 Rongrong Wei1
Rongrong Wei1 Fan Yang1
Fan Yang1 Thomas J. Urban2 Lang Li3 Naga Chalasani4
Thomas J. Urban2 Lang Li3 Naga Chalasani4 David A. Flockhart5
David A. Flockhart5 Wanqing Liu1,4*
Wanqing Liu1,4*- 1Department of Medicinal Chemistry and Molecular Pharmacology, College of Pharmacy, Purdue University, West Lafayette, IN, USA
- 2Center for Human Genome Variation, School of Medicine, Duke University, Durham, NC, USA
- 3Division of Medical and Molecular Genetics, School of Medicine, Indiana University, Indianapolis, IN, USA
- 4Division of Gastroenterology and Hepatology, School of Medicine, Indiana University, Indianapolis, IN, USA
- 5Division of Clinical Pharmacology, Department of Medicine, Indiana University School of Medicine, Indianapolis, IN, USA
Genetic variation in the expression of human xenobiotic metabolism enzymes and transporters (XMETs) leads to inter-individual variability in metabolism of therapeutic agents as well as differed susceptibility to various diseases. Recent expression quantitative traits loci (eQTL) mapping in a few human cells/tissues have identified a number of single nucleotide polymorphisms (SNPs) significantly associated with mRNA expression of many XMET genes. These eQTLs are therefore important candidate markers for pharmacogenetic studies. However, questions remain about whether these SNPs are causative and in what mechanism these SNPs may function. Given the important role of microRNAs (miRs) in gene transcription regulation, we hypothesize that those eQTLs or their proxies in strong linkage disequilibrium (LD) altering miR targeting are likely causative SNPs affecting gene expression. The aim of this study is to identify eQTLs potentially regulating major XMETs via interference with miR targeting. To this end, we performed a genome-wide screening for eQTLs for 409 genes encoding major drug metabolism enzymes, transporters and transcription factors, in publically available eQTL datasets generated from the HapMap lymphoblastoid cell lines and human liver and brain tissue. As a result, 308 eQTLs significantly (p < 10−5) associated with mRNA expression of 101 genes were identified. We further identified 7,869 SNPs in strong LD (r2 ≥ 0.8) with these eQTLs using the 1,000 Genome SNP data. Among these 8,177 SNPs, 27 are located in the 3′-UTR of 14 genes. Using two algorithms predicting miR-SNP interaction, we found that almost all these SNPs (26 out of 27) were predicted to create, abolish, or change the target site for miRs in both algorithms. Many of these miRs were also expressed in the same tissue that the eQTL were identified. Our study provides a strong rationale for continued investigation for the functions of these eQTLs in pharmacogenetic settings.
Introduction
Xenobiotic metabolizing enzymes and transporters (XMETs) are involved in biotransformation and detoxification of carcinogens, environmental toxins, and therapeutic drugs (Carlsten et al., 2008; Korkina et al., 2009). In humans, the process of biotransformation and detoxification of xenobiotics by XMETs can be divided into three phases: modification (phase I) primarily by enzymes of the cytochromes P450 superfamily; conjugation (phase II), e.g., glucuronidation by UDP-glucuronosyl transferase; and excretion (phase III) mainly by membrane transporters. XMETs are expressed in almost all tissue types, centrally and locally protecting the entire body against the damages caused by various natural and synthetic compounds. XMETs are highly expressed in digestive tract and especially in the liver, the most important organ for central metabolism (Conde-Vancells et al., 2010). Variations in the expression and activity of these XMETs lead to significant inter-individual difference in the disposition of exogenous chemicals including absorption, distribution, metabolism, and excretion (ADME) of pharmaceutical drugs. On the other hand, many XMETs are also found to be very abundant in non-digestive tract tissues/cells, e.g., brain, lung, bladder, and blood (Pavek and Dvorak, 2008). These XMETs could affect the local response to certain drugs at the site of action. Meanwhile, due to the crucial role of XMETs in detoxification of carcinogens and toxins, genetic variation in XMETs function in specific tissues/organs is also an important mechanism underlying genetic susceptibility to certain diseases, e.g., those XMETs expressed in lung and bladder may modify cancer risk. Recent genome-wide association studies have identified polymorphisms at the UGT1A locus strongly associated with urinary bladder cancer risk (Selinski et al., 2012). XMETs are sensitively regulated by various nuclear receptors (NRs) and transcription factors (TFs). These trans-acting regulators play a pivotal role in mediating cellular response to exposure to xenobiotics by modulating the transcription of XMETs, thus significantly contributing to the variability in the function of XMETs (Bourgine et al., 2012).
Identifying the DNA polymorphisms leading to the variations in XMET function is a major area of interest in pharmacogenetic and genomic research. To date, numerous studies focused on individual XMET genes have discovered a large number of sequence variations, many of which alter protein coding sequence and consequently affecting the activity of XMETs (Adjei et al., 2003; Hildebrandt et al., 2004; Ji et al., 2005; Moyer et al., 2007; Mrozikiewicz et al., 2011). Meanwhile, even more variants were suggested to quantitatively modulate gene transcription (Pavek and Dvorak, 2008). Recently, genome-wide mapping for gene expression quantitative trait loci (eQTLs) in a few human tissues/cells offered unprecedented opportunities to identify the most influential single nucleotide polymorphisms (SNPs) determining gene expression level of XMETs (Gamazon et al., 2010). However, unlike the variants located in the protein coding sequences for which the causality for altered enzyme activity can be more easily understood, how eQTLs affect gene transcription is largely unknown. Understanding the underlying mechanisms will lead to identification of novel causative DNA variants for XMET function as well as reliable pharmacogenetic markers.
MicroRNAs (miRs) are single stranded, about 22-nucleotides (nt) long, evolutionarily conserved, and function as important posttranscriptional regulators of mRNA expression by binding to the 3′-UTR of target mRNAs (Ambros, 2004; Bartel, 2004). MiRs are involved in various developmental and physiological processes by negatively regulating gene expression (Zhang et al., 2007). Over 30% of all protein-coding genes were estimated to be regulated by miRs (Brennecke et al., 2005; Krek et al., 2005; Lewis et al., 2005; Lim et al., 2005). Due to the conservation of the miR target site, SNPs located in 3′-UTR sequences may abolish or create a miR target, thus significantly affecting the mRNA expression (Saunders et al., 2007). Previous studies have suggested that many XMETs are regulated by miRs (Tsuchiya et al., 2006; Takagi et al., 2010; Patron et al., 2012). Several studies also demonstrated that SNPs in XMET gene 3′-UTRs led to different levels of enzyme activity (Saunders et al., 2007; Chin et al., 2008). Hence, we hypothesized that it may be an important mechanism that common SNPs or their linkage disequilibrium (LD) proxies located in the XMET gene 3′-UTR sequences alter mRNA expression via interference with miR targeting. In order to identify these candidate SNPs that may significantly modulate XMET expression, in this study we used multiple published human eQTL datasets to perform an in silico screening for SNPs that highly correlated with mRNA level of 409 major XMET genes. The significant SNPs and/or their LD proxies located in the gene 3′-UTRs were selected to predict a potential interference with miRs. We found that 27 SNPs located in the 3′-UTR of 14 XMET genes are likely associated with gene expression via altering miR binding.
Materials and Methods
Selection of eQTLs
The general strategy for the data analysis was presented in Figure 1. We used the published eQTLs datasets generated from the HapMap lymphoblastoid cell lines (LCLs; Montgomery et al., 2010), human liver (Schadt et al., 2008), and human brain (Gibbs et al., 2010). Although additional eQTL datasets in human LCLs are also available, we chose to use the one by Montgomery et al. (2010) which utilized high-throughput sequencing for the quantification of gene expression, as this technology has been suggested to produce more accurate gene expression data. To our knowledge, all datasets were collected from tissue/cells derived from individuals of Caucasian in origin. We used the online tool1 to search statistically significant eQTLs. As our study was focused on cis-acting eQTLs, we used a cut-off of p = 10−5 for significance, considering the window for genomic region (500 kb) of each gene and the potential number of SNPs (1 in every 100–1,000 bp).
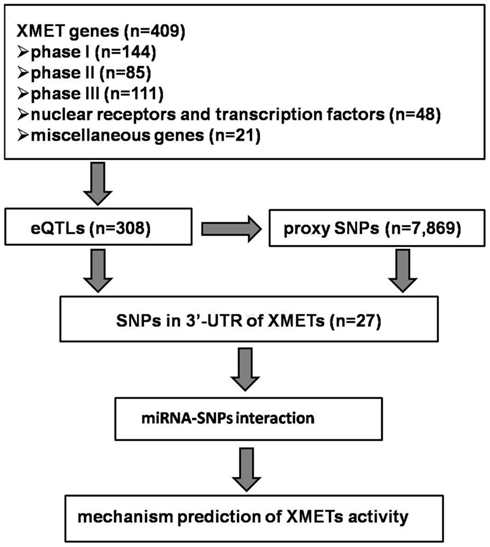
Figure 1. Schematic of the search for miRNAs and the associated SNPs from XMET genes.
Search for SNPs in LD with eQTLs
To search SNPs in LD with significant eQTLs, we used the SNAP2 program to screen the 1,000 Genome SNP data within 500 kb range of the eQTLs of interest in the CEU population with a LD level cut-off of R2 = 0.8. Annotation for the location of eQTLs and their proxies relative to the gene structure was also collected with the program. Only SNPs and/or their proxies located within the 3′-UTR of the studied genes of interest were retained for further analyses.
Prediction of SNP-miR Interaction
In order to predict the potential SNP-miR interaction, two programs, MicroSNiPer3 and PolymiRTS4 were used. The major difference between the two programs is the algorithm used to predict the target site of miRs. The PolymiRTS program used the TargetScan5; Lewis et al., 2005; Friedman et al., 2009) algorithm (Bao et al., 2007). In contrast, the MicroSNiPer program used the FASTA (Pearson and Lipman, 1988) alignment program to determine if a change in a nucleotide in 3′-UTR sequence would change the miR binding capability, based on the requirement of perfect Watson–Crick match to the seed 2–7 nt of miRs (Lewis et al., 2005). To be conservative, we used 7-mers match as the cut-off value for a positive prediction.
Results
Genome-Wide eQTL Analysis of XMETs
Expression quantitative traits loci were screened for all 409 major XMET genes, including 144 phase I, 85 phase II and 111 phase III genes, 48 NRs, and transcription factor genes as well as another 21 genes related to drug ADME (Table A1 in Appendix). As a result, a total of 308 significant (p < 10−5) eQTLs were identified from 101 XMET genes. These include nine in LCL, 83 in liver, and 221 in brain tissues. Five SNPs were found as eQTLs shared in two tissue types: rs1023252 in both LCL and brain tissues, rs11101992, rs156697, rs2071474, and rs241440 in both liver and brain tissues (Figure 2). Among the total of 308 eQTLs, 20 SNPs were found to be located in the 3′-UTR region; 3 SNPs were in the 5′-UTRs; 171 SNPs were intronic; 8 and 6 SNPs were synonymous and non-synonymous coding variants, respectively; and 12 and 15 SNPs were located in the upstream and downstream flanking region of the genes, respectively. The remaining 73 SNPs were located in intergenic regions.
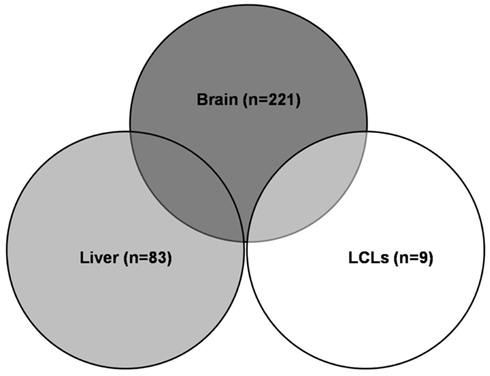
Figure 2. Significant eQTLs in different tissues. A total of 308 significant eQTLs were identified, including 9 eQTLs in LCL, 83 in liver, and 221 in brain tissues. Five eQTLs were shared in two tissue types.
eQTLs and Their LD Proxies
We chose to screen the 1,000 Genome SNP dataset as this would produce the most comprehensive coverage for the SNPs that may be in LD with a given eQTL. A total of 7,869 SNPs with significant LD with 260 eQTLs were identified. Combined with the remaining 48 eQTLs which had no reliable proxies in the 1,000 Genome dataset, a total of 8,177 SNPs (308 eQTLs and 7,869 proxy SNPs) were included in the subsequent analyses.
Prediction of miR-SNPs Interaction
Of the 112 eQTLs and proxies located in the 3′-UTR sequences, 27 SNPs were found in the 3′-UTR of 14 genes of interest. The remaining SNPs were located in nearby genes thus were excluded from the subsequent analysis. These SNPs were all common SNPs with their minor allele frequency (MAF) ≥0.067. Among the 27 SNPs, 12 were found in liver, and 15 were identified in brain tissue. More detailed information for these SNPs was listed in Table A2 in Appendix.
We focused our study on the association between miRs and these 27 SNPs in the 14 genes. After screened with the two algorithms, MicroSNiPer (Barenboim et al., 2010) and PolymiRTs (Gong et al., 2012), all the 27 SNPs apart from rs11807 (which is not predicted to be in a target site in PolymiRTs database) were found to potentially create, abolish, or alter the target site for miRs in both algorithms. Notably, 34 miRs were predicted by both algorithms to interact with 19 of these SNPs (Table A2 in Appendix). Of these 34 overlap miRs, except for rs2480256 of CYP2E1 which is not located in the seed sequence of hsa-miR-570-3p, all the remaining SNPs were found to be located in the seed sequence of miR targets.
To further validate the interaction between miRs and SNPs, we investigated whether the identified miRs were expressed in the same tissue as the identified eQTL. We used the GEO datasets (GSE21279 and GSE26545) to screen miR expression in liver and brain tissues, respectively (Hou et al., 2011; Hu et al., 2011). Since many predicted miRs were new and not probed by the published platforms, we thus only concentrate on the list of miRs probed in the platforms. Overall, over 74% (20 out of 27) of the identified miR-SNPs were found to have at least one predicted miR co-expressed with the gene of interest in the same tissue.
We further aimed to investigate whether these 27 SNPs are more likely to be targeted by miRs especially by the co-expressed miR in liver and brain tissues, compared to random-selected 3′-UTR SNPs with similar MAF. No statistical significance were found, possibly due to the limited power caused by the small number (n = 27) of SNPs involved (data not shown).
Discussion
Although a large number of DNA variants affecting the function of XMETs have been identified, and many of them have been well linked with clinical response to pharmacotherapy or disease susceptibility (Motsinger-Reif et al., 2010), genetic variations in the activity of most XMETs remain incompletely explained. Recent studies continue to discover novel functional variants in XMET genes (Ramsey et al., 2012). Meanwhile, genome-wide association studies have found a number of XMET SNPs without previously known function significantly associated with different phenotypes in humans (Teichert et al., 2009; Estrada et al., 2012). These studies consistently suggested that additional sequence variants with fundamental role in XMET function have not been identified. Recent eQTL mapping in human tissues provided an opportunity to discover functional XMET polymorphisms at the genome-wide level. However, questions remain whether the identified eQTLs are causal for the altered gene expression and via what mechanism. Our study provides a comprehensive evaluation for this question in major human XMET genes, and generated a list of candidate SNPs that may modulate XMET genes via interference with miR targeting in multiple human tissue types.
Single nucleotide polymorphisms located in the gene 3′-UTRs could have great impact on miR targeting. It has been demonstrated that the entire 3′-UTR sequence could play important roles in miR function in addition to miR target sites (Hu and Bruno, 2011). In particular, negative selection in humans is stronger on computationally predicted conserved miR binding sites than on other conserved sequence motifs in 3′-UTRs, and polymorphisms in predicted miR binding sites are highly likely to be deleterious (Chen and Rajewsky, 2006). Gong et al. (2012) mapped SNPs to the 3′-UTRs of all human protein coding genes. Their results showed that among the 225,759 SNPs identified in 3′-UTRs, over 25% of SNPs potentially abolished 90,784 original miR target sites, while another 25% created a similar number of putative miRNA target sites. Besides these in silico studies, a number of SNPs altering miR targeting have been experimentally demonstrated to be associated with multiple diseases as well as drug metabolism and environmental procarcinogen detoxification (Abelson et al., 2005; Tan et al., 2007; Yu et al., 2007; Yokoi and Nakajima, 2011). Although the seed sequences for miR binding are critical and highly conserved, recent studies have also suggested that 3′-UTR sequences outside of the seed sequences, e.g., flanking sequences may be equally important for miR targeting by controlling the accessibility of the miR or local RNA structure (Grimson et al., 2007). For example, a SNP (829C > T) located 14 bp downstream of a miR-24 binding site in the 3′-UTR of human dihydrofolate reductase gene (DHFR) was demonstrated to affect DHFR expression by interfering with miR-24 function, resulting in DHFR over expression and methotrexate resistance (Mishra et al., 2007). By using two algorithms predicting potential SNP-miR interaction, we suggested that 27 eQTLs or their proxies in high LD for 14XMET genes may function through interference with one or more miRs, with most of the SNPs located in the seed sequences. Meanwhile, the majority (20 out of 27) of the identified miR-SNPs were found to have predicted miR co-expressed with the gene of interest in the same tissue. Although no statistically significant enrichment of miR targeting for these SNPs, the strong trends observed here warrants further experimental validations.
Our findings may also provide useful information in addition to the previous observations on the function of these SNPs. Previous studies demonstrated that SNP rs2480256 in the CYP2E1 gene was significantly associated with systemic lupus erythematosus (Liao et al., 2011). Another study showed that cyclosporine A concentration in serum was significantly correlated with the genotype of the CYP3A5 rs15524 polymorphism (Onizuka et al., 2011). In addition, a GSTM3 haplotype including rs1537236 was significantly associated with a decreased growth for maximum mid-expiratory flow rate (MMEF) in a large population-based lung function study (Breton et al., 2009). SNP rs11807 in the 3′ region of GSTM5 was found to be associated with hypertension (Delles et al., 2008). Our results thus may help further elucidate the mechanism(s) by which the SNPs are involved in the susceptibility to these specific phenotypes.
In conclusion, our study summarized the potentially interacting SNP-miRs that may affect the expression of major XMET gene, which may ultimately facilitate to elucidate the mechanism how these genes are regulated as well as how they are involved in the genetic variations in drug metabolism and disease pathogenesis. Further investigations are necessary to corroborate the hypotheses generated in this study.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work is partly supported by the 2012 Ralph W. and Grace M. Showalter Research Trust Award (Wanqing Liu) and the start-up fund (to Wanqing Liu) from the Department of Medicinal Chemistry and Molecular Pharmacology, Purdue University.
Footnotes
References
Abelson, J. F., Kwan, K. Y., O’Roak, B. J., Baek, D. Y., Stillman, A. A., Morgan, T. M., et al. (2005). Sequence variants in SLITRK1 are associated with Tourette’s syndrome. Science 310, 317–320.
Adjei, A. A., Thomae, B. A., Prondzinski, J. L., Eckloff, B. W., Wieben, E. D., and Weinshilboum, R. M. (2003). Human estrogen sulfotransferase (SULT1E1) pharmacogenomics: gene resequencing and functional genomics. Br. J. Pharmacol. 139, 1373–1382.
Bao, L., Zhou, M., Wu, L., Lu, L., Goldowitz, D., Williams, R. W., et al. (2007). PolymiRTS Database: linking polymorphisms in microRNA target sites with complex traits. Nucleic Acids Res. 35, D51–D54.
Barenboim, M., Zoltick, B. J., Guo, Y., and Weinberger, D. R. (2010). MicroSNiPer: a web tool for prediction of SNP effects on putative microRNA targets. Hum. Mutat. 31, 1223–1232.
Bourgine, J., Billaut-Laden, I., Happillon, M., Lo-Guidice, J. M., Maunoury, V., Imbenotte, M., et al. (2012). Gene expression profiling of systems involved in the metabolism and the disposition of xenobiotics: comparison between human intestinal biopsy samples and colon cell lines. Drug Metab. Dispos. 40, 694–705.
Brennecke, J., Stark, A., Russell, R. B., and Cohen, S. M. (2005). Principles of microRNA-target recognition. PLoS Biol. 3, e85. doi:10.1371/journal.pbio.0030085
Breton, C. V., Vora, H., Salam, M. T., Islam, T., Wenten, M., Gauderman, W. J., et al. (2009). Variation in the GST mu locus and tobacco smoke exposure as determinants of childhood lung function. Am. J. Respir. Crit. Care Med. 179, 601–607.
Carlsten, C., Sagoo, G. S., Frodsham, A. J., Burke, W., and Higgins, J. P. (2008). Glutathione S-transferase M1 (GSTM1) polymorphisms and lung cancer: a literature-based systematic HuGE review and meta-analysis. Am. J. Epidemiol. 167, 759–774.
Chen, K., and Rajewsky, N. (2006). Natural selection on human microRNA binding sites inferred from SNP data. Nat. Genet. 38, 1452–1456.
Chin, L. J., Ratner, E., Leng, S., Zhai, R., Nallur, S., Babar, I., et al. (2008). A SNP in a let-7 microRNA complementary site in the KRAS 3′ untranslated region increases non-small cell lung cancer risk. Cancer Res. 68, 8535–8540.
Conde-Vancells, J., Gonzalez, E., Lu, S. C., Mato, J. M., and Falcon-Perez, J. M. (2010). Overview of extracellular microvesicles in drug metabolism. Expert Opin. Drug Metab. Toxicol. 6, 543–554.
Delles, C., Padmanabhan, S., Lee, W. K., Miller, W. H., McBride, M. W., McClure, J. D., et al. (2008). Glutathione S-transferase variants and hypertension. J. Hypertens. 26, 1343–1352.
Estrada, K., Styrkarsdottir, U., Evangelou, E., Hsu, Y. H., Duncan, E. L., Ntzani, E. E., et al. (2012). Genome-wide meta-analysis identifies 56 bone mineral density loci and reveals 14 loci associated with risk of fracture. Nat. Genet. 44, 491–501.
Friedman, R. C., Farh, K. K., Burge, C. B., and Bartel, D. P. (2009). Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 19, 92–105.
Gamazon, E. R., Huang, R. S., Cox, N. J., and Dolan, M. E. (2010). Chemotherapeutic drug susceptibility associated SNPs are enriched in expression quantitative trait loci. Proc. Natl. Acad. Sci. U.S.A. 107, 9287–9292.
Gibbs, J. R., Van Der Brug, M. P., Hernandez, D. G., Traynor, B. J., Nalls, M. A., Lai, S. L., et al. (2010). Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet. 6, e1000952. doi:10.1371/journal.pgen.1000952
Gong, J., Tong, Y., Zhang, H. M., Wang, K., Hu, T., Shan, G., et al. (2012). Genome-wide identification of SNPs in microRNA genes and the SNP effects on microRNA target binding and biogenesis. Hum. Mutat. 33, 254–263.
Grimson, A., Farh, K. K., Johnston, W. K., Garrett-Engele, P., Lim, L. P., and Bartel, D. P. (2007). MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol. Cell 27, 91–105.
Hildebrandt, M. A., Salavaggione, O. E., Martin, Y. N., Flynn, H. C., Jalal, S., Wieben, E. D., et al. (2004). Human SULT1A3 pharmacogenetics: gene duplication and functional genomic studies. Biochem. Biophys. Res. Commun. 321, 870–878.
Hou, J., Lin, L., Zhou, W., Wang, Z., Ding, G., Dong, Q., et al. (2011). Identification of miRNomes in human liver and hepatocellular carcinoma reveals miR-199a/b-3p as therapeutic target for hepatocellular carcinoma. Cancer Cell 19, 232–243.
Hu, H. Y., Guo, S., Xi, J., Yan, Z., Fu, N., Zhang, X., et al. (2011). MicroRNA expression and regulation in human, chimpanzee, and macaque brains. PLoS Genet. 7, e1002327. doi:10.1371/journal.pgen.1002327
Hu, Z., and Bruno, A. E. (2011). The Influence of 3′UTRs on MicroRNA function inferred from human SNP data. Comp. Funct. Genomics 2011, 910769.
Ji, Y., Salavaggione, O. E., Wang, L., Adjei, A. A., Eckloff, B., Wieben, E. D., et al. (2005). Human phenylethanolamine N-methyltransferase pharmacogenomics: gene re-sequencing and functional genomics. J. Neurochem. 95, 1766–1776.
Korkina, L., Scordo, M. G., Deeva, I., Cesareo, E., and De Luca, C. (2009). The chemical defensive system in the pathobiology of idiopathic environment-associated diseases. Curr. Drug Metab. 10, 914–931.
Krek, A., Grun, D., Poy, M. N., Wolf, R., Rosenberg, L., Epstein, E. J., et al. (2005). Combinatorial microRNA target predictions. Nat. Genet. 37, 495–500.
Lewis, B. P., Burge, C. B., and Bartel, D. P. (2005). Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120, 15–20.
Liao, L. H., Zhang, H., Lai, M. P., Chen, S. L., Wu, M., and Shen, N. (2011). Single-nucleotide polymorphisms and haplotype of CYP2E1 gene associated with systemic lupus erythematosus in Chinese population. Arthritis Res. Ther. 13, R11.
Lim, L. P., Lau, N. C., Garrett-Engele, P., Grimson, A., Schelter, J. M., Castle, J., et al. (2005). Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 433, 769–773.
Mishra, P. J., Humeniuk, R., Longo-Sorbello, G. S., Banerjee, D., and Bertino, J. R. (2007). A miR-24 microRNA binding-site polymorphism in dihydrofolate reductase gene leads to methotrexate resistance. Proc. Natl. Acad. Sci. U.S.A. 104, 13513–13518.
Montgomery, S. B., Sammeth, M., Gutierrez-Arcelus, M., Lach, R. P., Ingle, C., Nisbett, J., et al. (2010). Transcriptome genetics using second generation sequencing in a Caucasian population. Nature 464, 773–777.
Motsinger-Reif, A. A., Jorgenson, E., Relling, M. V., Kroetz, D. L., Weinshilboum, R., Cox, N. J., et al. (2010). Genome-wide association studies in pharmacogenomics: successes and lessons. Pharmacogenet. Genomics. 10.1097/FPC.0b013e32833d7b45
Moyer, A. M., Salavaggione, O. E., Hebbring, S. J., Moon, I., Hildebrandt, M. A., Eckloff, B. W., et al. (2007). Glutathione S-transferase T1 and M1: gene sequence variation and functional genomics. Clin. Cancer Res. 13, 7207–7216.
Mrozikiewicz, P. M., Grzeskowiak, E., Seremak-Mrozikiewicz, A., Bogacz, A., Barlik, M., Semczuk, A., et al. (2011). Importance of CYP1A1 polymorphism and its transcriptional regulation in ovarian and endometrial cancer. Ginekol. Pol. 82, 925–932.
Onizuka, M., Kunii, N., Toyosaki, M., Machida, S., Ohgiya, D., Ogawa, Y., et al. (2011). Cytochrome P450 genetic polymorphisms influence the serum concentration of calcineurin inhibitors in allogeneic hematopoietic SCT recipients. Bone Marrow Transplant. 46, 1113–1117.
Patron, J. P., Fendler, A., Bild, M., Jung, U., Muller, H., Arntzen, M. O., et al. (2012). MiR-133b targets antiapoptotic genes and enhances death receptor-induced apoptosis. PLoS ONE 7, e35345. doi:10.1371/journal.pone.0035345
Pavek, P., and Dvorak, Z. (2008). Xenobiotic-induced transcriptional regulation of xenobiotic metabolizing enzymes of the cytochrome P450 superfamily in human extrahepatic tissues. Curr. Drug Metab. 9, 129–143.
Pearson, W. R., and Lipman, D. J. (1988). Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. U.S.A. 85, 2444–2448.
Ramsey, L. B., Bruun, G. H., Yang, W., Trevino, L. R., Vattathil, S., Scheet, P., et al. (2012). Rare versus common variants in pharmacogenetics: SLCO1B1 variation and methotrexate disposition. Genome Res. 22, 1–8.
Saunders, M. A., Liang, H., and Li, W. H. (2007). Human polymorphism at microRNAs and microRNA target sites. Proc. Natl. Acad. Sci. U.S.A. 104, 3300–3305.
Schadt, E. E., Molony, C., Chudin, E., Hao, K., Yang, X., Lum, P. Y., et al. (2008). Mapping the genetic architecture of gene expression in human liver. PLoS Biol. 6, e107. doi:10.1371/journal.pbio.0060107
Selinski, S., Lehmann, M. L., Blaszkewicz, M., Ovsiannikov, D., Moormann, O., Guballa, C., et al. (2012). Rs11892031[A] on chromosome 2q37 in an intronic region of the UGT1A locus is associated with urinary bladder cancer risk. Arch. Toxicol. 86, 1369–1378.
Takagi, S., Nakajima, M., Kida, K., Yamaura, Y., Fukami, T., and Yokoi, T. (2010). MicroRNAs regulate human hepatocyte nuclear factor 4alpha, modulating the expression of metabolic enzymes and cell cycle. J. Biol. Chem. 285, 4415–4422.
Tan, Z., Randall, G., Fan, J., Camoretti-Mercado, B., Brockman-Schneider, R., Pan, L., et al. (2007). Allele-specific targeting of microRNAs to HLA-G and risk of asthma. Am. J. Hum. Genet. 81, 829–834.
Teichert, M., Eijgelsheim, M., Rivadeneira, F., Uitterlinden, A. G., Van Schaik, R. H., Hofman, A., et al. (2009). A genome-wide association study of acenocoumarol maintenance dosage. Hum. Mol. Genet. 18, 3758–3768.
Tsuchiya, Y., Nakajima, M., Takagi, S., Taniya, T., and Yokoi, T. (2006). MicroRNA regulates the expression of human cytochrome P450 1B1. Cancer Res. 66, 9090–9098.
Yokoi, T., and Nakajima, M. (2011). Toxicological implications of modulation of gene expression by microRNAs. Toxicol. Sci. 123, 1–14.
Yu, Z., Li, Z., Jolicoeur, N., Zhang, L., Fortin, Y., Wang, E., et al. (2007). Aberrant allele frequencies of the SNPs located in microRNA target sites are potentially associated with human cancers. Nucleic Acids Res. 35, 4535–4541.
Zhang, B., Wang, Q., and Pan, X. (2007). MicroRNAs and their regulatory roles in animals and plants. J. Cell. Physiol. 210, 279–289.
Appendix
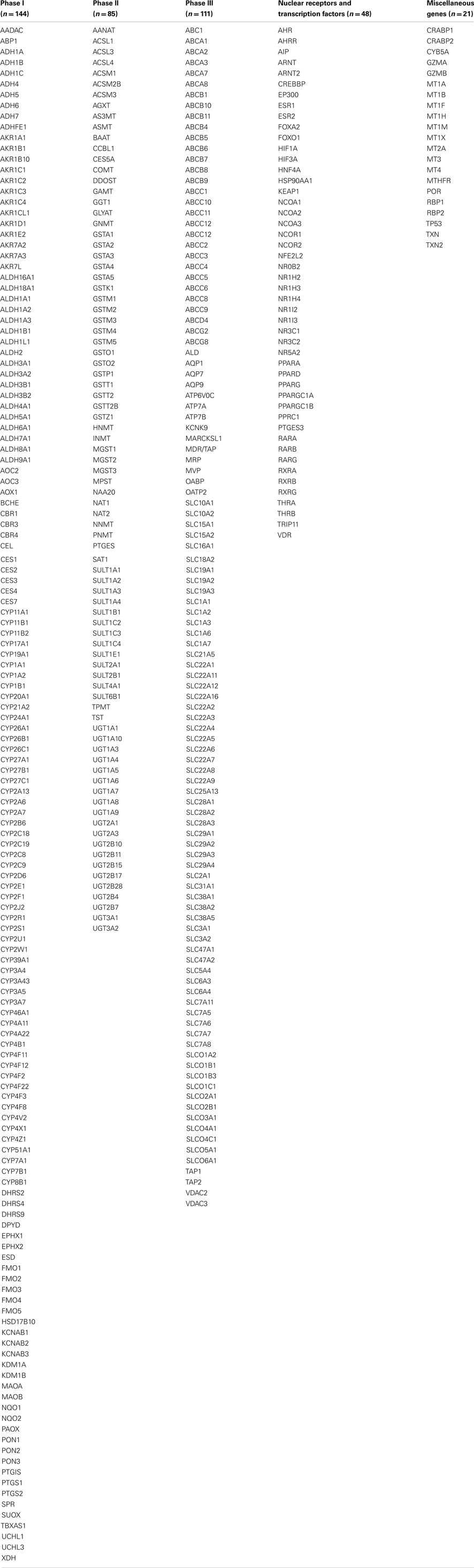
Table A1. Major XMETs and related genes investigated in this study.
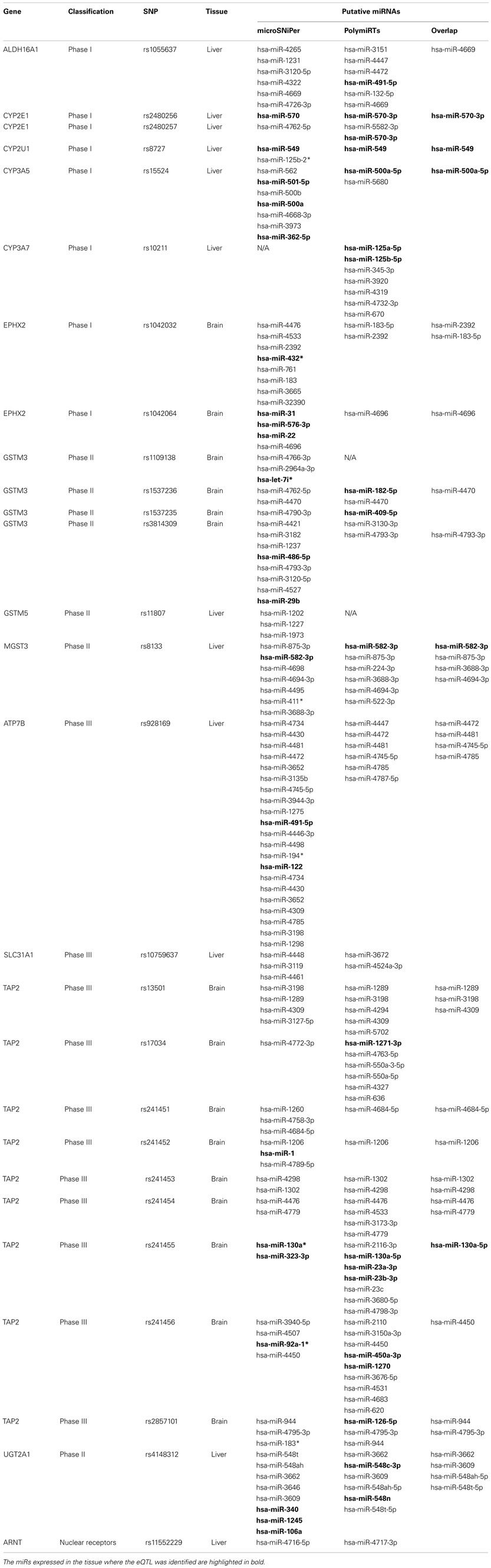
Table A2. Putative miRNAs associated with SNPs in the 3′-UTR region.
Keywords: eQTL, xenobiotic metabolism enzyme and transporter, microRNA, pharmacogenetics, 3′-UTR
Citation: Wei R, Yang F, Urban TJ, Li L, Chalasani N, Flockhart DA and Liu W (2012) Impact of the interaction between 3′-UTR SNPs and microRNA on the expression of human xenobiotic metabolism enzyme and transporter genes. Front. Gene. 3:248. doi: 10.3389/fgene.2012.00248
Received: 31 August 2012; Accepted: 25 October 2012;
Published online: 21 November 2012.
Edited by:
Kathrin Klein, Dr. Margarete Fischer-Bosch-Institute of Clinical Pharmacology, GermanyReviewed by:
Wichittra Tassaneeyakul, Khon Kaen University, ThailandTsuyoshi Yokoi, Kanazawa University, Japan
Copyright: © 2012 Wei, Yang, Urban, Li, Chalasani, Flockhart and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Wanqing Liu, Department of Medicinal Chemistry and Molecular Pharmacology, College of Pharmacy, Purdue University, 575 Stadium Mall Drive, West Lafayette, IN 47907, USA. e-mail:liu781@purdue.edu