 Timothy J. Pullen*
Timothy J. Pullen* Guy A. Rutter*
Guy A. Rutter*- Section of Cell Biology, Department of Medicine, Imperial Centre for Translational and Experimental Medicine, Imperial College London, London, UK
Type 2 diabetes usually ensues from the inability of pancreatic beta cells to compensate for incipient insulin resistance. The loss of beta cell mass, function, and potentially beta cell identity contribute to this dysfunction to extents which are debated. In recent years, long non-coding RNAs (lncRNAs) have emerged as potentially providing a novel level of gene regulation implicating critical cellular processes such as pluripotency and differentiation. With over 1000 lncRNAs now identified in beta cells, there is growing evidence for their involvement in the above processes in these cells. While functional evidence on individual islet lncRNAs is still scarce, we discuss how lncRNAs could contribute to type 2 diabetes susceptibility, particularly at loci identified through genome-wide association studies as affecting disease risk.
Introduction
Diabetes is a major and growing health problem affecting 347 m people worldwide (Danaei et al., 2011). Type 2 diabetes (T2D) accounts for 90% of affected individuals, and is a complex, progressive disease affected by a range of genetic and environmental risk factors. While insulin resistance plays a part in disease progression, pancreatic beta cell failure lies at the heart of T2D (Kahn, 2003).
Beta cells are the body’s sole source of circulating insulin, and as such are critical for maintaining blood glucose within healthy limits. The beta cell’s tightly regulated secretion of insulin in response to glucose is dependent on a highly specialized metabolic sensing system, underpinned by a specific pattern of gene expression. This includes specific genes required for glucose-sensing (e.g., Gck and Glut2), insulin production and processing (e.g., insulin, prohormone convertase 1/3 [PC1/3]) and regulated secretion. Equally important is the specific repression of genes which interfere with proper regulation of insulin secretion (e.g., Slc16a1 and Ldha) (Zhao et al., 2001; Pullen et al., 2012). Indeed, we (Pullen et al., 2010) and others (Thorrez et al., 2011) have recently identified ∼40 “forbidden” or “disallowed” genes specifically repressed in islets despite widespread expression across other tissues. While a unique pattern of transcription factors (e.g., Pdx1, Pax6, MafA, Nkx2.2) likely underlie the sustained expression of beta cell selective genes, epigenetic mechanisms are vital for the mitotically stable maintenance of cellular identity and probably the suppression of forbidden genes.
The tipping point from pre-diabetes to overt T2D is usually reached when beta cells can no longer compensate sufficiently for incipient insulin resistance. The nature of the progressive loss of functional beta cell mass has been the source of some debate (Weir and Bonner-Weir, 2013), with both decreases in overall beta cell mass (Butler et al., 2003) and impaired glucose-stimulated insulin secretion (GSIS) from the remaining islets being reported in T2D subjects (Del Guerra et al., 2005). Indeed, recent work has suggested that decreases in beta cell mass are modest (Rahier et al., 2008) and may even have been over-estimated (Marselli et al., 2014).
There has also been growing interest in a third option: that a progressive loss of beta cell identity underlies the development of T2D. Results from two rat models (Tokuyama et al., 1995; Jonas, 1999), and the db/db mouse model (Kjorholt et al., 2005) of T2D produce similar results showing a loss of differentiated beta cells accompanied by similar changes in gene expression: decreased expression of GSIS genes (e.g., Gck); increased expression of normally repressed genes (e.g., Ldha and Hk1); and decreases in islet transcription factors (e.g., Isl1, Neurod1, Pdx1, and Pax6) (Tokuyama et al., 1995; Jonas, 1999). The importance of these transcription factors in maintaining beta cell identity is underscored by the transformation of beta to alpha-like cells following Pdx1 deletion (Gao et al., 2014). In FoxO1 knockout mouse islets, beta cells dedifferentiate a step further, acquiring expression of immature endocrine cell markers (Ngn3, Oct4, Nanog, and Mycl) (Talchai et al., 2012). However, the relevance of this strain to human diabetic islets is as yet unestablished.
While the relative importance of these three factors (impaired beta cell mass, function and identity) in T2D is not fully understood, mechanisms which increase beta cell function, proliferation and identity are all sought to tackle the disease. Furthermore, increased understanding of the molecular defects underlying T2D will enable novel therapies to address the underlying causes of the disease, rather than just treat the symptoms.
A novel layer of gene regulation, acting partly through epigenetic mechanisms, was uncovered with the discovery that thousands of long non-coding RNAs (lncRNAs) are expressed from the genome (Guttman et al., 2009). lncRNAs are a diverse group of transcripts defined by a negative property: the lack of protein-coding potential. They are differentiated from short ncRNAs such as miRNAs (which also play an important role: see Poy et al., 2004; Guay et al., 2011; Pullen et al., 2011) by a minimum length threshold of 200 nt. lncRNAs are often capped, spliced and polyadenylated like mRNAs although both unspliced and non-polyadenylated variants are also common. As a class, lncRNAs are enriched in the nucleus relative to protein-coding transcripts, although there remains a population of cytoplasmic-enriched lncRNAs, and non-nuclear functions have been demonstrated for a number of lncRNAs (Derrien et al., 2012). Indeed lncRNAs have been demonstrated to regulate gene expression through a number of mechanisms at the transcriptional, post-transcriptional and translational level.
The expression patterns of lncRNAs are significantly more cell-type specific than protein-coding genes (Guttman et al., 2009; Djebali et al., 2012), making them well placed to play highly cell-type specific roles. However, the fact that lncRNAs are less evolutionarily conserved at the primary sequence level than protein-coding genes and the fact the numerous lncRNAs appear to be species-specific raise questions over their functions. lncRNAs may depend more on secondary structure for their function, and thus be more tolerant of mutations than protein-coding genes. Indeed, lncRNA exons show evidence of low but clear sequence conservation compared to other intergenic regions (Guttman et al., 2009). The advent of next generation sequencing technologies has led to an explosion of novel lncRNA discovery in diverse tissues, including pancreatic islets and beta cells. The discovery of lncRNAs with the potential to regulate gene expression and cellular identity in T2D-relevant tissues offers the opportunity for greater understanding of disease etiology and novel targets for treatment.
Mechanisms of lncRNA Action
miRNAs are a major class of short non-coding RNA which primarily act via a single, well-characterized mechanism to downregulate gene expression through mRNA degradation and translational inhibition. In contrast, lncRNAs have been shown to regulate gene expression through a bewildering array of mechanisms (reviewed extensively in Wang and Chang, 2011; Kung et al., 2013). On average, lncRNAs are less abundant than protein-coding transcripts, although some (e.g., H19) are expressed at comparable levels (Djebali et al., 2012). lncRNAs are a heterogeneous group of transcripts, and greater functional characterization will hopefully allow clearer classification into distinct groups acting through distinct mechanisms.
One of the first identified modes of action of lncRNAs was via alterations in chromatin modifications through recruiting histone modifying complexes to particular genomic loci (Rinn et al., 2007). This plays a major part in epigenetic silencing during X-inactivation (through the lncRNA Xist (Zhao et al., 2008 Pontier and Gribnau, 2011)) and at imprinted loci (e.g., Air at the Slc22a3 locus (Nagano et al., 2008)). Indeed, this appears to be a widespread function of lncRNAs, with around 20% of intergenic lncRNAs identified in one study interacting with the chromatin-modifying complex Polycomb Repressive Complex 2 (PRC2) (Khalil et al., 2009). In addition to affecting histone modifications, lncRNAs can also act on the other major branch of epigenetic regulation, DNA methylation, through interaction with DNA methylation machinery (Mohammad et al., 2010; Di Ruscio et al., 2013). lncRNAs can further regulate gene expression by mediating DNA looping (Lai et al., 2013). This affects the three-dimensional conformation of chromosomes which regulates gene expression by bringing distal enhancers physically adjacent to gene promoters.
lncRNAs also act through a number of mechanisms to regulate gene expression at the post-transcriptional level. Natural antisense transcripts (NATs) are lncRNAs transcribed in the opposite orientation to mRNAs with overlapping exons. They have the potential for direct interaction with mRNAs through complimentary base-pairing, which can increase mRNA stability (e.g., BASE1-AS and BASE1) (Faghihi et al., 2008). In contrast, lncRNAs can also lead to mRNA degradation through the recruitment of Staufen1 (STAU1) (Gong and Maquat, 2011). lncRNAs can also influence mRNAs indirectly by acting as “sponges” for miRNAs, preventing them from downregulating gene expression (Kallen et al., 2013). It is interesting to note that this last example refers to the highly expressed H19 lncRNA, and these post-transcriptional mechanisms presumably require lncRNAs in comparable abundance to their mRNA or miRNA targets. Given the lower expression of most lncRNAs relative to mRNAs these mechanisms requiring abundant lncRNAs are perhaps the exception rather than the norm.
lncRNAs and Beta Cell Identity
As the expression of many lncRNAs is highly cell-type specific, early catalogs of lncRNAs mainly from other cell lines provided little information relevant to beta cells. However, a number of studies have since identified lncRNAs genome-wide in human and mouse, islets and beta cells.
Morán et al. (2012) used combination of ChIP-Seq (to identify sites of active transcription), and RNA-Seq (to directly detect transcripts) to produce a comprehensive catalog of >1100 lncRNAs expressed in human islets. In agreement with studies in other tissues, these lncRNAs proved to be significantly more cell-type specific than protein-coding genes. Indeed, 55% of intergenic lncRNAs and 40% of antisense lncRNAs identified in this study were specific to islets. Importantly, Morán et al. (2012) identified several lncRNAs that were dysregulated in islets from T2D subjects.
One challenge to the theory of widespread functions for lncRNAs is the low level of sequence conservation between species. To address this, orthologous mouse genomic regions were detected for 70% of human lncRNA loci, and RNA-Seq revealed that 47% of these were actively transcribed in mouse islets (Morán et al., 2012). Indeed this may be an underestimate as several lncRNAs were detected by qPCR despite falling below the threshold for RNA-Seq detection. An independent transcriptomic study of mouse islets identified a similar number (>1000) of intergenic lncRNAs (Ku et al., 2012). Further dissection of islet cell-types was performed by Bramswig et al. (2013), with the identification of five alpha cell-specific and twelve beta cell-specific lncRNAs. The scarcity of lncRNAs identified in this study may be due to highly stringent removal of any transcripts overlapping repeat regions.
Previous studies have identified both lncRNAs that regulate the maintenance of pluripotency, and lncRNAs required for neuronal differentiation (Ng et al., 2012). Indeed, the regulation of differentiation and cell identity appears to be a major function of lncRNAs. Supporting this role for islet lncRNAs, Morán et al. (2012) identified dynamic regulation of lncRNAs during in vitro differentiation of human embryonic stem cells towards pancreatic endocrine cells. As such protocols have thus far failed to produce fully functional beta cells without an in vivo maturation stage in mice, perhaps the most tantalizing discovery was six lncRNAs whose expression was only activated during this last stage. Understanding regulators of this critical maturation stage, and the ability to recapitulate it without passage through animals would be essential for any therapeutic generation of beta cells from human stem cells. However, islet lncRNAs are not restricted to developmental roles, as depletion of a beta cell-specific lncRNA was also shown to influence gene expression in mature beta cells. Depletion of the lncRNA HI-LNC25 decreased expression of the transcription factor and monogenic diabetes gene, GLIS3 (Table 1) (Morán et al., 2012). The location of HI-LNC25 and GLIS3 on separate chromosomes (20 and 9, respectively) indicates that the lncRNA regulates GLIS3 in trans, although whether this is through a direct interaction with this locus has not been explored.

TABLE 1. The main lncRNAs discussed in this paper along with their tissue and species specificity.
lncRNAs Implicated in T2D Susceptibility
Within the last decade, genome-wide association studies (GWAS) have been a major focus of work to identify the genetic variants underlying susceptibility to T2D and related metabolic traits. Through identifying single nucleotide polymorphisms (SNPs) which correlate with diabetic phenotypes, investigators aim to single out genes which influence disease susceptibility. While much of the interpretation of GWAS hits has focussed on protein-coding genes, there are good reasons to indicate that lncRNAs are responsible for the effects of some of these SNPs.
It is striking how few SNPs identified through GWAS for T2D, indeed for most diseases, result in changes to protein sequences. SLC30A8 is one of the few examples from early studies where a SNP (rs13266634) resulted in an amino acid substitution (R325W) affecting the zinc transporter located on insulin granules (Sladek et al., 2007). Interestingly, expression of SLC30A8 is largely restricted to pancreatic islets, so the effects of any mutations are expected to be limited to the endocrine pancreas. In contrast, genes at most other GWAS loci are more widely expressed. Missense mutations at these widely expressed loci are far more likely to cause defects in multiple tissues and possibly embryonic lethality, which could account for absence from GWAS specific for T2D and related traits. In a more recent large-scale meta-analysis which aimed to finely map the causal SNPs, only two of the 65 T2D susceptibility loci examined had a lead SNP resulting in a missense mutation (PPARG [rs1801282] and KCNJ11 [rs5215]) (Morris et al., 2012). In both cases, rare severe mutations have previously been identified which cause monogenic forms of diabetes (Barroso et al., 1999; Gloyn et al., 2004). Most SNPs instead map to intronic or intergenic regions and thus likely act through altering gene expression and/or splicing.
While the effects of changes to protein sequences are relatively straightforward to investigate, it is often unclear which genes are affected by SNPs in non-coding regions. Whereas proximal promoters are directly adjacent to the genes they regulate, enhancer elements can influence the expression of genes hundreds of kilobases away, and can be interspersed between and even within other genes (Ilnytska et al., 2009). Interestingly, SNPs associated with T2D and fasting glycaemia are enriched in these distal regulatory regions (Pasquali et al., 2014). The gene affected by SNPs falling outside proximal promoters may not be clear, although the closest gene is often used as a starting point for further investigation. With the identification and annotation of increasing numbers of lncRNAs it has become apparent that a number of the GWAS hits fall close to, or within lncRNAs.
Mapping SNPs at these 65 loci to current Ensembl annotations shows five lead SNPs are within the exons or introns of lncRNAs (Table 2). Two of these loci (KCNQ1 and CDKN2A/CDKN2B) contain well characterized lncRNAs for which studies have started to reveal the mechanisms through which they function. At the PROX1 and PSMD6 loci, in both cases the lead SNP (rs2075423 and rs12497268) falls within the exon of an annotated antisense orientated lncRNA. At the CCND2 locus, the lead SNP (rs11063069) is approximately 9 kb upstream of the protein-coding gene, yet within the intron of two antisense lncRNAs (CCND2-AS1 and CCND2-AS2). Finally, as further islet-expressed lncRNAs are identified more of these SNPs are found to fall closer to lncRNAs than protein-coding genes (e.g., WFS1 locus (Morán et al., 2012)).
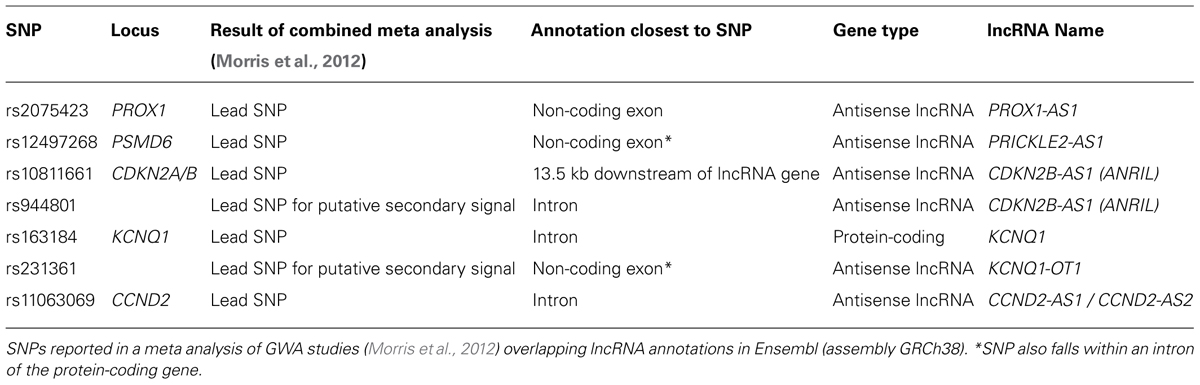
TABLE 2. Type 2 diabetes susceptibility loci where a lead SNP falls within a lncRNA gene.
KCNQ1
The KCNQ1 locus contains a number of genes with the potential to affect beta cell function and proliferation. Within this locus KCNQ1, KCNQ1OT1, CDKN1C, PHLDA2, SLC22A18, and SLC22A18AS are all expressed in both fetal pancreas and adult islets (Travers et al., 2013). KCNQ1 encodes a voltage-gated potassium channel subunit which is expressed in pancreatic beta cells although its effect on the regulation of insulin secretion is somewhat unclear. siRNA knockdown of KCNQ1 in human islets enhanced depolarization-induced exocytosis (Rosengren et al., 2012), whereas pharmacological inhibition in INS-1 cells did not affect basal, tolbutamide or GSIS (Ullrich et al., 2005). CDKN1C encodes the p57KIP2 cyclin-dependent kinase inhibitor which is a negative regulator of cell proliferation. It is expressed in islets, and loss of p57KIP2 expression is associated with increased beta cell proliferation in focal congenital hyperinsulinism (Kassem et al., 2001; Henquin et al., 2011). PHLDA2 also exerts a negative effect on cell proliferation, with increased expression in the placenta being associated with intrauterine growth retardation though uteroplacental insufficiency (McMinn et al., 2006). In contrast, decreased expression of PHLDA2 was detected in neuroendocrine tumors relative to normal islet controls, as a downstream effect of losing the tumor suppressor gene MEN1 resulting in Multiple Endocrine Neoplasia type 1 (Dilley et al., 2005).
The lead SNP at this locus is within the intron of the KCNQ1 protein-coding gene, yet the lead SNP for a putative secondary association signal is in the exon of an antisense lncRNA (KCNQ1OT1) (Morris et al., 2012). KCNQ1OT1 is a 91 kb long transcript encoded by RNA polymerase II and localized exclusively in the nucleus (Pandey et al., 2008). In early development, KCNQ1OT1 is expressed in a monoallelic fashion from the paternal allele and has been linked to silencing of nearby genes on the same chromosome, resulting in these genes being expressed exclusively from the maternal allele. This is mediated through the interaction of KCNQ1OT1 with PRC2 components and G9a histone methyltransferase resulting in enrichment of repressive histone modifications H3K27me3 and H3K9me3 at this locus (Pandey et al., 2008). However, this pattern is complicated by the lineage-specific loss of imprinting at some genes. Specifically, KCNQ1 and KCNQ1OT1 showed developmental loss of imprinting with biallelic expression in adult islets (Travers et al., 2013). Interestingly, in the developing mouse heart, Kncq1ot1 is required for paternal silencing of Cdkn1c and Slc22a18, but not Kcnq1 indicating that Kcnq1ot1 may be more directly involved in the regulation of these two genes (Korostowski et al., 2012).
One proposed model to explain the effect of SNPs at this locus is that risk alleles reduce KCNQ1OT1 expression, thereby decreasing repressive histone modifications, increasing CDKN1C expression and thereby impairing islet proliferation or development (Travers et al., 2013). In contrast to this model, KCNQ1OT1 was reported to be upregulated in T2D islets (Morán et al., 2012). However, this apparent contradiction may be explained by risk SNPs having an effect on imprinting early in development, whereas the KCNQ1OT1 upregulation in T2D adult islets may be part of the compensatory proliferative response of islets to hyperglycaemia.
ANRIL
CDKN2A/CDKN2B is another T2D GWAS locus containing a well-characterized lncRNA. This locus encodes three tumor-suppressors: p16INK4A, p14ARF, and p15INK4B. The expression of these genes is regulated by polycomb-mediated silencing through H3K27me3. Of particular relevance to diabetes, p16INK4A up-regulation has been shown to be responsible age-dependent decline in beta cell proliferative capacity (Krishnamurthy et al., 2006). The histone methyltransferase and PRC2 component, Ezh2, represses p16INK4A expression in young beta cells, permitting proliferation. However, declining Ezh2 levels in aging beta cells lead to derepression of p16INK4A expression, and consequent inhibition of proliferation (Chen et al., 2009).
The lead SNP (rs10811661) at this locus is downstream of, and closest to the lncRNA ANRIL (CDKN2B-AS1). The lead SNP for a putative secondary association signal (rs944801) is located within an intron of ANRIL (Morris et al., 2012). This region (chromosome 9p21) has been strongly associated with susceptibility to a number of diseases. SNPs associated with coronary disease, stroke, melanoma and glioma are all located close to ANRIL and correlated with ANRIL expression (Cunnington et al., 2010; Pasmant et al., 2011). Although no direct mouse ortholog of ANRIL has been reported, a lncRNA of unknown function has been detected at the orthologous region of the mouse genome. Deletion of part of the “9p21” orthologous region in mice (including a section of this lncRNA) increased mortality and affected expression of neighboring genes (Visel et al., 2010), indicating that gene regulatory sequences and possibly this lncRNA perform roles conserved between human and mouse.
ANRIL interacts with both PRC1 (CBX7) and PRC2 (SUZ12) components to affect the epigenetic regulation of gene expression at this locus (Yap et al., 2010; Kotake et al., 2011). Competitive inhibition of ANRIL was reported to increase p16INK4A expression resulting in decreased proliferation in fibroblasts (Yap et al., 2010). A separate depletion of ANRIL in lung fibroblasts also reported decreased proliferation, although in this case primarily through upregulation of p15INK4B. The direction of this effect fits with reports of T2D SNPs being associated with decreased ANRIL expression, although not in all populations (Cunnington et al., 2010). It therefore appears that ANRIL recruits both PRC1 and PRC2 to this locus in a coordinated manner to repress these proliferation inhibitors. Disruption of ANRIL expression or function by SNPs could impair silencing at this locus decreasing the proliferative capacity of beta cell.
One should caution that ANRIL may not be limited to cis-acting effects, as overexpression of ANRIL in HeLa cells has also been reported to produce genome-wide effects on gene expression (Sato et al., 2010). An interesting direction for future research may thus be to determine how regulation of this locus by ANRIL integrates with the PDGF pathway responsible for age-dependent decline in beta cell proliferative capacity (Chen et al., 2011). Whereas ectopic expression of Ezh2 was sufficient to increase beta cell proliferation in young mice, it was unable to repress p16INK4A in older mice without combined inhibition of trithorax group proteins (Zhou et al., 2013). It would be of particular interest to discover whether manipulation of ANRIL in adult beta cells can reverse this block on proliferation, or whether its effects are primarily in developing beta cells.
It is interesting to note that ANRIL and KCNQ1OT1 are both expressed in numerous tissues, yet certain SNPS within them are associated specifically with T2D susceptibility. It may be that beta cell replication during compensation to insulin resistance is particularly sensitive to ANRIL function meaning that these cells are most severely affected by ANRIL variants. Alternatively, ANRIL may be involved in beta cell-specific interactions which are uniquely affected by T2D SNPs. While the former proposition appears more likely, functional studies from one cell-type may not be directly applicable to others. The lack of a clear mouse ortholog of ANRIL has also prevented the use of mouse transgenics for investigating its function. Further investigation into the relationship between lncRNAs expressed in human and mouse at this locus would provide valuable insight into whether mouse experiments could be used to study ANRIL function.
lncRNAs as Therapeutic Targets
The discovery of lncRNAs implicated in susceptibility to, and the progression of, T2D raises the question of whether lncRNAs can be therapeutically manipulated to ameliorate this disease. lncRNAs offer a number of advantages as therapeutic targets. Firstly, the highly cell-type specific expression pattern of many lncRNAs is likely because they are involved in the cell-type specific regulation of genes which themselves are more widely expressed. Manipulating lncRNAs could therefore allow the effects to be specifically targeted to a single cell-type, with few side effects.
Furthermore, lncRNAs are involved in the epigenetic regulation of fundamental cellular processes such as pluripotency and cell identity. Much of the protein machinery regulating the epigenetic landscape is widely expressed, making targeting any intervention difficult. By acting as scaffolds to bring together particular combinations of chromatin-modifying complexes, transcription factors, etc. and target them to specific genomic loci, lncRNAs can provide specificity to this system. As such, lncRNAs may also provide a specific target to influence the epigenetic landscape underlying beta cell identity, and the potential to reinforce beta cell identity when this is challenged by T2D.
In contrast to lncRNAs, the more clearly defined mechanisms of miRNA processing and function have allowed greater progress to be made towards therapeutic targeting of these short non-coding RNAs. The most successful route has been to inhibit miRNAs in vivo by administrating modified oligonucleotides systemically (Montgomery and van Rooij, 2011). Although the short length of miRNAs makes them easy to target using short complementary oligonucleotides, a similar approach termed “antagoNATs” uses short antisense oligonucleotides to target lncRNAs. Brain-derived neurotrophic factor (BDNF-AS) is a brain expressed NAT which inhibits the BDNF gene. Continuous in vivo delivery to mouse brain via an osmotic minipump stably increased BDNF expression in a highly locus-specific effect, with a corresponding increase in neurite outgrowth and differentiation (Modarresi et al., 2012). While this form of delivery may be of limited use in clinics, advances in delivery of other modified oligonucleotides, such as siRNAs, will likely increase the feasibility of using this approach more widely (Gavrilov and Saltzman, 2012). Whether similar approaches could be used to target diabetic beta cells relies on the identification and characterization of islet-specific lncRNAs with negative impacts on beta cell identity, function or proliferation.
Therapeutic increases in miRNA function have been possible using chemically synthesized miRNA mimics. While the mimics themselves are highly effective, targeting their delivery to a particular cell type is not. The use of adeno-associated viral (AAV) vectors offers the possibility to target miRNA expression to particular tissues through both viral tropism and the use of tissue-specific promoters (Montgomery and van Rooij, 2011). Synthetic lncRNA mimics would not be practical due to the greater length, but viral expression approaches used for miRNAs could well be adapted for lncRNA delivery. However, as some models propose lncRNAs function through recruiting proteins to the nascent transcript, the site of expression may be critical for their functions. Ectopic expression may not be able to recapitulate the full effects of endogenous lncRNAs. Another concern is the stage of development at which lncRNAs act. T2D susceptibility at the KCNQ1OT1 locus involves the imprinting, which is lost in adult beta cells, suggesting that this lncRNA plays a role in beta cell development. It is therefore possible that manipulation of KCNQ1OT1 in adult beta cells would be unable to ameliorate any developmental defects. To counter this view, there are a large number of lncRNAs expressed in adult beta cells, including some only found in mature beta cells. Furthermore, the regulation of GLIS3 by one islet lncRNA demonstrates that significant diabetes genes are regulated in adult beta cells by lncRNAs (Morán et al., 2012).
Conclusion
The large number of highly cell-type specific lncRNAs identified in recent years provides the potential for significant regulation of gene expression and cell phenotype in pancreatic beta cells. However, the functional characterization of these lncRNAs has inevitably lagged behind their discovery. So far there is evidence for the epigenetic regulation of two highly significant T2D loci by lncRNAs. In addition there is more circumstantial evidence that a number of further lncRNAs are associated with beta cell development and function. Whether lncRNA expression changes during beta cell compensation to insulin resistance, potentially playing a role in beta cell expansion under these conditions, is also an important area for investigation, and may reveal the potential for therapeutic targeting. Further detailed investigation of the specific lncRNAs involved in this process are required to reveal the true extent to which lncRNAs regulate beta cell identity, proliferation and function.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Supported by a Diabetes Research and Wellness Fund Non-clinical Fellowship to Timothy J. Pullen and Wellcome Trust Senior Investigator (WT098424AIA), MRC Programme (MR/J0003042/1), Diabetes UK Project Grant (11/0004210) and Royal Society Wolfson Research Merit Awards to Guy A. Rutter. The work leading to this publication has received support from the Innovative Medicines Initiative Joint Undertaking under grant agreement no. 155005 (IMIDIA), resources of which are composed of financial contribution from the European Union’s Seventh Framework Programme (FP7/2007-2013) and EFPIA companies’ in kind contribution (Guy A. Rutter). We thank Prof. Jorge Ferrer for useful discussion.
References
Barroso, I., Gurnell, M., Crowley, V. E., Agostini, M., Schwabe, J. W., Soos, M. A.,et al. (1999). Dominant negative mutations in human PPARgamma associated with severe insulin resistance, diabetes mellitus and hypertension. Nature 402, 880–883. doi: 10.1038/47254
Bramswig, N. C., Everett, L. J., Schug, J., Dorrell, C., Liu, C., Luo, Y.,et al. (2013). Epigenomic plasticity enables human pancreatic α to β cell reprogramming. J. Clin. Invest. 123, 1275–1284. doi: 10.1172/JCI66514
Butler, A. E., Janson, J., Bonner-Weir, S., Ritzel, R., Rizza, R. A., and Butler, P. C. (2003). Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes Metab. Res. Rev. 52, 102–110. doi: 10.2337/diabetes.52.1.102
Chen, H., Gu, X., Liu, Y., Wang, J., Wirt, S. E., Bottino, R.,et al. (2011). PDGF signalling controls age-dependent proliferation in pancreatic β-cells. Nature 478, 349–355. doi: 10.1038/nature10502
Chen, H., Gu, X., Su, I., Bottino, R., Contreras, J. L., Tarakhovsky, A.,et al. (2009). Polycomb protein Ezh2 regulates pancreatic beta-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes Dev. 23, 975–85. doi: 10.1101/gad.1742509
Cunnington, M. S., Santibanez Koref, M., Mayosi, B. M., Burn, J., and Keavney, B. (2010). Chromosome 9p21 SNPs associated with multiple disease phenotypes correlate with ANRIL expression. PLoS Genet. 6:e1000899. doi: 10.1371/journal.pgen.1000899
Danaei, G., Finucane, M. M., Lu, Y., Singh, G. M., Cowan, M. J., Paciorek, C. J.,et al. (2011). National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: systematic analysis of health examination surveys and epidemiological studss with 370 country-years and 2⋅7 million participants. Lancet 378, 31–40. doi: 10.1016/S0140-6736(11)60679-X
Del Guerra, S., Lupi, R., Marselli, L., Masini, M., Bugliani, M., Sbrana, S.,et al. (2005). Functional and molecular defects of pancreatic islets in human type 2 diabetes. Diabetes Metab. Res. Rev 54, 727–735. doi: 10.2337/diabetes.54.3.727
Derrien, T., Johnson, R., Bussotti, G., Tanzer, A., Djebali, S., Tilgner, H.,et al. (2012). The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 22, 1775–1789. doi: 10.1101/gr.132159.111
Dilley, W., Kalyanaraman, S., Verma, S., Cobb, J. P., Laramie, J., and Lairmore, T. (2005). Global gene expression in neuroendocrine tumors from patients with the MEN1 syndrome. Mol. Cancer 4:9. doi: 10.1186/1476-4598-4-9
Di Ruscio, A., Ebralidze, A. K., Benoukraf, T., Amabile, G., Goff, L. A., Terragni, J.,et al. (2013). DNMT1-interacting RNAs block gene-specific DNA methylation. Nature 503, 371–376. doi: 10.1038/nature12598
Djebali, S., Davis, C. A., Merkel, A., Dobin, A., Lassmann, T., Mortazavi, A.,et al. (2012). Landscape of transcription in human cells. Nature 489, 101–108. doi: 10.1038/nature11233
Faghihi, M. A., Modarresi, F., Khalil, A. M., Wood, D. E., Sahagan, B. G., Morgan, T. E.,et al. (2008). Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of beta-secretase. Nat. Med. 14, 723–730. doi: 10.1038/nm1784
Gao, T., McKenna, B., Li, C., Reichert, M., Nguyen, J., Singh, T.,et al. (2014). Pdx1 maintains β cell identity and function by repressing an α cell program. Cell Metab. 19, 259–271. doi: 10.1016/j.cmet.2013.12.002
Gavrilov, K., and Saltzman, W. M. (2012). Therapeutic siRNA: principles, challenges, and strategies. Yale J. Biol. Med. 85, 187–200.
Gloyn, A. L., Pearson, E. R., Antcliff, J. F., Proks, P., Bruining, G. J., Slingerland, A. S.,et al. (2004). Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N. Engl. J. Med. 350, 1838–1849. doi: 10.1056/NEJMoa032922
Gong, C., and Maquat, L. E. (2011). lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3′ UTRs via Alu elements. Nature 470, 284–288. doi: 10.1038/nature09701
Guay, C., Roggli, E., Nesca, V., Jacovetti, C., and Regazzi, R. (2011). Diabetes mellitus, a microRNA-related disease? Transl. Res. 157, 253–264. doi: 10.1016/j.trsl.2011.01.009
Guttman, M., Amit, I., Garber, M., French, C., Lin, M. F., Feldser, D.,et al. (2009). Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458, 223–227. doi: 10.1038/nature07672
Henquin, J.-C., Nenquin, M., Sempoux, C., Guiot, Y., Bellanné-Chantelot, C., Otonkoski, T.,et al. (2011). In vitro insulin secretion by pancreatic tissue from infants with diazoxide-resistant congenital hyperinsulinism deviates from model predictions. J. Clin. Invest. 121, 3932–3942. doi: 10.1172/JCI58400
Ilnytska, O., Sözen, M. A., Dauterive, R., and Argyropoulos, G. (2009). Control elements in the neighboring ATPase gene influence spatiotemporal expression of the human agouti-related protein. J. Mol. Biol. 388, 239–251. doi: 10.1016/j.jmb.2009.03.017
Jonas, J.-C. (1999). Chronic hyperglycemia triggers loss of pancreatic beta cell differentiation in an animal model of diabetes. J. Biol. Chem. 274, 14112–14121. doi: 10.1074/jbc.274.20.14112
Kahn, S. E. (2003). The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of type 2 diabetes. Diabetologia 46, 3–19. doi: 10.1007/s00125-002-1009-0
Kallen, A. N., Zhou, X.-B., Xu, J., Qiao, C., Ma, J., Yan, L.,et al. (2013). The imprinted H19 lncRNA antagonizes let-7 microRNAs. Mol. Cell 52, 101–112. doi: 10.1016/j.molcel.2013.08.027
Kassem, S. A., Ariel, I., Thornton, P. S., Hussain, K., Smith, V., Lindley, K. J.,et al. (2001). p57KIP2 expression in normal islet cells and in hyperinsulinism of infancy. Diabetes Metab. Res. Rev. 50, 2763–2769. doi: 10.2337/diabetes.50.12.2763
Khalil, A. M., Guttman, M., Huarte, M., Garber, M., Raj, A., Rivea Morales, D.,et al. (2009). Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. U.S.A. 106, 11667–11672. doi: 10.1073/pnas.0904715106
Kjorholt, C., Akerfeldt, M. C., Biden, T. J., and Laybutt, D. R. (2005). Chronic hyperglycemia, independent of plasma lipid levels, is sufficient for the loss of cell differentiation and secretory function in the db/db mouse model of diabetes. Diabetes Metab. Res. Rev. 54, 2755–2763. doi: 10.2337/diabetes.54.9.2755
Korostowski, L., Sedlak, N., and Engel, N. (2012). The Kcnq1ot1 long non-coding RNA affects chromatin conformation and expression of Kcnq1, but does not regulate its imprinting in the developing heart. PLoS Genet. 8:e1002956. doi: 10.1371/journal.pgen.1002956
Kotake, Y., Nakagawa, T., Kitagawa, K., Suzuki, S., Liu, N., Kitagawa, M.,et al. (2011). Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15(INK4B) tumor suppressor gene. Oncogene 30, 1956–1962. doi: 10.1038/onc.2010.568
Krishnamurthy, J., Ramsey, M. R., Ligon, K. L., Torrice, C., Koh, A., Bonner-Weir, S.,et al. (2006). p16INK4a induces an age-dependent decline in islet regenerative potential. Nature 443, 453–457. doi: 10.1038/nature05092
Ku, G. M., Kim, H., Vaughn, I. W., Hangauer, M. J., Myung Oh, C., German, M. S.,et al. (2012). Research resource: RNA-Seq reveals unique features of the pancreatic β-cell transcriptome. Mol. Endocrinol. 26, 1783–1792. doi: 10.1210/me.2012-1176
Kung, J. T. Y., Colognori, D., and Lee, J. T. (2013). Long noncoding RNAs: past, present, and future. Genetics 193, 651–669. doi: 10.1534/genetics.112.146704
Lai, F., Orom, U. A., Cesaroni, M., Beringer, M., Taatjes, D. J., Blobel, G. A.,et al. (2013). Activating RNAs associate with mediator to enhance chromatin architecture and transcription. Nature 494, 497–501. doi: 10.1038/nature11884
Marselli, L., Suleiman, M., Masini, M., Campani, D., Bugliani, M., Syed, F.,et al. (2014). Are we overestimating the loss of beta cells in type 2 diabetes? Diabetologia 57, 362–365. doi: 10.1007/s00125-013-3098-3
McMinn, J., Wei, M., Schupf, N., Cusmai, J., Johnson, E. B., Smith, A. C.,et al. (2006). Unbalanced placental expression of imprinted genes in human intrauterine growth restriction. Placenta 27, 540–549. doi: 10.1016/j.placenta.2005.07.004
Modarresi, F., Faghihi, M. A., Lopez-Toledano, M. A., Fatemi, R. P., Magistri, M., Brothers, S. P.,et al. (2012). Inhibition of natural antisense transcripts in vivo results in gene-specific transcriptional upregulation. Nat. Biotechnol. 30, 453–459. doi: 10.1038/nbt.2158
Mohammad, F., Mondal, T., Guseva, N., Pandey, G. K., and Kanduri, C. (2010). Kcnq1ot1 noncoding RNA mediates transcriptional gene silencing by interacting with Dnmt1. Development 137, 2493–2499. doi: 10.1242/dev.048181
Montgomery, R. L., and van Rooij, E. (2011). Therapeutic advances in microRNA targeting. J. Cardiovasc. Pharmacol. 57, 1–7. doi: 10.1097/FJC.0b013e3181f603d0
Morán, I., Akerman, I., van de Bunt, M., Xie, R., Benazra, M., Nammo, T.,et al. (2012). Human β cell transcriptome analysis uncovers lncRNAs that are tissue-specific, dynamically regulated, and abnormally expressed in type 2 diabetes. Cell Metab. 16, 435–448. doi: 10.1016/j.cmet.2012.08.010
Morris, A. P., Voight, B. F., Teslovich, T. M., Ferreira, T., Segrè, A. V., Steinthorsdottir, V.,et al. (2012). Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat. Genet. 44, 981–990. doi: 10.1038/ng.2383
Nagano, T., Mitchell, J. A., Sanz, L. A., Pauler, F. M., Ferguson-Smith, A. C., Feil, R.,et al. (2008). The air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science 322, 1717–1720. doi: 10.1126/science.1163802
Ng, S.-Y., Johnson, R., and Stanton, L. W. (2012). Human long non-coding RNAs promote pluripotency and neuronal differentiation by association with chromatin modifiers and transcription factors. EMBO J. 31, 522–533. doi: 10.1038/emboj.2011.459
Pandey, R. R., Mondal, T., Mohammad, F., Enroth, S., Redrup, L., Komorowski, J.,et al. (2008). Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol. Cell 32, 232–246. doi: 10.1016/j.molcel.2008.08.022
Pasmant, E., Sabbagh, A., Vidaud, M., and Bièche, I. (2011). ANRIL, a long, noncoding RNA, is an unexpected major hotspot in GWAS. FASEB J. 25, 444–448. doi: 10.1096/fj.10-172452
Pasquali, L., Gaulton, K. J., Rodríguez-Seguí, S. A., Mularoni, L., Miguel-Escalada, I., Akerman, I.,et al. (2014). Pancreatic islet enhancer clusters enriched in type 2 diabetes risk-associated variants. Nat. Genet. 46, 136–143. doi: 10.1038/ng.2870
Pontier, D. B., and Gribnau, J. (2011). Xist regulation and function explored. Hum. Genet. 130, 223–236. doi: 10.1007/s00439-011-1008-7
Poy, M. N., Eliasson, L., Krutzfeldt, J., Kuwajima, S., Ma, X., Macdonald, P. E.,et al. (2004). A pancreatic islet-specific microRNA regulates insulin secretion. Nature 432, 226–230. doi: 10.1038/nature03076
Pullen, T. J., da Silva Xavier, G., Kelsey, G., and Rutter, G. A. (2011). miR-29a and miR-29b contribute to pancreatic beta-cell-specific silencing of monocarboxylate transporter 1 (Mct1). Mol. Cell. Biol. 31, 3182–3194. doi: 10.1128/MCB.01433-1410
Pullen, T. J., Khan, A. M., Barton, G., Butcher, S. A., Sun, G., and Rutter, G. A. (2010). Identification of genes selectively disallowed in the pancreatic islet. Islets 2, 89–95. doi: 10.4161/isl.2.2.11025
Pullen, T. J., Sylow, L., Sun, G., Halestrap, A. P., Richter, E. A., and Rutter, G. A. (2012). Overexpression of monocarboxylate transporter-1 (Slc16a1) in mouse pancreatic β-cells leads to relative hyperinsulinism during exercise. Diabetes 61, 1719–1725. doi: 10.2337/db11-1531
Rahier, J., Guiot, Y., Goebbels, R. M., Sempoux, C., and Henquin, J. C. (2008). Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes. Metab. 10(Suppl. 4), 32–42. doi: 10.1111/j.1463-1326.2008.00969.x
Rinn, J. L., Kertesz, M., Wang, J. K., Squazzo, S. L., Xu, X., Brugmann, S. A.,et al. (2007). Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129, 1311–1323. doi: 10.1016/j.cell.2007.05.022
Rosengren, A. H., Braun, M., Mahdi, T., Andersson, S. A., Travers, M. E., Shigeto, M.,et al. (2012). Reduced insulin exocytosis in human pancreatic β-cells with gene variants linked to type 2 diabetes. Diabetes Metab. Res. Rev. 61, 1726–1733. doi: 10.2337/db11-1516
Sato, K., Nakagawa, H., Tajima, A., Yoshida, K., and Inoue, I. (2010). ANRIL is implicated in the regulation of nucleus and potential transcriptional target of E2F1. Oncol. Rep. 24, 701–707. doi: 10.3892/or_00000910
Sladek, R., Rocheleau, G., Rung, J., Dina, C., Shen, L., Serre, D.,et al. (2007). A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 445, 881–885. doi: 10.1038/nature05616
Talchai, C., Xuan, S., Lin, H. V., Sussel, L., and Accili, D. (2012). Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell 150, 1223–1234. doi: 10.1016/j.cell.2012.07.029
Thorrez, L., Laudadio, I., Van Deun, K., Quintens, R., Hendrickx, N., Granvik, M.,et al. (2011). Tissue-specific disallowance of housekeeping genes: the other face of cell differentiation. Genome Res. 21, 95–105. doi: 10.1101/gr.109173.110
Tokuyama, Y., Sturis, J., DePaoli, A. M., Takeda, J., Stoffel, M., Tang, J.,et al. (1995). Evolution of beta-cell dysfunction in the male zucker diabetic fatty rat. Diabetes Metab. Res. Rev. 44, 1447–1457. doi: 10.2337/diab.44.12.1447
Travers, M. E., Mackay, D. J. G., Nitert, M. D., Morris, A. P., Lindgren, C. M., Berry, A.,et al. (2013). Insights into the molecular mechanism for type 2 diabetes susceptibility at the KCNQ1 locus from temporal changes in imprinting status in human islets. Diabetes Metab. Res. Rev. 62, 987–992. doi: 10.2337/db12-0819
Ullrich, S., Su, J., Ranta, F., Wittekindt, O. H., Ris, F., Rösler, M.,et al. (2005). Effects of I(Ks) channel inhibitors in insulin-secreting INS-1 cells. Pflugers Arch. 451, 428–436. doi: 10.1007/s00424-005-1479-2
Visel, A., Zhu, Y., May, D., Afzal, V., Gong, E., Attanasio, C.,et al. (2010). Targeted deletion of the 9p21 non-coding coronary artery disease risk interval in mice. Nature 464, 409–412. doi: 10.1038/nature08801
Wang, K. C., and Chang, H. Y. (2011). Molecular mechanisms of long noncoding RNAs. Mol. Cell 43, 904–914. doi: 10.1016/j.molcel.2011.08.018
Weir, G. C., and Bonner-Weir, S. (2013). Islet β cell mass in diabetes and how it relates to function, birth, and death. Ann. N. Y. Acad. Sci. 1281, 92–105. doi: 10.1111/nyas.12031
Yap, K. L., Li, S., Muñoz-Cabello, A. M., Raguz, S., Zeng, L., Mujtaba, S.,et al. (2010). Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol. Cell 38, 662–674. doi: 10.1016/j.molcel.2010.03.021
Zhao, C., Wilson, M. C., Schuit, F., Halestrap, A. P., and Rutter, G. A. (2001). Expression and distribution of lactate/monocarboxylate transporter isoforms in pancreatic islets and the exocrine pancreas. Diabetes Metab. Res. Rev. 50, 361–366. doi: 10.2337/diabetes.50.2.361
Zhao, J., Sun, B. K., Erwin, J. A., Song, J.-J., and Lee, J. T. (2008). Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 322, 750–756. doi: 10.1126/science.1163045
Keywords: type 2 diabetes, genome-wide association studies, lncRNAs, beta cell, islets of Langerhans, cell identity
Citation: Pullen TJ and Rutter GA (2014) Roles of lncRNAs in pancreatic beta cell identity and diabetes susceptibility. Front. Genet. 5:193. doi: 10.3389/fgene.2014.00193
Received: 06 May 2014; Accepted: 12 June 2014;
Published online: 01 July 2014.
Edited by:
Romano Regazzi, University of Lausanne, SwitzerlandReviewed by:
Scot J. Matkovich, Washington University School of Medicine, USALorenzo Pasquali, Institut d’Investigacions Biomèdiques August Pi i Sunyer, Spain
Copyright © 2014 Pullen and Rutter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Timothy J. Pullen and Guy A. Rutter, Section of Cell Biology, Department of Medicine, Imperial Centre for Translational and Experimental Medicine, Imperial College London, Du Cane Road, London W12 0NN, UK e-mail:dC5wdWxsZW5AaW1wZXJpYWwuYWMudWs=;Zy5ydXR0ZXJAaW1wZXJpYWwuYWMudWs=