 Reut Suliman
Reut Suliman Eyal Ben-David
Eyal Ben-David Sagiv Shifman
Sagiv Shifman
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
HYPOTHESIS AND THEORY article
Front. Genet., 10 April 2014
Sec. Neurogenomics
Volume 5 - 2014 | https://doi.org/10.3389/fgene.2014.00081
Though extensively characterized clinically, the causes of autism spectrum disorder (ASD) remain a mystery. ASD is known to have a strong genetic basis, but it is genetically very heterogeneous. Recent studies have estimated that de novo disruptive mutations in hundreds of genes may contribute to ASD. However, it is unclear how it is possible for mutations in so many different genes to contribute to ASD. Recent findings suggest that many of the mutations disrupt genes involved in transcription regulation that are expressed prenatally in the developing brain. De novo disruptive mutations are also more frequent in girls with ASD, despite the fact that ASD is more prevalent in boys. In this paper, we hypothesize that loss of robustness may contribute to ASD. Loss of phenotypic robustness may be caused by mutations that disrupt capacitors that operate in the developing brain. This may lead to the release of cryptic genetic variation that contributes to ASD. Reduced robustness is consistent with the observed variability in expressivity and incomplete penetrance. It is also consistent with the hypothesis that the development of the female brain is more robust, and it may explain the higher rate and severity of disruptive de novo mutations in girls with ASD.
The genetic architecture of ASD is very complex and hard to dissect. Recently, there have been several breakthroughs in our understanding of this heterogeneous disease, including increased appreciation of the importance of de novo mutations (Sebat et al., 2007). We now know that ASD can be triggered by different types of genetic variations in many different genes, a phenomenon termed non-allelic genetic heterogeneity. It is estimated that mutations that are likely to disrupt gene function (such as nonsense, splice site and frameshift mutations) in hundreds of different genes may contribute to ASD (Iossifov et al., 2012; Neale et al., 2012). The involvement of rare mutations was clear from the finding that ASD is associated with genetic syndromes that show Mendelian inheritance (Cook et al., 1997; Manning et al., 2004; Splawski et al., 2004; Abrahams and Geschwind, 2008). In addition, rare and de novo large chromosomal rearrangements are also found more frequently in ASD, currently estimated to account for 2–7% of cases (Marshall et al., 2008; Shen et al., 2010).
Recently, with the advent of molecular tools that enable to study the genome in high resolution and unbiased way, de novo variations, both copy number variations (CNVs) and single nucleotide variations (SNVs), have been found to be associated with ASD, currently estimated to account for 6 and 10% of the cases, respectively (Sebat et al., 2007; Pinto et al., 2010; Sanders et al., 2011; Iossifov et al., 2012; Neale et al., 2012; O'Roak et al., 2012b; Sanders et al., 2012). Based on the rate of de novo SNVs it has been estimated that hundreds of genes can lead to ASD when disrupted (Iossifov et al., 2012; Neale et al., 2012; O'Roak et al., 2012b; Sanders et al., 2012). Consequently, each gene could account for only a small proportion of cases.
The current view of the genetic architecture of ASD is that it is similar to other complex diseases. ASD risk is attributed to both rare genetic variants and combinations of common variants that act together with environmental risk factors (Huguet et al., 2013). It is assumed that people with ASD have a set of genetic variants that predispose them to abnormal development of specialized brain structures involved in social information processing (the “social brain”). Thus, it is puzzling that there is no unifying pathophysiology in ASD, in addition to the findings of mutations in many different genes, involved in different functions.
A possible solution to this puzzle would be if shared mechanisms between some of those genes could be identified. First attempts to identify such mechanisms focused on genes within rare copy-number variations (de novo or inherited) (Pinto et al., 2010; Gai et al., 2011; Gilman et al., 2011). One such effort found enrichment in rare deletions for genes involved in neuronal development and function, as well as for GTPase/Ras signaling (Pinto et al., 2010). Other studies found enrichment in CNVs for genes involved in synaptic transmission (Gai et al., 2011) and synaptogenesis (Gilman et al., 2011). It is important to note, however, that enrichment studies in CNVs have several limitations. First, the power of enrichment analyses based on CNVs is limited by their large size, which in many cases encompasses a large number of genes. Furthermore, in such analyses gene size may also be an important confounding factor, especially when dealing with disorders of the brain (Raychaudhuri et al., 2010). Another approach was to study genes with single base mutations; including mutations that lead to ASD associated genetic syndromes (Sakai et al., 2011; Ben-David and Shifman, 2012). However, many of these genes have been discovered in studies which were at least partly hypothesis driven, and used previous knowledge to identify the causal gene in large inherited chromosomal aberrations or segregating loci in families with ASD.
The advent of next generation sequencing, and with it the recent sequencing of four large ASD cohorts of families for de novo exonic mutations (Iossifov et al., 2012; Neale et al., 2012; O'Roak et al., 2012b; Sanders et al., 2012), has provided an unprecedented sample of de novo disruptions which have been discovered in an unbiased genome-wide manner, each affecting a specific gene. When combining the mutations found in these cohorts and performing a functional enrichment analysis, an enrichment was found for genes involved in transcription regulation, and in particular chromatin regulators that control chromatin structure and function (Table 1) (Ben-David and Shifman, 2013). This enrichment is apparent when concentrating on genes with mutations highly likely to be functional—nonsense, frame shift or splice site mutations (Ben-David and Shifman, 2013). When looking at the temporal expression pattern of these genes during brain development, they were found to be strongly expressed prenatally, with much lower expression after birth (Ben-David and Shifman, 2013). The specificity of the enrichment of transcription regulators to ASD was demonstrated by comparing the distribution of mutations between cases and a large set of controls, and to unaffected siblings. Furthermore, by comparing the distribution of disruptive vs. silent mutations in the same subjects it was shown that the enrichment of chromatin regulators is unlikely to be a result of detection or mutation bias (Ben-David and Shifman, 2013). Still it is not clear how disruptive mutations in chromatin regulators and other genes involved in regulation of transcription link to ASD. We suggest below that mutation in chromatin regulators causes loss of the robustness of brain development that together with other ASD specific risk factors may lead to ASD.
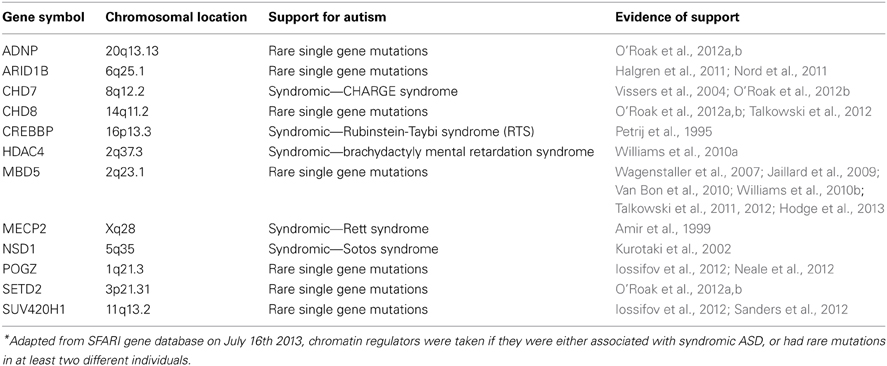
Table 1. Chromatin regulators associated with autism spectrum disorders*.
Phenotypic robustness is the insensitivity of the phenotype to genetic or environmental perturbations. An equivalent concept is developmental stability, which is defined as the ability of the individual to produce a robust phenotype even if faced with genetic and environmental perturbations during development. A further related term is canalization, which refers to the selection during evolution for more stable phenotypes, or in other words, adaptive robustness (Gibson and Wagner, 2000). In the following, we will use the term phenotypic robustness without the distinction between adaptive and intrinsic forms of robustness (Gibson and Wagner, 2000). Loss of phenotypic robustness has been recently proposed to explain the missing heritability in complex disease (Gibson, 2009; McGrath et al., 2011; Queitsch et al., 2012). Based on this proposal, the degree of robustness varies between individuals and it influences the probability of developing a disease or disorder. It was also argued that findings in ASD and schizophrenia may be consistent with decreased robustness, a phenomenon termed decanalization (Woolf, 1997; McGrath et al., 2011; Queitsch et al., 2012). Furthermore, it was speculated that brain structures (such as the neocortex) that have undergone profound evolution in the recent lineage leading to humans, may be more vulnerable to loss of robustness because there has been insufficient time to evolve sufficient buffering capacity (McGrath et al., 2011). The brain may be also more vulnerable than other organs, because it is a complex organ that develops later in life and mainly composed of terminally differentiated cells. In the first years of life, the brain may be particularly sensitive to reduced robustness because the social development depends on signals from the environment.
The development of the brain is generally robust, so it may seem surprising that de novo mutations in the heterozygote state contribute to ASD and other neurodevelopmental disorders. Many genes can be inhibited in mice without producing a phenotype. Moreover, heterozygous knockout (KO) mice rarely show a clear phenotype; In fact, in many experiments heterozygous KO mice are often used as a control for homozygote KO mice. While neurodevelopmental systems have evolved to be robust, they may be vulnerable to perturbations in a specific subset of genes, termed phenotypic capacitors (Rutherford and Lindquist, 1998; Levy and Siegal, 2008). Phenotypic capacitors are genes that buffer against perturbations and therefore contribute to the robustness of the phenotype. So, phenotypic capacitors, when operating normally, may prevent the development of disorders like ASD even in individuals carrying or exposed to genetic and environmental risk factors.
The most studied capacitor is Hsp90 (Rutherford and Lindquist, 1998). Hsp90 was termed “genetic capacitor” because the reduction in its activity increased phenotypic variation. It was first suggested that its chaperone activity, that facilitate the correct folding of proteins with destabilizing mutations, is how Hsp90 can buffer mutations (Tokuriki and Tawfik, 2009). However, more recent work has suggested that Hsp90 prevents phenotypic variation by suppressing transposon activity (Specchia et al., 2010). Besides Hsp90, yeast and worm studies strongly implicate chromatin regulators to be phenotypic capacitors (Lehner et al., 2006; Levy and Siegal, 2008; Tirosh et al., 2010). In yeast, more than 300 genes are known to be important for robustness. Very similar to the type of genes implicated in ASD, the phenotypic capacitors in yeast tend to be highly connected regulatory genes, many of which are involved in transcription and chromatin regulation (Levy and Siegal, 2008). A major source of variation during development is stochastic fluctuations in gene expression (McAdams and Arkin, 1997). Gene expression shows significant variation, even between genetically identical cells (Lehner, 2008). In this setting, chromatin regulators and other transcription factors are likely critical to the ability of a cell to buffer the fluctuations in gene expression against cryptic genetic variations (Tirosh et al., 2010). Cryptic genetic variations are hidden genetic variations that do not affect the trait in a given genetic background or environmental condition, but may be expressed after environmental, genetic, or epigenetic perturbations (Gibson and Dworkin, 2004).
The chromatin regulators that were found to be disrupted by de novo mutations in ASD may be involved in multiple cellular processes, such as regulation of transcription, cell cycle regulation, genomic stability, and DNA damage repair. Still it is not clear how mutations in chromatin regulators lead to ASD. While collectively de novo mutations in chromatin regulators account for a small proportion of cases, understanding how they operate may shed light on the biological mechanisms of ASD. Following the models of loss of robustness, we suggest that mutations that affect chromatin regulators may lead to ASD because they are “global regulators,” namely they are genes that are placed at the very top of a regulatory hierarchy. Chromatin regulators interact with many genes and pathways, and so perturbation in them can affect multiple target genes simultaneously. Furthermore, we propose that these genes may also act as phenotypic capacitors, protecting the developmental processes by buffering genetic and environmental perturbations. Similarly, other regulators (e.g., transcription factors and genes involved in translation regulation) may be associated with ASD because their disruption leads to loss of robustness. New examples for regulators that are involved in ASD are topoisomerases. One study found that inhibiting topoisomerase 1 (TOP1) reduces the expression of large number of very long genes, which are particularly expressed in the brain (King et al., 2013). Two other studies provided evidence that Top3β is an RNA topoisomerase that interacts with the fragile X mental retardation protein (FMRP) to regulate the expression of multiple mRNAs that are crucial for neurodevelopment (Stoll et al., 2013; Xu et al., 2013).
The proposed model is that individuals, in whom the robustness of brain development has been impaired by genetic or environmental insults, will have an increased risk of developing a neurodevelopmental disorder (Figure 1). The specific phenotype will depend on the combination of other genetic and environmental factors including stochastic events. According to this model, individuals with disruptive mutations in regulators, such as chromatin regulators, will have reduced phenotypic robustness (or developmental instability) which may lead to a number of different conditions. In fact, some of the genes found to be disrupted in individuals with ASD were also found in congenital heart disease (Zaidi et al., 2013). Similarly, we propose that being a male is a risk factor for different neurodevelopmental disorders because of reduced phenotypic robustness. Other factors, including the environment, may as well lead to loss of robustness. Furthermore, an individual may carry a mutation that decreases robustness but will have typical development under specific environmental and genetic conditions, which are more stable. Aside from the genetic evidence linking chromatin and other regulators to autism, this model is also attractive as it has the potential to elucidate a large set of puzzling observations in ASD. We highlight some of the evidence for this theory and the evidence for the more classical view in Table 2. We also propose in Table 2 several experiments that could be used to test the loss of robustness theory and its connection to chromatin and other regulators in ASD.
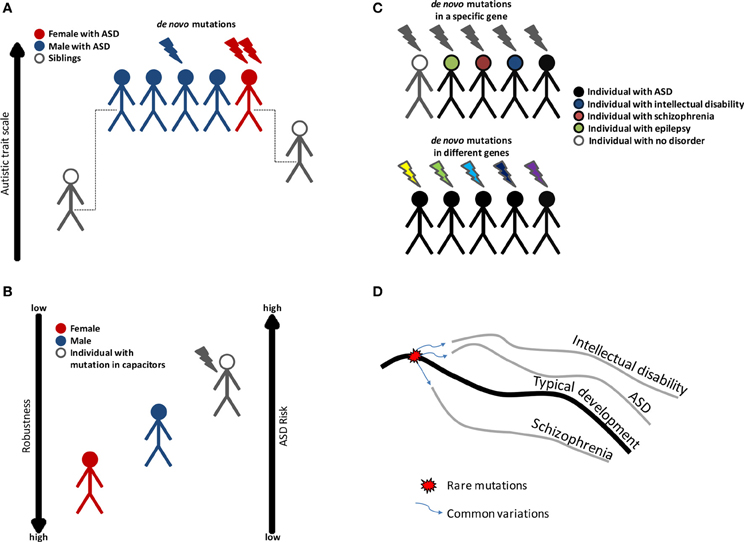
Figure 1. Variation in ASD risk may be linked to phenotypic robustness. (A) Evidence to show that females are protected against ASD. ASD is more prevalent in males; however females exhibit a higher rate of genes disrupted by de novo mutation. On average, family members of females with ASD have higher autistic trait scores than the family members of males with ASD. (B) ASD risk may be associated with phenotypic robustness, and decreased robustness leads to a higher risk of ASD. For this reason, males are more predisposed to ASD than females, and individuals with mutations in phenotypic capacitors have the highest risk. (C) ASD is characterized by genetic heterogeneity, incomplete penetrance and variable expressivity. Variations in phenotypic robustness may explain some of the genetic complexity in ASD. (D) A model of how rare and common variations cause different neurodevelopmental disorders. A genetic loss of robustness (decanalization) is caused by mutation in regulators that act as capacitors, followed by an abnormal development of the brain. Common variations direct the abnormal development toward a specific trajectory (a specific disorder).
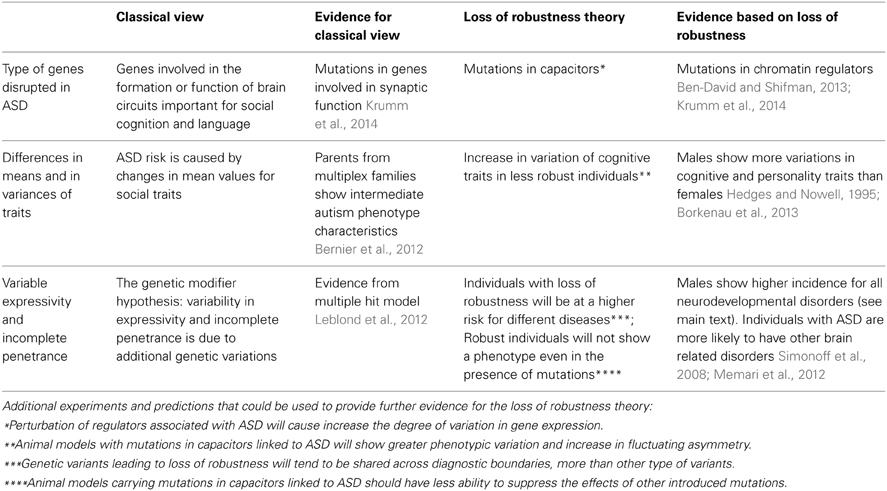
Table 2. Evidence for the classical view and the loss of robustness theory in ASD.
One finding that remains consistent in ASD research is that boys are more likely to be diagnosed with autism than girls. The ratio between males and females is 4:1, but it may increase up to 15:1 for the high functioning end of the spectrum (Wing, 1981). This means that girls are less likely to develop ASD, but when they do, they tend to show a more severe phenotype. There are several explanations for this major difference in prevalence between the sexes. From early prenatal development, through adolescence, males and females have different levels of sex hormones, such as testosterone and estrogen. These hormones are known to have a major influence on brain development, structure and function (Berenbaum and Beltz, 2011). It has been proposed that different hormone levels can increase or decrease the risk of developing ASD (Baron-Cohen et al., 2005).
A different hypothesis that has gained recent support is that there are unknown factors that “protect” females from ASD (Werling and Geschwind, 2013). Recent genome-wide studies showed that on average females with ASD have more de novo mutations than males, including CNVs and SNVs (Levy et al., 2011; Iossifov et al., 2012; Neale et al., 2012). For example, in the exome sequencing study of Iossifov et al. they observe, on average, twice the number of de novo disruptive mutations in females relative to males with ASD (9 in 29 females [31%] vs. 50 in 314 males [16%]). In addition, de novo CNVs in females were larger and included significantly more genes than CNVs in males (Levy et al., 2011). A recent study addressed this hypothesis directly by examining dizygotic twin pairs, comparing autistic traits in siblings of female and male probands that were at the top 90 and 95th percentiles of the distribution of the autistic trait score (Robinson et al., 2013). They showed that siblings of females with ASD had higher scores in autistic traits scale than siblings of males with the disorder. This result, replicated across two nationally-representative samples, implies that in girls a greater load of genetic risk factors is required to cause ASD. Although other studies did not find the same effect (Ozonoff et al., 2011), this was the first study to examine representative community samples without ascertainment bias, and using quantitative measurements that may be more sensitive.
The female protection theory fits with the model of loss of robustness. According to this model, besides the variation between individuals, there is a difference in the average degree of robustness of the brain between males and females. Supporting this suggestion is the higher rate of boys relative to girls in other neurodevelopmental disorders such as attention-deficit–hyperactivity disorder (ADHD), dyslexia, specific language impairment, Tourette Syndrome, and other learning difficulties (Shaywitz et al., 1990; Kadesjo and Gillberg, 2000; Rutter et al., 2003; Simon et al., 2009). There are several possible genetic explanations for the difference in robustness between males and females. The early genetic hypothesis was that some genes involved in ASD are located on the X chromosome (Skuse, 2000). Females inherit two X chromosomes from both parents, while males inherit only one maternal copy. Consequently, females are less vulnerable to mutations in genes on the X-chromosome since they have a second copy that can compensate. The robustness model generalizes this concept of genetic protection in females, by stating that females may have a more robust development of the brain and therefore require larger perturbations to develop ASD and other neurodevelopmental disorders. Robustness may lead to lower prevalence, but it also means that females require a greater genetic insult that may go along with a more severe phenotype.
Genetic findings from recent years show that mutations associated with ASD have low specificity (Figure 1C). Not only are there probably hundreds of genes that may contribute to ASD, many of the genes that were identified until now have also been found to be associated with other neuropsychiatric conditions, including mental retardation, epilepsy, ADHD, and schizophrenia (Betancur, 2011; Devlin and Scherer, 2012; Kirov et al., 2013). This phenomenon is known as variable expressivity, whereby individuals with a certain mutation exhibit differences in phenotypic expression, such as disease severity or symptoms. Variable expressivity among different individuals may arise from individual-specific genetic and environmental factors. It coincides with the finding that around 70% of the individuals with ASD also suffer from other psychiatric conditions (Simonoff et al., 2008; Memari et al., 2012). In fact, the authors cannot state a single example of a gene that causes only ASD when disrupted. Furthermore, some of the rare variants associated with ASD were observed in individuals with normal development, showing that those variants may not be sufficient to cause ASD (Zhao et al., 2007; Ben-Shachar et al., 2009; Beunders et al., 2010; Schaaf et al., 2011; Kirov et al., 2013). The observation that not all people who carry a mutation develop the disease is termed incomplete penetrance. A recent study showed that CNVs associated with ASD or schizophrenia affect cognition in control individuals. The CNVs were considered to have incomplete penetrance; however, the results show that the differences between cases and controls are in fact due to variable expressivity (Stefansson et al., 2014).
One explanation for the variable expressivity and incomplete penetrance is that other genetic variations may modify the phenotype (Schaaf et al., 2011). Evidence for this explanation comes from mouse models of ASD. In mouse models, it is very common to observe a large phenotypic variation dependent on the genetic background. For example, when the FMR1 (the gene for fragile X syndrome) knockout mouse was crossed with six different inbred strains, only one line (B6.D2 F1 hybrid) showed both social and communication deficits along with repetitive behaviors (Spencer et al., 2011). This means that there are genetic variations between the strains that modify the effect of the mutation without a direct effect on phenotype. In other words, these effects are due to epistatic interactions (Epistasis is when the phenotypic effect of one genotype depends on the genotype of other genes).
In ASD, we suggest to attribute the incomplete penetrance to differences in robustness among individuals. It was demonstrated in Caenorhabditis elegans that incomplete penetrance is a consequence of stochastic variation in gene expression, which is influenced by chromatin regulators and other capacitors (Raj et al., 2010; Burga et al., 2011). Individuals in whom brain development is more robust are expected to show low variation in gene expression and lower penetrance for mutations (Queitsch et al., 2012). Furthermore, in robust individuals some of the genetic variations will be cryptic. In less robust individuals, such as individuals with mutations in phenotypic capacitors, genetic variations that were compensated for will be revealed and contribute to the disease. Different genetic backgrounds, including common and rare variations, could explain the phenotypic variations observed in humans with mutations in the same gene.
As with many other common complex diseases, one of the main hypotheses about the genetic architecture of ASD was that it is influenced by common variants (Risch and Merikangas, 1996; Risch et al., 1999). In addition to past studies looking at candidate genes, genome-wide association studies were performed in recent years to identify single nucleotide polymorphism (SNPs) associated with ASD (Ma et al., 2009; Wang et al., 2009; Weiss et al., 2009; Anney et al., 2010). While some studies were able to identify SNPs with genome-wide significance (P < 5 × 10−8), none of those were replicated across studies. Until now, the sample size in GWAS has been modest (487–1558 families), therefore the failure of those studies could not be used as evidence against the contribution of common variants to ASD risk. Although efforts to identify specific SNP associated with ASD have not been very successful, several studies have shown that common SNPs collectively contribute to ASD (Voineagu et al., 2011; Ben-David and Shifman, 2012; Klei et al., 2012), estimated to explain 17–40% of ASD liability (Klei et al., 2012; Lee et al., 2013). The association of rare variants with ASD, on the other hand, has had a long history of success, as described above. But, do common and rare variants operate together? A support for a model that combines rare and common variations in ASD risk comes from a study that showed that both typse of variants affect that same kind of neuronal genes (Ben-David and Shifman, 2012). By dividing the genes in the human genome into functionally related groups, based on pattern of gene co-expression in the human brain, it was shown that genetic risk variants are most enriched in a group of synaptic genes expressed in adulthood, and in another group that contains genes involved in neurogenesis (genes active both during development and adulthood). The “synaptic group” was more enriched with common risk variants, while the “neurogenesis group” with rare variants. Whereas the relative contribution of rare and common variants is now starting to be explored at the population level, we still know very little about how common and rare variations at the individual level act together to cause ASD.
Adaptive robustness can also be achieved through negative epistatic interactions among alleles of common variants (Gibson, 2009). In the presence of risk alleles, selection that reduces the additive genetic effects will result in a more robust phenotype, since the effect of risk alleles is suppressed by other loci. It was suggested that dramatic changes in the environment, such as the ones that accrued in modern times in human urban societies, may result in changes in the epistatic interactions among alleles and reduced robustness (decanalization) (Gibson, 2009). This may explain the increase in incidence for different common diseases, including ASD.
As stated above, loss of robustness may increase the risk of neurodevelopmental disorders, but the specific phenotype may be modified by common genetic variations (Figure 1D). Thus, based on this model, genetic variation associated with ASD could be divided to two types: (1) genetic variation, mainly rare mutations, that reduce the phenotypic robustness of the brain, and (2) genetic variation, such as common variants, that influence brain functions involved specifically with social cognition. These latter variations are cryptic, since they are exposed only in individuals with loss of robustness. The genetic evidence for such a model comes from the low specificity of rare mutations, the disruption of genes that could be considered as master regulators in several diseases, and the high specificity of common variations to ASD. The enrichment of genes involved in transcription regulation was found in ASD, schizophrenia and other neurodevelopmental disorders (Najmabadi et al., 2011; Ben-David and Shifman, 2013; Gulsuner et al., 2013), suggesting that loss of robustness might be a more general cause of neurodevelopmental disorders. Hence, a single rare mutation may be associated with different disorders. In contrast, there is a very modest or non-significant genetic correlation between ASD and other psychiatric disorders, based on polygenic risk scores calculated from common variations (Cross-Disorder Group of the Psychiatric Genomics Consortium, 2013; Lee et al., 2013). While larger GWAS in ASD are needed to validate the low correlation, the current findings suggest that common variations may contribute to the specific phenotypes of ASD in individuals with rare mutations.
The classical genetic view of ASD and many other neuropsychiatric disorders is focused on genes that code for neural components that are essential for brain function. In the field of ASD, a great deal of research was focused on genes and proteins that are expressed in the adult brain and function at the synapse, or are regulated by neuronal activity (Zoghbi, 2003). The notion is that ASD is caused by mutations that interfere with brain regions or functions that are involved in the, so called, “social brain.” The recent developments of new techniques, allowing us to examine the autism genome in a genome-wide, unbiased manner, have brought to light another class of genes as associated with ASD risk. These genes are chromatin regulators, which are active during brain development. Based on this finding, and together with the observation of variable expressivity of mutations and the recent evidence for a female protective effect, we propose a unifying framework that connects phenotypic robustness theories with ASD risk (Figure 1). The implication of this theory, if proved true, is that the prevalence of ASD and other neurodevelopmental disorders may be associated with environmental factors that decrease robustness.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by a grant from the Simons Foundation (SFARI) (award no. 266554 to Sagiv Shifman), and from the Legacy Heritage Fund program of the Israel Science Foundation (grant no. 940/13 to Sagiv Shifman). Eyal Ben-David is supported by the Dennis Weatherstone Pre-doctoral Fellowship from Autism Speaks (grant no. 8595).
Abrahams, B. S., and Geschwind, D. H. (2008). Advances in autism genetics: on the threshold of a new neurobiology. Nat. Rev. Genet. 9, 341–355. doi: 10.1038/nrg2346
Amir, R. E., Van Den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188. doi: 10.1038/13810
Anney, R., Klei, L., Pinto, D., Regan, R., Conroy, J., Magalhaes, T. R., et al. (2010). A genome-wide scan for common alleles affecting risk for autism. Hum. Mol. Genet. 19, 4072–4082. doi: 10.1093/hmg/ddq307
Baron-Cohen, S., Knickmeyer, R. C., and Belmonte, M. K. (2005). Sex differences in the brain: implications for explaining autism. Science 310, 819–823. doi: 10.1126/science.1115455
Ben-David, E., and Shifman, S. (2012). Networks of neuronal genes affected by common and rare variants in autism spectrum disorders. PLoS Genet. 8:e1002556. doi: 10.1371/journal.pgen.1002556
Ben-David, E., and Shifman, S. (2013). Combined analysis of exome sequencing points toward a major role for transcription regulation during brain development in autism. Mol. Psychiatry 18, 1054–1056. doi: 10.1038/mp.2012.148
Ben-Shachar, S., Lanpher, B., German, J. R., Qasaymeh, M., Potocki, L., Nagamani, S. C., et al. (2009). Microdeletion 15q13.3: a locus with incomplete penetrance for autism, mental retardation, and psychiatric disorders. J. Med. Genet. 46, 382–388. doi: 10.1136/jmg.2008.064378
Berenbaum, S. A., and Beltz, A. M. (2011). Sexual differentiation of human behavior: effects of prenatal and pubertal organizational hormones. Front. Neuroendocrinol. 32, 183–200. doi: 10.1016/j.yfrne.2011.03.001
Bernier, R., Gerdts, J., Munson, J., Dawson, G., and Estes, A. (2012). Evidence for broader autism phenotype characteristics in parents from multiple-incidence autism families. Autism Res. 5, 13–20. doi: 10.1002/aur.226
Betancur, C. (2011). Etiological heterogeneity in autism spectrum disorders: more than 100 genetic and genomic disorders and still counting. Brain Res. 1380, 42–77. doi: 10.1016/j.brainres.2010.11.078
Beunders, G., Van De Kamp, J. M., Veenhoven, R. H., Van Hagen, J. M., Nieuwint, A. W., and Sistermans, E. A. (2010). A triplication of the Williams-Beuren syndrome region in a patient with mental retardation, a severe expressive language delay, behavioural problems and dysmorphisms. J. Med. Genet. 47, 271–275. doi: 10.1136/jmg.2009.070490
Borkenau, P., McCrae, R. R., and Terracciano, A. (2013). Do Men Vary more than women in personality? A study in 51 cultures. J. Res. Pers. 47, 135–144. doi: 10.1016/j.jrp.2012.12.001
Burga, A., Casanueva, M. O., and Lehner, B. (2011). Predicting mutation outcome from early stochastic variation in genetic interaction partners. Nature 480, 250–253. doi: 10.1038/nature10665
Cook, E. H. Jr., Lindgren, V., Leventhal, B. L., Courchesne, R., Lincoln, A., Shulman, C., et al. (1997). Autism or atypical autism in maternally but not paternally derived proximal 15q duplication. Am. J. Hum. Genet. 60, 928–934.
Cross-Disorder Group of the Psychiatric Genomics Consortium. (2013). Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 381, 1371–1379. doi: 10.1016/S0140-6736(12)62129-1
Devlin, B., and Scherer, S. W. (2012). Genetic architecture in autism spectrum disorder. Curr. Opin. Genet. Dev. 22, 229–237. doi: 10.1016/j.gde.2012.03.002
Gai, X., Xie, H. M., Perin, J. C., Takahashi, N., Murphy, K., Wenocur, A. S., et al. (2011). Rare structural variation of synapse and neurotransmission genes in autism. Mol. Psychiatry 17, 402–411. doi: 10.1038/mp.2011.10
Gibson, G. (2009). Decanalization and the origin of complex disease. Nat. Rev. Genet. 10, 134–140. doi: 10.1038/nrg2502
Gibson, G., and Dworkin, I. (2004). Uncovering cryptic genetic variation. Nat. Rev. Genet. 5, 681–690. doi: 10.1038/nrg1426
Gibson, G., and Wagner, G. (2000). Canalization in evolutionary genetics: a stabilizing theory? Bioessays 22, 372–380. doi: 10.1002/(SICI)1521-1878(200004)22:4%3C372::AID-BIES7%3E3.3.CO;2-A
Gilman, S. R., Iossifov, I., Levy, D., Ronemus, M., Wigler, M., and Vitkup, D. (2011). Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron 70, 898–907. doi: 10.1016/j.neuron.2011.05.021
Gulsuner, S., Walsh, T., Watts, A. C., Lee, M. K., Thornton, A. M., Casadei, S., et al. (2013). Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell 154, 518–529. doi: 10.1016/j.cell.2013.06.049
Halgren, C., Kjaergaard, S., Bak, M., Hansen, C., El-Schich, Z., Anderson, C. M., et al. (2011). Corpus callosum abnormalities, intellectual disability, speech impairment, and autism in patients with haploinsufficiency of ARID1B. Clin. Genet. 82, 248–255. doi: 10.1111/j.1399-0004.2011.01755.x
Hedges, L. V., and Nowell, A. (1995). Sex differences in mental test scores, variability, and numbers of high-scoring individuals. Science 269, 41–45. doi: 10.1126/science.7604277
Hodge, J. C., Mitchell, E., Pillalamarri, V., Toler, T. L., Bartel, F., Kearney, H. M., et al. (2013). Disruption of MBD5 contributes to a spectrum of psychopathology and neurodevelopmental abnormalities. Mol. Psychiatry 19, 368–379. doi: 10.1038/mp.2013.42
Huguet, G., Ey, E., and Bourgeron, T. (2013). The genetic landscapes of autism spectrum disorders. Annu. Rev. Genomics Hum. Genet. 14, 191–213. doi: 10.1146/annurev-genom-091212-153431
Iossifov, I., Ronemus, M., Levy, D., Wang, Z., Hakker, I., Rosenbaum, J., et al. (2012). De novo gene disruptions in children on the autistic spectrum. Neuron 74, 285–299. doi: 10.1016/j.neuron.2012.04.009
Jaillard, S., Dubourg, C., Gerard-Blanluet, M., Delahaye, A., Pasquier, L., Dupont, C., et al. (2009). 2q23.1 microdeletion identified by array comparative genomic hybridisation: an emerging phenotype with Angelman-like features? J. Med. Genet. 46, 847–855. doi: 10.1136/jmg.2008.058156
Kadesjo, B., and Gillberg, C. (2000). Tourette's disorder: epidemiology and comorbidity in primary school children. J. Am. Acad. Child Adolesc. Psychiatry 39, 548–555. doi: 10.1097/00004583-200005000-00007
King, I. F., Yandava, C. N., Mabb, A. M., Hsiao, J. S., Huang, H. S., Pearson, B. L., et al. (2013). Topoisomerases facilitate transcription of long genes linked to autism. Nature 501, 58–62. doi: 10.1038/nature12504
Kirov, G., Rees, E., Walters, J. T., Escott-Price, V., Georgieva, L., Richards, A. L., et al. (2013). The penetrance of copy number variations for schizophrenia and developmental delay. Biol. Psychiatry 75, 378–385. doi: 10.1016/j.biopsych.2013.07.022
Klei, L., Sanders, S. J., Murtha, M. T., Hus, V., Lowe, J. K., Willsey, A. J., et al. (2012). Common genetic variants, acting additively, are a major source of risk for autism. Mol. Autism 3, 9. doi: 10.1186/2040-2392-3-9
Krumm, N., O'roak, B. J., Shendure, J., and Eichler, E. E. (2014). A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci. 37, 95–105. doi: 10.1016/j.tins.2013.11.005
Kurotaki, N., Imaizumi, K., Harada, N., Masuno, M., Kondoh, T., Nagai, T., et al. (2002). Haploinsufficiency of NSD1 causes Sotos syndrome. Nat. Genet. 30, 365–366. doi: 10.1038/ng863
Leblond, C. S., Heinrich, J., Delorme, R., Proepper, C., Betancur, C., Huguet, G., et al. (2012). Genetic and functional analyses of SHANK2 mutations suggest a multiple hit model of autism spectrum disorders. PLoS Genet. 8:e1002521. doi: 10.1371/journal.pgen.1002521
Lee, S. H., Ripke, S., Neale, B. M., Faraone, S. V., Purcell, S. M., Perlis, R. H., et al. (2013). Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat. Genet. 45, 984–994. doi: 10.1038/ng.2711
Lehner, B. (2008). Selection to minimise noise in living systems and its implications for the evolution of gene expression. Mol. Syst. Biol. 4, 170. doi: 10.1038/msb.2008.11
Lehner, B., Crombie, C., Tischler, J., Fortunato, A., and Fraser, A. G. (2006). Systematic mapping of genetic interactions in Caenorhabditis elegans identifies common modifiers of diverse signaling pathways. Nat. Genet. 38, 896–903. doi: 10.1038/ng1844
Levy, D., Ronemus, M., Yamrom, B., Lee, Y.-H., Leotta, A., Kendall, J., et al. (2011). Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron 70, 886–897. doi: 10.1016/j.neuron.2011.05.015
Levy, S. F., and Siegal, M. L. (2008). Network hubs buffer environmental variation in Saccharomyces cerevisiae. PLoS Biol. 6:e264. doi: 10.1371/journal.pbio.0060264
Ma, D., Salyakina, D., Jaworski, J. M., Konidari, I., Whitehead, P. L., Andersen, A. N., et al. (2009). A genome-wide association study of autism reveals a common novel risk locus at 5p14.1. Ann. Hum. Genet. 73, 263–273. doi: 10.1111/j.1469-1809.2009.00523.x
Manning, M. A., Cassidy, S. B., Clericuzio, C., Cherry, A. M., Schwartz, S., Hudgins, L., et al. (2004). Terminal 22q deletion syndrome: a newly recognized cause of speech and language disability in the autism spectrum. Pediatrics 114, 451–457. doi: 10.1542/peds.114.2.451
Marshall, C. R., Noor, A., Vincent, J. B., Lionel, A. C., Feuk, L., Skaug, J., et al. (2008). Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 82, 477–488. doi: 10.1016/j.ajhg.2007.12.009
McAdams, H. H., and Arkin, A. (1997). Stochastic mechanisms in gene expression. Proc. Natl. Acad. Sci. U.S.A. 94, 814–819. doi: 10.1073/pnas.94.3.814
McGrath, J. J., Hannan, A. J., and Gibson, G. (2011). Decanalization, brain development and risk of schizophrenia. Transl. Psychiatry 1, e14. doi: 10.1038/tp.2011.16
Memari, A., Ziaee, V., Mirfazeli, F., and Kordi, R. (2012). Investigation of autism comorbidities and associations in a school-based community sample. J. Child Adolesc. Psychiatr. Nurs. 25, 84–90. doi: 10.1111/j.1744-6171.2012.00325.x
Najmabadi, H., Hu, H., Garshasbi, M., Zemojtel, T., Abedini, S. S., Chen, W., et al. (2011). Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature 478, 57–63. doi: 10.1038/nature10423
Neale, B. M., Kou, Y., Liu, L., Ma'ayan, A., Samocha, K. E., Sabo, A., et al. (2012). Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242–245. doi: 10.1038/nature11011
Nord, A. S., Roeb, W., Dickel, D. E., Walsh, T., Kusenda, M., O'Connor, K. L., et al. (2011). Reduced transcript expression of genes affected by inherited and de novo CNVs in autism. Eur. J. Hum. Genet. 19, 727–731. doi: 10.1038/ejhg.2011.24
O'Roak, B. J., Vives, L., Fu, W., Egertson, J. D., Stanaway, I. B., Phelps, I. G., et al. (2012a). Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 338, 1619–1622. doi: 10.1126/science.1227764
O'Roak, B. J., Vives, L., Girirajan, S., Karakoc, E., Krumm, N., Coe, B. P., et al. (2012b). Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250. doi: 10.1038/nature10989
Ozonoff, S., Young, G. S., Carter, A., Messinger, D., Yirmiya, N., Zwaigenbaum, L., et al. (2011). Recurrence risk for autism spectrum disorders: a baby siblings research consortium study. Pediatrics 128, e488–495. doi: 10.1542/peds.2010-2825
Petrij, F., Giles, R. H., Dauwerse, H. G., Saris, J. J., Hennekam, R. C., Masuno, M., et al. (1995). Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature 376, 348–351. doi: 10.1038/376348a0
Pinto, D., Pagnamenta, A. T., Klei, L., Anney, R., Merico, D., Regan, R., et al. (2010). Functional impact of global rare copy number variation in autism spectrum disorders. Nature 466, 368–372. doi: 10.1038/nature09146
Queitsch, C., Carlson, K. D., and Girirajan, S. (2012). Lessons from model organisms: phenotypic robustness and missing heritability in complex disease. PLoS Genet. 8:e1003041. doi: 10.1371/journal.pgen.1003041
Raj, A., Rifkin, S. A., Andersen, E., and Van Oudenaarden, A. (2010). Variability in gene expression underlies incomplete penetrance. Nature 463, 913–918. doi: 10.1038/nature08781
Raychaudhuri, S., Korn, J. M., McCarroll, S. A., Altshuler, D., Sklar, P., Purcell, S., et al. (2010). Accurately assessing the risk of schizophrenia conferred by rare copy-number variation affecting genes with brain function. PLoS Genet. 6:e1001097. doi: 10.1371/journal.pgen.1001097
Risch, N., and Merikangas, K. (1996). The future of genetic studies of complex human diseases. Science 273, 1516–1517. doi: 10.1126/science.273.5281.1516
Risch, N., Spiker, D., Lotspeich, L., Nouri, N., Hinds, D., Hallmayer, J., et al. (1999). A genomic screen of autism: evidence for a multilocus etiology. Am. J. Hum. Genet. 65, 493–507. doi: 10.1086/302497
Robinson, E. B., Lichtenstein, P., Anckarsater, H., Happe, F., and Ronald, A. (2013). Examining and interpreting the female protective effect against autistic behavior. Proc. Natl. Acad. Sci. U.S.A. 110, 5258–5262. doi: 10.1073/pnas.1211070110
Rutherford, S. L., and Lindquist, S. (1998). Hsp90 as a capacitor for morphological evolution. Nature 396, 336–342. doi: 10.1038/24550
Rutter, M., Caspi, A., and Moffitt, T. E. (2003). Using sex differences in psychopathology to study causal mechanisms: unifying issues and research strategies. J. Child Psychol. Psychiatry 44, 1092–1115. doi: 10.1111/1469-7610.00194
Sakai, Y., Shaw, C. A., Dawson, B. C., Dugas, D. V., Al-Mohtaseb, Z., Hill, D. E., et al. (2011). Protein interactome reveals converging molecular pathways among autism disorders. Sci. Transl. Med. 3, 86–86. doi: 10.1126/scitranslmed.3002166
Sanders, S. J., Ercan-Sencicek, A. G., Hus, V., Luo, R., Murtha, M. T., Moreno-De-Luca, D., et al. (2011). Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 70, 863–885. doi: 10.1016/j.neuron.2011.05.002
Sanders, S. J., Murtha, M. T., Gupta, A. R., Murdoch, J. D., Raubeson, M. J., Willsey, A. J., et al. (2012). De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237–241. doi: 10.1038/nature10945
Schaaf, C. P., Sabo, A., Sakai, Y., Crosby, J., Muzny, D., Hawes, A., et al. (2011). Oligogenic heterozygosity in individuals with high-functioning autism spectrum disorders. Hum. Mol. Genet. 20, 3366–3375. doi: 10.1093/hmg/ddr243
Sebat, J., Lakshmi, B., Malhotra, D., Troge, J., Lese-Martin, C., Walsh, T., et al. (2007). Strong association of de novo copy number mutations with autism. Science 316, 445–449. doi: 10.1126/science.1138659
Shaywitz, S. E., Shaywitz, B. A., Fletcher, J. M., and Escobar, M. D. (1990). Prevalence of reading disability in boys and girls. Results of the connecticut longitudinal study. JAMA 264, 998–1002. doi: 10.1001/jama.1990.03450080084036
Shen, Y., Dies, K. A., Holm, I. A., Bridgemohan, C., Sobeih, M. M., Caronna, E. B., et al. (2010). Clinical genetic testing for patients with autism spectrum disorders. Pediatrics 125, e727–e735. doi: 10.1542/peds.2009-1684
Simon, V., Czobor, P., Balint, S., Meszaros, A., and Bitter, I. (2009). Prevalence and correlates of adult attention-deficit hyperactivity disorder: meta-analysis. Br. J. Psychiatry 194, 204–211. doi: 10.1192/bjp.bp.107.048827
Simonoff, E., Pickles, A., Charman, T., Chandler, S., Loucas, T., and Baird, G. (2008). Psychiatric disorders in children with autism spectrum disorders: prevalence, comorbidity, and associated factors in a population-derived sample. J. Am. Acad. Child Adolesc. Psychiatry 47, 921–929. doi: 10.1097/CHI.0b013e318179964f
Skuse, D. H. (2000). Imprinting, the X-chromosome, and the male brain: explaining sex differences in the liability to autism. Pediatr. Res. 47, 9–16. doi: 10.1203/00006450-200001000-00006
Specchia, V., Piacentini, L., Tritto, P., Fanti, L., D'alessandro, R., Palumbo, G., et al. (2010). Hsp90 prevents phenotypic variation by suppressing the mutagenic activity of transposons. Nature 463, 662–665. doi: 10.1038/nature08739
Spencer, C. M., Alekseyenko, O., Hamilton, S. M., Thomas, A. M., Serysheva, E., Yuva-Paylor, L. A., et al. (2011). Modifying behavioral phenotypes in Fmr1KO mice: genetic background differences reveal autistic-like responses. Autism Res. 4, 40–56. doi: 10.1002/aur.168
Splawski, I., Timothy, K. W., Sharpe, L. M., Decher, N., Kumar, P., Bloise, R., et al. (2004). Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119, 19–31. doi: 10.1016/j.cell.2004.09.011
Stefansson, H., Meyer-Lindenberg, A., Steinberg, S., Magnusdottir, B., Morgen, K., Arnarsdottir, S., et al. (2014). CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature 505, 361–366. doi: 10.1038/nature12818
Stoll, G., Pietilainen, O. P., Linder, B., Suvisaari, J., Brosi, C., Hennah, W., et al. (2013). Deletion of TOP3beta, a component of FMRP-containing mRNPs, contributes to neurodevelopmental disorders. Nat. Neurosci. 16, 1228–1237. doi: 10.1038/nn.3484
Talkowski, M. E., Mullegama, S. V., Rosenfeld, J. A., Van Bon, B. W., Shen, Y., Repnikova, E. A., et al. (2011). Assessment of 2q23.1 microdeletion syndrome implicates MBD5 as a single causal locus of intellectual disability, epilepsy, and autism spectrum disorder. Am. J. Hum. Genet. 89, 551–563. doi: 10.1016/j.ajhg.2011.09.011
Talkowski, M. E., Rosenfeld, J. A., Blumenthal, I., Pillalamarri, V., Chiang, C., Heilbut, A., et al. (2012). Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell 149, 525–537. doi: 10.1016/j.cell.2012.03.028
Tirosh, I., Reikhav, S., Sigal, N., Assia, Y., and Barkai, N. (2010). Chromatin regulators as capacitors of interspecies variations in gene expression. Mol. Syst. Biol. 6, 435. doi: 10.1038/msb.2010.84
Tokuriki, N., and Tawfik, D. S. (2009). Chaperonin overexpression promotes genetic variation and enzyme evolution. Nature 459, 668–673. doi: 10.1038/nature08009
Van Bon, B. W., Koolen, D. A., Brueton, L., McMullan, D., Lichtenbelt, K. D., Ades, L. C., et al. (2010). The 2q23.1 microdeletion syndrome: clinical and behavioural phenotype. Eur. J. Hum. Genet. 18, 163–170. doi: 10.1038/ejhg.2009.152
Vissers, L. E., Van Ravenswaaij, C. M., Admiraal, R., Hurst, J. A., De Vries, B. B., Janssen, I. M., et al. (2004). Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat. Genet. 36, 955–957. doi: 10.1038/ng1407
Voineagu, I., Wang, X., Johnston, P., Lowe, J. K., Tian, Y., Horvath, S., et al. (2011). Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474, 380–384. doi: 10.1038/nature10110
Wagenstaller, J., Spranger, S., Lorenz-Depiereux, B., Kazmierczak, B., Nathrath, M., Wahl, D., et al. (2007). Copy-number variations measured by single-nucleotide-polymorphism oligonucleotide arrays in patients with mental retardation. Am. J. Hum. Genet. 81, 768–779. doi: 10.1086/521274
Wang, K., Zhang, H., Ma, D., Bucan, M., Glessner, J. T., Abrahams, B. S., et al. (2009). Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature 459, 528–533. doi: 10.1038/nature07999
Weiss, L. A., Arking, D. E., Daly, M. J., and Chakravarti, A. (2009). A genome-wide linkage and association scan reveals novel loci for autism. Nature 461, 802–808. doi: 10.1038/nature08490
Werling, D. M., and Geschwind, D. H. (2013). Sex differences in autism spectrum disorders. Curr. Opin. Neurol. 26, 146–153. doi: 10.1097/WCO.0b013e32835ee548
Williams, S. R., Aldred, M. A., Der Kaloustian, V. M., Halal, F., Gowans, G., McLeod, D. R., et al. (2010a). Haploinsufficiency of HDAC4 causes brachydactyly mental retardation syndrome, with brachydactyly type E, developmental delays, and behavioral problems. Am. J. Hum. Genet. 87, 219–228. doi: 10.1016/j.ajhg.2010.07.011
Williams, S. R., Mullegama, S. V., Rosenfeld, J. A., Dagli, A. I., Hatchwell, E., Allen, W. P., et al. (2010b). Haploinsufficiency of MBD5 associated with a syndrome involving microcephaly, intellectual disabilities, severe speech impairment, and seizures. Eur. J. Hum. Genet. 18, 436–441. doi: 10.1038/ejhg.2009.199
Wing, L. (1981). Sex ratios in early childhood autism and related conditions. Psychiatry Res. 5, 129–137. doi: 10.1016/0165-1781(81)90043-3
Woolf, C. M. (1997). Does the genotype for schizophrenia often remain unexpressed because of canalization and stochastic events during development? Psychol. Med. 27, 659–668.
Xu, D., Shen, W., Guo, R., Xue, Y., Peng, W., Sima, J., et al. (2013). Top3beta is an RNA topoisomerase that works with fragile X syndrome protein to promote synapse formation. Nat. Neurosci. 16, 1238–1247. doi: 10.1038/nn.3479
Zaidi, S., Choi, M., Wakimoto, H., Ma, L., Jiang, J., Overton, J. D., et al. (2013). De novo mutations in histone-modifying genes in congenital heart disease. Nature 498, 220–223. doi: 10.1038/nature12141
Zhao, X., Leotta, A., Kustanovich, V., Lajonchere, C., Geschwind, D. H., Law, K., et al. (2007). A unified genetic theory for sporadic and inherited autism. Proc. Natl. Acad. Sci. U.S.A. 104, 12831–12836. doi: 10.1073/pnas.0705803104
Keywords: autism spectrum disorder, de novo mutation, chromatin regulators, phenotypic robustness, common genetic variants
Citation: Suliman R, Ben-David E and Shifman S (2014) Chromatin regulators, phenotypic robustness, and autism risk. Front. Genet. 5:81. doi: 10.3389/fgene.2014.00081
Received: 07 December 2013; Accepted: 25 March 2014;
Published online: 10 April 2014.
Edited by:
Michael F. Miles, Virginia Commonwealth University, USAReviewed by:
Bradley Webb, Virginia Commonwealth University, USACopyright © 2014 Suliman, Ben-David and Shifman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sagiv Shifman, Department of Genetics, The Alexander Silberman Institute of Life Sciences, The Hebrew University of Jerusalem, Edmond J. Safra Campus, Jerusalem 91904, Israel e-mail:c2FnaXZAdm1zLmh1amkuYWMuaWw=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.