


94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet., 19 November 2012
Sec. Behavioral and Psychiatric Genetics
Volume 3 - 2012 | https://doi.org/10.3389/fgene.2012.00250
Glyoxalase 1 (GLO1) is a ubiquitous cellular enzyme that participates in the detoxification of methylglyoxal (MG), a cytotoxic byproduct of glycolysis that induces protein modification (advanced glycation end-products, AGEs), oxidative stress, and apoptosis. The concentration of MG is elevated under high-glucose conditions, such as diabetes. As such, GLO1 and MG have been implicated in the pathogenesis of diabetic complications. Recently, findings have linked GLO1 to numerous behavioral phenotypes, including psychiatric diseases (anxiety, depression, schizophrenia, and autism) and pain. This review highlights GLO1's association with behavioral phenotypes, describes recent discoveries that have elucidated the underlying mechanisms, and identifies opportunities for future research.
Glyoxalase 1 (GLO1) is an enzyme in the glyoxalase system, a metabolic pathway that detoxifies α-oxoaldehydes, particularly methylglyoxal (MG) (Thornalley, 1990, 1993, 2003a; Mannervik, 2008). MG is primarily formed by the degradation of the glycolytic intermediates, dihydroxyacetone phosphate, and glyceraldehyde-3-phosphate (Thornalley, 1993). MG levels rise under high-glucose conditions, such as diabetes (Brownlee, 2001). At supra-physiological levels, MG induces protein and nucleotide modification (advanced glycation end-products, AGEs), reactive oxygen species (ROS), and apoptosis (Brownlee, 2001; Thornalley, 2003b). AGEs cause dysfunction of affected proteins (Brownlee, 2001) and ligate the receptor for advanced glycation end-products (RAGE), triggering the production of ROS (Schleicher and Friess, 2007) and apoptosis (Loh et al., 2006). To combat MG's cytotoxic effects, GLO1 enzymatically converts MG into the less reactive substance, d-lactate (Thornalley, 2003b). In vitro, overexpression of Glo1 prevents MG accumulation (Shinohara et al., 1998); conversely, GLO1 inhibition increases MG accumulation and decreased cellular viability (Kuhla et al., 2006). Given their roles in AGE formation and cytotoxicity, GLO1 and MG have been implicated in diseases where these effects are relevant to pathogenesis, such as diabetic complications (i.e., micro- and macro-vascular disease), cancer, and aging (Brownlee, 2001; Thornalley, 2003a; Ahmed and Thornalley, 2007; Morcos et al., 2008; Fleming et al., 2010; Thornalley and Rabbani, 2011).
Surprisingly, an increasing number of studies have identified associations between GLO1 and behavioral phenotypes. In mice, several lines of evidence suggest that Glo1 expression is associated with anxiety-like behavior (Hovatta et al., 2005; Reiner-Benaim et al., 2007; Loos et al., 2009; Benton et al., 2011; Distler et al., 2012), depression (Benton et al., 2011), and neuropathic pain (Jack et al., 2011, 2012; Bierhaus et al., 2012). New observations in rats using oxidative stress models (Salim et al., 2010a,b) and the sleep-deprivation model of psychological stress (Vollert et al., 2011), both suggest role of GLO1 in the anxious phenotype of rats (Salim et al., 2011). Additional associations have been identified with habituation, locomotor activity, motor coordination, exploratory behavior, and learning and memory (Williams et al., 2009). In humans, genetic studies have implicated polymorphisms in GLO1 in panic disorder (Politi et al., 2006), depression (Fujimoto et al., 2008), autism (Junaid et al., 2004; Barua et al., 2011), schizophrenia (Arai et al., 2010; Toyosima et al., 2011), and restless legs syndrome (RLS) (Stefansson et al., 2007; Winkelmann et al., 2007, 2011; Kemlink et al., 2009). While the studies of RLS identify a haplotype block that includes GLO1, more attention has been paid to the neighboring gene BTBD9. The other human genetic studies utilized a candidate gene approach and had small sample sizes; in most cases, those results have not been replicated. Aside from genome-wide association studies (GWAS) of RLS, GLO1 has not been identified in GWAS for other psychiatric or neurological traits.
Recently, two major studies have shed light on the mechanisms underlying GLO1's behavioral effects. One identified MG as an agonist at GABAA receptors (Distler et al., 2012), while the other identified MG modification of voltage-gated sodium channels (Bierhaus et al., 2012). This article will focus on GLO1's behavioral correlates and the potential underlying mechanisms. The basic biochemistry of GLO1 has been reviewed elsewhere (Thornalley, 1990, 1993, 2003b), as have its roles in cancer (Thornalley, 2003a; Thornalley and Rabbani, 2011) and diabetic complications (Brownlee, 2001; Ahmed and Thornalley, 2007; Schleicher and Friess, 2007; Jack and Wright, 2012; Rabbani and Thornalley, 2012).
Among the behavioral phenotypes associated with GLO1, anxiety-like behavior is the most commonly reported and widely studied. In mice, numerous genetic studies have identified associations between Glo1 expression and anxiety-like behavior (Hovatta et al., 2005; Kromer et al., 2005; Williams et al., 2009). Hovatta et al. analyzed gene expression profiles in a panel of six inbred mouse strains, which were scored for anxiety-like behavior in the open field and light-dark box tests (Hovatta et al., 2005). They found a positive correlation between Glo1 expression and anxiety-like behavior. Several subsequent studies also identified positive correlations between Glo1 expression and anxiety-like behavior among inbred mouse strains (Reiner-Benaim et al., 2007; Loos et al., 2009; Benton et al., 2011).
Importantly, Hovatta et al. established a causal role for Glo1 in anxiety-like behavior by using viral vectors to overexpress or knock down Glo1 in the anterior cingulate cortex (Hovatta et al., 2005). Local Glo1 overexpression increased anxiety-like behavior, while local Glo1 knockdown decreased anxiety-like behavior (Hovatta et al., 2005). This experimental manipulation supported a causal role for Glo1 in anxiety-like behavior. However, this study was limited by the use of viral vectors: expression was manipulated only in one brain region during adulthood. As such, it might not have accurately modeled Glo1's normal physiological role in anxiety-like behavior. Furthermore, viral vectors often produce variable expression levels between animals (Kirik and Bjorklund, 2003).
Kromer et al. also identified a correlation between GLO1 and anxiety-like behavior in mice (Kromer et al., 2005). They selectively bred mice for high anxiety-like behavior (HAB) or low anxiety-like behavior (LAB) and subsequently inbred the mice to generate two lines. The LAB line had higher GLO1 protein levels than the HAB line (Kromer et al., 2005). Therefore, Kromer et al. proposed that increased GLO1 was a biomarker for low anxiety-like behavior, a result directionally opposite from studies that used panels of inbred mouse strains (Hovatta et al., 2005; Reiner-Benaim et al., 2007; Loos et al., 2009; Benton et al., 2011) and those that used viral vectors to manipulate Glo1 expression (Hovatta et al., 2005). In fact, the discrepancy among these studies was used as grounds to dispute a role for Glo1 in anxiety-like behavior (Thornalley, 2006).
A subsequent study identified a common copy number variant (CNV) among inbred mice that caused a duplication of four genes, including Glo1 (Williams et al., 2009). Of the 72 inbred strains examined, 23 carried the duplication. The duplication was positively correlated with increased Glo1 expression (it was an expression QTL; eQTL) and increased anxiety-like behavior (Williams et al., 2009). Increased Glo1 copy number was also associated with other behavioral phenotypes that were available from public databases, including locomotor habituation in a novel environment (negative correlation), total locomotor activity (positive correlation), motor coordination (positive correlation), rearing (positive correlation), anxiety-like behavior in the elevated plus maze and light-dark box tests (positive correlation), stretch-attends in tests of anxiety-like behavior (negative correlation), and measures of learning and memory (Williams et al., 2009). These findings suggested a role for Glo1 copy number in a broad range of behavioral phenotypes.
The discovery of this CNV helped to explain the previously reported differences in Glo1 expression among inbred strains (Hovatta et al., 2005). In addition, it explained the differential GLO1 protein expression reported in the HAB and LAB lines: LAB mice were subsequently shown to have the duplication, while HAB mice do not (Hambsch et al., 2010). Although it is somewhat surprising that selection did not have the opposite result, other factors, including differences in initial allelic frequencies, linked alleles, and drift before or during inbreeding could have contributed to the fixation of the duplication in LAB but not HAB mice. The effect of the Glo1 duplication on anxiety-like behavior was likely offset by numerous other alleles that collectively contributed to anxiety-like behavior in the selected lines. HAB and LAB lines are known to harbor many genetic differences due to selection and inbreeding (Bunck et al., 2009; Ditzen et al., 2010; Czibere et al., 2011; Filiou et al., 2011; Tasan et al., 2011; Zhang et al., 2011). Indeed, selection studies are prone to such confounding forces, especially when the selected lines are not replicated.
The discovery of the Glo1 CNV also raised the possibility that Glo1 expression could simply be a marker for the presence of the CNV rather than directly regulating anxiety-like behavior. Specifically, the CNV contained four genes, any of which could have affected behavioral phenotypes. Further, the CNV could have been linked to unidentified causal alleles or could have disrupted neighboring genes. As such, the discovery of the CNV complicated observed associations between Glo1 expression and behavior.
Therefore, in order to test the hypothesis that increased Glo1 expression was sufficient to replicated the behavioral differences associated with the CNV, targeted genetic manipulation of Glo1 copy number performed in which mice with a transgenic bacterial artificial chromosome (BAC) containing Glo1 were created to model the CNV (Distler et al., 2012). To isolate Glo1's effect, the BAC was engineered so that only Glo1 was expressed. BAC transgenic mice overexpressed Glo1 and displayed increased anxiety-like behavior. This effect was observed in multiple founder lines and on two genetic backgrounds: C57BL/6J (B6) and FVB/NJ (FVB). The effect of Glo1 overexpression on anxiety-like behavior was strongest in the lines with the highest copy numbers. Unlike the viral vector studies (Hovatta et al., 2005), this study demonstrated the physiological relevance of Glo1 in behavior, because Glo1 was expressed under its endogenous promoter elements, in its natural expression pattern, and throughout the animal's life. Therefore, this study demonstrated that increased Glo1 copy number increased Glo1 expression and anxiety-like behavior.
In contrast to mouse studies, human genetic studies have not identified robust associations between GLO1 and anxiety disorders. In humans, there are two common GLO1 alleles (Kompf et al., 1975) that result from a single nucleotide polymorphism (SNP) at position 419 (Kim et al., 1995). Adenine (419A) encodes a glutamate at amino acid 111 (111E), and cytosine (419C) encodes an alanine (111A). Politi et al. (2006) identified a weak, but significant increase in the risk of panic disorder without agoraphobia among people carrying the 419A allele. Limitations, such as the small sample size and potential population stratification, made this result preliminary. A subsequent study found no correlation between GLO1 expression and susceptibility to panic attacks (Eser et al., 2011), but suffered from the same limitations, so cannot be said to disprove the original observation. Similarly, no published GWAS studies of panic disorder have identified GLO1 (Erhardt et al., 2011). Despite disappointing findings from human genetic studies, there remains a need for large, carefully controlled genetic studies in order to address whether differences in GLO1 affect anxiety among humans.
There is strong evidence for shared genetic vulnerability to anxiety and depression (Cerda et al., 2010). Among a panel of inbred mice, Benton et al. found a positive correlation between GLO1 protein levels and baseline depression-like behavior using the tail-suspension test (Benton et al., 2011), suggesting that Glo1 could contribute to the common genetic etiology of anxiety and depression. However, a small human study of GLO1 and depression reported apparently conflicting data. Fujimoto et al. reported a negative correlation between GLO1 expression and depression in humans (Fujimoto et al., 2008). GLO1 mRNA expression was reduced in peripheral white blood cells (WBC) of patients with major depressive and bipolar disorders (Fujimoto et al., 2008). GLO1 mRNA expression did not significantly differ between patients in remission and healthy controls, suggesting that reduced GLO1 expression was a state-dependent marker for depression (Fujimoto et al., 2008). However, this study did not establish whether reduced GLO1 expression affected GLO1 protein levels or enzymatic activity, which would reflect the functional significance of differences in mRNA expression. Further, WBC mRNA may not adequately reflect mRNA expression in the brain (Mehta et al., 2010). Corroborating findings from post-mortem brain samples would significantly strengthen the association between GLO1 expression and depression (Mehta et al., 2010). Therefore, although data from mice implicate Glo1 in increasing depression-like behavior, further experiments are required to resolve the discrepancy with human data.
Human studies have proposed an association between a coding SNP in GLO1 (419A/C; rs2736654) and autism. Junaid et al. reported that the 419A allele was 1.5 times more common among autistic patients than controls (Junaid et al., 2004). However, this study was limited by its small sample size and potential confounding variables (e.g., population stratification). Indeed, subsequent studies have not replicated this finding: there was no association between SNPs in GLO1 and autism among Han Chinese patients (Wu et al., 2008), nor was there significant linkage or association between SNPs in GLO1 and autism among Finnish patients (Rehnstrom et al., 2008). Furthermore, a family-based and case-control study found that the 419A allele was more common among unaffected siblings of autistic patients, indicating that it might be protective (Sacco et al., 2007). Finally, several human genome-wide association studies (GWAS) have failed to identify associations between GLO1 polymorphisms and autism. At the very least, this suggests that the study of Junaid et al. overestimated the effect size of this allele, which must have been small enough to escape detection by larger GWAS.
Nevertheless, Junaid et al. suggested that the 419A GLO1 allele was associated with a functional change in GLO1 enzymatic activity. They reported that post-mortem brain tissue from autistic patients had reduced GLO1 enzymatic activity and increased AGE content compared to that from control patients (Junaid et al., 2004). In a later study, Baura et al. reported a correlation between the 111E GLO1 isoform and reduced enzymatic activity (Barua et al., 2011). However, Barua et al. did not directly examine enzymatic activity of the isoforms. Rather, they utilized cell extracts derived from autistic and control patients. As such, differences in enzymatic activity and MG concentration could have resulted from differences in GLO1 expression or other genetic differences. Therefore, further studies will help determine the functional significance of the 419A/C SNP and its contribution to autism.
Experimental studies in mice may help determine whether GLO1 is related to autism-like behaviors. Various behavioral assays have been proposed for measuring autism-like phenotypes in mice, including social interaction tests, ultrasonic vocalization measurements, assessment of repetitive and stereotyped behavior, and reversal learning tests (Silverman et al., 2010). In future studies, these assays may be used to study mutant mice with increased or decreased Glo1 expression.
Recently, two studies have suggested a role for GLO1 in schizophrenia (Arai et al., 2010; Toyosima et al., 2011). In a single schizophrenic patient, a frameshift mutation in GLO1 was correlated with reduced GLO1 enzymatic activity (Toyosima et al., 2011). In a separate study, schizophrenic patients were found to have increased AGE accumulation compared to control subjects (Arai et al., 2010), suggesting reduced GLO1 function. Nevertheless, small sample sizes and confounding variables made these associations preliminary. Therefore, additional human genetic studies and studies of GLO1 in mice will help establish an association with schizophrenia. Several behavioral assays of schizophrenia-like behavior have been proposed (Young et al., 2010), which could be used for studying mice with aberrant Glo1 expression.
Multiple genome-wide association studies have implicated a haplotype block that includes GLO1 in restless legs syndrome (Stefansson et al., 2007; Winkelmann et al., 2007, 2011; Kemlink et al., 2009). These studies have focused on BTBD9, another gene that, like GLO1, is in this largely non-recombinant haplotype, but this association could also reflect regulatory changes that impact GLO1.
Neuropathic pain is a feature of neuropathy, a common sequela of diabetes (Vinik et al., 2000; Edwards et al., 2008). There is an extensive literature on the relationship between Glo1 and neuropathy which has been recently reviewed (Jack and Wright, 2012). Recent studies have identified associations between the Glo1 CNV in mice and behavioral measures of neuropathic pain (Jack et al., 2011, 2012). After diabetes induction, A/J mice, which carry a triplication of the Glo1 allele, were less sensitive to mechanical pain than B6 mice, which have only a single copy of Glo1 (Jack et al., 2011). Similarly, Glo1 copy number was associated with mechanical pain sensitivity after induction of diabetes in two closely related inbred strains, BALB/cJ and BALB/cByJ. BALB/cByJ mice carry the Glo1 duplication, while BALB/cJ mice do not (Williams et al., 2009). Similar to the previous report, increased levels of Glo1 protected against diabetic hyperalgesia in BALB/cByJ mice compared to BALB/cJ mice (Jack et al., 2012). These results suggested that Glo1 mediates behavioral responses to pain associated with diabetic neuropathy. A recent study has confirmed that GLO1 protects against neuropathic pain associated with diabetes. Overexpression of human GLO1 reduced thermal hyperalgesia in diabetic mice (Bierhaus et al., 2012). Conversely, pharmacological inhibition of GLO1 and Glo1 knockdown exacerbated both mechanical and thermal hyperalgesia in diabetic mice (Bierhaus et al., 2012). Together, these findings suggest a role for GLO1 in mediating behavioral measures of pain, particularly that related to diabetic neuropathy. To date, no study has investigated the role of GLO1 in other types of pain, such as nociceptive and central pain, which is an important area of future investigation.
There is robust evidence that GLO1 activity regulates MG concentration (Thornalley, 1990, 1993, 2003b). Recent studies have suggested this activity is critical to GLO1's effects on behavior. In vitro, Glo1 overexpression prevented MG accumulation under conditions of high glucose (Shinohara et al., 1998), and in C. elegans, Glo1 overexpression reduced basal MG concentration (Morcos et al., 2008). Similarly, Glo1 overexpression reduced baseline MG concentration in the brains of mice (Distler et al., 2012) and plasma MG concentration in diabetic mice (Bierhaus et al., 2012).
Given the strong association between GLO1 and MG concentration, it was hypothesized that GLO1 affects behavior by controlling levels of MG. This hypothesis suggested that direct administration of MG would alter behavior. Recent reports have demonstrated MG's behavioral effects, including those related to anxiety-like behavior (Hambsch et al., 2010; Distler et al., 2012), depression (Zhang et al., 2011), motor coordination (Distler et al., 2012), and pain (Bierhaus et al., 2012). Hambsch et al. administered MG to mice by intra-cereberoventricular injection daily for 6 days; MG-treated mice displayed reduced anxiety-like behavior in the elevated plus maze compared to vehicle-treated mice (Hambsch et al., 2010). Chronic MG treatment also reduced immobility in the tail suspension test, reflecting an anti-depressant effect of MG (Hambsch et al., 2010). Distler et al. identified a similar anxiolytic effect of MG within minutes of a single intra-peritoneal injection (Distler et al., 2012), suggesting that MG acutely affects behavior. At high doses, acute MG administration was also shown to have sedative effects, including locomotor depression, ataxia, hypothermia, and lethargy (Distler et al., 2012). Acute MG administration also caused hyperalgesia equivalent to that caused by diabetes in mice (Bierhaus et al., 2012). Together, these studies indicated that MG concentration regulates multiple behavioral phenotypes and that MG's effects are opposite to those of Glo1 overexpression.
The behavioral experiments described above utilized crude MG preparations, which may contain contaminants, such as formaldehyde and methanol (Pourmotabbed and Creighton, 1986; McLellan and Thornalley, 1992). We recently tested the behavioral effects of pure MG and found that it elicited similar pharmacological effects as crude MG. At a low dose, pure MG was anxiolytic in the open field test, and at a high dose, it caused locomotor depression, ataxia, and hypothermia (Distler et al., 2012). Notably, the doses of pure MG required for sedation, locomotor depression, ataxia, and hypothermia were similar to the doses of crude MG, while pure MG elicited an anxiolytic effect at a lower dose than crude MG. It remains to be determined whether pure MG affects other reported behavioral effects, such as depression and hyperalgesia.
MG is best characterized as a cytotoxic agent that forms AGEs. This molecular action has been attributed to the pathogenesis of several diseases characterized by increased MG concentrations, including diabetic complications. For instance, increased MG levels and subsequent AGE formation have been well-established pathogenic mechanisms in diabetes-associated microvascular disease (e.g., diabetic retinopathy) and macrovascular disease (e.g., atherosclerosis) (Brownlee, 2001).
Recently, AGEs have been attributed to the development of diabetic neuropathy and neuropathic pain (Jack and Wright, 2012). Induction of diabetes in rats increased AGEs in the extracellular matrix of sensory nerves (Duran-Jimenez et al., 2009). In particular, laminin and fibronectin were found to be modified by MG (Duran-Jimenez et al., 2009). Among the numerous proteins likely modified by MG in diabetes (Rabbani and Thornalley, 2012), a voltage-gated sodium channel, Nav1.8, was recently reported to be modified by MG and it was suggested that this modification contribute to neuropathic pain (Bierhaus et al., 2012). Nav1.8 was MG-modified in peripheral sensory neurons of mice subjected to diabetes, MG administration, or Glo1 knock down. Knock down of Nav1.8 reduced MG-induced thermal hyperalgesia. Finally, MG-modification of Nav1.8 dysregulated electrical signaling in sensory neurons. Specifically, prolonged incubation with MG (3–14 h) modified Nav1.8 and altered resting membrane potential, current threshold, and voltage threshold. These findings suggested that AGEs promote protein dysfunction under conditions of high MG concentrations, thus contributing to neuropathic pain.
Because MG's role in AGE formation is well characterized, it was thought that this mechanism was relevant to other behavioral phenotypes, including anxiety-like behavior and depression (Hambsch, 2011). Hambsch et al. proposed that MG-dependent AGE formation contributed to MG's anxiolytic action (Thornalley, 2006; Hambsch et al., 2010). They found increased AGEs in the brains of mice treated chronically with MG by intracerebroventricular administration. Hambsch et al. utilized a high dose of MG, corresponding to an average concentration of 1.6 mM throughout the brain (assuming a brain volume of 0.45 mL). This study did not establish a causal relationship between AGE formation and anxiety-like behavior. Therefore, a role for AGE formation in anxiety-like behavior remains uncertain. Further, the discovery that MG activates GABAA receptors (discussed below) provides a compelling alternative mechanism for MG's anxiolytic effect.
A novel role for MG-independent of AGE formation was recently identified. MG was found to activate GABAA receptors at physiological concentrations (Distler et al., 2012). In vivo, MG's pharmacodynamic profile was similar to that of known GABAA receptor agonists, which are anxiolytic at low doses and have sedative effects at higher doses (Rudolph and Mohler, 2004; Carter et al., 2009; Kumar et al., 2009). In vitro, MG selectively activated GABAA receptors and was characterized as a competitive partial agonist (Distler et al., 2012). There is a wealth of evidence that GABAA receptors play key roles in anxiety (Kent et al., 2002; Kalueff and Nutt, 2007). As such, MG's activity at GABAA receptors can account its anxiolytic effect.
This mechanism is distinct from MG's previously reported cellular effects, specifically AGE formation and cytotoxicity. A low dose of MG was anxiolytic within minutes of peripheral administration (Distler et al., 2012). This time course differs from that of AGE formation, which requires hours to days (Lo et al., 1994). Therefore, AGE formation is unlikely to mediate MG's effect on anxiety-like behavior. MG's anxiolytic effect was also independent of cytotoxicity. In vitro, MG concentrations of 100–1000 μM have been reported to induce neuronal cell death (Di Loreto et al., 2004, 2008; Li et al., 2010). In contrast, an anxiolytic dose of MG only increased the concentration of MG in the brain to approximately 6 μM (Distler et al., 2012). In addition, MG requires hours to days to induce apoptosis (Di Loreto et al., 2008), which is inconsistent with the acute time course (~10 min) of MG's behavioral effects. Finally, there was no evidence of apoptosis in mice treated with an anxiolytic dose of MG (Distler et al., 2012). Therefore, MG's effect at GABAA receptors occurs at lower concentrations and is temporally dissociable for its better-known effects, such as AGE formation and cytotoxicity. MG's GABAergic action may explain previous observations of MG's electrophysiological effects. For example, Hambsch et al. found that incubation with 10 μM MG reduced the magnitude of long-term potentiation (LTP) in hippocampal brain slices (Hambsch et al., 2010). This is characteristic of GABAA receptor activation, which decreases excitatory post-synaptic potentials and suppresses LTP in CA1 slices (Seabrook et al., 1997).
MG's role in activating GABAA receptors has important physiological implications. MG production increases during high glycolytic activity. MG can then be metabolized by GLO1, can form AGEs or can activate GABAA receptors (Figure 1). Importantly, the effects at GABAA receptors occur at physiological concentrations of MG. Similarly, changes in MG concentration that are associated with diabetes can lead to changes in AGE formation at a variety of targets, including Nav1.8, which appears to modulate pain sensitivity.
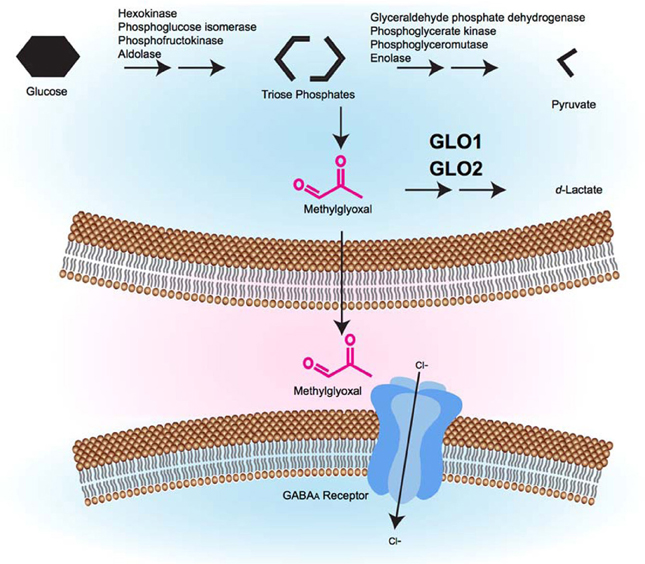
Figure 1. A model of the glyoxalase system's role in GABAergic signaling. During glycolysis, triose phosphate intermediates can be converted to MG by non-enzymatic fragmentation. Excess MG is catabolized by the glyoxalase system (GLO1 and GLO2) to form d-lactate. MG that is not catabolized can cross the plasma membrane, where it accesses pre- or post-synaptic GABAA receptors where it causes GABAA receptor activation, inward Cl− current, and membrane hyperpolarization, which is hypothesized to alter behavior.
One physiological function of MG's activity at GABAA receptors may be to maintain inhibitory tone under conditions of low GABA concentrations. In the synaptic cleft, the concentration of GABA is in the millimolar range during synaptic release (Farrant and Nusser, 2005); in the extracellular space, GABA concentrations are in the submicromolar range (Vithlani et al., 2010). The concentration of MG in the brain is approximately 5 μM (Distler et al., 2012), and these levels are likely similar in both the synapse and extracellular space. Therefore, MG is less abundant than GABA at the synapse; however, in the extracellular space MG is more abundant than GABA and likely acts as a partial agonist (Distler et al., 2012). Thus, while it has yet to be well explored MG may have a complex role in the regulation of GABAergic signaling.
While there are no data to directly support the possibility, another physiological role for MG may be as a negative regulator of excitatory signaling. Excitatory synaptic transmission is associated with increased glycolysis (Barros and Deitmer, 2010). In fact, 60–75% of neuronal glucose utilization is estimated to be in glutamatergic neurons (Shulman et al., 2004). Since neuronal excitation is coupled with glycolysis, MG concentrations may increase in excitatory neurons. MG would then activate local GABAA receptors and thus serve as a negative feedback signal. In addition, it would also provide inhibitory tone to neighboring cells. Such an action could be neuroprotective, since excessive N-methyl-D-aspartate (NMDA) receptor activation by glutamate triggers excitotoxicity and apoptosis (Naegele, 2007; Henshall and Murphy, 2008).
At the organismal level, MG concentration increases under conditions of high glucose load. Accordingly, it is possible that MG concentration may link metabolic state to neuronal inhibitory tone and behavior, including anxiety. Specifically, high levels of glucose may be coupled to reduced anxiety through increased levels of MG. In humans, consumption of foods high in fat and sugar is associated with emotional reward (Jacquier et al., 2012). In rats, fructose consumption increased levels of MG in rats (Wang et al., 2008), which could provide a direct link between the intake of high-sugar foods and emotional satiety. One notable exception to the association between increased metabolic load and reduced anxiety may be in diabetic patients, where elevated MG level have consistently been observed (Shinohara et al., 1998; Brownlee, 2001; Masterjohn et al., 2011). There is an increased prevalence of anxiety among diabetic patients (Anderson et al., 2002), whereas higher concentrations of MG would be expected to decrease anxiety. Nevertheless, it remains possible that diabetic patients experience increased anxiety in spite of increased MG concentration, since diabetes is clearly associated with numerous physiological changes in addition to changes in MG concentration and is correlated with numerous environmental and epidemiological factors. Thus, the presence of increased anxiety in diabetes patients is difficult to interpret.
Glo1's effect on anxiety-like behavior has not been assessed in females. Virtually all of the studies that identified associations between Glo1 expression and anxiety-like behavior utilized only male mice (Hovatta et al., 2005; Loos et al., 2009; Benton et al., 2011), although there are isolated exceptions (Williams et al., 2009). Studies that directly manipulated Glo1 overexpression by viral vectors (Hovatta et al., 2005) and BAC transgenes (Distler et al., 2012) studied behavioral effects in male mice only. It would not be unprecedented if the behavioral effects of Glo1 are sex-specific; sex differences in anxiety have been described in both humans and animal models (Palanza, 2001; Hamann, 2005; McLean and Anderson, 2009). The etiology underlying differential anxiety between males and females is not well understood, but it likely includes environmental and hormonal factors (McLean and Anderson, 2009; Solomon and Herman, 2009; Nillni et al., 2011).
Another unresolved issue is that only the BAC transgenic lines with high Glo1 copy numbers, and correspondingly high Glo1 expression, exhibited increased anxiety-like behavior (Distler et al., 2012). The initial association between Glo1 copy number and anxiety-like behavior involved a duplication. In contrast, B6 and FVB BAC transgenic mice with two additional copies of Glo1 (equivalent to a homozygous duplication) did not significantly differ from wild-type mice in anxiety-like behavior (Distler et al., 2012). This negative result could reflect inadequate power to detect a difference or could reflect a true lack of effect of a Glo1 duplication on anxiety-like behavior. The latter possibility could be due to differences in Glo1 expression despite equal copy numbers at the genomic level. Another possibility is that Glo1 overexpression has more robust anxiogenic effects on specific genetic backgrounds. Consistent with this explanation, Hovatta et al. reported that viral-vector-mediated Glo1 overexpression robustly increased anxiety-like behavior in 129S6/SvEvTac mice but not B6 mice (Hovatta et al., 2005).
The evolutionary significance of the Glo1 CNV is unknown. Glo1 CNVs are present in both laboratory and wild -caught mice, indicating that the duplicated allele arose prior to the domestication of mice by mouse fanciers and later scientists (Williams et al., 2009). While Redon et al. (2006) reported CNVs that included GLO1 in humans, the underlying data for those particular CNVs are unconvincing. We are not aware of any other evidence that GLO1 CNVs are common among humans or any other species; although it is possible they have simply escaped detection.
MG is byproduct of glycolysis that is generated in every organism. Homologs of GLO1 exist in bacteria, fungi, plants, and animals (Cooper, 1984; Yadav et al., 2008; Inoue et al., 2011), highlighting its ancient physiological importance. Because MG accumulation is cytotoxic, GLO1's conservation is likely tied to its role in cellular survival. The extent to which MG's action at GABAA receptors is conserved is unknown. GABA exists in bacteria, fungi, plants, and animals (Guthrie and Nicholson-Guthrie, 1989; Rauh et al., 1990; Kumar and Punekar, 1997; Bouche and Fromm, 2004), though it is only thought to serve as a neurotransmitter among animals; GABAA receptors are prominent among animals, including invertebrates (Rauh et al., 1990). Therefore, an important future direction will be to define the evolution of the relationship between MG and GABAA receptors in other species. It is possible that MG acts at GABAA receptors in invertebrates; application of 10–150 μM MG had an excitatory effect on the sixth abdominal ganglion of the cockroach (Davies et al., 1986). Although GABA is an inhibitory neurotransmitter in insects, it is also excitatory in some cell types (Beg and Jorgensen, 2003; Gisselmann et al., 2004).
The discovery that MG activates GABAA receptors has created an exciting new direction of research. Specifically, studying MG's biophysiocal interactions with GABAA receptors is liable to yield important results. Although MG and GABA are both small molecules, they are not structurally similar (Figure 2). Therefore, it is unknown whether they share a binding site on GABAA receptors or whether binding of one prevents binding of the other. Structural studies are necessary to resolve this question.
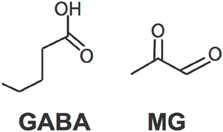
Figure 2. Molecular structures of GABA and MG.
Similarly, GABAA receptors are pentamers comprised of various combinations of at least 16 possible subunits: α1–6, β1–3, γ1–3, δ, ε, θ, and π (Bollan et al., 2003). Future studies will be required to identify MG's receptor subtype selectivity. This is relevant to MG's behavioral effects, because different subunits and subunit compositions mediate different GABAergic effects. For instance, α2 subunits have been reported to mediate the anxiolytic effects of benzodiazepines, and α5 subunits are thought to mediate their sedative effects (Rudolph and Knoflach, 2011). MG's activity at different receptor subtypes may contribute to its endogenous role in the CNS. Future studies will be critical in identifying the pharmacological and biophysical responses of specific GABAA receptor compositions to MG.
Future studies will also be important for assessing MG's activity at synaptic and extra-synaptic GABAA receptors. Synaptic GABAA receptors regulate phasic inhibitory signaling, while extra-synaptic GABAA receptors contribute to tonic inhibitory signaling (Hablitz et al., 2009) and anxiety (Maguire et al., 2005). Future studies must directly measure MG's activity at synaptic and extra-synaptic receptors in order to determine MG's role in phasic and tonic inhibitory signaling.
As reviewed above, two major mechanisms have been ascribed to GLO1's behavioral effects: regulating AGE formation and GABAA receptor activation. These mechanisms are proposed to mediate GLO1's role in pain and anxiety, respectively. However, the extent to which each mechanism underlies the many other behavioral correlates of GLO1 remains an important area of future investigation. For instance, it is possible that GLO1 regulates depression through its role in modulating GABAergic tone, given evidence that deficits in the GABA system have been implicated in depression (Gerner and Hare, 1981; Petty and Schlesser, 1981; Petty and Sherman, 1984; Crestani et al., 1999; Earnheart et al., 2007; Shen et al., 2010; Hasler and Northoff, 2011; Luscher et al., 2011). Similarly, the mechanism underlying GLO1's putative involvement in autism remains unknown. Autism has been associated with high levels of MG and AGEs (Junaid et al., 2004); however, few studies have examined whether AGEs contribute to autism (Boso et al., 2006). Alternatively, it is possible that GLO1's role in GABAergic signaling could contribute to autism since disruptions in GABAergic signaling have been identified in autism (Di Cristo, 2007; Rubenstein, 2011). Therefore, additional studies in humans and mice are required to elucidate the mechanisms underlying GLO1's associations with additional behavioral phenotypes.
GLO1 may be a useful target for pharmacological intervention. For instance, GLO1 inhibition was shown to increase MG concentration and reduce anxiety-like behavior in vivo (Distler et al., 2012). GLO1 inhibition would represent a novel mechanism of action among psychiatric drugs. Most current anxiolytic therapies target neurotransmitter systems, including the serotonin and GABA systems (Pollack, 2009). In contrast, GLO1 inhibitors would increase the production of MG, an endogenous GABAA receptor agonist. This could have a different and possibly more favorable side-effect profile compared to agents that allosterically activate GABAA receptors. For instance, endogenous MG might have receptor subtype specificity, thus preventing off-target effects. Similarly, GLO1 inhibition may not increase MG levels sufficiently to cause sedation or have abuse potential, which are common concerns of current GABAA receptor agonists. Development of better GLO1 inhibitors and characterization of the side-effects of GLO1 inhibition are thus important goals for future studies.
Studies of the adverse effects of GLO1 inhibition in vivo must also pay particular attention to cytotoxicity. In vitro studies have demonstrated that GLO1 inhibition can cause cell death (Kuhla et al., 2006). Over a short period of time, this is unlikely to occur in vivo, since GLO1 inhibition was shown to only modestly increase MG levels. Nevertheless, chronic GLO1 inhibition may increase MG to more toxic levels, causing AGE formation or cell death. Since AGEs play a prominent role in diabetic complications, GLO1 inhibitors may be a poor choice for patients with diabetes (Brownlee, 2001). In particular, GLO1 inhibition was recently shown to exacerbate diabetic hyperalgesia (Bierhaus et al., 2012), which could limit the use of GLO1 inhibitors, especially in diabetic patients.
Despite a controversial history, recent studies have demonstrated roles for GLO1 in behavioral phenotypes, including anxiety-like behavior and pain. Although animal studies support a role for GLO1 in behavioral phenotypes, human genetic studies are less convincing. As such, the extent to which polymorphisms in GLO1 regulate human diseases remains an important direction for future studies. MG can induce protein modification and acts as an endogenous GABAA receptor agonist. These cellular functions likely contribute to MG's behavioral effects. GLO1 inhibitors may provide novel therapeutic tools for the treatment of CNS disorders.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ahmed, N., and Thornalley, P. J. (2007). Advanced glycation endproducts: what is their relevance to diabetic complications? Diabetes Obes. Metab. 9, 233–245.
Anderson, R. J., Grigsby, A. B., Freedland, K. E., de Groot, M., McGill, J. B., Clouse, R. E., et al. (2002). Anxiety and poor glycemic control: a meta-analytic review of the literature. Int. J. Psychiatry Med. 32, 235–247.
Arai, M., Yuzawa, H., Nohara, I., Ohnishi, T., Obata, N., Iwayama, Y., et al. (2010). Enhanced carbonyl stress in a subpopulation of schizophrenia. Arch. Gen. Psychiatry 67, 589–597.
Barros, L. F., and Deitmer, J. W. (2010). Glucose and lactate supply to the synapse. Brain Res. Rev. 63, 149–159.
Barua, M., Jenkins, E. C., Chen, W., Kuizon, S., Pullarkat, R. K., and Junaid, M. A. (2011). Glyoxalase I polymorphism rs2736654 causing the Ala111Glu substitution modulates enzyme activity–implications for autism. Autism Res. 4, 262–270.
Beg, A. A., and Jorgensen, E. M. (2003). EXP-1 is an excitatory GABA-gated cation channel. Nat. Neurosci. 6, 1145–1152.
Benton, C. S., Miller, B. H., Skwerer, S., Suzuki, O., Schultz, L. E., Cameron, M. D., et al. (2011). Evaluating genetic markers and neurobiochemical analytes for fluoxetine response using a panel of mouse inbred strains. Psychopharmacology (Berl.) 221, 297–315.
Bierhaus, A., Fleming, T., Stoyanov, S., Leffler, A., Babes, A., Neacsu, C., et al. (2012). Methylglyoxal modification of Na(v)1.8 facilitates nociceptive neuron firing and causes hyperalgesia in diabetic neuropathy. Nat. Med. 18, 926–933.
Bollan, K., Robertson, L. A., Tang, H., and Connolly, C. N. (2003). Multiple assembly signals in gamma-aminobutyric acid (type A) receptor subunits combine to drive receptor construction and composition. Biochem. Soc. Trans. 31, 875–879.
Boso, M., Emanuele, E., Minoretti, P., Arra, M., Politi, P., Ucelli di Nemi, S., et al. (2006). Alterations of circulating endogenous secretory RAGE and S100A9 levels indicating dysfunction of the AGE-RAGE axis in autism. Neurosci. Lett. 410, 169–173.
Brownlee, M. (2001). Biochemistry and molecular cell biology of diabetic complications. Nature 414, 813–820.
Bunck, M., Czibere, L., Horvath, C., Graf, C., Frank, E., Kessler, M. S., et al. (2009). A hypomorphic vasopressin allele prevents anxiety-related behavior. PLoS ONE 4:e5129. doi: 10.1371/journal.pone.0005129
Carter, L. P., Koek, W., and France, C. P. (2009). Behavioral analyses of GHB: receptor mechanisms. Pharmacol. Ther. 121, 100–114.
Cerda, M., Sagdeo, A., Johnson, J., and Galea, S. (2010). Genetic and environmental influences on psychiatric comorbidity: a systematic review. J. Affect. Disord. 126, 14–38.
Cooper, R. A. (1984). Metabolism of methylglyoxal in microorganisms. Annu. Rev. Microbiol. 38, 49–68.
Crestani, F., Lorez, M., Baer, K., Essrich, C., Benke, D., Laurent, J. P., et al. (1999). Decreased GABAA-receptor clustering results in enhanced anxiety and a bias for threat cues. Nat. Neurosci. 2, 833–839.
Czibere, L., Baur, L. A., Wittmann, A., Gemmeke, K., Steiner, A., Weber, P., et al. (2011). Profiling trait anxiety: transcriptome analysis reveals cathepsin B (ctsb) as a novel candidate gene for emotionality in mice. PLoS ONE 6:e23604. doi: 10.1371/journal.pone.0023604
Davies, M. G., Chambers, P. L., and Rowan, M. J. (1986). Effects of methylglyoxal on central and peripheral cholinergic responses. Arch. Toxicol. Suppl. 9, 46–50.
Di Cristo, G. (2007). Development of cortical GABAergic circuits and its implications for neurodevelopmental disorders. Clin. Genet. 72, 1–8.
Di Loreto, S., Caracciolo, V., Colafarina, S., Sebastiani, P., Gasbarri, A., and Amicarelli, F. (2004). Methylglyoxal induces oxidative stress-dependent cell injury and up-regulation of interleukin-1beta and nerve growth factor in cultured hippocampal neuronal cells. Brain Res. 1006, 157–167.
Di Loreto, S., Zimmitti, V., Sebastiani, P., Cervelli, C., Falone, S., and Amicarelli, F. (2008). Methylglyoxal causes strong weakening of detoxifying capacity and apoptotic cell death in rat hippocampal neurons. Int. J. Biochem. Cell Biol. 40, 245–257.
Distler, M. G., Plant, L. D., Sokoloff, G., Hawk, A. J., Aneas, I., Wuenschell, G. E., et al. (2012). Glyoxalase 1 increases anxiety by reducing GABAA receptor agonist methylglyoxal. J. Clin. Invest. 122, 2306–2315.
Ditzen, C., Varadarajulu, J., Czibere, L., Gonik, M., Targosz, B. S., Hambsch, B., et al. (2010). Proteomic-based genotyping in a mouse model of trait anxiety exposes disease-relevant pathways. Mol. Psychiatry 15, 702–711.
Duran-Jimenez, B., Dobler, D., Moffatt, S., Rabbani, N., Streuli, C. H., Thornalley, P. J., et al. (2009). Advanced glycation end products in extracellular matrix proteins contribute to the failure of sensory nerve regeneration in diabetes. Diabetes 58, 2893–2903.
Earnheart, J. C., Schweizer, C., Crestani, F., Iwasato, T., Itohara, S., Mohler, H., et al. (2007). GABAergic control of adult hippocampal neurogenesis in relation to behavior indicative of trait anxiety and depression states. J. Neurosci. 27, 3845–3854.
Edwards, J. L., Vincent, A. M., Cheng, H. T., and Feldman, E. L. (2008). Diabetic neuropathy: mechanisms to management. Pharmacol. Ther. 120, 1–34.
Erhardt, A., Czibere, L., Roeske, D., Lucae, S., Unschuld, P. G., Ripke, S., et al. (2011). TMEM132D, a new candidate for anxiety phenotypes: evidence from human and mouse studies. Mol. Psychiatry 16, 647–663.
Eser, D., Uhr, M., Leicht, G., Asmus, M., Langer, A., Schule, C., et al. (2011). Glyoxalase-I mRNA expression and CCK-4 induced panic attacks. J. Psychiatr. Res. 45, 60–63.
Farrant, M., and Nusser, Z. (2005). Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat. Rev. Neurosci. 6, 215–229.
Filiou, M. D., Zhang, Y., Teplytska, L., Reckow, S., Gormanns, P., Maccarrone, G., et al. (2011). Proteomics and metabolomics analysis of a trait anxiety mouse model reveals divergent mitochondrial pathways. Biol. Psychiatry 70, 1074–1082.
Fleming, T. H., Humpert, P. M., Nawroth, P. P., and Bierhaus, A. (2010). Reactive metabolites and AGE/RAGE-mediated cellular dysfunction affect the aging process - a mini-review. Gerontology 57, 435–443.
Fujimoto, M., Uchida, S., Watanuki, T., Wakabayashi, Y., Otsuki, K., Matsubara, T., et al. (2008). Reduced expression of glyoxalase-1 mRNA in mood disorder patients. Neurosci. Lett. 438, 196–199.
Gerner, R. H., and Hare, T. A. (1981). CSF GABA in normal subjects and patients with depression, schizophrenia, mania, and anorexia nervosa. Am. J. Psychiatry 138, 1098–1101.
Gisselmann, G., Plonka, J., Pusch, H., and Hatt, H. (2004). Drosophila melanogaster GRD and LCCH3 subunits form heteromultimeric GABA-gated cation channels. Br. J. Pharmacol. 142, 409–413.
Guthrie, G. D., and Nicholson-Guthrie, C. S. (1989). gamma-Aminobutyric acid uptake by a bacterial system with neurotransmitter binding characteristics. Proc. Natl. Acad. Sci. U.S.A. 86, 7378–7381.
Hablitz, J. J., Mathew, S. S., and Pozzo-Miller, L. (2009). GABA vesicles at synapses: are there 2 distinct pools? Neuroscientist 15, 218–224.
Hamann, S. (2005). Sex differences in the responses of the human amygdala. Neuroscientist 11, 288–293.
Hambsch, B. (2011). Altered glyoxalase 1 expression in psychiatric disorders: cause or consequence? Semin. Cell Dev. Biol. 22, 302–308.
Hambsch, B., Chen, B. G., Brenndorfer, J., Meyer, M., Avrabos, C., Maccarrone, G., et al. (2010). Methylglyoxal-mediated anxiolysis involves increased protein modification and elevated expression of glyoxalase 1 in the brain. J. Neurochem. 113, 1240–1251.
Hasler, G., and Northoff, G. (2011). Discovering imaging endophenotypes for major depression. Mol. Psychiatry 16, 604–619.
Henshall, D. C., and Murphy, B. M. (2008). Modulators of neuronal cell death in epilepsy. Curr. Opin. Pharmacol. 8, 75–81.
Hovatta, I., Tennant, R. S., Helton, R., Marr, R. A., Singer, O., Redwine, J. M., et al. (2005). Glyoxalase 1 and glutathione reductase 1 regulate anxiety in mice. Nature 438, 662–666.
Inoue, Y., Maeta, K., and Nomura, W. (2011). Glyoxalase system in yeasts: structure, function, and physiology. Semin. Cell Dev. Biol. 22, 278–284.
Jack, M. M., Ryals, J. M., and Wright, D. E. (2011). Characterisation of glyoxalase I in a streptozocin-induced mouse model of diabetes with painful and insensate neuropathy. Diabetologia 54, 2174–2182.
Jack, M. M., Ryals, J. M., and Wright, D. E. (2012). Protection from diabetes-induced peripheral sensory neuropathy–a role for elevated glyoxalase I? Exp. Neurol. 234, 62–69.
Jack, M., and Wright, D. (2012). Role of advanced glycation endproducts and glyoxalase I in diabetic peripheral sensory neuropathy. Transl. Res. 159, 355–365.
Jacquier, C., Bonthoux, F., Baciu, M., and Ruffieux, B. (2012). Improving the effectiveness of nutritional information policies: assessment of unconscious pleasure mechanisms involved in food-choice decisions. Nutr. Rev. 70, 118–131.
Junaid, M. A., Kowal, D., Barua, M., Pullarkat, P. S., Sklower Brooks, S., and Pullarkat, R. K. (2004). Proteomic studies identified a single nucleotide polymorphism in glyoxalase I as autism susceptibility factor. Am. J. Med. Genet. A 131, 11–17.
Kalueff, A. V., and Nutt, D. J. (2007). Role of GABA in anxiety and depression. Depress. Anxiety 24, 495–517.
Kemlink, D., Polo, O., Frauscher, B., Gschliesser, V., Hogl, B., Poewe, W., et al. (2009). Replication of restless legs syndrome loci in three European populations. J. Med. Genet. 46, 315–318.
Kent, J. M., Mathew, S. J., and Gorman, J. M. (2002). Molecular targets in the treatment of anxiety. Biol. Psychiatry 52, 1008–1030.
Kim, N. S., Sekine, S., Kiuchi, N., and Kato, S. (1995). cDNA cloning and characterization of human glyoxalase I isoforms from HT-1080 cells. J. Biochem. 117, 359–361.
Kirik, D., and Bjorklund, A. (2003). Modeling CNS neurodegeneration by overexpression of disease-causing proteins using viral vectors. Trends Neurosci. 26, 386–392.
Kompf, J., Bissbort, S., Gussmann, S., and Ritter, H. (1975). Polymorphism of red cell glyoxalase I (EI: 4.4.1.5); a new genetic marker in man. Investigation of 169 mother-child combinations. Humangenetik 27, 141–143.
Kromer, S. A., Kessler, M. S., Milfay, D., Birg, I. N., Bunck, M., Czibere, L., et al. (2005). Identification of glyoxalase-I as a protein marker in a mouse model of extremes in trait anxiety. J. Neurosci. 25, 4375–4384.
Kuhla, B., Luth, H. J., Haferburg, D., Weick, M., Reichenbach, A., Arendt, T., et al. (2006). Pathological effects of glyoxalase I inhibition in SH-SY5Y neuroblastoma cells. J. Neurosci. Res. 83, 1591–1600.
Kumar, S., and Punekar, N. (1997). The metabolism of 4-aminobutyrate (GABA) in fungi. Mycol. Res. 101, 403–409.
Kumar, S., Porcu, P., Werner, D. F., Matthews, D. B., Diaz-Granados, J. L., Helfand, R. S., et al. (2009). The role of GABA(A) receptors in the acute and chronic effects of ethanol: a decade of progress. Psychopharmacology (Berl.) 205, 529–564.
Li, G., Chang, M., Jiang, H., Xie, H., Dong, Z., and Hu, L. (2010). Proteomics analysis of methylglyoxal-induced neurotoxic effects in SH-SY5Y cells. Cell Biochem. Funct. 29, 30–35.
Loh, K. P., Huang, S. H., De Silva, R., Tan, B. K., and Zhu, Y. Z. (2006). Oxidative stress: apoptosis in neuronal injury. Curr. Alzheimer Res. 3, 327–337.
Loos, M., van der Sluis, S., Bochdanovits, Z., van Zutphen, I. J., Pattij, T., Stiedl, O., et al. (2009). Activity and impulsive action are controlled by different genetic and environmental factors. Genes Brain Behav. 8, 817–828.
Lo, T. W., Westwood, M. E., McLellan, A. C., Selwood, T., and Thornalley, P. J. (1994). Binding and modification of proteins by methylglyoxal under physiological conditions. A kinetic and mechanistic study with N alpha-acetylarginine, N alpha-acetylcysteine, and N alpha-acetyllysine, and bovine serum albumin. J. Biol. Chem. 269, 32299–32305.
Luscher, B., Shen, Q., and Sahir, N. (2011). The GABAergic deficit hypothesis of major depressive disorder. Mol. Psychiatry 16, 383–406.
Maguire, J. L., Stell, B. M., Rafizadeh, M., and Mody, I. (2005). Ovarian cycle-linked changes in GABA(A) receptors mediating tonic inhibition alter seizure susceptibility and anxiety. Nat. Neurosci. 8, 797–804.
Mannervik, B. (2008). Molecular enzymology of the glyoxalase system. Drug Metabol. Drug Interact. 23, 13–27.
Masterjohn, C., Mah, E., Guo, Y., Koo, S. I., and Bruno, R. S. (2011). Gamma-Tocopherol abolishes postprandial increases in plasma methylglyoxal following an oral dose of glucose in healthy, college-aged men. J. Nutr. Biochem. 23, 292–298.
McLean, C. P., and Anderson, E. R. (2009). Brave men and timid women? A review of the gender differences in fear and anxiety. Clin. Psychol. Rev. 29, 496–505.
McLellan, A. C., and Thornalley, P. (1992). Synthesis and chromatography of 1, 2-diamino-4, 5dimethoxybenzene, 6, 7-dimethoxy-2-methylquinoxaline and 6, 7-dimethoxy-2, 3 dimethylquinoxaline for use in a liquid chromatographic fluorimetric assay of methylglyoxal. Anal. Chim. Acta 263, 137–142.
Mehta, D., Menke, A., and Binder, E. B. (2010). Gene expression studies in major depression. Curr. Psychiatry Rep. 12, 135–144.
Morcos, M., Du, X., Pfisterer, F., Hutter, H., Sayed, A. A., Thornalley, P., et al. (2008). Glyoxalase-1 prevents mitochondrial protein modification and enhances lifespan in Caenor-habditis elegans. Aging Cell 7, 260–269.
Naegele, J. R. (2007). Neuroprotective strategies to avert seizure-induced neurodegeneration in epilepsy. Epilepsia 48, 107–117.
Nillni, Y. I., Toufexis, D. J., and Rohan, K. J. (2011). Anxiety sensitivity, the menstrual cycle, and panic disorder: a putative neuroendocrine and psychological interaction. Clin. Psychol. Rev. 31, 1183–1191.
Palanza, P. (2001). Animal models of anxiety and depression: how are females different? Neurosci. Biobehav. Rev. 25, 219–233.
Petty, F., and Schlesser, M. A. (1981). Plasma GABA in affective illness. A preliminary investigation. J. Affect. Disord 3, 339–343.
Petty, F., and Sherman, A. D. (1984). Plasma GABA levels in psychiatric illness. J. Affect. Disord. 6, 131–138.
Politi, P., Minoretti, P., Falcone, C., Martinelli, V., and Emanuele, E. (2006). Association analysis of the functional Ala111Glu polymorphism of the glyoxalase I gene in panic disorder. Neurosci. Lett. 396, 163–166.
Pourmotabbed, T., and Creighton, D. J. (1986). Substrate specificity of bovine liver formaldehyde dehydrogenase. J. Biol. Chem. 261, 14240–14244.
Rabbani, N., and Thornalley, P. J. (2012). Methylglyoxal, glyoxalase 1 and the dicarbonyl proteome. Amino Acids 42, 1133–1142.
Rauh, J. J., Lummis, S. C., and Sattelle, D. B. (1990). Pharmacological and biochemical properties of insect GABA receptors. Trends Pharmacol. Sci. 11, 325–329.
Redon, R., Ishikawa, S., Fitch, K. R., Feuk, L., Perry, G. H., Andrews, T. D., et al. (2006). Global variation in copy number in the human genome. Nature 444, 444–454.
Rehnstrom, K., Ylisaukko-Oja, T., Vanhala, R., von Wendt, L., Peltonen, L., and Hovatta, I. (2008). No association between common variants in glyoxalase 1 and autism spectrum disorders. Am. J. Med. Genet. B Neuropsychiatr. Genet. 147B, 124–127.
Reiner-Benaim, A., Yekutieli, D., Letwin, N. E., Elmer, G. I., Lee, N. H., Kafkafi, N., et al. (2007). Associating quantitative behavioral traits with gene expression in the brain: searching for diamonds in the hay. Bioinformatics 23, 2239–2246.
Rubenstein, J. L. (2011). Annual research review: development of the cerebral cortex: implications for neurodevelopmental disorders. J. Child Psychol. Psychiatry. 52, 339–355.
Rudolph, U., and Knoflach, F. (2011). Beyond classical benzodiazepines: novel therapeutic potential of GABAA receptor subtypes. Nat. Rev. Drug Discov. 10, 685–697.
Rudolph, U., and Mohler, H. (2004). Analysis of GABAA receptor function and dissection of the pharmacology of benzodiazepines and general anesthetics through mouse genetics. Annu. Rev. Pharmacol. Toxicol. 44, 475–498.
Sacco, R., Papaleo, V., Hager, J., Rousseau, F., Moessner, R., Militerni, R., et al. (2007). Case-control and family-based association studies of candidate genes in autistic disorder and its endophenotypes: TPH2 and GLO1. BMC Med. Genet. 8:11. doi: 10.1186/1471-2350-8-11
Salim, S., Asghar, M., Chugh, G., Taneja, M., Xia, Z., and Saha, K. (2010a). Oxidative stress: a potential recipe for anxiety, hypertension and insulin resistance. Brain Res. 1359, 178–185.
Salim, S., Sarraj, N., Taneja, M., Saha, K., Tejada-Simon, M. V., and Chugh, G. (2010b). Moderate treadmill exercise prevents oxidative stress-induced anxiety-like behavior in rats. Behav. Brain Res. 208, 545–552.
Salim, S., Asghar, M., Taneja, M., Hovatta, I., Chugh, G., Vollert, C., et al. (2011). Potential contribution of oxidative stress and inflammation to anxiety and hypertension. Brain Res. 1404, 63–71.
Schleicher, E., and Friess, U. (2007). Oxidative stress, AGE, and atherosclerosis. Kidney Int. Suppl. 106, S17–S26.
Seabrook, G. R., Easter, A., Dawson, G. R., and Bowery, B. J. (1997). Modulation of long-term potentiation in CA1 region of mouse hippocampal brain slices by GABAA receptor benzodiazepine site ligands. Neuropharmacology 36, 823–830.
Shen, Q., Lal, R., Luellen, B. A., Earnheart, J. C., Andrews, A. M., and Luscher, B. (2010). gamma-Aminobutyric acid-type A receptor deficits cause hypothalamic-pituitary-adrenal axis hyperactivity and antidepressant drug sensitivity reminiscent of melancholic forms of depression. Biol. Psychiatry 68, 512–520.
Shinohara, M., Thornalley, P. J., Giardino, I., Beisswenger, P., Thorpe, S. R., Onorato, J., et al. (1998). Overexpression of glyoxalase-I in bovine endothelial cells inhibits intracellular advanced glycation endproduct formation and prevents hyperglycemia-induced increases in macromolecular endocytosis. J. Clin. Invest. 101, 1142–1147.
Shulman, R. G., Rothman, D. L., Behar, K. L., and Hyder, F. (2004). Energetic basis of brain activity: implications for neuroimaging. Trends Neurosci. 27, 489–495.
Silverman, J. L., Yang, M., Lord, C., and Crawley, J. N. (2010). Behavioural phenotyping assays for mouse models of autism. Nat. Rev. Neurosci. 11, 490–502.
Solomon, M. B., and Herman, J. P. (2009). Sex differences in psychopathology: of gonads, adrenals and mental illness. Physiol. Behav. 97, 250–258.
Stefansson, H., Rye, D. B., Hicks, A., Petursson, H., Ingason, A., Thorgeirsson, T. E., et al. (2007). A genetic risk factor for periodic limb movements in sleep. N. Engl. J. Med. 357, 639–647.
Tasan, R. O., Bukovac, A., Peterschmitt, Y. N., Sartori, S. B., Landgraf, R., Singewald, N., et al. (2011). Altered GABA transmission in a mouse model of increased trait anxiety. Neuroscience 183, 71–80.
Thornalley, P. J. (1990). The glyoxalase system: new developments towards functional characterization of a metabolic pathway fundamental to biological life. Biochem. J. 269, 1–11.
Thornalley, P. J. (1993). The glyoxalase system in health and disease. Mol. Aspects Med. 14, 287–371.
Thornalley, P. J. (2003a). Protecting the genome: defence against nucleotide glycation and emerging role of glyoxalase I overexpression in multidrug resistance in cancer chemotherapy. Biochem. Soc. Trans. 31, 1372–1377.
Thornalley, P. J. (2003b). Glyoxalase I–structure, function and a critical role in the enzymatic defence against glycation. Biochem. Soc. Trans. 31, 1343–1348.
Thornalley, P. J. (2006). Unease on the role of glyoxalase 1 in high-anxiety-related behaviour. Trends Mol. Med. 12, 195–199.
Thornalley, P. J., and Rabbani, N. (2011). Glyoxalase in tumourigenesis and multidrug resistance. Semin. Cell Dev. Biol. 22, 318–325.
Toyosima, M., Maekawa, M., Toyota, T., Iwayama, Y., Arai, M., Ichikawa, T., et al. (2011). Schizophrenia with the 22q11.2 deletion and additional genetic defects: case history. Br. J. Psychiatry 199, 245–246.
Vinik, A. I., Park, T. S., Stansberry, K. B., and Pittenger, G. L. (2000). Diabetic neuropathies. Diabetologia 43, 957–973.
Vithlani, M., Terunuma, M., and Moss, S. J. (2010). The dynamic modulation of GABA(A) receptor trafficking and its role in regulating the plasticity of inhibitory synapses. Physiol. Rev. 91, 1009–1022.
Vollert, C., Zagaar, M., Hovatta, I., Taneja, M., Vu, A., Dao, A., et al. (2011). Exercise prevents sleep deprivation-associated anxiety-like behavior in rats: potential role of oxidative stress mechanisms. Behav. Brain Res. 224, 233–240.
Wang, X., Jia, X., Chang, T., Desai, K., and Wu, L. (2008). Attenuation of hypertension development by scavenging methylglyoxal in fructose-treated rats. J. Hypertens. 26, 765–772.
Williams, R. T., Lim, J. E., Harr, B., Wing, C., Walters, R., Distler, M. G., et al. (2009). A common and unstable copy number variant is associated with differences in Glo1 expression and anxiety-like behavior. PLoS ONE 4:e4649. doi: 10.1371/journal.pone.0004649
Winkelmann, J., Czamara, D., Schormair, B., Knauf, F., Schulte, E. C., Trenkwalder, C., et al. (2011). Genome-wide association study identifies novel restless legs syndrome susceptibility loci on 2p14 and 16q12.1. PLoS Genet. 7:e1002171. doi: 10.1371/journal.pgen.1002171
Winkelmann, J., Schormair, B., Lichtner, P., Ripke, S., Xiong, L., Jalilzadeh, S., et al. (2007). Genome-wide association study of restless legs syndrome identifies common variants in three genomic regions. Nat. Genet. 39, 1000–1006.
Wu, Y. Y., Chien, W. H., Huang, Y. S., Gau, S. S., and Chen, C. H. (2008). Lack of evidence to support the glyoxalase 1 gene (GLO1) as a risk gene of autism in Han Chinese patients from Taiwan. Prog. Neuropsychopharmacol. Biol. Psychiatry 32, 1740–1744.
Yadav, S. K., Singla-Pareek, S. L., and Sopory, S. K. (2008). An overview on the role of methylglyoxal and glyoxalases in plants. Drug Metabol. Drug Interact. 23, 51–68.
Young, J. W., Zhou, X., and Geyer, M. A. (2010). Animal models of schizophrenia. Curr. Top. Behav. Neurosci. 4, 391–433.
Keywords: anxiety-like behavior, pain, depression, restless-leg syndrome, GABA, CNV, advanced glycation end-products, AGE
Citation: Distler MG and Palmer AA (2012) Role of Glyoxalase 1 (Glo1) and methylglyoxal (MG) in behavior: recent advances and mechanistic insights. Front. Gene. 3:250. doi: 10.3389/fgene.2012.00250
Received: 22 September 2012; Accepted: 26 October 2012;
Published online: 19 November 2012.
Edited by:
Maria Grigoroiu-Serbanescu, Medical University Bucharest, RomaniaReviewed by:
Sabine Spijker, Research Institute of Neurosciences, NetherlandsCopyright © 2012 Distler and Palmer. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Abraham A. Palmer, Department of Human Genetics, University of Chicago, 920 E. 58th Street, CLSC 507D, Chicago, IL 60637, USA. e-mail:YWFwQHVjaGljYWdvLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.