
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 12 May 2014
Sec. Applied Genetic Epidemiology
Volume 5 - 2014 | https://doi.org/10.3389/fgene.2014.00096
This article is part of the Research Topic Genetics Research in Electronic Health Records Linked to DNA Biobanks View all 21 articles
 Berta Almoguera1*
Berta Almoguera1* Lyam Vazquez1
Lyam Vazquez1 John J. Connolly1
John J. Connolly1 Jonathan Bradfield1
Jonathan Bradfield1 Patrick Sleiman1,2
Patrick Sleiman1,2 Brendan Keating1,2
Brendan Keating1,2 Hakon Hakonarson1,2
Hakon Hakonarson1,2Background: The activity of thiopurine methyltransferase (TPMT) is subject to genetic variation. Loss-of-function alleles are associated with various degrees of myelosuppression after treatment with thiopurine drugs, thus genotype-based dosing recommendations currently exist. The aim of this study was to evaluate the potential utility of leveraging genomic data from large biorepositories in the identification of individuals with TPMT defective alleles.
Material and methods: TPMT variants were imputed using the 1000 Genomes Project reference panel in 87,979 samples from the biobank at The Children's Hospital of Philadelphia. Population ancestry was determined by principal component analysis using HapMap3 samples as reference. Frequencies of the TPMT imputed alleles, genotypes and the associated phenotype were determined across the different populations. A sample of 630 subjects with genotype data from Sanger sequencing (N = 59) and direct genotyping (N = 583) (12 samples overlapping in the two groups) was used to check the concordance between the imputed and observed genotypes, as well as the sensitivity, specificity and positive and negative predictive values of the imputation.
Results: Two SNPs (rs1800460 and rs1142345) that represent three TPMT alleles (*3A, *3B, and *3C) were imputed with adequate quality. Frequency for the associated enzyme activity varied across populations and 89.36–94.58% were predicted to have normal TPMT activity, 5.3–10.31% intermediate and 0.12–0.34% poor activities. Overall, 98.88% of individuals (623/630) were correctly imputed into carrying no risk alleles (553/553), heterozygous (45/46) and homozygous (25/31). Sensitivity, specificity and predictive values of imputation were over 90% in all cases except for the sensitivity of imputing homozygous subjects that was 80.64%.
Conclusion: Imputation of TPMT alleles from existing genomic data can be used as a first step in the screening of individuals at risk of developing serious adverse events secondary to thiopurine drugs.
Thiopurine S-methyltransferase (TPMT) is an enzyme involved in the metabolism of purine analogs such as azathioprine, 6-mercaptopurine and thioguanine, drugs that are used as chemotherapeutic and immunosuppressant agents in diseases such as lymphoid malignancies, leukemias, inflammatory bowel disease, and other immune conditions (Relling et al., 2011; Appell et al., 2013). TPMT maps to chromosome 6p22.3. It is subject to genetic variation and, to date, 34 alleles have been identified and characterized, most of which are associated with reduced activity in vitro (Relling et al., 2011). Alleles *2 (rs1800462), *3A (rs1800460 and rs1142345), *3B (rs1800460), and *3C (rs1142345) account for 95% of all defective alleles and all four involve missense mutations: allele *2 results in the p.Ala80Pro change (chr6:18143955), allele *3A contains two missense changes: p.Ala154Thr (chr6: 6:18139228) and p.Tyr240Cys (chr6:18130918), and alleles *3B and *3C are defined by p.Ala154Thr and p.Tyr240Cys, respectively (reference sequence NP_000358.1). The frequencies of these alleles vary significantly across ethnic populations (Appell et al., 2013): while *3A is the most frequently found in Caucasians (4.5%) (Schaeffeler et al., 2004), *3C is more prevalent in Africans or Asians, with 5.4–7.6% and 0.3–3%, respectively (reviewed in Teml et al., 2007).
TPMT enzymatic activity exhibits a trimodal distribution and approximately 0.3% of the population carry two defective alleles (associated with negligible activity), about 10% are heterozygous (intermediate activity), and 89% have normal activity (Weinshilboum and Sladek, 1980; Schaeffeler et al., 2004). Therefore, both heterozygous and homozygous individuals are at higher risk of developing myelosuppression within a few weeks after starting treatment with conventional doses that can be lethal if unrecognized, independent of the underlying disease being treated (Sim et al., 2013).
Due to the potential cytotoxicity and narrow therapeutic index of thiopurines, the US Food and Drug Administration (FDA) recommends TPMT testing prior to starting treatment with thiopurine drugs, and TPMT genotype-guided dosing recommendations are currently in use (Relling et al., 2011, 2013).
Results of genetic tests are potentially relevant over a patient's lifetime and having that information incorporated into patients' medical records may be useful in the improvement and guidance of drug treatments, if ever needed. With electronic medical records (EMR) currently widely implemented at academic hospitals and other treatment institutions, pharmacogenetic actionable variants can be integrated to the already available patient's information, helping optimize clinical decision making and care planning (Gottesman et al., 2013). Moreover, genome-wide data is increasingly accessible due to decreasing costs of genomic technologies and the development of methods that allow for accurate imputation of genotypes not directly probed by specific arrays could influence health care decisions (Marchini and Howie, 2010). Genomic data is frequently stored within large biorepositories where DNA samples are linked with phenotypic data. These biorepositories have been efficient and successful in studies of genotype-phenotype associations and they can be used as a model for the implementation and evaluation of pharmacogenomics in routine clinical practice (Gottesman et al., 2013).
In the present study, we leverage existing genome-wide genotyping data to impute common defective TPMT alleles with the aim of identifying individuals carrying high-risk genotypes for thiopurines-induced adverse events.
This study was approved by the institutional review board and the ethics committee of The Children's Hospital of Philadelphia (CHOP). Written informed consent was obtained from each participant in accordance with institutional requirements and the Declaration of Helsinki Principles. Subjects were selected from the biorepository at the Center for Applied Genomics at CHOP. The CHOP biobank has a collection of over 160,000 samples including 60,000 internal pediatric samples and over 100,000 adult and pediatric samples from external collaborators genotyped using standard GWAS arrays from Illumina and Affymetrix (summarized in Gottesman et al., 2013).
Figure 1 illustrates the study process. For TPMT imputation, we selected a total of 87,979 samples genotyped with either InfiniumII HumanHap550 (550; N = 45,893) or Human610-Quad version 1 (Quad; N = 42,086) arrays (Illumina, San Diego, CA). Genotyping data were used to impute sex using PLINK (http://pngu.mgh.harvard.edu/purcell/plink/) (Purcell et al., 2007); population ancestry was determined by principal component analysis (Eigenstrat 3.0) (Price et al., 2006), and samples were grouped into populations using nearest neighbors analysis and the HapMap3 samples (https://www.sanger.ac.uk/resources/downloads/human/hapmap3.html) as a reference.
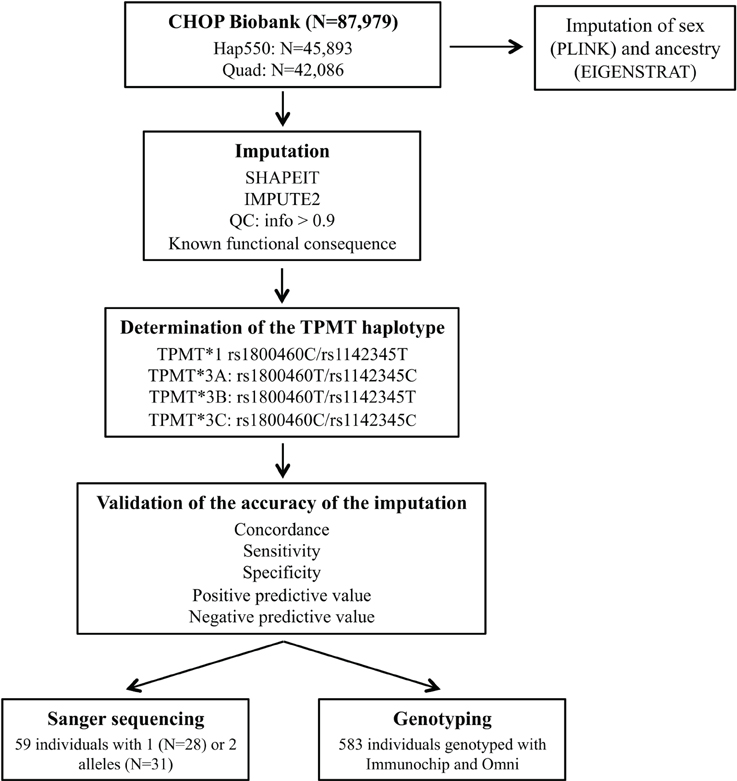
Figure 1. General schema for the study process. Hap550 is the InfiniumII HumanHap550 array, Quad is Human610-Quad version 1 array, Immunochip is the Illumina Infinium Immunochip array and Omni is the HumanOmni1-Quad version 1.
Imputation of unobserved genotypes in TPMT gene locus (chr6:18,128,545–18,155,374) was carried out with the IMPUTE2 package (http://mathgen.stats.ox.ac.uk/impute/impute_v2.html) (Howie et al., 2009) with the 1,000 Genomes Project reference panel, after prephasing chromosome 6 haplotypes with SHAPEIT version 2 (http://www.shapeit.fr/) (Delaneau et al., 2013). Since rs1800460 is probed on the Illumina HH610 Quad array, prephasing and imputation was performed for each chip type separately. Quality control filters were applied and only SNPs with an info score >0.9 were kept.
To determine the accuracy of imputation, TPMT imputed haplotypes were compared to those obtained by other genotyping platforms covering TPMT variation. Of the 87,979 samples, 583 also had genotyping data on Illumina Infinium Immunochip (Immunochip), and HumanOmni1-Quad version 1 (Omni), which captured both rs1800460 and rs1142345. Additionally, Sanger sequencing of rs1800460 in exon 7 and rs1142345 in exon 10 was used to validate the imputation results (primers previously described in Schaeffeler et al., 2001). The sample selected for Sanger sequencing consisted of 59 individuals predicted to carry one or two defective alleles by imputation that had been exposed to a TPMT medication based on the EMR.
We determined the concordance of the imputation as the number of imputed genotypes that correspond with output from direct genotyping or sequencing (expressed as percentage). Sensitivity, specificity and positive and negative predictive values of the imputation in the discrimination of subjects carrying the *1/*1 genotype, and one and two TPMT defective alleles were determined as shown in Figure 2.
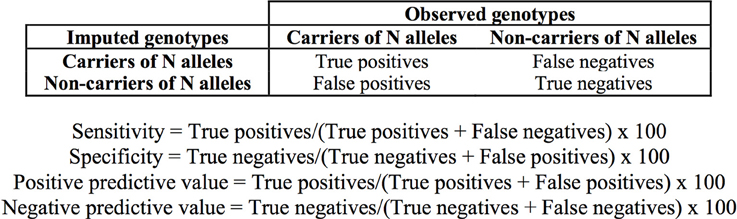
Figure 2. Definition of true and false positive and negative values and formulae used for the determination of the sensitivity, specificity, and predictive values of the imputation.
Ancestry, sex estimation and imputation of TPMT genotypes for the 89,797 individuals were performed in eleven batches of ~8,000 samples. An average of 174,911 SNPs with r2 < 0.2 were used for principal components calculation.
There were 50.04% males out of the 89,797 individuals (imputed sex for 0.87% of the individuals was undetermined). Principal component analysis classified 72.74% of individuals as Caucasians, 18.78% with African ancestry, 6.55% Hispanics, and 1.93% Asians (Table 1). There were also data on self-reported ethnicity for 24,527 out of the 89,797 individuals, with 50.15% Caucasians (N = 12,304), 41.81% African Americans (N = 10,263), 1.6% Asians (N = 385), 0.08% American Indians (N = 21), 0.03% Native Hawaiians (N = 7), 0.02% Indians (N = 5), and 6.29% were considered as “Other” (N = 1542). Concordance between self-reported and imputed ancestry was >80% for Caucasians, African Americans, and Asians (93.92, 98.34, and 81.30%, respectively). For the remaining groups, 57.58% were classified as Hispanics (19 out of 33), 15.15% as Asians (N = 5) and Caucasians (N = 5) and 12.12% as African Americans (N = 4).
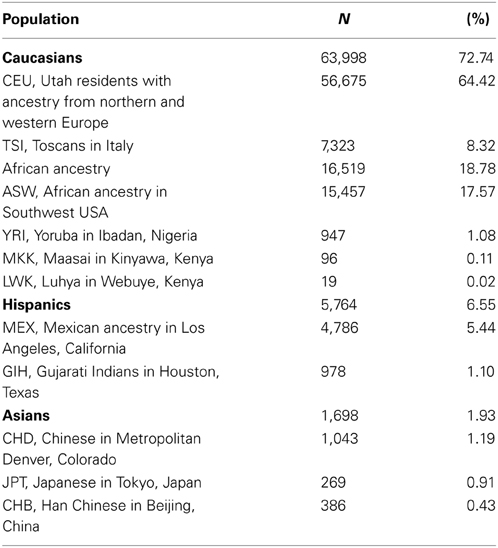
Table 1. Frequencies of the different ethnicities in the sample investigated based on principal component analysis of imputed genotype data.
Three hundred and fifty four variants were imputed in the TPMT gene, including 322 SNPs and 33 insertion/deletion polymorphisms (indels). Out of these, only 117 had an info value ≥0.9 (103 SNPs and 14 indels). However for the subsequent analysis only those with a known functional significance were considered: rs1800460 and rs1142345, which define alleles *3A, *3B, and *3C. The loss-of-function variant rs1800462 that defines allele *2 was also imputed but did not pass the quality filters, thus it was excluded from the further analyses.
TPMT alleles were assigned as *1 when the rs1800460 C>T/rs1142345 T>C diplotype was CT, *3B when TT, and *3C when CC. For allele *3A, given the high linkage disequilibrium between rs1800460 (*3B) and rs1142345 (*3C) and the low minor allele frequency, whenever an individual carried both variants (rs1800460T and rs1142345C), the allele was assigned as *3A.
TPMT allelic, genotypic and associated phenotypic frequencies for *3A, *3B, and *3C across ethnic groups are illustrated in Tables 2, 3 and 4, respectively. As shown in Table 2, the distribution of the three defective alleles varied largely across populations: *3A was more represented among the Caucasians, *3B in the Hispanics and *3C in both Asians and African Americans, being the latter the group with the highest frequency of carriers of TPMT defective alleles (5.49 vs. 4.07% in Caucasians, 4.41% in Hispanics and 2.77% in Asians). According to the genotype-associated enzymatic activity, Asians harbored the lowest rates of poor metabolizers with only 0.12% whereas Caucasians, African Americans and Hispanics have a frequency close to 0.33%.
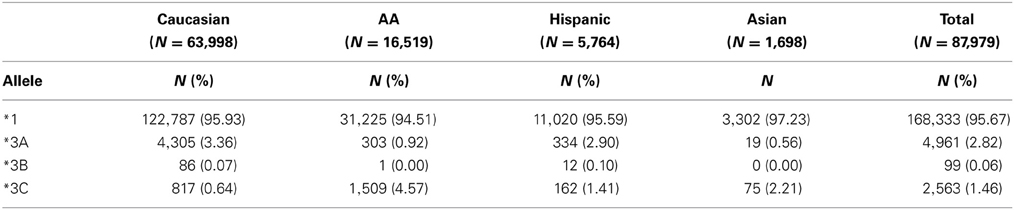
Table 2. Distribution of allele frequencies for TPMT alleles *3A, *3B, and *3C across the different ethnic groups.
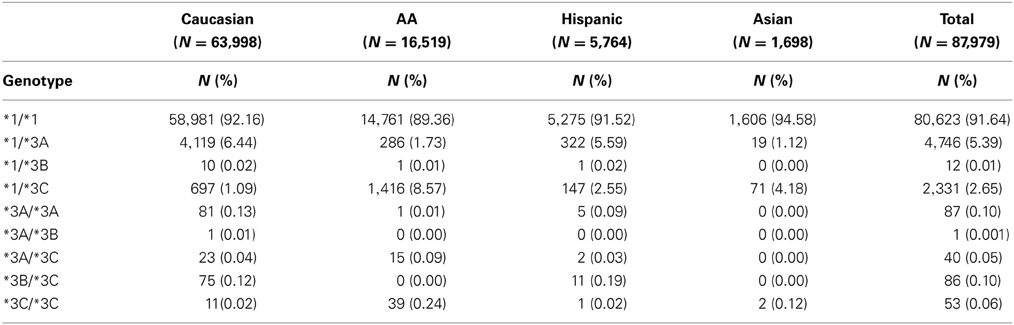
Table 3. Distribution of genotypes for TPMT alleles *3A, *3B, and *3C across the different ethnic groups.
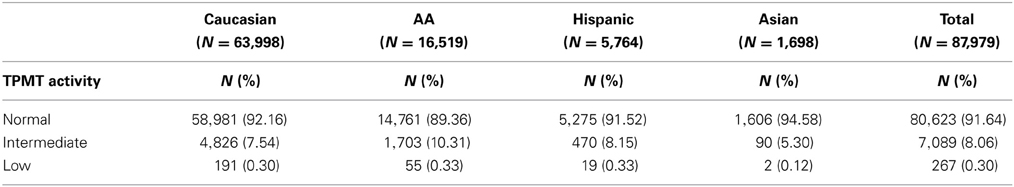
Table 4. TPMT genotype-associated phenotypic frequencies across the different ethnic groups.
Out of the 87,979 samples used for imputation, 583 had genotyping data on both rs1800460 and rs1142345: 94.8% of them carried the genotype *1/*1, 4.5% *1/*3A, and 0.7% the genotype *1/*3C. Concordance of the imputed haplotypes compared to those determined by genotyping was 99.8% (Table 5).
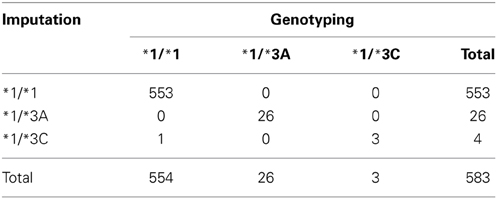
Table 5. Concordance between the imputed genotypes and genotypes determined by genotyping using Immunochip (Illumina Infinium Immunochip array) and Omni (HumanOmni1-Quad version 1) (N = 583).
Sanger sequencing was performed in a subset of 59 samples predicted to carry 1 (N = 28) or 2 (N = 31) defective alleles (*3A, or *3C). Twelve of the 59 samples also had genotype data and results were consistent across the two methods and with the imputation. The overall concordance was 84.7% for the total 59 samples. Table 6 illustrates the concordance of the imputed genotypes after validation with Sanger sequencing.
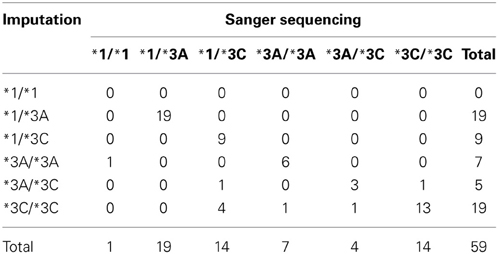
Table 6. Concordance between imputed genotypes and genotypes determined by Sanger sequencing (N = 59).
When taking into account the number of defective alleles, 98.88% of individuals (623 of the 630 individuals—excluding the 12 in the two groups-) were accurately imputed. All of the samples identified as carrying no risk alleles (*1/*1) were confirmed by direct genotyping or sequencing (553/553) and for heterozygous and homozygous individuals, the concordance was lower, with 97.8% (45/46) and 80.64% (25/31), respectively (Table 7). Sensitivity, specificity and positive and negative predictive values of imputation of TPMT genotypes are summarized in Table 7. Since the importance of TPMT genotyping lies in the discrimination of individuals carrying defective alleles, these metrics were determined for the discrimination of individuals carrying the *1/*1 genotype, individuals with one defective allele and individuals with two defective alleles. Sensitivity, specificity, and predictive values were close to 100% for all cases except for the positive predictive value of identifying homozygous carriers, which was 80.64%.

Table 7. Concordance between imputed and observed genotypes according to the number of defective alleles and characteristics of the imputation in terms of sensitivity (S), specificity (SP), positive predictive value (PPV) and negative predictive value (NPV) (N = 630).
A growing number of drug-gene interactions, affecting routinely prescribed drugs, are being validated (Relling and Klein, 2011). The FDA has to date recommended the inclusion of pharmacogenetic markers in the labels of more than a 100 drugs (http://www.fda.gov/drugs/scienceresearch/researchareas/pharmacogenetics/ucm083378.htm) (Shuldiner et al., 2013) and initiatives such as Pharmacogenomics Knowledge Base (PharmGKB) and the Clinical Pharmacogenetics Implementation Consortium (CPIC) (Relling and Klein, 2011) provide essential pharmacogenetic information and play a major role in establishing recommendations to aid clinicians in guiding therapies. If one considers the report by Schildcrout et al, who demonstrated that up to 65% of patients were exposed to at least one medication with an established drug-gene association within 5 years (Schildcrout et al., 2012), then pharmacogenetics integration into individuals' medical records for clinical use is becoming an urgent need. Availability of pharmacogenetic information prior to patients' treatment has the opportunity to identify individuals potentially benefiting from a given therapy, select adequate medications and doses, in order to ultimately administer the most effective treatment to each patient, with the lower incidence of adverse events.
Using existing genomic data from the CHOP biobank repository we have been able to impute three of the most common defective TPMT alleles *3A, *3B, *3C in a cohort of 87,979 individuals. The sensitivity, specificity and positive and negative predictive values of the imputation were sufficiently high to allow discrimination of patients carrying one or two defective alleles from those with a *1/*1 genotype. Concordance between observed and imputed genotypes was 100% for individuals with *1/*1 genotype, and for carriers of TPMT alleles, discordant results were essentially cases of individuals predicted to be heterozygous by imputation that were found to be homozygous by genotyping or sequencing. Additionally, probably because of the rarity of SNPs rs1800460 and rs1142345 and the increase in imputation errors as minor allele frequency decreases, alleles *3A and *3C were frequently switched, and allele *3B was only identified in the subset of samples genotyped with the Quad array. The rarity of allele *2 may also be the explanation for the inability of imputing with adequate quality.
TPMT deficiency exhibits an extensive interethnic variability (Wang et al., 2010; Appell et al., 2013). The population investigated in this study is characterized for being largely admixed with African Americans, Asians and Hispanics accounting for almost 30% of all individuals. As previously described, frequency of alleles *3A, *3B and *3C is population-specific. Whereas *3A and *3B were predominantly found in Caucasians and Hispanics (3.36 and 2.90%, for *3A and 0.07 and 0.1%, for *3B, respectively), the most prevalent defective allele in African Americans and Asians was *3C (4.57 and 2.21%, respectively). These results were similar to frequencies previously reported for those populations (Oliveira et al., 2007; Taja-Chayeb et al., 2008; Appell et al., 2013). Regarding TPMT associated-phenotypes, African Americans had the highest proportion of intermediate (10.31%) and low methylators (0.33%), being the ethnic group with the highest risk of developing adverse events derived from TPMT treatment. Conversely, Asians are the lowest risk group, with only 5.4% of individuals carrying one (5.30%) or two alleles (0.12%). Caucasians and Hispanics had a similar percentage of individuals with TPMT intermediate (7.54 and 8.15%, respectively) and low activity (0.30 and 0.33%, respectively). Other population-specific alleles not imputed in the current study, such as *2 that is almost restricted to Caucasians, or *6 (rs75543815) and *8 (rs56161402) that occur at frequencies between 1.5 and 3.5% in some African and Asian populations (Oliveira et al., 2007), are also important contributors of TPMT deficiency. These rare TPMT alleles or novel variants will not be detected with this approach and can only be identified by direct genotyping or sequencing. Thus, the frequency of intermediate and low methylators in this study may be slightly underestimated.
It is worth mentioning that approximately 1 in 10 individuals tested from our biobank were found to carry at least one high-risk TPMT allele. There are currently over 2.5 million children enrolled in the CHOP healthcare system, and if one extrapolates the results yielded from this study to the entire population at CHOP, then more than 170,000 patients would be expected to be TPMT deficient. Identification of such carriers is especially important in the pediatric population, as thiopurines are commonly prescribed drugs in children. Thiopurines are the backbone drugs for maintenance of acute lymphoblastic leukemia (ALL), which is the most common childhood malignancy (Pui and Evans, 2006), and are also frequently used as chronic immunosuppressive therapy after organ transplantation and in inflammatory bowel disease (Dubinsky, 2004; Relling et al., 2011; Appell et al., 2013). A major limitation of their use is their narrow therapeutic index and the severe myelosuppression they cause, a life threating adverse event highly associated with TPMT deficiency (Relling et al., 1999). In a study by Relling and coworkers in 180 children with ALL receiving conventional doses of 6-mercaptopurin, the authors found that the cumulative incidence of toxicity was 100% for homozygous TPMT deficiency, 35% for heterozygous, and 7% for patients homozygous for allele *1 (Relling et al., 1999). This association has been widely replicated, so genotyping of TPMT is recommended in US FDA-approved labeling and currently genotype-based dosing recommendations exist (Relling et al., 2011, 2013). The high genotype-phenotype correlation existent for TPMT and the large interethnic variability in the susceptibility to thiopurine hematopoietic toxicity, sustain the need of availing such genetic information to prospectively identify individuals where thiopurine therapy may need to be modified or changed.
Biorepositories where DNA samples are linked to the EMR of patients, such as the CHOP biobank, offer the ideal platform for screening and identification of individuals with high-risk genotypes that may require a modification in the therapy if a given drug is prescribed. CHOP is part of the Electronic Medical Records and Genomics (eMERGE) consortium that is actively working on large-scale testing and integration of information on actionable pharmacogenetic variants, such as TPMT alleles, into clinical practice using EMR technologies (Gottesman et al., 2013). One of the goals of eMERGE is the creation of SPHINX (Sequence, Phenotype, and pHarmacogenomics INtegration eXchange http://www.emergesphinx.org/), a web accessible repository of genomic variants derived from a panel of 84 genes involved in the pharmacogenetics of a large number of drugs, designed by the NIH-supported Pharmacogenetics Research Network (PGRN), and linked to clinical information. To date, SPHINX contains data on 2000 of the nearly 9000 subjects that are planned to be enrolled in the project. Interestingly, so far SPHINX lists 174 variants in the TPMT gene, including known and novel variants, and the minor allele frequency information. Variant repositories such as SPHINX allow the advance in the knowledge of pharmacogenetics through the exploration of new hypotheses and the further integration of this information into the EMR.
The results yielded from this study demonstrate that imputation of TPMT alleles from existing genomic data is feasible and may be used as a first step in the screening of high-risk individuals for thiopurine drugs toxicity. Sensitivity, specificity, and predictive values of the imputation were over 90% in all cases, except for the positive predictive value of the imputation of homozygous subjects. Given that around 90% of the population is expected to have two fully functional TPMT alleles, being able to accurately identify such individuals based on existing genomic data yields 10% of the population to be screened for high-risk genotypes with direct genotyping methods. The positive and negative predictive values of 100 and 97.40%, respectively, obtained for the discrimination of individuals with the *1/*1 genotype supports the potential utility of imputation in narrowing the target population where TPMT genotypes need to be determined. Further integration of such pharmacogenetic information into the EMR, with clinical decision support, may be used to aid clinicians prescribe therapies with the maximum risk-benefit ratio based on each individual's information.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was funded by institutional support from the Children's Hospital of Philadelphia and the National Human Genome Research Institute (# U01 HG006830).
Appell, M. L., Berg, J., Duley, J., Evans, W. E., Kennedy, M. A., Lennard, L., et al. (2013). Nomenclature for alleles of the thiopurine methyltransferase gene. Pharmacogenet. Genomics 23, 242–248. doi: 10.1097/FPC.0b013e32835f1cc0
Delaneau, O., Zagury, J. F., and Marchini, J. (2013). Improved whole-chromosome phasing for disease and population genetic studies. Nat. Methods 10, 5–6. doi: 10.1038/nmeth.2307
Dubinsky, M. C. (2004). Azathioprine, 6-mercaptopurine in inflammatory bowel disease: pharmacology, efficacy, and safety. Clin. Gastroenterol. Hepatol. 2, 731–743. doi: 10.1016/S1542-3565(04)00344-1
Gottesman, O., Kuivaniemi, H., Tromp, G., Faucett, W. A., Li, R., Manolio, T. A., et al. (2013). The Electronic Medical Records and Genomics (eMERGE) Network: past, present, and future. Genet. Med. 15, 761–771. doi: 10.1038/gim.2013.72
Howie, B. N., Donnelly, P., and Marchini, J. (2009). A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 5:e1000529. doi: 10.1371/journal.pgen.1000529
Marchini, J., and Howie, B. (2010). Genotype imputation for genome-wide association studies. Nat. Rev. Genet. 11, 499–511. doi: 10.1038/nrg2796
Oliveira, E., Quental, S., Alves, S., Amorim, A., and Prata, M. J. (2007). Do the distribution patterns of polymorphisms at the thiopurine S-methyltransferase locus in sub-Saharan populations need revision? Hints from Cabinda and Mozambique. Eur. J. Clin. Pharmacol. 63, 703–706. doi: 10.1007/s00228-007-0310-8
Price, A. L., Patterson, N. J., Plenge, R. M., Weinblatt, M. E., Shadick, N. A., and Reich, D. (2006). Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909. doi: 10.1038/ng1847
Pui, C. H., and Evans, W. E. (2006). Treatment of acute lymphoblastic leukemia. N. Engl. J. Med. 354, 166–178. doi: 10.1056/NEJMra052603
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A., Bender, D., et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. doi: 10.1086/519795
Relling, M. V., Gardner, E. E., Sandborn, W. J., Schmiegelow, K., Pui, C. H., Yee, S. W., et al. (2013). Clinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing: 2013 update. Clin. Pharmacol. Ther. 93, 324–325. doi: 10.1038/clpt.2013.4
Relling, M. V., Gardner, E. E., Sandborn, W. J., Schmiegelow, K., Pui, C. H., Yee, S. W., et al. (2011). Clinical Pharmacogenetics Implementation Consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin. Pharmacol. Ther. 89, 387–391. doi: 10.1038/clpt.2010.320
Relling, M. V., Hancock, M. L., Rivera, G. K., Sandlund, J. T., Ribeiro, R. C., Krynetski, E. Y., et al. (1999). Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J. Natl. Cancer Inst. 91, 2001–2008.
Relling, M. V., and Klein, T. E. (2011). CPIC: Clinical Pharmacogenetics Implementation Consortium of the Pharmacogenomics Research Network. Clin. Pharmacol. Ther. 89, 464–467. doi: 10.1038/clpt.2010.279
Schaeffeler, E., Fischer, C., Brockmeier, D., Wernet, D., Moerike, K., Eichelbaum, M., et al. (2004). Comprehensive analysis of thiopurine S-methyltransferase phenotype-genotype correlation in a large population of German-Caucasians and identification of novel TPMT variants. Pharmacogenetics 14, 407–417. doi: 10.1097/01.fpc.0000114745.08559.db
Schaeffeler, E., Lang, T., Zanger, U. M., Eichelbaum, M., and Schwab, M. (2001). High-throughput genotyping of thiopurine S-methyltransferase by denaturing HPLC. Clin. Chem. 47, 548–555.
Schildcrout, J. S., Denny, J. C., Bowton, E., Gregg, W., Pulley, J. M., Basford, M. A., et al. (2012). Optimizing drug outcomes through pharmacogenetics: a case for preemptive genotyping. Clin. Pharmacol. Ther. 92, 235–242. doi: 10.1038/clpt.2012.66
Shuldiner, A. R., Relling, M. V., Peterson, J. F., Hicks, J. K., Freimuth, R. R., Sadee, W., et al. (2013). The pharmacogenomics research network translational pharmacogenetics program: overcoming challenges of real-world implementation. Clin. Pharmacol. Ther. 94, 207–210. doi: 10.1038/clpt.2013.59
Sim, S. C., Kacevska, M., and Ingelman-Sundberg, M. (2013). Pharmacogenomics of drug-metabolizing enzymes: a recent update on clinical implications and endogenous effects. Pharmacogenomics J. 13, 1–11. doi: 10.1038/tpj.2012.45
Taja-Chayeb, L., Vidal-Millan, S., Gutierrez, O., Ostrosky-Wegman, P., Duenas-Gonzalez, A., and Candelaria, M. (2008). Thiopurine S-methyltransferase gene (TMPT) polymorphisms in a Mexican population of healthy individuals and leukemic patients. Med. Oncol. 25, 56–62. doi: 10.1007/s12032-007-9002-6
Teml, A., Schaeffeler, E., Herrlinger, K. R., Klotz, U., and Schwab, M. (2007). Thiopurine treatment in inflammatory bowel disease: clinical pharmacology and implication of pharmacogenetically guided dosing. Clin. Pharmacokinet. 46, 187–208. doi: 10.2165/00003088-200746030-00001
Wang, L., Pelleymounter, L., Weinshilboum, R., Johnson, J. A., Hebert, J. M., Altman, R. B., et al. (2010). Very important pharmacogene summary: thiopurine S-methyltransferase. Pharmacogenet. Genomics 20, 401–405. doi: 10.1097/FPC.0b013e3283352860
Keywords: TPMT, genotype imputation, DNA biobank, pharmacogenetics, Electronic Medical Records
Citation: Almoguera B, Vazquez L, Connolly JJ, Bradfield J, Sleiman P, Keating B and Hakonarson H (2014) Imputation of TPMT defective alleles for the identification of patients with high-risk phenotypes. Front. Genet. 5:96. doi: 10.3389/fgene.2014.00096
Received: 14 January 2014; Accepted: 04 April 2014;
Published online: 12 May 2014.
Edited by:
Helena Kuivaniemi, Geisinger Health System, USAReviewed by:
Peter A. Kanetsky, Moffitt Cancer Center, USACopyright © 2014 Almoguera, Vazquez, Connolly, Bradfield, Sleiman, Keating and Hakonarson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Berta Almoguera, Center for Applied Genomics, The Children's Hospital of Philadelphia, 3615 Civic Center Boulevard, Abramson Building, Philadelphia, PA 19104, USA e-mail:Y2FzdGlsbG9iQGVtYWlsLmNob3AuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.