 Tiziana Lodi
Tiziana Lodi Cristina Dallabona
Cristina Dallabona Cecilia Nolli
Cecilia Nolli Paola Goffrini
Paola Goffrini Claudia Donnini
Claudia Donnini Enrico Baruffini
Enrico Baruffini- Department of Life Sciences, University of Parma, Parma, Italy
Mip1 is the Saccharomyces cerevisiae DNA polymerase γ (Pol γ), which is responsible for the replication of mitochondrial DNA (mtDNA). It belongs to the family A of the DNA polymerases and it is orthologs to human POLGA. In humans, mutations in POLG(1) cause many mitochondrial pathologies, such as progressive external ophthalmoplegia (PEO), Alpers' syndrome, and ataxia-neuropathy syndrome, all of which present instability of mtDNA, which results in impaired mitochondrial function in several tissues with variable degrees of severity. In this review, we summarize the genetic and biochemical knowledge published on yeast mitochondrial DNA polymerase from 1989, when the MIP1 gene was first cloned, up until now. The role of yeast is particularly emphasized in (i) validating the pathological mutations found in human POLG and modeled in MIP1, (ii) determining the molecular defects caused by these mutations and (iii) finding the correlation between mutations/polymorphisms in POLGA and mtDNA toxicity induced by specific drugs. We also describe recent findings regarding the discovery of molecules able to rescue the phenotypic defects caused by pathological mutations in Mip1, and the construction of a model system in which the human Pol γ holoenzyme is expressed in yeast and complements the loss of Mip1.
Introduction
DNA polymerase γ (or Pol γ) is the only DNA replicase identified in animal and fungal mitochondria. Although four other DNA polymerase activities were recently identified in Saccharomyces cerevisiae mitochondria (DNA polymerase ζ, Rev1, DNA polymerase η, and DNA polymerase α) (Zhang et al., 2006; Chatterjee et al., 2013; Lasserre et al., 2013), Mip1, the yeast Pol γ, is the only enzyme to be able to fully replicate mitochondrial DNA (mtDNA). Deletion of MIP1 makes the strain rho0, i.e., devoid of mtDNA.
S. cerevisiae is a facultative anaerobe yeast and is able to survive in the absence of oxidative phosphorylation (OXPHOS) activity and of mitochondrial genome, provided that a fermentative carbon source is available. The non-essentiality of respiration for viability makes this organism an excellent model for studying mitochondrial biogenesis as well as nucleo-mitochondrial interactions. Respiratory deficient (RD) cells efficiently generate the ATP needed to sustain the growth by alcoholic fermentation. On media containing a limiting amount of fermentative carbon sources, colonies generated by RD cells display the so called “petite phenotype”, since they are smaller than the respiratory proficient wild type colonies because they are unable to produce biomass through respiration of ethanol, the end product of fermentation. Since both nuclear and mitochondrial genomes participate to OXPHOS functions, the petite phenotype can derive from mutations in nuclear genes that encode mitochondrial functions (pet mutants, Mendelian inherited, Sherman, 1963) or from mtDNA mutations (petite mutants, non-Mendelian inherited, Ephrussi and Slonimski, 1955). Cytoplasmic petite mutants, which arise spontaneously with high frequency (around 1–10% depending on the strains), are completely devoid of mtDNA (rho0 cells) or carry extensive deletions of mtDNA and regular repetitions of the conserved sequences (rho− cells). Cells containing functional mitochondrial genomes are termed rho+ (Dujon, 1981). S. cerevisiae is sensitive to antibiotics that inhibit the mitochondrial translation, such as erythromycin, when grown on oxidative carbon sources. Erythromycin resistant mutants arise spontaneously with low frequency (10−7 to 10−8) as a result of mutations in specific nucleotides of the mitochondrial gene encoding the 21S rRNA (Sor and Fukuhara, 1984). For this reason, in yeast strains mutated in MIP1 the frequency of mutants resistant to erythromycin (EryR mutants) is an in vivo index of the replication fidelity of Mip1.
Each yeast cell contains 10–50 copies of mtDNA per nuclear genome, depending on the growth condition. Recombination between these molecules is highly active (Dujon et al., 1974). A strong link between erroneous recombination and rho− transmission has been postulated, which would explain the molecular mechanism that generates this class of cytoplasmic petite. In particular, it has been suggested that mtDNA deletions giving rise to rho− genomes can occur through homologs recombination between imperfect repeats (Slonimski and Lazowska, 1977; Gaillard et al., 1980). Rho− mtDNA genomes are not very stable and may then evolve into rho0 status. Rho0 clones can also be generated directly by treatment with different molecules, such as ethidium bromide (EtBr), which inhibits both mitochondrial transcription and mtDNA replication, either directly or indirectly (Slonimski et al., 1968; Richardson and Parker, 1973). Human tissues, in case of mtDNA mutations, are mostly heteroplasmic, whereas yeast is homoplasmic: if mtDNA point mutations or mtDNA rearrangements occur, after a few generations two populations of cells will be produced, one bearing only wild type mtDNA molecules and the other bearing only mutated mtDNA molecules.
mtDNA is packaged within protein-DNA structures called nucleoids, which are anchored to the inner mitochondrial membranes (Chen et al., 2005). A variable number of nucleoids are present in each mitochondrion and each nucleoid contains several copies of mtDNA. Different proteins make up nucleoid structures, and are all involved in maintaining the integrity of the mtDNA and are responsible for its replication, recombination, repair and transmission to the bud (Chen and Butow, 2005; Kucej et al., 2008). Mutants which have been altered or are devoid of these functions are heavily affected with respect to mtDNA stability. Therefore, depending on the nuclear background, the level of cytoplasmic petite mutants can increase, in some cases reaching the entire cell population.
The mechanism of mtDNA replication in yeast is not completely known. In contrast to what has been assumed for decades, mtDNA is not always circular, as in the case of animals. The majority of mtDNA is present as concatenamers of linear tandem arrays of several genome units and only a small proportion of mtDNA has a circular shape (Maleszka et al., 1991). In particular, concatenamers are mainly present in mother cells whereas circular mtDNA is found in the buds (Ling and Shibata, 2002). Two theories were proposed regarding the replication of yeast mitochondrial DNA. (i) According to the most accepted theory, the replication starts at several origins called Ori sites, and it is RNA-primed and bidirectional like that of chromosomal DNA (reviewed in Lecrenier and Foury, 2000). While Ori sites have not been identified in other fungi, it is known that Ori sites of S. cerevisiae are 300-bp-long sequences composed of three repeated GC-rich clusters separated by an AT-reach region and preceded by a transcription site called r (Baldacci et al., 1984; Foury et al., 1998). (ii) According to the second hypothesis, the replication occurs via a “rolling circle” mechanism, which produces long tandemly repeated mtDNA molecules, which are then converted into circular monomers (Ling and Shibata, 2002; Ling et al., 2007). Recent findings also demonstrate that homologs recombination and strand invasion could account for initiating replication in yeasts (Ling and Shibata, 2002; Ling et al., 2007). Therefore, the mtDNA replication mechanism in yeast is different from the best-known mechanism in mammalians, in which both the H and the L strands are continuously synthesized from OriH and OriL, respectively, which are located far from each other. In this case, two models have been proposed. (i) According to the most accepted hypothesis, i.e., the asynchronous strand displacement model, at the beginning the H strand is replicated by single-stranded replication starting from the OriH, with displacement of the D-loop. This synthesis proceeds until OriL is exposed. In the OriL site, synthesis of the L-strand is initiated in the opposite direction and is primed by RNA synthesis (Shadel and Clayton, 1997). (ii) Alternatively, the strand-coupled bidirectional replication model has been proposed, in which bidirectional replication is initiated from a region near OriH, followed by progression of the two forks around the mtDNA circle (Holt et al., 2000). More recently other mechanisms have been proposed (reviewed in McKinney and Oliveira, 2013). A limited number of proteins are involved in mtDNA replication, and most of them are conserved in yeast and humans, which suggests that the molecular mechanisms of the replication are partially similar (reviewed in Lecrenier and Foury, 2000).
Mip1 Milestones: A Historical Point of View
The first information on yeast DNA polymerase γ dates back to 1970, when a mitochondrial DNA polymerase was proven to be resistant to aphidicolin and highly sensitive to ddNTPs, such as DNA polymerase γ of higher eukaryotes (Wintersberger and Wintersberger, 1970; Wintersberger and Blutsch, 1976). The MIP1 gene was cloned by functional complementation of a thermosensitive (ts) mutant, called mip1-1, identified in a screening of mutants able to grow on glycerol at 25°C but not at 36°C, due to massive production of petite mutants at high temperatures (Genga et al., 1986). In particular, in these conditions the mip1-1 mutant was completely deficient of both mtDNA replication and mtDNA polymerase activity (Foury, 1989). The gene was mapped to chromosome 15 and sequenced, resulting in an open reading frame encoding a 1254 amino acid long protein. The deletion of MIP1, as expected, produced a strain that was unable to grow on respiratory carbon sources and devoid of mtDNA. Cloning MIP1 paved the way for the isolation of genes encoding the DNA polymerase γ of Schizosaccharomyces pombe (Ropp and Copeland, 1995) and, subsequently, of humans and Drosophila (Ropp and Copeland, 1996).
Based on the alignment with bacterial DNA polymerases of family A, which includes bacterial and bacteriophage polymerases which share significant similarity to E. coli polymerase I, three highly conserved motifs were identified in the exonuclease domain and called exo1, exo2, and exo3 (Bernad et al., 1989). A mutation in exo1, a mutation in exo2 and two single mutations in exo3 were introduced in Mip1 (Foury and Vanderstraeten, 1992). These mutations determined a decrease in proofreading activity and consequently a mtDNA mutator phenotype, characterized by an increased frequency of EryR mutants, which demonstrates the functional role of the exo motifs. However, in these mutants an increase of mtDNA extended mutability, accompanied by a decrease in the gap-filling activity and processivity, was also observed, especially at 35°C. This suggests that the two catalytic domains do not act independently of each other in Pol γ. The discovery that the D230A mutant displayed a significant reduction of exonuclease activity was the basis for the creation of a mouse model lacking Pol γ exonuclease activity. In this model, an increase in mtDNA point mutability and deletions, a reduction in the life span and the onset of premature aging were observed (Trifunovic et al., 2004).
Thanks to its ability to grow even in the absence of mitochondrial DNA and to the sequence conservation among eukaryotic polymerase γ, yeast was considered to be the organism of choice to study the effects of pathological mutations in human Pol γ, starting from 2006 (Stuart et al., 2006), a few years after the first identification of pathological mutation in the POLG gene (Van Goethem et al., 2001) Several works have been published since then, in which mip1 alleles carrying substitutions corresponding to pathological mutations were expressed in mutant strains devoid of mtDNA polymerase, as explained later. Besides studies designed to validate the pathogenicity of human mutations and to understand the molecular mechanisms responsible for the associated diseases, studies performed in yeast led to the discovery of the genetic and chemical rescue of the effects of mutations in MIP1, through ribonucleotide reductase overexpression and the administration of antioxidants, respectively (Baruffini et al., 2006). These observations led to several studies on human cells or murine organisms. At the same time, with the help of yeast, pharmacogenetic research was performed in order to study the correlation between toxicity due to sodium valproate or NRTIs, and polymorphisms in POLG (Baruffini and Lodi, 2010; Stewart et al., 2010). The milestones described in this chapter are illustrated in Figure 1.
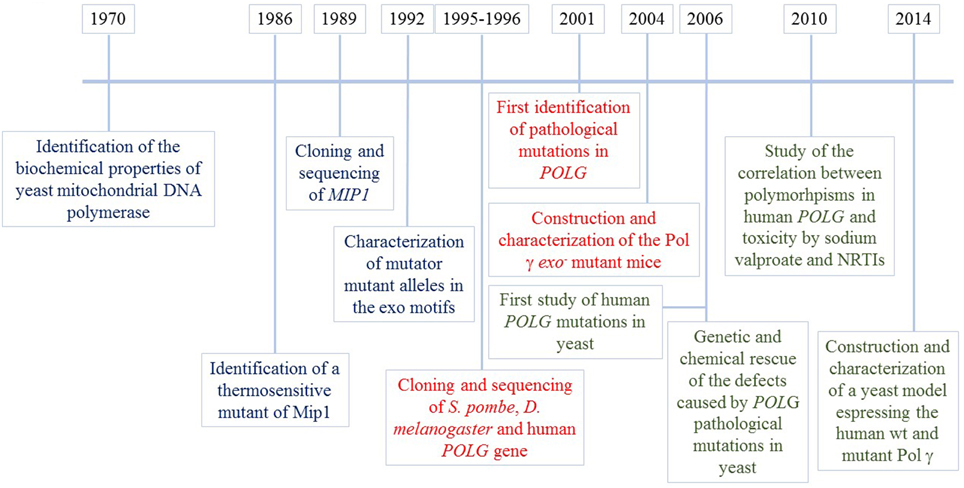
Figure 1. Mip1 milestones. Information regarding yeast Mip1 is shown in blue, information on other eukaryotic Pol γ obtained thanks to the use of yeast Mip1 is in red, information on human POLGA mutations/polymorphisms obtained by modeling and studying the mutations in yeast is in green.
Mip1 Biochemical Properties
Mip1 is a protein of approximately 140 kDa, it belongs to the subclass γ of the family A polymerases to which several bacterial and viral DNA polymerases belong, and is divided into four domains: besides a mitochondrial targeting signal (MTS) motif, which is necessary for the import into the mitochondrial matrix, it possesses an exonucleasic (exo) domain, a spacer or linker region, a polymerase (Pol) domain and a C-terminal extension (CTE) specific to fungi polymerase γ (Figure 2).
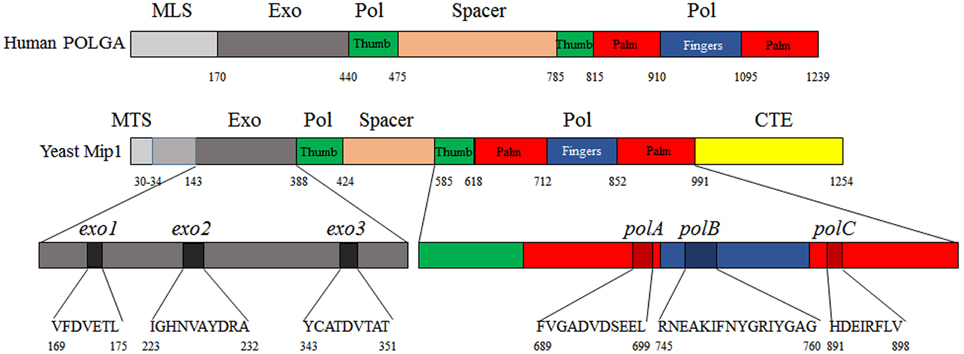
Figure 2. Domains of human POLGA and yeast Mip1 shown in linear form. Linear form of human POLGA according to its 3D structure. The linear form of Mip1 was constructed following the alignment of yeast Mip1 and human POLGA. MLS, mitochondrial localization signal; Exo, exonuclease domain; Spacer, spacer or linker region; Pol, polymerase domain; MTS, mitochondrial targeting sequence; CTE, C-terminal extension.
Thanks to the purification of the native protein from S. cerevisiae (Sen et al., 1994), it has been possible to determine the kinetic properties of Mip1 polymerase. Mip1 has a Km for dNTPs lower than 1 μM (Sen et al., 1994), while the kpol is about 60 nt/s and the processivity is about 500 nt per one binding event (Viikov et al., 2011). It can use different types of DNA as substrates, including the poly [dA-dT] and poly[dA]-oligo[dT], but not the poly [rA]-oligo [dT]. The inability to use this product as a substrate distinguished Mip1 from Pol γ of higher eukaryotes. The following molecules act as inhibitors of the catalytic activity: the Mn2+ ion, whose in vivo administration increases up to 100 times the mtDNA point mutability (Putrament et al., 1975), EtBr, ddTTP, and ddCTP as well as various nucleoside reverse transcriptase inhibitors (NRTIs) which are also used in antiretroviral therapy, such as 3′-fluoro-TTP and didehydro(d4)CTP (Sen et al., 1994; Eriksson et al., 1995). Furthermore, Mip1, similarly to prokaryotic polymerases of family A, is also able to perform strand displacements with rates of about 30 nt/s (Viikov et al., 2011), in accordance with the observation that human Pol γ can be stimulated to perform strand displacement by the human DNA2 protein (reviewed in Copeland and Longley, 2008).
The polymerase domain is divided into three subdomains: thumb, palm and fingers. Three motifs, which are highly conserved in all eukaryotic Pol γ, are located in the Pol domain and are called polA, polB, and polC. The first mutants in the catalytic domain were constructed by random mutagenesis in 1995 (Hu et al., 1995). Three mutations in the fingers subdomain, T716I, E724K, and P851L, were associated both with increased point mutability and increased extended mutability (Table 1), indicating that the catalytic domain is involved both in the mtDNA polymerization and in replication fidelity. Moreover, these mutations are partially dominant, as are most of the pathological substitutions located in the fingers subdomain identified in patients with adPEO.
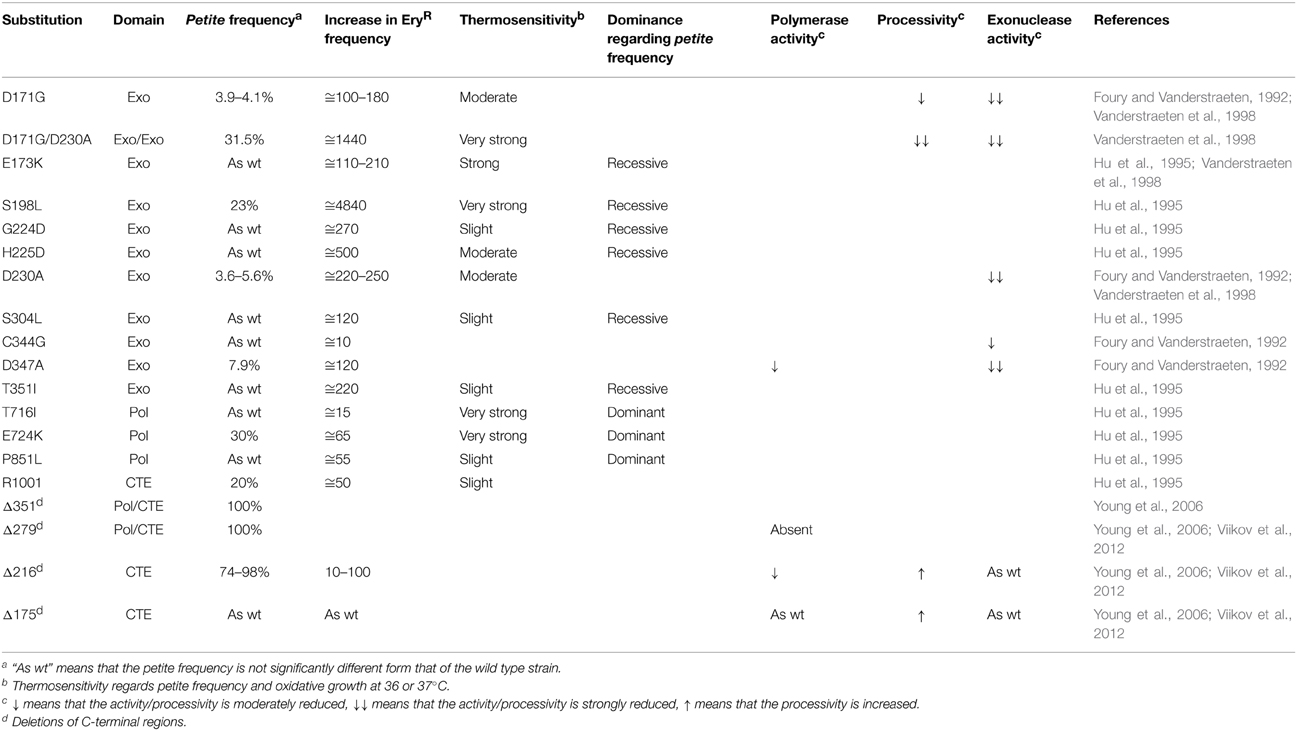
Table 1. Substitutions which alter in vivo mtDNA mutability and/or in vitro biochemical properties.
The 3′–5′ exonuclease domain has a proofreading activity which, in its human counterpart, contributes at least 20-fold to the fidelity of replication (Longley et al., 2001). Specific mutations in exo1, exo2, and exo3 drastically reduce the exonuclease activity and consequently, the fidelity of replication (Foury and Vanderstraeten, 1992; Hu et al., 1995; Vanderstraeten et al., 1998) (Table 1). In addition, some of these mutations, such as D171A and the double mutant D171A/D230A, not only decrease the proofreading activity, but also determine an increase of the mismatch extension, with a double negative effect on the replication fidelity. The analysis of these mutants showed that the Mip1 proofreading activity mainly corrects transversions, whereas the protein Msh1, which is involved in the post-replication mismatch repair, mainly corrects transitions. In addition, many mutations in the exo domain, as previously mentioned, determine an increase in the percentage of petite colonies in the clonal population. This suggests that the two catalytic domains do not carry out their activities independently of each other and, in particular, that mutations in the exo domain decrease the polymerase activity.
Human DNA polymerase γ consists of a catalytic subunit (POLGA) and two accessory subunits (POLGB) (Yakubovskaya et al., 2006). POLGB is a small subunit which binds the catalytic subunit, binds tightly the DNA and increases the processivity of the whole holoenzyme (Lim et al., 1999). The spacer domain of POLGA is divided into two subdomains: an intrinsic processivity (IP) subdomain and a long accessory-interacting determinant (AID) subdomain (Lee et al., 2009). The first one is responsible for the intrinsic processivity of the catalytic subunit, whereas the second, which interacts with the accessory subunits, is responsible for enhanced processivity. On the contrary, Mip1 is a monomeric protein (Lucas et al., 2004; Viikov et al., 2011). The absence of the accessory subunit explains why the Mip1 spacer is shorter than that of POLGA of higher eukaryotes. Indeed, most of the sequence that corresponds to the AID subdomain is absent in Mip1, and the IP subdomain has a shorter sequence than the human counterpart (Lee et al., 2009). Nevertheless, Mip1 is highly processive: this probably indicates that the mechanisms responsible for processivity in Mip1 and in human Pol γ are partially different (Viikov et al., 2011).
The CTE is a specific region of fungal Pol γ and its length is variable from species to species, reaching its maximum in Saccharomycetales (Young et al., 2006). In Mip1, the CTE corresponds to residues 975–1254. Deletion of the last 216 amino acids, among which the most conserved residues are located, leads to a sharp increase of extended mutability (petite frequency of 98% after 24 h) and point mutability (10–100-fold increase in EryR mutants), while the deletion of the last 175 poorly conserved amino acids leads to a wild type phenotype: this indicates that some residues between 1038 and 1079 are critical for the activity and replication fidelity of Mip1 (Young et al., 2006). In addition, arginine 1001, a very conserved residue in fungal polymerases, is important for the polymerase activity, since replacing it with isoleucine causes a petite frequency of 20% and a 50-fold higher point mutability compared to the wild type (Hu et al., 1995). In vitro analysis showed that the region 1038–1079, despite containing several positively charged residues, is important for efficient polymerization but not for processivity, which is increased in the absence of this region (Viikov et al., 2012). However, the exonuclease activity of the mutant Mip1 lacking the last 216 amino acids is similar to that of wild type Mip1, so that in the presence of low concentrations of dNTPs the balance between the exonuclease activity and the polymerase activity is tipped in favor of the first. Similarly, the strand displacement activity is halved (Viikov et al., 2012).
Polymorphisms in Mip1
Alignment of Mip1 sequences from several strains obtained by the Sanger Saccharomyces genome resequencing project (https://www.sanger.ac.uk/research/projects/genomeinformatics/sgrp.html) and by the Saccharomyces genome database (www.yeastgenome.org) reveals the presence of 28 amino acid substitutions (Baruffini et al., 2007a) (Supplementary Table 1). The strains with the greatest differences are the reference strain S288C, which possesses the allele named MIP1[S], and strain Sigma1278b, which possesses the allele MIP1[Σ] (Baruffini et al., 2007a). Most of the substitutions are semi-conservative (Supplementary Table 1, green amino acids) or map in positions which are poorly conserved in Pol γ from different fungal and animal organisms (yellow amino acids). Two exceptions are the amino acid 357, which is E in S288C-derived strains and is K in all the other strains, and the amino acid 661, which is A in the former strains but is T in the latter ones. Amino acids E357 and A661 were transmitted to S288C by its ancestor EM93, a heterozygous diploid strain which contains one K357-T661 allele, and one E357-A661 allele. The latter allele is also present in strains BY474X, which are isogenic to S288C and which were used to construct the deletant strain collections, and in the W303-1B strain, which shares a common origin with S288C and is one of the most used strains in the analysis of mitochondrial phenotypes (Schacherer et al., 2007). We and others have shown that the presence of alanine at position 661 results both in higher extended mtDNA mutability (2–2.5% petite frequency for strain W303-1B compared to 0.5–1% for the strains with T661) and in increased thermosensitivity (5–20% petite frequency at 36°C, 25–40% at 37°C, 60–70% at 38°C for strain W303-1B compared to 0.5–3%) (Baruffini et al., 2007a; Young and Court, 2009). This substitution is then responsible for about one fourth of the petite mutability in strains BY474X (Dimitrov et al., 2009). A661 also determines a 3-4-fold increase of mtDNA point mutability (Baruffini et al., 2007a). The amino acid E357 is responsible for a 2-3-fold increase of the mtDNA point mutability, but has no effects on the mtDNA extended mutability.
Due to the presence of amino acid substitutions affecting the mitochondrial mutability, it is critical to choose correctly the genetic background for evaluating the effects of substitutions in Mip1. The use of a strain containing the high mutator allele MIP1[S], with increased basal mitochondrial mutability, allows one to note even small effects consequent to amino acid substitutions, since the gap resulting from the introduction of pathological mutations is larger. On the other hand, the use of a strain containing the low mutator alleles MIP1[Σ] or MIP1[S]A661T allows the study of highly deleterious mutations, which in the MIP1[S] background could lead to a total loss of mtDNA (Baruffini et al., 2007b).
Mip1 Interactions
Physical or genetic interactions between Mip1 and a few mitochondrial and non-mitochondrial proteins have been reported. Physical interaction with Sed1 was demonstrated by co-immunoprecipitation experiments (Phadnis and Sia, 2004). Sed1 is located both on the cell surface and within the inner mitochondrial membrane (IMM). Deletion of SED1 leads to a 3.2-fold increase of EryR mutability and to a 4.3-fold increase of petite mutability, and to a decrease of Cox3 protein levels. Furthermore, Mip1 levels are reduced 3-fold. On the basis of these observations, it has been hypothesized that Sed1 could assist other proteins, including Mip1, in the mitochondrial import, leading to a reduction of their levels in its absence. Sed1p could also be associated with the mitochondrial replication machinery (MRM), at the site of mtDNA binding to the inner membrane, leading to a stabilization of the proteins of this complex, such as Mip1 (Phadnis and Sia, 2004). Mip1 is also part of a complex which includes Abf2, Mgm101 and the mtDNA. Thanks to the Mgm101 interaction with transmembrane protein Mmm1 in the outer membrane, this complex takes part to a two-membrane-spanning replisome, which is essential for mtDNA maintenance (Meeusen and Nunnari, 2003). This complex could also include Pif1, the mitochondrial helicase involved in recombination (Cheng and Ivessa, 2010).
At a high temperature (>37°C), the maintenance of mitochondrial genome is partially impaired. It has been demonstrated that at a non-permissive temperature Mip1 is partially misfolded, thus explaining the increase of petite frequency. Most Mip1 molecules are misfolded in the strain mutated in MDJ1, which encodes a mitochondrial co-chaperone which has a role in maintaining the active conformation of several proteins, including Mip1, at elevated temperatures (Duchniewicz et al., 1999). Moreover, after a heat shock at 46–48°C, Mip1 can partially reactivate its active conformation only in the presence of the cochaperone Mdj1 and the bichaperones Ssc1(Hsp70)-Hsp78 (Germaniuk et al., 2002; Lewandowska et al., 2006).
The first identified genetic interactor of MIP1 was RNR1, which encodes the large subunit of the ribonucleotide reductase, which catalyzes the reduction of rNDPs to dNDPs (Elledge and Davis, 1990; reviewed in Nordlund and Reichard, 2006). Foury and coworkers found that RNR1 is a multicopy suppressor of the thermosensitive mutation mip1-1 capable of restoring the growth of the mutant strain at 36°C; moreover, in the diploid strain MIP1/mip1Δ, which has a petite frequency of about 45% at 37°C, overexpression of RNR1 reduces the extended mtDNA mutability approximately 2-fold (Lecrenier and Foury, 1995). Subsequently, it was found that the overexpression of RNR1 reduced the detrimental effects on extended mtDNA mutability caused by different pathological mutations modeled in Mip1 (Baruffini et al., 2006; Stumpf et al., 2010). The rescue by RNR1 overexpression is probably due to the increase of the dNTP pools, which are 3-8-fold higher. The same levels of rescue can be achieved by deletion of SML1, which encodes a protein that inhibits the ribonucleotide reductase activity by binding and sequestering Rnr1 (Chabes et al., 1999; Zhao et al., 2000). The deletion of SML1 had the same effect of RNR1 overexpression both on the mip1-1 mutant and on the pathological Mip1 variants (Zhao et al., 1998; Baruffini et al., 2006, 2012).
A third genetic interactor is MSH1, which encodes the only mitochondrial protein responsible for the mismatch repair. Deletion or overexpression of MSH1 affects both the point and the extended mtDNA mutability. In fact, msh1Δ cells become petite within a few generations (Reenan and Kolodner, 1992), whereas strains overexpressing MSH1 display an increased petite frequency (Dzierzbicki et al., 2004). The deletion of a single MSH1 copy in a diploid strain increases 7-fold the point mutability (Chi and Kolodner, 1994), whereas a moderate overexpression reduces the EryR mutability (Koprowski et al., 2002). Deletion of MSH1 in hyper- or hypomutator mip1 mutant strains affects the point mutability, as described for the mip1R233W mutant strain, where the effects due to the two mutations were additive (Foury and Szczepanowska, 2011).
The last genetic interactors known so far are REV3 and REV7. These encode the two subunits of Pol ζ, which is involved in the error-prone translesion synthesis (TLS), and REV1. REV1 encodes a deoxycytidyl transferase involved in the repair through TLS of abasic sites and adducted guanines in damaged DNA and forms a complex with Pol ζ. These three proteins have also been detected in mitochondria (Zhang et al., 2006). The deletion of each of these three genes reduced the frequency of spontaneous or UV-induced -1 frameshift mutations in the mtDNA about 5-20-fold, but at the same time it increased 2-30-fold the spontaneous or UV-induced point mutability (Zhang et al., 2006; Kalifa and Sia, 2007). This suggests that mitochondrial TLS system is more error-prone than that of Pol ζ, and may be represented by Mip1 itself (Kalifa and Sia, 2007). Furthermore, the deletion of REV3, REV7, or REV1 in a mip1Δ strain unable to lose the mtDNA showed how Pol ζ and Pol γ belong to the same epistatic group, while Rev1 belongs to a different group. In contrast, overexpression of REV3 and REV1, as discussed in more detail later, is able to reduce the petite mutability due to pathological substitutions in Mip1 (Baruffini et al., 2012).
Validation of Pol γ Pathological Mutations in Yeast
In humans, mutations in POLG cause many mitochondrial pathologies, like PEO, Alpers' syndrome and ataxia-neuropathy syndrome. These are all characterized by instability of mtDNA, i.e., mtDNA depletion and/or large scale deletions, which result in impaired mitochondrial function in several tissues with different degrees of severity (reviewed in Stumpf and Copeland, 2011). From the first observation that a disease associated with multiple deletions of mtDNA was caused by a mutation in a nuclear gene (Zeviani et al., 1989) later identified as POLG and from the first identification of mutations in POLG as the cause of PEO (Van Goethem et al., 2001), almost 250 disease-associated mutations have been documented. These are compiled at the Human DNA Polymerase γ Mutation Database (http://tools.niehs.nih.gov/polg/), which reports the genetics related to the different mutations and a description of the associated phenotypes. Most of the mutations were identified in compound heterozygosity, i.e., a mutation was present in the maternal allele and a different mutation in the paternal allele. Some patients carry only a single mutation in each allele, but most patients carry two mutations in one or both the alleles. In these cases, it is unclear how the two substitutions in the same allele contribute to the pathological phenotype. In other cases, patients carry both a putative pathological mutation and an amino acid substitution considered as a neutral polymorphism in the same allele, and determining whether the polymorphism can influence the severity of the disease should be of great interest. Moreover, it is often not possible to assess the dominance/recessivity of pathological mutations, especially when the family history is incomplete or absent.
Yeast has been proven to be an ideal genetic system to obtain such information and to validate in vivo the pathogenicity of Pol γ mutations, i.e., to establish relations between a mutation and the associated phenotypes. For validation in yeast, the MIP1 wild type residue is substituted with the corresponding residue found in patients. When the human mutation involves a conserved residue, the corresponding yeast MIP1 codon can be directly mutated, thus producing the “pathological” allele. When the Pol γ mutated residue is not conserved, but the surrounding stretch is, it is possible to produce a “humanized wild type allele” by replacing the Mip1 amino acid with the wild type amino acid present at the equivalent position of Pol γ. The “humanized wild type allele” is then mutagenized to obtain the “pathological allele.” In order to correctly substitute the orthologs residue, the sequence alignment between human and yeast mtDNA polymerase has to be done unambiguously. However, not all residues involved in diseases are located in conserved regions. Another great advantage of yeast is the possibility of working in the same genetic background, in which the mutant strains differ from each other only for the mip1 mutant allele. For the validation of disease-related mutations, mip1 mutant alleles are introduced in a mip1Δ strain. Since the deletion of the chromosomal MIP1 gene makes the cells immediately and irreversibly rho0, the deletion of MIP1 has to be carried out in cells containing a plasmid-borne wild type MIP1 allele, which can be removed by plasmid shuffling only after its replacement with the mutant allele under analysis (as in Baruffini et al., 2006, 2007a,b, 2010, 2011; Spinazzola et al., 2009; Stricker et al., 2009). Another strategy involves first disrupting the MIP1 wild type gene thus obtaining a mip1Δrho0 strain, and then introducing the mutant allele at the chromosomal locus or in a plasmid. Functional mtDNA is subsequently reintroduced either by crossing with wild type rho+ cells and sporulation (as in Stuart et al., 2006; Stumpf et al., 2010) or by the cytoduction techniques (as in Szczepanowska and Foury, 2010). However, it must be underlined that the use of yeast Mip1 to model pathological mutations also has some shortcomings. Amino acids which are not conserved and are not in a conserved stretch cannot be studied. In addition, the validation is based on the general assumption that if an amino acid is conserved between Mip1 and human Pol γ, the substitution of that amino acid in one protein can predict the effect of the mutation in the second protein. However, this assumption is not applicable to all amino acids, especially if the amino acid lies far from the active site or has a second function specific to human POLGA, such as the binding of POLGB, as in the case of the amino acid A467 (Chan et al., 2005a).
The obtained mutant strains are then analyzed to assess the effects of the mip1 mutation on: (i) oxidative growth, by spot assays on media supplemented with non-fermentative carbon sources; (ii) mtDNA extended mutability, measured as frequency of petite mutants; (iii) mtDNA point mutability measured as frequency of EryR mutants, which arise from specific point mutations in the mitochondrial 21S rRNA encoding gene. These analyses are carried out at the optimal growth temperature, which in yeast is 28–30°C, but also at a higher temperature (37°C), which is more stressful for mitochondrial metabolism, in order to assess thermosensitivity of mutant Mip1.
It is also possible to determine whether the pathological mutation is responsible for mtDNA deletions or complete depletion by analyzing the nature of petite mutants produced (rho− or rho0). For this purpose, different techniques have been applied. The most frequently used method is crossing a large number of defined petite mutants (tested strains) with several mit− strains (tester strains) harboring point mutations in mitochondrial genes encoding respiratory proteins. The method is based on the capacity of rho− genomes to retrieve mit− genomes to wild type through homologs recombination. If restoration of respiratory competence in diploids is observed, it means that the rho− genome has retained a DNA fragment encompassing the mit− mutation (as in Baruffini et al., 2006, 2007b, 2011; Szczepanowska and Foury, 2010). This method was also validated by Southern-blot analysis (Baruffini et al., 2006). Petite mutants can also be examined by confocal fluorescence microscopy following 40,60-diamidino-2-phenylindole hydrochloride (DAPI) staining of cytoplasm, which in yeast also stains mtDNA (as in Stuart et al., 2006; Qian et al., 2014). In both rho− cells and mit− cells stained with DAPI, several small spots can be observed under the surface of the cells, whereas these spots are absent in rho0 due to the lack of mtDNA.
To examine the quantity and integrity of the mitochondrial genomes in petite mutants, quantitative PCR (qPCR) methods were used (as in Stuart et al., 2006; Qian et al., 2014). The copy number of mitochondrial genomes was determined by qPCR of short mitochondrial targets: no detectable PCR products indicate the absence of mtDNA. The integrity of mtDNAs was determined by qPCR of long mitochondrial targets. The relative amplification of the long mtDNA fragment in petite cells, compared to that of the reference wild type controls, provides information about the presence of damaged DNA that blocks the PCR polymerase (as in Stuart et al., 2006). Altogether, these experimental approaches provide relevant information about the molecular mechanism of the replication defect associated with the disease.
Besides these analyses, other biochemical parameters regarding specific Mip1 activity can also be analyzed. For example mtDNA polymerase activity can be analyzed by gap filling experiments, processivity, DNA binding affinity, exonuclease activity, dNTP misincorporation (as in Szczepanowska and Foury, 2010).
In Supplementary Table 2, a list of results concerning validation in yeast of several POLG mutations is presented. All the mutations involve amino acids that are conserved between human and yeast mtDNA polymerase or located in a conserved region, except human A467T, which lies in the linker domain. In humans, this mutation was often found in compound with other mutations. However, homozygous subjects were also described, which indicates that a specific pathological defect is associated with this mutation.
Validation results obtained by different authors are in general coherent, except for a few cases, probably due to the different (non-isogenic) background of the strains used, which results in a higher variability and thus in a non-statistically significant difference. Considering only mutations for which a pathological role has been demonstrated or postulated on the basis of several observations, the prediction capability of the yeast model systems fluctuates from 70% (as in Stumpf et al., 2010) to 100% (as in Baruffini et al., 2007b, 2011). An example of discrepancy concerns human mutation R574W (yeast R467W). Two independent laboratories have obtained similar results using different yeast strains, showing that the presence of the yR467W mutation increases both extended and point mtDNA mutability (Szczepanowska and Foury, 2010; Baruffini et al., 2011); on the contrary, on the basis of results reported by Stumpf et al. (2010), the yR467W mutation has to be considered a neutral change in yeast. This discrepancy can be explained by the onset of a second mutation, capable of suppressing the phenotypic effect of the yR467W mutation, or by the use of a genetic background with a high basal petite frequency (approximately 10%). Indeed, in our experience, a higher basal petite frequency is associated with a higher standard deviation between experiments, so that it is more difficult to demonstrate that a small difference is statistically significant.
One of the advantages offered by yeast is the possibility of obtaining accurate information on the dominance/recessivity of mutations, which is not always easy and straightforward in patients. The dominance/recessivity is determined analyzing mtDNA mutability in the so-called “heteroallelic” strains, in which both a wild type MIP1 copy and a mip1 mutant allele are present, thus mimicking the human diploid condition. For instance, yeast studies have been crucial in establishing the dominance of the extremely severe mutation hE895G (yE698G), found in a child dead 36 h after birth. Due to unclear family history, it could not be established whether the hE895G mutation was inherited as a dominant or recessive trait, as well as whether it was transmitted from the reportedly normal proband father, whose DNA was not available for the study, or was the result of a de novo event. The latter hypothesis was supported by the results obtained in the heteroallelic MIP1/mip1E698G yeast strain, which demonstrated the negative dominant character of the hE698G mutation (Spinazzola et al., 2009). The dominant character of hH932Y (yH734Y) and the recessive character of hG1051R (yG807R) were also clearly defined through yeast studies (Baruffini et al., 2007b).
Determination of mtDNA mutability in heteroallelic strains not only allows one to determine the dominance/recessivity of a mutation, but also to understand whether the mutation leads to a gain of function (i.e., dominant negative) or a loss of function (i.e., null allele). In fact an increase of petite accumulation was observed in a strain carrying a single copy (hemiallelic strain), compared to a strain carrying two copies of wild type MIP1 (homoallelic strain), indicating haploinsufficiency (Baruffini et al., 2006). So if the heteroallelic MIP1/mip1 strain shows a petite frequency higher than the homoallelic MIP1/MIP1 but similar to the hemiallelic MIP1/mip1Δ, the mutation determines loss of function and is dominant by haploinsufficiency. On the other hand, if the mutant strain shows a petite frequency higher than that of the hemiallelic strain, the mutation is considered dominant negative. It is necessary to underline that, so far, almost all the putative dominant mutations in humans are dominant negative in yeast, whereas mutations that cause a total loss of function in yeast are recessive in humans and often associated with severe diseases.
Yeast is not only suitable for validating mutations and clarifying the pathogenicity of mutations. In some cases, it can also have a “predictive role.” This was the case of mutation hS305R, found in two related subjects. One patient carried the hS305R mutation in compound heterozygous with a second mutation, whereas the second one was heterozygous for the hS305R mutation alone, suggesting that the mutation, which was inherited from the mother, behaves as dominant. In yeast, the corresponding yC261R mutation displayed a recessive phenotype that was then divergent from the clinical feature of the second patient. These data obtained in yeast prompted the authors to look for another mutation. Additional analysis on the patient's DNA was not feasible, but further examination of parents' DNA allowed to detect an additional pathogenic mutation, hP1073L, which was identified in the father and was missed in the first genetic analysis. This confirmed that the second patient was also compound heterozygous for two allelic mutations (Baruffini et al., 2011). Therefore, the discrepancy between the results obtained in yeast and the clinical features may in some case predict the presence of a cryptic, undetected mutation in the patients second allele, as postulated in the case of mutation hR386H (yI334H).
Patients often carry more than one POLG mutation, in cis or in trans, and the specific contribution of each mutation to the disease cannot be established. In yeast, mutations can be studied alone or in combination, so it is possible to clearly evaluate the contribution of each mutation to the pathology. Functional interactions of mip1 mutations in cis were studied by introducing both mutations in the same mip1 allele, and in trans by transforming the mip1Δ strain with the two plasmid-borne mutant alleles (Supplementary Table 3). For instance, in the case of mutation hH932Y (yH734Y), found in trans with hG1051R (yG807R), the comparison of the severity of the yH734Y and yG807R mutations, alone or in compound, strongly suggested that the major contributor to the disease was hH932Y, even though a strong synergistic effect of the two mutations, when associated, was confirmed (Baruffini et al., 2007b).
A different degree of severity of Alpers' disease has been described in patients carrying the common A467T mutation in compound with mutation G303R, R574W or P625R, suggesting that the phenotypic differences observed are likely to be ascribed at least in part to the presence of these additional mutations. Results in yeast demonstrated that hG303R (yG259R) is highly detrimental, hR574W (yR467W) produces an intermediate phenotype which is also very severe but less damaging than G303R, and hP625R (P513R) induces a mild defect at 28°C but a strong increase of mtDNA mutability at 37°C. The detrimental effects observed in yeast correlated to the severity of this phenotype in humans, thus explaining the phenotypic modulation of the observed clinical features.
In some cases pathological mutations are present in compound with single nucleotide polymorphisms (SNP). This raises the question of whether these substitutions are neutral or can act as phenotypic modifiers which lead to more severe clinical phenotypes. This was the case of the hA889T (yA692T) mutation present in cis-compound with the SNP E1143G (E900G). Comparing the effect of the mip1A692T+E900G allele, which carries the two mutations in compound, and the effect of mip1A692T or mip1E900G mutant alleles, demonstrated that the presence of the two substitutions in cis exerts a deleterious synergistic effect on the mtDNA mutability, thus indicating that E900G is not a neutral polymorphism if in compound with another mutation (Baruffini et al., 2007b).
Besides Validation: Molecular Defects Associated with Mip1 Mutations
Studies on Pol γ mutations in yeast, animal models, cell culture, in vitro and in silico analysis have clarified some of the molecular defects associated with different mutations.
In yeast it has been observed that the mutations L210P (human L244P), G651S (hG848S), A692T (hA889T), H734Y (hY932Y), G807R (hG1051R), and E900G (hE1143G) lead to a 50 to 90%, reduction in mitochondrial protein levels. Reduced protein levels may account, at least in part, for the increase of petite mutability associated with these mutations (Baruffini et al., 2007b; Szczepanowska and Foury, 2010). Since the expression levels of such Mip1 variants are normal the decrease of soluble mitochondrial proteins suggests either that the mutant proteins are not transported properly into the mitochondria or that proteins are misfolded and/or degraded in mitochondria. Although the analyses performed on patients' cells are limited, it was observed that a decrease of Pol γ holoenzyme levels occurs in fibroblasts from patients bearing mutations R232H and G848S in trans (75% reduction), and mutations A467T and T914P in trans (45% reduction) (Taanman et al., 2008). A reduction in Pol γ catalytic subunit was also observed in a patient bearing the mutation T9214P (Roos et al., 2013) and, in another patient, the complete lack of POLGA containing the stop mutation E873X, due to nonsense-mediated mRNA decay, was reported (Chan et al., 2005b).
Most of the mutations in POLG determine alterations of the mtDNA polymerase biochemical properties, i.e., decrease of catalytic activity, processivity, DNA binding affinity, and/or binding affinity for the incoming dNTP (reviewed in Chan and Copeland, 2009; Stumpf and Copeland, 2011). Regarding the Mip1 mutant, the following was observed: (1) decrease in the gap-filling DNA synthesis associated with mutations L260R (hL304R), R265L (hR309L), R265H (hR309H), F268R (hW312R), R467W (hR574W), G651S (hG848S), A692T (hA889T), H734Y (hY932Y), G807R (hG1051R); (2) decrease in processivity and of DNA binding affinity in the case of mutations L360R, R265L, R265H, R467W, and F268R; (3) increase in dNTP misincorporation in the case of mutations L260R and R265H; (4) increase in Exo/Pol ratio in the case of mutations L260R, R265L, R265H, and F268R (Baruffini et al., 2007b; Szczepanowska and Foury, 2010). These data were confirmed for human Pol γ harboring mutation G848S, showing less than 1% of polymerase activity and a 5-fold increase in Kd(DNA) (Kasiviswanathan et al., 2009).
In contrast, the exonuclease activity is only slightly or not at all reduced, even in the case of mutations in the exo domain, suggesting that the defects of mtDNA replication are not due to defects of proofreading activity but to deficiency of polymerase activity (Szczepanowska and Foury, 2010). In yeast, the absence of the Mip1 exonuclease activity results in a 160-fold increase of the frequency of deletions between 21 bp direct repeats (Phadnis et al., 2005; Stumpf and Copeland, 2013). Seven mutations in the exo domain did not cause an increase of this type of deletions, further indicating that the exonuclease activity is scarcely or not at all affected (Stumpf and Copeland, 2013).
Regarding the dominant negative mutations, two main hypotheses have been proposed to explain this dominance. Some mutations completely inhibit the polymerase activity of Pol γ, but not the DNA binding affinity. Thus, the mutant polymerase that binds the DNA with similar affinity to that of wild type Pol γ may block the replication but may also prevent the binding of the wild type enzyme. For example, the Y955C mutation, which is dominant in both yeast and humans, prevents the polymerase from synthesizing DNA, especially in the case of the incorporation of dATP:T, but it does not change the Kd(DNA) (Graziewicz et al., 2004; Estep and Johnson, 2011). On the other hand, some mutant Pol γ variants may directly cause lesions to DNA. In the diploid strain containing a wild type MIP1 allele and a mip1 allele harboring mutation Y757C, which corresponds to human Y955C, an accumulation of mtDNA lesions was found (Stuart et al., 2006). In addition, in mice expressing a cardiac targeted Y955C variant, an accumulation of the oxidized nucleotide 8-hydroxy-2-deoxyguanosine (8-OHdG) in the mtDNA (Lewis et al., 2007) was observed, probably because the Y955C Pol γ displays a reduced discrimination for incorporation of 8-oxo-dGTP or for translesion synthesis opposite to 8-oxo-dG (Graziewicz et al., 2007). However, more recently it was shown that physiological effects of the Y955C mutation might not be due to an increased incorporation of 8-oxo-dG, since the incorporation rate of 8-oxo-dG is low in wt Pol γ and further decreased 500-fold in Y955C Pol γ (Hanes and Johnson, 2007; Estep and Johnson, 2011).
On the basis of information obtained from several studies, including studies in yeast and an in silico analysis, the majority of mutations in POLG were included in five clusters, depending on the defect associated with the mtDNA polymerase (Euro et al., 2011; Farnum et al., 2014): cluster 1, mutations in the polymerase domain, which are mostly dominant and which affect the polymerase activity; cluster 2, recessive mutations in the spacer domain which affect the upstream DNA-binding channel; cluster 3, recessive mutations in the exo domain and in the fingers subdomain which are associated with a Pol γ specific functional module involved in the partitioning of the DNA substrate between the exo and the pol catalytic sites; cluster 4, recessive mutations in the exo domain which influence the bond with one of the accessory subunit; cluster 5, recessive mutations of the IP subdomain which affect the binding to other proteins of the replisome.
Besides Validation: Chemical and Genetic Rescue of the Phenotype
By using yeast, it is possible to easily identify, in short times, molecules or genes able to reduce the effects of pathological substitutions.
Regarding the chemical rescue, active molecules were identified by analyzing their ability to restore oxidative growth and/or to reduce petite frequency of mip1 mutants grown in their presence. The identification of these molecules has a double significance. Firstly, on the basis of the mechanisms by which the drugs act, it is possible to deduce information on the molecular defects caused by the Mip1 mutation. Secondly, identification of such drugs can pave the way for studies on their therapeutic potential. It has been demonstrated in yeast that the administration of lipoic acid or MitoQ, two mitochondrial antioxidant molecules, reduces the petite mutability due to some mip1 mutations (Baruffini et al., 2006, 2011, 2012). Out of all the studied mutations, the dominant mutations in the polymerase domain, in particular the mutation Y757C (hY955C), are particularly sensitive. This observation is consistent with the fact that, as reported in the previous section, human Pol γ harboring Y955C leads to increased incorporation of 8-OHdG. Thus, it is possible that supplementation with antioxidants reduces the levels of oxidized bases, which would consequently be incorporated into the mtDNA.
Regarding the genetic rescue, both the overexpression of RNR1 and the deletion of SML1 reduce the petite mutability in the ts mutant mip1-1 by increasing the concentration of dNTP. This was also observed for most of the pathological mutations introduced in MIP1 (Baruffini et al., 2006, 2011, 2012; Stumpf et al., 2010), although at a different extent. It is reasonable to think that the rescue is greater when the ability of the Mip1 mutant to bind the incoming dNTP is more compromised. This was demonstrated for the H734Y mutation, for which RNR1 overexpression causes a 5-fold decrease of petite frequency. In vitro analysis on human Pol γ containing the corresponding H932Y mutation showed that the affinity for the incoming dNTP is reduced approximately 200-fold compared to the wild type (Stumpf et al., 2010). The increase of the dNTP pools as a rescue mechanism was also demonstrated by the observation that the processivity defect of human DNA polymerase γ harboring the Y955C mutation was overcome by increasing the dATP or dTTP concentration during in vitro synthesis of mtDNA fragments (Atanassova et al., 2011). Supplementation of dNTPs precursors as a potential therapeutic agent was shown on differentiating myotubes of a patient harboring mutations in POLG, in which the addition of dAMP and dGMP slightly increased the levels of mtDNA (Bulst et al., 2009). In addition, supplementing myotubes of patients harboring mutations in POLG with specific combinations of the four dNMPs showed near normalization of the mtDNA levels (Bulst et al., 2012). However, it is necessary to emphasize that the alteration of the dNTPs concentration may also have deleterious consequences (Xu et al., 2008).
Finally, the extended and point mtDNA mutability due to Mip1 mutations is rescued by overexpression of Pol ζ and Rev1 that, in yeast, are also located in the mitochondria (Zhang et al., 2006; Baruffini et al., 2012). Intriguingly, the Mip1 mutations which are rescued by Pol ζ overexpression are not recovered by antioxidant treatment, and vice versa, suggesting that the rescue is exerted through two alternative mechanisms. The first one is most likely related to the ability of Pol ζ to replace Mip1 containing mutations which mainly reduce the catalytic activity. The second one is likely linked to the ability of antioxidant molecules to reduce the concentration of oxidized bases which can be incorporated by Mip1 bearing specific mutations such as Y757C.
Beyond Validation: Study of Correlation between Mutations/Polymorphisms in Pol γ and Mitochondrial Toxicity Caused by Sodium Valproate, NRTIs and Environmental Mutagens
Sodium valproate (valproic acid, or VPA) is a drug used as an anticonvulsant, and for migraine, bipolar disorder and chronic headache. However, the administration of VPA in some individuals can lead to a fulminant liver failure. The frequency of this adverse effect is very high in patients with Alpers' syndrome bearing mutations in POLG, and also in subjects not affected by Alpers' syndrome but carrying Pol γ polymorphisms. Through a combined approach performed on cells of subjects showing episodes of VPA-induced liver toxicity and harboring polymorphisms in POLG, and on yeast cells bearing the corresponding mip1 mutations, it was demonstrated that patients heterozygous for polymorphisms E1143G and Q1236H have a higher probability of developing VPA-induced toxicity (Stewart et al., 2010).
Nucleoside reverse transcriptase inhibitors (NRTIs) are used in the highly active antiretroviral therapy (HAART), which has significantly increased the life expectancy of HIV patients. However, in some patients a prolonged treatment induces side effects, most of which are due to interference of the NRTIs or of their triphosphorylated form with the mitochondrial function. NRTIs are nucleosides analogs in which the hydroxyl moiety in 3′ was substituted with a group that blocks the formation of the 3′-5′ bond in the nascent strand. Several observations suggest that mitochondrial toxicity depends on NRTIs interference with Pol γ activity, resulting in decreased mtDNA levels, especially in the case of pyrimidine analogs, such as stavudine (2′,3′-didehydro-2′,3′-dideoxythymidine, or d4T) and zalcitabine (2′,3′-dideoxycytidine, or ddC) (reviewed in Lee et al., 2003 and Koczor and Lewis, 2010). In addition, some triphosphorylated NRTIs can inhibit the activity of human Pol γ in vitro (Johnson et al., 2001; Lim and Copeland, 2001). It has also been demonstrated by in vitro experiments that substitutions in Pol γ may alter the mtDNA polymerase discrimination between the correct dNTP and the corresponding dNRTI-TP. This results either in a decreased ability to incorporate the dNTPs, for example in the case of E895G, Y951F, and Y955F substitutions (Lim et al., 2003), or in an increased ability to incorporate the dNRTI-TP, such as in the case of mutation R964C, which causes a 33% decrease of dTTP incorporation efficiency and a 3-fold lower d4TTP discrimination compared to wild type Pol γ (Bailey et al., 2009). Mitochondrial toxicity was observed in a patient homozygous for the R964C mutation after treatment with D4T (Yamanaka et al., 2007), and a correlation between treatment with D4T and mitochondrial toxicity was proposed for subjects heterozygous for the widespread SNP E1143G (Chiappini et al., 2009). We have previously constructed and validated two yeast models useful for studying the correlation between mutations or polymorphisms in POLG and mtDNA depletion after treatment with D4T or ddC, (Baruffini and Lodi, 2010; Baruffini et al., 2015). These models were constructed by cloning and introducing in yeast the human ENT1 gene, which encodes a membrane nucleoside transporter, and the herpes simplex virus TK1 gene, which encodes a thymidine kinase, or the human DCK gene, which encodes a deoxycytidine kinase, in order to allow the proper transport and phosphorylation of the analog. Our results show that, besides R964C and E1143G, other polymorphisms in POLG might cause mtDNA instability and depletion following treatment with these NRTIs. However, further studies are needed in human cells.
Alkylating agents, such as methyl methanesulfonate (MMS), also increase mtDNA point mutability besides nuclear DNA mutability. It was shown that administration of MMS in strains harboring mutator mip1 heterozygous mutations corresponding to human pathological ones further increased the EryR point mutability due to CG transversions (Stumpf and Copeland, 2014). MMS induced mutability in mip1 mutant strains seems to be due to an active mechanism, in which the mutant polymerase binds to the DNA and stalls mtDNA replication, resulting in ssmtDNA which can be alkylated by MMS. Trace amounts of CdCl2 are also associated with an increase in the petite frequency and a decrease in the mtDNA content both in homozygous and MIP1/mip1 heterozygous strains (Stumpf and Copeland, 2014).
Recent Findings and Future Directions in Modeling Human Mutations and Drug Discovery
In recent years, research on MIP1 focused on genetic and molecular suppressors able to reduce point and extended mtDNA mutability due to mtDNA polymerase dysfunctions. By random mutagenesis, a MIP1 mutation, A256T, was recently identified. This behaves like an antimutator, decreasing 2.2-fold the frequency of EryR mutants (Foury and Szczepanowska, 2011). The A256 residue is conserved in humans (A300) and is part of a conserved region, suggesting that Pol γ A300T can behave as an antimutator allele. Further experiments in animal models should be performed to prove this hypothesis.
In 2014, Qian and coauthors constructed a “humanized” yeast model in which yeast Mip1 was replaced by human Pol γ. They deleted most of the endogenous MIP1 gene and cloned both POLG(1) and POLG2 under the MIP1 promoter and in frame with the MIP1 MLS region. The mip1Δ “humanized” strain, containing both POLGA and POLGB, was able to grow on oxidative carbon sources at a similar rate to MIP1 wild type strain, indicating that the human holoenzyme can complement the loss of Mip1 and can replicate yeast mtDNA. The complementation was partial, since in the humanized strain the mtDNA levels were reduced to 50% compared to the wild type strain, and both petite and EryR frequencies were doubled. Nevertheless, this system has proven useful to model human Pol γ mutations in yeast. Four mutations were introduced in POLGA (S305R, H932Y, Y951N, and Y955C). The behavior of mutant POLGA strains is very similar to strains harboring the corresponding mutations in MIP1 (Table 2). In both systems, S305R strongly increases the petite frequency and is recessive, whereas H932Y, Y951N, and Y955C make the strain rho0 (and thus unable to grow on oxidative carbon sources) and are dominant. In both systems, EryR mutant frequency, which is measured in the diploid strain, increases when each mutation is present in heterozygosis, though at different extents (Qian et al., 2014). Overall, the construction of this “humanized” strain indicates that MIP1, used until now in validation studies, is a good model to study the phenotypic consequences of mutations, at least for the conserved amino acids. Furthermore, it provides a novel and invaluable tool to assess the physiological effects of disease-associated mutations directly in human Pol γ itself, thus also allowing to overcome the weaknesses associated to the use of Mip1 discussed above.
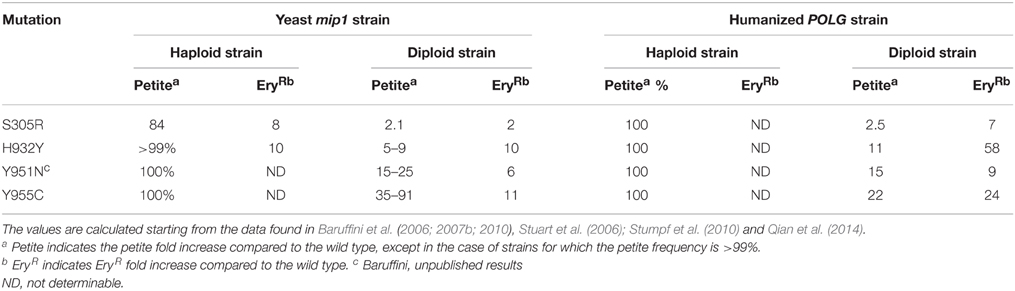
Table 2. Comparison between four mutations studied in yeast mip1 strain and in humanized yeast POLG strain.
Recently, yeast has proven to be an excellent tool for drug discovery, in particular in the case of mitochondrial disorders. This is of paramount importance since no established treatment for these pathologies is available so far. Molecules acting as potential therapeutics in disorders associated with primary deficiencies in the mitochondrial ATP synthase were successfully found in yeast by high throughput approaches aimed at identifying chemical suppressors of pathological phenotypes (Couplan et al., 2011; Aiyar et al., 2014). In such a screen, the fmc1 null mutant, unable to assemble the F1 sector of ATP synthase at high temperatures, was used as yeast model of these pathologies. Taking advantage of the RD thermosensitive phenotype of this mutant, a yeast-based assay was developed in which thousands of chemical compounds from several chemical libraries were tested for their ability to suppress the respiratory growth defect at 37°C. By such a screen, drugs effective on a fmc1 yeast mutant were successfully found. These drugs were also active on strains mutated in ATP6, which, if mutated in humans, caused neuropathy, ataxia, and retinitis pigmentosa (NARP) syndrome. Moreover, they have proven to be effective in human cybrids derived from NARP patients, thus validating the yeast-based approach and demonstrating that yeast can be used as a pharmacological model for the study of mitochondrial diseases.
We used a similar high throughput approach, in collaboration with A. Delahodde (I2BC, Université Paris-Sud), to find drugs that could potentially be used in POLG disease therapy. About 1600 molecules included in two chemical libraries were assayed for their ability to restore the oxidative growth of thermosensitive mip1 strains harboring mutations equivalent to the human pathological ones. Six rescuing molecules were identified, three of which also decreased the frequency of cells depleted of mtDNA at 28°C. The rescue has also been proven effective in a C. elegans POLG model. Studies on fibroblasts from patients bearing mutations in POLG and experiments aimed at gaining more insight into the molecular mechanism behind the rescue are in progress.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We wish to thank Beatrice Martinelli (kelimatranslations.com, UK) for proofreading the manuscript and Agnès Delahodde for the suggestions in writing the last section. This work was supported by Telethon (GP11011).
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fgene.2015.00106/abstract
References
Aiyar, R. S., Bohnert, M., Duvezin-Caubet, S., Voisset, C., Gagneur, J., Fritsch, E. S., et al. (2014). Mitochondrial protein sorting as a therapeutic target for ATP synthase disorders. Nat. Commun. 5, 5585. doi: 10.1038/ncomms6585
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Atanassova, N., Fusté, J. M., Wanrooij, S., Macao, B., Goffart, S., Bäckström, S., et al. (2011). Sequence-specific stalling of DNA polymerase γ and the effects of mutations causing progressive ophthalmoplegia. Hum. Mol. Genet. 20, 1212–12123. doi: 10.1093/hmg/ddq565
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bailey, C. M., Kasiviswanathan, R., Copeland, W. C., and Anderson, K. S. (2009). R964C mutation of DNA polymerase gamma imparts increased stavudine toxicity by decreasing nucleoside analog discrimination and impairing polymerase activity. Antimicrob. Agents Chemother. 53, 2610–2612. doi: 10.1128/AAC.01659-08
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Baldacci, G., Chérif-Zahar, B., and Bernardi, G. (1984). The initiation of DNA replication in the mitochondrial genome of yeast. EMBO J. 3, 2115–2120.
Baruffini, E., Ferrari, J., Dallabona, C., Donnini, C., and Lodi, T. (2015). Polymorphisms in DNA polymerase γ affect the mtDNA stability and the NRTI-induced mitochondrial toxicity in Saccharomyces cerevisiae. Mitochondrion 20, 52–63. doi: 10.1016/j.mito.2014.11.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Baruffini, E., Ferrero, I., and Foury, F. (2007b). Mitochondrial DNA defects in Saccharomyces cerevisiae caused by functional interactions between DNA polymerase gamma mutations associated with disease in human. Biochim. Biophys. Acta 1772, 1225–1235. doi: 10.1016/j.bbadis.2007.10.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Baruffini, E., Ferrero, I., and Foury, F. (2010). In vivo analysis of mtDNA replication defects in yeast. Methods 51, 426–436. doi: 10.1016/j.ymeth.2010.02.023
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Baruffini, E., Horvath, R., Dallabona, C., Czermin, B., Lamantea, E., Bindoff, L., et al. (2011). Predicting the contribution of novel POLG mutations to human disease through analysis in yeast model. Mitochondrion 11, 182–190. doi: 10.1016/j.mito.2010.09.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Baruffini, E., and Lodi, T. (2010). Construction and validation of a yeast model system for studying in vivo the susceptibility to nucleoside analogues of DNA polymerase gamma allelic variants. Mitochondrion 10, 183–187. doi: 10.1016/j.mito.2009.10.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Baruffini, E., Lodi, T., Dallabona, C., and Foury, F. (2007a). A single nucleotide polymorphism in the DNA polymerase gamma gene of Saccharomyces cerevisiae laboratory strains is responsible for increased mitochondrial DNA mutability. Genetics 177, 1227–1231. doi: 10.1534/genetics.107.079293
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Baruffini, E., Lodi, T., Dallabona, C., Puglisi, A., Zeviani, M., and Ferrero, I. (2006). Genetic and chemical rescue of the Saccharomyces cerevisiae phenotype induced by mitochondrial DNA polymerase mutations associated with progressive external ophthalmoplegia in humans. Hum. Mol. Genet. 15, 2846–2855. doi: 10.1093/hmg/ddl219
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Baruffini, E., Serafini, F., Ferrero, I., and Lodi, T. (2012). Overexpression of DNA polymerase zeta reduces the mitochondrial mutability caused by pathological mutations in DNA polymerase gamma in yeast. PLoS ONE 7:e34322. doi: 10.1371/journal.pone.0034322
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bernad, A., Blanco, L., Lázaro, J. M., Martín, G., and Salas, M. (1989). A conserved 3′-5′ exonuclease active site in prokaryotic and eukaryotic DNA polymerases. Cell 59, 219–228. doi: 10.1016/0092-8674(89)90883-0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bulst, S., Abicht, A., Holinski-Feder, E., Müller-Ziermann, S., Koehler, U., Thirion, C., et al. (2009). In vitro supplementation with dAMP/dGMP leads to partial restoration of mtDNA levels in mitochondrial depletion syndromes. Hum. Mol. Genet. 18, 1590–1599. doi: 10.1093/hmg/ddp074
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bulst, S., Holinski-Feder, E., Payne, B., Abicht, A., Krause, S., Lochmüller, H., et al. (2012). In vitro supplementation with deoxynucleoside monophosphates rescues mitochondrial DNA depletion. Mol. Genet. Metab. 107, 95–103. doi: 10.1016/j.ymgme.2012.04.022
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chabes, A., Domkin, V., and Thelander, L. (1999). Yeast Sml1, a protein inhibitor of ribonucleotide reductase. J. Biol. Chem. 17, 36679–36683. doi: 10.1074/jbc.274.51.36679
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chan, S. S., and Copeland, W. C. (2009). DNA polymerase gamma and mitochondrial disease: understanding the consequence of POLG mutations. Biochim. Biophys. Acta 1787, 312–319. doi: 10.1016/j.bbabio.2008.10.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chan, S. S. L., Longley, M. J., Naviaux, R. K., and Copeland, W. C. (2005b). Mono-allelic POLG expression resulting from nonsense-mediated decay and alternative splicing in a patient with Alpers syndrome. DNA Repair 4, 1381–1389. doi: 10.1016/j.dnarep.2005.08.010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chan, S. S., Longley, M. J., and Copeland, W. C. (2005a). The common A467T mutation in the human mitochondrial DNA polymerase (POLG) compromises catalytic efficiency and interaction with the accessory subunit. J. Biol. Chem. 280, 31341–31346. doi: 10.1074/jbc.M506762200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chatterjee, N., Pabla, R., and Siede, W. (2013). Role of polymerase η in mitochondrial mutagenesis of Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 431, 270–273. doi: 10.1016/j.bbrc.2012.12.119
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, X. J., and Butow, R. A. (2005). The organization and inheritance of the mitochondrial genome. Nat. Rev. Genet. 6, 815–825. doi: 10.1038/nrg1708
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, X. J., Wang, X. W., Kaufman, B. A., and Butow, R. A. (2005). Aconitase couples metabolic regulation to mitochondrial DNA maintenance. Science 307, 714–717. doi: 10.1126/science.1106391
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cheng, X., and Ivessa, A. S. (2010). Association of the yeast DNA helicase Pif1p with mitochondrial membranes and mitochondrial DNA. Eur. J. Cell. Biol. 89, 742–747. doi: 10.1016/j.ejcb.2010.06.008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chi, N. W., and Kolodner, R. D. (1994). Purification and characterization of MSH1, a yeast mitochondrial protein that binds to DNA mismatches. J. Biol. Chem. 269, 29984–29992.
Chiappini, F., Teicher, E., Saffroy, R., Debuire, B., Vittecoq, D., and Lemoine, A. (2009). Relationship between polymerase gamma (POLG) polymorphisms and antiretroviral therapy-induced lipodystrophy in HIV-1 infected patients: a case-control study. Curr. HIV Res. 7, 244–253. doi: 10.2174/157016209787581409
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Copeland, W. C., and Longley, M. J. (2008). DNA2 resolves expanding flap in mitochondrial base excision repair. Mol. Cell. 32, 457–458. doi: 10.1016/j.molcel.2008.11.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Couplan, E., Aiyar, R. S., Kucharczyk, R., Kabala, A., Ezkurdia, N., Gagneur, J., et al. (2011). A yeast-based assay identifies drugs active against human mitochondrial disorders. Proc. Natl. Acad. Sci. U.S.A. 108, 11989–11994. doi: 10.1073/pnas.1101478108
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dimitrov, L. N., Brem, R. B., Kruglyak, L., and Gottschling, D. E. (2009). Polymorphisms in multiple genes contribute to the spontaneous mitochondrial genome instability of Saccharomyces cerevisiae S288C strains. Genetics 183, 365–383. doi: 10.1534/genetics.109.104497
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Duchniewicz, M., Germaniuk, A., Westermann, B., Neupert, W., Schwarz, E., and Marszalek, J. (1999). Dual role of the mitochondrial chaperone Mdj1p in inheritance of mitochondrial DNA in yeast. Mol. Cell. Biol. 19, 8201–8210.
Dujon, B. (1981). “Mitochondrial genetics and functions,” in The Molecular Biology of the Yeast Saccharomyces: Life Cycle and Inheritance, eds J. N. Strathern, E. W. Jones and J. R. Broach (Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press), 505–635.
Dujon, B., Slonimski, P. P., and Weill, L. (1974). Mitochondrial genetics IX: a model for recombination and segregation of mitochondrial genomes in Saccharomyces cerevisiae. Genetics 78, 415–437.
Dzierzbicki, P., Koprowski, P., Fikus, M. U., Malc, E., and Ciesla, Z. (2004). Repair of oxidative damage in mitochondrial DNA of Saccharomyces cerevisiae: involvement of the MSH1-dependent pathway. DNA Repair (Amst) 3, 403–411. doi: 10.1016/j.dnarep.2003.12.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Elledge, S. J., and Davis, R. W. (1990). Two genes differentially regulated in the cell cycle and by DNA-damaging agents encode alternative regulatory subunits of ribonucleotide reductase. Genes Dev. 4, 740–751. doi: 10.1101/gad.4.5.740
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ephrussi, B., and Slonimski, P. P. (1955). Yeast mitochondria. Subcellular units involved in the synthesis of respiratory enzymes in yeast. Nature 176, 1207–1208. doi: 10.1038/1761207b0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Eriksson, S., Xu, B., and Clayton, D. A. (1995). Efficient incorporation of anti-HIV deoxynucleotides by recombinant yeast mitochondrial DNA polymerase. J. Biol. Chem. 270, 18929–18934. doi: 10.1074/jbc.270.32.18929
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Estep, P. A., and Johnson, K. A. (2011). Effect of the Y955C mutation on mitochondrial DNA polymerase nucleotide incorporation efficiency and fidelity. Biochemistry 50, 6376–6386. doi: 10.1021/bi200280r
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Euro, L., Farnum, G. A., Palin, E., Suomalainen, A., and Kaguni, L. S. (2011). Clustering of Alpers disease mutations and catalytic defects in biochemical variants reveal new features of molecular mechanism of the human mitochondrial replicase, Pol γ. Nucleic Acids Res. 39, 9072–9084. doi: 10.1093/nar/gkr618
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Farnum, G. A., Nurminen, A., and Kaguni, L. S. (2014). Mapping 136 pathogenic mutations into functional modules in human DNA polymerase γ establishes predictive genotype-phenotype correlations for the complete spectrum of POLG syndromes. Biochim. Biophys. Acta. 1837, 1113–11121. doi: 10.1016/j.bbabio.2014.01.021
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Foury, F. (1989). Cloning and sequencing of the nuclear gene MIP1 encoding the catalytic subunit of the yeast mitochondrial DNA polymerase. J. Biol. Chem. 264, 20552–20560.
Foury, F., Roganti, T., Lecrenier, N., and Purnelle, B. (1998). The complete sequence of the mitochondrial genome of Saccharomyces cerevisiae. FEBS Lett. 440, 325–331. doi: 10.1016/S0014-5793(98)01467-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Foury, F., and Szczepanowska, K. (2011). Antimutator alleles of yeast DNA polymerase gamma modulate the balance between DNA synthesis and excision. PLoS ONE 6:e27847. doi: 10.1371/journal.pone.0027847
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Foury, F., and Vanderstraeten, S. (1992). Yeast mitochondrial DNA mutators with deficient proofreading exonucleolytic activity. EMBO J. 11, 2717–2726.
Gaillard, C., Strauss, F., and Bernardi, G. (1980). Excision sequences in the mitochondrial genome of yeast. Nature 283, 218–220. doi: 10.1038/283218a0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Genga, A., Bianchi, L., and Foury, F. (1986). A nuclear mutant of Saccharomyces cerevisiae deficient in mitochondrial DNA replication and polymerase activity. J. Biol. Chem. 261, 9328–9332.
Gerhold, J. M., Aun, A., Sedman, T., Jõers, P., and Sedman, J. (2010). Strand invasion structures in the inverted repeat of Candida albicans mitochondrial DNA reveal a role for homologous recombination in replication. Mol. Cell. 39, 851–861. doi: 10.1016/j.molcel.2010.09.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Germaniuk, A., Liberek, K., and Marszalek, J. (2002). A bichaperone (Hsp70-Hsp78) system restores mitochondrial DNA synthesis following thermal inactivation of Mip1p polymerase. J. Biol. Chem. 277, 27801–27808. doi: 10.1074/jbc.M201756200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Graziewicz, M. A., Bienstock, R. J., and Copeland, W. C. (2007). The DNA polymerase gamma Y955C disease variant associated with PEO and parkinsonism mediates the incorporation and translesion synthesis opposite 7,8-dihydro-8-oxo-2′-deoxyguanosine. Hum. Mol. Genet. 16, 2729–2739. doi: 10.1093/hmg/ddm227
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Graziewicz, M. A., Longley, M. J., Bienstock, R. J., Zeviani, M., and Copeland, W. C. (2004). Structure-function defects of human mitochondrial DNA polymerase in autosomal dominant progressive external ophthalmoplegia. Nat. Struct. Mol. Biol. 11, 770–776. doi: 10.1038/nsmb805
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hanes, J. W., and Johnson, K. A. (2007). A novel mechanism of selectivity against AZT by the human mitochondrial DNA polymerase. Nucleic Acids Res. 35, 6973–6983 doi: 10.1093/nar/gkm695
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Holt, I. J., Lorimer, H. E., and Jacobs, H. T. (2000). Coupled leading- and lagging-strand synthesis of mammalian mitochondrial DNA. Cell 100, 515–524. doi: 10.1016/S0092-8674(00)80688-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hu, J. P., Vanderstraeten, S., and Foury, F. (1995). Isolation and characterization of ten mutator alleles of the mitochondrial DNA polymerase-encoding MIP1 gene from Saccharomyces cerevisiae. Gene 160, 105–110. doi: 10.1016/0378-1119(95)00215-R
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Johnson, A. A., Ray, A. S., Hanes, J., Suo, Z., Colacino, J. M., Anderson, K. S., et al. (2001). Toxicity of antiviral nucleoside analogs and the human mitochondrial DNA polymerase. J. Biol. Chem. 276, 40847–40857. doi: 10.1074/jbc.M106743200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kalifa, L., and Sia, E. A. (2007). Analysis of Rev1p and Pol zeta in mitochondrial mutagenesis suggests an alternative pathway of damage tolerance. DNA Repair (Amst) 6, 1732–1739. doi: 10.1016/j.dnarep.2007.06.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kasiviswanathan, R., Longley, M. J., Chan, S. S., and Copeland, W. C. (2009). Disease mutations in the human mitochondrial DNA polymerase thumb subdomain impart severe defects in mitochondrial DNA replication. J. Biol. Chem. 284, 19501–19510. doi: 10.1074/jbc.M109.011940
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Koczor, C. A., and Lewis, W. (2010). Nucleoside reverse transcriptase inhibitor toxicity and mitochondrial DNA. Expert. Opin. Drug. Metab. Toxicol. 6, 1493–1504. doi: 10.1517/17425255.2010.526602
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Koprowski, P., Fikus, M. U., Mieczkowski, P., and Ciesla, Z. (2002). A dominant mitochondrial mutator phenotype of Saccharomyces cerevisiae conferred by msh1 alleles altered in the sequence encoding the ATP-binding domain. Mol. Genet. Genomics 266, 988–994. doi: 10.1007/s00438-001-0621-x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kucej, M., Kucejova, B., Subramanian, R., Chen, X. J., and Butow, R. A. (2008). Mitochondrial nucleoids undergo remodeling in response to metabolic cues. J. Cell. Sci. 121, 1861–1868. doi: 10.1242/jcs.028605
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lasserre, J. P., Plissonneau, J., Velours, C., Bonneu, M., Litvak, S., Laquel, P., et al. (2013). Biochemical, cellular and molecular identification of DNA polymerase α in yeast mitochondria. Biochimie 95, 759–771. doi: 10.1016/j.biochi.2012.11.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lecrenier, N., and Foury, F. (1995). Overexpression of the RNR1 gene rescues Saccharomyces cerevisiae mutants in the mitochondrial DNA polymerase encoding MIP1 gene. Mol. Gen. Genet. 249, 1–7. doi: 10.1007/BF00290229
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lecrenier, N., and Foury, F. (2000). New features of mitochondrial DNA replication system in yeast and man. Gene 246, 37–48. doi: 10.1016/S0378-1119(00)00107-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lee, H., Hanes, J., and Johnson, K. A. (2003). Toxicity of Nucleoside Analogs Used to Treat AIDS and the Selectivity of the Mitochondrial DNA Polymerase. Biochemistry 42, 14711–14719. doi: 10.1021/bi035596s
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lee, Y. S., Kennedy, W. D., and Yin, Y. W. (2009). Structural insight into processive human mitochondrial DNA synthesis and disease-related polymerase mutations. Cell 139, 312–324. doi: 10.1016/j.cell.2009.07.050
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lewandowska, A., Gierszewska, M., Marszalek, J., and Liberek, K. (2006). Hsp78 chaperone functions in restoration of mitochondrial network following heat stress. Biochim. Biophys. Acta 1763, 141–151. doi: 10.1016/j.bbamcr.2006.01.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lewis, W., Day, B. J., Kohler, J. J., Hosseini, S. H., Chan, S. S., Green, E. C., et al. (2007). Decreased mtDNA, oxidative stress, cardiomyopathy, and death from transgenic cardiac targeted human mutant polymerase gamma. Lab. Invest. 87, 326–335. doi: 10.1038/labinvest.3700523
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lim, S. E., and Copeland, W. C. (2001). Differential incorporation and removal of antiviral deoxynucleotides by human DNA polymerase gamma. J. Biol. Chem. 276, 23616–23623. doi: 10.1074/jbc.M101114200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lim, S. E., Longley, M. J., and Copeland, W. C. (1999). The mitochondrial p55 accessory subunit of human DNA polymerase gamma enhances DNA binding, promotes processive DNA synthesis, and confers N-ethylmaleimide resistance. J. Biol. Chem. 274, 38197–38203. doi: 10.1074/jbc.274.53.38197
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lim, S. E., Ponamarev, M. V., Longley, M. J., and Copeland, W. C. (2003). Structural determinants in human DNA polymerase gamma account for mitochondrial toxicity from nucleoside analogs. J. Mol. Biol. 23, 45–57. doi: 10.1016/S0022-2836(03)00405-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ling, F., Hori, A., and Shibata, T. (2007). DNA recombination-initiation plays a role in the extremely biased inheritance of yeast [rho−] mitochondrial DNA that contains the replication origin ori5. Mol. Cell. Biol. 27, 1133–1145. doi: 10.1128/MCB.00770-06
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ling, F., and Shibata, T. (2002). Recombination-dependent mtDNA partitioning: in vivo role of Mhr1p to promote pairing of homologous DNA. EMBO J. 21, 4730–4740. doi: 10.1093/emboj/cdf466
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Longley, M. J., Nguyen, D., Kunkel, T. A., and Copeland, W. C. (2001). The fidelity of human DNA polymerase gamma with and without exonucleolytic proofreading and the p55 accessory subunit. J. Biol. Chem. 276, 38555–38562. doi: 10.1074/jbc.M105230200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lucas, P., Lasserre, J. P., Plissonneau, J., and Castroviejo, M. (2004). Absence of accessory subunit in the DNA polymerase gamma purified from yeast mitochondria. Mitochondrion 4, 13–20. doi: 10.1016/j.mito.2004.04.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Maleszka, R., Skelly, P. J., and Clark-Walker, G. D. (1991). Rolling circle replication of DNA in yeast mitochondria. EMBO J. 10, 3923–3929.
McKinney, E. A., and Oliveira, M. T. (2013). Replicating animal mitochondrial DNA. Genet. Mol. Biol. 36, 308–315. doi: 10.1590/S1415-47572013000300002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Meeusen, S., and Nunnari, J. (2003). Evidence for a two membrane-spanning autonomous mitochondrial DNA replisome. J. Cell. Biol. 163, 503–510. doi: 10.1083/jcb.200304040
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nordlund, P., and Reichard, P. (2006). Ribonucleotide reductases. Annu. Rev. Biochem. 75, 681–706. doi: 10.1146/annurev.biochem.75.103004.142443
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Phadnis, N., and Sia, E. A. (2004). Role of the putative structural protein Sed1p in mitochondrial genome maintenance. J. Mol. Biol. 342, 1115–1129. doi: 10.1016/j.jmb.2004.07.096
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Phadnis, N., Sia, R. A., and Sia, E. A. (2005). Analysis of repeat-mediated deletions in the mitochondrial genome of Saccharomyces cerevisiae. Genetics 171, 1549–1559. doi: 10.1534/genetics.105.047092
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Putrament, A., Branowska, H., Ejchart, A., and Prazmo, W. (1975). Manganese mutagenesis in yeast. A practical application of manganese for the induction of mitochondrial antibiotic-resistant mutations. J. Gen. Microbiol. 90, 265–270. doi: 10.1099/00221287-90-2-265
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Qian, Y., Kachroo, A. H., Yellman, C. M., Marcotte, E. M., and Johnson, K. A. (2014). Yeast cells expressing the human mitochondrial DNA polymerase reveal correlations between polymerase fidelity and human disease progression. J. Biol. Chem. 289, 5970–5985. doi: 10.1074/jbc.M113.526418
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Reenan, R. A., and Kolodner, R. D. (1992). Characterization of insertion mutations in the Saccharomyces cerevisiae MSH1 and MSH2 genes: evidence for separate mitochondrial and nuclear functions. Genetics 132, 975–985.
Richardson, J. P., and Parker, S. R. (1973). Letter: effect of ethidium bromide on transcription of linear and circular DNA templates. J. Mol. Biol. 78, 715–720. doi: 10.1016/0022-2836(73)90291-X
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Roos, S., Macao, B., Fuste, J. M., Lindberg, C., Jemt, E., Hoome, E., et al. (2013). Subnormal levels of Pol gamma A cause inefficeint initiation of light-strand DNA synthesis and lead to mitochondrial DNA deletions and autosomal dominant progressive external ophthalmoplegia. Hum. Mol. Genet. 22, 2411–2422. doi: 10.1093/hmg/ddt094
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ropp, P. A., and Copeland, W. C. (1995). Characterization of a new DNA polymerase from Schizosaccharomyces pombe: a probable homologue of the Saccharomyces cerevisiae DNA polymerase gamma. Gene 165, 103–107. doi: 10.1016/0378-1119(95)00412-Y
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ropp, P. A., and Copeland, W. C. (1996). Cloning and characterization of the human mitochondrial DNA polymerase, DNA polymerase gamma. Genomics 36, 449–458. doi: 10.1006/geno.1996.0490
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schacherer, J., Ruderfer, D. M., Gresham, D., Dolinski, K., Botstein, D., and Kruglyak, L. (2007). Genome-wide analysis of nucleotide-level variation in commonly used Saccharomyces cerevisiae strains. PLoS ONE 2:e322. doi: 10.1371/journal.pone.0000322
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sen, S., Mukhopadhyay, S., Wetzel, J., and Biswas, T. K. (1994). Characterization of the mitochondrial DNA polymerase from Saccharomyces cerevisiae. Acta Biochim Pol. 41, 79–86.
Shadel, G. S., and Clayton, D. A. (1997). Mitochondrial DNA maintenance in vertebrates. Annu. Rev. Biochem. 66, 409–435. doi: 10.1146/annurev.biochem.66.1.409
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Slonimski, P. P., and Lazowska, J. (1977). “Transposable segments of mitochondrial DNA: a unitary hypothesis for the mechanism of mutation, recombination, sequence reiteration and suppressiveness of yeast “petite colony” mutants,” in Mitochondria 1977: Genetics and Biogenesis of Mitochondria, eds W. Bandlow, R. J. Schweyen, K. Wolf, and F. Kaudewitz (Berlin: Walter de Gruyter & Co.), 39–52.
Slonimski, P. P., Perrodin, G., and Croft, J. H. (1968). Ethidium bromide induced mutation of yeast mitochondria: complete transformation of cells into respiratory deficient non-chromosomal “petites.” Biochem. Biophys. Res. Comm. 30, 232–239. doi: 10.1016/0006-291X(68)90440-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sor, F., and Fukuhara, H. (1984). Erythromycin and spiramycin resistance mutations of yeast mitochondria: nature of the rib2 locus in the large ribosomal RNA gene. Nucleic Acids Res. 12, 8313–8318. doi: 10.1093/nar/12.22.8313
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Spinazzola, A., Invernizzi, F., Carrara, F., Lamantea, E., Donati, A., Dirocco, M., et al. (2009). Clinical and molecular features of mitochondrial DNA depletion syndromes. J. Inherit. Metab. Dis. 32, 143–158. doi: 10.1007/s10545-008-1038-z
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stewart, J. D., Horvath, R., Baruffini, E., Ferrero, I., Bulst, S., Watkins, P. B., et al. (2010). Polymerase γ gene POLG determines the risk of sodium valproate-induced liver toxicity. Hepatology 52, 1791–1796. doi: 10.1002/hep.23891
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stricker, S., Prüss, H., Horvath, R., Baruffini, E., Lodi, T., Siebert, E., et al. (2009). A variable neurodegenerative phenotype with polymerase gamma mutation. J. Neurol. Neurosurg. Psychiatry 80, 1181–1182. doi: 10.1136/jnnp.2008.166066
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stuart, G. R., Santos, J. H., Strand, M. K., Van Houten, B., and Copeland, W. C. (2006). Mitochondrial and nuclear DNA defects in Saccharomyces cerevisiae with mutations in DNA polymerase gamma associated with progressive external ophthalmoplegia. Hum. Mol. Genet. 15, 363–374. doi: 10.1093/hmg/ddi454
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stumpf, J. D., Bailey, C. M., Spell, D., Stillwagon, M., Anderson, K. S., and Copeland, W. C. (2010). mip1 containing mutations associated with mitochondrial disease causes mutagenesis and depletion of mtDNA in Saccharomyces cerevisiae. Hum. Mol. Genet. 19, 2123–2133. doi: 10.1093/hmg/ddq089
Stumpf, J. D., and Copeland, W. C. (2011). Mitochondrial DNA replication and disease: insights from DNA polymerase γ mutations. Cell. Mol. Life. Sci. 68, 219–233. doi: 10.1007/s00018-010-0530-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stumpf, J. D., and Copeland, W. C. (2013). The exonuclease activity of the yeast mitochondrial DNA polymerase γ suppresses mitochondrial DNA deletions between short direct repeats in Saccharomyces cerevisiae. Genetics 194, 519–522. doi: 10.1534/genetics.113.150920
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stumpf, J. D., and Copeland, W. C. (2014). MMS Exposure Promotes Increased MtDNA Mutagenesis in the Presence of Replication-Defective Disease-Associated DNA Polymerase γ Variants. PLoS Genet. 10:e1004748. doi: 10.1371/journal.pgen.1004748
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Szczepanowska, K., and Foury, F. (2010). A cluster of pathogenic mutations in the 3′-5′ exonuclease domain of DNA polymerase gamma defines a novel module coupling DNA synthesis and degradation. Hum. Mol. Genet. 19, 3516–3529. doi: 10.1093/hmg/ddq267
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Taanman, J. W., Rahman, S., Pagnamenta, A. T., Morris, A. A. M., Bitner-Glindzicz, M., Wolf, N. I., et al. (2008). Analysis of mutant DNA Polymerase gamma in patients with mitochondrial DNA depletion. Hum. Mutat. 30, 248–254. doi: 10.1002/humu.20852
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Trifunovic, A., Wredenberg, A., Falkenberg, M., Spelbrink, J. N., Rovio, A. T., Bruder, C. E., et al. (2004). Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429, 417–423. doi: 10.1038/nature02517
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vanderstraeten, S., Van den Brûle, S., Hu, J., and Foury, F. (1998). The role of 3′-5′ exonucleolytic proofreading and mismatch repair in yeast mitochondrial DNA error avoidance. J. Biol. Chem. 273, 23690–23697. doi: 10.1074/jbc.273.37.23690
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Van Goethem, G., Dermaut, B., Lofgren, A., Martin, J. J., and Van Broeckhoven, C. (2001). Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat. Genet. 28, 211–212. doi: 10.1038/90034
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Viikov, K., Jasnovidova, O., Tamm, T., and Sedman, J. (2012). C-terminal extension of the yeast mitochondrial DNA polymerase determines the balance between synthesis and degradation. PLoS ONE 7:e33482. doi: 10.1371/journal.pone.0033482
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Viikov, K., Väljamäe, P., and Sedman, J. (2011). Yeast mitochondrial DNA polymerase is a highly processive single-subunit enzyme. Mitochondrion 11, 119–126. doi: 10.1016/j.mito.2010.08.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wintersberger, U., and Blutsch, H. (1976). DNA-dependent DNA polymerase from yeast mitochondria. Dependence of enzyme activity on conditions of cell growth, and properties of the highly purified polymerase. Eur. J. Biochem. 68, 199–207. doi: 10.1111/j.1432-1033.1976.tb10779.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wintersberger, U., and Wintersberger, E. (1970). Studies on deoxyribonucleic acid polymerases from yeast. 2. Partial purification and characterization of mitochondrial DNA polymerase from wild type and respiration-deficient yeast cells. Eur. J. Biochem. 13, 20–27. doi: 10.1111/j.1432-1033.1970.tb00894.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Xu, X., Page, J. L., Surtees, J. A., Liu, H., Lagedrost, S., Lu, Y., et al. (2008). Broad overexpression of ribonucleotide reductase genes in mice specifically induces lung neoplasms. Cancer Res. 68, 2652–2660. doi: 10.1158/0008-5472.CAN-07-5873
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yakubovskaya, E., Chen, Z., Carrodeguas, J. A., Kisker, C., and Bogenhagen, D. F. (2006). Functional human mitochondrial DNA polymerase gamma forms a heterotrimer. J. Biol. Chem. 281, 374–382. doi: 10.1074/jbc.M509730200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yamanaka, H., Gatanaga, H., Kosalaraksa, P., Matsuoka-Aizawa, S., Takahashi, T., Kimura, S., et al. (2007). Novel mutation of human DNA polymerase gamma associated with mitochondrial toxicity induced by anti-HIV treatment. J. Infect. Dis. 195, 1419–1425. doi: 10.1086/513872
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Young, M. J., and Court, D. A. (2009). Effects of the S288c genetic background and common auxotrophic markers on mitochondrial DNA function in Saccharomyces cerevisiae. Yeast 25, 903–912. doi: 10.1002/yea.1644
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Young, M. J., Theriault, S. S., Li, M., and Court, D. A. (2006). The carboxyl-terminal extension on fungal mitochondrial DNA polymerases: identification of a critical region of the enzyme from Saccharomyces cerevisiae. Yeast 23, 101–116. doi: 10.1002/yea.1344
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zeviani, M., Servidei, S., Gellera, C., Bertini, E., DiMauro, S., and DiDonato, S. (1989). An autosomal dominant disorder with multiple deletions of mitochondrial DNA starting at the D-loop region. Nature 339, 309–311. doi: 10.1038/339309a0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhang, H., Chatterjee, A., and Singh, K. K. (2006). Saccharomyces cerevisiae polymerase zeta functions in mitochondria. Genetics 172, 2683–2688. doi: 10.1534/genetics.105.051029
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhao, X., Georgieva, B., Chabes, A., Domkin, V., Ippel, J. H., Schleucher, J., et al. (2000). Mutational and structural analyses of the ribonucleotide reductase inhibitor Sml1 define its Rnr1 interaction domain whose inactivation allows suppression of mec1 and rad53 lethality. Mol. Cell. Biol. 20, 9076–9083. doi: 10.1128/MCB.20.23.9076-9083.2000
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhao, X., Muller, E. G., and Rothstein, R. (1998). A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol Cell. 2, 329–340. doi: 10.1016/S1097-2765(00)80277-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: DNA polymerase γ, Mip1, Mip1 interactions, Pol γ mutations, yeast model
Citation: Lodi T, Dallabona C, Nolli C, Goffrini P, Donnini C and Baruffini E (2015) DNA polymerase γ and disease: what we have learned from yeast Front. Genet. 6:106. doi: 10.3389/fgene.2015.00106
Received: 17 December 2014; Paper pending published: 27 January 2015;
Accepted: 02 March 2015; Published: 17 March 2015.
Edited by:
Ming Qi, Zhejiang University Medical School, ChinaReviewed by:
Qingfeng Yan, Zhejiang University, ChinaYufeng Qian, University of Texas at Austin, USA
Kenneth Johnson, University of Texas at Austin, USA
Monique Bolotin-Fukuhara, Université Paris Sud, France
William Copeland, National Institutes of Health, USA
Jean-Paul Di Rago, CNRS–Bordeaux University, France
Copyright © 2015 Lodi, Dallabona, Nolli, Goffrini, Donnini and Baruffini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Enrico Baruffini, Department of Life Sciences, University of Parma, Parco Area delle Scienze 11/A, 43124 Parma, ItalyZW5yaWNvLmJhcnVmZmluaUB1bmlwci5pdA==
†These authors have contributed equally to this work.