
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 16 April 2013
Sec. Behavioral and Psychiatric Genetics
Volume 4 - 2013 | https://doi.org/10.3389/fgene.2013.00054
This article is part of the Research Topic Ion Channels and Mental Illness: Exploring etiology and pathophysiology in major psychiatric disorders View all 9 articles
 Mette Gilling1,2 Hanne B. Rasmussen3
Mette Gilling1,2 Hanne B. Rasmussen3 Kirstine Calloe3 Ana F. Sequeira4,5 Marta Baretto4,5 Guiomar Oliveira6 Joana Almeida6 Marlene B. Lauritsen7 Reinhard Ullmann8
Kirstine Calloe3 Ana F. Sequeira4,5 Marta Baretto4,5 Guiomar Oliveira6 Joana Almeida6 Marlene B. Lauritsen7 Reinhard Ullmann8 Susanne E. Boonen9 Karen Brondum-Nielsen9
Susanne E. Boonen9 Karen Brondum-Nielsen9 Vera M. Kalscheuer8
Vera M. Kalscheuer8 Zeynep Tümer1,9 Astrid M. Vicente4,5
Zeynep Tümer1,9 Astrid M. Vicente4,5 Nicole Schmitt3* and Niels Tommerup1
Nicole Schmitt3* and Niels Tommerup1
Heterozygous mutations in the KCNQ3 gene on chromosome 8q24 encoding the voltage-gated potassium channel KV7.3 subunit have previously been associated with rolandic epilepsy and idiopathic generalized epilepsy (IGE) including benign neonatal convulsions. We identified a de novo t(3;8) (q21;q24) translocation truncating KCNQ3 in a boy with childhood autism. In addition, we identified a c.1720C > T [p.P574S] nucleotide change in three unrelated individuals with childhood autism and no history of convulsions. This nucleotide change was previously reported in patients with rolandic epilepsy or IGE and has now been annotated as a very rare SNP (rs74582884) in dbSNP. The p.P574S KV7.3 variant significantly reduced potassium current amplitude in Xenopus laevis oocytes when co-expressed with KV7.5 but not with KV7.2 or KV7.4. The nucleotide change did not affect trafficking of heteromeric mutant KV7.3/2, KV7.3/4, or KV7.3/5 channels in HEK 293 cells or primary rat hippocampal neurons. Our results suggest that dysfunction of the heteromeric KV7.3/5 channel is implicated in the pathogenesis of some forms of autism spectrum disorders, epilepsy, and possibly other psychiatric disorders and therefore, KCNQ3 and KCNQ5 are suggested as candidate genes for these disorders.
Autism spectrum disorders (ASD, OMIM 209850) are neurodevelopmental disorders with early childhood onset and a lifelong persistence. They are characterized by severe impairments in reciprocal social interaction and communication as well as by stereotypic behavior or interests. The prevalence of ASDs is estimated to be 9 in 1000 (Autism and Developmental Disabilities Monitoring Network, 2009; Baron-Cohen et al., 2009) with a male to female ratio of 4:1 (Folstein and Rosen-Sheidley, 2001). Intellectual disability (ID) occurs in approximately 50% of ASD individuals; approximately 20–30% have comorbid epilepsy (State, 2010; Tuchman et al., 2010; Kohane et al., 2012) Furthermore, 72% of children diagnosed with ASD have at least one additional psychiatric disorder such as attention deficit hyperactivity disorder (ADHD), major depression, schizophrenia, phobia, obsessive compulsive disorder (OCD) (Stahlberg et al., 2004; Leyfer et al., 2006; Abdallah et al., 2011). Common genetic etiologies have been identified for ASD, ID, epilepsy and various psychiatric disorders which emphasizes the likely overlap in pathogenesis for these disorders (Moreno-De-Luca et al., 2010; Gregor et al., 2011; Duong et al., 2012). Twin and family studies show that genetic factors are of profound importance for the development of ASD (Folstein and Rutter, 1977; Bailey et al., 1995), but only few genes have been accepted as ASD susceptibility genes. Likely explanations are the apparently high degree of locus heterogeneity rendering it difficult to identify mutations in a gene in a convincing number of patients, the pleiotropic effects of many neuronal disease genes making the connection between genotype and phenotype less obvious (Duong et al., 2012; Iossifov et al., 2012; O’Roak et al., 2012), and the likely substantial contribution of de novo mutations (Sebat, 2007; Levy et al., 2011; Iossifov et al., 2012; Neale et al., 2012). In addition, point mutations (Jamain et al., 2003; Feng et al., 2006; Berkel et al., 2010; O’Roak et al., 2012), rare genomic copy number variants (CNVs), and recurrent CNVs (Ullmann et al., 2007; de Kovel et al., 2010; Moreno-De-Luca et al., 2010) that increase the risk of ASD and/or epilepsy, ID, and psychiatric disorders may be transmitted from apparently normal parents.
Mutations in the genes KCNQ2 and KCNQ3 cause idiopathic generalized epilepsy (IGE) (Neubauer et al., 2008). These include benign neonatal epilepsy (Biervert et al., 1998; Charlier et al., 1998) as well as benign childhood epilepsy with centrotemporal spikes (rolandic epilepsy) (Neubauer et al., 2008) consistent with dysregulation of neuronal excitability. Intriguingly, >20% of patients with rolandic epilepsy have cognitive deficits and >10% have behavioral problems (ADHD, anxiety, depression, and pervasive developmental disorder (PDD) (Tovia et al., 2011). Furthermore, 40% of patients with benign familial neonatal convulsions show delayed psychomotor development or ID (Steinlein et al., 2007) and >25% of patients with IGE have comorbid mental disorders (Akanuma et al., 2008). KCNQ3 is one of five KCNQ genes (KCNQ1-5) encoding the KV7 family of voltage-gated potassium channels (Brown and Passmore, 2009). Four of these genes (KCNQ2-5) are expressed in the central nervous system both on RNA and protein level (Brown and Passmore, 2009) and are therefore excellent candidate susceptibility genes for a wide range of neuronal disorders. KV7.3 forms heterotetrameric channels with KV7.2 (Schroeder et al., 1998), KV7.4 (Kubisch et al., 1999), and KV7.5 (Schroeder et al., 2000). KV7.2/KV7.3 heteromeric channels primarily localize at the axon initial segment (AIS) and underlie the M-current involved in regulation of neuronal excitability (Wang et al., 1998; Schroeder et al., 2000).
In this study we have investigated KCNQ3 gene variability in two independent ASD cohorts from Portugal and Denmark.
Patient A is a Danish boy who carries a de novo balanced translocation t(3;8) (q21;q24). He was born in 1998 as the second child of non-consanguineous, Caucasian, healthy parents aged 37 (mother) and 38 (father) at the time of birth. Both parents have academic degrees. According to the parents the older sister is both intellectually and socially very well-functioning. The pregnancy was normal and the delivery at gestational age 40 + 2 was uncomplicated with Apgar scores 9/1 and 10/5. The birth weight was 3900 g; birth length 53 cm; and head circumference 36 cm. Growth parameters are currently still normal. No dysmorphic features were noted at birth and hearing was normal.
In the neonatal period the parents noticed an abnormal social interaction with him. Later on it was apparent that both verbal and social development was delayed; however motor milestones were achieved normally. Genetic testing for the fragile X syndrome was negative and metabolic screening showed no abnormalities. At 2 years of age he was diagnosed with childhood autism (Autism Diagnostic Observation Schedule type G (ADOS-G); Communication score: 5, Social score: 14). At 8 years of age a WISC-III test showed a very uneven profile with specific non-verbal visio-spatial difficulties (verbal IQ = 103, performance IQ = 60, global IQ = 79). Currently, he attends a class for children with special needs in a normal primary school. Verbally he is highly skilled in both Danish and English.
Periodic idiopathic trembling was noted from the age of 2 days and according to the mother it persisted for the first 5 weeks. This description is in accordance with a diagnosis of benign neonatal convulsions but this was never diagnosed. Currently, he has no epilepsy diagnosis; however, according to the parents he has brief episodes of non-responsiveness resembling absence seizures. Consequently, electroencephalographic (EEG) examination was carried out at ages 5 and 9 years during sleep, hyperventilation, photo-stimulation, and during periods of non-responsiveness but no abnormalities were observed. Cerebral magnetic resonance (MR) scanning of the brain at age 8 years was normal.
The National Ethics Committees and the Danish Data Protection Agency approved the study, and informed consent was obtained from the parents.
Mutation screening of KCNQ3 was performed in two steps. As a first step DNA from a cohort comprising 100 Portuguese and 48 Danish ASD patients were screened for KCNQ3 mutations by direct sequencing. The Portuguese ASD patients were recruited at the Hospital Pediátrico de Coimbra and all originated from mainland Portugal and the Azorean islands. The male-female ratio was 4.8:1, and the ages ranged between 2 and 18 years (mean age 6.8 years). Idiopathic subjects were included after clinical assessment and screening for known medical and genetic conditions associated with autism, including testing for Fragile X mutations (FRAXA and FRAXE), chromosomal abnormalities, neurocutaneous syndromes, endocrine (thyroid function screening), and metabolic disorders. About 35 of the 48 Danish ASD patients were recruited at child psychiatric hospitals in the western part of Denmark (Jutland) (age range 3–30 years, with mean age of 10 years and male-female ratio of 3:1). Seven autistic patients were ascertained at the Kennedy Center (Glostrup, Denmark) (age range 13–37 years, mean age 20.4 and male-female ratio of 2.5:1). These patients were unrelated and part of the IMGSAC group and accordingly some of the patients had siblings and some even additional relatives with a diagnosis of pervasive developmental disorder. These patients were screened for chromosomal abnormalities and fragile X syndrome and a physical examination included a careful search for phakamatoses to rule out Tuberous Sclerosis (TSC). Four patients diagnosed within the autism spectrum were collected at the Psychiatric Hospital in Hillerød (Frederiksborg Amt, Denmark). In addition, two DNA samples (one male, one female) from individuals diagnosed within the ASD spectrum and with chromosomal rearrangements were included in the screening. These samples were collected at the Wilhelm Johannsen Centre for Functional Genome Research, University of Copenhagen (Denmark). In all of the above ASD patients diagnosis was made in accordance with DSM-IV or ICD-10 criteria using ADI-R in addition to ADOS or the Childhood Autism Rating Scale.
The c.1720C > T variant in KCNQ3 was first identified in one Portuguese ASD patient (patient B, Table 1) by direct sequencing. As a next step 271 additional Portuguese ASD patients fulfilling the same criteria as the first cohort were specifically screened for the c.1720C > T variant by a PCR/enzyme cleavage assay whereby two additional patients (patient C and D) were identified as carriers of the c.1720C > T variant. Hence, a total of 419 ASD patients (371 Portuguese- and 48 Danish ASD patients) were screened for the c.1720C > T variant. The three male Portuguese patients (Patient B, C, D in Table 1) were diagnosed with childhood autism using the Autism Diagnostic Interview-Revised (ADI-R) and ADOS (Lord et al., 1994) and had no history of convulsions. The inheritance pattern of the c.1720C > T variant was ascertained both by direct sequencing of exon 13 of KCNQ3 and by the PCR/enzyme cleavage assay in patient B, C, and D and their parents. As controls 96 Caucasian individuals from the Human Random Control DNA panel (HRC-1, Sigma-Aldrich, St. Louis, USA), and 100 Portuguese individuals without neuropsychiatric disease (self reported) from blood donor centers throughout Portugal were included.
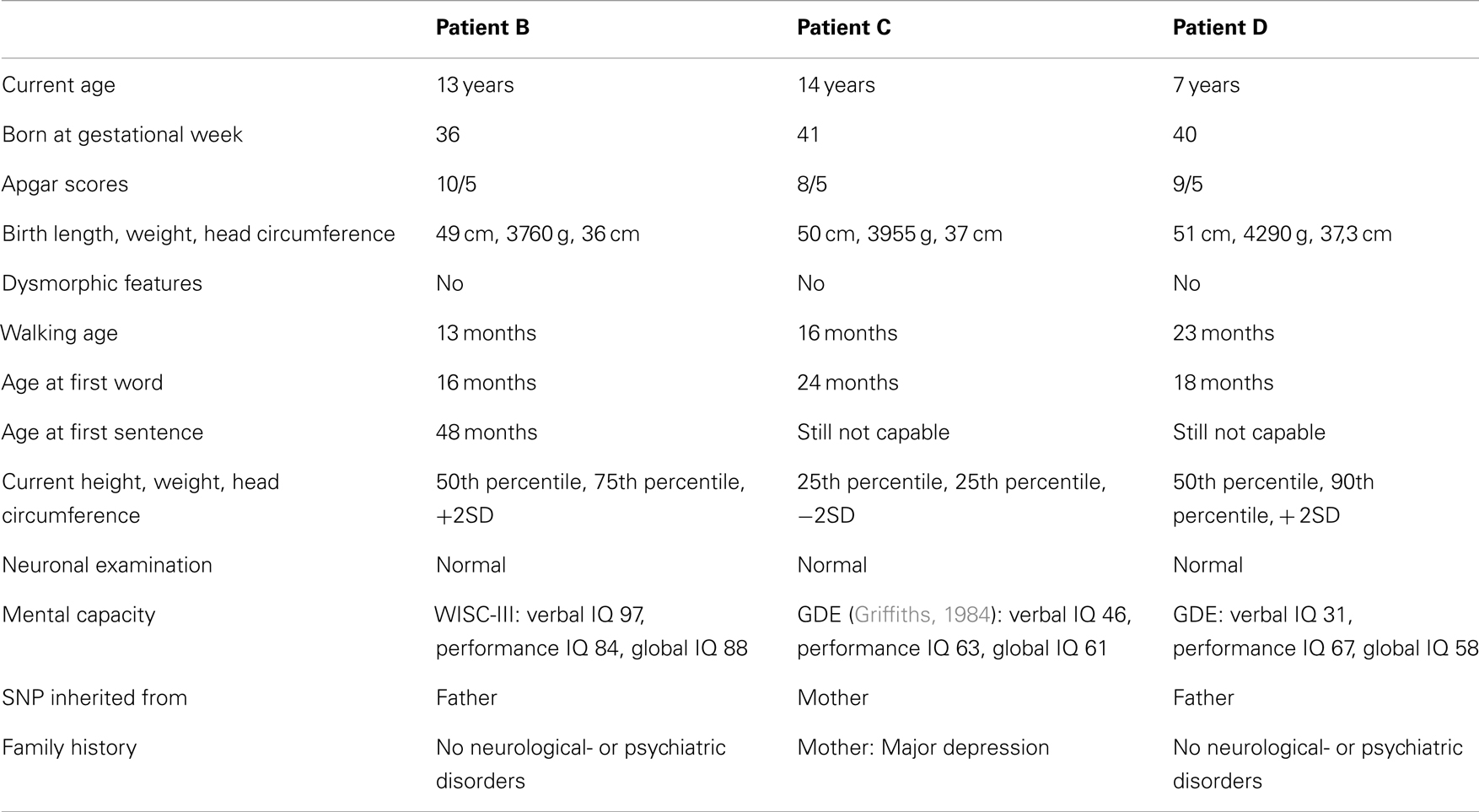
Table 1. Clinical description of three Portuguese individuals carrying a c.1720C > T variant and diagnosed with childhood autism.
Cytogenetic analysis and fluorescence in situ hybridization (FISH) was performed according to standard protocols, and array-based comparative genome hybridization was performed as previously described (Erdogan et al., 2006).
When necessary, genomic DNA was uniformly amplified using GenomiPhi™ DNA Amplification Kit (GE Healthcare, Buckinghamshire, UK).
All 15 coding exons and intron-exon boundaries of KCNQ3 (NM_004519.3) were amplified by PCR. Sequencing reactions were carried out using BigDye® Terminator v 1.1 Cycle Sequencing Kit (Life Technologies, California, USA) and analyzed by an ABI 3100 AVANT Genetic Analyzer (Life Technologies, California, USA). ChromasPro version 1.33 (Technelysium Pty Ltd, Australia) was used to visualize the data. Nucleotide changes were verified by a second PCR amplification of non-genome amplified patient DNA, sequencing and restriction cleavage.
A PCR product of 461 base pairs (bp) encompassing exon 13 of KCNQ3 was amplified using primers KCNQ3_13a: TATTCCAAACCCTTATCTCAT and KCNQ3_13b: AAACAGGTGGGG CTATTA. PCR fragments amplified from the WT allele were digested into two fragments with lengths 438 and 23 bp by the restriction enzyme Hpy188III, whereas PCR fragments amplified from the c.1720C > T allele were digested into three fragments with lengths 337, 101, and 23 bp.
The plasmids hKV7.2-hKV7.5 in pXOOM or pXOON, hKV7.3-flag in pNS2z, and hKV7.2-cmyc in pNS2z used in this study have been described previously (Bentzen et al., 2006; Rasmussen et al., 2007). KV7.4 cDNA was amplified with PCR and inserted into pNS2z to generate C-terminally myc-tagged KV7.4. To generate the extracellularly tagged expression plasmid hKV7.5-3xHA in pXOOM, 3 HA-tags were inserted into the TM3-TM4 linker of hKV7.5 by PCR using the primers 5′-CCAGATTACGCGTACCC TTACGACGTTCCAGATTACGCTGGTAATATTTTTGCCAC-3′ and 5′-GACATCGTAT GGGTAAGCGTAGTCTGGGACGTCGTATGGGTACTGAGTTTTTGCAGAAAC-3′. Human CD4-WT in pcDNA3.1 was a kind gift from James Trimmer (University of California Davis, CA, USA) and has been described earlier (Gu et al., 2003). The chimera hCD4-hKV7.3CT in pcDNA3.1 was generated using standard PCR and in-frame insertion of cDNA corresponding to KV7.3 amino acids 358–873 into NotI and XhoI sites of the wild-type (WT) construct. The point mutation c.1720C > T leading to the amino acid exchange P574S was introduced using mutated oligonucleotide extension (PfuTurbo Polymerase, Stratagene, La Jolla, CA, USA) from the plasmid template harboring the cDNA of interest, digested with DpnI (Fermentas, St. Leon, Germany) and transformed into E. coli XL1 Blue cells. All plasmids were verified by complete DNA sequencing of the cDNA insert (Macrogen Inc., Seoul, Rep. of Korea). The Gene Bank Accession numbers of the human cDNAs are: NM_004519 (KV7.3), NM_004518 (KV7.3, isoform c), NM_004700 (KV7.4), and NM_019842 (KV7.5). Protein accession number for KV7.3 is NP_004510.
The cRNA were prepared from linearized hKV7.2, hKV7.3 WT and mutant, KV7.4, and KV7.5 constructs in pXOOM or pXOON using the Ambion T7 m-Message Machine kit according to the manufacturer’s instructions (Ambion, Austin, TX, USA). RNA concentrations were determined by UV spectroscopy, integrity was confirmed by gel electrophoresis. cRNAs were stored at −80°C until injection.
Female Xenopus laevis frogs were anesthetized with Tricain (2 g/l, Sigma, Brøndby, Denmark) and ovarian lobes were removed. Oocytes were defolliculated enzymatically in 1% collagenase (Boehringer Mannheim/Roche, Hvidovre, Denmark) and 0.1% trypsin inhibitor (Sigma) in Kulori’s solution for 1 h followed by wash in Kulori’s solution (in mM: 90 NaCl, 4 KCl, 1 MgCl2, 1 CaCl2, 5 Hepes, pH 7.4) containing 0.1% BSA (Sigma). Oocytes were injected using a Nanoject microinjector (Drummond Scientific, Broomall, PA, USA) with 1 ng hKV7.2, 7.4, or 7.5 mixed with hKV7.3 WT or hKV7.3_P574S cRNA (in a 1:1 molar ratio) diluted in 50 nl diethylpyrocarbonate treated water. Oocytes were kept in Kulori’s solution at 19°C.
Currents were recorded at room temperature by two-electrode voltage-clamp (TEVC) 2 days after injection using a Dagan 2B amplifier (Clampator 1, Dagan, Chicago, IL, USA). The oocytes were perfused with Kulori’s solution and pipettes were pulled from borosilicate glass and had a final tip resistance of 0.5–2.5 MΩ when filled with 2 M KCl. Data were acquired using Pulse software (HEKA electronics, Germany) and analyzed with Igor (WaveMetrics, Lake Oswego, OR, USA) and GraphPad Prism (GraphPad Software, San Diego, CA, USA). All experiments were performed in 3–4 different batches of oocytes.
Data are presented as mean ± standard error of the mean. For statistical analyses ANOVA combined with Student-Newman-Keuls post test was used and p < 0.05 was considered significant (*).
HEK 293 cells were grown in DMEM (Invitrogen, Glostrup, Denmark) supplemented with 100 U/ml penicillin, 100 mg/ml streptomycin and 10% FCS (Sigma-Aldrich, Copenhagen, Denmark) at 37°C in a humidified atmosphere with 5% CO2. Transfections were carried out using the Lipofectamine-Plus Reagent system (Invitrogen) according to the manufacturer’s instructions. Hippocampal cultures were prepared as previously described (Rasmussen et al., 2007) and transfected at 7–8 days in vitro (DIV) using the lipofectamine 2000 method (Invitrogen) with a total of 0.9 μg of DNA per cover-slip. Transfection was carried out for 1 h at 37°C, 5% CO2 after which the neurons were transferred back to the dishes containing the glial cell layer. Neurons were left for expression for 48 h.
HEK 293 cells or primary hippocampal neurons were fixed in 3% paraformaldehyde in PBS for 15–20 min at room temperature. Blocking and permeabilization was performed by a 30 min incubation with 0.2% fish skin gelatin in phosphate buffered saline supplemented with 0.1% Triton X-100 (PBST). The cells were then incubated for 1 h in primary antibodies diluted in PBST. Primary antibodies employed were: rabbit anti-c-myc (A-14, 1:50 dilution, Santa Cruz Biotechnology, Heidelberg, Germany), mouse anti-FLAG (M2, 1:250 dilution, Sigma-Aldrich, Copenhagen, Denmark), and rat anti-HA (3F10, 1:50 dilution, Santa Cruz Biotechnology). For immunofluorescent detection, Alexa Fluor®–coupled secondary antibodies were diluted in PBST and applied for 45 min. The cover-slips were mounted in Prolong Gold (Invitrogen, Glostrup, Denmark).
Laser scanning confocal microscopy was performed using the Leica TCS SP2 system equipped with argon and helium-neon lasers. Images were acquired using a 63× water immersion objective, NA 1.2 with a pinhole size of 0.8-1 and a pixel format of 1024 × 1024. Line averaging was used to reduce noise. For double- and triple-labeling experiments sequential scanning was employed to allow the separation of signals from the individual channels. Acquired images were treated using Adobe Photoshop CS4 and Adobe Illustrator CS4.
By cytogenetic analysis a de novo t(3;8) (q21;q24) translocation was identified in Patient A. The breakpoints of this translocation were mapped using FISH. The breakpoint on chromosome 3 was delineated to a 26 kb region (chr3:131.189.991-131.216.096; NCBI36/hg18) at 3q21.3 located 10.5 kb downstream of the thyrotropin-releasing hormone (THR) gene (Figure 1). No annotated human genes or mRNAs are located within this breakpoint region. On chromosome 8q24.22 the breakpoint was localized in a 25 kb region (chr8:133.318.034-133.343.450; NCBI36/hg18) within exon 1 of the KCNQ3 gene (Figure 1). No disease-related copy number variations were identified by array comparative genomic hybridization (CGH) in this patient.
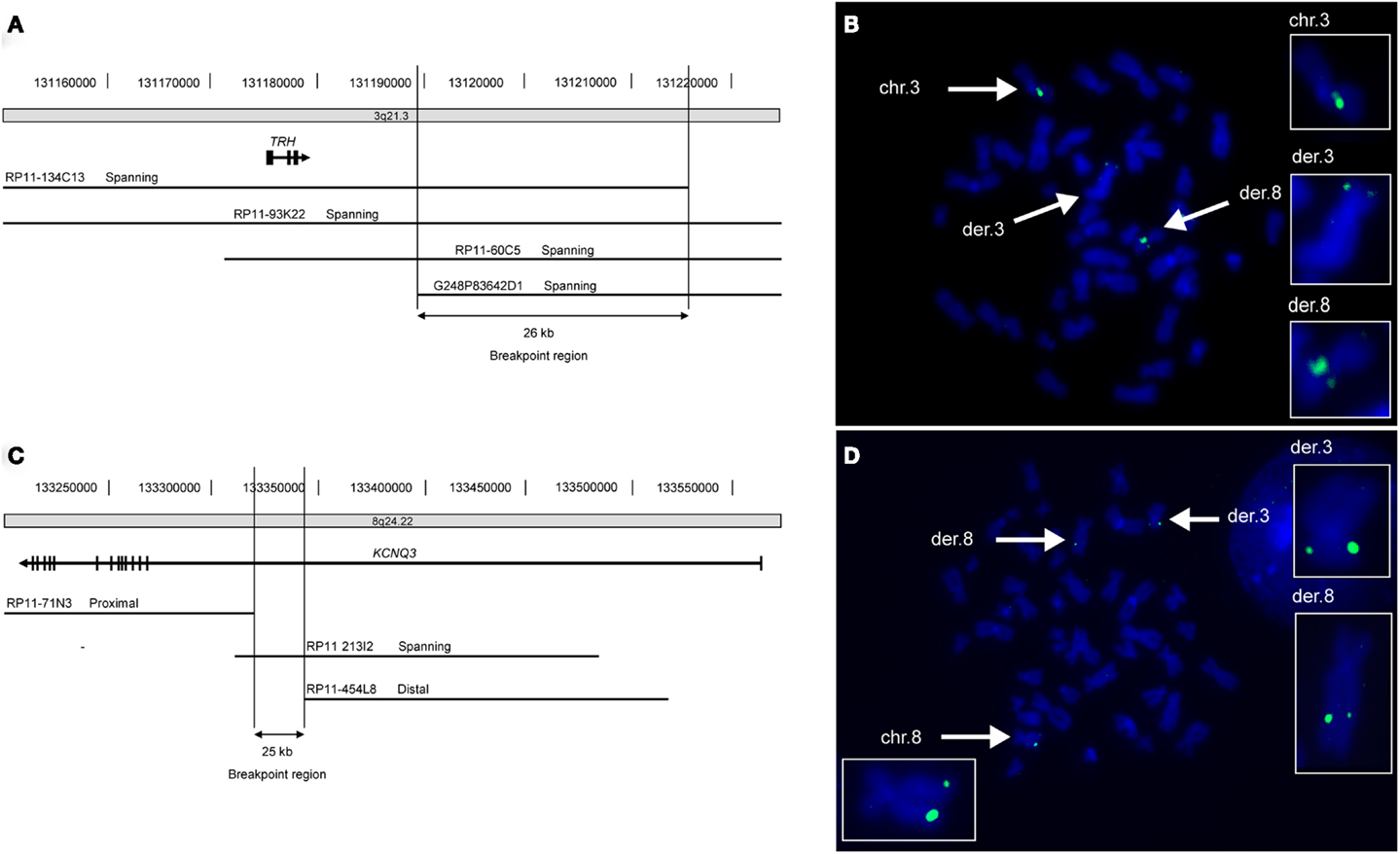
Figure 1. FISH mapping of translocation breakpoints reveals truncated KCNQ3 gene in patient A. (A) Schematic depiction of the 26 kb breakpoint region within chromosomal region 3q21.3 of patient A. The breakpoint region is located 10.5 kb downstream of the TRH gene and contains no genes. (B) Picture showing metaphase chromosomes from patient A (blue) hybridized with the chromosome 3 specific probe RP11-93K22 (green) that spans the breakpoint. The normal chromosome 3 as well as both derivative chromosomes are marked with white arrows and enlarged in the boxes to the right. (C) The 25 kb breakpoint region on chromosome 8 lies within intron 1 of the KCNQ3 gene. (D) The chromosome 8 specific probe RP11-213I2 (green) spans the breakpoint. The normal chromosome 8 as well as the two derivative chromosomes are marked with white arrows and enlarged.
A paternally inherited c.1720C > T missense mutation in exon 13 of KCNQ3 was identified in patient B by direct sequencing (Figure 2A). There is no history of psychiatric- or neurological disorders in this family. The mutation results in an amino acid change at position 574 replacing proline by serine (p.P574S). By restriction enzyme assay the c.1720C > T (p.P574S) variant in KCNQ3 was identified in two additional ASD patients (patient C and D) (Figure 2B) and was confirmed in patient B. Patient C inherited the variant from the mother who suffers from major depression and patient D inherited the variant from the father who does not suffer from any psychiatric- or neurological disorders (Figure 2B). The c.1720C > T mutation in patients C and D was confirmed by direct sequencing of a second PCR product from non-amplified DNA. No c.1720C > T mutations were identified in 96 UK Caucasian and 100 Portuguese controls.
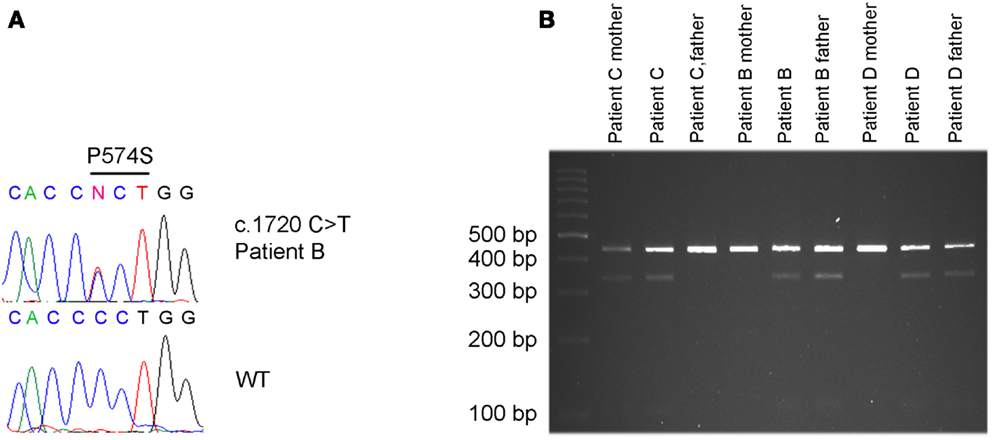
Figure 2. c.1720C > T [p.P574S] variant detected in patient B, C and D. (A) The c.1720 C > T mutation in patient B was identified by Sanger sequencing. (B) The same mutation was identified in patient C and his mother, in patient D and his father and confirmed in patient B and his father by restriction enzyme cleavage of a PCR product encompassing the mutation.
To address effects of the mutation P574S on ion channel function, we heterologously expressed mutant channels in Xenopus laevis oocytes. Since KV7.3 does not form functional channels on its own (Schwake et al., 2000), we investigated whether the KV7.3_P574S mutation could affect the function of other neuronal members of the KV7 family. KV7.3_P574S or KV7.3 WT was co-expressed with KV7.2, KV7.4, or KV7.5 in Xenopus laevis oocytes and currents were recorded by TEVC. In agreement with Neubauer et al. (2008), we found that current levels for KV7.2/KV7.3_P574S were similar to those of KV7.2/KV7.3 (Figure 3A). Similarly, the function of KV7.4 channels did not appear to be affected by the mutation, as oocytes expressing KV7.3_P574S/KV7.4 had similar current levels as KV7.3/KV7.4 (Figure 3B). In a final set of experiments, we tested the effect of KV7.3_P574S on KV7.5 currents. In line with previous work by Lerche et al. (2000), co-expression of KV7.5 with KV7.3 dramatically increased current levels compared to KV7.5 alone (Figure 4). Expression of KV7.3_P574S also enhanced KV7.5 current levels but to a significantly lesser extent than WT KV7.3. These results suggest that KV7.3_P574S has not lost its ability to interact with KV7.5. Since both patients B and C were heterozygous for the KV7.3_P574S mutation, we mimicked the heterozygous state by co-expressing KV7.5 with KV7.3 and KV7.3_P574S in a 2:1:1 ratio. The resulting current levels were intermediate of that of KV7.3/KV7.5 and KV7.3_P574S/KV7.5, indicating that (1) KV7.3_P574S is not dominant-negative, and (2) co-expression of WT does not rescue the KV7.3_P574S phenotype. The difference in current levels between the heterozygote and WT KV7.3/KV7.5 is statistically significant as indicated by the asterisk in Figure 4B. Meticulous inspection of the curves in Figure 4B reveals that for KV7.5 expressed alone, there is a tendency for the current-voltage relationship to flatten out at higher voltages and this tendency appears to be removed by co-expression with KV7.3, making the current-voltage curve more linear.
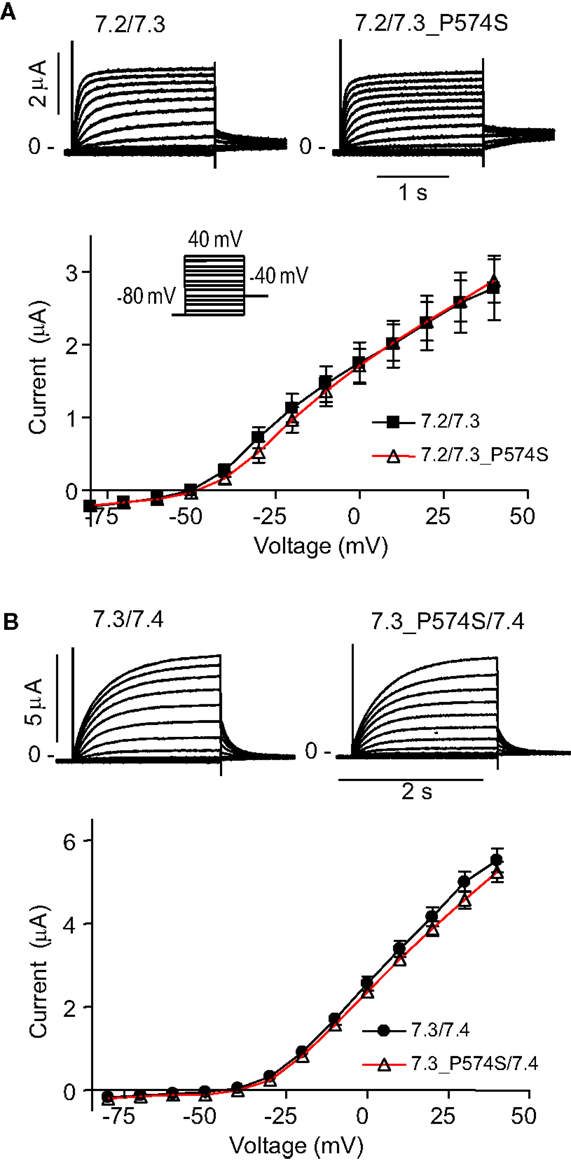
Figure 3. Effect of KV7.3_P574S on KV7.2 and KV7.4 in X. laevis oocytes. Currents were activated by voltage-steps from −80 mV to +40 mV in 10 mV increments. Representative currents are shown as well as steady-state current plotted as a function of voltage. (A) KV7.2 was co-expressed with either KV7.3 WT (n = 6) or P574S (n = 5). (B) Effect of KV7.3_P574S on KV7.4. KV7.4 was co-expressed with either KV7.3 WT (n = 10) or P574S (n = 13) in X. laevis oocytes.
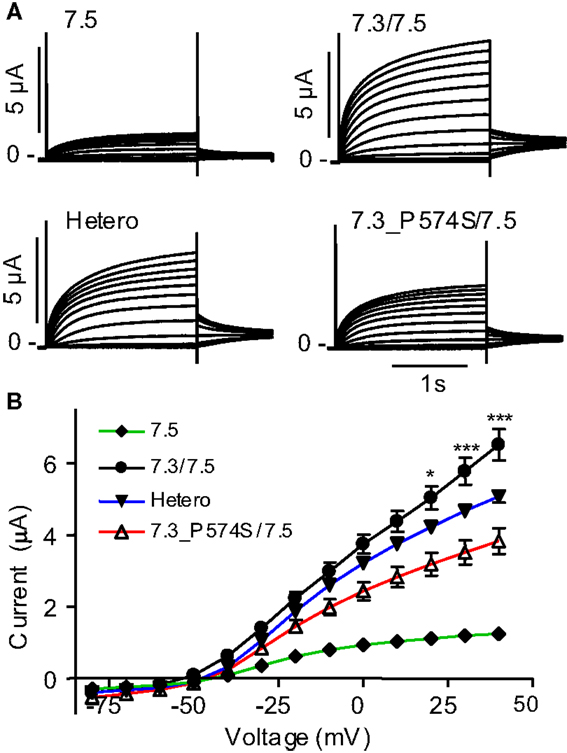
Figure 4. Effect of KV7.3_P574S on KV7.5. KV7.5 was expressed alone (n = 8) or co-expressed with either KV7.3 WT (n = 7), P547S (n = 6), or KV7.5 mixed with KV7.3 WT or KV7.3_P574S (in a 1:1 molar ratio, Hetero, n = 6) in X. laevis oocytes. Currents were activated by voltage-steps from −80 to +40 mV in 10 mV increments. (A) Representative currents are shown. (B) Steady-state current plotted as a function of voltage. Asterisks indicate statistical difference between Hetero and KV7.3/KV7.5. Comparison of the other points were left out for clarity and for voltages higher than 0 mV, all points were statistically different.
Since the P574S mutation reportedly is without effect upon the current characteristics of the KV7.2/KV7.3 complex (Miceli et al., 2009), we decided to investigate whether the P574S mutation could affect the localization of the heteromeric KV7.2/KV7.3 complex. We first analyzed the localization of KV7.2 and KV7.3 upon co-expression in HEK 293 cells. As illustrated in Figure 5, both channel subunits displayed a primarily intracellular staining pattern. The subunits appeared to co-localize to a large degree in the intracellular structures and only weak staining could be detected in association with the cell surface. Importantly, co-expression of KV7.2 and KV7.3_P574S resulted in a staining pattern that was indistinguishable from the co-expression of the WT channels. Thus, KV7.3_P574S does not appear to have an impact on the localization of the KV7.2/KV7.3 heteromeric complex in HEK 293 cells.
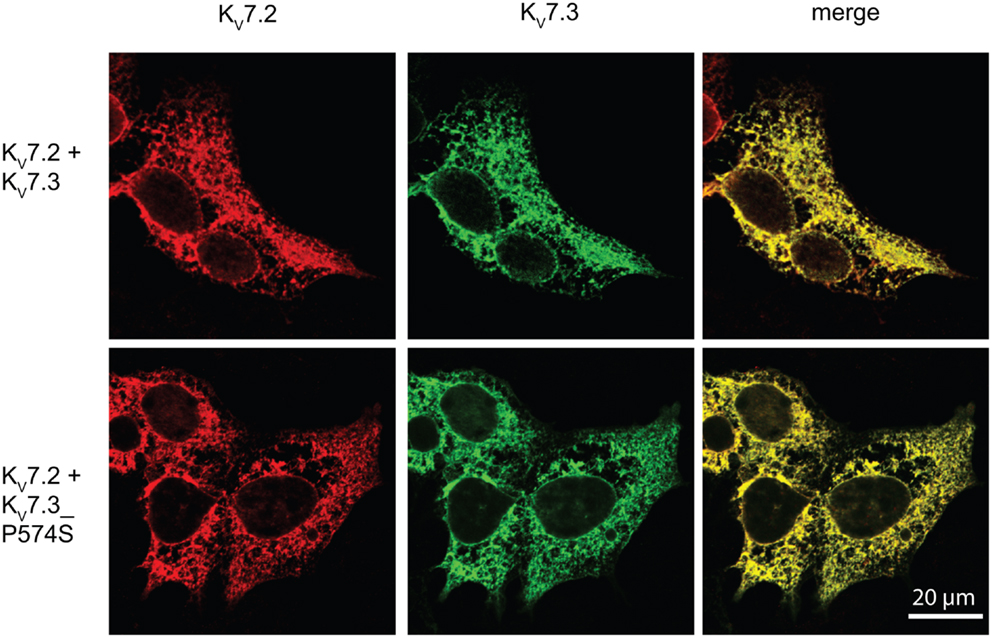
Figure 5. No effect of KV7.3_P574S on localization of the KV7.2/KV7.3 complex in HEK 293 cells. KV7.2 was transiently expressed in HEK 293 cells together with KV7.3 WT or KV7.3_P574S, and the localization of the complex was analyzed by immunocytochemistry and confocal microscopy. As illustrated, the P574S mutation is without effect on the localization of the complex that displays a primarily intracellular localization pattern. Scale bar 20 μm.
In neurons, the KV7.2/KV7.3 complex is localized to the AIS (Devaux et al., 2004; Chung et al., 2006; Rasmussen et al., 2007). We therefore speculated that the specific localization of the complex to the AIS could be disturbed by the P574S mutation. To address this question, we first examined the localization of KV7.3 and KV7.3_P574S upon exogenous expression in cultured rat hippocampal neurons. As previously reported, singly expressed KV7.3 was primarily observed intracellularly with no significant enrichment in the AIS (Rasmussen et al., 2007). Likewise, KV7.3_P574S demonstrated a primarily intracellular staining pattern similar to the WT subunit.
Upon co-expression of KV7.2 and KV7.3, the channel complex appears in the AIS (Rasmussen et al., 2007). To investigate whether the KV7.3_P574S mutation perturbed the localization of the complex to the AIS, we transiently expressed KV7.2 with either WT KV7.3 or KV7.3_P574S in cultured hippocampal neurons. As illustrated in Figure 6B, the P574S mutation did not impair the localization of the KV7.2/KV7.3_P574S complex as it localized to the AIS similar to the WT complex. These results were further emphasized by experiments using chimeric constructs of the transmembrane protein CD4 and KV7.3/KV7.3_P574S. We have previously demonstrated that the ability of KV7.3 to direct the KV7.2/KV7.3 complex to the AIS critically depends on an ankyrin-G binding sequence in the C-terminal tail of KV7.3 (Rasmussen et al., 2007). We therefore attached the C-terminal tail of KV7.3 to a truncated version of the CD4 receptor to examine whether this part of KV7.3 would be sufficient to redirect the otherwise non-polarized protein CD4 to the AIS (Figure 7, left panel). As expected, the truncated version of CD4 displayed a non-polarized localization pattern upon expression in cultured hippocampal neurons (Figure 7, top panel). Attachment of the KV7.3 C-terminus was sufficient to drive an AIS localization of the chimera (Figure 7, middle panel). As illustrated, introduction of the P574S mutation into the chimera was without effect as the mutated chimera was still able to target efficiently to the AIS (Figure 7, lower panels).
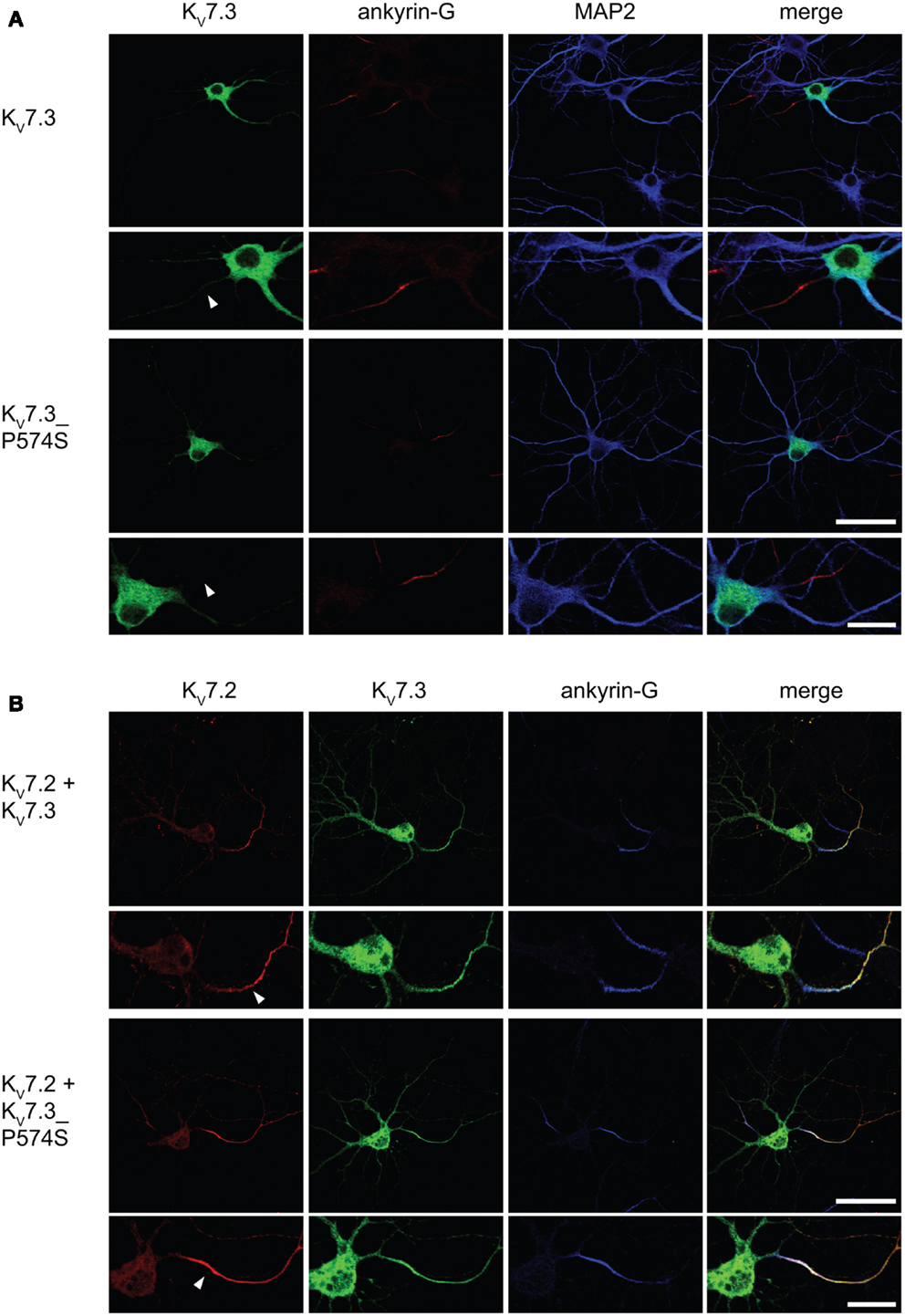
Figure 6. KV7.2/KV7.3_P574 complexes still target to the AIS of cultured hippocampal neurons. Confocal images of cultured rat hippocampal neurons (10 DIV) expressing KV7.3 WT or KV7.3_P574S (A) and co-transfected with KV7.2 (B). (A) KV7.3 is primarily intracellularly expressed when expressed on its own. No significant KV7.3 expression is observed at the AIS. (B) When co-expressing KV7.3 with KV7.2, both channel subunits are observed at the AIS. KV7.3_P574S displays the same localization characteristics. White arrowhead points to the location of the AIS. Ankyrin-G: marker of the axon initial segment, MAP2: marker of the somatodendritic region of neurons. Scale bars 50 and 20 μm, respectively.
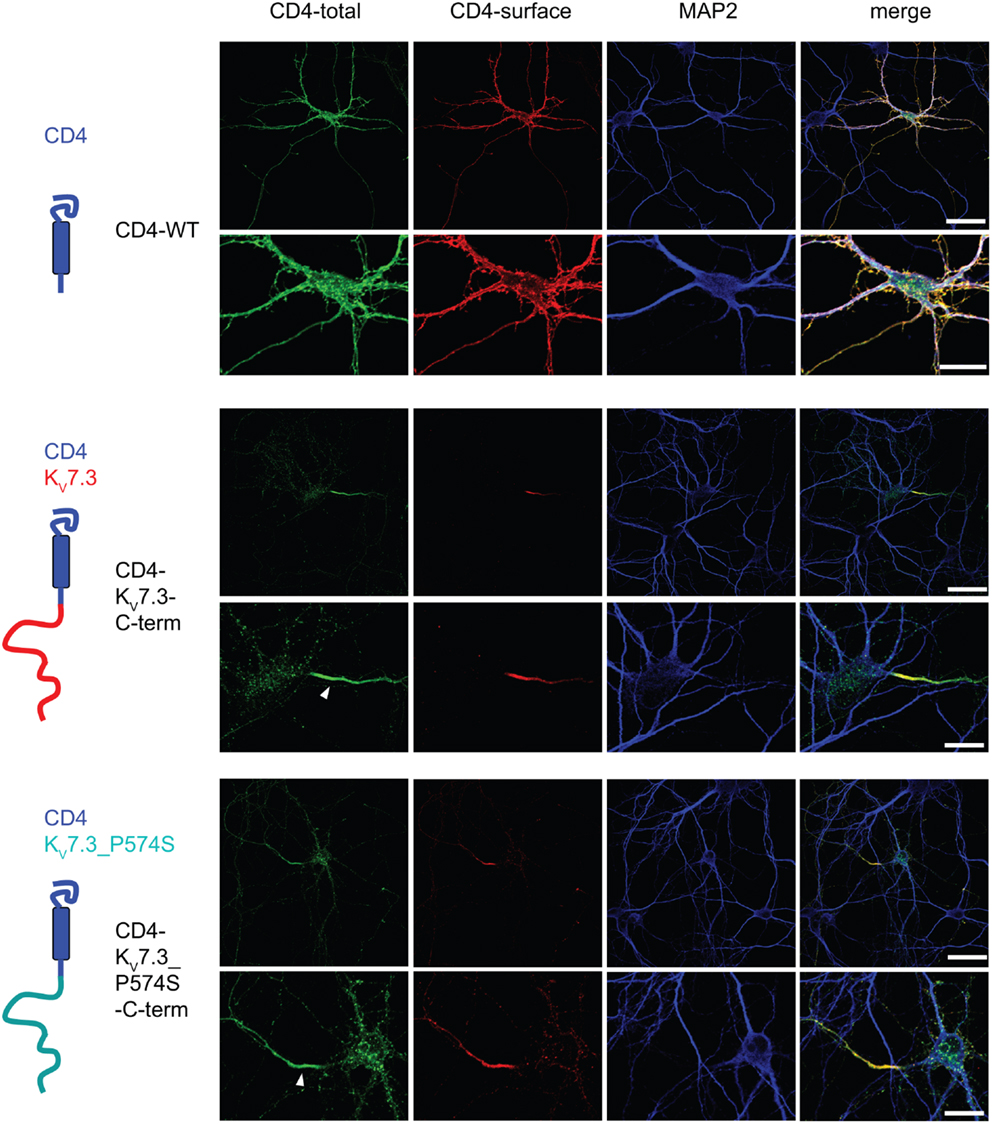
Figure 7. CD4-KV7.3Cterm_P574 can target to the AIS. Confocal images of cultured rat hippocampal neurons (10 DIV) transfected with CD4 (two upper panels), CD4-KV7.3 C-term (two middle panels) and CD4-KV7.3_P574S C-term (two lower panels). The panels to the left illustrate the structure of the chimeric constructs analyzed. CD4-total reflects total CD4 staining in permeabilized cells. CD4-surface is a surface staining of the same cells where the CD4 antibody was applied before permeabilization. The somatodendritic marker MAP2 was included to indicate the location of the AIS (neurite which is MAP2 negative). As expected, CD4 distributes in a non-polarized manner on the surface of axon, soma and dendrites. When the C-terminal of KV7.3 is attached to CD4, the reporter redistributes to the axon initial segment, which illustrates that the KV7.3 C-terminal contains information for AIS localization. The C-terminus of KV7.3_P574S still has the ability to direct CD4 to the AIS. White arrowhead points to the location of the AIS. Scale bars 50 and 20 μm, respectively.
Since the P574S mutation did not affect the localization of the classical KV7.2/KV7.3 complex, we investigated the impact of the mutation on the localization of heteromeric channels including the KV7.4 or KV7.5 subunits. We transiently co-expressed WT KV7.3 or the mutant P574S with either the KV7.4 or the KV7.5 subunit in HEK 293 cells and analyzed the localization of the subunits by confocal microscopy. As illustrated in Figure 8, KV7.3/KV7.4 and KV7.3/KV7.5 complexes demonstrated a mixed surface and intracellular staining pattern demonstrating that both KV7.4 and KV7.5 subunits can pull a fraction of KV7.3 subunits to the cell surface. However, the same localization pattern was observed when analyzing the mutant complexes. Thus, the P574S mutation was without significant effect on the localization of KV7.4 and KV7.5 containing complexes.
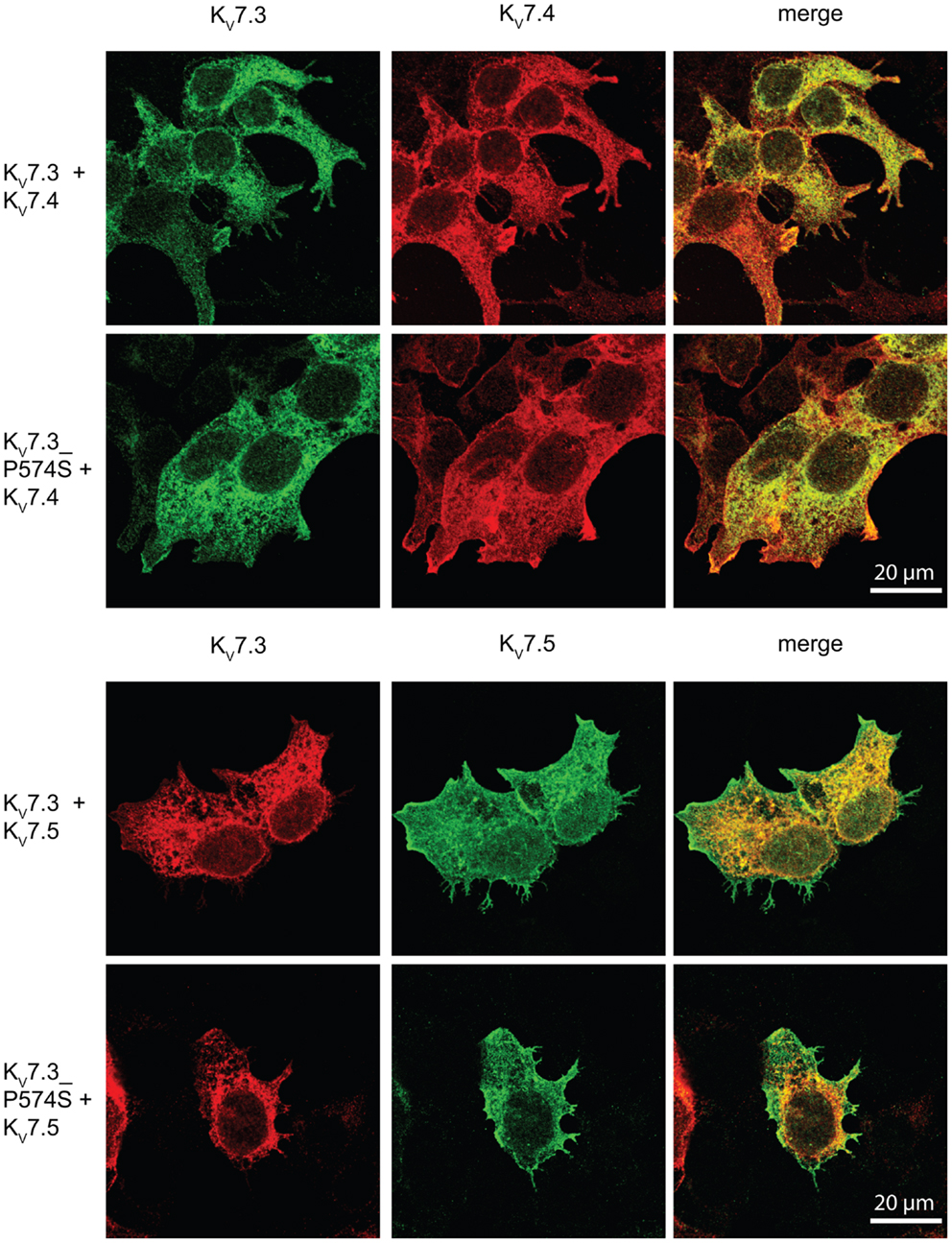
Figure 8. No effect of KV7.3_P574S on the localization of KV7.4 and KV7.5 containing complexes in HEK 293 cells. KV7.4 and KV7.5 were transiently expressed in HEK 293 cells together with KV7.3 or KV7.3_P574S and the localization of the expressed subunits analyzed by immunocytochemistry and confocal microscopy. As illustrated, the P574S mutation is without effect on the localization pattern of the complexes that displays a mixed surface and intracellular staining pattern. Scale bar 20 μm.
Mutations in KCNQ3 (and KCNQ2) have been previously described in patients with rolandic epilepsy and IGE (Neubauer et al., 2008) including benign neonatal convulsions. A considerable proportion of patients with these types of epilepsies also have ID and/or behavioral problems (ADHD, ASD, anxiety, depression) (Borgatti et al., 2004; Steinlein et al., 2007; Akanuma et al., 2008; Tovia et al., 2011) which supports a common genetic etiology and accordingly suggest KCNQ3 (and KCNQ2) as candidate susceptibility genes for ID and various psychiatric disorders. This is substantiated by a KCNQ2 knock-out mouse model that shows spontaneous seizures and behavioral hyperactivity (Peters et al., 2005); by finding of two patients with psychomotor retardation and convulsions with a 8,35 Mb deletion encompassing KCNQ3 (Verheij et al., 2009); and by the association of markers close to KCNQ3 with bipolar disorder (Avramopoulos et al., 2004; Zandi et al., 2008; Zhang et al., 2010).
In line with this hypothesis we here demonstrate different KCNQ3 alterations (truncating mutation, rare SNP with abnormal electrophysiological profile) in four patients with childhood autism and in one transmitting parent with major depression.
The c.1720C > T [p.P574S] nucleotide change was identified in three unrelated Portuguese patients with childhood autism. In two cases (patients B and D) the variant was inherited from an apparently normal parent and in the third case (patient C) transmitted from a mother with major depression. This nucleotide change is now annotated as a rare SNP in dbSNP (rs74582884, Minor Allele Frequency A = 0,012) and was previously reported in 2 of 62 patients with rolandic epilepsy and in 8 of 455 patients with IGE but not in 454 healthy controls (Neubauer et al., 2008). Both patients with rolandic epilepsy inherited the mutation from a healthy parent. This raises the possibility that the rs74582884 SNP conveys liability for general psychopathology but at the same time suggests that additional genetic and/or environmental factors may have an impact on the phenotypical outcome of carriers. Indeed, the same SNP was reported in a patient with benign familial neonatal seizures who, in addition, carried a de novo mutation in KCNQ2 that changed channel gating. Since the SNP in KCNQ3 was inherited from a father and a paternal grandmother without neurological abnormalities the authors suggested that the SNP was not responsible for the observed epilepsy (Miceli et al., 2009). However, our data suggest the c.1720C > T nucleotide change as a contributing factor. Combining our data with all published sequencing studies of KCNQ3 shows that rs74582884 is a rare variant as it is absent in a total of 700 controls (Neubauer et al., 2008; Miceli et al., 2009).
The proline P574 is located in the linker region between two subunit interaction domains in the C-terminal region of KV7.3 (Figure 9A). This part of the protein is involved in subunit assembly, maturation, and transport of channels (Schwake et al., 2006). The amino acid residue P574 is evolutionally conserved (Figure 9B), but it is not conserved among the KV7 family members of voltage-gated potassium channels (Figure 9C). This indicates that P574 is important for KV7.3 function and this function is probably not shared by the other KV7 members. Since KV7.3 is the most promiscuous of the KV7 proteins forming heterotetrameric channels with KV7.2 (Schroeder et al., 1998), KV7.4 (Kubisch et al., 1999), and KV7.5 (Schroeder et al., 2000) we investigated the effect of the P574S change on the localization of and current through heteromeric KV7.2/KV7.3_P574S, KV7.4/KV7.3_P574S, and KV7.5/KV7.3_P574S channels.
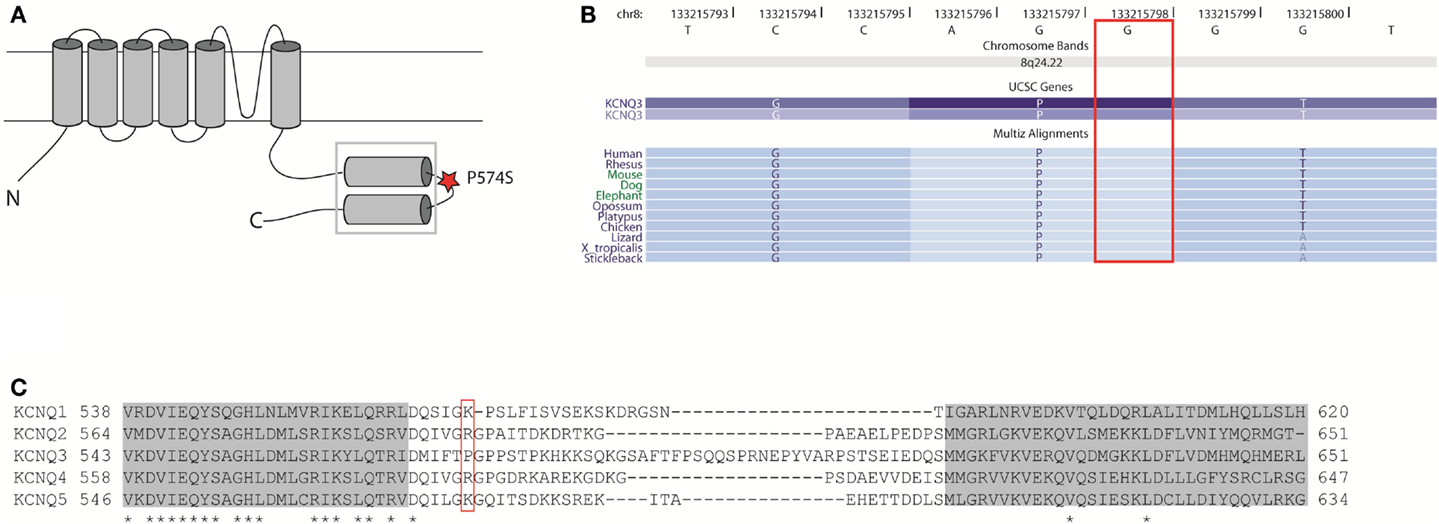
Figure 9. Localization and conservation of the SNP c.1720C > T [p.P574S] in KCNQ3. (A) The KV7 subunits consist of six transmembrane domains and a long intracellular carboxy-terminal that contains the subunit interaction domain (sid) (within the square). The P574 amino acid is located in the linker region between two coiled-coil regions. Adapted from (Wehling et al., 2007). (B) The c.1720C nucleotide is conserved between species. Adapted from the UCSC genome browser hg18. (C) Amino acid alignment of the si domains from all KV7 channels. The two coiled-coil domains are depicted as gray boxes and conserved amino acids are marked with an asterisk. The P574 amino acid is located in the linker region between two coiled-coil regions and is not conserved between KV7 members. Adapted from Wehling et al. (2007).
There was no significant effect of the P574S amino acid change on the localization of KV7.3 containing channel complexes neither in HEK 293 cells (Figures 5 and 8) nor in cultured rat hippocampal neurons (Figure 6). The data further show that subunit assembly as well as AIS localization were unaffected by the mutation (Figure 7).
Currents elicited in X. laevis oocytes upon expression of KV7.2/KV7.3_P574S did not differ from WT currents (Figure 3A) in agreement with previous reports (Neubauer et al., 2008; Miceli et al., 2009). Likewise, we did not observe any changes in the current mediated by KV7.4/KV7.3_P574S complexes (Figure 3B). However, co-expressing KV7.3_P574S with KV7.5 reduced the current significantly compared to WT KV7.3 (Figure 4), possibly due to altered inactivation properties. Mimicking the heterozygous state of the patients showed intermediate current amplitudes indicating that KV7.3_P574S does not have a dominant-negative effect; however, the effect of the mutation is not rescued by co-expression of WT KV7.3. These results show, for the first time, how the rs74582884 SNP in KCNQ3 identified in patients with ASD, ID, major depression or various types of epilepsy functionally impairs the function of a channel complex formed by KV7.3/KV7.5 complexes. Accordingly, KCNQ3 and KCNQ5 (OMIM 607357) are suggestive susceptibility genes for ASD, ID, major depression, epilepsy, and due to the considerable overlap in etiologies also for other psychiatric disorders like ADHD, bipolar disorder, and anxiety disorder. To our knowledge, this is the first report associating KV7.5 with a disease.
The physiological relevance of this finding lies in the major impact these channel complexes underlying the M-current have for controlling neuronal excitability (Wang et al., 1998; Schroeder et al., 2000; Cooper and Jan, 2003) and generation of theta oscillations which are involved in memory formation and spatial navigation (Hu et al., 2002; Peters et al., 2005; Wang, 2010). Theta oscillations are not only observed in the hippocampus but also in the surrounding limbic structures as well as in the prefrontal cortex (Wang, 2010). These areas of the brain are involved in memory storage (Morgado-Bernal, 2011), emotional processing (Adolphs, 2010), behavioral monitoring, and valuation of response outcomes (Wang, 2010) which are all aspects of normal brain functioning that appear to be affected in individuals with ASD, ID, or psychiatric disorders and accordingly might contribute to the observed phenotypes of the patients presented here. Since several neurotransmitters, neuromodulators, and pharmacological drugs can influence the properties of M-channels (Cooper and Jan, 2003) it can thus be speculated whether any of these modulators could improve the quality of life for the patients described here and potentially other psychiatric patients.
In conclusion, we here present four unrelated ASD patients with variations in KCNQ3. One patient has a truncating de novo mutation whereas the other three patients have inherited a c.1720C > T [p.P574S] nucleotide change (rs74582884). One transmitting parent suffers from major depression whereas the other two are phenotypically normal. This SNP was previously reported in patients with rolandic epilepsy, IGE, or benign neonatal convulsions and accordingly, shows varying expressivity and reduced penetrance. The p.P574S change in the KV7.3 channel protein significantly reduces currents when co-expressed with KV7.5 but not KV7.2 or KV7.4 in a heterologous expression system. This suggests that specific dysfunction of the KV7.3/KV7.5 channel may be associated with some forms of ASD, ID, major depression, epilepsy, and possibly other psychiatric disorders and accordingly KCNQ5 should also be considered a candidate gene for these disorders.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the patients and families for participating in this study and the Wellcome Trust Sanger Institute for providing BAC clones. This study was supported by the University of Copenhagen and the Danish National Research Foundation who established the Wilhelm Johannsen Centre for Functional Genome Research and the Danish National Research Foundation Centre for Cardiac Arrhythmia; the Lundbeck foundation (R67-A6206); the Danish Medical Research Council (HBR and NS grant 271-08-0531), the Novo Nordisk Foundation, and the German Mental Retardation Network (MRNET) funded through a grant from the German Ministry of Research and Education (01GS08161); and the European Union’s Seventh Framework Program under grant agreement number 241995, project GENCODYS. The authors have no conflict of interest to declare.
Abdallah, M. W., Greaves-Lord, K., Grove, J., Norgaard-Pedersen, B., Hougaard, D. M., and Mortensen, E. L. (2011). Psychiatric comorbidities in autism spectrum disorders: findings from a Danish historic birth cohort. Eur. Child. Adolesc. Psychiatry 20, 599–601.
Adolphs, R. (2010). What does the amygdala contribute to social cognition? Ann. N. Y. Acad. Sci. 1191, 42–61.
Akanuma, N., Hara, E., Adachi, N., Hara, K., and Koutroumanidis, M. (2008). Psychiatric comorbidity in adult patients with idiopathic generalized epilepsy. Epilepsy Behav. 13, 248–251.
Autism and Developmental Disabilities Monitoring Network. (2009). Prevalence of autism spectrum disorders – Autism and developmental disabilities monitoring network, United States, 2006. MMWR Surveill. Summ. 58, 1–20.
Avramopoulos, D., Willour, V. L., Zandi, P. P., Huo, Y., MacKinnon, D. F., Potash, J. B., et al. (2004). Linkage of bipolar affective disorder on chromosome 8q24: follow-up and parametric analysis. Mol. Psychiatry 9, 191–196.
Bailey, A., Le, C. A., Gottesman, I., Bolton, P., Simonoff, E., Yuzda, E., et al. (1995). Autism as a strongly genetic disorder: evidence from a British twin study. Psychol. Med. 25, 63–77.
Baron-Cohen, S., Scott, F. J., Allison, C., Williams, J., Bolton, P., Matthews, F. E., et al. (2009). Prevalence of autism-spectrum conditions: UK school-based population study. Br. J. Psychiatry 194, 500–509.
Bentzen, B. H., Schmitt, N., Calloe, K., Dalby, B. W., Grunnet, M., and Olesen, S. P. (2006). The acrylamide (S)-1 differentially affects Kv7 (KCNQ) potassium channels. Neuropharmacology 51, 1068–1077.
Berkel, S., Marshall, C. R., Weiss, B., Howe, J., Roeth, R., Moog, U., et al. (2010). Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nat. Genet. 42, 489–491.
Biervert, C., Schroeder, B. C., Kubisch, C., Berkovic, S. F., Propping, P., Jentsch, T. J., et al. (1998). A potassium channel mutation in neonatal human epilepsy. Science 279, 403–406.
Borgatti, R., Zucca, C., Cavallini, A., Ferrario, M., Panzeri, C., Castaldo, P., et al. (2004). A novel mutation in KCNQ2 associated with BFNC, drug resistant epilepsy, and mental retardation. Neurology 63, 57–65.
Brown, D. A., and Passmore, G. M. (2009). Neural KCNQ (Kv7) channels. Br. J. Pharmacol. 156, 1185–1195.
Charlier, C., Singh, N. A., Ryan, S. G., Lewis, T. B., Reus, B. E., Leach, R. J., et al. (1998). A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family. Nat. Genet. 18, 53–55.
Chung, H. J., Jan, Y. N., and Jan, L. Y. (2006). Polarized axonal surface expression of neuronal KCNQ channels is mediated by multiple signals in the KCNQ2 and KCNQ3 C-terminal domains. Proc. Natl. Acad. Sci. U.S.A. 103, 8870–8875.
Cooper, E. C., and Jan, L. Y. (2003). M-channels: neurological diseases, neuromodulation, and drug development. Arch. Neurol. 60, 496–500.
de Kovel, C. G., Trucks, H., Helbig, I., Mefford, H. C., Baker, C., Leu, C., et al. (2010). Recurrent microdeletions at 15q11.2 and 16p13.11 predispose to idiopathic generalized epilepsies. Brain 133, 23–32.
Devaux, J. J., Kleopa, K. A., Cooper, E. C., and Scherer, S. S. (2004). KCNQ2 is a nodal K+ channel. J. Neurosci. 24, 1236–1244.
Duong, L., Klitten, L. L., Moller, R. S., Ingason, A., Jakobsen, K. D., Skjodt, C., et al. (2012). Mutations in NRXN1 in a family multiply affected with brain disorders: NRXN1 mutations and brain disorders. Am. J. Med. Genet. B Neuropsychiatr. Genet. 159B, 354–358.
Erdogan, F., Chen, W., Kirchhoff, M., Kalscheuer, V. M., Hultschig, C., Muller, I., et al. (2006). Impact of low copy repeats on the generation of balanced and unbalanced chromosomal aberrations in mental retardation. Cytogenet. Genome Res. 115, 247–253.
Feng, J., Schroer, R., Yan, J., Song, W., Yang, C., Bockholt, A., et al. (2006). High frequency of neurexin 1beta signal peptide structural variants in patients with autism. Neurosci. Lett. 409, 10–13.
Folstein, S., and Rutter, M. (1977). Infantile autism: a genetic study of 21 twin pairs. J. Child. Psychol. Psychiatry 18, 297–321.
Folstein, S. E., and Rosen-Sheidley, B. (2001). Genetics of autism: complex aetiology for a heterogeneous disorder. Nat. Rev. Genet. 2, 943–955.
Gregor, A., Albrecht, B., Bader, I., Bijlsma, E. K., Ekici, A. B., Engels, H., et al. (2011). Expanding the clinical spectrum associated with defects in CNTNAP2 and NRXN1. BMC Med. Genet. 12:106. doi:10.1186/1471-2350-12-106
Gu, C., Jan, Y. N., and Jan, L. Y. (2003). A conserved domain in axonal targeting of Kv1 (Shaker) voltage-gated potassium channels. Science 301, 646–649.
Hu, H., Vervaeke, K., and Storm, J. F. (2002). Two forms of electrical resonance at theta frequencies, generated by M-current, h-current and persistent Na+ current in rat hippocampal pyramidal cells. J. Physiol. (Lond.) 545, 783–805.
Iossifov, I., Ronemus, M., Levy, D., Wang, Z., Hakker, I., Rosenbaum, J., et al. (2012). De novo gene disruptions in children on the autistic spectrum. Neuron 74, 285–299.
Jamain, S., Quach, H., Betancur, C., Rastam, M., Colineaux, C., Gillberg, I. C., et al. (2003). Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 34, 27–29.
Kohane, I. S., McMurry, A., Weber, G., MacFadden, D., Rappaport, L., Kunkel, L., et al. (2012). The co-morbidity burden of children and young adults with autism spectrum disorders. PLoS ONE 7:e33224. doi:10.1371/journal.pone.0033224
Kubisch, C., Schroeder, B. C., Friedrich, T., Lutjohann, B., El-Amraoui, A., Marlin, S., et al. (1999). KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell 96, 437–446.
Lerche, C., Scherer, C. R., Seebohm, G., Derst, C., Wei, A. D., Busch, A. E., et al. (2000). Molecular cloning and functional expression of KCNQ5, a potassium channel subunit that may contribute to neuronal M-current diversity. J. Biol. Chem. 275, 22395–22400.
Levy, D., Ronemus, M., Yamrom, B., Lee, Y. H., Leotta, A., Kendall, J., et al. (2011). Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron 70, 886–897.
Leyfer, O. T., Folstein, S. E., Bacalman, S., Davis, N. O., Dinh, E., Morgan, J., et al. (2006). Comorbid psychiatric disorders in children with autism: interview development and rates of disorders. J. Autism Dev. Disord. 36, 849–861.
Lord, C., Rutter, M., and Le, C. A. (1994). Autism Diagnostic Interview-Revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J. Autism Dev. Disord. 24, 659–685.
Miceli, F., Soldovieri, M. V., Lugli, L., Bellini, G., Ambrosino, P., Migliore, M., et al. (2009). Neutralization of a unique, negatively-charged residue in the voltage sensor of K V 7.2 subunits in a sporadic case of benign familial neonatal seizures. Neurobiol. Dis. 34, 501–510.
Moreno-De-Luca, D., Mulle, J. G., Kaminsky, E. B., Sanders, S. J., Myers, S. M., Adam, M. P., et al. (2010). Deletion 17q12 is a recurrent copy number variant that confers high risk of autism and schizophrenia. Am. J. Hum. Genet. 87, 618–630.
Morgado-Bernal, I. (2011). Learning and memory consolidation: linking molecular and behavioral data. Neuroscience 176, 12–19.
Neale, B. M., Kou, Y., Liu, L., Ma’Ayan, A., Samocha, K. E., Sabo, A., et al. (2012). Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242–245.
Neubauer, B. A., Waldegger, S., Heinzinger, J., Hahn, A., Kurlemann, G., Fiedler, B., et al. (2008). KCNQ2 and KCNQ3 mutations contribute to different idiopathic epilepsy syndromes. Neurology 71, 177–183.
O’Roak, B. J., Vives, L., Girirajan, S., Karakoc, E., Krumm, N., Coe, B. P., et al. (2012). Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250.
Peters, H. C., Hu, H., Pongs, O., Storm, J. F., and Isbrandt, D. (2005). Conditional transgenic suppression of M channels in mouse brain reveals functions in neuronal excitability, resonance and behavior. Nat. Neurosci. 8, 51–60.
Rasmussen, H. B., Frokjaer-Jensen, C., Jensen, C. S., Jensen, H. S., Jorgensen, N. K., Misonou, H., et al. (2007). Requirement of subunit co-assembly and ankyrin-G for M-channel localization at the axon initial segment. J. Cell. Sci. 120, 953–963.
Schroeder, B. C., Hechenberger, M., Weinreich, F., Kubisch, C., and Jentsch, T. J. (2000). KCNQ5, a novel potassium channel broadly expressed in brain, mediates M-type currents. J. Biol. Chem. 275, 24089–24095.
Schroeder, B. C., Kubisch, C., Stein, V., and Jentsch, T. J. (1998). Moderate loss of function of cyclic-AMP-modulated KCNQ2/KCNQ3 K+ channels causes epilepsy. Nature 396, 687–690.
Schwake, M., Athanasiadu, D., Beimgraben, C., Blanz, J., Beck, C., Jentsch, T. J., et al. (2006). Structural determinants of M-type KCNQ (Kv7) K+ channel assembly. J. Neurosci. 26, 3757–3766.
Schwake, M., Pusch, M., Kharkovets, T., and Jentsch, T. J. (2000). Surface expression and single channel properties of KCNQ2/KCNQ3, M-type K+ channels involved in epilepsy. J. Biol. Chem. 275, 13343–13348.
Sebat, J. (2007). Major changes in our DNA lead to major changes in our thinking. Nat. Genet. 39, S3–S5.
Stahlberg, O., Soderstrom, H., Rastam, M., and Gillberg, C. (2004). Bipolar disorder, schizophrenia, and other psychotic disorders in adults with childhood onset AD/HD and/or autism spectrum disorders. J. Neural Transm. 111, 891–902.
State, M. W. (2010). The genetics of child psychiatric disorders: focus on autism and Tourette syndrome. Neuron 68, 254–269.
Steinlein, O. K., Conrad, C., and Weidner, B. (2007). Benign familial neonatal convulsions: always benign? Epilepsy Res. 73, 245–249.
Tovia, E., Goldberg-Stern, H., Ben, Z. B., Heyman, E., Watemberg, N., Fattal-Valevski, A., et al. (2011). The prevalence of atypical presentations and comorbidities of benign childhood epilepsy with centrotemporal spikes. Epilepsia 52, 1483–1488.
Tuchman, R., Cuccaro, M., and Alessandri, M. (2010). Autism and epilepsy: historical perspective. Brain Dev. 32, 709–718.
Ullmann, R., Turner, G., Kirchhoff, M., Chen, W., Tonge, B., Rosenberg, C., et al. (2007). Array CGH identifies reciprocal 16p13.1 duplications and deletions that predispose to autism and/or mental retardation. Hum. Mutat. 28, 674–682.
Verheij, J. B., de Munnik, S. A., Dijkhuizen, T., de, L. N., Olde, W. D., van den Hoek, G. J., et al. (2009). An 8.35 Mb overlapping interstitial deletion of 8q24 in two patients with coloboma, congenital heart defect, limb abnormalities, psychomotor retardation and convulsions. Eur. J. Med. Genet. 52, 353–357.
Wang, H. S., Pan, Z., Shi, W., Brown, B. S., Wymore, R. S., Cohen, I. S., et al. (1998). KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science 282, 1890–1893.
Wang, X. J. (2010). Neurophysiological and computational principles of cortical rhythms in cognition. Physiol. Rev. 90, 1195–1268.
Wehling, C., Beimgraben, C., Gelhaus, C., Friedrich, T., Saftig, P., Grotzinger, J., et al. (2007). Self-assembly of the isolated KCNQ2 subunit interaction domain. FEBS Lett. 581, 1594–1598.
Zandi, P. P., Zollner, S., Avramopoulos, D., Willour, V. L., Chen, Y., Qin, Z. S., et al. (2008). Family-based SNP association study on 8q24 in bipolar disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 147B, 612–618.
Keywords: Autism, KCNQ3, KCNQ5, KV7.3, KV7.5, translocation, SNP
Citation: Gilling M, Rasmussen HB, Calloe K, Sequeira AF, Baretto M, Oliveira G, Almeida J, Lauritsen MB, Ullmann R, Boonen SE, Brondum-Nielsen K, Kalscheuer VM, Tümer Z, Vicente AM, Schmitt N and Tommerup N (2013) Dysfunction of the heteromeric KV7.3/KV7.5 potassium channel is associated with autism spectrum disorders. Front. Genet. 4:54. doi: 10.3389/fgene.2013.00054
Received: 06 December 2012; Paper pending published: 30 December 2012;
Accepted: 25 March 2013; Published online: 16 April 2013.
Edited by:
Kathleen D. Askland, Brown University, USAReviewed by:
Hung-Teh Kao, Brown University, USACopyright: © 2013 Gilling, Rasmussen, Calloe, Sequeira, Baretto, Oliveira, Almeida, Lauritsen, Ullmann, Boonen, Brondum-Nielsen, Kalscheuer, Tümer, Vicente, Schmitt and Tommerup. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Nicole Schmitt, Danish National Research Foundation Centre for Cardiac Arrhythmia, Department of Biomedical Sciences, University of Copenhagen, The Panum Institute 12.5.14, Blegdamsvej 3, 2200 Copenhagen N, Denmark. e-mail:bnNjaG1pdHRAc3VuZC5rdS5kaw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.