool. However, from an evolutionary perspective, it carries no natural meaning or independent reality. [“Reduced population tree showing four separation events”] (Zhivotovsky et al., 2003). Figure reproduced with permission from Cell Press/Elsevier.
It is noteworthy that Rosenberg et al. used Structure to study the same data set of (STRs)/microsatellite markers as Zhivotovsky et al. (Rosenberg et al., 2002; Zhivotovsky et al., 2003). But Structure identified six clusters, with the 6th comprising the Kalash from Pakistan. In 2005, the 6th cluster sometimes subdivides Native American populations across runs (Rosenberg et al., 2005). In 2011, the Oceania cluster was fully identified only at K = 6 (Rosenberg, 2011). That is, the number of K-clusters that allegedly corresponds to major geographic regions varies from 5 to 6 with the same data set and computer algorithm. Yet although the features of the SHO hypothesis fully emerged in Rosenberg's work (Rosenberg, 2011), this study was in short of addressing the emerging finding about the complex genetic diversity between African populations and non-African population. Complex genetic diversity which, in the meantime, also received support from Y-chromosome studies (Cavalli-Sforza, 2000; Ingman et al., 2000; Yu et al., 2002; Underhill and Kivisild, 2007; Kalinowski, 2010; Henn et al., 2011; Maglo, 2011).
In Tishkoff et al.'s (2009) study, Structure identified 14 clusters within our species (Tishkoff et al., 2009). That is, there are presumably fourteen DL-2 meta-populations worldwide rather than 5. Interestingly, in this study, at K = 5, most populations in S1-Metapopulations split from S2-Metapopulations (Tishkoff et al., 2009). In 2011, Rosenberg also reported the same split but at K = 6 in the ninth run (Rosenberg, 2011), indicating that the continental cluster storyline, together with the SHO hypothesis, may be falling apart. Actually, the authors of Structure warned against the dogmatization of any given value of K for geographically dispersed populations like humans by explaining that estimations of K work well only for “data sets with a small number of discrete populations” and that in the case of “real-world data sets” structured by IBD or inbreeding “there may not be a natural answer to what is the “correct” value of K” (Pritchard et al., 2007).
There is, however, more. As explained above, our evaluation focuses primarily on studies using the method of the software Structure. Yet, in current population genomic research, methods such as Principal Component Anlysis (PCA) are frequently used to study population substructure and to determine ancestry (Li et al., 2008; Crosslin et al., 2014). PCA, like the Baysian approach of Structure, is a powerful tool in styuding ancestry and population structure. Generally speaking, population structure analysis plays a very useful role in understanding the relatedness among humans and their ancestral origins as well as for designing disease genetic studies (Baye, 2011; Baye et al., 2011). Ancestry informative markers (AIMs) are tremendously important in this respect because they help understand population history and probe genetic and non-genetic disease susceptibility factors and treatment response determinants (Galanter et al., 2012; Ricks-Santi et al., 2012; Hollenbach et al., 2015). However, it is important to note that the selection of AIMs will influence the clusters generated with common variants detecting more continental ancestry while rare variants detect different patterns (Baye et al., 2011). Thus, when racial categories are predicated on ancestral membership identifications, it is necessary that the issue pertaining to their putative objective natural reality be assessed based on the evolutionary criteria of common ancestry and degree of similarity regardless of the method used to apportion genetic diversity and determine ancestral groupings.
Over the past two decades, genomic research has increasingly supplied various types of empirical evidence which, within an evolutionary framework, unambiguously refutes claims about the natural reality of human races. For instance, by 2002, it had become clear, as discussed above, that genetic distance is greater among sub-Saharan Africans than between some sub-Saharan African meta-populations and “Eurasians” regardless of whether one uses mtDNA, X-linked or autosome genetic markers (Ingman et al., 2000; Yu et al., 2002; Zhivotovsky et al., 2003; Maglo, 2011). Cavalli-Sforza referred to the pattern of genetic distance between Africa and Europe as an “anomaly.” Actually, the Africa-Europe anomaly (hereafter AE-A), consists of: AE-A1—an unexpected shortness of genetic distance between sub-Saharan Africa and Europe compared to sub-Saharan Africa and other continents (Cavalli-Sforza, 2000; Tishkoff and Kidd, 2004); AE-A2—some meta-populations in sub-Saharan Africa being genetically more similar to Europeans than to neighboring meta-populations on the same side of the Sahara (Ingman et al., 2000; Yu et al., 2002; Kalinowski, 2010; Henn et al., 2011). Yu et al. enunciated what we construe as AE-A2 with the revealing title “Larger Genetic Differences within Africans than between Africans and Eurasians” (Yu et al., 2002).
A point that needs to be emphasized here is that not only are there many sub-Saharan African DL-2 meta-populations at a global level (Figure 2B) but also that S2-Metapopulations are evolutionary closer than their neighbor S1-Metapopulations, to “non-Africans” (Figure 2A). An evolutionary-based grouping of world populations attempts to summarize the complex human population history (Figures 2A,B) while an instrumental grouping lumps pragmatically world populations into five continental groups reducing evolutionary relations (Figure 3). It is this instrumentally engineered clustered picture of human evolutionary history that is misleadingly construed as corresponding to socially defined races in countries such as the US. Although these socially defined races and continental genetic clusters do not actually match, the alleged correspondence has generated its own sets of debates (Maglo, 2010; Maglo et al., 2014).
Yet perhaps, spatiotemporal correlates (IBD and divergence time) are the factors underpinning continental clusters because they appear to comprise populations whose ancestral origins are close in space and time regardless of genetic dissimilarity. Simulations using Structure suggest that, at a constant degree of differentiation, cluster membership varies with sample size and divergence times (Kalinowski, 2010). Spatiotemporality is crucial to the evolutionary ordering of living things. Descent with modification (generation) and adaptation (environment) are just some of the familiar evolutionary concepts that implicitly deploy spatiotemporal parameters (Mayr and Bock, 2002). Yet the spatiotemporal proximity of ancestral origins of continentally dwelling subpopulations simply reveals the storing functionality of the cluster construct instead of the ontological order of a natural classification.
By natural classification, we mean, in Duhem's sense, the theoretical organization of experimental laws in a given scientific domain such that they reflect “real relations among things” (Duhem, 1991). Duhem considered scientific theories as a means to logically classify experimental laws. These laws depict symbolical relations between phenomena but not the intrinsic nature of things (Duhem, 1991). Nonetheless, “the more complete” a scientific theory “becomes,” Duhem wrote, “the more we apprehend that the logical order in which theory orders experimental laws is the reflection of an ontological order, the more we suspect that the relations it establishes among the data of observation correspond to real relations among things, and the more we feel that theory tends to be a natural classification” (Duhem, 1991: 26-7). Duhem's model of natural classification is zoological classification. The zoologist, he maintains, considers the genealogical relations established among animals to reflect a natural order in such a way that even if evolutionary theory happens to be proven false s/he will “continue to believe that the plan drawn by his classification depicts real relations among animals; he would admit being deceived about the nature of these relations but not about their existence” (Duhem, 1991).
That said, we distinguish questions of “reality” from questions of “utility” (Maglo, 2007, 2010, 2012). In the not too distant past, determining continental ancestral origin was an astounding achievement. Today, however, the geographic origin of an individual can be determined within just a few hundred kilometers (Novembre et al., 2008). Indeed, with genetic data we are able to subdivide even relatively homogeneous countries into sub-national genomic groups corresponding to linguistic affiliations, e.g., Switzerland (Novembre et al., 2008) or to North-Central-South geographic location, e.g., Sweden (Salmela et al., 2011). However, in a rational classification of biological organisms, the computational possibility to determine group membership (Edwards, 2003; Edge and Rosenberg, 2015) does not imply that these groups are meaningful according to biological systematic and evolutionary classification criteria (Cavalli-Sforza, 2000; Bolnick, 2008; Maglo, 2011). Thus, it is essential that the utility of these classifications are carefully evaluated in well-controlled epidemiological and clinical contexts (Maglo, 2010, 2011, 2012; Maglo and Martin, 2012; Mersha and Abebe, 2015).
As we are transitioning from the universalist clinical concept of “one dose fits all” toward personalized precision medicine, attention has been called to the implications of the phenomenon of phenotypic plasticity in stratified medicine because of the environmental correlates of epidemiological and pharmacogenomic profiles (Maglo and Martin, 2012). Studies have indeed shown great variability in the distribution and expression of clinically relevant genetic variants across subpopulations within continents due to various evolutionary and environmental mechanisms, including ecological and socio-cultural factors (Wilson et al., 2001; Burroughs et al., 2002; Bains et al., 2013). As an increasing number of researchers have shown, it is important for the success of personalized precision medicine that human genetic diversity be considered (Lu et al., 2014; Petersen et al., 2014). But a continental level substructure or race may very well be a confounder in epidemiologic and clinical research. For instance, race accounts for 14.2% of the variance in warfarin dosing when not considering other factors. Yet when pharmacogenomic and relevant biomarkers are taken into account, the statistical value of race was markedly attenutated, 0.3% (Kahn, 2013). This indicates that, from a clinical genomic perspective just as from evolutionary and population genomic perspectives, race is a notion that has at best a contextual instrumental value (Maglo, 2010, 2011).
Despites its lack of natural ontological character, cluster may well be a useful subsidiary notion alongside the foundational concept “cline” in human population genomics. Moreover, because of ecological and environmental variability, a wastebasket taxon may be useful in population health studies. Genetic polymorphisms influencing disease incidence and drug response in humans vary among individuals but they also show patterns of geographic distributions (Baye et al., 2009; Bains et al., 2013). Examples of functional variants exhibiting clear geographic distributions include the ABO blood system (Cavalli-Sforza et al., 1994), sickle cell anemia (Piel et al., 2010) and cystic fibrosis (Bobadilla et al., 2002). However, continental genetic clusters are determined primarily by use of large sets of neutral markers. While a single neutral marker may be in linkage disequilibrium with disease susceptibility or treatment response locus, this single marker is not however sufficient to define populations. Furthermore, susceptibility variants and drug metabolizing enzymes are not the focus of genomic diversity studies, thus making it difficult to generalize the relationship between population diversity on a genomic scale (e.g., across many markers) and risk variants. Nonetheless, as mentioned in the introduction, the frequency of many health-related phenotypes exhibit variation by race. For example the rates of breast cancer are highest in individuals of European ancestry and lowest in individuals of Asian ancestry, with African American in between (Howlader et al., 2014b). However, diagnosis of breast cancer at an early stage age was less common in African Americans while survival was lowest in African Americans and highest in Asian Americans (Iqbal et al., 2015). These differences in outcomes may be due in part to underlying genetic differences as African Americans have higher rates of triple negative tumors, which are known to be more aggressive and require different therapeutic approaches (Howlader et al., 2014a). As such, epidemiologic evidence showing racial differences in health outcomes means that public health and clinical interventions need to consider race.
What this suggests is that researchers and clinicians need to approach race with caution both in the lab and in clinics. The extent to which continental genomic clusters provide useful actionable information to biomedicine remains an open question. Recent studies showed that ancestry mapping has been successfully applied for disease in which prevalence is significantly different between the ancestral populations to identify genomic regions harboring diseases susceptibility loci for cardiovascular disease (Tang et al., 2005), multiple sclerosis (Reich et al., 2005), prostate cancer (Freedman et al., 2006), obesity (Cheng et al., 2009), and asthma (Vergara et al., 2009). Yet, the problem is that even when self-report racial/ethnic identity are said to correspond generally speaking to continental genetic ancestry, racial/ethnic descriptors in the US, for instance, are not necessary good indicators of the complex history of an individual's genetic ancestry (Mersha and Abebe, 2015). The genetic make-up of individuals are highly variable but can however be captured with large dimensional genomic data. Emerging technologies now make it possible to genotype hundreds of thousands of genetic variations in individuals, across the genome with great potential in biomedical research. It is the understanding of the complexity of human individual genetic ancestry that is bringing us closer to personalized medicine (Madore et al., 2007; Bielinski et al., 2014).
It is also important to note, that race is a construct with social and cultural underpinnings that may have biological and biomedical implications. In the United States there are for example marked differences between African Americans and European Americans with respect to economic factors such as education, income, rates of poverty, and rates of being on public insurance (Elster et al., 2003; Williams, 2005; Adler et al., 2012; Cheng et al., 2015). In addition, there is still marked segregation nationally and even within communities (Cable, 2013) which may contribute to health disparities. Thus, simply attributing differences in population groupings to differences in underlying biology may be short sided and may actually cause more harm than good. For example, African Americans have higher rates of obesity than whites (Ogden et al., 2012; Romero et al., 2012). However, there are many potential reasons for increased risk of obesity which may be separate from underlying biology, including local environment and availability of healthy food options and cultural food preferences. Indeed, in Africa the rates of obesity are much lower than what is reported in African Americans in the US (Maglo and Martin, 2012; Mersha and Abebe, 2015). Without understanding the complex dynamic between biologic differences and socio-cultural factors, the optimal strategies for reducing obesity risk cannot be determined and may be missed. Thus, the authors caution against the generalization of the importance of race without by neglecting other factors which may be at play.
It is important in race debate for researchers to distinguish between pragmatically useful and natural biological classifications. Just because we can identify continental ancestral membership computationally does not necessarily imply that race as defined by continental ancestry is meaningful in biological systematics and evolutionary classification. In fact, the genomic and statistical evidence currently available shows that phylogenetic and genetic similarity-based concepts of race fail to be applicable to humans even under minimal rational theoretical principles currently accepted in population genetics/genomics. Although spatiotemporal parameters connect evolutionary and environmental medicine, continental clusters may not necessary be the most relevant partitions in biomedicine. Awareness of the mere instrumental function of cluster and race by researchers and practitioners is necessary to avoid the reification and naturalization of these notions in the lab and clinic. Epidemiological research and pharmacogenomics indicate indeed that biomedical and statistical values of race are generally vanishing when relevant covariates are controlled for.
All authors listed, have made substantial, direct, and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported in part by the National Institutes of Health grant K01 HL103165 and Diversity and Health Disparities Award of the Cincinnati Children's Research Foundation to TM.
SHO, Sahara, Himalayas and Oceans; DLs, Divisionary Levels; DL-1, Divisionary Level 1; DL-2, Divisionary Level 2; IBD, Isolation by Distance; S1-Metapopulations, First Series of (sub-Saharan African) Meta-populations; S2-Metapopulations, Second Series of (sub-Saharan African) Meta-populations.
Adler, N., Bush, N. R., and Pantell, M. S. (2012). Rigor, vigor, and the study of health disparities. Proc. Natl. Acad. Sci. U.S.A. 109 (Suppl. 2), 17154–17159. doi: 10.1073/pnas.1121399109
Al Sweih, N., Al Hashem, G., Jamal, W., and Rotimi, V. (2010). National surveillance of antimicrobial susceptibility of CTX-M-positive and -negative clinical isolates of Escherichia coli from Kuwait government hospitals. J. Chemother. 22, 254–258. doi: 10.1179/joc.2010.22.4.254
Bains, R. K., Kovacevic, M., Plaster, C. A., Tarekegn, A., Bekele, E., Bradman, N. N., et al. (2013). Molecular diversity and population structure at the Cytochrome P450 3A5 gene in Africa. BMC Genet. 14:34. doi: 10.1186/1471-2156-14-34
Balloux, F., and Lugon-Moulin, N. (2002). The estimation of population differentiation with microsatellite markers. Mol. Ecol. 11, 155–165. doi: 10.1046/j.0962-1083.2001.01436.x
Bamshad, M. J., Wooding, S., Watkins, W. S., Ostler, C. T., Batzer, M. A., and Jorde, L. B. (2003). Human population genetic structure and inference of group membership. Am. J. Hum. Genet. 72, 578–589. doi: 10.1086/368061
Bamshad, M., Kivisild, T., Watkins, W. S., Dixon, M. E., Ricker, C. E., Rao, B. B., et al. (2001). Genetic evidence on the origins of Indian caste populations. Genome Res. 11, 994–1004. doi: 10.1101/gr.GR-1733RR
Bamshad, M., Wooding, S., Salisbury, B. A., and Stephens, J. C. (2004). Deconstructing the relationship between genetics and race. Nat. Rev. Genet. 5, U598–U592. doi: 10.1038/nrg1401
Barbujani, G., Magagni, A., Minch, E., and Cavallisforza, L. L. (1997). An apportionment of human DNA diversity. Proc. Natl. Acad. Sci. U.S.A. 94, 4516–4519. doi: 10.1073/pnas.94.9.4516
Baye, T. M. (2011). Inter-chromosomal variation in the pattern of human population genetic structure. Hum. Genomics 5, 220–240. doi: 10.1186/1479-7364-5-4-220
Baye, T. M., He, H., Ding, L., Kurowski, B. G., Zhang, X., and Martin, L. J. (2011). Population structure analysis using rare and common functional variants. BMC Proc. 5 (Suppl. 9):S8. doi: 10.1186/1753-6561-5-s9-s8
Baye, T. M., Wilke, R. A., and Olivier, M. (2009). Genomic and geographic distribution of private SNPs and pathways in human populations. Per. Med. 6, 623–641. doi: 10.2217/pme.09.54
Bhatia, G., Patterson, N., Sankararaman, S., and Price, A. L. (2013). Estimating and interpreting FST: the impact of rare variants. Genome Res. 23, 1514–1521. doi: 10.1101/gr.154831.113
Bielinski, S. J., Olson, J. E., Pathak, J., Weinshilboum, R. M., Wang, L., Lyke, K. J., et al. (2014). Preemptive genotyping for personalized medicine: design of the right drug, right dose, right time-using genomic data to individualize treatment protocol. Mayo Clin. Proc. 89, 25–33. doi: 10.1016/j.mayocp.2013.10.021
Bliss, C. (2013). Translating racial genomics: passages in and beyond the lab. Qual. Sociol. 36, 423–443. doi: 10.1007/s11133-013-9257-5
Bobadilla, J. L., Macek, M. Jr. Fine, J. P., and Farrell, P. M. (2002). Cystic fibrosis: a worldwide analysis of CFTR mutations–correlation with incidence data and application to screening. Hum. Mutat. 19, 575–606. doi: 10.1002/humu.10041
Bolnick, D. (2008). “Individual ancestry inference and the reification of race as a biological phenomenon,” in Revisiting Race in A Genomic Age, eds B. Koenig, S. S.-J. Lee, and S. S. Richardson (New Brunswick, NJ: Rutgers University Press), 70–85.
Bryc, K., Auton, A., Nelson, M. R., Oksenberg, J. R., Hauser, S. L., Williams, S., et al. (2010). Genome-wide patterns of population structure and admixture in West Africans and African Americans. Proc. Natl. Acad. Sci. U.S.A. 107, 786–791. doi: 10.1073/pnas.0909559107
Burchard, E. G., Ziv, E., Coyle, N., Gomez, S. L., Tang, H., Karter, A. J., et al. (2003). The importance of race and ethnic background in biomedical research and clinical practice. N. Engl. J. Med. 348, 1170–1175. doi: 10.1056/NEJMsb025007
Burroughs, V. J., Maxey, R. W., and Levy, R. A. (2002). Racial and ethnic differences in response to medicines: towards individualized pharmaceutical treatment. J. Natl. Med. Assoc. 94, 1–26.
Cable, D. (2013). The Racial Dot Map [Online]. Weldon Cooper Center for Public Service: University of Virginia. Available online at: http://www.coopercenter.org/demographics/Racial-Dot-Map (Accessed June 1 2015).
Caspari, R. (2009). 1918: three perspectives on race and human variation. Am. J. Phys. Anthropol. 139, 5–15. doi: 10.1002/ajpa.20975
Cavalli-Sforza, L. L., Menozzi, P., and Piazza, A. (1994). The History and Geography of Human Genes. Princeton, NJ: Princeton University Press.
Cheng, C. Y., Kao, W. H., Patterson, N., Tandon, A., Haiman, C. A., Harris, T. B., et al. (2009). Admixture mapping of 15,280 African Americans identifies obesity susceptibility loci on chromosomes 5 and X. PLoS Genet. 5:e1000490. doi: 10.1371/journal.pgen.1000490
Cheng, T. L., Goodman, E., and Committee on Pediatric, R. (2015). Race, ethnicity, and socioeconomic status in research on child health. Pediatrics 135, e225–e237. doi: 10.1542/peds.2014-3109
Claridge, M. (2010). “Species are real biological entities,” in Comtemporary Debates in Philosophy of Biology, eds Francisco J. A. and A. Robert (Hoboken, NJ: Wiley-Blackwell), 91–109.
Cooper, R. S., Kaufman, J. S., and Ward, R. (2003). Race and genomics. N. Engl. J. Med. 348, 1166–1170. doi: 10.1056/NEJMsb022863
Crosslin, D. R., Tromp, G., Burt, A., Kim, D. S., Verma, S. S., Lucas, A. M., et al. (2014). Controlling for population structure and genotyping platform bias in the eMERGE multi-institutional biobank linked to electronic health records. Front. Genet. 5:352. doi: 10.3389/fgene.2014.00352
Dobzhansky, T. (1973). Nothing in biology makes sense except in light of evolution. Am. Biol. Teach. 35, 125–129. doi: 10.2307/4444260
Dupré, J. (1993). The Disorder of Things: Metaphysical Foundations of the Disunity of Science. Cambridge, MA: Harvard University Press.
Duster, T. (2005). Medicine. Race and reification in science. Science 307, 1050–1051. doi: 10.1126/science.1110303
Duster, T. (2015). A post-genomic surprise. The molecular reinscription of race in science, law and medicine. Br. J. Sociol. 66, 1–27. doi: 10.1111/1468-4446.12118
Edge, M. D., and Rosenberg, N. A. (2015). Implications of the apportionment of human genetic diversity for the apportionment of human phenotypic diversity. Stud. Hist. Philos. Biol. Biomed. Sci. 52, 32–45. doi: 10.1016/j.shpsc.2014.12.005
Edwards, A. W. (2003). Human genetic diversity: lewontin's fallacy. Bioessays 25, 798–801. doi: 10.1002/bies.10315
Elhaik, E. (2012). Empirical distributions of F(ST) from large-scale human polymorphism data. PLoS ONE 7:e49837. doi: 10.1371/journal.pone.0049837
Elster, A., Jarosik, J., Vangeest, J., and Fleming, M. (2003). Racial and ethnic disparities in health care for adolescents: a systematic review of the literature. Arch. Pediatr. Adolesc. Med. 157, 867–874. doi: 10.1001/archpedi.157.9.867
Evanno, G., Regnaut, S., and Goudet, J. (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x
Falush, D., Stephens, M., and Pritchard, J. K. (2003). Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164, 1567–1587.
Frank, R. (2014). The molecular reinscription of race: a comment on “Genetic bio-ancestry and social construction of racial classification in social surveys in the contemporary United States”. Demography 51, 2333–2336. doi: 10.1007/s13524-014-0342-5
Freedman, M. L., Haiman, C. A., Patterson, N., Mcdonald, G. J., Tandon, A., Waliszewska, A., et al. (2006). Admixture mapping identifies 8q24 as a prostate cancer risk locus in African-American men. Proc. Natl. Acad. Sci. U.S.A. 103, 14068–14073. doi: 10.1073/pnas.0605832103
Fujimura, J. H., and Rajagopalan, R. (2011). Different differences: the use of ‘genetic ancestry’ versus race in biomedical human genetic research. Soc. Stud. Sci. 41, 5–30. doi: 10.1177/0306312710379170
Fullwiley, D. (2011). The Enculturated Gene: Sickle Cell Health Politics and Biological Difference in West Africa. Princeton, NJ: Princeton University Press.
Galanter, J. M., Fernandez-Lopez, J. C., Gignoux, C. R., Barnholtz-Sloan, J., Fernandez-Rozadilla, C., Via, M., et al. (2012). Development of a panel of genome-wide ancestry informative markers to study admixture throughout the Americas. PLoS Genet. 8:e1002554. doi: 10.1371/journal.pgen.1002554
Gannett, L. (2004). The biological reification of race. Br. J. Philos. Sci. 55, 323–345. doi: 10.1093/bjps/55.2.323
Gannett, L. (2014). Biogeographical ancestry and race. Stud. Hist. Philos. Biol. Biomed. Sci. 47A, 173–184. doi: 10.1016/j.shpsc.2014.05.017
Graves, J. L. (2011). “Evolutionary versus Racial Medicine,” in Race and the Genetic Revolution: Science, Myth, and Culture, eds S. Krimsky, K. Sloan, and Council for Responsible Genetics (New York, NY: Columbia University Press), 142–170.
Guo, G., Fu, Y., Lee, H., Cai, T., Mullan Harris, K., and Li, Y. (2014). Genetic bio-ancestry and social construction of racial classification in social surveys in the contemporary United States. Demography 51, 141–172. doi: 10.1007/s13524-013-0242-0
Handley, L. J. L., Manica, A., Goudet, J., and Balloux, F. (2007). Going the distance: human population genetics in a clinal world. Trends Genet. 23, 432–439. doi: 10.1016/j.tig.2007.07.002
Henn, B. M., Cavalli-Sforza, L. L., and Feldman, M. W. (2012). The great human expansion. Proc. Natl. Acad. Sci. U.S.A. 109, 17758–17764. doi: 10.1073/pnas.1212380109
Henn, B. M., Gignoux, C. R., Jobin, M., Granka, J. M., Macpherson, J. M., Kidd, J. M., et al. (2011). Hunter-gatherer genomic diversity suggests a southern African origin for modern humans. Proc. Natl. Acad. Sci. U.S.A. 108, 5154–5162. doi: 10.1073/pnas.1017511108
Hochman, A. (2013). Against the new racial naturalism. J. Philos. 110, 331–351. doi: 10.5840/jphil2013110625
Hollenbach, J. A., Saperstein, A., Albrecht, M., Vierra-Green, C., Parham, P., Norman, P. J., et al. (2015). Race, ethnicity and ancestry in unrelated transplant matching for the national marrow donor program: a comparison of multiple forms of self-identification with genetics. PLoS ONE 10:e0135960. doi: 10.1371/journal.pone.0135960
Howlader, N., Altekruse, S. F., Li, C. I., Chen, V. W., Clarke, C. A., Ries, L. A., et al. (2014a). US incidence of breast cancer subtypes defined by joint hormone receptor and HER2 status. J. Natl. Cancer Inst. 106. doi: 10.1093/jnci/dju055
Howlader, N., Noone, A. M., Krapcho, M., Garshell, J., Miller, D., Altekruse, S. F., et al. (2014b). SEER Cancer Statistics Review, 1975-2011, ed National Cancer Institute. Bethesda, MD: National Cancer Institute.
Hunley, K. L., Healy, M. E., and Long, J. C. (2009). The global pattern of gene identity variation reveals a history of long-range migrations, bottlenecks, and local mate exchange: implications for biological race. Am. J. Phys. Anthropol. 139, 35–46. doi: 10.1002/ajpa.20932
Ingman, M., Kaessmann, H., Paabo, S., and Gyllensten, U. (2000). Mitochondrial genome variation and the origin of modern humans. Nature 408, 708–713. doi: 10.1038/35047064
Iqbal, J., Ginsburg, O., Rochon, P. A., Sun, P., and Narod, S. A. (2015). Differences in breast cancer stage at diagnosis and cancer-specific survival by race and ethnicity in the United States. JAMA 313, 165–173. doi: 10.1001/jama.2014.17322
Jin, W., Wang, S., Wang, H., Jin, L., and Xu, S. (2012). Exploring population admixture dynamics via empirical and simulated genome-wide distribution of ancestral chromosomal segments. Am. J. Hum. Genet. 91, 849–862. doi: 10.1016/j.ajhg.2012.09.008
Jorde, L. B., and Wooding, S. P. (2004). Genetic variation, classification and ‘race’. Nat. Genet. 36, S28–S33. doi: 10.1038/ng1435
Kahn, J. (2013). Race in a Bottle: The Story of BiDil and Racialized Medicine in a Post-Genomic Age. New York, NY: Columbia University Press.
Kalinowski, S. T. (2010). The computer program STRUCTURE does not reliably identify the main genetic clusters within species: simulations and implications for human population structure. Heredity 106, 625–632. doi: 10.1038/hdy.2010.95
Keet, C. A., Mccormack, M. C., Pollack, C. E., Peng, R. D., Mcgowan, E., and Matsui, E. C. (2015). Neighborhood poverty, urban residence, race/ethnicity, and asthma: rethinking the inner-city asthma epidemic. J. Allergy Clin. Immunol. 135, 655–662. doi: 10.1016/j.jaci.2014.11.022
Keita, S. O. Y., Kittles, R. A., Royal, C. D. M., Bonney, G. E., Furbert-Harris, P., Dunston, G. M., et al. (2004). Conceptualizing human variation. Nat. Genet. 36, S17–S20. doi: 10.1038/ng1455
Lewontin, R. (1972). “The apportionment of human diversity,” in Evolutionary Biology, eds T. Dobzhansky, M. K. Hecht, and W. C. Steere (New York, NY: Appleton-Century-Crofts), 381–398.
Li, J. Z., Absher, D. M., Tang, H., Southwick, A. M., Casto, A. M., Ramachandran, S., et al. (2008). Worldwide human relationships inferred from genome-wide patterns of variation. Science 319, 1100–1104. doi: 10.1126/science.1153717
Long, J. C., Li, J., and Healy, M. E. (2009). Human DNA sequences: more variation and less race. Am. J. Phys. Anthropol. 139, 23–34. doi: 10.1002/ajpa.21011
Lu, Y.-F., Goldstein, D. B., Angrist, M., and Cavalleri, G. (2014). Personalized medicine and human genetic diversity. Cold Spring Harb. Perspect. Med. 4:a008581. doi: 10.1101/cshperspect.a008581
Madore, A. M., Houde, L., Vezina, H., Vohl, M. C., Perusse, L., Mior, N., et al. (2007). Contribution of hierarchical clustering techniques to the modeling of the geographic distribution of genetic polymorphisms associated with chronic inflammatory diseases in the Quebec population. Community Genet. 10, 218–226. doi: 10.1159/000106560
Maglo, K. N. (2007). Force, mathematics, and physics in Newton's Principia: a new approach to enduring issues. Sci. Context 20, 571–600. doi: 10.1017/S0269889707001457
Maglo, K. N. (2010). Genomics and the conundrum of race: some epistemic and ethical considerations. Perspect. Biol. Med. 53, 357–372. doi: 10.1353/pbm.0.0171
Maglo, K. N. (2011). The case against biological realism about race: from Darwin to the post-genomic era. Perspect. Sci. 19, 361–390. doi: 10.1162/POSC_a_00048
Maglo, K. N. (2012). Group-based and personalized care in an age of genomic and evidence-based medicine: a reappraisal. Perspect. Biol. Med. 55, 137–154. doi: 10.1353/pbm.2012.0006
Maglo, K. N., and Martin, L. (2012). Researching vs. reifying race: the case of obesity research. Humana Mente J. Philos. Stud. 22, 111–143.
Maglo, K. N., Rubinstein, J., Huang, B., and Ittenbach, R. F. (2014). BiDil in the Clinic: an interdisciplinary investigation of physicians' prescription patterns of a race-based therapy. AJOB Empir. Bioeth. 5, 37–52. doi: 10.1080/23294515.2014.907371
Mayr, E., and Bock, W. J. (2002). Classifications and other ordering systems. J. Zool. System. Evol. Res. 40, 169–194. doi: 10.1046/j.1439-0469.2002.00211.x
Mersha, T. B., and Abebe, T. (2015). Self-reported race/ethnicity in the age of genomic research: its potential impact on understanding health disparities. Hum. Genomics 9:1. doi: 10.1186/s40246-014-0023-x
Mishler, B. (2010). “Species are Not Uniquely Real Biological Entities,” in Comtemporary Debates in Philosophy of Biology, eds J. Francisco and A. Robert (Hoboken, NJ: Wiley-Blackwell), 110–122.
Nelis, M., Esko, T., Magi, R., Zimprich, F., Zimprich, A., Toncheva, D., et al. (2009). Genetic structure of Europeans: a view from the North-East. PLoS ONE 4:e5472. doi: 10.1371/journal.pone.0005472
Novembre, J., Johnson, T., Bryc, K., Kutalik, Z., Boyko, A. R., Auton, A., et al. (2008). Genes mirror geography within Europe. Nature 456, U98–U95. doi: 10.1038/nature07331
Ogden, C. L., Carroll, M. D., Kit, B. K., and Flegal, K. M. (2012). Prevalence of obesity and trends in body mass index among US children and adolescents, 1999-2010. JAMA 307, 483–490. doi: 10.1001/jama.2012.40
Ossorio, P., and Duster, T. (2005). Race and genetics: controversies in biomedical, behavioral, and forensic sciences. Am. Psychol. 60, 115–128. doi: 10.1037/0003-066X.60.1.115
Parfrey, L. W., Barbero, E., Lasser, E., Dunthorn, M., Bhattacharya, D., Patterson, D. J., et al. (2006). Evaluating support for the current classification of eukaryotic diversity. PLoS Genet. 2:e220. doi: 10.1371/journal.pgen.0020220
Paschou, P., Drineas, P., Lewis, J., Nievergelt, C. M., Nickerson, D. A., Smith, J. D., et al. (2008). Tracing sub-structure in the European American population with PCA-informative markers. PLoS Genet. 4:e1000114. doi: 10.1371/journal.pgen.1000114
Petersen, K. E., Prows, C. A., Martin, L. J., and Maglo, K. N. (2014). Personalized medicine, availability, and group disparity: an inquiry into how physicians perceive and rate the elements and barriers of personalized medicine. Public Health Genomics 17, 209–220. doi: 10.1159/000362359
Piel, F. B., Patil, A. P., Howes, R. E., Nyangiri, O. A., Gething, P. W., Williams, T. N., et al. (2010). Global distribution of the sickle cell gene and geographical confirmation of the malaria hypothesis. Nat. Commun. 1:104. doi: 10.1038/ncomms1104
Pritchard, J. K., Stephens, M., and Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959.
Pritchard, J., Wen, X., and Falush, D. (2007). Documentation for Structure Software: Version 2.2. Department of Human Genetics, University of Chicago; Department of Statistics, University of Oxford. Available online at: http://pritch.bsd.uchicago.edu/software/structure22/readme.pdf
Ramachandran, S., Deshpande, O., Roseman, C. C., Rosenberg, N. A., Feldman, M. W., and Cavalli-Sforza, L. L. (2005). Support from the relationship of genetic and geographic distance in human populations for a serial founder effect originating in Africa. Proc. Natl. Acad. Sci. U.S.A. 102, 15942–15947. doi: 10.1073/pnas.0507611102
Reich, D., Patterson, N., De Jager, P. L., Mcdonald, G. J., Waliszewska, A., Tandon, A., et al. (2005). A whole-genome admixture scan finds a candidate locus for multiple sclerosis susceptibility. Nat. Genet. 37, 1113–1118. doi: 10.1038/ng1646
Ricks-Santi, L. J., Apprey, V., Mason, T., Wilson, B., Abbas, M., Hernandez, W., et al. (2012). Identification of genetic risk associated with prostate cancer using ancestry informative markers. Prostate Cancer Prostatic Dis. 15, 359–364. doi: 10.1038/pcan.2012.19
Risch, N., Burchard, E., Ziv, E., and Tang, H. (2002). Categorization of humans in biomedical research: genes, race and disease. Genome Biol. 3:comment2007. doi: 10.1186/gb-2002-3-7-comment2007
Romero, C. X., Romero, T. E., Shlay, J. C., Ogden, L. G., and Dabelea, D. (2012). Changing trends in the prevalence and disparities of obesity and other cardiovascular disease risk factors in three racial/ethnic groups of USA adults. Adv. Prev. Med. 2012:172423. doi: 10.1155/2012/172423
Rosenberg, N. A. (2011). A population-genetic perspective on the similarities and differences among worldwide human populations. Hum. Biol. 83, 659–684. doi: 10.3378/027.083.0601
Rosenberg, N. A., Mahajan, S., Ramachandran, S., Zhao, C., Pritchard, J. K., and Feldman, M. W. (2005). Clines, clusters, and the effect of study design on the inference of human population structure. PLoS Genet. 1:e70. doi: 10.1371/journal.pgen.0010070
Rosenberg, N. A., Pritchard, J. K., Weber, J. L., Cann, H. M., Kidd, K. K., Zhivotovsky, L. A., et al. (2002). Genetic structure of human populations. Science 298, 2381–2385. doi: 10.1126/science.1078311
Salmela, E., Lappalainen, T., Liu, J., Sistonen, P., Andersen, P. M., Schreiber, S., et al. (2011). Swedish population substructure revealed by genome-wide single nucleotide polymorphism data. PLoS ONE 6:e16747. doi: 10.1371/journal.pone.0016747
Schuh, R. T., and Brower, A. V. Z. (2009). Biological Systematics: Principles and Applications. Ithaca, NY: Cornell University Press.
Schwartz, M. K., and McKelvey, K. S. (2009). Why sampling scheme matters: the effect of sampling scheme on landscape genetic results. Conserv. Genet. 10, 441–452. doi: 10.1007/s10592-008-9622-1
Serre, D., and Paabo, S. P. (2004). Evidence for gradients of human genetic diversity within and among continents. Genome Res. 14, 1679–1685. doi: 10.1101/gr.2529604
Smedley, A., and Smedley, B. D. (2005). Race as biology is. fiction, racism as a social problem is real - Anthropological and historical perspectives on the social construction of race. Am. Psychol. 60, 16–26. doi: 10.1037/0003-066X.60.1.16
Stevens, J. (2003). Racial meanings and scientific methods: changing policies for NIH-sponsored publications reporting human variation. J. Health Polit. Policy Law 28, 1033–1087. doi: 10.1215/03616878-28-6-1033
Tang, H., Quertermous, T., Rodriguez, B., Kardia, S. L. R., Zhu, X. F., Brown, A., et al. (2005). Genetic structure, self-identified race/ethnicity, and confounding in case-control association studies. Am. J. Hum. Genet. 76, 268–275. doi: 10.1086/427888
Templeton, A. R. (1998). Human races: a genetic and evolutionary perspective. Am. Anthropol. 100, 632–650. doi: 10.1525/aa.1998.100.3.632
Templeton, A. R. (2013). Biological races in humans. Stud. Hist. Philos. Biol. Biomed. Sci. 44, 262–271. doi: 10.1016/j.shpsc.2013.04.010
Tishkoff, S. A., and Kidd, K. K. (2004). Implications of biogeography of human populations for ‘race’ and medicine. Nat. Genet. 36, 21–27. doi: 10.1038/ng1438
Tishkoff, S. A., Reed, F. A., Friedlaender, F. R., Ehret, C., Ranciaro, A., Froment, A., et al. (2009). The genetic structure and history of Africans and African Americans. Science 324, 1035–1044. doi: 10.1126/science.1172257
Tishkoff, S. A., and Verrelli, B. C. (2003). Patterns of human genetic diversity: implications for human evolutionary history and disease. Annu. Rev. Genomics Hum. Genet. 4, 293–340. doi: 10.1146/annurev.genom.4.070802.110226
Underhill, P. A., and Kivisild, T. (2007). Use of y chromosome and mitochondrial DNA population structure in tracing human migrations. Annu. Rev. Genet. 41, 539–564. doi: 10.1146/annurev.genet.41.110306.130407
Veeramah, K. R., Wegmann, D., Woerner, A., Mendez, F. L., Watkins, J. C., Destro-Bisol, G., et al. (2012). An early divergence of khoesan ancestors from those of other modern humans is supported by an ABC-Based analysis of autosomal resequencing data. Mol. Biol. Evol. 29, 617–630. doi: 10.1093/molbev/msr212
Vergara, C., Caraballo, L., Mercado, D., Jimenez, S., Rojas, W., Rafaels, N., et al. (2009). African ancestry is associated with risk of asthma and high total serum IgE in a population from the Caribbean Coast of Colombia. Hum. Genet. 125, 565–579. doi: 10.1007/s00439-009-0649-2
Wiley, E. O., and Lieberman, B. S. (2011). “Phylogenetic Classification,” in Phylogenetics. (Hoboken, NJ: John Wiley & Sons, Inc.), 229–259. doi: 10.1002/9781118017883
Williams, D. R. (2005). The health of U.S. racial and ethnic populations. J. Gerontol B Psychol. Sci. Soc. Sci. 60(Spec No 2), 53–62. doi: 10.1093/geronb/60.Special_Issue_2.S53
Wilson, J. F., Weale, M. E., Smith, A. C., Gratrix, F., Fletcher, B., Thomas, M. G., et al. (2001). Population genetic structure of variable drug response. Nat. Genet. 29, 265–269. doi: 10.1038/ng761
Wright, S. (1978). Variability within and Among Natural Populations. Chicago, IL: University of Chicago Press.
Yu, N., Chen, F. C., Ota, S., Jorde, L. B., Pamilo, P., Patthy, L., et al. (2002). Larger genetic differences within Africans than between Africans and Eurasians. Genetics 161, 269–274.
Keywords: ancestry, cline, cluster analysis, Darwinian classification, genomic medicine, pair-wise Fst, phylogenomics, population structure
Citation: Maglo KN, Mersha TB and Martin LJ (2016) Population Genomics and the Statistical Values of Race: An Interdisciplinary Perspective on the Biological Classification of Human Populations and Implications for Clinical Genetic Epidemiological Research. Front. Genet. 7:22. doi: 10.3389/fgene.2016.00022
Received: 08 June 2015; Accepted: 02 February 2016;
Published: 17 February 2016.
Edited by:
Karen T. Cuenco, Genentech, USAReviewed by:
Yiran Guo, The Children's Hospital of Philadelphia, USACopyright © 2016 Maglo, Mersha and Martin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Koffi N. Maglo, bWFnbG9rbkB1Y21haWwudWMuZWR1;
Lisa J. Martin, bGlzYS5tYXJ0aW5AY2NobWMub3Jn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.