 Ya-Wen Lu
Ya-Wen Lu Steven M. Claypool
Steven M. Claypool
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet., 03 February 2015
Sec. Genetics of Common and Rare Diseases
Volume 6 - 2015 | https://doi.org/10.3389/fgene.2015.00003
This article is part of the Research TopicImpact of nuclear genetic variants on mitochondrial pathophysiologyView all 20 articles
The human nuclear and mitochondrial genomes co-exist within each cell. While the mitochondrial genome encodes for a limited number of proteins, transfer RNAs, and ribosomal RNAs, the vast majority of mitochondrial proteins are encoded in the nuclear genome. Of the multitude of mitochondrial disorders known to date, only a fifth are maternally inherited. The recent characterization of the mitochondrial proteome therefore serves as an important step toward delineating the nosology of a large spectrum of phenotypically heterogeneous diseases. Following the identification of the first nuclear gene defect to underlie a mitochondrial disorder, a plenitude of genetic variants that provoke mitochondrial pathophysiology have been molecularly elucidated and classified into six categories that impact: (1) oxidative phosphorylation (subunits and assembly factors); (2) mitochondrial DNA maintenance and expression; (3) mitochondrial protein import and assembly; (4) mitochondrial quality control (chaperones and proteases); (5) iron–sulfur cluster homeostasis; and (6) mitochondrial dynamics (fission and fusion). Here, we propose that an additional class of genetic variant be included in the classification schema to acknowledge the role of genetic defects in phospholipid biosynthesis, remodeling, and metabolism in mitochondrial pathophysiology. This seventh class includes a small but notable group of nuclear-encoded proteins whose dysfunction impacts normal mitochondrial phospholipid metabolism. The resulting human disorders present with a diverse array of pathologic consequences that reflect the variety of functions that phospholipids have in mitochondria and highlight the important role of proper membrane homeostasis in mitochondrial biology.
The mitochondrion is the primary generator of adenosine triphosphate (ATP) in eukaryotes. In addition to oxidative phosphorylation (OXPHOS), the mitochondrion is involved in a wide range of essential cellular processes. The organelle is the home for the tricarboxylic acid cycle, fatty acid beta-oxidation, iron–sulfur cluster biogenesis, a portion of the urea cycle, and steps in the porphyrin and pyrimidine biosynthetic pathways. Moreover, mitochondria have important roles in Ca2+ buffering and thus, Ca2+ signaling, are a major producer (a consequence of OXPHOS) and scavenger of reactive oxygen species (ROS), and are intimately involved in programmed cell death (Kroemer et al., 1995; Green and Kroemer, 2004). Finally, the mitochondrion directly contributes to cellular phospholipid metabolism by hosting machinery that can produce at least four distinct phospholipids: phosphatidic acid (PA), phosphatidylglycerol (PG), cardiolipin (CL), and phosphatidylethanolamine (PE). Given the myriad of roles the mitochondrion plays in maintaining cellular homeostasis, it is no surprise that defects in mitochondrial function lead to a broad spectrum of diseases (Wong, 2007).
Mitochondrial disorders, first recognized in Luft et al. (1962), are the most common source of inborn errors of metabolism (Naviaux, 2004; Schaefer et al., 2008; Foundation, 2014). Primary mitochondrial disorders can be caused by mutation of genes encoded by either mitochondrial (mt)DNA or nuclear (n)DNA (but whose protein product resides in the mitochondrion). Thus, while the discovery of pathogenic mtDNA mutations in the 1980s greatly facilitated understanding of maternally-inherited OXPHOS disease (Holt et al., 1988; Wallace et al., 1988), the clinical and genetic heterogeneity of mitochondrial disease still complicates efforts to diagnose, manage, and treat affected patients (Wong, 2007; Calvo and Mootha, 2010; DaRe et al., 2013).
Human mtDNA encodes 13 OXPHOS subunits, 22 transfer RNAs, and two ribosomal RNAs (Anderson et al., 1981). In contrast, 99% of the mitochondrial proteome, which consists of over 1000 proteins (Mootha et al., 2003; Taylor et al., 2003; Zahedi et al., 2006; Johnson et al., 2007; Pagliarini et al., 2008; Calvo and Mootha, 2010; Calvo et al., 2012; Rhee et al., 2013; Hung et al., 2014), is encoded by the nuclear genome. Thus, not surprisingly, disorders that are caused by pathogenic mtDNA mutations [identified in 30 of the 37 mtDNA-encoded proteins (Ye et al., 2014b)] only comprise a fifth of the known mitochondrial diseases (Dimauro and Davidzon, 2005).
Mitochondrial dysfunction is not only defined by specific defects in mtDNA or in OXPHOS-associated processes. Mutations in enzymes involved in any number of the mitochondrion’s essential functions can lead to mitochondrial disease. Genetic variants that drive mitochondrial pathophysiology have been broadly classified into six categories: (1) OXPHOS-related (subunits and assembly factors; Ugalde et al., 2009); (2) mtDNA maintenance and expression (Jacobs and Turnbull, 2005); (3) mitochondrial biogenesis (Perez-Martinez et al., 2008); (4) mitochondrial quality control (chaperones and proteases; Koppen and Langer, 2007); (5) iron–sulfur cluster homeostasis (Lill et al., 2012); and (6) mitochondrial dynamics (fission and fusion; Chen and Chan, 2009). These categories are delineated by the pathways in which the impacted proteins partake but do not predict in any way known, the clinical presentation caused by any given genetic mutation.
Here, we propose that an additional class of genetic variants be included in the classification schema to acknowledge the role of defects in mitochondrial phospholipid metabolism as a cause of mitochondrial disease. This seventh class will encompass a small but notable group of Mendelian-inherited disorders that specifically impact normal mitochondrial phospholipid metabolism and thus highlight the important role of proper membrane homeostasis in mitochondrial physiology. PA, PG, CL, and PE have parts, if not all, of their biosynthetic pathways localized to the mitochondrion. Combined with the essential import of extra-mitochondrial phosphatidylcholine, phosphatidylserine and phosphatidylinositol, the mitochondrion requires and maintains a highly articulated lipid trafficking network. Therefore, it is no surprise that disruption of mitochondrial phospholipid metabolism can lead to mitochondrial dysfunction.
While the mitochondrion hosts a major PE biosynthetic pathway, to date, an inherited disease directly impinging on mitochondrial PE metabolism has not been demonstrated. As such, the biology of this important phospholipid will not be covered here. Instead, this review will only focus on those phospholipids which are impacted by mutations in genes encoded by the nuclear genome with defined or emerging roles in mitochondrial phospholipid metabolism.
Each mitochondrion has two highly specialized membranes, the outer mitochondrial membrane (OMM) and inner mitochondrial membrane (IMM), delineating two aqueous compartments, a dense internal matrix and an intermembrane space (IMS). Characteristic to mitochondrial membranes is a low phospholipid to protein ratio relative to other organelle membranes, high PC and PE content, cumulatively accounting for up to 80% of total lipid phosphorous, low amounts of sterols and sphingolipids, and enrichment of a unique phospholipid, CL (Horvath and Daum, 2013).
Commonly known as the signature phospholipid of mitochondria, CL is a phospholipid dimer that consists of a pair of PAs, each with two fatty acyl chains, bridged by a central glycerol moiety (Lecocq and Ballou, 1964; Schlame et al., 2000). The presence of four acyl chains per molecule provides the opportunity for an incredible diversity of CL molecular forms while the two phosphate groups confer upon the lipid a net –1 charge at physiological pH (Haines and Dencher, 2002). CL, like PE and PA, is a non-bilayer forming phospholipid; these lipids have much smaller hydrophilic head group diameters than their hydrophobic acyl chains, making them cone-shaped. Non-bilayer lipids participate in membrane fusion events (Verkleij et al., 1984), facilitate membrane bending (Gruner, 1985), and can impart order to surrounding lipids (Zeczycki et al., 2014). This distinguishes them from bilayer-forming lipids that have head groups and acyl chains that are of similar diameter.
Each mitochondrial membrane has a distinct protein population and phospholipid composition (Colbeau et al., 1971; Zinser et al., 1991; Zinser and Daum, 1995; Daum and Vance, 1997; Mootha et al., 2003; Taylor et al., 2003; Zahedi et al., 2006; Johnson et al., 2007; Pagliarini et al., 2008; Gebert et al., 2009; Rhee et al., 2013; Hung et al., 2014). In fact, the protein to phospholipid ratio differs significantly between the IMM and OMM, with the IMM being significantly more proteinaceous (Sperka-Gottlieb et al., 1988; Ardail et al., 1990; Simbeni et al., 1991). When purified, OMM and IMM vesicles have different shapes, structures, and lipid compositions (Hovius et al., 1990; Gebert et al., 2009). Notably, the OMM has relatively less CL than the IMM (Daum and Vance, 1997; de Kroon et al., 1997). Even within a membrane bilayer, there is asymmetry in the lipid composition between the two leaflets (Krebs et al., 1979; Harb et al., 1981; Cheneval et al., 1985; Sperka-Gottlieb et al., 1988; Hovius et al., 1993; Gallet et al., 1997). For instance, most of the associated PE and all of the low amounts of CL are on the cytoplasmic-facing side of OMM (Hovius et al., 1993).
The high protein to phospholipid ratio in the IMM reflects the sheer magnitude of essential processes that occur in its context. Embedded in the IMM is the OXPHOS system, three distinct translocation machineries (TIM22, TIM23, and OXA1), carrier proteins that mediate the flux of metabolites across the IMM, quality control proteases, and phospholipid metabolizing enzymes. In spite of this protein density, the IMM is an intact diffusion barrier that enforces the proton gradient generated by the electron transport chain. The stored power of the electrochemical gradient across the IMM is central to mitochondrial biology. Not only is it used as the source of energy for ATP production and harnessed to drive numerous transport processes, but in fact, the electrochemical gradient is required for mitochondrial biogenesis itself (Chacinska et al., 2009). Therefore, maintaining a proper lipid composition is likely not only required to support the functionality of the numerous proteins and protein complexes embedded in the IMM but also to maintain its crucial barrier function.
Cardiolipin biosynthesis begins with PA, a common substrate in triacylglycerol and glycerolipid metabolism. Reflecting this central role, PA can be made in a multitude of ways. Worth mentioning from the outset is that the source of PA used for CL biosynthesis has yet to be experimentally established; given the apparent redundancy, we anticipate that more than one PA-producing pathway will be at play.
De novo biosynthesis of PA is catalyzed by glycerophosphate acyltransferases (GPATs) that are localized to the mitochondrial OMM (GPAT1/2; Yet et al., 1993; Lewin et al., 2004; Wang et al., 2007) or ER (GPAT3/4; Chen et al., 2008; Nagle et al., 2008). GPATs transfer acyl groups from acyl-CoA donors to the sn-1 position of glycerol-3-phosphate generating lyso-PA (LPA). Whether and how the mitochondrial and ER GPAT isoforms contribute to mitochondrial phospholipid metabolism is currently unresolved. The LPA that is generated on either the OMM or within the mitochondria-associated membrane (MAM, Vance, 1990; Gaigg et al., 1995) subcompartment of the ER can be acylated by LPA acyltransferases (LPAATs). In mammals, there are four LPAAT isoforms that differ in their tissue distribution and acyl-CoA specificities (some LPAATs can additionally acylate other lyso-lipids; West et al., 1997; Aguado and Campbell, 1998; Lu et al., 2005; Schmidt et al., 2010; Prasad et al., 2011; Eto et al., 2014). In addition to the defined LPAATs, other lyso-phospholipid acyltransferases are able to esterify LPA to some degree (Agarwal, 2012).
Another potential source of PA is mitochondrial phospholipase D (MitoPLD), a divergent member of the PLD superfamily localized to the OMM that can hydrolyze CL to PA in vitro (Choi et al., 2006). Similar to yeast, mammals can also produce PA via the dihydroxyacetone phosphate pathway in the peroxisome (Hajra and Bishop, 1982). All of the discussed pathways of PA synthesis are located outside of, or on the outside of, mitochondria. However, the recent identification of acylglycerol kinase (AGK) in the IMS suggests that PA may be produced inside the mitochondrion (Hung et al., 2014). AGK, previously termed MuLK for multi-substrate lipid kinase (Waggoner et al., 2004), phosphorylates diacylglycerol (DAG) and monoacylglycerol generating PA and LPA (Waggoner et al., 2004; Bektas et al., 2005). AGK activity is modulated by surface charge (via Mg2+) and stimulated, in a dose-dependent manner, by CL (Waggoner et al., 2004). Moreover, overexpression of AGK in the human prostate cancer PC-3 model results in increased mitochondrial PA although CL levels are unchanged (Bektas et al., 2005). Thus, whether PA made by AGK inside of the mitochondrion can and does access the downstream CL biosynthetic machinery is unclear.
Upon gaining access to the matrix side of the IMM (discussed later), PA is converted to cytidine diphosphate-DAG (CDP-DAG) and pyrophosphate upon reaction with cytidine triphosphate (CTP; Figure 1). The reaction is catalyzed by CDP-DAG synthases (CDSs), whose activities have been demonstrated in yeast ER and mitochondria (Kuchler et al., 1986; Shen et al., 1996). It has been recently established that the conserved IMM resident Tam41p is the mitochondrial CDP-DAG synthase whose activity provides CDP-DAG for CL biosynthesis (Tamura et al., 2006, 2013; Kutik et al., 2008). Accordingly, CDS proteins in the ER provide CDP-DAG that is used for phospholipid biosynthesis therein while TAMM41 supplies the CL pathway (Horvath and Daum, 2013).
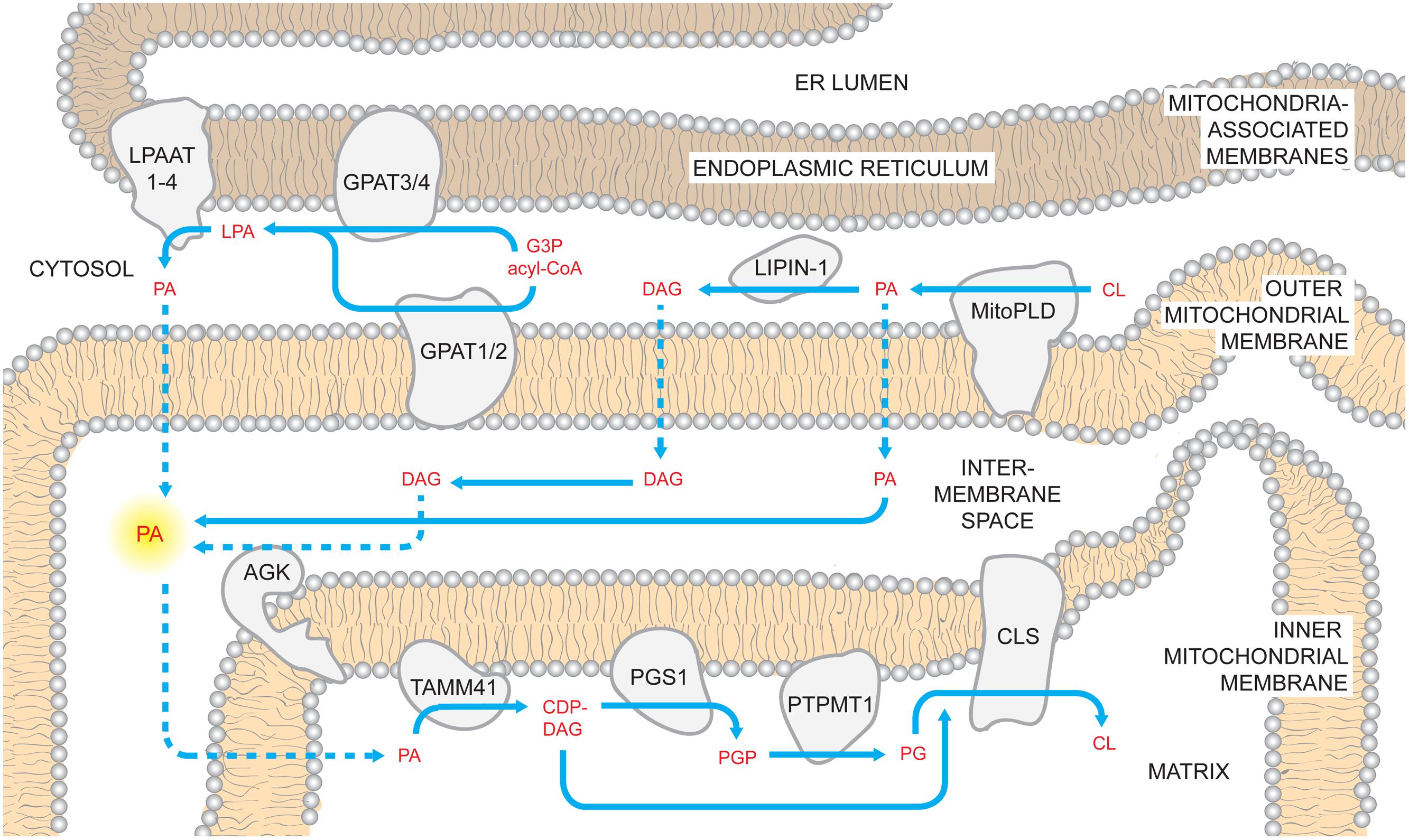
FIGURE 1. Mammalian cardiolipin biosynthesis. CL biosynthesis likely involves PA sourced from multiple pathways. PA can be generated by LPAATs which acylate LPA; LPA can be made using glycerol 3-phosphate (G3P) and acyl-CoAs by mitochondrial and ER GPATs. Additionally, PA can be made on the OMM through the hydrolysis of CL by MitoPLD. In the OMM, PA recruits the phosphatase lipin-1 which dephosphorylates PA into DAG. In turn, DAG may traffic across the OMM and be phosphorylated by AGK forming PA in the context of the IMS-side of the IMM. Regardless of where it is made, PA must reach the matrix side of the IMM to gain access to the core CL biosynthetic machinery. Here, PA is converted to CDP-DAG by TAMM41, thus providing the precursor for the committed step in CL biosynthesis, PGP synthesis by PGS1. PGP is rapidly dephosphorylated by PTPMT1 and the produced PG is condensed with CDP-DAG by CLS generating CL. Dashed arrows describe uncharacterized steps and pathways.
Also localized on the matrix-facing leaflet of the membrane are downstream enzymes in PG and CL biosynthesis. The committed step of this pathway is catalyzed by phosphatidylglycerol phosphate (PGP) synthase (PGS1), which forms PGP from CDP-DAG and glycerol 3-phosphate (Chang et al., 1998a). After PGP is synthesized, it is rapidly dephosphorylated to PG by PTPMT1 (Xiao et al., 2011; Zhang et al., 2011) of the protein tyrosine phosphatase family that shares no primary sequence similarity to the yeast PGP phosphatase, Gep4p (Osman et al., 2010). Notably, at steady state, PG is present at extremely low levels (1–2%) relative to the other major mitochondrial phospholipids (Daum, 1985), indicating that newly synthesized PG is quickly consumed by downstream pathways. Finally, cardiolipin synthase (CLS), an integral IMM protein with its active site facing the matrix (Schlame and Haldar, 1993), condenses PG with another molecule of CDP-DAG to generate nascent unremodeled CL (Chang et al., 1998b; Chen et al., 2006; Lu et al., 2006).
Cardiolipin biosynthetic enzymes exhibit no or only limited acyl chain specificity (Chang et al., 1998b; Chen et al., 2006; Houtkooper et al., 2006). The general lack of acyl chain specificity in CL biosynthesis is significant as it is in direct contrast to the observation that in any given tissue [except the brain (Cheng et al., 2008)], there exists a dominant homogeneous molecular form of CL that is characterized by the incorporation of unsaturated fatty acyl chains (Schlame et al., 1993) and molecular symmetry with respect to the two chiral centers of CL (Schlame et al., 2005). In mammals, CL is predominantly composed of unsaturated 18-carbon linoleic acid (18:2). The enrichment in this particular species is exemplified by human heart where 18:2-linoleic acid constitutes up to 90% of the acyl chains in CL, yielding the abundant and stereotypical tetralinoleoyl-CL species (Schlame and Otten, 1991; Schlame et al., 2005). However, it is notable that the final acyl chain composition of CL in different tissues is not the same; this observation has led to the hypothesis that the final acyl chain composition of CL is tailored to the unique demands of its host tissue (Schlame et al., 2005; Han et al., 2006; Cheng et al., 2008).
As such, the generation of CL molecular species that accumulate at steady state requires the active remodeling of CL shortly after its de novo synthesis (Eichberg, 1974; Schlame and Rustow, 1990). To initiate the remodeling process, a lipase removes an acyl chain from CL generating monolyso-CL (MLCL) that is subsequently re-acylated by one of several enzymes (Figure 2). Through a series of such reactions at each position in CL, a tissue-specific homogeneous pool of CL is generated that is characterized by molecular symmetry and a higher degree of acyl chain unsaturation.
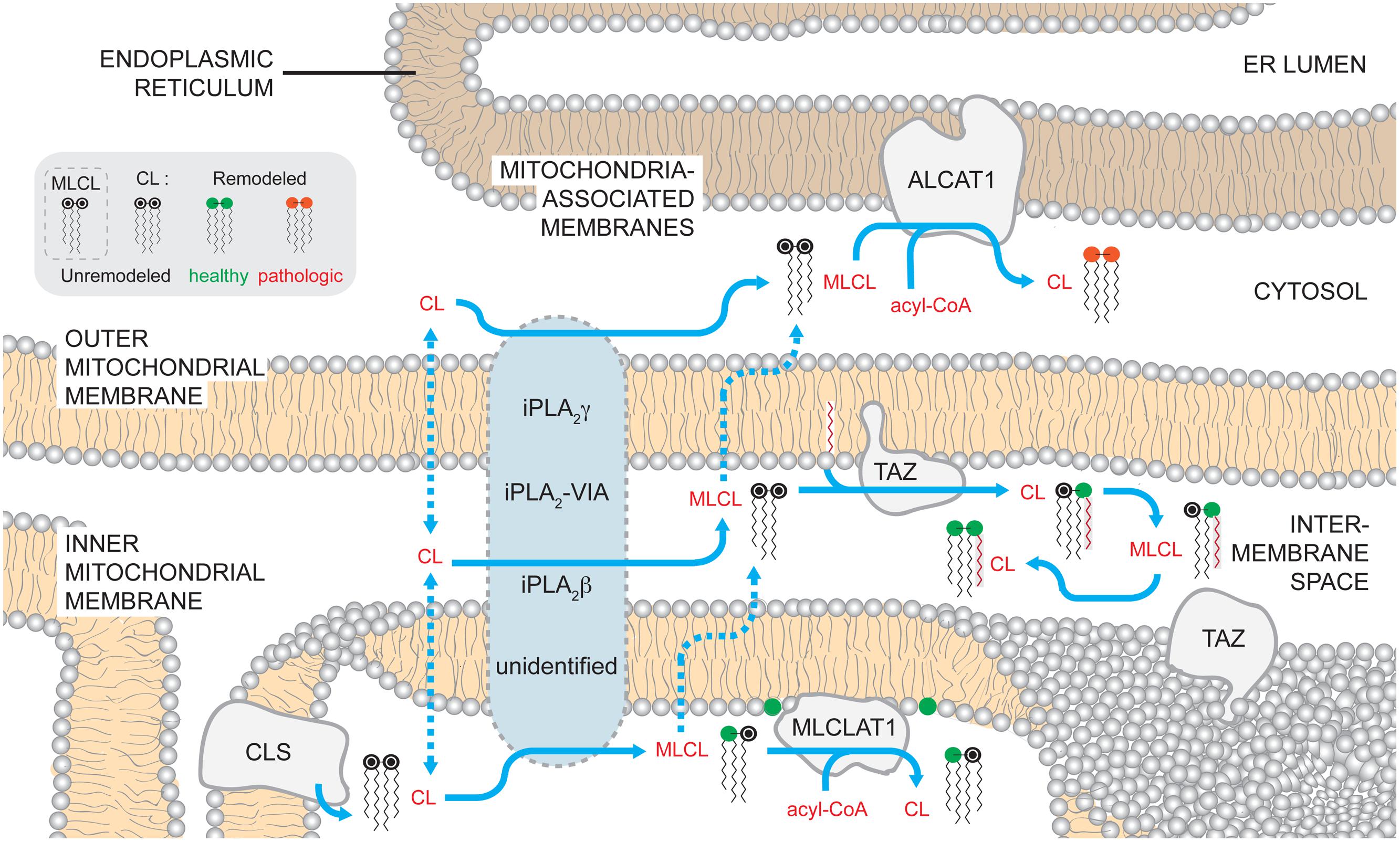
FIGURE 2. Mammalian cardiolipin remodeling. CL that is synthesized by CLS on the matrix-facing leaflet of the IMM can by remodeled by three different pathways. While no single enzyme has been demonstrated to be required to initiate CL remodeling in mammals, several phospholipases of the iPLA2 family have been demonstrated to have a role in the process. Additionally, the submitochondrial localization of the phospholipases and the mechanisms by which CL gains access to these enzymes are unknown. After a fatty acyl chain is hydrolyzed from CL, generating MLCL, MLCL can be acylated back to CL by acyltransferases or transacylases. MLCLAT1 on the matrix-leaflet of the IMM, TAZ on the IMS-facing leaflets of the OMM and IMM, and ALCAT1 on the ER MAM all have the capacity to re-acylate MLCL. The acyltransferases, ALCAT1 and MLCLAT1, use acyl-CoAs as an acyl chain donor to acylate MLCL. In contrast, the transacylase TAZ uses acyl chains donated from other phospholipids. The activity of TAZ is required to establish the steady state physiological CL molecular form. In contrast, the CL formed by ALCAT1 is more sensitive to oxidative damage and associated with pathologic states. Dashed arrows describe uncharacterized steps and pathways. In the phospholipid key, unremodeled CL corresponds to the newly synthesized CL that enters the pathway at the point of CLS (black head group). CL can undergo either physiologically relevant CL remodeling (green head group) or pathological remodeling (red head group).
In yeast, the recently identified cardiolipin deacylase, Cld1p, initiates CL remodeling and preferentially catalyzes the hydrolysis of palmitic acid (16:0) from newly synthesized CL, forming MLCL (Beranek et al., 2009; Baile et al., 2014b). In the absence of Cld1p, the acyl chain composition of CL shifts to palmitic acid (16:0) residues at the expense of palmitoleic (16:1) and oleic (18:1) acid moieties (Beranek et al., 2009; Baile et al., 2014b). Cld1p associates with the matrix-facing leaflet of the IMM and lacks any membrane spanning segments (Baile et al., 2013), placing the initiation of CL remodeling on the same side of the IMM as its biosynthesis.
Although there are no orthologs of Cld1p in higher eukaryotes, evidence points to Ca2+-independent phospholipases A2 (iPLA2) as being involved in mammalian CL remodeling. The PLA2 family of enzymes catalyzes the hydrolysis of membrane glycerophospholipids at the sn-2 position, generating free fatty acids and lyso-lipids. Interestingly, all of the PLA2 isoforms capable of hydrolyzing CL in vitro have different specificities for CL molecular species (Hsu et al., 2013).
iPLA2γ is membrane-bound, dually localized to the mitochondrion and peroxisome, and participates in CL metabolism (Mancuso et al., 2000). In the hearts and skeletal muscle of ipla2γ-/- mice, CL levels are reduced and the acyl chain pattern is altered (Mancuso et al., 2007; Yoda et al., 2010). However, the absence of iPLA2γ in TAZ knockdown mice does not prevent the accumulation of MLCL as predicted if iPLA2γ functions immediately upstream of TAZ (Kiebish et al., 2013), the major physiological CL remodeling enzyme.
Ablation of another mitochondrially localized iPLA2 family member, iPLA2-VIA, in taz-/- flies, partially restores MLCL:CL ratios, a biochemical hallmark of TAZ dysfunction, and rescues male sterility (Houtkooper et al., 2009a,b; Malhotra et al., 2009a). Still, the CL acyl chain pattern in single ipla2-via-/- flies is not significantly different from wild type (wt) flies, suggesting that like murine iPLA2γ, iPLA2-VIA activity is not obligately required to initiate the remodeling process. Thus, which phospholipase(s) functions upstream of mammalian TAZ remains an open and important question. Once identified, basic biochemical and cell biological characterization will further establish the topology of CL remodeling. For instance, it is anticipated that the lipase(s) that initiates CL remodeling will reside in the same compartment as the enzyme that subsequently re-acylates MLCL; it is worthwhile to note that although iPLA2γ localizes to the mitochondrion, its submitochondrial distribution has not been formally demonstrated. If the lipase and its substrate do not co-localize, then appropriate trafficking steps of the lipid substrate (CL or MLCL) will be inferred. A consideration of the basic cell biology of the lipase(s) required for CL remodeling is particularly relevant given that all three enzymes that can re-acylate MLCL reside in distinct cellular compartments.
In mammals, there are at least three enzymes that have the capacity to re-acylate MLCL: TAZ, MLCL acyltransferase 1 (MLCLAT1), and acyl-CoA:lysocardiolipin acyltransferase-1 (ALCAT1), of which only TAZ is evolutionarily conserved from yeast to higher eukaryotes (Taylor and Hatch, 2003, 2009; Xu et al., 2006b; Claypool and Koehler, 2012). All evidence to date indicates that the MLCL transacylase, TAZ, is responsible for the vast majority of physiological CL remodeling. TAZ deficiency is biochemically characterized by increased MLCL, the remodeling intermediate, decreased CL, both the substrate and product, and an abnormal acyl chain pattern of the remaining CL (Vaz et al., 2003; Gu et al., 2004; Valianpour et al., 2005; Claypool et al., 2006, 2011; Houtkooper et al., 2009a). However, the mechanism by which TAZ establishes the steady state CL acyl chain composition is unresolved; TAZ catalyzes a reversible transacylation that exhibits no intrinsic acyl chain specificity, acts on acyl chains at both sn-1 and sn-2 positions, and can use any number of phospholipids and their lyso-derivatives as fatty acyl donors and acceptors, respectively (Xu et al., 2006b; Malhotra et al., 2009b). Notably, heterologous expression of human TAZ in taz-/- flies or Δtaz1 yeast generates CL with acyl chain patterns typical of Drosophila melanogaster and yeast, respectively, and not humans (Vaz et al., 2003; Xu et al., 2009). This suggests that the characteristic fatty acid profile of CL may not be conferred by the substrate specificity of TAZ. Interestingly, recombinant fly TAZ can generate the physiologically relevant tetralinoleoyl-CL species from MLCL but only under experimental conditions that promote non-bilayer membranes (Schlame et al., 2012a). This suggests that unique membrane physical states such as regions of high curvature, can confer upon TAZ acyl chain specificity. Other mechanisms that are hypothesized to play a role in the TAZ-based establishment of CL molecular species include the specificity of enzymes immediately upstream of TAZ that initiate the remodeling process and thus dictate which substrates are available to TAZ (Baile et al., 2014b), as well as the dietary intake of fatty acids (Stavrovskaya et al., 2013).
Presently, the submitochondrial localization of endogenous mammalian TAZ has not been documented. Yeast Taz1p associates with the IMS-facing leaflets of the OMM and the IMM as an interfacial protein (Brandner et al., 2005; Claypool et al., 2006; Gebert et al., 2009). This is in sharp contrast to the deacylase, Cld1p that initiates CL remodeling and is localized to the matrix-facing IMM leaflet (Baile et al., 2013). Topologically, this means that MLCL produced on the matrix side of the IMM in yeast has to flip across the membrane and/or be transported to the OMM, to access TAZ for remodeling (Baile et al., 2014a). As TAZ is the only CL remodeling enzyme that is conserved in eukaryotes, it is anticipated that mammalian TAZ will be similarly localized in the organelle. Therefore, determining the submitochondrial localization of the upstream phospholipase in mammals will establish the CL and MLCL trafficking steps needed to access TAZ for remodeling (Baile et al., 2014a).
In mammals, MLCLAT1, which associates with the matrix side of the IMM (Carpenter et al., 1992), does not encounter the same problem of substrate access as TAZ. MLCLAT1 was originally identified in porcine liver mitochondria as a 74-kDa protein (Taylor and Hatch, 2003) that corresponds to the COOH terminus of the human trifunctional protein (TFP) alpha (α) (Taylor and Hatch, 2009). The TFP complex consists of α- and β-subunits that catalyze the last three steps of mitochondrial long-chain fatty acid beta-oxidation thus providing a significant source of cellular energy (Carpenter et al., 1992; Uchida et al., 1992). Pathogenic mutations in either TFP subunit are associated with beta-oxidation disorders, where patients suffer from cardiomyopathy and skeletal myopathy (IJlst et al., 1994; Sims et al., 1995; Ushikubo et al., 1996). Interestingly, recombinant TFPα is soluble when expressed alone, in contrast to its partner TFPβ, suggesting that it may contain a function independent of its association with TFPβ that may be described by MLCLAT1 (Fould et al., 2010). Presently, the gene encoding MLCLAT1 has not been definitively determined. That MLCLAT1 is likely a splice variant of TFPα is suggested by the fact that knockdown using RNAi targeting the NH2-terminal portion of TFPα, absent in MLCLAT1, reduces TFPα, but not MLCLAT1 mRNA levels (Taylor et al., 2012). However, whether RNAi targeting a shared region between TFPα and MLCLAT1 can reduce the expression of both genes has not been established.
Both recombinant human TFPα and MLCLAT1 can bind MLCL in vitro and are able to incorporate linoleoyl-(18:2), oleoyl- (18:1), and palmitoyl-(16:1) CoA into MLCL (Taylor and Hatch, 2009; Fould et al., 2010; Taylor et al., 2012). When either protein is overexpressed in Barth syndrome (BTHS) lymphoblasts, there is increased incorporation of linoleic acid in CL (Taylor and Hatch, 2009; Taylor et al., 2012). However, TFPα overexpression does not generate a CL profile that reflects its in vitro specificity. Thus, while TFPα and/or MLCLAT1 can participate in CL remodeling, especially in the absence of TAZ function when the levels of MLCL are high, the exact contribution of this acyltransferase(s) to physiological CL remodeling is unclear.
Finally, ALCAT1 acylates MLCL and dilyso-CL in vivo and can use a number of lyso-phospholipids as acyl acceptors in vitro (Cao et al., 2004, 2009; Zhao et al., 2009). Depending on the acyl acceptor, tissue type, and species, ALCAT1 has been described to preferentially incorporate long-chain unsaturated fatty acyl chains or promiscuously accept all acyl-CoA derivatives (Cao et al., 2004; Zhao et al., 2009). As such, ALCAT1 lacks the specificity expected of an enzyme with a critical role in physiological CL remodeling. Notably, ALCAT1 resides in the ER MAMs (Cao et al., 2004; Zhao et al., 2009). Therefore, for ALCAT1 to participate in CL metabolism, CL and/or MLCL must travel from the IMM to at least the external leaflet of the OMM, if the active site of ALCAT1, which has not been determined, faces the cytosol.
The absence of TAZ causes alterations in CL molecular species in every model tested to date (Vreken et al., 2000; Bissler et al., 2002; Valianpour et al., 2002, 2005; Schlame et al., 2003, 2005; Vaz et al., 2003; Gu et al., 2004; Xu et al., 2005, 2006a,b; van Werkhoven et al., 2006; Acehan et al., 2009, 2011b; Houtkooper et al., 2009a,b; Dudek et al., 2013; Gonzalvez et al., 2013; Baile et al., 2014b; Wang et al., 2014a). In contrast, the loss of MLCLAT1/TFPα or ALCAT1 does not consistently result in changes in the steady state acyl chain composition of CL (Li et al., 2010; Schlame et al., 2012b; Richter-Dennerlein et al., 2014). Thus, it is unlikely that either MLCLAT1/TFPα or ALCAT1 significantly contributes to the steady state CL acyl chain profile under normal conditions. These results further underscore the predominant role of TAZ in physiological CL remodeling.
Once made, PA must travel to the matrix side of the IMM to gain access to the CL biosynthetic machinery. If made in the ER, this requires movement of PA from ER to OMM, flipping between OMM leaflets, movement from the OMM to the IMM, and finally flipping to the matrix-facing IMM leaflet. Inter-organelle contacts have recently emerged as being critically important for mitochondrial phospholipid metabolism. In yeast, there are at least two distinct structures that contribute to the physical association of ER and mitochondria (Figure 3). The first described complex is the ER-mitochondria encounter structure (ERMES; Kornmann et al., 2009). A second ER-mitochondria tether, the ER-membrane protein complex (EMC), that is distinct from ERMES, was recently identified in yeast (Lahiri et al., 2014). When either EMC or ERMES subunits are missing, the number and length of ER-mitochondria contacts are reduced and mitochondria are unable to support growth on respiratory media (Kornmann et al., 2009, 2011; Tamura et al., 2012; Lahiri et al., 2014). Also, a synthetic mitochondria-ER tether similarly rescues the defects caused by the absence of EMC or ERMES. While EMC may play a more direct role in phospholipid trafficking (Lahiri et al., 2014), EMC and ERMES complexes likely have both overlapping and distinct functions in phospholipid transfer between the ER and mitochondria (Nguyen et al., 2012; Lahiri et al., 2014).
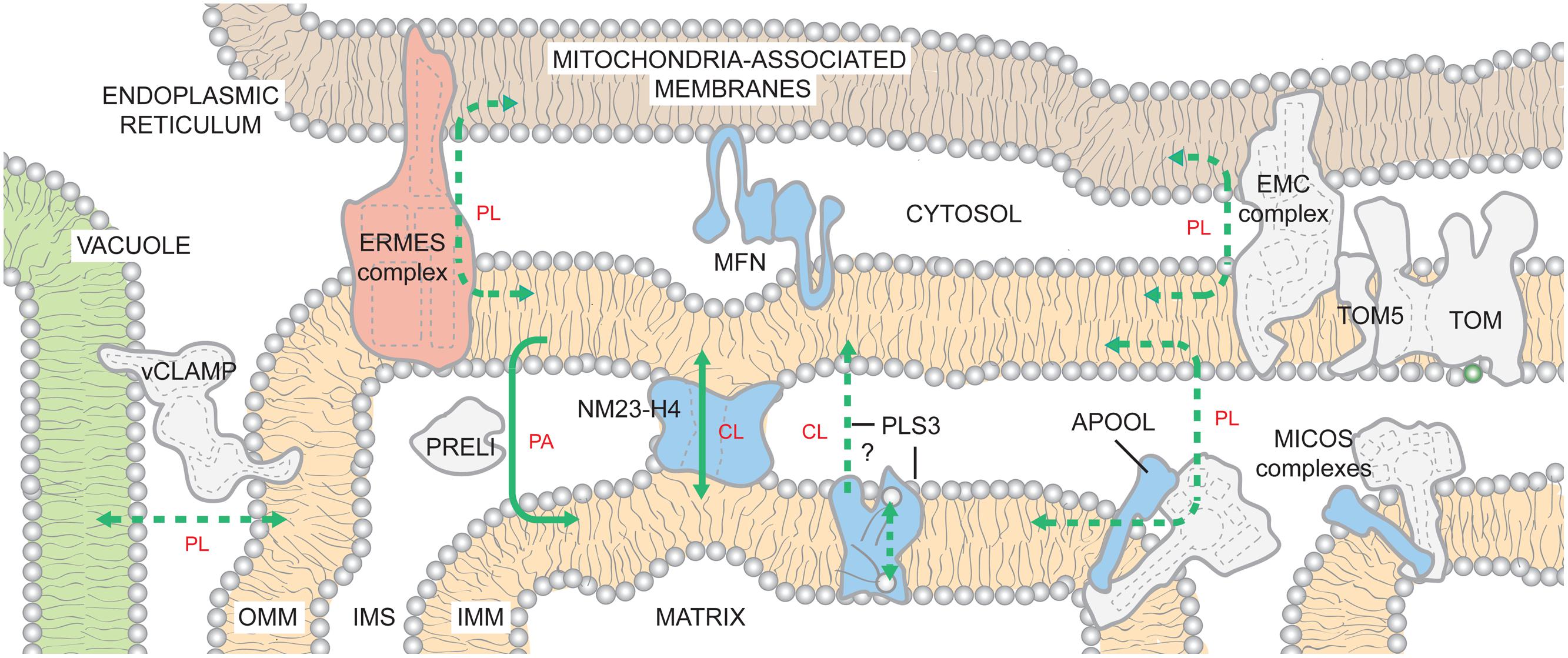
FIGURE 3. Inter-organelle and intra-organelle phospholipid trafficking. The existence of ER- and vacuole-mitochondria contacts is highly conserved from yeast to humans. By generating closely appositioned membranes, the inter-organelle and intra-organelle tethers are hypothesized to promote movement of lipids across the aqueous cytosol and IMS, respectively. Within the mitochondrion, phospholipid trafficking may involve contacts between the OMM and the IMM mediated by MICOS complexes or NM23-H4. In addition, PRELI transports PA from the OMM to the IMM. PLS3 activity stimulates CL externalization on OMM. It may directly transport CL from the IMM to the OMM or instead function as a scramblase that redistributes CL between both leaflets of the IMM. CL now exposed to IMS-side of IMM would then be transported to the OMM by other mechanisms. With the possible exception of EMC, it is presently unclear if any of the known tethers has specificity for a defined phospholipid(s); as such, they are shown to promote the flux of phospholipids (PLs) in bulk. If a specific phospholipid is impacted by mutations in a complex/protein (levels and/or composition), the lipid is indicated. Solid lines indicate known transport mechanisms. Dashed lines describe possible trafficking routes and/or highlight transport events whose mechanisms have not been resolved. The ERMES complex is found only in yeast and color-coded pink. For the remaining proteins/complexes, those found only in mammals are in blue and those that are likely to be conserved across species are colored gray. See text for additional details.
The ERMES subunits are conserved in fungi but not in metazoans. In contrast, the close apposition of ER and mitochondria is conserved (Csordas et al., 2006). In mammals, IP3 receptor/GRP75/VDAC1-containing complexes (Matsuzaki et al., 2013), mitofusin-2 (de Brito and Scorrano, 2008), and several other proteins have roles in ER-mitochondria tethering (Merkwirth and Langer, 2008; Csordas et al., 2010). Moreover, the newly described EMC is conserved but its role in ER-mitochondrion tethering has only been tested in yeast. The diversity of players implicated in this inter-organelle association provides strong evidence of the physiological importance of such contacts.
The existence of vacuolar and mitochondrial contacts, termed vCLAMPs (vacuole and mitochondrial patch), recently identified in yeast, further highlights the importance of inter-organelle associations for mitochondrial phospholipid metabolism (Elbaz-Alon et al., 2014; Honscher et al., 2014). Simultaneous deletion of ERMES and vCLAMP components is synthetically lethal. Deletion of vCLAMP and repression of ERMES impairs PA trafficking into mitochondria and results in a 40% reduction in CL levels (Elbaz-Alon et al., 2014). A recent report describing mitofusin-2-mediated contacts between mouse melanosomes (a lysosomal-like compartment in pigment cells) and mitochondria (Daniele et al., 2014), suggests that there may be similar vacuolar-tethering mechanisms in mammals.
Analogous to inter-organelle tethers, regions of close physical apposition between the IMM and OMM are important for lipid trafficking across the IMS (Ardail et al., 1990; Schlattner et al., 2014), although the underlying mechanism(s) is unknown. In addition, IMM-OMM contact sites have roles in energy transduction from the mitochondrial matrix to the cytosol (Nicolay et al., 1990) and precursor protein import (Vogel et al., 2006). MICOS (mitochondrial contact site and cristae organizing system) is a hetero-oligomeric protein complex that is embedded in the IMM but interacts with several distinct OMM proteins (Harner et al., 2011; Hoppins et al., 2011; von der Malsburg et al., 2011; van der Laan et al., 2012; Pfanner et al., 2014). Depletion of the conserved mitofilin/Fcj1p (yeast formation of cristae junction 1) subunit, which constitutes the MICOS core, results in an expanded IMM surface, dramatic loss of cristae junctions, and the remaining cristae are stacked, lamellar, and aberrantly disconnected from the OMM (John et al., 2005; Mun et al., 2010; Harner et al., 2011; von der Malsburg et al., 2011). Recently, apolipoprotein O (APOO) and APOO-like protein (APOOL) were identified as potential subunits of the bovine MICOS complex (Weber et al., 2013). The IMS-facing APOOL specifically binds CL in vitro and its knockdown results in morphological phenotypes similar to yeast MICOS mutants (Weber et al., 2013). Thus, MICOS complexes are key determinants of cristae morphology that contain subunits capable of binding CL (Harner et al., 2011; Hoppins et al., 2011; von der Malsburg et al., 2011). Whether these two properties are leveraged in the context of phospholipid trafficking between the IMM and OMM is presently unclear although it is interesting to note that biochemically isolated IMM-OMM contact sites are enriched in CL (Ardail et al., 1990; Simbeni et al., 1991).
The movement of PA from the OMM to the IMM is mediated by the IMS-resident Ups1p/PRELI-like proteins (Connerth et al., 2012; Potting et al., 2013). In vitro, the yeast Ups1p/Mdm35p dimer (Potting et al., 2010; Tamura et al., 2010) binds numerous anionic phospholipids but only transports PA (Connerth et al., 2012). The directionality of PA transport is likely conferred by the fact that Ups1p remains tightly bound to membranes containing physiological amounts of CL, leading to Ups1p’s subsequent degradation. These observations suggest a mechanism for regulating CL biosynthesis by limiting precursor trafficking between the two membranes when CL levels are bountiful (Connerth et al., 2012). Whether transport of PA by the Ups1p/PRELI-like proteins utilizes in some manner MICOS or is instead mechanistically distinct is at present unknown.
Once PA is transported to the IMM, or if synthesized in this compartment by AGK, the transbilayer movement of PA to the matrix-leaflet is required for PA to gain access to the CL biosynthetic enzymes. How this is achieved is presently not known but could involve a specific protein or protein complex. However, the requirement for a specific PA transporter does not seem obligate as a transmembrane pH gradient is sufficient to disseminate PA across both IMM leaflets (Hope et al., 1989; Gallet et al., 1999).
Since CL is made in the context of the matrix-leaflet and can eventually be exposed on the OMM, mechanisms must be present to facilitate the movement of CL intra-mitrochondrially. Scramblases are Ca2+-dependent, ATP-independent bidirectional transporters that equilibrate lipids unevenly distributed across a bilayer (Contreras et al., 2010). As such, a scramblase could serve to redistribute CL made on the matrix side, between IMM leaflets. Alternatively, phospholipid translocation between membrane leaflets may not be mediated by specific proteins, but instead facilitated by the presence of numerous transmembrane proteins (especially in the context of the IMM) in a non-specific manner, as suggested for bacterial and ER membranes (Kol et al., 2001, 2004).
Albeit minor, CL is a normal constituent of the OMM and can traffic to the OMM following certain stimuli (Gonzalvez et al., 2008; Chu et al., 2013). Phospholipid scramblase 3 (PLS3) is the only known mitochondrial scramblase (Zhou et al., 1998; Liu et al., 2003a; Van et al., 2007) and in vitro, murine and human PLS3 catalyze the Ca2+-dependent flip-flop of CL in proteoliposomes (Zhou et al., 1998). However, it is unclear whether PLS3 functions in vivo as a CL scramblase or instead mediates the movement of CL from the IMM to the OMM. PLS3 overexpression increases mitochondrial mass, CLS transcription, CL synthesis, and CL externalization to the OMM (Liu et al., 2003b; Van et al., 2007). Conversely, overexpression of a catalytically dead pls3 allele or PLS3 knockdown reduces CL externalization following UV irradiation or rotenone poisoning, respectively (Liu et al., 2003b; Chu et al., 2013). How does PLS3 activity contribute to movement of CL to the OMM? If PLS3 is a true scramblase, then the equilibration of CL between the leaflets of the IMM may be required for the subsequent ability of CL to traffic to the OMM. Alternatively, PLS3 may instead directly participate in the movement of CL between mitochondrial membranes. Future studies are needed to clarify the role of PLS3 in this process.
Another potential mechanism by which lipids can be transferred from the IMM to the OMM involves the NM23-H4/NDPK-D (nucleotide diphosphate kinase isoform D; Milon et al., 2000). NM23-H4 is the only mitochondrially targeted member of a family of NDPKs whose role in phosphotransfer is well-established. NM23-H4 has been additionally implicated in the trafficking of anionic phospholipids (in particular CL) between the IMM and the OMM (Tokarska-Schlattner et al., 2008). Interestingly, the levels of CL can functionally switch NM23-H4 between phosphotransfer and lipid transfer modes (Schlattner et al., 2013). Normally, the lipid transfer mode is inhibited by anionic lipids, including CL, and the protein operates as a nucleotide kinase. However, when CL levels are low (due to mitochondrial dysfunction), NM23-H4’s lipid transfer function is de-repressed and the protein cross-links the IMS-facing leaflets of the IMM and OMM. Subsequently, NM23-H4 facilitates the thermodynamically unfavorable movement of negatively charged lipids across the aqueous IMS.
While a role for both NM23-H4 and PLS3 in the stimulated externalization of CL on the OMM is clearly emerging, whether either or both enzymes participate in the routine processes of CL biosynthesis and remodeling is not known. Given its MAM-residence, it is tempting to speculate that NM23-H4 and/or PLS3 may be involved in ALCAT1-based CL remodeling.
Phospholipids play a myriad of roles in cellular and mitochondrial physiology that are beyond the scope of any single review. The following section is focused on recently discovered roles and guided by those physiological functions of mitochondrial lipids, that when disturbed, may contribute to human disease. Of note, the diversity of functions attributed to the discussed mitochondrial phospholipids is reflected by the vast array of pathogenic mechanisms that underlie this cohort of mitochondrial diseases.
The dynamic appearance and disappearance of PA on the OMM is a recently established determinant of mitochondrial fusion and fission. Overexpressed MitoPLD generates PA on the OMM, promoting mitochondrial fusion and subsequent aggregation (Choi et al., 2006), while loss of MitoPLD leads to fragmented mitochondria (Huang et al., 2011). PA generated by MitoPLD recruits the PA phosphatase, lipin-1. Lipin-1 dephosphorylates PA to DAG which stimulates mitochondrial fission while simultaneously removing the pro-fusogenic accumulation of PA on the OMM (Reue and Dwyer, 2009; Huang et al., 2011). Interestingly, mitopld-/- flies (Pane et al., 2007) and mice (Huang et al., 2011; Watanabe et al., 2011) have defects in the biogenesis of Piwi-interacting (pi)RNAs that have a role in providing a germline-specific defense against retrotransposon activity (Gunawardane et al., 2007). Male mitopld-/- flies are sterile, typical of flies lacking piRNAs (He et al., 2009), lose nuages (Russell and Frank, 1978), sites where piRNA production and processing is thought to occur, and have de-repressed retrotransposons in their testes (Pane et al., 2007; Watanabe et al., 2011). Moreover, lipin-1-/- mice have elevated PA on the mitochondrial surface and significantly increased nuage formation (Huang et al., 2011). Genetic evidence therefore strongly supports a role for PA and/or DAG at the OMM in piRNA production (Huang et al., 2011). However, the exact role of MitoPLD with respect to the mitochondrial phospholipid pool is unclear.
The importance of the dynamic regulation of PA on the OMM is further substantiated by the recent characterization of PA-PLA1 (Baba et al., 2014). In vitro, PA-PLA1 preferentially deacylates PA to LPA (Higgs and Glomset, 1994). PA-PLA1 overexpression or depletion in HeLa cells causes mitochondrial fragmentation and elongation, respectively (Baba et al., 2014). Interestingly, co-expression of PA-PLA1 and MitoPLD prevents the accumulation of PA on the OMM surface and the morphological defects associated with MitoPLD overexpression alone (Baba et al., 2014). Similar to mitopld-/-and lipin-1-/- mice, pa-pla1-/- mice have a defect in spermatogenesis that correlates with mitochondrial disorganization (Baba et al., 2014). Finally, diminution of ddhd2, a related iPLA1 family member with a similar specificity for PA as PA-PLA1, causes mitochondrial elongation in mouse embryonic fibroblasts (Baba et al., 2014).
Besides being a required intermediate in CL biosynthesis, PG is also a precursor for bis(monoacylglycerol)phosphate (BMP; Hullin-Matsuda et al., 2007), a class of phospholipid that is highly enriched in late endosomes and lysosomes (Wherrett and Huterer, 1972; Poorthuis and Hostetler, 1976; Mobius et al., 2003). While BMP is found in many tissues and cells, it is usually present at less than 1% of the total phospholipid mass (Simon and Rouser, 1969; Mason et al., 1972). BMP is a structural isomer of PG and is thought to function in the maintenance and regulation of endosomal/lysosomal membrane dynamics and cholesterol trafficking (Kobayashi et al., 1998, 1999; Hullin-Matsuda et al., 2009; Gallala and Sandhoff, 2011). However, its exact biological role(s) is unresolved.
The absolute requirement of PG and/or CL for life is underscored by the observation that ptpmt1-/- mice die in utero before embryonic day 8.5 (Zhang et al., 2011). Reflecting this importance, CL has a multitude of functional roles in mitochondria (Figure 4). CL is highly enriched in cardiac tissues making up 15–20% of the total phospholipid phosphorus mass of the heart (Pangborn, 1942; Hostetler et al., 1975). Its relative abundance in cells and tissues with high energetic demands point to CL as being intimately involved in maintaining mitochondrial structure and function. Indeed, it interacts with numerous mitochondrial proteins, including all OXPHOS complexes and most mitochondrial solute carriers, and is often required for their functional reconstitution in liposomes (Cheneval et al., 1983; Beyer and Klingenberg, 1985; Eble et al., 1990; Shinzawa-Itoh et al., 2007; Claypool et al., 2008; Claypool, 2009; Schwall et al., 2012). In addition, CL is proposed to function as a proton trap that helps funnel pumped protons toward the ATP synthase to generate ATP (Haines and Dencher, 2002). An association with CL promotes the assembly of membrane proteins into oligomeric complexes (Zhang et al., 2002; Pfeiffer et al., 2003; Claypool et al., 2008; Strauss et al., 2008; Acehan et al., 2011a; Althoff et al., 2011). Indeed, CL is important for the assembly and function of IMM and OMM translocases and thus, mitochondrial biogenesis (Jiang et al., 2000; van der Laan et al., 2007; Gebert et al., 2009). CL is also critically important for stabilizing respiratory supercomplexes (SCs), supramolecular assemblies built from respiratory complexes I, III, and IV (Schagger and Pfeiffer, 2000). These SCs are thought to increase the efficiency of electron transfer between the respiratory chain components by substrate channeling mechanisms (Acin-Perez et al., 2008; Lapuente-Brun et al., 2013), thereby maximizing OXPHOS. Further, SC assembly shortens the distance traveled by mobile electron carriers, minimizing ROS leakage and reducing oxidative damage. Finally, CL-binding stimulates the activity of dynamin-related GTPases with pivotal roles in IMM fusion and mitochondrial fission (Ban et al., 2010; Montessuit et al., 2010; Bustillo-Zabalbeitia et al., 2014). All of these CL-supported functions have been discussed extensively in several excellent reviews (Chicco and Sparagna, 2007; Houtkooper and Vaz, 2008; Joshi et al., 2009; Lewis and McElhaney, 2009; Sparagna and Lesnefsky, 2009; Osman et al., 2011; Claypool and Koehler, 2012).
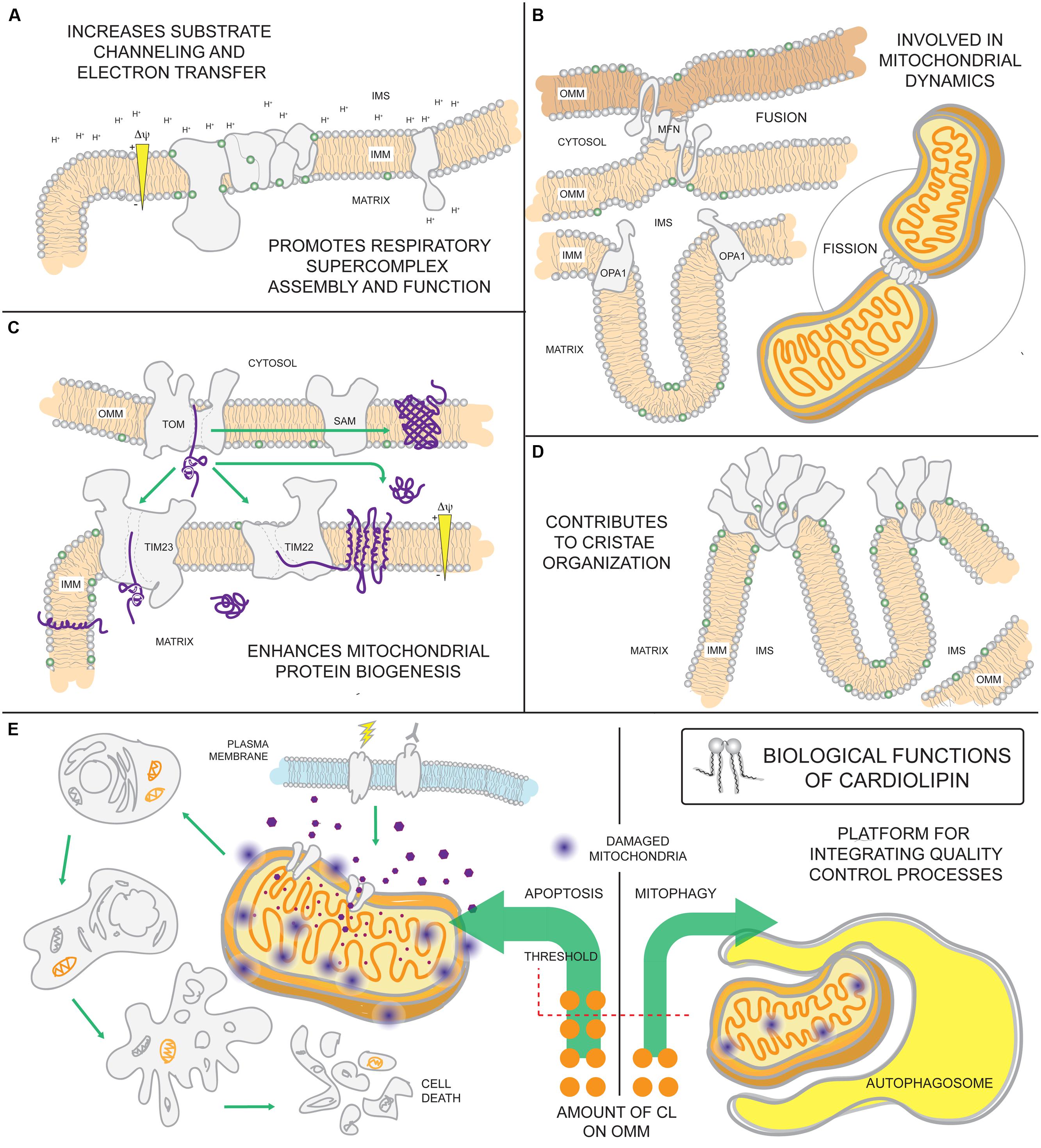
FIGURE 4. Biological functions of cardiolipin. As the signature phospholipid of the mitochondrion, CL is intimately involved in a number of mitochondrial processes. (A) Anionic CL on the IMM can function as a proton trap by attracting (and providing) a local pool of protons that can be funneled towards the ATP synthase. Moreover, CL is associated with every OXPHOS component and can promote their assembly into respiratory supercomplexes. Such supramolecular assemblies are thought to enhance electron transfer and reduce ROS leakage from the electron transport chain. (B) CL associates with dynamin-related GTPases that are intimately involved in fusion and fission and (C) contributes to the assembly and function of IMM and OMM translocases vital for mitochondrial biogenesis. (D) Besides enhancing OXPHOS by stabilizing SCs, CL also promotes the assembly of ATP synthase oligomers that provide a structural scaffold required for establishing the characteristic shape of mitochondrial cristae. (E) Externalization of CL on the surface of the mitochondrion is involved in signaling the execution of either mitophagy or apoptotic cell death.
Both apoptosis and mitophagy, a macro-autophagic process that is pre-emptive to cell death via apoptosis, are signaled in part by the externalization of CL to the OMM. The movement of CL to the OMM is thought to involve the scramblase PLS3 and/or NM23-H4 and may occur preferentially at IMM-OMM contact sites thought to be enriched in CL (Ardail et al., 1990; Simbeni et al., 1991). Overexpression of NM23-H4, but not a CL-binding mutant, increases apoptotic markers that may be ascribed to increased CL externalization on the OMM (Schlattner et al., 2013). CL on the OMM attracts and activates caspase-8 (Gonzalvez et al., 2008) and aids in pro-apoptotic Bax insertion into and permeabilization of the OMM (Kuwana et al., 2002; Lucken-Ardjomande et al., 2008). In preparations of giant unilamellar vesicles lacking CL, caspase-8 is unable to interact with vesicle membranes, a step necessary for caspase-8 activation and subsequent recruitment of tBid (Jalmar et al., 2010, 2013). Yeast mitochondria lacking CL are protected from the bioenergetic perturbations normally induced upon incubation with tBid (Gonzalvez et al., 2005) underscoring the importance of CL for tBid function. Furthermore, CL peroxidation by cytochrome c promotes the release of a number of pro-apoptotic factors, including cytochrome c, following OMM permeabilization (Kagan et al., 2005). Under conditions of mild mitochondrial dysfunction, CL is externalized on the OMM where it promotes the specific destruction of the mitochondrion by mitophagy (Chu et al., 2013). siRNA knockdown of the scramblase PLS3 or CL synthase CLS in neuronal cells, diminishes CL externalization and the number of mitochondrially-associated autophagic markers, and attenuates chemically-induced mitophagy (Chu et al., 2013). As CL externalization is important for both apoptosis and mitophagy, there is likely to be a threshold level of mitochondrial damage (severity of insult and percentage of mitochondrial pool impacted) above which apoptosis is executed and below which the affected mitochondria are selectively removed. In addition, qualitative and/or quantitative differences in the CL exposed on the OMM may influence how this lipid signal is interpreted by the cell.
While the role and functional consequence of MLCAT1-based remodeling is presently unresolved, TAZ- and ALCAT1-mediated CL remodeling are associated with very different physiologic outcomes. In the absence of TAZ, the acyl chain composition of CL is significantly diversified and molecular symmetry is lost (Schlame et al., 2005). Thus, TAZ has a preeminent role in dictating the final collection of acyl chains attached to CL under physiological conditions. The final acyl chain pattern of CL, which is tissue-specific, is thought to be critical for normal mitochondrial physiology by supporting some combination of the functions attributed to CL. However, Δcld1 yeast, which fail to initiate CL remodeling and accumulate normal amounts of unremodeled CL, have wt OXPHOS activity and normal mitochondrial morphology (Baile et al., 2014b; Ye et al., 2014a). These results question the idea that TAZ-based CL remodeling produces “optimized” CL species that promote mitochondrial fitness, and instead suggests that CL remodeling may actually accomplish other physiologically important functions that either were not tested (Baile et al., 2014b; Ye et al., 2014a) or have not yet been discovered.
In contrast to TAZ-based CL remodeling, CL remodeled by ALCAT1 predisposes mitochondria to damage. In mouse myoblasts, ALCAT1 overexpression increases the amount of CL enriched with docosahexaenoic acid (22:6) at the expense of linoleic acid (18:2; Li et al., 2010), consistent with alterations in CL species that are observed in aging rat hearts [the latter additionally contain 20:4 arachidonic acid (Paradies et al., 2009)]. The increased acyl chain unsaturation in these CL forms makes them peroxidation-prone and increases the susceptibility of mitochondria to undergo apoptosis (Watkins et al., 1998; Paradies et al., 2009; Li et al., 2010). As such, ALCAT1 seems to perform “pathogenic” remodeling of CL. Consistent with this, ALCAT1 overexpression, also noted in mouse models of metabolic disease, increases the rate of ATP, and consequently, ROS production during oxidative stress (Li et al., 2010). Conversely, ablation of ALCAT1 elevates tetralinoleoyl-CL content in the heart (Li et al., 2010), prevents the onset of disease, increases insulin resistance, mitigates OXPHOS dysfunction by increasing complex I activity, restores mtDNA fidelity, alleviates fusion defects and associated mitochondrial fragmentation, and re-establishes mitochondrial quality control (Li et al., 2010, 2012; Liu et al., 2012; Wang et al., 2014b). Combined, this suggests that CL remodeled by ALCAT1 may exacerbate and/or signal mitochondrial dysfunction in disease pathogenesis (Wang et al., 2014b).
With the recent application of next generation sequencing methodologies, new disease-causing genes are being implicated in mitochondrial disorders each year. For the remainder of this review, we describe a new category of mitochondrial disorder that is caused by nuclear defects that specifically alter mitochondrial phospholipid metabolism. We anticipate that the diseases discussed below represent the tip of the iceberg and that more disorders that impinge on mitochondrial phospholipid metabolism will be identified in the near future.
Mutations in the gene that encodes the MLCL transacylase, TAZ, lead to BTHS (Barth et al., 1983, 2004; Bione et al., 1996). BTHS is the founding member of this new class of mitochondrial disease and thus not surprisingly, is the best characterized. This X-linked multisystem disorder presents with cardiomyopathy, skeletal muscle weakness, neutropenia, growth retardation, and 3-methylglutaconic aciduria (3-MGA), and can be fatal if not diagnosed early [Barth et al., 1983; Kelley et al., 1991; Johnston et al., 1997; one isolated case of BTHS in a female patient has been reported (Cosson et al., 2012)]. 3-MGA is a heterogeneous group of syndromes characterized by an increased excretion of 3-methylglutaconic and 3-methylglutaric acids, breakdown products of leucine catabolism (Wysocki et al., 1976). Additional features such as isolated left ventricular non-compaction, ventricular arrhythmia, motor delay, exercise intolerance, poor appetite, fatigue, hypoglycemia, lactic acidosis, and hyperammonemia have also been described in BTHS patients (Ichida et al., 2001; Steward et al., 2010). Patient heart, liver, and skeletal muscle biopsies contain malformed mitochondria with tightly stacked or circular bundles of cristae (Barth et al., 1983; Hodgson et al., 1987; Orstavik et al., 1998; Bissler et al., 2002). In patient-derived lymphoblasts, mitochondria have dramatically reduced inner membranes, collapsed cristae, and are often fragmented (Xu et al., 2005; Acehan et al., 2007). TAZ (G4.5) contains 11 exons, is localized on a gene-rich region on Xq28, and is highly expressed in cardiac and skeletal tissues (Bione et al., 1996). Pathogenic taz variants identified to date encompass splice site mutations, insertions, deletions, as well as missense and nonsense mutations (Johnston et al., 1997; a current list of known genetic variants is maintained by the BTHS foundation; https://www.barthsyndrome.org/science).
Efforts to understand BTHS pathogenesis are complicated by the complete lack of genotype–phenotype correlations. Patients with the same mutation, or even siblings sharing the same mutation, can manifest with extremely disparate symptoms. A case in point is that of a 51 years old proband, the oldest BTHS patient reported, and his 3 years old grandnephew (Ronvelia et al., 2012). Although they harbor the same mutation, the boy presented with cardinal manifestations of BTHS (including congestive heart failure that required a heart transplant at 11 months of age) while the great uncle was 43 years old when he was diagnosed with myopathy. These observations highlight the importance of modifying factors as key determinants in BTHS disease progression.
Furthermore, links between different mutations and severity of disease have not been established for BTHS. Model organisms can help bridge this gap. Using a yeast BTHS model, 21 distinct pathogenic missense mutations have been modeled in yeast Taz1p and their loss-of-function mechanism defined. This effort has identified seven classes of BTHS mutants defined by their loss-of-function mechanisms (Claypool et al., 2006, 2011; Whited et al., 2013). Briefly, the seven classes comprise of variants that are (1) non-functional truncated products resulting from frameshifts or aberrant splicing, (2) mislocalized within mitochondria and aggregation-prone, (3) aberrantly assembled, (4) catalytically dead, (5) hypomorphic alleles with residual transacylase activity, (6) unable to engage in stable productive assemblies, or (7) temperature-sensitive. Systematic analyses of pathogenic variants in this manner can provide important mechanistic insight into the clinical heterogeneity of BTHS. The next step in attaining this goal is to verify that the defined loss-of-function mechanisms are conserved in an appropriate mammalian model. Such a model is additionally needed to characterize pathogenic alleles that cannot be modeled in yeast due to a lack of conservation.
There are a number of BTHS models presently available. Importantly, every BTHS model has the characteristic biochemical defects that underlie BTHS – increased MLCL, decreased CL, and an abnormal acyl chain composition of the remaining CL. In addition to yeast, other cellular BTHS models include patient-derived fibroblasts (Barth et al., 1996), lymphocytes (Schlame et al., 2005; Xu et al., 2005), and iPSCs (Dudek et al., 2013; Wang et al., 2014a), and TAZ-depleted rodent (Acehan et al., 2009; He, 2010; He et al., 2014) and human cell lines (Gonzalvez et al., 2008). Animal models of BTHS include TAZ-depleted zebrafish (Khuchua et al., 2006) and mice (Acehan et al., 2011b; Soustek et al., 2011; Phoon et al., 2012), and taz-/- flies (Xu et al., 2006a).
Cellular models of BTHS show dysmorphic changes in mitochondrial morphology and energetic defects (Acehan et al., 2007, 2009; Gonzalvez et al., 2013). In patient-derived lymphoblasts (Xu et al., 2005; Gonzalvez et al., 2013), iPSCs (Dudek et al., 2013), and fibroblasts (Barth et al., 1996), there is low basal respiration, reduced membrane potential, and compromised coupling of OXPHOS. Respiratory SCs are decreased and there is a shift in SC assembly from large “respirasomes” to smaller, and presumably, less efficient SCs (McKenzie et al., 2006; Dudek et al., 2013; Gonzalvez et al., 2013). Therefore, it is postulated that these alterations in respiratory chain assembly diminish respiratory efficiency and consequently, augment ROS production. While basal respiration is unaffected in the shRNA-inducible TAZ knockdown mouse, maximal uncoupled respiration is reduced; whether this reflects changes in respiratory SC stability has not been demonstrated (Powers et al., 2013). Enzymatic analyses of respiratory chain complexes using cardiomyocytes derived from the TAZ knockdown mouse indicate that complex III is impaired (Powers et al., 2013), suggestive of a similar bioenergetic dysfunction as noted in the cellular models. In TAZ-depleted mice (Acehan et al., 2011b; Soustek et al., 2011; Phoon et al., 2012) and zebrafish (Khuchua et al., 2006), cardiac defects are observed that recapitulate many of the relevant cardiac parameters noted in BTHS patients. With respect to zebrafish, TAZ knockdown severely impairs zebrafish development and the degree of cardiac dysmorphology is proportional to the morpholino dose. In addition to impaired cardiac function, TAZ knockdown mice have abnormal skeletal muscle ultrastructure (Xu et al., 2006a; Acehan et al., 2011b) consistent with taz-/- flies that also have impaired muscle functions (Xu et al., 2006a; Acehan et al., 2009). Recently, cardiomyocytes differentiated from BTHS patient iPSCs have been generated (Dudek et al., 2013; Wang et al., 2014a). In these cells, there is structural destabilization of respiratory SCs that correlates with reduced respiratory complex activities (Dudek et al., 2013). When wt cells are seeded onto engineered chips, they form sarcomeres and contract; in contrast, the ability of BTHS-derived cardiomyocytes to form organized sarcomeric arrays is severely impacted as is their contractility (Wang et al., 2014a). Therefore, results from the myriad of BTHS models indicate that the lipid abnormalities that occur in the absence of TAZ result in OXPHOS dysfunction associated with SC destabilization. OXPHOS dysfunction increases the production of ROS and in sum, these impairments compromise heart development and function.
As already discussed, the stimulated externalization of CL onto the OMM is an important event that can alternatively trigger apoptosis or mitophagy. It is thus notable that BTHS-derived lymphocytes are resistant to mitochondrial-dependent apoptosis due to an impaired ability to recruit and activate caspase-8 (Gonzalvez et al., 2008, 2013). Interestingly, caspase-8-/- mice die in utero and the embryos have heart abnormalities that include thin and disorganized trabeculae (Varfolomeev et al., 1998), phenotypes also observed upon TAZ depletion in utero (Phoon et al., 2012). Thus, defects in caspase-8 activity may contribute to the cardiomyopathy in BTHS. The relative capacity of BTHS mitochondria to be consumed by mitophagy has not been reported. However, BTHS lymphocytes have more mitochondria that are individually less functional (Gonzalvez et al., 2013) suggesting that mitochondrial homeostasis may indeed be perturbed in BTHS and contribute to disease progression.
Importantly, the detailed biochemical and cell biologic characterization of the numerous BTHS models have begun to identify potential avenues for therapeutic intervention. For instance, suppression of mitochondrial ROS attenuates the energetic and functional decline caused by TAZ-depletion in rodent cardiac myocytes (He et al., 2014) and corrects the sarcomere organization and contractile function of induced BTHS cardiomyocytes (Wang et al., 2014a). Genetic and/or pharmacologic targeting of the lipase that initiates CL remodeling in yeast, flies, and patient lymphoblasts prevents, to varying degrees, the mitochondrial dysfunction caused by the absence of TAZ and can additionally rescue the sterility of male taz-/- flies (Malhotra et al., 2009a; Baile et al., 2014b; Ye et al., 2014a). These results suggest that the mitochondrial dysfunction stemming from TAZ deficiency is not likely due to reduced remodeled CL, but instead caused by the increased abundance of MLCL and/or the low total amounts of CL. As such, drugs that can scavenge mitochondrial ROS or prevent the accumulation of MLCL (inhibit the activity of the upstream deacylase(s); enhance the activity of other putative MLCL remodelers; augment a pathway that degrades MLCL) could be used to the potential therapeutic benefit of BTHS patients.
To date, only two mutations have been identified in DNAJC19 (DnaJ/Hsp40 homolog, subfamily C, member 19) that are associated with dilated cardiomyopathy with ataxia (DCMA; Davey et al., 2006; Ojala et al., 2012), an autosomal-recessive disorder that presents with early onset dilated cardiomyopathy, non-progressive cerebellar ataxia leading to motor delays, testicular dysgenesis, growth failure, and elevated levels of 3-MGA. Additional features include microcytic anemia, mild to borderline non-progressive mental retardation, hepatic steatosis, and occasional optic atrophy (Davey et al., 2006; Sparkes et al., 2007; Ojala et al., 2012). DCMA shares with BTHS certain clinical features including left ventricular non-compaction with spongey and trabeculated myocardium (Sparkes et al., 2007). However, unlike in BTHS (Barth et al., 2004), DCMA patients do not exhibit neutropenia or skeletal myopathy.
DNAJC19 is on chromosome 3q26.33 (Davey et al., 2006). The gene consists of six exons and is ubiquitously expressed as a 525 bp transcript with a minor 435 bp form, lacking exon 4, in all tissues tested and control fibroblasts (Davey et al., 2006). The consanguineous Canadian Dariusleut Hutterite families all have an exon4 splicing defect while two Finnish brothers have a frameshift mutation resulting in a truncated protein (Davey et al., 2006; Ojala et al., 2012). Consistent with these mutations, only the shorter transcript is expressed in fibroblasts from one Hutterite patient (Davey et al., 2006) and DNAJC19 protein is not detected in fibroblasts derived from the Finnish siblings (Ojala et al., 2012). Since there are only two genetic variants of DNAJC19 linked to DCMA, it is not possible to describe genotype–phenotype correlations in this disorder. Still, in a retrospective study of the Hutterite patients that all share the same mutation (Sparkes et al., 2007), 13 of the 17 patients developed dilated cardiomyopathy and 10 later died. Interestingly, three patients had resolved or stabilized cardiomyopathy and another four did not present with cardiac defects at all. Thus, there is significant clinical variability with respect to this particular DCMA allele.
DNAJC19 is associated with the matrix-facing leaflet of the IMM via a predicted NH2-terminal transmembrane region (Richter-Dennerlein et al., 2014) and contains a conserved DNAJ domain at the COOH terminus, in contrast to other conventional DNAJ-proteins with an NH2-terminal J-domain. DNAJ domain-containing proteins typically act as molecular chaperones for Hsp70/Hsp40s and prevent protein aggregation by aiding in the folding and assembly of newly synthesized proteins (Ohtsuka and Hata, 2000; Mayer and Bukau, 2005). Sequence alignment indicates that DNAJC19 is orthologous to yeast Pam18p, a constituent of the TIM23 import machinery (Mokranjac et al., 2003). In yeast, the Pam18p and Mdj2p proteins are essential components of the TIM23 translocation machinery (Mokranjac et al., 2005), interacting with mitochondrial Hsp70p, Pam16p, and Tim44p to form the presequence translocase-associated motor complex (Schneider et al., 1994; Moro et al., 2002; Truscott et al., 2003; Frazier et al., 2004; Kozany et al., 2004; D’Silva et al., 2008). This subcomplex associates with the core TIM23 translocon (consisting of Tim23p, Tim17p, Tim50p and variably, Tim21p) and mediates import of precursors destined for the matrix in an ATP- and membrane potential-dependent manner (Bomer et al., 1997; Moro et al., 1999; Geissler et al., 2002; Yamamoto et al., 2002; Chacinska et al., 2005; van der Laan et al., 2007). Thus, DNAJC19 may, like yeast Pam18p, stimulate the ATPase activity of mtHsp70 and stabilize mtHsp70 binding to incoming peptides. For an excellent review on mitochondrial protein translocation, refer to (Chacinska et al., 2009).
Consistent with a role in protein import, DNAJC19 interacts with MAGMAS, the conserved mammalian ortholog of yeast Pam16p that is essential for development (Gonczy et al., 2000; Jubinsky et al., 2001, 2003; D’Silva et al., 2005). MAGMAS is structurally similar to DNAJC19 but peripherally associated with the matrix-leaflet of the IMM. The interaction between DNAJC19 and MAGMAS occurs via their reciprocal J-domains and is required to recruit DNAJC19 to the core TIM23 translocon (Sinha et al., 2010). MAGMAS then associates with TIM17, a subunit of the TIM23 core, to form the TIM23 translocation machinery (Mehawej et al., 2014; Sinha et al., 2014). Interestingly, there are three distinct forms of TIM23 translocon that incorporate different TIM17 isoforms and associate with either DNAJC15 (TIM17a; translocase A), another co-chaperone of the same Hsp40-type (Hatle et al., 2013; Schusdziarra et al., 2013), or DNAJC19 (TIM17b1 and TIM17b2; translocase Bs). Of the three versions of TIM23, translocase Bs are critical for basal mitochondrial biogenesis (e.g., OXPHOS, iron–sulfur cluster biogenesis, mtDNA copy number, and maintenance of mitochondrial membrane potential) while translocase A plays a dispensable, albeit supportive role when translocase Bs are absent (Sinha et al., 2014). In light of these results, defects in mitochondrial presequence protein import and consequently, mitochondrial biogenesis, may represent the mechanism of DCMA pathogenesis.
Recently, however, human and murine DNAJC19 were found to additionally interact with prohibitin (PHB complexes) (Richter-Dennerlein et al., 2014). PHB complexes are large hetero-oligomeric complexes composed of PHB1 and PHB2 subunits (Tatsuta et al., 2005) that are involved in cristae morphogenesis (Merkwirth et al., 2008) and modeled to function as lipid scaffolds in the IMM, redistributing lipids such as CL (Christie et al., 2011) and delineating functional membrane domains (Osman et al., 2009b). In yeast, Δphb1 is synthetically lethal with Δcrd1 highlighting the importance of conserved PHB complexes in CL metabolism (Osman et al., 2009a). Interestingly, DNAJC19-depletion in HEK293T cells shifts the CL acyl profile to longer and less saturated chains; however, CL levels are unaffected and MLCL does not accumulate (Richter-Dennerlein et al., 2014). Moreover, while a functional DNAJ-domain is not required for DNAJC19 to interact with PHB complexes, it is necessary to rescue the changes in CL molecular composition that occur upon DNAJC19 knockdown (Richter-Dennerlein et al., 2014). In contrast, PHB2 knockdown results in the same three biochemical alterations that characterize BTHS (increased MLCL, decreased CL, and changes in CL acyl chain composition), although the increase in MLCL is significantly less than in TAZ knockdown cells (Richter-Dennerlein et al., 2014). Concomitant knockdown of PHB2 or DNAJC19 with TAZ does not alter the accumulation of MLCL that occurs upon depletion of TAZ alone, indicating that neither PHB complexes nor DNAJC19 are required to generate the substrate, MLCL, used by TAZ. Combined, these results suggest unexpected roles for both PHB complexes and DNAJC19 in CL remodeling and further indicate that their functions in this regard, are at least partially distinct. TAZ does not interact with PHB2 or DNAJC19 directly (Richter-Dennerlein et al., 2014). Thus, the ability of PHB/DNAJC19 complexes to define specific membrane domains, such as those with negative curvature, is postulated to confer acyl chain specificity to TAZ (Schlame et al., 2012a; Richter-Dennerlein et al., 2014). In the absence of such privileged domains, TAZ remodeling still occurs, it just lacks acyl chain specificity.
In sum, it appears that DNAJC19 has dual functions in the regulation of CL remodeling and mitochondrial protein biogenesis (Figure 5). A key question that remains unresolved is whether both activities contribute to DCMA disease pathogenesis. Presently lacking is any information regarding the lipid profile and protein import functionality of mitochondria isolated from actual DCMA patients; such data is needed to better understand the underlying pathogenic mechanism. If similar alterations in CL are detected in DCMA patient cells, future studies will be needed to define how DNAJC19/PHB complexes regulate the collection of acyl chains attached to CL and relate these changes to mitochondrial dysfunction. This latter question is all the more interesting and relevant given that in yeast, flies, and mammalian cells, genetically and/or pharmacologically preventing production of MLCL by targeting the lipase that begins the remodeling cascade rescues the multitude of phenotypes attributed to TAZ deficiency (Malhotra et al., 2009a; Baile et al., 2014b; Ye et al., 2014a).
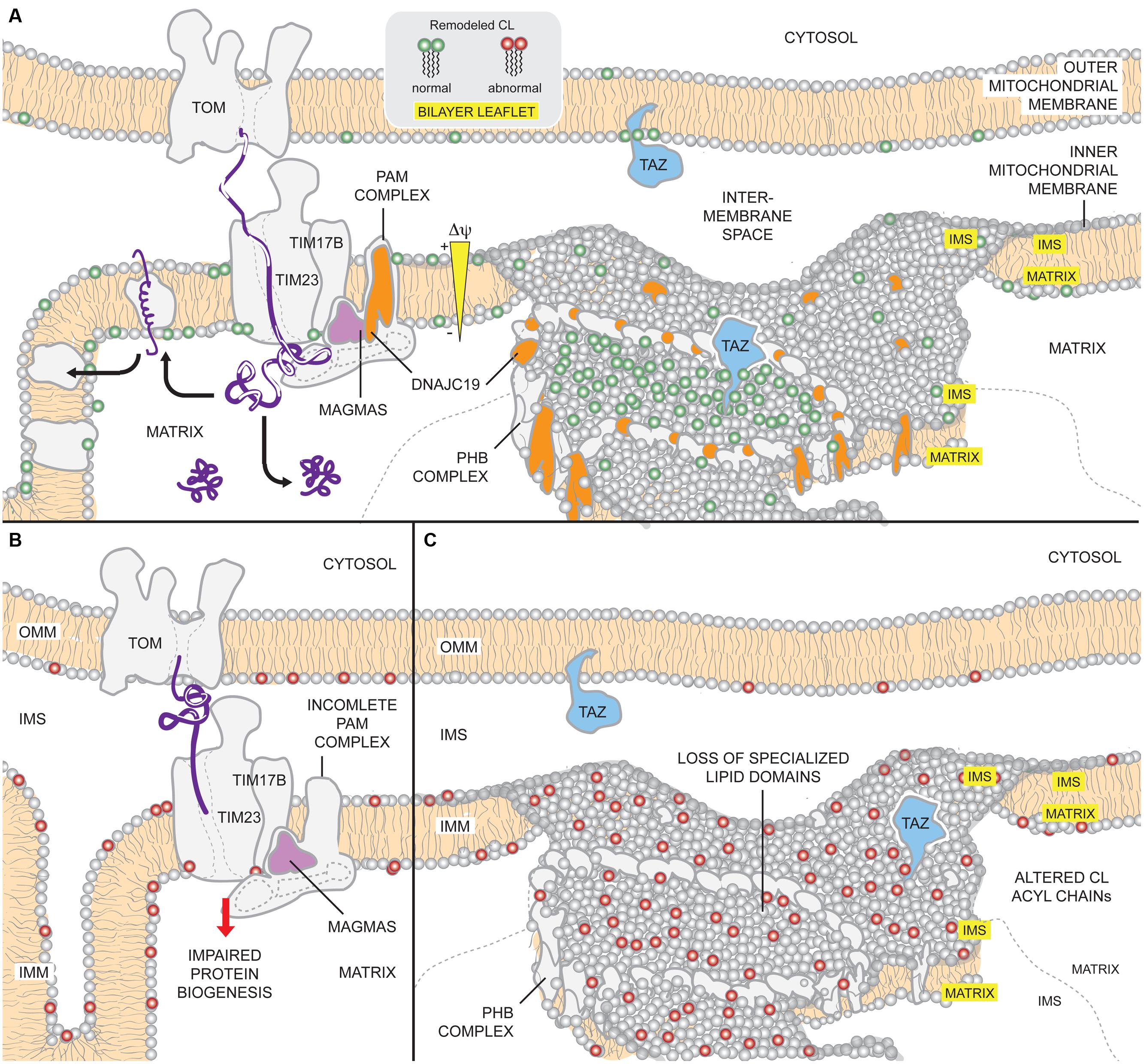
FIGURE 5. Potential mechanisms of DCMA mitochondrial dysfunction. (A) Under physiological conditions, DNAJC19 is targeted to TIM17B by MAGMAS and associates with components of the TIM23 translocation machinery, forming translocase B. DNAJC19 is homologous to yeast Pam18p which stimulates the mtHsp70 activity of the PAM (presequence-associated motor) complex and stabilizes binding of incoming precursors. Moreover, DNAJC19 also interacts with prohibitin-2 (PHB) of the PHB complexes. PHB1/PHB2 oligomers form ring-like complexes that are modeled to delineate specialized membrane domains. Functional segregation of CL and TAZ in such domains may confer acyl chain specificity to TAZ, allowing it to perform physiologically relevant CL remodeling (green). Thus, DNAJC19 may participate in both mitochondrial presequence protein import as well as formation of membrane domains that are important for TAZ-based CL remodeling. (B) In the absence of DNAJC19, the ability of the TIM23 machinery to import proteins across the IMM may be compromised. Consequently, the biogenesis of mitochondrial proteins, such as subunits of respiratory complexes, may be reduced. (C) Further, loss of DNAJC19 prevents the PHB complex-based generation of privileged membrane domains. In the absence of such domains, TAZ remodeling, which may still occur, lacks specificity (red). For clarity, not all components of the TOM, TIM, and PAM complexes are depicted.
Evidence that PG, like CL, undergoes physiologically important remodeling was recently provided by the identification of serine active site containing 1 (SERAC1) mutations that cause autosomal-recessive MEGDEL syndrome [3-MGA, sensorineural deafness, encephalopathy, and neuroradiological evidence of progressive Leigh-like syndrome (Wortmann et al., 2006, 2012)]. In addition to classical MEGDEL symptoms, an increasing list of clinical phenotypes have been associated with SERAC1 mutations; infantile mitochondrial hepatopathy, psychomotor and developmental delay, bilateral optic nerve atrophy, myoclonic epilepsy, and microcephaly (Sarig et al., 2013; Lumish et al., 2014; Vilarinho et al., 2014; Wedatilake et al., 2014). Similar to BTHS and DCMA, MEGDEL patients have 3-MGA and variable mitochondrial dysfunction.
SERAC1 is located on chromosome 6q25.3 and the encoded protein resides in the MAM (Wortmann et al., 2012). SERAC1 is a predicted single-pass transmembrane protein that is 654 amino acids long and translated from 17 exons into three isoforms (Wortmann et al., 2012). The protein is a member of the PGAP (post-GPI attachment to protein 1)-like protein domain family and contains an α/β-hydrolase fold and a highly conserved serine-lipase domain (Wortmann et al., 2012). To date, 18 different mutations have been described in 24 patients, many of which are frameshift, nonsense, or missense mutations within or upstream of the lipase domain (Karkucinska-Wieckowska et al., 2011; Wortmann et al., 2012; Sarig et al., 2013; Tort et al., 2013; Dweikat et al., 2014; Lumish et al., 2014; Vilarinho et al., 2014; Wedatilake et al., 2014). Of note, a patient recently described with severe, early onset MEGDEL symptoms harbors compound frameshift and stop-gain heterozygous mutations upstream of the lipase domain (Lumish et al., 2014). In contrast to patients with variants within the lipase domain, which may allow for a protein with residual activity, the production of truncated SERAC1 completely lacking the lipase domain from both alleles may account for the severity of the particular patient’s phenotype.
SERAC1 is implicated in changing the acyl chain composition of CL’s precursor, PG. Specifically, MEGDEL patient fibroblasts have elevated concentrations of PG-34:1 and lower concentrations of PG-36:1; the acyl chain compositions of the other major phospholipid classes, with the notable exception of CL, are normal (Wortmann et al., 2012). The inability to convert PG-34:1 to PG-36:1 results in the accumulation of PG-34:1 and subsequent incorporation of PG with these acyl chain species into CL. Thus, in MEGDEL patients, CL levels are normal but the acyl chain composition of CL is altered (Wortmann et al., 2012). As in the case of DNAJC19 deficiency, if and how normal levels of CL of abnormal acyl chain composition affects mitochondrial function is at present unclear. Notably, most MEGDEL patient tissues and fibroblasts exhibit OXPHOS dysfunction (Wortmann et al., 2006, 2012; Karkucinska-Wieckowska et al., 2011; Sarig et al., 2013; Tort et al., 2013; Dweikat et al., 2014; Wedatilake et al., 2014), though the degree of impairment and the affected respiratory chain component(s) varies. Moreover, ROS production is increased and catalase levels decreased in fibroblasts derived from at least one MEGDEL patient (Karkucinska-Wieckowska et al., 2011); therefore, similar to BTHS, defective OXPHOS function may cause an imbalance in redox homeostasis in MEGDEL patients. However, additional basic work is required to define whether increased ROS production and reduced scavenging is a consistent feature associated with SERAC1 dysfunction, exactly how defective SERAC1 variably impairs OXPHOS, and if the changes noted in the acyl chain composition of CL in MEGDEL patients are detrimental to mitochondrial functionality.
It is curious to note that endogenous TAZ, which is presumably functional in MEGDEL patients, is unable to correct the unusual CL acyl chains that accumulate in the absence of SERAC1 function. PG is a precursor of CL and therefore contributes directly to the collection of acyl chains associated with pre-remodeled CL. Since other phospholipids have normal acyl chain compositions in MEGDEL patients (Wortmann et al., 2012), in theory, TAZ should still be able to generate “normal” CL. One possible explanation for this discrepancy is that perhaps the CL that accumulates in MEGDEL patients is not a substrate for the lipase that functions upstream of TAZ to initiate CL remodeling.
While SERAC1 defects do not affect the abundance or acyl chain pattern of phosphatidylcholine, phosphatidylserine, or PE, it does significantly reduce levels of BMP (Wortmann et al., 2012), a lipid implicated in endosomal/lysosomal homeostasis and function (Hullin-Matsuda et al., 2009; Figure 6). BMP levels regulate cholesterol trafficking with low intracellular BMP causing accumulation of free cholesterol in late endosomes. Indeed, SERAC1 patient fibroblasts accumulate unesterified cholesterol (Sarig et al., 2013; Tort et al., 2013) and patient biopsies show dramatically disorganized striated muscle ultrastructure with abnormal mitochondria and lysosomal accumulation of neutral fat droplets (Wortmann et al., 2012; Wedatilake et al., 2014). It is presently not clear how a change in the acyl chain composition of PG translates into lower amounts of BMP especially since the molecular composition of BMP in MEGDEL patients is not altered relative to controls (Wortmann et al., 2012). Also unclear is how PG produced on the matrix side of the IMM gains access to SERAC1, which resides in the MAM. As our understanding of how SERAC1 regulates the acyl chain composition of CL and the production of BMP is in its infancy, future basic work is needed to develop assorted models designed to better understand these processes and how their disturbance cause the numerous MEGDEL syndrome phenotypes.
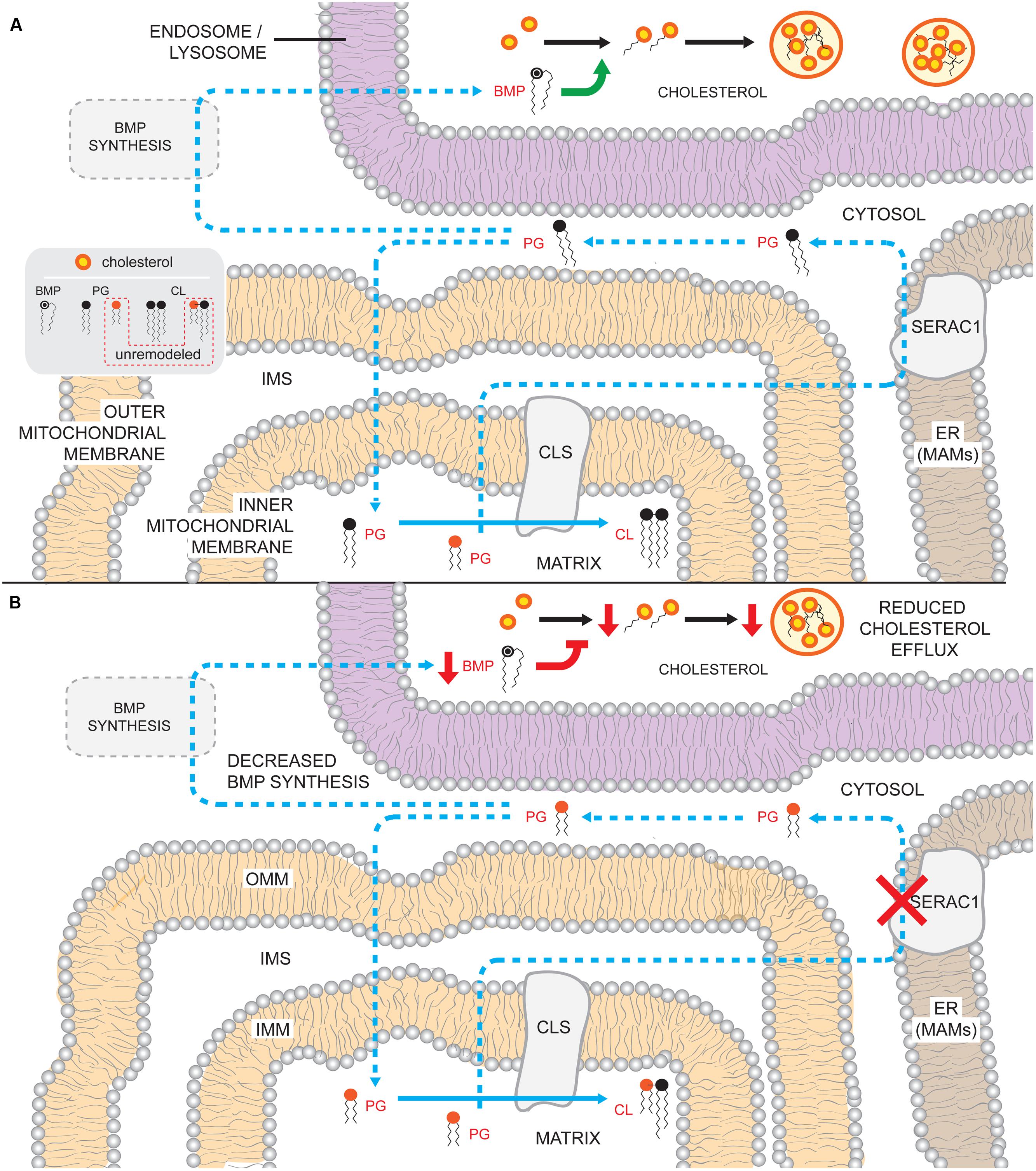
FIGURE 6. Potential consequences of the absence of SERAC1 activity. (A) PG that is generated on the matrix-leaflet of the IMM is trafficked out of the mitochondrion and remodeled by SERAC1 on the ER MAMs. Remodeled PG can subsequently serve as substrate for BMP and CL synthesis. BMP in the endosomsal/lysosomal compartments regulates (green arrow) cholesterol esterification and trafficking out of the compartment. (B) In the absence of a functional SERAC1, PG is not remodeled and accumulates shorter acyl chain moieties. The decrease in acyl chain length somehow reduces overall BMP levels, perhaps because the unremodeled PG is a poor precursor for BMP synthesis. Reduced BMP levels in the endosome/lysosome impairs cholesterol esterification (inhibition in red) and leads to decreased (red arrows) cholesterol efflux. The lack of SERAC1 also changes the steady state composition of CL acyl chains. Blue arrows denote enzymatic reactions and lipid movements. Dashed arrows describe uncharacterized steps and pathways.
Sengers syndrome is caused by the absence of the IMS-residing AGK (Calvo et al., 2012; Mayr et al., 2012; Haghighi et al., 2014). Symptoms of Sengers syndrome may present at birth, childhood, or early adulthood and its clinical manifestations range from being severe, causing death in infancy, to mild, allowing survival into adulthood (van Ekeren et al., 1993; Mayr et al., 2012). This autosomal-recessive disorder is characterized by congenital cataracts, hypertrophic cardiomyopathy, skeletal myopathy, exercise intolerance, lactic acidosis, and increased urinary 3-MGA; though motor development is delayed, mental development of the affected individuals are normal (Sengers et al., 1975). Hence, the phenotypic spectrum associated with Sengers syndrome substantially overlaps with both Barth and DCMA syndromes. Sengers syndrome was originally thought to be due to deficiencies in the level and/or function of adenine nucleotide translocase 1 (ANT1; Jordens et al., 2002). ANT1 is the heart/muscle isoform of the mitochondrial ADP/ATP exchanger that plays a fundamental role in OXPHOS by mediating the flux of ADP and ATP across the IMM (Li et al., 1989). However, genetic analyses excluded mutations in the ANT1 gene; as such, it was subsequently proposed that transcriptional, translational, or post-translational events might be responsible for the lower amounts of ANT1 observed in patients (Jordens et al., 2002). Recently, two groups localized the defect via exome sequencing, not in or around ANT1, but instead to the AGK locus on chromosome 7q34 (Calvo et al., 2012; Mayr et al., 2012).
The AGK gene contains 17 exons and encodes for a 422 amino acid protein; it is a mitochondrial membrane-associated, multi-substrate lipid kinase that contains domains that are highly homologous to sphingosine kinase-2 as well as DAG kinase (Topham and Prescott, 1999; Waggoner et al., 2004; Bektas et al., 2005). When phylogenetic analyses of AGK were undertaken, sequence comparisons to other lipid kinases such as DAG, ceramide, and sphingosine kinases revealed that despite containing a highly conserved DAG kinase catalytic domain, human and murine AGK segregate to a unique branch and are not members of any previously described lipid kinase family (Waggoner et al., 2004).
Like Barth and DCMA syndromes, genotype–phenotype correlations in Sengers syndrome are not readily apparent. For instance, there are Sengers syndrome patients who are non-syndromic or do not develop lactic acidosis (Aldahmesh et al., 2012), and siblings who share the same mutation but present with very different disease courses [one had the severe form of disease and died at 15 months of age while the other brother, at 3 years old, was without skeletal myopathy or physical limitations (Siriwardena et al., 2013)]. Thus, the etiology of Sengers syndrome is also likely to involve genetic modifiers that can significantly affect disease progression and severity.
However, there is evidence that a genotype–phenotype correlation for Sengers syndrome patients potentially exists (Haghighi et al., 2014). Two forms of the syndrome have been described, a severe neonatal form that leads to infantile death (Smeitink et al., 1989) and a more benign, but chronic, form that has allowed survival into the fourth decade (van Ekeren et al., 1993). The former disease type is always associated with homozygous AGK nonsense mutations while in the latter, patients usually harbor at least one splice site variant or a start codon mutation, but not doubly null/missense alleles (Haghighi et al., 2014). Presumably, aberrant splicing of the COOH terminal exons (in or after the DAG kinase domain) produces proteins with residual AGK activity while inactivation of the canonical start site allows for initiation at an alternative/cryptic start site (Starck et al., 2012). In support of this possible genotype–phenotype relation, the oldest surviving patients (>35 years) carry either heterozygous start site and stop codon mutations or a homozygous mutation affecting exon 15–16 splicing (Lalive D’Epinay et al., 1986; van Ekeren et al., 1993).
A role for AGK in the assembly, stability and/or regulation of OXPHOS components such as ANT1 and complex I has been speculated (Pitkanen et al., 1996; Haghighi et al., 2014). OXPHOS defects in patient samples have been variable (Pitkanen et al., 1996; Jordens et al., 2002; Morava et al., 2004; Aldahmesh et al., 2012; Calvo et al., 2012; Mayr et al., 2012; Siriwardena et al., 2013; Haghighi et al., 2014). In the more severe cases, ANT1 and complex I levels are low and the mtDNA copy number reduced. However, in the milder cases, OXPHOS function is spared. Curiously, ANT1 levels are normal in undifferentiated myoblasts but are dramatically reduced upon myoblast differentiation (Mayr et al., 2012). This observation is notable as it suggests that ANT1 stability is compromised during muscle fiber differentiation in Sengers syndrome patients. While the exact molecular function of AGK is not known, its potential role in CL metabolism is consistent with the varied bioenergetic impairments, structurally abnormal mitochondria, and lipid and glycogen deposits in patient-derived skeletal and cardiac muscle (Sengers et al., 1975; Siriwardena et al., 2013). A comprehensive phospholipid analysis has not been performed for Sengers syndrome cells and/or tissues. Therefore, whether the absence of AGK function correlates with disturbances in phospholipid metabolism has not yet been established. If AGK generates PA that contributes substantially to TAMM41’s substrate pool (Figure 1), then the absence of AGK function should significantly impact downstream PG and CL synthesis. Given the aforementioned results concerning the relative stability of ANT1 in undifferentiated versus differentiated myoblasts, the choice of cell/tissue to analyze is likely to be critically important in uncovering if and how AGK participates in mitochondrial phospholipid metabolism. Such information is anticipated to shed crucial light onto Sengers syndrome disease pathogenesis.
Mutations in the phospholipase A1 family members DDHD1 (Bouslam et al., 2005; Tesson et al., 2012; Liguori et al., 2014) and DDHD2 (Schuurs-Hoeijmakers et al., 2012; Gonzalez et al., 2013; Citterio et al., 2014; Doi et al., 2014; Magariello et al., 2014) have recently been linked to autosomal-recessive forms of hereditary spastic paraplegia (HSP). HSPs are an extremely heterogeneous group of inherited neurological disorders that are classified by patients who present with (complex HSP) or without (uncomplicated or pure HSP) neurological defects. Pure HSP is characterized by progressive spasticity and weakness of the lower limbs. In complex HSP, spastic paraplegia is compounded with neurological and systemic abnormalities such as dementia, intellectual disability, ataxia, and neuropathy, amongst others [see (Fink, 2013) for comprehensive discussion of HSP and spastic paraplegia variants]. Furthermore, there is varied disease onset, progression, and “functional plateaus” (a point at which there is very little further disability) between the HSPs and as such, there is very little genotype–phenotype correlation in this heterogeneous disorder.
Hereditary spastic paraplegia has been linked to over 50 genetic loci, covering all chromosomes, of which only 22 genes have been identified so far (Finsterer et al., 2012; Fink, 2013). Two of the identified disease-associated genes are DDHD1 and DDHD2 that encode protein products with PA-phospholipase A1 activity. DDHD1 is localized to chromosome 14q21.1, contains 12 exons (Tesson et al., 2012) and encodes for the 872 amino acid protein, PA-PLA1. Initial characterization identified substantial PA-PLA1 activity in bovine brain and testis that corresponds to the two tissues with the highest DDHD1 mRNA expression (Higgs and Glomset, 1994; Higgs et al., 1998). In humans, these two tissues, along with heart and skeletal muscle, additionally contain high levels of DDHD2, a more ubiquitously expressed DDHD family member (Nakajima et al., 2002; Morikawa et al., 2009; Sato et al., 2010). DDHD2 is both cytosolic and membrane-associated (Sato et al., 2010; Baba et al., 2013). Located on chromosome 8p11.23 and containing 18 exons, DDHD2 is expressed as two major mRNA transcripts that correspond to the full length 711 amino acid protein and another that lacks the COOH terminal DDHD phospholipase domain. When patient ddhd2 mutants were overexpressed in and partially purified from HEK293T cells, PA-PLA1 activity was either low or absent compared to overexpressed wt DDHD2 (Doi et al., 2014). Notably, depletion of PA-PLA1 activity (DDHD1 or DDHD2) in some, but not all cells, can cause mitochondrial elongation (Schuurs-Hoeijmakers et al., 2012; Baba et al., 2013, 2014), presumably due to unopposed fusion. Thus, one might expect that the balance between fission and fusion may be disturbed in some cells in patients with ddhd1 or ddhd2 mutations with an overall shift toward too much fusion. As active fission is a crucial process for clearance of damaged mitochondria by mitophagy, unopposed fusion could drive the accumulation of dysfunctional mitochondria (Twig et al., 2008). Consistent with the possible accretion of sub-optimal mitochondria, respiration and ATP levels are decreased and cytosolic ROS increased in ddhd1 patient-derived lymphoblasts (Tesson et al., 2012). Unfortunately, PA levels and mitochondrial morphology have not been documented in either ddhd1 or ddhd2 patient-derived cells (Tesson et al., 2012) but there are no major changes in phospholipids in ddhd2-/- mouse brains (Inloes et al., 2014).
It is unclear whether or not depletion of DDHD1 or DDHD2 can directly affect mitochondrial dynamics by accumulating pro-fusogenic PA on the OMM. Given their similar substrate specificity, they may have redundant roles in this regard. Still, there might be functional differences for these two PA phospholipases that are dictated by their expression patterns and subcellular distributions. As such, future studies are needed to investigate the potential causative link between dysregulation of PA metabolism on the OMM with disease pathogenesis for these subtypes of HSP.
Phospholipids are not simply cellular barriers that delineate cells and organelles thus allowing biochemical pathways to be compartmentalized and cross-regulated. Although predominantly synthesized in the ER, some phospholipids, such as PG and CL, are exclusively made in the mitochondrion, while others, such as PE, involve a pathway that begins in the ER but is completed in the cell’s powerhouse. In mitochondria, phospholipids are important for an impressive array of functions. Indeed, when phospholipid levels, molecular species, or distribution within the mitochondrion is affected, as is the case for the diseases discussed herein, mitochondrial dysfunction ensues. BTHS was the first known inborn error of mitochondrial phospholipid metabolism. Since the identification of TAZ as the causative gene for BTHS, exome sequencing and basic research have both contributed to the recent addition of new diseases that impinge upon mitochondrial lipid metabolism. We propose to classify these disorders as a new category of mitochondrial disease that specifically impacts phospholipid homeostasis. The clinical heterogeneity of patient phenotypes reflects the variety of functions that phospholipids partake in and their fundamental importance for mitochondrial physiology. Future work needs to focus on developing appropriate models for these diseases. As exemplified by BTHS, the availability and in-depth characterization of such models will contribute enormously to our understanding of each disease process and identify potential therapeutic strategies. Further, such basic work should help us better understand disease mechanisms and provide explanations for shared and distinct phenotypes.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the Claypool lab for critical reading of the manuscript. This work was supported by National Institutes of Health Grants R00HL089185 and R01GM111548 (Steven M. Claypool), and a pre-doctoral fellowship from the American Heart Association (Ya-Wen Lu).
Acehan, D., Khuchua, Z., Houtkooper, R. H., Malhotra, A., Kaufman, J., Vaz, F. M.,et al. (2009). Distinct effects of tafazzin deletion in differentiated and undifferentiated mitochondria. Mitochondrion 9, 86–95. doi: 10.1016/j.mito.2008.12.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Acehan, D., Malhotra, A., Xu, Y., Ren, M., Stokes, D. L., and Schlame, M. (2011a). Cardiolipin affects the supramolecular organization of ATP synthase in mitochondria. Biophys. J. 100, 2184–2192. doi: 10.1016/j.bpj.2011.03.031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Acehan, D., Vaz, F., Houtkooper, R. H., James, J., Moore, V., Tokunaga, C.,et al. (2011b). Cardiac and skeletal muscle defects in a mouse model of human Barth syndrome. J. Biol. Chem. 286, 899–908. doi: 10.1074/jbc.M110.171439
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Acehan, D., Xu, Y., Stokes, D. L., and Schlame, M. (2007). Comparison of lymphoblast mitochondria from normal subjects and patients with Barth syndrome using electron microscopic tomography. Lab. Invest. 87, 40–48. doi: 10.1038/labinvest.3700480
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Acin-Perez, R., Fernandez-Silva, P., Peleato, M. L., Perez-Martos, A., and Enriquez, J. A. (2008). Respiratory active mitochondrial supercomplexes. Mol. Cell 32, 529–539. doi: 10.1016/j.molcel.2008.10.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Agarwal, A. K. (2012). Lysophospholipid acyltransferases: 1-acylglycerol-3-phosphate O-acyltransferases. From discovery to disease. Curr. Opin. Lipidol. 23, 290–302. doi: 10.1097/MOL.0b013e328354fcf4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Aguado, B., and Campbell, R. D. (1998). Characterization of a human lysophosphatidic acid acyltransferase that is encoded by a gene located in the class III region of the human major histocompatibility complex. J. Biol. Chem. 273, 4096–4105. doi: 10.1074/jbc.273.7.4096
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Aldahmesh, M. A., Khan, A. O., Mohamed, J. Y., Alghamdi, M. H., and Alkuraya, F. S. (2012). Identification of a truncation mutation of acylglycerol kinase (AGK) gene in a novel autosomal recessive cataract locus. Hum. Mutat. 33, 960–962. doi: 10.1002/humu.22071
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Althoff, T., Mills, D. J., Popot, J. L., and Kuhlbrandt, W. (2011). Arrangement of electron transport chain components in bovine mitochondrial supercomplex I1III2IV1. EMBO J. 30, 4652–4664. doi: 10.1038/emboj.2011.324
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Anderson, S., Bankier, A. T., Barrell, B. G., De Bruijn, M. H., Coulson, A. R., Drouin, J.,et al. (1981). Sequence and organization of the human mitochondrial genome. Nature 290, 457–465. doi: 10.1038/290457a0
Ardail, D., Privat, J. P., Egret-Charlier, M., Levrat, C., Lerme, F., and Louisot, P. (1990). Mitochondrial contact sites. Lipid composition and dynamics. J. Biol. Chem. 265, 18797–18802.
Baba, T., Kashiwagi, Y., Arimitsu, N., Kogure, T., Edo, A., Maruyama, T.,et al. (2014). Phosphatidic acid (PA)-preferring phospholipase A1 regulates mitochondrial dynamics. J. Biol. Chem. 289, 11497–11511. doi: 10.1074/jbc.M113.531921
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Baba, T., Yamamoto, A., Tagaya, M., and Tani, K. (2013). A lysophospholipid acyltransferase antagonist, CI-976, creates novel membrane tubules marked by intracellular phospholipase A1 KIAA0725p. Mol. Cell. Biochem. 376, 151–161. doi: 10.1007/s11010-013-1563-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Baile, M. G., Lu, Y. W., and Claypool, S. M. (2014a). The topology and regulation of cardiolipin biosynthesis and remodeling in yeast. Chem. Phys. Lipids 179, 25–31. doi: 10.1016/j.chemphyslip.2013.10.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Baile, M. G., Sathappa, M., Lu, Y. W., Pryce, E., Whited, K., Mccaffery, J. M.,et al. (2014b). Unremodeled and remodeled cardiolipin are functionally indistinguishable in yeast. J. Biol. Chem. 289, 1768–1778. doi: 10.1074/jbc.M113.525733
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Baile, M. G., Whited, K., and Claypool, S. M. (2013). Deacylation on the matrix side of the mitochondrial inner membrane regulates cardiolipin remodeling. Mol. Biol. Cell 24, 2008–2020. doi: 10.1091/mbc.E13-03-0121
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ban, T., Heymann, J. A., Song, Z., Hinshaw, J. E., and Chan, D. C. (2010). OPA1 disease alleles causing dominant optic atrophy have defects in cardiolipin-stimulated GTP hydrolysis and membrane tubulation. Hum. Mol. Genet. 19, 2113–2122. doi: 10.1093/hmg/ddq088
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barth, P. G., Scholte, H. R., Berden, J. A., Van Der Klei-Van Moorsel, J. M., Luyt-Houwen, I. E., Van ‘T Veer-Korthof, E. T.,et al. (1983). An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J. Neurol. Sci. 62, 327–355. doi: 10.1016/0022-510X(83)90209-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barth, P. G., Valianpour, F., Bowen, V. M., Lam, J., Duran, M., Vaz, F. M.,et al. (2004). X-linked cardioskeletal myopathy and neutropenia (Barth syndrome): an update. Am. J. Med. Genet. A 126A, 349–354. doi: 10.1002/ajmg.a.20660
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barth, P. G., Van Den Bogert, C., Bolhuis, P. A., Scholte, H. R., Van Gennip, A. H., Schutgens, R. B.,et al. (1996). X-linked cardioskeletal myopathy and neutropenia (Barth syndrome): respiratory-chain abnormalities in cultured fibroblasts. J. Inherit. Metab. Dis. 19, 157–160. doi: 10.1007/BF01799418
Bektas, M., Payne, S. G., Liu, H., Goparaju, S., Milstien, S., and Spiegel, S. (2005). A novel acylglycerol kinase that produces lysophosphatidic acid modulates cross talk with EGFR in prostate cancer cells. J. Cell Biol. 169, 801–811. doi: 10.1083/jcb.200407123
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Beranek, A., Rechberger, G., Knauer, H., Wolinski, H., Kohlwein, S. D., and Leber, R. (2009). Identification of a cardiolipin-specific phospholipase encoded by the gene CLD1 (YGR110W) in yeast. J. Biol. Chem. 284, 11572–11578. doi: 10.1074/jbc.M805511200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Beyer, K., and Klingenberg, M. (1985). ADP/ATP carrier protein from beef heart mitochondria has high amounts of tightly bound cardiolipin, as revealed by 31P nuclear magnetic resonance. Biochemistry 24, 3821–3826. doi: 10.1021/bi00336a001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bione, S., D’adamo, P., Maestrini, E., Gedeon, A. K., Bolhuis, P. A., and Toniolo, D. (1996). A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat. Genet. 12, 385–389. doi: 10.1038/ng0496-385
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bissler, J. J., Tsoras, M., Goring, H. H., Hug, P., Chuck, G., Tombragel, E.,et al. (2002). Infantile dilated X-linked cardiomyopathy, G4.5 mutations, altered lipids, and ultrastructural malformations of mitochondria in heart, liver, and skeletal muscle. Lab. Invest. 82, 335–344. doi: 10.1038/labinvest.3780427
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bomer, U., Meijer, M., Maarse, A. C., Honlinger, A., Dekker, P. J., Pfanner, N.,et al. (1997). Multiple interactions of components mediating preprotein translocation across the inner mitochondrial membrane. EMBO J. 16, 2205–2216. doi: 10.1093/emboj/16.9.2205
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bouslam, N., Benomar, A., Azzedine, H., Bouhouche, A., Namekawa, M., Klebe, S.,et al. (2005). Mapping of a new form of pure autosomal recessive spastic paraplegia (SPG28). Ann. Neurol. 57, 567–571. doi: 10.1002/ana.20416
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brandner, K., Mick, D. U., Frazier, A. E., Taylor, R. D., Meisinger, C., and Rehling, P. (2005). Taz1, an outer mitochondrial membrane protein, affects stability and assembly of inner membrane protein complexes: implications for Barth syndrome. Mol. Biol. Cell 16, 5202–5214. doi: 10.1091/mbc.E05-03-0256
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bustillo-Zabalbeitia, I., Montessuit, S., Raemy, E., Basanez, G., Terrones, O., and Martinou, J. C. (2014). Specific interaction with cardiolipin triggers functional activation of Dynamin-Related Protein 1. PLoS ONE 9:e102738. doi: 10.1371/journal.pone.0102738
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Calvo, S. E., Compton, A. G., Hershman, S. G., Lim, S. C., Lieber, D. S., Tucker, E. J.,et al. (2012). Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci. Transl. Med. 4:118ra110. doi: 10.1126/scitranslmed.3003310
Calvo, S. E., and Mootha, V. K. (2010). The mitochondrial proteome and human disease. Annu. Rev. Genomics Hum. Genet. 11, 25–44. doi: 10.1146/annurev-genom-082509-141720
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cao, J., Liu, Y., Lockwood, J., Burn, P., and Shi, Y. (2004). A novel cardiolipin-remodeling pathway revealed by a gene encoding an endoplasmic reticulum-associated acyl-CoA:lysocardiolipin acyltransferase (ALCAT1) in mouse. J. Biol. Chem. 279, 31727–31734. doi: 10.1074/jbc.M402930200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cao, J., Shen, W., Chang, Z., and Shi, Y. (2009). ALCAT1 is a polyglycerophospholipid acyltransferase potently regulated by adenine nucleotide and thyroid status. Am. J. Physiol. Endocrinol. Metab. 296, E647–E653. doi: 10.1152/ajpendo.90761.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Carpenter, K., Pollitt, R. J., and Middleton, B. (1992). Human liver long-chain 3-hydroxyacyl-coenzyme A dehydrogenase is a multifunctional membrane-bound beta-oxidation enzyme of mitochondria. Biochem. Biophys. Res. Commun. 183, 443–448. doi: 10.1016/0006-291X(92)90501-B
Chacinska, A., Koehler, C. M., Milenkovic, D., Lithgow, T., and Pfanner, N. (2009). Importing mitochondrial proteins: machineries and mechanisms. Cell 138, 628–644. doi: 10.1016/j.cell.2009.08.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chacinska, A., Lind, M., Frazier, A. E., Dudek, J., Meisinger, C., Geissler, A.,et al. (2005). Mitochondrial presequence translocase: switching between TOM tethering and motor recruitment involves Tim21 and Tim17. Cell 120, 817–829. doi: 10.1016/j.cell.2005.01.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chang, S. C., Heacock, P. N., Clancey, C. J., and Dowhan, W. (1998a). The PEL1 gene (renamed PGS1) encodes the phosphatidylglycero-phosphate synthase of Saccharomyces cerevisiae. J. Biol. Chem. 273, 9829–9836. doi: 10.1074/jbc.273.16.9829
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chang, S. C., Heacock, P. N., Mileykovskaya, E., Voelker, D. R., and Dowhan, W. (1998b). Isolation and characterization of the gene (CLS1) encoding cardiolipin synthase in Saccharomyces cerevisiae. J. Biol. Chem. 273, 14933–14941. doi: 10.1074/jbc.273.24.14933
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, D., Zhang, X. Y., and Shi, Y. (2006). Identification and functional characterization of hCLS1, a human cardiolipin synthase localized in mitochondria. Biochem. J. 398, 169–176. doi: 10.1042/BJ20060303
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, H., and Chan, D. C. (2009). Mitochondrial dynamics – fusion, fission, movement, and mitophagy – in neurodegenerative diseases. Hum. Mol. Genet. 18, R169–R176. doi: 10.1093/hmg/ddp326
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, Y. Q., Kuo, M. S., Li, S., Bui, H. H., Peake, D. A., Sanders, P. E.,et al. (2008). AGPAT6 is a novel microsomal glycerol-3-phosphate acyltransferase. J. Biol. Chem. 283, 10048–10057. doi: 10.1074/jbc.M708151200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cheneval, D., Muller, M., and Carafoli, E. (1983). The mitochondrial phosphate carrier reconstituted in liposomes is inhibited by doxorubicin. FEBS Lett. 159, 123–126. doi: 10.1016/0014-5793(83)80429-3
Cheneval, D., Muller, M., Toni, R., Ruetz, S., and Carafoli, E. (1985). Adriamycin as a probe for the transversal distribution of cardiolipin in the inner mitochondrial membrane. J. Biol. Chem. 260, 13003–13007.
Cheng, H., Mancuso, D. J., Jiang, X., Guan, S., Yang, J., Yang, K.,et al. (2008). Shotgun lipidomics reveals the temporally dependent, highly diversified cardiolipin profile in the mammalian brain: temporally coordinated postnatal diversification of cardiolipin molecular species with neuronal remodeling. Biochemistry 47, 5869–5880. doi: 10.1021/bi7023282
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chicco, A. J., and Sparagna, G. C. (2007). Role of cardiolipin alterations in mitochondrial dysfunction and disease. Am. J. Physiol. Cell Physiol. 292, C33–C44. doi: 10.1152/ajpcell.00243.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Choi, S. Y., Huang, P., Jenkins, G. M., Chan, D. C., Schiller, J., and Frohman, M. A. (2006). A common lipid links Mfn-mediated mitochondrial fusion and SNARE-regulated exocytosis. Nat. Cell Biol. 8, 1255–1262. doi: 10.1038/ncb1487
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Christie, D. A., Lemke, C. D., Elias, I. M., Chau, L. A., Kirchhof, M. G., Li, B.,et al. (2011). Stomatin-like protein 2 binds cardiolipin and regulates mitochondrial biogenesis and function. Mol. Cell. Biol. 31, 3845–3856. doi: 10.1128/MCB.05393-11
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chu, C. T., Ji, J., Dagda, R. K., Jiang, J. F., Tyurina, Y. Y., Kapralov, A. A.,et al. (2013). Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 15, 1197–1205. doi: 10.1038/ncb2837
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Citterio, A., Arnoldi, A., Panzeri, E., D’angelo, M. G., Filosto, M., Dilena, R.,et al. (2014). Mutations in CYP2U1, DDHD2 and GBA2 genes are rare causes of complicated forms of hereditary spastic paraparesis. J. Neurol. 261, 373–381. doi: 10.1007/s00415-013-7206-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Claypool, S. M. (2009). Cardiolipin, a critical determinant of mitochondrial carrier protein assembly and function. Biochim. Biophys. Acta 1788, 2059–2068. doi: 10.1016/j.bbamem.2009.04.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Claypool, S. M., and Koehler, C. M. (2012). The complexity of cardiolipin in health and disease. Trends Biochem. Sci. 37, 32–41. doi: 10.1016/j.tibs.2011.09.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Claypool, S. M., Mccaffery, J. M., and Koehler, C. M. (2006). Mitochondrial mislocalization and altered assembly of a cluster of Barth syndrome mutant tafazzins. J. Cell Biol. 174, 379–390. doi: 10.1083/jcb.200605043
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Claypool, S. M., Oktay, Y., Boontheung, P., Loo, J. A., and Koehler, C. M. (2008). Cardiolipin defines the interactome of the major ADP/ATP carrier protein of the mitochondrial inner membrane. J. Cell Biol. 182, 937–950. doi: 10.1083/jcb.200801152
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Claypool, S. M., Whited, K., Srijumnong, S., Han, X., and Koehler, C. M. (2011). Barth syndrome mutations that cause tafazzin complex lability. J. Cell Biol. 192, 447–462. doi: 10.1083/jcb.201008177
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Colbeau, A., Nachbaur, J., and Vignais, P. M. (1971). Enzymic characterization and lipid composition of rat liver subcellular membranes. Biochim. Biophys. Acta 249, 462–492. doi: 10.1016/0005-2736(71)90123-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Connerth, M., Tatsuta, T., Haag, M., Klecker, T., Westermann, B., and Langer, T. (2012). Intramitochondrial transport of phosphatidic acid in yeast by a lipid transfer protein. Science 338, 815–818. doi: 10.1126/science.1225625
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Contreras, F. X., Sanchez-Magraner, L., Alonso, A., and Goni, F. M. (2010). Transbilayer (flip-flop) lipid motion and lipid scrambling in membranes. FEBS Lett. 584, 1779–1786. doi: 10.1016/j.febslet.2009.12.049.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cosson, L., Toutain, A., Simard, G., Kulik, W., Matyas, G., Guichet, A.,et al. (2012). Barth syndrome in a female patient. Mol. Genet. Metab. 106, 115–120. doi: 10.1016/j.ymgme.2012.01.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Csordas, G., Renken, C., Varnai, P., Walter, L., Weaver, D., Buttle, K. F.,et al. (2006). Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 174, 915–921. doi: 10.1083/jcb.200604016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Csordas, G., Varnai, P., Golenar, T., Roy, S., Purkins, G., Schneider, T. G.,et al. (2010). Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol. Cell 39, 121–132. doi: 10.1016/j.molcel.2010.06.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Daniele, T., Hurbain, I., Vago, R., Casari, G., Raposo, G., Tacchetti, C.,et al. (2014). Mitochondria and melanosomes establish physical contacts modulated by Mfn2 and involved in organelle biogenesis. Curr. Biol. 24, 393–403. doi: 10.1016/j.cub.2014.01.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
DaRe, J. T., Vasta, V., Penn, J., Tran, N. T., and Hahn, S. H. (2013). Targeted exome sequencing for mitochondrial disorders reveals high genetic heterogeneity. BMC Med. Genet. 14:118. doi: 10.1186/1471-2350-14-118
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Daum, G. (1985). Lipids of mitochondria. Biochim. Biophys. Acta 822, 1–42. doi: 10.1016/0304-4157(85)90002-4
Daum, G., and Vance, J. E. (1997). Import of lipids into mitochondria. Prog. Lipid Res. 36, 103–130. doi: 10.1016/S0163-7827(97)00006-4
Davey, K. M., Parboosingh, J. S., Mcleod, D. R., Chan, A., Casey, R., Ferreira, P.,et al. (2006). Mutation of DNAJC19, a human homologue of yeast inner mitochondrial membrane co-chaperones, causes DCMA syndrome, a novel autosomal recessive Barth syndrome-like condition. J. Med. Genet. 43, 385–393. doi: 10.1136/jmg.2005.036657
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
de Brito, O. M., and Scorrano, L. (2008). Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610. doi: 10.1038/nature07534
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
de Kroon, A. I., Dolis, D., Mayer, A., Lill, R., and De Kruijff, B. (1997). Phospholipid composition of highly purified mitochondrial outer membranes of rat liver and Neurospora crassa. Is cardiolipin present in the mitochondrial outer membrane? Biochim. Biophys. Acta 1325, 108–116. doi: 10.1016/S0005-2736(96)00240-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dimauro, S., and Davidzon, G. (2005). Mitochondrial DNA and disease. Ann. Med. 37, 222–232. doi: 10.1080/07853890510007368
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Doi, H., Ushiyama, M., Baba, T., Tani, K., Shiina, M., Ogata, K.,et al. (2014). Late-onset spastic ataxia phenotype in a patient with a homozygous DDHD2 mutation. Sci. Rep. 4:7132. doi: 10.1038/srep07132
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
D’Silva, P. R., Schilke, B., Hayashi, M., and Craig, E. A. (2008). Interaction of the J-protein heterodimer Pam18/Pam16 of the mitochondrial import motor with the translocon of the inner membrane. Mol. Biol. Cell 19, 424–432. doi: 10.1091/mbc.E07-08-0748
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
D’Silva, P. R., Schilke, B., Walter, W., and Craig, E. A. (2005). Role of Pam16’s degenerate J domain in protein import across the mitochondrial inner membrane. Proc. Natl. Acad. Sci. U.S.A. 102, 12419–12424. doi: 10.1073/pnas.0505969102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dudek, J., Cheng, I. F., Balleininger, M., Vaz, F. M., Streckfuss-Bomeke, K., Hubscher, D.,et al. (2013). Cardiolipin deficiency affects respiratory chain function and organization in an induced pluripotent stem cell model of Barth syndrome. Stem Cell Res. 11, 806–819. doi: 10.1016/j.scr.2013.05.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dweikat, I. M., Abdelrazeq, S., Ayesh, S., and Jundi, T. (2014). MEGDEL syndrome in a child from palestine: report of a novel mutation in SERAC1 gene. J. Child Neurol. doi: 10.1177/0883073814541474 [Epub ahead of print].
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eble, K. S., Coleman, W. B., Hantgan, R. R., and Cunningham, C. C. (1990). Tightly associated cardiolipin in the bovine heart mitochondrial ATP synthase as analyzed by 31P nuclear magnetic resonance spectroscopy. J. Biol. Chem. 265, 19434–19440.
Eichberg, J. (1974). The reacylation of deacylated derivatives of diphosphatidylglycerol by microsomes and mitochondria from rat liver. J. Biol. Chem. 249, 3423–3429.
Elbaz-Alon, Y., Rosenfeld-Gur, E., Shinder, V., Futerman, A. H., Geiger, T., and Schuldiner, M. (2014). A dynamic interface between vacuoles and mitochondria in yeast. Dev. Cell 30, 95–102. doi: 10.1016/j.devcel.2014.06.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eto, M., Shindou, H., and Shimizu, T. (2014). A novel lysophosphatidic acid acyltransferase enzyme (LPAAT4) with a possible role for incorporating docosahexaenoic acid into brain glycerophospholipids. Biochem. Biophys. Res. Commun. 443, 718–724. doi: 10.1016/j.bbrc.2013.12.043
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fink, J. K. (2013). Hereditary spastic paraplegia: clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol. 126, 307–328. doi: 10.1007/s00401-013-1115-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Finsterer, J., Loscher, W., Quasthoff, S., Wanschitz, J., Auer-Grumbach, M., and Stevanin, G. (2012). Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. J. Neurol. Sci. 318, 1–18. doi: 10.1016/j.jns.2012.03.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fould, B., Garlatti, V., Neumann, E., Fenel, D., Gaboriaud, C., and Arlaud, G. J. (2010). Structural and functional characterization of the recombinant human mitochondrial trifunctional protein. Biochemistry 49, 8608–8617. doi: 10.1021/bi100742w
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Foundation, T. U. M. D. (2014). The United Mitochondrial Disease Foundation. Available at: http://www.umdf.org/ [accessed October 17, 2014].
Frazier, A. E., Dudek, J., Guiard, B., Voos, W., Li, Y., Lind, M.,et al. (2004). Pam16 has an essential role in the mitochondrial protein import motor. Nat. Struct. Mol. Biol. 11, 226–233. doi: 10.1038/nsmb735
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gaigg, B., Simbeni, R., Hrastnik, C., Paltauf, F., and Daum, G. (1995). Characterization of a microsomal subfraction associated with mitochondria of the yeast, Saccharomyces cerevisiae. Involvement in synthesis and import of phospholipids into mitochondria. Biochim. Biophys. Acta 1234, 214–220. doi: 10.1016/0005-2736(94)00287-Y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gallala, H. D., and Sandhoff, K. (2011). Biological function of the cellular lipid BMP-BMP as a key activator for cholesterol sorting and membrane digestion. Neurochem. Res. 36, 1594–1600. doi: 10.1007/s11064-010-0337-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gallet, P. F., Petit, J. M., Maftah, A., Zachowski, A., and Julien, R. (1997). Asymmetrical distribution of cardiolipin in yeast inner mitochondrial membrane triggered by carbon catabolite repression. Biochem. J. 324(Pt 2), 627–634.
Gallet, P. F., Zachowski, A., Julien, R., Fellmann, P., Devaux, P. F., and Maftah, A. (1999). Transbilayer movement and distribution of spin-labelled phospholipids in the inner mitochondrial membrane. Biochim. Biophys. Acta 1418, 61–70. doi: 10.1016/S0005-2736(99)00022-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gebert, N., Joshi, A. S., Kutik, S., Becker, T., Mckenzie, M., Guan, X. L.,et al. (2009). Mitochondrial cardiolipin involved in outer-membrane protein biogenesis: implications for Barth syndrome. Curr. Biol. 19, 2133–2139. doi: 10.1016/j.cub.2009.10.074
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Geissler, A., Chacinska, A., Truscott, K. N., Wiedemann, N., Brandner, K., Sickmann, A.,et al. (2002). The mitochondrial presequence translocase: an essential role of Tim50 in directing preproteins to the import channel. Cell 111, 507–518. doi: 10.1016/S0092-8674(02)01073-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gonczy, P., Echeverri, C., Oegema, K., Coulson, A., Jones, S. J., Copley, R. R.,et al. (2000). Functional genomic analysis of cell division in C. elegans using RNAi of genes on chromosome III. Nature 408, 331–336. doi: 10.1038/35042526
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gonzalez, M., Nampoothiri, S., Kornblum, C., Oteyza, A. C., Walter, J., Konidari, I.,et al. (2013). Mutations in phospholipase DDHD2 cause autosomal recessive hereditary spastic paraplegia (SPG54). Eur. J. Hum. Genet. 21, 1214–1218. doi: 10.1038/ejhg.2013.29
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gonzalvez, F., D’Aurelio, M., Boutant, M., Moustapha, A., Puech, J. P., Landes, T.,et al. (2013). Barth syndrome: cellular compensation of mitochondrial dysfunction and apoptosis inhibition due to changes in cardiolipin remodeling linked to tafazzin (TAZ) gene mutation. Biochim. Biophys. Acta 1832, 1194–1206. doi: 10.1016/j.bbadis.2013.03.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gonzalvez, F., Pariselli, F., Dupaigne, P., Budihardjo, I., Lutter, M., Antonsson, B.,et al. (2005). tBid interaction with cardiolipin primarily orchestrates mitochondrial dysfunctions and subsequently activates Bax and Bak. Cell Death Differ. 12, 614–626. doi: 10.1038/sj.cdd.4401571
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gonzalvez, F., Schug, Z. T., Houtkooper, R. H., Mackenzie, E. D., Brooks, D. G., Wanders, R. J.,et al. (2008). Cardiolipin provides an essential activating platform for caspase-8 on mitochondria. J. Cell Biol. 183, 681–696. doi: 10.1083/jcb.200803129
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Green, D. R., and Kroemer, G. (2004). The pathophysiology of mitochondrial cell death. Science 305, 626–629. doi: 10.1126/science.1099320
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gruner, S. M. (1985). Intrinsic curvature hypothesis for biomembrane lipid composition: a role for nonbilayer lipids. Proc. Natl. Acad. Sci. U.S.A. 82, 3665–3669. doi: 10.1073/pnas.82.11.3665
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gu, Z., Valianpour, F., Chen, S., Vaz, F. M., Hakkaart, G. A., Wanders, R. J.,et al. (2004). Aberrant cardiolipin metabolism in the yeast taz1 mutant: a model for Barth syndrome. Mol. Microbiol. 51, 149–158. doi: 10.1046/j.1365-2958.2003.03802.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gunawardane, L. S., Saito, K., Nishida, K. M., Miyoshi, K., Kawamura, Y., Nagami, T.,et al. (2007). A slicer-mediated mechanism for repeat-associated siRNA 5’ end formation in Drosophila. Science 315, 1587–1590. doi: 10.1126/science.1140494
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Haghighi, A., Haack, T. B., Atiq, M., Mottaghi, H., Haghighi-Kakhki, H., Bashir, R. A.,et al. (2014). Sengers syndrome: six novel AGK mutations in seven new families and review of the phenotypic and mutational spectrum of 29 patients. Orphanet J. Rare Dis. 9, 119. doi: 10.1186/s13023-014-0119-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Haines, T. H., and Dencher, N. A. (2002). Cardiolipin: a proton trap for oxidative phosphorylation. FEBS Lett. 528, 35–39. doi: 10.1016/S0014-5793(02)03292-1
Hajra, A. K., and Bishop, J. E. (1982). Glycerolipid biosynthesis in peroxisomes via the acyl dihydroxyacetone phosphate pathway. Ann. N. Y. Acad. Sci. 386, 170–182. doi: 10.1111/j.1749-6632.1982.tb21415.x
Han, X., Yang, K., Yang, J., Cheng, H., and Gross, R. W. (2006). Shotgun lipidomics of cardiolipin molecular species in lipid extracts of biological samples. J. Lipid Res. 47, 864–879. doi: 10.1194/jlr.D500044-JLR200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Harb, J. S., Comte, J., and Gautheron, D. C. (1981). Asymmetrical orientation of phospholipids and their interactions with marker enzymes in pig heart mitochondrial inner membrane. Arch. Biochem. Biophys. 208, 305–318. doi: 10.1016/0003-9861(81)90153-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Harner, M., Korner, C., Walther, D., Mokranjac, D., Kaesmacher, J., Welsch, U.,et al. (2011). The mitochondrial contact site complex, a determinant of mitochondrial architecture. EMBO J. 30, 4356–4370. doi: 10.1038/emboj.2011.379
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hatle, K. M., Gummadidala, P., Navasa, N., Bernardo, E., Dodge, J., Silverstrim, B.,et al. (2013). MCJ/DnaJC15, an endogenous mitochondrial repressor of the respiratory chain that controls metabolic alterations. Mol. Cell. Biol. 33, 2302–2314. doi: 10.1128/MCB.00189-13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
He, Q. (2010). Tafazzin knockdown causes hypertrophy of neonatal ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 299, H210–H216. doi: 10.1152/ajpheart.00098.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
He, Q., Harris, N., Ren, J., and Han, X. (2014). Mitochondria-targeted antioxidant prevents cardiac dysfunction induced by tafazzin gene knockdown in cardiac myocytes. Oxid. Med. Cell. Longev. 2014, 654198. doi: 10.1155/2014/654198
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
He, Z., Kokkinaki, M., Pant, D., Gallicano, G. I., and Dym, M. (2009). Small RNA molecules in the regulation of spermatogenesis. Reproduction 137, 901–911. doi: 10.1530/REP-08-0494
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Higgs, H. N., and Glomset, J. A. (1994). Identification of a phosphatidic acid-preferring phospholipase A1 from bovine brain and testis. Proc. Natl. Acad. Sci. U.S.A. 91, 9574–9578. doi: 10.1073/pnas.91.20.9574
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Higgs, H. N., Han, M. H., Johnson, G. E., and Glomset, J. A. (1998). Cloning of a phosphatidic acid-preferring phospholipase A1 from bovine testis. J. Biol. Chem. 273, 5468–5477. doi: 10.1074/jbc.273.10.5468
Hodgson, S., Child, A., and Dyson, M. (1987). Endocardial fibroelastosis: possible X linked inheritance. J. Med. Genet. 24, 210–214. doi: 10.1136/jmg.24.4.210
Holt, I. J., Harding, A. E., and Morgan-Hughes, J. A. (1988). Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 331, 717–719. doi: 10.1038/331717a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Honscher, C., Mari, M., Auffarth, K., Bohnert, M., Griffith, J., Geerts, W.,et al. (2014). Cellular metabolism regulates contact sites between vacuoles and mitochondria. Dev. Cell 30, 86–94. doi: 10.1016/j.devcel.2014.06.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hope, M. J., Redelmeier, T. E., Wong, K. F., Rodrigueza, W., and Cullis, P. R. (1989). Phospholipid asymmetry in large unilamellar vesicles induced by transmembrane pH gradients. Biochemistry 28, 4181–4187. doi: 10.1021/bi00436a009
Hoppins, S., Collins, S. R., Cassidy-Stone, A., Hummel, E., Devay, R. M., Lackner, L. L.,et al. (2011). A mitochondrial-focused genetic interaction map reveals a scaffold-like complex required for inner membrane organization in mitochondria. J. Cell Biol. 195, 323–340. doi: 10.1083/jcb.201107053
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Horvath, S. E., and Daum, G. (2013). Lipids of mitochondria. Prog. Lipid Res. 52, 590–614. doi: 10.1016/j.plipres.2013.07.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hostetler, K. Y., Galesloot, J. M., Boer, P., and Van Den Bosch, H. (1975). Further studies on the formation of cardiolipin and phosphatidylglycerol in rat liver mitochondria. Effect of divalent cations and the fatty acid composition of CDP-diglyceride. Biochim. Biophys. Acta 380, 382–389. doi: 10.1016/0005-2760(75)90106-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Houtkooper, R. H., Akbari, H., Van Lenthe, H., Kulik, W., Wanders, R. J., Frentzen, M.,et al. (2006). Identification and characterization of human cardiolipin synthase. FEBS Lett. 580, 3059–3064. doi: 10.1016/j.febslet.2006.04.054
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Houtkooper, R. H., Rodenburg, R. J., Thiels, C., Van Lenthe, H., Stet, F., Poll-The, B. T.,et al. (2009a). Cardiolipin and monolysocardiolipin analysis in fibroblasts, lymphocytes, and tissues using high-performance liquid chromatography-mass spectrometry as a diagnostic test for Barth syndrome. Anal. Biochem. 387, 230–237. doi: 10.1016/j.ab.2009.01.032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Houtkooper, R. H., Turkenburg, M., Poll-The, B. T., Karall, D., Perez-Cerda, C., Morrone, A.,et al. (2009b). The enigmatic role of tafazzin in cardiolipin metabolism. Biochim. Biophys. Acta 1788, 2003–2014. doi: 10.1016/j.bbamem.2009.07.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Houtkooper, R. H., and Vaz, F. M. (2008). Cardiolipin, the heart of mitochondrial metabolism. Cell. Mol. Life Sci. 65, 2493–2506. doi: 10.1007/s00018-008-8030-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hovius, R., Lambrechts, H., Nicolay, K., and De Kruijff, B. (1990). Improved methods to isolate and subfractionate rat liver mitochondria. Lipid composition of the inner and outer membrane. Biochim. Biophys. Acta 1021, 217–226. doi: 10.1016/0005-2736(90)90036-N
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hovius, R., Thijssen, J., van der Linden, P., Nicolay, K., and De Kruijff, B. (1993). Phospholipid asymmetry of the outer membrane of rat liver mitochondria. Evidence for the presence of cardiolipin on the outside of the outer membrane. FEBS Lett. 330, 71–76. doi: 10.1016/0014-5793(93)80922-H
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hsu, Y. H., Dumlao, D. S., Cao, J., and Dennis, E. A. (2013). Assessing phospholipase A2 activity toward cardiolipin by mass spectrometry. PLoS ONE 8:e59267. doi: 10.1371/journal.pone.0059267
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Huang, H., Gao, Q., Peng, X., Choi, S. Y., Sarma, K., Ren, H.,et al. (2011). piRNA-associated germline nuage formation and spermatogenesis require MitoPLD profusogenic mitochondrial-surface lipid signaling. Dev. Cell 20, 376–387. doi: 10.1016/j.devcel.2011.01.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hullin-Matsuda, F., Kawasaki, K., Delton-Vandenbroucke, I., Xu, Y., Nishijima, M., Lagarde, M.,et al. (2007). De novo biosynthesis of the late endosome lipid, bis(monoacylglycero)phosphate. J. Lipid Res. 48, 1997–2008. doi: 10.1194/jlr.M700154-JLR200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hullin-Matsuda, F., Luquain-Costaz, C., Bouvier, J., and Delton-Vandenbroucke, I. (2009). Bis(monoacylglycero)phosphate, a peculiar phospholipid to control the fate of cholesterol: implications in pathology. Prostaglandins Leukot. Essent. Fatty Acids 81, 313–324. doi: 10.1016/j.plefa.2009.09.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hung, V., Zou, P., Rhee, H. W., Udeshi, N. D., Cracan, V., Svinkina, T.,et al. (2014). Proteomic mapping of the human mitochondrial intermembrane space in live cells via ratiometric APEX tagging. Mol. Cell 55, 332–341. doi: 10.1016/j.molcel.2014.06.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ichida, F., Tsubata, S., Bowles, K. R., Haneda, N., Uese, K., Miyawaki, T.,et al. (2001). Novel gene mutations in patients with left ventricular noncompaction or Barth syndrome. Circulation 103, 1256–1263. doi: 10.1161/01.CIR.103.9.1256
IJlst, L., Wanders, R. J., Ushikubo, S., Kamijo, T., and Hashimoto, T. (1994). Molecular basis of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: identification of the major disease-causing mutation in the alpha-subunit of the mitochondrial trifunctional protein. Biochim. Biophys. Acta 1215, 347–350. doi: 10.1016/0005-2760(94)90064-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Inloes, J. M., Hsu, K. L., Dix, M. M., Viader, A., Masuda, K., Takei, T.,et al. (2014). The hereditary spastic paraplegia-related enzyme DDHD2 is a principal brain triglyceride lipase. Proc. Natl. Acad. Sci. U.S.A. 111, 14924–14929. doi: 10.1073/pnas.1413706111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jacobs, H. T., and Turnbull, D. M. (2005). Nuclear genes and mitochondrial translation: a new class of genetic disease. Trends Genet. 21, 312–314. doi: 10.1016/j.tig.2005.04.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jalmar, O., Francois-Moutal, L., Garcia-Saez, A. J., Perry, M., Granjon, T., Gonzalvez, F.,et al. (2013). Caspase-8 binding to cardiolipin in giant unilamellar vesicles provides a functional docking platform for bid. PLoS ONE 8:e55250. doi: 10.1371/journal.pone.0055250
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jalmar, O., Garcia-Saez, A. J., Berland, L., Gonzalvez, F., and Petit, P. X. (2010). Giant unilamellar vesicles (GUVs) as a new tool for analysis of caspase-8/Bid-FL complex binding to cardiolipin and its functional activity. Cell Death Dis. 1:e103. doi: 10.1038/cddis.2010.81
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jiang, F., Ryan, M. T., Schlame, M., Zhao, M., Gu, Z., Klingenberg, M.,et al. (2000). Absence of cardiolipin in the crd1 null mutant results in decreased mitochondrial membrane potential and reduced mitochondrial function. J. Biol. Chem. 275, 22387–22394. doi: 10.1074/jbc.M909868199
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
John, G. B., Shang, Y., Li, L., Renken, C., Mannella, C. A., Selker, J. M.,et al. (2005). The mitochondrial inner membrane protein mitofilin controls cristae morphology. Mol. Biol. Cell 16, 1543–1554. doi: 10.1091/mbc.E04-08-0697
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Johnson, D. T., Harris, R. A., French, S., Blair, P. V., You, J., Bemis, K. G.,et al. (2007). Tissue heterogeneity of the mammalian mitochondrial proteome. Am. J. Physiol. Cell Physiol. 292, C689–C697. doi: 10.1152/ajpcell.00108.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Johnston, J., Kelley, R. I., Feigenbaum, A., Cox, G. F., Iyer, G. S., Funanage, V. L.,et al. (1997). Mutation characterization and genotype-phenotype correlation in Barth syndrome. Am. J. Hum. Genet. 61, 1053–1058. doi: 10.1086/301604
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jordens, E. Z., Palmieri, L., Huizing, M., Van Den Heuvel, L. P., Sengers, R. C., Dorner, A.,et al. (2002). Adenine nucleotide translocator 1 deficiency associated with Sengers syndrome. Ann. Neurol. 52, 95–99. doi: 10.1002/ana.10214
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Joshi, A. S., Zhou, J., Gohil, V. M., Chen, S., and Greenberg, M. L. (2009). Cellular functions of cardiolipin in yeast. Biochim. Biophys. Acta 1793, 212–218. doi: 10.1016/j.bbamcr.2008.07.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jubinsky, P. T., Messer, A., Bender, J., Morris, R. E., Ciraolo, G. M., Witte, D. P.,et al. (2001). Identification and characterization of Magmas, a novel mitochondria-associated protein involved in granulocyte-macrophage colony-stimulating factor signal transduction. Exp. Hematol. 29, 1392–1402. doi: 10.1016/S0301-472X(01)00749-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jubinsky, P. T., Short, M. K., Mutema, G., and Witte, D. P. (2003). Developmental expression of Magmas in murine tissues and its co-expression with the GM-CSF receptor. J. Histochem. Cytochem. 51, 585–596. doi: 10.1177/002215540305100504
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kagan, V. E., Tyurin, V. A., Jiang, J., Tyurina, Y. Y., Ritov, V. B., Amoscato, A. A.,et al. (2005). Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat. Chem. Biol. 1, 223–232. doi: 10.1038/nchembio727
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Karkucinska-Wieckowska, A., Lebiedzinska, M., Jurkiewicz, E., Pajdowska, M., Trubicka, J., Szymanska-Debinska, T.,et al. (2011). Increased reactive oxygen species (ROS) production and low catalase level in fibroblasts of a girl with MEGDEL association (Leigh syndrome, deafness, 3-methylglutaconic aciduria). Folia Neuropathol. 49, 56–63.
Kelley, R. I., Cheatham, J. P., Clark, B. J., Nigro, M. A., Powell, B. R., Sherwood, G. W.,et al. (1991). X-linked dilated cardiomyopathy with neutropenia, growth retardation, and 3-methylglutaconic aciduria. J. Pediatr. 119, 738–747. doi: 10.1016/S0022-3476(05)80289-6
Khuchua, Z., Yue, Z., Batts, L., and Strauss, A. W. (2006). A zebrafish model of human Barth syndrome reveals the essential role of tafazzin in cardiac development and function. Circ. Res. 99, 201–208. doi: 10.1161/01.RES.0000233378.95325.ce
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kiebish, M. A., Yang, K., Liu, X., Mancuso, D. J., Guan, S., Zhao, Z.,et al. (2013). Dysfunctional cardiac mitochondrial bioenergetic, lipidomic, and signaling in a murine model of Barth syndrome. J. Lipid Res. 54, 1312–1325. doi: 10.1194/jlr.M034728
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kobayashi, T., Beuchat, M. H., Lindsay, M., Frias, S., Palmiter, R. D., Sakuraba, H.,et al. (1999). Late endosomal membranes rich in lysobisphosphatidic acid regulate cholesterol transport. Nat. Cell Biol. 1, 113–118. doi: 10.1038/10084
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kobayashi, T., Stang, E., Fang, K. S., De Moerloose, P., Parton, R. G., and Gruenberg, J. (1998). A lipid associated with the antiphospholipid syndrome regulates endosome structure and function. Nature 392, 193–197. doi: 10.1038/32440
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kol, M. A., De Kroon, A. I., Killian, J. A., and De Kruijff, B. (2004). Transbilayer movement of phospholipids in biogenic membranes. Biochemistry 43, 2673–2681. doi: 10.1021/bi036200f
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kol, M. A., De Kroon, A. I., Rijkers, D. T., Killian, J. A., and De Kruijff, B. (2001). Membrane-spanning peptides induce phospholipid flop: a model for phospholipid translocation across the inner membrane of E. coli. Biochemistry 40, 10500–10506. doi: 10.1021/bi010627+
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Koppen, M., and Langer, T. (2007). Protein degradation within mitochondria: versatile activities of AAA proteases and other peptidases. Crit. Rev. Biochem. Mol. Biol. 42, 221–242. doi: 10.1080/10409230701380452
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kornmann, B., Currie, E., Collins, S. R., Schuldiner, M., Nunnari, J., Weissman, J. S.,et al. (2009). An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science 325, 477–481. doi: 10.1126/science.1175088
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kornmann, B., Osman, C., and Walter, P. (2011). The conserved GTPase Gem1 regulates endoplasmic reticulum-mitochondria connections. Proc. Natl. Acad. Sci. U.S.A. 108, 14151–14156. doi: 10.1073/pnas.1111314108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kozany, C., Mokranjac, D., Sichting, M., Neupert, W., and Hell, K. (2004). The J domain-related cochaperone Tim16 is a constituent of the mitochondrial TIM23 preprotein translocase. Nat. Struct. Mol. Biol. 11, 234–241. doi: 10.1038/nsmb734
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Krebs, J. J., Hauser, H., and Carafoli, E. (1979). Asymmetric distribution of phospholipids in the inner membrane of beef heart mitochondria. J. Biol. Chem. 254, 5308–5316.
Kroemer, G., Petit, P., Zamzami, N., Vayssiere, J. L., and Mignotte, B. (1995). The biochemistry of programmed cell death. FASEB J. 9, 1277–1287.
Kuchler, K., Daum, G., and Paltauf, F. (1986). Subcellular and submitochondrial localization of phospholipid-synthesizing enzymes in Saccharomyces cerevisiae. J. Bacteriol. 165, 901–910.
Kutik, S., Rissler, M., Guan, X. L., Guiard, B., Shui, G., Gebert, N.,et al. (2008). The translocator maintenance protein Tam41 is required for mitochondrial cardiolipin biosynthesis. J. Cell Biol. 183, 1213–1221. doi: 10.1083/jcb.200806048
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kuwana, T., Mackey, M. R., Perkins, G., Ellisman, M. H., Latterich, M., Schneiter, R.,et al. (2002). Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell 111, 331–342. doi: 10.1016/S0092-8674(02)01036-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lahiri, S., Chao, J. T., Tavassoli, S., Wong, A. K., Choudhary, V., Young, B. P.,et al. (2014). A conserved endoplasmic reticulum membrane protein complex (EMC) facilitates phospholipid transfer from the ER to mitochondria. PLoS Biol. 12:e1001969. doi: 10.1371/journal.pbio.1001969
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lalive D’Epinay, S., Rampini, S., Arbenz, U., Steinmann, B., and Gitzelmann, R. (1986). [Infantile cataract, hypertrophic cardiomyopathy and lactic acidosis following minor muscular exertion – a little known metabolic disease]. Klin. Monbl. Augenheilkd. 189, 482–485. doi: 10.1055/s-2008-1050858
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lapuente-Brun, E., Moreno-Loshuertos, R., Acin-Perez, R., Latorre-Pellicer, A., Colas, C., Balsa, E.,et al. (2013). Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science 340, 1567–1570. doi: 10.1126/science.1230381
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lecocq, J., and Ballou, C. E. (1964). On the structure of cardiolipin. Biochemistry 3, 976–980. doi: 10.1021/bi00895a023
Lewin, T. M., Schwerbrock, N. M., Lee, D. P., and Coleman, R. A. (2004). Identification of a new glycerol-3-phosphate acyltransferase isoenzyme, mtGPAT2, in mitochondria. J. Biol. Chem. 279, 13488–13495. doi: 10.1074/jbc.M314032200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lewis, R. N., and McElhaney, R. N. (2009). The physicochemical properties of cardiolipin bilayers and cardiolipin-containing lipid membranes. Biochim. Biophys. Acta 1788, 2069–2079. doi: 10.1016/j.bbamem.2009.03.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, J., Liu, X., Wang, H., Zhang, W., Chan, D. C., and Shi, Y. (2012). Lysocardiolipin acyltransferase 1 (ALCAT1) controls mitochondrial DNA fidelity and biogenesis through modulation of MFN2 expression. Proc. Natl. Acad. Sci. U.S.A. 109, 6975–6980. doi: 10.1073/pnas.1120043109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, J., Romestaing, C., Han, X., Li, Y., Hao, X., Wu, Y.,et al. (2010). Cardiolipin remodeling by ALCAT1 links oxidative stress and mitochondrial dysfunction to obesity. Cell Metab. 12, 154–165. doi: 10.1016/j.cmet.2010.07.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, K., Warner, C. K., Hodge, J. A., Minoshima, S., Kudoh, J., Fukuyama, R.,et al. (1989). A human muscle adenine nucleotide translocator gene has four exons, is located on chromosome 4, and is differentially expressed. J. Biol. Chem. 264, 13998–14004.
Liguori, R., Giannoccaro, M. P., Arnoldi, A., Citterio, A., Tonon, C., Lodi, R.,et al. (2014). Impairment of brain and muscle energy metabolism detected by magnetic resonance spectroscopy in hereditary spastic paraparesis type 28 patients with DDHD1 mutations. J. Neurol. 261, 1789–1793. doi: 10.1007/s00415-014-7418-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lill, R., Hoffmann, B., Molik, S., Pierik, A. J., Rietzschel, N., Stehling, O.,et al. (2012). The role of mitochondria in cellular iron-sulfur protein biogenesis and iron metabolism. Biochim. Biophys. Acta 1823, 1491–1508. doi: 10.1016/j.bbamcr.2012.05.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, J., Chen, J., Dai, Q., and Lee, R. M. (2003a). Phospholipid scramblase 3 is the mitochondrial target of protein kinase C delta-induced apoptosis. Cancer Res. 63, 1153–1156.
Liu, J., Dai, Q., Chen, J., Durrant, D., Freeman, A., Liu, T.,et al. (2003b). Phospholipid scramblase 3 controls mitochondrial structure, function, and apoptotic response. Mol. Cancer Res. 1, 892–902.
Liu, X., Ye, B., Miller, S., Yuan, H., Zhang, H., Tian, L.,et al. (2012). Ablation of ALCAT1 mitigates hypertrophic cardiomyopathy through effects on oxidative stress and mitophagy. Mol. Cell. Biol. 32, 4493–4504. doi: 10.1128/MCB.01092-12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lu, B., Jiang, Y. J., Zhou, Y., Xu, F. Y., Hatch, G. M., and Choy, P. C. (2005). Cloning and characterization of murine 1-acyl-sn-glycerol 3-phosphate acyltransferases and their regulation by PPARalpha in murine heart. Biochem. J. 385, 469–477. doi: 10.1042/BJ20041348
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lu, B., Xu, F. Y., Jiang, Y. J., Choy, P. C., Hatch, G. M., Grunfeld, C.,et al. (2006). Cloning and characterization of a cDNA encoding human cardiolipin synthase (hCLS1). J. Lipid Res. 47, 1140–1145. doi: 10.1194/jlr.C600004-JLR200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lucken-Ardjomande, S., Montessuit, S., and Martinou, J. C. (2008). Contributions to Bax insertion and oligomerization of lipids of the mitochondrial outer membrane. Cell Death Differ. 15, 929–937. doi: 10.1038/cdd.2008.9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Luft, R., Ikkos, D., Palmieri, G., Ernster, L., and Afzelius, B. (1962). A case of severe hypermetabolism of nonthyroid origin with a defect in the maintenance of mitochondrial respiratory control: a correlated clinical, biochemical, and morphological study. J. Clin. Invest. 41, 1776–1804. doi: 10.1172/JCI104637
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lumish, H. S., Yang, Y., Xia, F., Wilson, A., and Chung, W. K. (2014). The expanding MEGDEL phenotype: optic nerve atrophy, microcephaly, and myoclonic epilepsy in a child with SERAC1 mutations. JIMD Rep. 16, 75–79. doi: 10.1007/8904_2014_322
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Magariello, A., Citrigno, L., Zuchner, S., Gonzalez, M., Patitucci, A., Sofia, V.,et al. (2014). Further evidence that DDHD2 gene mutations cause autosomal recessive hereditary spastic paraplegia with thin corpus callosum. Eur. J. Neurol. 21, e25–e26. doi: 10.1111/ene.12305
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Malhotra, A., Edelman-Novemsky, I., Xu, Y., Plesken, H., Ma, J., Schlame, M.,et al. (2009a). Role of calcium-independent phospholipase A2 in the pathogenesis of Barth syndrome. Proc. Natl. Acad. Sci. U.S.A. 106, 2337–2341. doi: 10.1073/pnas.0811224106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Malhotra, A., Xu, Y., Ren, M., and Schlame, M. (2009b). Formation of molecular species of mitochondrial cardiolipin. 1. A novel transacylation mechanism to shuttle fatty acids between sn-1 and sn-2 positions of multiple phospholipid species. Biochim. Biophys. Acta 1791, 314–320. doi: 10.1016/j.bbalip.2009.01.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mancuso, D. J., Jenkins, C. M., and Gross, R. W. (2000). The genomic organization, complete mRNA sequence, cloning, and expression of a novel human intracellular membrane-associated calcium-independent phospholipase A(2). J. Biol. Chem. 275, 9937–9945. doi: 10.1074/jbc.275.14.9937
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mancuso, D. J., Sims, H. F., Han, X., Jenkins, C. M., Guan, S. P., Yang, K.,et al. (2007). Genetic ablation of calcium-independent phospholipase A2gamma leads to alterations in mitochondrial lipid metabolism and function resulting in a deficient mitochondrial bioenergetic phenotype. J. Biol. Chem. 282, 34611–34622. doi: 10.1074/jbc.M707795200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mason, R. J., Stossel, T. P., and Vaughan, M. (1972). Lipids of alveolar macrophages, polymorphonuclear leukocytes, and their phagocytic vesicles. J. Clin. Invest. 51, 2399–2407. doi: 10.1172/JCI107052
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Matsuzaki, H., Fujimoto, T., Tanaka, M., and Shirasawa, S. (2013). Tespa1 is a novel component of mitochondria-associated endoplasmic reticulum membranes and affects mitochondrial calcium flux. Biochem. Biophys. Res. Commun. 433, 322–326. doi: 10.1016/j.bbrc.2013.02.099
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mayer, M. P., and Bukau, B. (2005). Hsp70 chaperones: cellular functions and molecular mechanism. Cell. Mol. Life Sci. 62, 670–684. doi: 10.1007/s00018-004-4464-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mayr, J. A., Haack, T. B., Graf, E., Zimmermann, F. A., Wieland, T., Haberberger, B.,et al. (2012). Lack of the mitochondrial protein acylglycerol kinase causes Sengers syndrome. Am. J. Hum. Genet. 90, 314–320. doi: 10.1016/j.ajhg.2011.12.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McKenzie, M., Lazarou, M., Thorburn, D. R., and Ryan, M. T. (2006). Mitochondrial respiratory chain supercomplexes are destabilized in Barth syndrome patients. J. Mol. Biol. 361, 462–469. doi: 10.1016/j.jmb.2006.06.057
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mehawej, C., Delahodde, A., Legeai-Mallet, L., Delague, V., Kaci, N., Desvignes, J. P.,et al. (2014). The impairment of MAGMAS function in human is responsible for a severe skeletal dysplasia. PLoS Genet. 10:e1004311. doi: 10.1371/journal.pgen.1004311
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Merkwirth, C., Dargazanli, S., Tatsuta, T., Geimer, S., Lower, B., Wunderlich, F. T.,et al. (2008). Prohibitins control cell proliferation and apoptosis by regulating OPA1-dependent cristae morphogenesis in mitochondria. Genes Dev. 22, 476–488. doi: 10.1101/gad.460708
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Merkwirth, C., and Langer, T. (2008). Mitofusin 2 builds a bridge between ER and mitochondria. Cell 135, 1165–1167. doi: 10.1016/j.cell.2008.12.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Milon, L., Meyer, P., Chiadmi, M., Munier, A., Johansson, M., Karlsson, A.,et al. (2000). The human nm23-H4 gene product is a mitochondrial nucleoside diphosphate kinase. J. Biol. Chem. 275, 14264–14272. doi: 10.1074/jbc.275.19.14264
Mobius, W., Van Donselaar, E., Ohno-Iwashita, Y., Shimada, Y., Heijnen, H. F., Slot, J. W.,et al. (2003). Recycling compartments and the internal vesicles of multivesicular bodies harbor most of the cholesterol found in the endocytic pathway. Traffic 4, 222–231. doi: 10.1034/j.1600-0854.2003.00072.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mokranjac, D., Sichting, M., Neupert, W., and Hell, K. (2003). Tim14, a novel key component of the import motor of the TIM23 protein translocase of mitochondria. EMBO J. 22, 4945–4956. doi: 10.1093/emboj/cdg485
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mokranjac, D., Sichting, M., Popov-Celeketic, D., Berg, A., Hell, K., and Neupert, W. (2005). The import motor of the yeast mitochondrial TIM23 preprotein translocase contains two different J proteins, Tim14 and Mdj2. J. Biol. Chem. 280, 31608–31614. doi: 10.1074/jbc.M502397200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Montessuit, S., Somasekharan, S. P., Terrones, O., Lucken-Ardjomande, S., Herzig, S., Schwarzenbacher, R.,et al. (2010). Membrane remodeling induced by the dynamin-related protein Drp1 stimulates Bax oligomerization. Cell 142, 889–901. doi: 10.1016/j.cell.2010.08.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mootha, V. K., Bunkenborg, J., Olsen, J. V., Hjerrild, M., Wisniewski, J. R., Stahl, E.,et al. (2003). Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell 115, 629–640. doi: 10.1016/S0092-8674(03)00926-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Morava, E., Sengers, R., Ter Laak, H., Van Den Heuvel, L., Janssen, A., Trijbels, F.,et al. (2004). Congenital hypertrophic cardiomyopathy, cataract, mitochondrial myopathy and defective oxidative phosphorylation in two siblings with Sengers-like syndrome. Eur. J. Pediatr. 163, 467–471. doi: 10.1007/s00431-004-1465-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Morikawa, R. K., Aoki, J., Kano, F., Murata, M., Yamamoto, A., Tsujimoto, M.,et al. (2009). Intracellular phospholipase A1gamma (iPLA1gamma) is a novel factor involved in coat protein complex I- and Rab6-independent retrograde transport between the endoplasmic reticulum and the Golgi complex. J. Biol. Chem. 284, 26620–26630. doi: 10.1074/jbc.M109.038869
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moro, F., Okamoto, K., Donzeau, M., Neupert, W., and Brunner, M. (2002). Mitochondrial protein import: molecular basis of the ATP-dependent interaction of MtHsp70 with Tim44. J. Biol. Chem. 277, 6874–6880. doi: 10.1074/jbc.M107935200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moro, F., Sirrenberg, C., Schneider, H. C., Neupert, W., and Brunner, M. (1999). The TIM17.23 preprotein translocase of mitochondria: composition and function in protein transport into the matrix. EMBO J. 18, 3667–3675. doi: 10.1093/emboj/18.13.3667
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mun, J. Y., Lee, T. H., Kim, J. H., Yoo, B. H., Bahk, Y. Y., Koo, H. S.,et al. (2010). Caenorhabditis elegans mitofilin homologs control the morphology of mitochondrial cristae and influence reproduction and physiology. J. Cell. Physiol. 224, 748–756. doi: 10.1002/jcp.22177
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nagle, C. A., Vergnes, L., Dejong, H., Wang, S., Lewin, T. M., Reue, K.,et al. (2008). Identification of a novel sn-glycerol-3-phosphate acyltransferase isoform, GPAT4, as the enzyme deficient in Agpat6–/– mice. J. Lipid Res. 49, 823–831. doi: 10.1194/jlr.M700592-JLR200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nakajima, K., Sonoda, H., Mizoguchi, T., Aoki, J., Arai, H., Nagahama, M.,et al. (2002). A novel phospholipase A1 with sequence homology to a mammalian Sec23p-interacting protein, p125. J. Biol. Chem. 277, 11329–11335. doi: 10.1074/jbc.M111092200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Naviaux, R. K. (2004). Developing a systematic approach to the diagnosis and classification of mitochondrial disease. Mitochondrion 4, 351–361. doi: 10.1016/j.mito.2004.07.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nguyen, T. T., Lewandowska, A., Choi, J. Y., Markgraf, D. F., Junker, M., Bilgin, M.,et al. (2012). Gem1 and ERMES do not directly affect phosphatidylserine transport from ER to mitochondria or mitochondrial inheritance. Traffic 13, 880–890. doi: 10.1111/j.1600-0854.2012.01352.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nicolay, K., Rojo, M., Wallimann, T., Demel, R., and Hovius, R. (1990). The role of contact sites between inner and outer mitochondrial membrane in energy transfer. Biochim. Biophys. Acta 1018, 229–233. doi: 10.1016/0005-2728(90)90255-3
Ohtsuka, K., and Hata, M. (2000). Mammalian HSP40/DNAJ homologs: cloning of novel cDNAs and a proposal for their classification and nomenclature. Cell Stress Chaperones 5, 98–112. doi: 10.1379/1466-1268(2000)005<0098:MHDHCO>2.0.CO;2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ojala, T., Polinati, P., Manninen, T., Hiippala, A., Rajantie, J., Karikoski, R.,et al. (2012). New mutation of mitochondrial DNAJC19 causing dilated and noncompaction cardiomyopathy, anemia, ataxia, and male genital anomalies. Pediatr. Res. 72, 432–437. doi: 10.1038/pr.2012.92
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Orstavik, K. H., Orstavik, R. E., Naumova, A. K., D’Adamo, P., Gedeon, A., Bolhuis, P. A.,et al. (1998). X chromosome inactivation in carriers of Barth syndrome. Am. J. Hum. Genet. 63, 1457–1463. doi: 10.1086/302095
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Osman, C., Haag, M., Potting, C., Rodenfels, J., Dip, P. V., Wieland, F. T.,et al. (2009a). The genetic interactome of prohibitins: coordinated control of cardiolipin and phosphatidylethanolamine by conserved regulators in mitochondria. J. Cell Biol. 184, 583–596. doi: 10.1083/jcb.200810189
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Osman, C., Merkwirth, C., and Langer, T. (2009b). Prohibitins and the functional compartmentalization of mitochondrial membranes. J. Cell Sci. 122, 3823–3830. doi: 10.1242/jcs.037655
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Osman, C., Haag, M., Wieland, F. T., Brugger, B., and Langer, T. (2010). A mitochondrial phosphatase required for cardiolipin biosynthesis: the PGP phosphatase Gep4. EMBO J. 29, 1976–1987. doi: 10.1038/emboj.2010.98
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Osman, C., Voelker, D. R., and Langer, T. (2011). Making heads or tails of phospholipids in mitochondria. J. Cell Biol. 192, 7–16. doi: 10.1083/jcb.201006159
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pagliarini, D. J., Calvo, S. E., Chang, B., Sheth, S. A., Vafai, S. B., Ong, S. E.,et al. (2008). A mitochondrial protein compendium elucidates complex I disease biology. Cell 134, 112–123. doi: 10.1016/j.cell.2008.06.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pane, A., Wehr, K., and Schupbach, T. (2007). Zucchini and squash encode two putative nucleases required for rasiRNA production in the Drosophila germline. Dev. Cell 12, 851–862. doi: 10.1016/j.devcel.2007.03.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pangborn, M. (1942). Isolation and purification of a serologically active phospholipid from beef heart. J. Biol. Chem. 143, 247–256.
Paradies, G., Petrosillo, G., Paradies, V., and Ruggiero, F. M. (2009). Role of cardiolipin peroxidation and Ca2+ in mitochondrial dysfunction and disease. Cell Calcium 45, 643–650. doi: 10.1016/j.ceca.2009.03.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Perez-Martinez, X., Funes, S., Camacho-Villasana, Y., Marjavaara, S., Tavares-Carreon, F., and Shingu-Vazquez, M. (2008). Protein synthesis and assembly in mitochondrial disorders. Curr. Top. Med. Chem. 8, 1335–1350. doi: 10.2174/156802608786141124
Pfanner, N., van der Laan, M., Amati, P., Capaldi, R. A., Caudy, A. A., Chacinska, A.,et al. (2014). Uniform nomenclature for the mitochondrial contact site and cristae organizing system. J. Cell Biol. 204, 1083–1086. doi: 10.1083/jcb.201401006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pfeiffer, K., Gohil, V., Stuart, R. A., Hunte, C., Brandt, U., Greenberg, M. L.,et al. (2003). Cardiolipin stabilizes respiratory chain supercomplexes. J. Biol. Chem. 278, 52873–52880. doi: 10.1074/jbc.M308366200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Phoon, C. K., Acehan, D., Schlame, M., Stokes, D. L., Edelman-Novemsky, I., Yu, D.,et al. (2012). Tafazzin knockdown in mice leads to a developmental cardiomyopathy with early diastolic dysfunction preceding myocardial noncompaction. J. Am. Heart Assoc. 1:e000455. doi: 10.1161/JAHA.111.000455
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pitkanen, S., Merante, F., Mcleod, D. R., Applegarth, D., Tong, T., and Robinson, B. H. (1996). Familial cardiomyopathy with cataracts and lactic acidosis: a defect in complex I (NADH-dehydrogenase) of the mitochondria respiratory chain. Pediatr. Res. 39, 513–521. doi: 10.1203/00006450-199603000-00021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Poorthuis, B. J., and Hostetler, K. Y. (1976). Studies on the subcellular localization and properties of bis(monoacylglyceryl)phosphate biosynthesis in rat liver. J. Biol. Chem. 251, 4596–4602.
Potting, C., Tatsuta, T., Konig, T., Haag, M., Wai, T., Aaltonen, M. J.,et al. (2013). TRIAP1/PRELI complexes prevent apoptosis by mediating intramitochondrial transport of phosphatidic acid. Cell Metab. 18, 287–295. doi: 10.1016/j.cmet.2013.07.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Potting, C., Wilmes, C., Engmann, T., Osman, C., and Langer, T. (2010). Regulation of mitochondrial phospholipids by Ups1/PRELI-like proteins depends on proteolysis and Mdm35. EMBO J. 29, 2888–2898. doi: 10.1038/emboj.2010.169
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Powers, C., Huang, Y., Strauss, A., and Khuchua, Z. (2013). Diminished exercise capacity and mitochondrial bc1 complex deficiency in Tafazzin-knockdown mice. Front. Physiol. 4:74. doi: 10.3389/fphys.2013.00074
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Prasad, S. S., Garg, A., and Agarwal, A. K. (2011). Enzymatic activities of the human AGPAT isoform 3 and isoform 5: localization of AGPAT5 to mitochondria. J. Lipid Res. 52, 451–462. doi: 10.1194/jlr.M007575
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reue, K., and Dwyer, J. R. (2009). Lipin proteins and metabolic homeostasis. J. Lipid Res. 50(Suppl.), S109–S114. doi: 10.1194/jlr.R800052-JLR200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rhee, H. W., Zou, P., Udeshi, N. D., Martell, J. D., Mootha, V. K., Carr, S. A.,et al. (2013). Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 339, 1328–1331. doi: 10.1126/science.1230593
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Richter-Dennerlein, R., Korwitz, A., Haag, M., Tatsuta, T., Dargazanli, S., Baker, M.,et al. (2014). DNAJC19, a mitochondrial cochaperone associated with cardiomyopathy, forms a complex with prohibitins to regulate cardiolipin remodeling. Cell Metab. 20, 158–171. doi: 10.1016/j.cmet.2014.04.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ronvelia, D., Greenwood, J., Platt, J., Hakim, S., and Zaragoza, M. V. (2012). Intrafamilial variability for novel TAZ gene mutation: Barth syndrome with dilated cardiomyopathy and heart failure in an infant and left ventricular noncompaction in his great-uncle. Mol. Genet. Metab. 107, 428–432. doi: 10.1016/j.ymgme.2012.09.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Russell, L., and Frank, B. (1978). Ultrastructural characterization of nuage in spermatocytes of the rat testis. Anat. Rec. 190, 79–97. doi: 10.1002/ar.1091900108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sarig, O., Goldsher, D., Nousbeck, J., Fuchs-Telem, D., Cohen-Katsenelson, K., Iancu, T. C.,et al. (2013). Infantile mitochondrial hepatopathy is a cardinal feature of MEGDEL syndrome (3-methylglutaconic aciduria type IV with sensorineural deafness, encephalopathy and Leigh-like syndrome) caused by novel mutations in SERAC1. Am. J. Med. Genet. A 161, 2204–2215. doi: 10.1002/ajmg.a.36059
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sato, S., Inoue, H., Kogure, T., Tagaya, M., and Tani, K. (2010). Golgi-localized KIAA0725p regulates membrane trafficking from the Golgi apparatus to the plasma membrane in mammalian cells. FEBS Lett. 584, 4389–4395. doi: 10.1016/j.febslet.2010.09.047
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schaefer, A. M., Mcfarland, R., Blakely, E. L., He, L., Whittaker, R. G., Taylor, R. W.,et al. (2008). Prevalence of mitochondrial DNA disease in adults. Ann. Neurol. 63, 35–39. doi: 10.1002/ana.21217
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schagger, H., and Pfeiffer, K. (2000). Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. 19, 1777–1783. doi: 10.1093/emboj/19.8.1777
Schlame, M., Acehan, D., Berno, B., Xu, Y., Valvo, S., Ren, M.,et al. (2012a). The physical state of lipid substrates provides transacylation specificity for tafazzin. Nat. Chem. Biol. 8, 862–869. doi: 10.1038/nchembio.1064
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schlame, M., Blais, S., Edelman-Novemsky, I., Xu, Y., Montecillo, F., Phoon, C. K.,et al. (2012b). Comparison of cardiolipins from Drosophila strains with mutations in putative remodeling enzymes. Chem. Phys. Lipids 165, 512–519. doi: 10.1016/j.chemphyslip.2012.03.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schlame, M., Brody, S., and Hostetler, K. Y. (1993). Mitochondrial cardiolipin in diverse eukaryotes. Comparison of biosynthetic reactions and molecular acyl species. Eur. J. Biochem. 212, 727–735. doi: 10.1111/j.1432-1033.1993.tb17711.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schlame, M., and Haldar, D. (1993). Cardiolipin is synthesized on the matrix side of the inner membrane in rat liver mitochondria. J. Biol. Chem. 268, 74–79.
Schlame, M., Kelley, R. I., Feigenbaum, A., Towbin, J. A., Heerdt, P. M., Schieble, T.,et al. (2003). Phospholipid abnormalities in children with Barth syndrome. J. Am. Coll. Cardiol. 42, 1994–1999. doi: 10.1016/j.jacc.2003.06.015
Schlame, M., and Otten, D. (1991). Analysis of cardiolipin molecular species by high-performance liquid chromatography of its derivative 1,3-bisphosphatidyl-2-benzoyl-sn-glycerol dimethyl ester. Anal. Biochem. 195, 290–295. doi: 10.1016/0003-2697(91)90332-N
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schlame, M., Ren, M., Xu, Y., Greenberg, M. L., and Haller, I. (2005). Molecular symmetry in mitochondrial cardiolipins. Chem. Phys. Lipids 138, 38–49. doi: 10.1016/j.chemphyslip.2005.08.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schlame, M., Rua, D., and Greenberg, M. L. (2000). The biosynthesis and functional role of cardiolipin. Prog. Lipid Res. 39, 257–288. doi: 10.1016/S0163-7827(00)00005-9
Schlame, M., and Rustow, B. (1990). Lysocardiolipin formation and reacylation in isolated rat liver mitochondria. Biochem. J. 272, 589–595.
Schlattner, U., Tokarska-Schlattner, M., Ramirez, S., Tyurina, Y. Y., Amoscato, A. A., Mohammadyani, D.,et al. (2013). Dual function of mitochondrial Nm23-H4 protein in phosphotransfer and intermembrane lipid transfer: a cardiolipin-dependent switch. J. Biol. Chem. 288, 111–121. doi: 10.1074/jbc.M112.408633
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schlattner, U., Tokarska-Schlattner, M., Rousseau, D., Boissan, M., Mannella, C., Epand, R.,et al. (2014). Mitochondrial cardiolipin/phospholipid trafficking: the role of membrane contact site complexes and lipid transfer proteins. Chem. Phys. Lipids 179, 32–41. doi: 10.1016/j.chemphyslip.2013.12.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schmidt, J. A., Yvone, G. M., and Brown, W. J. (2010). Membrane topology of human AGPAT3 (LPAAT3). Biochem. Biophys. Res. Commun. 397, 661–667. doi: 10.1016/j.bbrc.2010.05.149
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schneider, H. C., Berthold, J., Bauer, M. F., Dietmeier, K., Guiard, B., Brunner, M.,et al. (1994). Mitochondrial Hsp70/MIM44 complex facilitates protein import. Nature 371, 768–774. doi: 10.1038/371768a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schusdziarra, C., Blamowska, M., Azem, A., and Hell, K. (2013). Methylation-controlled J-protein MCJ acts in the import of proteins into human mitochondria. Hum. Mol. Genet. 22, 1348–1357. doi: 10.1093/hmg/dds541
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schuurs-Hoeijmakers, J. H., Geraghty, M. T., Kamsteeg, E. J., Ben-Salem, S., De Bot, S. T., Nijhof, B.,et al. (2012). Mutations in DDHD2, encoding an intracellular phospholipase A(1), cause a recessive form of complex hereditary spastic paraplegia. Am. J. Hum. Genet. 91, 1073–1081. doi: 10.1016/j.ajhg.2012.10.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schwall, C. T., Greenwood, V. L., and Alder, N. N. (2012). The stability and activity of respiratory Complex II is cardiolipin-dependent. Biochim. Biophys. Acta 1817, 1588–1596. doi: 10.1016/j.bbabio.2012.04.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sengers, R. C., Trijbels, J. M., Willems, J. L., Daniels, O., and Stadhouders, A. M. (1975). Congenital cataract and mitochondrial myopathy of skeletal and heart muscle associated with lactic acidosis after exercise. J. Pediatr. 86, 873–880. doi: 10.1016/S0022-3476(75)80217-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shen, H., Heacock, P. N., Clancey, C. J., and Dowhan, W. (1996). The CDS1 gene encoding CDP-diacylglycerol synthase in Saccharomyces cerevisiae is essential for cell growth. J. Biol. Chem. 271, 789–795. doi: 10.1074/jbc.271.2.789
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shinzawa-Itoh, K., Aoyama, H., Muramoto, K., Terada, H., Kurauchi, T., Tadehara, Y.,et al. (2007). Structures and physiological roles of 13 integral lipids of bovine heart cytochrome c oxidase. EMBO J. 26, 1713–1725. doi: 10.1038/sj.emboj.7601618
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Simbeni, R., Pon, L., Zinser, E., Paltauf, F., and Daum, G. (1991). Mitochondrial membrane contact sites of yeast. Characterization of lipid components and possible involvement in intramitochondrial translocation of phospholipids. J. Biol. Chem. 266, 10047–10049.
Simon, G., and Rouser, G. (1969). Species variations in phospholipid class distribution of organs. II. Heart and skeletal muscle. Lipids 4, 607–614. doi: 10.1007/BF02531048
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sims, H. F., Brackett, J. C., Powell, C. K., Treem, W. R., Hale, D. E., Bennett, M. J.,et al. (1995). The molecular basis of pediatric long chain 3-hydroxyacyl-CoA dehydrogenase deficiency associated with maternal acute fatty liver of pregnancy. Proc. Natl. Acad. Sci. U.S.A. 92, 841–845. doi: 10.1073/pnas.92.3.841
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sinha, D., Joshi, N., Chittoor, B., Samji, P., and D’Silva, P. (2010). Role of magmas in protein transport and human mitochondria biogenesis. Hum. Mol. Genet. 19, 1248–1262. doi: 10.1093/hmg/ddq002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sinha, D., Srivastava, S., Krishna, L., and D’Silva, P. (2014). Unraveling the intricate organization of mammalian mitochondrial presequence translocases: existence of multiple translocases for maintenance of mitochondrial function. Mol. Cell. Biol. 34, 1757–1775. doi: 10.1128/MCB.01527-13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Siriwardena, K., Mackay, N., Levandovskiy, V., Blaser, S., Raiman, J., Kantor, P. F.,et al. (2013). Mitochondrial citrate synthase crystals: novel finding in Sengers syndrome caused by acylglycerol kinase (AGK) mutations. Mol. Genet. Metab. 108, 40–50. doi: 10.1016/j.ymgme.2012.11.282
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Smeitink, J. A., Sengers, R. C., Trijbels, J. M., Ruitenbeek, W., Daniels, O., Stadhouders, A. M.,et al. (1989). Fatal neonatal cardiomyopathy associated with cataract and mitochondrial myopathy. Eur. J. Pediatr. 148, 656–659. doi: 10.1007/BF00441527
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Soustek, M. S., Falk, D. J., Mah, C. S., Toth, M. J., Schlame, M., Lewin, A. S.,et al. (2011). Characterization of a transgenic short hairpin RNA-induced murine model of Tafazzin deficiency. Hum. Gene Ther. 22, 865–871. doi: 10.1089/hum.2010.199
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sparagna, G. C., and Lesnefsky, E. J. (2009). Cardiolipin remodeling in the heart. J. Cardiovasc. Pharmacol. 53, 290–301. doi: 10.1097/FJC.0b013e31819b5461
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sparkes, R., Patton, D., and Bernier, F. (2007). Cardiac features of a novel autosomal recessive dilated cardiomyopathic syndrome due to defective importation of mitochondrial protein. Cardiol. Young 17, 215–217. doi: 10.1017/S1047951107000042
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sperka-Gottlieb, C. D., Hermetter, A., Paltauf, F., and Daum, G. (1988). Lipid topology and physical properties of the outer mitochondrial membrane of the yeast, Saccharomyces cerevisiae. Biochim. Biophys. Acta 946, 227–234. doi: 10.1016/0005-2736(88)90397-5
Starck, S. R., Jiang, V., Pavon-Eternod, M., Prasad, S., Mccarthy, B., Pan, T.,et al. (2012). Leucine-tRNA initiates at CUG start codons for protein synthesis and presentation by MHC class I. Science 336, 1719–1723. doi: 10.1126/science.1220270
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stavrovskaya, I. G., Bird, S. S., Marur, V. R., Sniatynski, M. J., Baranov, S. V., Greenberg, H. K.,et al. (2013). Dietary macronutrients modulate the fatty acyl composition of rat liver mitochondrial cardiolipins. J. Lipid Res. 54, 2623–2635. doi: 10.1194/jlr.M036285
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Steward, C. G., Newbury-Ecob, R. A., Hastings, R., Smithson, S. F., Tsai-Goodman, B., Quarrell, O. W.,et al. (2010). Barth syndrome: an X-linked cause of fetal cardiomyopathy and stillbirth. Prenat. Diagn. 30, 970–976. doi: 10.1002/pd.2599
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Strauss, M., Hofhaus, G., Schroder, R. R., and Kuhlbrandt, W. (2008). Dimer ribbons of ATP synthase shape the inner mitochondrial membrane. EMBO J. 27, 1154–1160. doi: 10.1038/emboj.2008.35
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tamura, Y., Harada, Y., Nishikawa, S., Yamano, K., Kamiya, M., Shiota, T.,et al. (2013). Tam41 is a CDP-diacylglycerol synthase required for cardiolipin biosynthesis in mitochondria. Cell Metab. 17, 709–718. doi: 10.1016/j.cmet.2013.03.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tamura, Y., Harada, Y., Yamano, K., Watanabe, K., Ishikawa, D., Ohshima, C.,et al. (2006). Identification of Tam41 maintaining integrity of the TIM23 protein translocator complex in mitochondria. J. Cell Biol. 174, 631–637. doi: 10.1083/jcb.200603087
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tamura, Y., Iijima, M., and Sesaki, H. (2010). Mdm35p imports Ups proteins into the mitochondrial intermembrane space by functional complex formation. EMBO J. 29, 2875–2887. doi: 10.1038/emboj.2010.149
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tamura, Y., Onguka, O., Hobbs, A. E., Jensen, R. E., Iijima, M., Claypool, S. M.,et al. (2012). Role for two conserved intermembrane space proteins, Ups1p and Ups2p, [corrected] in intra-mitochondrial phospholipid trafficking. J. Biol. Chem. 287, 15205–15218. doi: 10.1074/jbc.M111.338665
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tatsuta, T., Model, K., and Langer, T. (2005). Formation of membrane-bound ring complexes by prohibitins in mitochondria. Mol. Biol. Cell 16, 248–259. doi: 10.1091/mbc.E04-09-0807
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Taylor, S. W., Fahy, E., Zhang, B., Glenn, G. M., Warnock, D. E., Wiley, S.,et al. (2003). Characterization of the human heart mitochondrial proteome. Nat. Biotechnol. 21, 281–286. doi: 10.1038/nbt793
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Taylor, W. A., and Hatch, G. M. (2003). Purification and characterization of monolysocardiolipin acyltransferase from pig liver mitochondria. J. Biol. Chem. 278, 12716–12721. doi: 10.1074/jbc.M210329200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Taylor, W. A., and Hatch, G. M. (2009). Identification of the human mitochondrial linoleoyl-coenzyme A monolysocardiolipin acyltransferase (MLCL AT-1). J. Biol. Chem. 284, 30360–30371. doi: 10.1074/jbc.M109.048322
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Taylor, W. A., Mejia, E. M., Mitchell, R. W., Choy, P. C., Sparagna, G. C., and Hatch, G. M. (2012). Human trifunctional protein alpha links cardiolipin remodeling to beta-oxidation. PLoS ONE 7:e48628. doi: 10.1371/journal.pone.0048628
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tesson, C., Nawara, M., Salih, M. A., Rossignol, R., Zaki, M. S., Al Balwi, M.,et al. (2012). Alteration of fatty-acid-metabolizing enzymes affects mitochondrial form and function in hereditary spastic paraplegia. Am. J. Hum. Genet. 91, 1051–1064. doi: 10.1016/j.ajhg.2012.11.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tokarska-Schlattner, M., Boissan, M., Munier, A., Borot, C., Mailleau, C., Speer, O.,et al. (2008). The nucleoside diphosphate kinase D (NM23-H4) binds the inner mitochondrial membrane with high affinity to cardiolipin and couples nucleotide transfer with respiration. J. Biol. Chem. 283, 26198–26207. doi: 10.1074/jbc.M803132200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Topham, M. K., and Prescott, S. M. (1999). Mammalian diacylglycerol kinases, a family of lipid kinases with signaling functions. J. Biol. Chem. 274, 11447–11450. doi: 10.1074/jbc.274.17.11447
Tort, F., Garcia-Silva, M. T., Ferrer-Cortes, X., Navarro-Sastre, A., Garcia-Villoria, J., Coll, M. J.,et al. (2013). Exome sequencing identifies a new mutation in SERAC1 in a patient with 3-methylglutaconic aciduria. Mol. Genet. Metab. 110, 73–77. doi: 10.1016/j.ymgme.2013.04.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Truscott, K. N., Voos, W., Frazier, A. E., Lind, M., Li, Y., Geissler, A.,et al. (2003). A J-protein is an essential subunit of the presequence translocase-associated protein import motor of mitochondria. J. Cell Biol. 163, 707–713. doi: 10.1083/jcb.200308004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Twig, G., Elorza, A., Molina, A. J., Mohamed, H., Wikstrom, J. D., Walzer, G.,et al. (2008). Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27, 433–446. doi: 10.1038/sj.emboj.7601963
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Uchida, Y., Izai, K., Orii, T., and Hashimoto, T. (1992). Novel fatty acid beta-oxidation enzymes in rat liver mitochondria. II. Purification and properties of enoyl-coenzyme A (CoA) hydratase/3-hydroxyacyl-CoA dehydrogenase/3-ketoacyl-CoA thiolase trifunctional protein. J. Biol. Chem. 267, 1034–1041.
Ugalde, C., Moran, M., Blazquez, A., Arenas, J., and Martin, M. A. (2009). Mitochondrial disorders due to nuclear OXPHOS gene defects. Adv. Exp. Med. Biol. 652, 85–116. doi: 10.1007/978-90-481-2813-6_7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ushikubo, S., Aoyama, T., Kamijo, T., Wanders, R. J., Rinaldo, P., Vockley, J.,et al. (1996). Molecular characterization of mitochondrial trifunctional protein deficiency: formation of the enzyme complex is important for stabilization of both alpha- and beta-subunits. Am. J. Hum. Genet. 58, 979–988.
Valianpour, F., Mitsakos, V., Schlemmer, D., Towbin, J. A., Taylor, J. M., Ekert, P. G.,et al. (2005). Monolysocardiolipins accumulate in Barth syndrome but do not lead to enhanced apoptosis. J. Lipid Res. 46, 1182–1195. doi: 10.1194/jlr.M500056-JLR200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Valianpour, F., Wanders, R. J., Overmars, H., Vreken, P., Van Gennip, A. H., Baas, F.,et al. (2002). Cardiolipin deficiency in X-linked cardioskeletal myopathy and neutropenia (Barth syndrome, MIM 302060): a study in cultured skin fibroblasts. J. Pediatr. 141, 729–733. doi: 10.1067/mpd.2002.129174
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Van, Q., Liu, J., Lu, B., Feingold, K. R., Shi, Y., Lee, R. M.,et al. (2007). Phospholipid scramblase-3 regulates cardiolipin de novo biosynthesis and its resynthesis in growing HeLa cells. Biochem. J. 401, 103–109. doi: 10.1042/BJ20060373
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vance, J. E. (1990). Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 265, 7248–7256.
van der Laan, M., Bohnert, M., Wiedemann, N., and Pfanner, N. (2012). Role of MINOS in mitochondrial membrane architecture and biogenesis. Trends Cell Biol. 22, 185–192. doi: 10.1016/j.tcb.2012.01.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
van der Laan, M., Meinecke, M., Dudek, J., Hutu, D. P., Lind, M., Perschil, I.,et al. (2007). Motor-free mitochondrial presequence translocase drives membrane integration of preproteins. Nat. Cell Biol. 9, 1152–1159. doi: 10.1038/ncb1635
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
van Ekeren, G. J., Stadhouders, A. M., Smeitink, J. A., and Sengers, R. C. (1993). A retrospective study of patients with the hereditary syndrome of congenital cataract, mitochondrial myopathy of heart and skeletal muscle and lactic acidosis. Eur. J. Pediatr. 152, 255–259. doi: 10.1007/BF01956157
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
van Werkhoven, M. A., Thorburn, D. R., Gedeon, A. K., and Pitt, J. J. (2006). Monolysocardiolipin in cultured fibroblasts is a sensitive and specific marker for Barth syndrome. J. Lipid Res. 47, 2346–2351. doi: 10.1194/jlr.D600024-JLR200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Varfolomeev, E. E., Schuchmann, M., Luria, V., Chiannilkulchai, N., Beckmann, J. S., Mett, I. L.,et al. (1998). Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity 9, 267–276. doi: 10.1016/S1074-7613(00)80609-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vaz, F. M., Houtkooper, R. H., Valianpour, F., Barth, P. G., and Wanders, R. J. (2003). Only one splice variant of the human TAZ gene encodes a functional protein with a role in cardiolipin metabolism. J. Biol. Chem. 278, 43089–43094. doi: 10.1074/jbc.M305956200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Verkleij, A. J., Leunissen-Bijvelt, J., De Kruijff, B., Hope, M., and Cullis, P. R. (1984). Non-bilayer structures in membrane fusion. Ciba Found. Symp. 103, 45–59.
Vilarinho, S., Choi, M., Jain, D., Malhotra, A., Kulkarni, S., Pashankar, D.,et al. (2014). Individual exome analysis in diagnosis and management of paediatric liver failure of indeterminate aetiology. J. Hepatol. 61, 1056–1063. doi: 10.1016/j.jhep.2014.06.038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vogel, F., Bornhövd, C., Neupert, W., and Reichert, A. S. (2006). Dynamic subcompartmentalization of the mitochondrial inner membrane. J. Cell Biol. 175, 237–247. doi: 10.1083/jcb.200605138
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
von der Malsburg, K., Muller, J. M., Bohnert, M., Oeljeklaus, S., Kwiatkowska, P., Becker, T.,et al. (2011). Dual role of mitofilin in mitochondrial membrane organization and protein biogenesis. Dev. Cell 21, 694–707. doi: 10.1016/j.devcel.2011.08.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vreken, P., Valianpour, F., Nijtmans, L. G., Grivell, L. A., Plecko, B., Wanders, R. J.,et al. (2000). Defective remodeling of cardiolipin and phosphatidylglycerol in Barth syndrome. Biochem. Biophys. Res. Commun. 279, 378–382. doi: 10.1006/bbrc.2000.3952
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Waggoner, D. W., Johnson, L. B., Mann, P. C., Morris, V., Guastella, J., and Bajjalieh, S. M. (2004). MuLK, a eukaryotic multi-substrate lipid kinase. J. Biol. Chem. 279, 38228–38235. doi: 10.1074/jbc.M405932200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wallace, D. C., Singh, G., Lott, M. T., Hodge, J. A., Schurr, T. G., Lezza, A. M.,et al. (1988). Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 242, 1427–1430. doi: 10.1126/science.3201231
Wang, G., Mccain, M. L., Yang, L., He, A., Pasqualini, F. S., Agarwal, A.,et al. (2014a). Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat. Med. 20, 616–623. doi: 10.1038/nm.3545
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, L., Liu, X., Nie, J., Zhang, J., Kimball, S. R., Zhang, H.,et al. (2014b). ALCAT1 controls mitochondrial etiology of fatty liver diseases, linking defective mitophagy to hepatosteatosis. Hepatology doi: 10.1002/hep.27420 [Epub ahead of print].
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, S., Lee, D. P., Gong, N., Schwerbrock, N. M., Mashek, D. G., Gonzalez-Baro, M. R.,et al. (2007). Cloning and functional characterization of a novel mitochondrial N-ethylmaleimide-sensitive glycerol-3-phosphate acyltransferase (GPAT2). Arch. Biochem. Biophys. 465, 347–358. doi: 10.1016/j.abb.2007.06.033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Watanabe, T., Chuma, S., Yamamoto, Y., Kuramochi-Miyagawa, S., Totoki, Y., Toyoda, A.,et al. (2011). MITOPLD is a mitochondrial protein essential for nuage formation and piRNA biogenesis in the mouse germline. Dev. Cell 20, 364–375. doi: 10.1016/j.devcel.2011.01.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Watkins, S. M., Carter, L. C., and German, J. B. (1998). Docosahexaenoic acid accumulates in cardiolipin and enhances HT-29 cell oxidant production. J. Lipid Res. 39, 1583–1588.
Weber, T. A., Koob, S., Heide, H., Wittig, I., Head, B., Van Der Bliek, A.,et al. (2013). APOOL is a cardiolipin-binding constituent of the Mitofilin/MINOS protein complex determining cristae morphology in mammalian mitochondria. PLoS ONE 8:e63683. doi: 10.1371/journal.pone.0063683
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wedatilake, Y., Plagnol, V., Anderson, G., Paine, S., Clayton, P., Jacques, T.,et al. (2014). Tubular aggregates caused by serine active site containing 1 (SERAC1) mutations in a patient with a mitochondrial encephalopathy. Neuropathol. Appl. Neurobiol. doi: 10.1111/nan.12190 [Epub ahead of print].
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
West, J., Tompkins, C. K., Balantac, N., Nudelman, E., Meengs, B., White, T.,et al. (1997). Cloning and expression of two human lysophosphatidic acid acyltransferase cDNAs that enhance cytokine-induced signaling responses in cells. DNA Cell Biol. 16, 691–701.
Wherrett, J. R., and Huterer, S. (1972). Enrichment of bis-(monoacylglyceryl) phosphate in lysosomes from rat liver. J. Biol. Chem. 247, 4114–4120.
Whited, K., Baile, M. G., Currier, P., and Claypool, S. M. (2013). Seven functional classes of Barth syndrome mutation. Hum. Mol. Genet. 22, 483–492. doi: 10.1093/hmg/dds447
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wong, L. J. (2007). Diagnostic challenges of mitochondrial DNA disorders. Mitochondrion 7, 45–52. doi: 10.1016/j.mito.2006.11.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wortmann, S., Rodenburg, R. J., Huizing, M., Loupatty, F. J., De Koning, T., Kluijtmans, L. A.,et al. (2006). Association of 3-methylglutaconic aciduria with sensori-neural deafness, encephalopathy, and Leigh-like syndrome (MEGDEL association) in four patients with a disorder of the oxidative phosphorylation. Mol. Genet. Metab. 88, 47–52. doi: 10.1016/j.ymgme.2006.01.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wortmann, S. B., Vaz, F. M., Gardeitchik, T., Vissers, L. E., Renkema, G. H., Schuurs-Hoeijmakers, J. H.,et al. (2012). Mutations in the phospholipid remodeling gene SERAC1 impair mitochondrial function and intracellular cholesterol trafficking and cause dystonia and deafness. Nat. Genet. 44, 797–802. doi: 10.1038/ng.2325
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wysocki, S. J., Wilkinson, S. P., Hahnel, R., Wong, C. Y., and Panegyres, P. K. (1976). 3-Hydroxy-3-methylglutaric aciduria, combined with 3-methylglutaconic aciduria. Clin. Chim. Acta 70, 399–406. doi: 10.1016/0009-8981(76)90353-3
Xiao, J., Engel, J. L., Zhang, J., Chen, M. J., Manning, G., and Dixon, J. E. (2011). Structural and functional analysis of PTPMT1, a phosphatase required for cardiolipin synthesis. Proc. Natl. Acad. Sci. U.S.A. 108, 11860–11865. doi: 10.1073/pnas.1109290108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xu, Y., Condell, M., Plesken, H., Edelman-Novemsky, I., Ma, J., Ren, M.,et al. (2006a). A Drosophila model of Barth syndrome. Proc. Natl. Acad. Sci. U.S.A. 103, 11584–11588. doi: 10.1073/pnas.0603242103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xu, Y., Malhotra, A., Ren, M., and Schlame, M. (2006b). The enzymatic function of tafazzin. J. Biol. Chem. 281, 39217–39224. doi: 10.1074/jbc.M606100200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xu, Y., Sutachan, J. J., Plesken, H., Kelley, R. I., and Schlame, M. (2005). Characterization of lymphoblast mitochondria from patients with Barth syndrome. Lab. Invest. 85, 823–830. doi: 10.1038/labinvest.3700274
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xu, Y., Zhang, S., Malhotra, A., Edelman-Novemsky, I., Ma, J., Kruppa, A.,et al. (2009). Characterization of tafazzin splice variants from humans and fruit flies. J. Biol. Chem. 284, 29230–29239. doi: 10.1074/jbc.M109.016642
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yamamoto, H., Esaki, M., Kanamori, T., Tamura, Y., Nishikawa, S., and Endo, T. (2002). Tim50 is a subunit of the TIM23 complex that links protein translocation across the outer and inner mitochondrial membranes. Cell 111, 519–528. doi: 10.1016/S0092-8674(02)01053-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ye, C., Lou, W., Li, Y., Chatzispyrou, I. A., Huttemann, M., Lee, I.,et al. (2014a). Deletion of the cardiolipin-specific phospholipase Cld1 rescues growth and life span defects in the tafazzin mutant: implications for Barth syndrome. J. Biol. Chem. 289, 3114–3125. doi: 10.1074/jbc.M113.529487
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ye, K., Lu, J., Ma, F., Keinan, A., and Gu, Z. (2014b). Extensive pathogenicity of mitochondrial heteroplasmy in healthy human individuals. Proc. Natl. Acad. Sci. U.S.A. 111, 10654–10659. doi: 10.1073/pnas.1403521111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yet, S. F., Lee, S., Hahm, Y. T., and Sul, H. S. (1993). Expression and identification of p90 as the murine mitochondrial glycerol-3-phosphate acyltransferase. Biochemistry 32, 9486–9491. doi: 10.1021/bi00087a029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yoda, E., Hachisu, K., Taketomi, Y., Yoshida, K., Nakamura, M., Ikeda, K.,et al. (2010). Mitochondrial dysfunction and reduced prostaglandin synthesis in skeletal muscle of Group VIB Ca2+-independent phospholipase A2gamma-deficient mice. J. Lipid Res. 51, 3003–3015. doi: 10.1194/jlr.M008060
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zahedi, R. P., Sickmann, A., Boehm, A. M., Winkler, C., Zufall, N., Schonfisch, B.,et al. (2006). Proteomic analysis of the yeast mitochondrial outer membrane reveals accumulation of a subclass of preproteins. Mol. Biol. Cell 17, 1436–1450. doi: 10.1091/mbc.E05-08–0740
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zeczycki, T. N., Whelan, J., Hayden, W. T., Brown, D. A., and Shaikh, S. R. (2014). Increasing levels of cardiolipin differentially influence packing of phospholipids found in the mitochondrial inner membrane. Biochem. Biophys. Res. Commun. 450, 366–371. doi: 10.1016/j.bbrc.2014.05.133
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, J., Guan, Z., Murphy, A. N., Wiley, S. E., Perkins, G. A., Worby, C. A.,et al. (2011). Mitochondrial phosphatase PTPMT1 is essential for cardiolipin biosynthesis. Cell Metab. 13, 690–700. doi: 10.1016/j.cmet.2011.04.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, M., Mileykovskaya, E., and Dowhan, W. (2002). Gluing the respiratory chain together. Cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. J. Biol. Chem. 277, 43553–43556. doi: 10.1074/jbc.C200551200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhao, Y., Chen, Y. Q., Li, S., Konrad, R. J., and Cao, G. (2009). The microsomal cardiolipin remodeling enzyme acyl-CoA lysocardiolipin acyltransferase is an acyltransferase of multiple anionic lysophospholipids. J. Lipid Res. 50, 945–956. doi: 10.1194/jlr.M800567-JLR200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhou, Q., Sims, P. J., and Wiedmer, T. (1998). Identity of a conserved motif in phospholipid scramblase that is required for Ca2+-accelerated transbilayer movement of membrane phospholipids. Biochemistry 37, 2356–2360. doi: 10.1021/bi972625o
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zinser, E., and Daum, G. (1995). Isolation and biochemical characterization of organelles from the yeast, Saccharomyces cerevisiae. Yeast 11, 493–536. doi: 10.1002/yea.320110602
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: mitochondrial disease, phospholipid metabolism, cardiolipin, Barth syndrome, MEGDEL, DCMA, Sengers syndrome, hereditary spastic paraplegia
Citation: Lu Y-W and Claypool SM (2015) Disorders of phospholipid metabolism: an emerging class of mitochondrial disease due to defects in nuclear genes. Front. Genet. 6:3. doi: 10.3389/fgene.2015.00003
Received: 26 November 2014; Accepted: 06 January 2015;
Published online: 03 February 2015.
Edited by:
Enrico Baruffini, University of Parma, ItalyReviewed by:
Patrice X. Petit, Centre National de la Recherche Scientifique, FranceCopyright © 2015 Lu and Claypool. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Steven M. Claypool, Department of Physiology, School of Medicine, Johns Hopkins University, 725 North Wolfe Street, Hunterian Building Room 205, Baltimore, MD 21205, USA e-mail:c2NsYXlwbzFAamhtaS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.