
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet. , 12 September 2012
Sec. Epigenomics and Epigenetics
Volume 3 - 2012 | https://doi.org/10.3389/fgene.2012.00171
This article is part of the Research Topic Administration and coordination of genetic expression by proteins structuring the genome View all 12 articles
 Julia A. Horsfield1*
Julia A. Horsfield1* Cristin G. Print2 and Maren Mönnich1
Cristin G. Print2 and Maren Mönnich1
The multi-subunit protein complex, cohesin, is responsible for sister chromatid cohesion during cell division. The interaction of cohesin with DNA is controlled by a number of additional regulatory proteins. Mutations in cohesin, or its regulators, cause a spectrum of human developmental syndromes known as the “cohesinopathies.” Cohesinopathy disorders include Cornelia de Lange Syndrome and Roberts Syndrome. The discovery of novel roles for chromatid cohesion proteins in regulating gene expression led to the idea that cohesinopathies are caused by dysregulation of multiple genes downstream of mutations in cohesion proteins. Consistent with this idea, Drosophila, mouse, and zebrafish cohesinopathy models all show altered expression of developmental genes. However, there appears to be incomplete overlap among dysregulated genes downstream of mutations in different components of the cohesion apparatus. This is surprising because mutations in all cohesion proteins would be predicted to affect cohesin’s roles in cell division and gene expression in similar ways. Here we review the differences and similarities between genetic pathways downstream of components of the cohesion apparatus, and discuss how such differences might arise, and contribute to the spectrum of cohesinopathy disorders. We propose that mutations in different elements of the cohesion apparatus have distinct developmental outcomes that can be explained by sometimes subtly different molecular effects.
The cohesin complex and proteins that regulate its interaction with chromatin have multiple roles in cell division, DNA damage repair, gene transcription, and chromosome architecture. Proteins that make up the cohesin complex have been characterized in several model systems (see Table 1). The mechanics of cell division has been well researched for decades, and the identity of the chromosome cohesion proteins that hold together sister chromatids after S phase and prior to mitosis has been known for 15 years (Guacci et al., 1997; Michaelis et al., 1997). Consequently, sister chromatid cohesion remains the best-characterized role for the cohesin complex and its regulators.
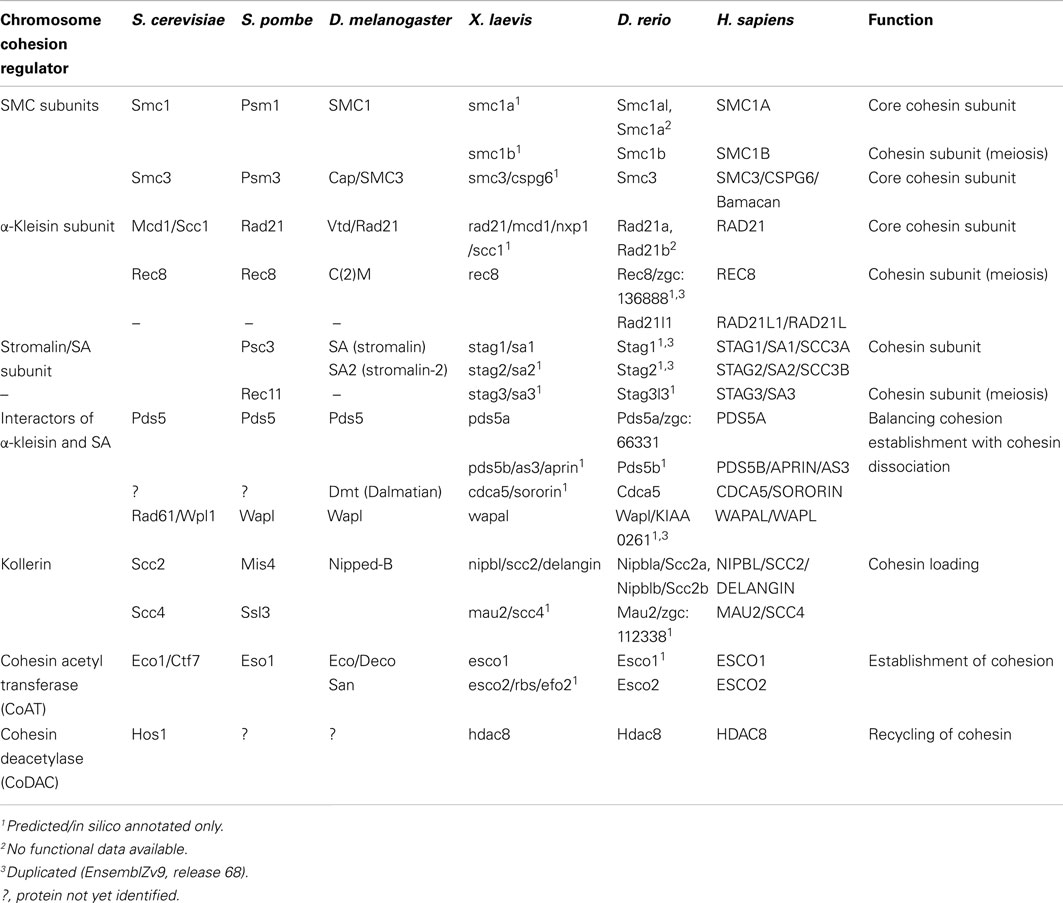
Table 1. Nomenclature and function of cohesin subunits and cohesin regulators.
The first evidence that a transcriptional function existed for chromosome cohesion proteins emerged in 1999, when the Nipped-B gene was identified in a genetic screen for modifiers of long-range enhancer-promoter communication regulating cut gene expression in the Drosophila wing margin (Rollins et al., 1999). Further evidence that gene transcription isone of cohesin’s crucial functions unfolded over the following years, and included a role for Scc1 in mating-type silencing in yeast (Lau et al., 2002), a transcriptional co-activation function for SA in mammalian cell lines (Lara-Pezzi et al., 2004), and complex long-range regulation of cut gene expression resulting from cohesin and Nipped-B Drosophila mutants (Rollins et al., 2004). Interest in the transcription function of cohesion proteins heightened when heterozygous mutations NIPBL, the human homolog of Nipped-B, were found to cause the human developmental disease, Cornelia de Lange Syndrome (CdLS; OMIM 122470; Krantz et al., 2004; Tonkin et al., 2004). Additional mutations causing CdLS were found in the cohesin subunits SMC1 (Musio et al., 2006; Deardorff et al., 2007) and SMC3 (Deardorff et al., 2007). Furthermore, homozygous mutations in ESCO2, which encodes a cohesion acetyltransferase (CoAT; Nasmyth, 2011; Higashi et al., 2012), were found to underlie a second human disorder, Robert’s Syndrome (RBS; OMIM 268300; Vega et al., 2005). More recently, mutations in RAD21 have been found to cause a related developmental disorder that partially overlaps with CdLS (Deardorff et al., 2012b).
After the causative genes for CdLS and RBS were found, a flood of new results in vertebrates, from fish (Horsfield et al., 2007; Muto et al., 2011), mouse (Zhang et al., 2007, 2009; Kawauchi et al., 2009), and human cell lines (Liu et al., 2009), supported the notion that these syndromes could be caused by dysregulated expression of multiple developmental genes. This suggested that cohesin-related developmental disorders have related pathologies, and led to use of the term “cohesinopathies” to describe these disorders (Liu and Krantz, 2008; McNairn and Gerton, 2008).
Although the idea that cohesinopathies have a common causal basis in dysregulated gene expression is a popular one, it is clear that the output of gene regulation is different for each disorder. Human syndromes caused by NIPBL, SMC1, SMC3, RAD21, and ESCO2 mutations share common features but appear to be clinically distinct. Here we revisit the theory that cohesinopathies result from dysregulated gene expression, and raise the question of whether subunits contributing to cohesin or its regulation can interact separately with distinct pathways leading to diverse phenotypic consequences.
The mitotic cohesin complex comprises two structural maintenance of chromosomes (SMC) subunits Smc1 and Smc3, which associate to form a tripartite ring incorporating an α-kleisin subunit, Mcd1/Scc1/Rad21. Smc1 and Smc3 are large rod-shaped proteins that dimerize at one end to form a “hinge” domain, and also interact at the other end via ATP-binding “heads,” which in turn interact with the α-kleisin subunit (Figure 1). The α-kleisin interacts with additional subunits Scc3/Stromalin (SA), Pds5, and Wapl (Nasmyth, 2011; see Table 1). The formation of cohesin subunits into a large ring structure led to the theory that cohesin topologically entraps sister chromatids inside a single ring (Haering et al., 2008). Alternative models have been proposed for how cohesin physically holds two molecules of DNA together (Huang et al., 2005; Zhang et al., 2008b; Skibbens, 2010), although most are not compatible with the single ring theory (reviewed in Nasmyth, 2011).
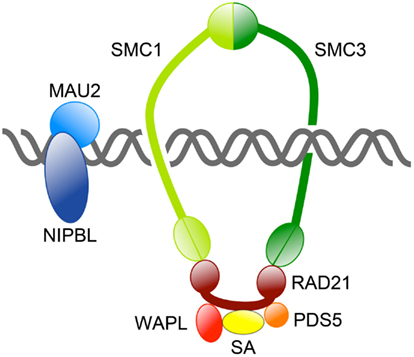
Figure 1. Overview of the cohesin complex and its associated proteins. The cohesin complex consists of four core subunits: SMC1, SMC3, RAD21, and SA. Together these subunits form a large ring capable of topologically encircling DNA strands. Other proteins regulate cohesin’s binding to DNA and its residency there. The NIPBL/MAU2 dimer loads cohesin onto DNA, whereas WAPL/PDS5 release cohesin from chromosomes by opening the SMC3-RAD21 interface.
The many functions of cohesin have been well described in recent reviews (Hirano, 2006; Nasmyth and Haering, 2009; Carretero et al., 2010; Nasmyth, 2011; Rhodes et al., 2011; Mehta et al., 2012). Cohesin turnover, recycling, loading onto chromosomes and residency there is controlled by several other proteins (Figure 2; Table 1). It was recently proposed that cohesin is loaded and unloaded from chromosomes by a “dual gate” mechanism (Nasmyth, 2011). The cohesin loading complex containing Scc2 (Nipped-B in Drosophila and NIPBL in human) and Scc4/MAU2, recently dubbed “kollerin” (Nasmyth, 2011), is responsible for loading cohesin onto chromosomes in G1 phase in yeast, and telophase in most other organisms. Kollerin directly loads cohesin onto the pre-replication complex (pre-RC) on chromatin in vitro in Xenopus extracts (Bermudez et al., 2012), indicating that it is likely to be necessary and sufficient for cohesin loading. Kollerin likely facilitates cohesin loading by enabling the transient opening of the Smc1-Smc3 hinge domains (Figure 1; Nasmyth, 2011). An opposing unloading activity is mediated by “releasin,” a cohesion disestablishment complex containing Pds5 and Wapl that interacts with SA to unlock the cohesin ring (Gandhi et al., 2006; Kueng et al., 2006; Shintomi and Hirano, 2009). Releasin allows exit of DNA via the Smc1-Smc3 head domains by opening the Smc3-kleisin interface. In theory, cohesin snaps onto DNA via opening of the hinge domains, and exits DNA via opening the ring at the opposite end (Nasmyth, 2011).
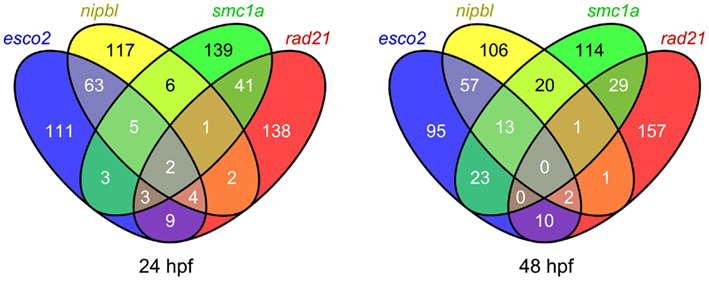
Figure 2. Comparison of the top 200 affected probe sets in zebrafish embryos depleted of different cohesin subunits. Venn diagrams showing the overlap of the top 200 probe sets affected in zebrafish esco2 and nipbl morphants, and smc1ahi1113a and rad21nz171 mutants at 24 and 48 hpf (q value < 0.05).
Once loaded onto chromosomes, cohesin binds DNA with variable modes of stability (Gerlich et al., 2006; Gause et al., 2010) and is mobile, having the ability to translocate along chromosome arms (Lengronne et al., 2004; Hu et al., 2011), or readily detach via interaction with releasin. However during S phase, cohesin becomes stably bound for long enough to fulfill its function in sister chromatid cohesion. Stabilization of cohesin binding happens during the process of DNA replication (Skibbens et al., 1999; Kenna and Skibbens, 2003; Moldovan et al., 2006), and is mediated via acetylation of Smc3 by cohesin acetyl transferase (CoAT; Nasmyth, 2011). The known CoATs for Smc3 are Ctf7/Eco1 (yeast), or Esco1/2 (vertebrates; Skibbens et al., 1999; Ivanov et al., 2002; Hou and Zou, 2005).
CoAT-mediated acetylation of Smc3 generates the cohesive form of cohesin that holds together the sister chromatids from G2 until M phase (Ben-Shahar et al., 2008; Unal et al., 2008; Zhang et al., 2008a). In humans, both ESCO1 and ESCO2 CoATs are necessary for proper sister chromatid cohesion (Hou and Zou, 2005). However, it appears that ESCO2 CoAT is primarily required for cohesion in heterochromatic regions, and RBS patients who lack ESCO2 exhibit heterochromatin repulsion and precocious sister chromatid separation, particularly at centromeric regions (Vega et al., 2005). In human and Drosophila (but not yeast), the Sororin protein is additionally required to establish and maintain cohesion (Rankin et al., 2005; Schmitz et al., 2007; Nishiyama et al., 2010).
Once cohesion has been established in G2, cohesion-promoting and cohesin-releasing activities compete during chromosome condensation in prophase. The releasing activity that removes cohesin from chromosomes prevails along chromosome arms in a process known as the “prophase pathway,” which involves phosphorylation of SA1/2 by Polo-like kinase (Plk) and Aurora B (Losada et al., 2002; Hauf et al., 2005) and complexing of SA and RAD21 by releasin (Gandhi et al., 2006; Kueng et al., 2006; Shintomi and Hirano, 2009). In the competing “establishment” activity, Sororin and CoAT function to antagonize releasin activity (Rowland et al., 2009; Sutani et al., 2009; Lafont et al., 2010; Nishiyama et al., 2010; Nasmyth, 2011) by a mechanism that also requires Pds5 (Vaur et al., 2012), and the phosphatase Ssu72 promotes cohesion by countering the phosphorylation of SA1/2 (Hauf et al., 2005; Kim et al., 2010b). By metaphase, most cohesin has been removed from chromosome arms, and the remaining, primarily centromeric cohesin, is protected from removal by Shugoshin (Wang and Dai, 2005).
At Anaphase, the remaining cohesin rings are opened, allowing chromosomes to separate (Craig and Choo, 2005). APC-mediated degradation of Securin (Salah and Nasmyth, 2000) releases the protease Separase, which cleaves the Rad21 subunit of cohesin (Waizenegger et al., 2000, 2002; Hornig et al., 2002). After telophase, Smc complexes can be recycled and reloaded onto chromatin. An important requirement for cohesin recycling is deacetylation of Smc3 by the class I histone deacetylase Hos1 (yeast) or HDAC8 (human; Beckouet et al., 2010; Borges et al., 2010; Xiong et al., 2010; Deardorff et al., 2012a). Thus, Smc3 deacetylation by Hos1 opposes Esco2’s acetylation activity.
Cohesin has a further important role in DNA double strand break repair (reviewed in Dorsett and Strom, 2012; Wu and Yu, 2012). To effect double strand break repair, the cohesive form of cohesin must be established at the location of the break (Ball and Yokomori, 2008). Stabilization of cohesin at double strand breaks in budding yeast depends on acetylation of the Rad21/Mcd1p subunit by Eco1p, plus antagonism of the releasin complex containing Wpl (Heidinger-Pauli et al., 2009). Cohesin is recruited de novo at double strand breaks in G2 phase (Strom et al., 2007) in a Scc2/kollerin-dependent manner (Strom et al., 2004), and in vertebrates, this association also involves another SMC complex: the Smc5/6 complex (Strom and Sjogren, 2007; De Piccoli et al., 2009).
Other molecular events contribute to cohesin function in DSB repair. In budding yeast, it was shown that the phosphorylation of Mcd1p (Rad21) through ATR and Chk1 pathway is important for cohesion and DSB repair (Heidinger-Pauli et al., 2008). In human cells, cohesive cohesin at DSBs also depends on the pro-establishment activity of Sororin (Schmitz et al., 2007). Cohesin, but not chromosome cohesion, is required for activation of G1, intra-S, and G2–M DNA damage checkpoints (Jessberger, 2009; Watrin and Peters, 2009). In cancer cells, cohesin binding through the genome is reinforced following ionizing radiation (IR), in a process that requires ATM and SMC3 phosphorylation, and SMC3 acetylation by ESCO1. Both ESCO1 and SMC3 acetylation are required for intra-S phase checkpoint and cellular survival after IR (Kim et al., 2010a).
Despite good evidence that cohesin regulates gene expression directly and independently of cell division (Pauli et al., 2010; Dorsett, 2011), the mechanism(s) of transcriptional regulation by cohesin are not well understood. Cohesin binds to many sites throughout the genome, sometimes in combination with the CCCTC-binding factor (CTCF) insulator protein, which is known to mediate chromatin loop formation (Gondor and Ohlsson, 2008). Previous studies demonstrated that cohesin colocalizes with CTCF along chromosome arms, and is likely to cooperate with this protein in the regulation of gene expression or chromatin structure (Parelho et al., 2008; Rubio et al., 2008; Wendt et al., 2008). As well as CTCF, cohesin colocates genome-wide with other transcriptional regulators, such as estrogen receptor-α (Schmidt et al., 2010), and Mediator (Kagey et al., 2010) in a cell type-specific manner. Likely in combination with other factors, cohesin selectively binds genes with paused RNA polymerase. Although it is not involved in RNA polymerase pausing itself, cohesin can regulate transcription by determining the amount of elongating RNA polymerase on genes (Fay et al., 2011).
Regulation of many genes by cohesin appears to involve the three-dimensional (3D) organization of chromatin (Merkenschlager, 2010; Dorsett, 2011). A direct role for cohesin in chromatin looping has been demonstrated for several loci (Hadjur et al., 2009; Mishiro et al., 2009; Nativio et al., 2009; Hou et al., 2010; Chien et al., 2011; Kim et al., 2011) by studies showing that long-range interactions between regulatory sequences are reduced by cohesin knockdown. It is likely that cohesin regulates spatiotemporal gene expression in combination with diverse tissue-specific transcription factors, and by distinct modes of transcription regulation (Dorsett, 2011).
An overlapping spectrum of human syndromes can be attributed to mutations in cohesin subunits, or regulators of cohesin loading and unloading from chromosomes. The best known cohesinopathy is CdLS (OMIM 122470) also termed Brachmann de Lange syndrome (BdLS), a broad spectrum disorder with multiple developmental and cognitive abnormalities (de Lange, 1933; Opitz, 1985; Ireland et al., 1993; Jackson et al., 1993). CdLS patients are small in size and have a characteristic facial appearance, including arched eyebrows, hirsutism, synophrys, ptosis, long eyelashes, an upturned nose, a long philtrum, thin upper lip, and micrognathia. Developmental anomalies range from mild to severe, with more severe cases having upper limb truncations or limb differences. CdLS patients also frequently present with hearing loss, gastrointestinal defects, pyloric stenosis, genital abnormality, congenital diaphragmatic hernias, cardiac septal defects, and autistic behaviors (Jackson et al., 1993). All patients within the CdLS spectrum have neurodevelopmental delay and highly variable mental retardation (Deardorff et al., 2007).
More than half of CdLS cases (∼65%) are dominantly inherited, and caused by mutations in the NIPBL gene (OMIM 608667; Krantz et al., 2004; Tonkin et al., 2004), which encodes a crucial component of kollerin. Heterozygous truncating or non-sense NIPBL mutations are haploinsufficient, and strikingly, NIPBL protein levels need only be reduced by 15–30% to give rise to a CdLS phenotype (Krantz et al., 2004; Tonkin et al., 2004). This implies that the remaining intact NIPBL allele is upregulated in an attempt to compensate, and also that certain cell types and/or developmental processes are exquisitely sensitive to the levels of NIPBL. Missense mutations in NIPBL were also identified that may interfere with the interaction of NIPBL with its partner, MAU2, or other proteins (Braunholz et al., 2012).
Mutations in SMC1A (OMIM 300040) and SMC3 (OMIM 606062) also give rise to syndromes that fall within the CdLS spectrum, and account for about 5% of CdLS cases (Musio et al., 2006; Deardorff et al., 2007; Mannini et al., 2010). SMC mutations are heterozygous missense mutations and are thought to interfere with the structure of the SMC subunits such that functional interactions of the cohesin complex are disturbed, causing the disease pathology (Deardorff et al., 2007). In some cases missense mutations were shown to interfere with cohesin binding to DNA (Revenkova et al., 2009). Human developmental phenotypes resulting from SMC mutations are inclined to be milder than for NIPBL mutations; these individuals have fuller eyebrows and a prominence of the nasal bridge, with fewer structural abnormalities; however, all patients had some degree of mental retardation (Deardorff et al., 2007; Rohatgi et al., 2010). This suggests that brain development is particularly sensitive to disruption of SMC subunits.
RAD21 (OMIM 606462) mutations also cause a cohesinopathy syndrome (Deardorff et al., 2012b). Heterozygous deletions of RAD21 and missense mutations, which included a dominant interfering mutation and one with essentially no function, gave rise to developmental anomalies with some overlap with CdLS. Patients with RAD21 mutations have an even milder phenotype than those with SMC mutations. They have some divergence in the facial features and, most notably, they have extremely mild cognitive and physical abnormalities (Deardorff et al., 2012b). Consistent with RAD21 having a role in DNA damage response, lymphoblastoid cell lines from patients with RAD21 mutations exhibited radiation sensitivity. A gene transcription assay in zebrafish showed that RAD21 missense mutations present in patients are not competent for proper regulation of gene expression (Deardorff et al., 2012b).
Homozygous recessive mutations in the ESCO2 gene, which encodes a CoAT, cause another cohesinopathy, RBS (OMIM 268300; Schule et al., 2005; Vega et al., 2005, 2010; Gordillo et al., 2008). RBS is characterized by mild to severe growth deficiency, limb malformations (in particular, symmetric tetraphocomelia), multiple craniofacial abnormalities including cleft lip and/or cleft palate, microcephaly, and mental retardation. Mortality is high among severely affected pregnancies and newborns (Gordillo et al., 1993). A milder disorder with less marked limb reduction and survival to adulthood is known as SC phocomelia, but since both disorders arise from ESCO2 mutations with no apparent genotype/phenotype correlation (Schule et al., 2005; Vega et al., 2010), it has been proposed all ESCO2 mutations be referred to as RBS (Vega et al., 2010). Unlike CdLS, cells from RBS patients exhibit precocious sister chromatid separation, particularly at heterochromatic regions of the chromosomes (Schule et al., 2005; Vega et al., 2005) leading to mitotic defects, lagging chromosomes, aneuploidy, and micronuclei formation. The acetyltransferase activity of ESCO2 appears to be crucial, since mutations in this domain are sufficient for the pathogenesis of RBS (Gordillo et al., 2008). While RBS features overlap with those of CdLS, there are appreciable differences. Whether gene regulation downstream of ESCO2 is responsible for RBS pathology is still under debate.
The wide spectrum of human developmental phenotypes owing to cohesin mutations characterized to date indicate that although these disorders have many features in common, there are also distinct differences. Gene expression and molecular studies in cells and in animal models have helped to uncover the common and divergent pathways that lie downstream of cohesinopathy mutations.
A comparison of the consequences of knocking down cohesin or its regulators in different animal model systems indicates there are a wide variety of outcomes for cell biology and gene expression. For mutations causing CdLS and similar cohesinopathies, it seems likely that specific developmental pathways are regulated downstream of the causative gene mutations. Several groups have conducted analyses of gene expression downstream of cohesinopathy mutations.
For some genes, it seems likely that small changes in the dose of cohesin or its regulators could have a large impact on transcription. In Drosophila, cohesin and Nipped-B bind to actively transcribed regions of the genome and are excluded from regions of polycomb group (PcG) silencing (Misulovin et al., 2008). For the rare genes where cohesin binding overlaps with PcG-mediated methylation of lysine 27 on histone 3 (H3K27me3), expression of those genes is hypersensitive to cohesin dose (Schaaf et al., 2009). In addition, cohesin ablation in post-mitotic neurons in the Drosophila mushroom body (Pauli et al., 2008; Schuldiner et al., 2008), or salivary glands (Pauli et al., 2010) affected the expression of specific loci including the gene encoding the ecdysone receptor. This suggests that some genes, perhaps in specific cell types, may dramatically change their transcriptional activity in response to a slight alteration of cohesin dose.
Intriguingly, it seems that the transcriptional response of some genes to cohesin or Nipped-B depletion is biphasic, and depends on the degree to which these proteins are depleted (Schaaf et al., 2009). The Enhancer of split gene complex (E(spl)-C) in Drosophila is exquisitely responsive to Rad21 and Nipped-B levels. Furthermore, when mRNA encoding these proteins is depleted in BG3 cells, the direction in which some E(spl)-C are regulated depends on the length of time of RNAi treatment, and the degree of Rad21 or Nipped-B knockdown. For example, E(spl)-C transcripts decrease after 3 days of Nipped-B RNAi, but increase by day 6 (Schaaf et al., 2009). These findings have implications for genome-wide gene expression studies in cohesinopathy models. Which genes are altered in expression is likely to depend on tissue type, developmental stage and degree to which cohesinopathy gene function has been knocked down.
On the other hand, loss of Nipbl also appears to result in low (≤2) fold changes in the expression of a great many genes. Liu et al. (2009) analyzed gene expression and genome-wide binding of cohesin in lymphoblastoid cell lines from CdLS probands with mutations in NIPBL or in the cohesin subunit SMC1A, and found that ∼1500 genes (FDR ≤ 0.05) were dysregulated compared with controls. Dysregulated gene expression in the mutant cell lines was conserved, and correlated with disease severity and cohesin binding at misexpressed genes (Liu et al., 2009). Significantly, a panel of 23 genes could differentiate NIPBL mutations from SMC1A and ESCO2 mutations indicating that NIPBL mutations have a distinguishable effect on gene expression.
Heterozygous mice carrying a gene-trap insertion into the Nipbl locus show many features overlapping with CdLS, and microarray analyses indicated that reducing Nipbl dose resulted in small changes in expression of a great many genes. These mice also had severe developmental phenotypes, including craniofacial dysmorphology and heart defects, resembling CdLS. Of note was the altered expression of genes involved in fat metabolism, which could account for the lean habitus observed in mice and in CdLS patients (Kawauchi et al., 2009).
Mice with mutations in Pds5a and 5b have also been generated. Mice homozygous null for Pds5b died shortly after birth, with multiple congenital anomalies, including heart defects, cleft palate, skeletal defects, gut defects, abnormal migration and axonal projections of sympathetic neurons, and germ cell depletion (Zhang et al., 2007). Mice null for Pds5a exhibit many of the same multiple abnormalities that were previously observed in Pds5b-deficient mice, plus additional abnormalities including renal agenesis (Zhang et al., 2009). Elimination of both Pds5a and 5b gave an additional lens phenotype not observed in single null mice, and resulted in embryonic lethality (Zhang et al., 2009). Gene expression studies in the Pds5 mice have not been published.
Most recently, significant knowledge about cohesin function was gained by generating mice deficient for cohesin subunit SA1 (Cuadrado et al., 2012; Remeseiro et al., 2012a,b). Loss of SA1 results in embryonic lethality, and heterozygous animals have shorter lifespan and increased aneuploidy as a result of chromosome segregation defects. Segregation defects arose from compromised telomere replication, which requires cohesion mediated specifically by cohesin-SA1. The resulting aneuploidy in SA1 heterozygotes is thought to lead to early onset of tumorigenesis in these animals (Remeseiro et al., 2012a).
Interestingly, gene expression and genome-wide distribution of cohesin binding are dramatically altered in SA1 null mice, with important implications for CdLS. Location of cohesin to gene promoters and CTCF binding sites appears to depend on SA1. Furthermore, SA1 ablation led to altered cohesin binding at particular gene clusters accompanied by dysregulation of their transcription (Remeseiro et al., 2012b). These studies highlight the function of SA1 in multiple processes, and identify a key transcriptional role that is distinct from the function of SA2 in centromeric chromosome cohesion.
Zebrafish models have also shed light on the role of cohesin and Nipbl in gene expression. In fact, the first published evidence that cohesin regulates gene expression in a vertebrate model system came from a forward genetic screen in zebrafish. This screen identified the Rad21 subunit as a tissue-specific regulator of runx1, which encodes a hematopoietic transcription factor (Horsfield et al., 2007). In rad21 mutants at 12 h post-fertilization (hpf), runx1 expression was retained in Rohon–Beard neurons, but was absent from a discrete population of cells in the hematopoietic mesoderm. Importantly, the hematopoietic mesoderm precursor cell population was still present in mutants, and expressed the dimerization partner for Runx1, cbfb, although not runx1 itself. Cohesin probably targets other runx genes in a cell type-specific manner, since rad21 mutants also lacked expression of runx3 in Rohon–Beard neurons and the lateral line primordia (Horsfield et al., 2007). Unfortunately, the onset of runx2 expression (∼48 hpf) in zebrafish embryos is too late to determine its involvement, since rad21 mutants arrest in development at 35 hpf. Like in Drosophila, cohesin is likely to regulate expression of genes in zebrafish brain; cohesin subunits are expressed in non-proliferating neurons of zebrafish brain implying a non-cell cycle role for cohesin in this tissue (Monnich et al., 2009).
A zebrafish model of NIPBL-mediated CdLS revealed much about the multifactorial origins of this developmental syndrome. Zebrafish have two copies of the nipbl gene, and depletion of both versions by morpholino oligonucleotides to create “morphants” also led to small-scale dysregulation of a large number of genes in early embryogenesis (up to 6 hpf; Muto et al., 2011). Because gene expression changes were measured at early gastrula stages it is likely that many are directly caused by reduced Nipbl function rather than by secondary effects. Interestingly, genes involved in endoderm development and left-right axial patterning including sox17 and foxa2, were specifically downregulated in endoderm. Dysregulation of the endoderm-specifying hierarchy of Sox32, Sox17, and Foxa2 by Nipbl depletion is likely to contribute to the heart looping defects and gut tube defects observed at later stages in Nipbl-depleted zebrafish embryos (Muto et al., 2011). The zebrafish pathologies recapitulate heart and gastrointestinal tract abnormalities observed in CdLS, thereby allowing insight into the etiology of CdLS developmental defects.
Our own group conducted Affymetrix microarray analyses at a later stages (24 and 48 hpf) of zebrafish development in rad21 mutants (Rhodes et al., 2010), esco2 morphants (Monnich et al., 2011), smc1a mutants (available as part of an insertion mutant collection; Amsterdam et al., 2004) and nipbl morphants (Maren Mönnich, Cristin G. Print, Julia A. Horsfield, unpublished data). Interestingly, we found that the eomes gene, a master regulator of endoderm formation, is consistently downregulated in rad21 and smc1a mutants, and nipbl morphants (FDR < 0.02), supporting a role for cohesin and Nipbl in endoderm formation. Eomes expression is regulated by pluripotency factors Nanog, Oct4, and Sox2 (Teo et al., 2011), all of which are transcriptional targets of cohesin and Nipbl in embryonic stem cells (Kagey et al., 2010). It is enticing to speculate that cohesin and Nipbl could participate in the initial specification of germ layers from stem cell precursors through modulating the expression of pluripotency factors.
We expected our microarray analyses of zebrafish cohesinopathy mutants and morphants to result in similar lists of up- or downregulated genes, since embryos were analyzed at similar stages and cohesinopathy genes would be predicted to have similar roles in gene expression. Therefore we were surprised to find only modest overlap between regulated gene sets (example in Figure 2).
Strikingly, a comparison of rad21 mutant microarray data with esco2 morphant microarray data revealed that there is scant overlap between genes regulated downstream of these mutations (Monnich et al., 2011). For example, the myca gene, which is downregulated in rad21 zebrafish mutants and other species as well, is actually slightly upregulated in esco2 morphants. Most of the genes regulated downstream of esco2 are involved in cell proliferation or apoptosis, whereas many genes affected by the rad21 mutation are developmental regulators (Monnich et al., 2011). What could be the reason for these differences? We concluded that while Esco2 and Rad21 have related roles in sister chromatid cohesion, they do not have the same input into the regulation of gene expression. We found that although esco2 depletion has mild effects on neural crest cell migration, it does not induce patterning defects. Instead, even modest esco2 depletion results in robust activation of caspases, p53/mdm2 upregulation, and massive cell death (Monnich et al., 2011). Loss of jaw elements and fin stunting in esco2 morphants, which resemble RBS features, are therefore likely to be due to insufficient cells to contribute to the affected structures. In agreement with results from a conditional mouse knockout of Esco2 (Whelan et al., 2012b), it appears that developmental defects observed in esco2 morphant zebrafish arise from problems with cell survival rather than dysregulation of developmental genes.
Our microarray data of nipbl morphants was conducted under very mild knock down conditions of both nipbl genes at 24 and 48 hpf (Table 2; Maren Mönnich, Cristin G. Print, Julia A. Horsfield, unpublished data). We observed regulation of different sets of genes than those found by Muto et al. (2011) at the earlier timepoint of 6 hpf, which is not unexpected due the different developmental stage at which embryos were analyzed. We did not find any Gene Ontology categories of significance other than elevated expression of a network of genes related to p53. It is possible that degree of nipbl gene knockdown could also contribute to differences observed in regulated genes as discussed above, since at least some gene expression is likely to be sensitive to the dose of Nipbl protein (Schaaf et al., 2009).
Many genes that have altered regulation in response to depletion of cohesinopathy genes are different, raising the possibility that cohesin subunits and regulators have different functions in various pathways. However, genome-wide analyses of gene expression identified some commonly regulated pathways/genes such as those involved in endoderm development (eomes, sox17, foxa3), the myc transcription factor (except in esco2 morphants), and downstream effectors of Notch signaling such as hey1, her4.2, and ascl1.
Despite varying outcomes for gene expression and development identified using animal models of the cohesinopathies, some pathways seem more likely to be affected than others downstream of cohesinopathy genes. Common themes of pathways regulated by cohesinopathy genes are outlined below.
Perhaps not surprisingly, several studies have found links between cohesin and its regulators, and the control of pathways that underpin cell growth and proliferation. Somewhat more surprisingly, the level at which cohesin regulates growth and metabolism includes transcriptional control of specific gene targets. For example, the Myc oncogene is positively regulated by Nipbl and all cohesin subunits investigated to date (Misulovin et al., 2008; Kawauchi et al., 2009; Liu et al., 2009; Rhodes et al., 2010; Remeseiro et al., 2012b). Myc is a pluripotency factor, and it is probably significant that genes encoding other pluripotency factors Oct4, Nanog, and Sox2, are also bound and regulated by cohesin (Kagey et al., 2010; Nitzsche et al., 2011). Interestingly, pluripotency factors, e.g., Oct4 (Kim et al., 2011) and Nanog (Nitzsche et al., 2011) in turn appear to combine with cohesin to both positively and negatively regulate other target genes. These findings raise the interesting possibility that cohesin-mediated transcription is pivotal to cell fate decisions that determine the balance between pluripotency and differentiation (Dorsett, 2010).
Cohesinopathy genes regulate other growth pathways as well. In yeast, cohesinopathy mutations, including an Eco1RBS mutation (W216G), block transcription of ribosomal RNA genes thereby directly influencing ribosome biogenesis, protein translation and the cell’s ability to grow (Bose et al., 2012). This finding links cohesin function to metabolism and growth through a role in rDNA transcription and translation regulation. Since Myc, a transcriptional target of cohesin, also regulates ribosome biogenesis (Eilers and Eisenman, 2008), cohesin appears to be a central regulator of growth by transcriptional control of multiple pathways. In Nipbl+/− mice, genes controlling fat metabolism are dysregulated (Kawauchi et al., 2009), indicating a direct involvement in regulation of another metabolic pathway. Consistent with dysregulated growth and metabolism, CdLS patients are small and lean (Liu and Krantz, 2009). It is possible that many of the large number of dysregulated genes in CdLS are targets of MYC, which regulates 10–20% of genes in the genome.
Transcriptional regulation of cell growth and proliferation pathways by cohesin could be elegantly intertwined with its role in the cell cycle, where it mediates sister chromatid cohesion. Transcriptional pathways promoting growth are tightly linked to cell division, and it is entirely possible that cohesin and its regulators have central roles in making these links.
Neurodevelopmental disorders are among the most conserved features of the cohesinopathies (Deardorff et al., 2007). It is possible that these neurodevelopmental pathologies have a common molecular basis. Several lines of evidence suggest that cohesinopathy proteins influence the Notch signaling pathway, although the exact mechanisms are unknown. A recent study suggested that Esco2 physically interacts with Notch to antagonize Notch signaling, suggesting that one possible mechanism includes direct interaction with Notch receptor(s; Leem et al., 2011).
Our microarray analyses of zebrafish “cohesinopathy” embryos depleted for Rad21, Smc1a, Nipbl, or Esco2 identified conserved regulation of selected gene targets of the Notch signaling pathway. Notably, we found that the ascl1 gene is downregulated in both rad21 mutants (Horsfield et al., 2007; Rhodes et al., 2010) and esco2 morphants (Monnich et al., 2011), as well as Nipbl-depleted embryos and smc1a mutants (Table 2; Maren Mönnich, Cristin G. Print, Julia A. Horsfield, unpublished data). In 48 hpf rad21 heterozygous embryos (which are phenotypically normal), ascl1 is significantly downregulated (Rhodes et al., 2010), indicating that ascl1 expression is highly sensitive to even a slight reduction of Rad21 (heterozygotes have 60–70% of wild type rad21 mRNA levels). Such sensitivity could have high functional significance. Ascl1 is a potent neuronal lineage-specifying gene, being one of three genes sufficient to convert fibroblasts into iPN cells (Vierbuchen et al., 2010). Furthermore, Pds5b depletion altered Ascl1 expression and blocked neuronal differentiation in a stem cell model (Denes et al., 2010).
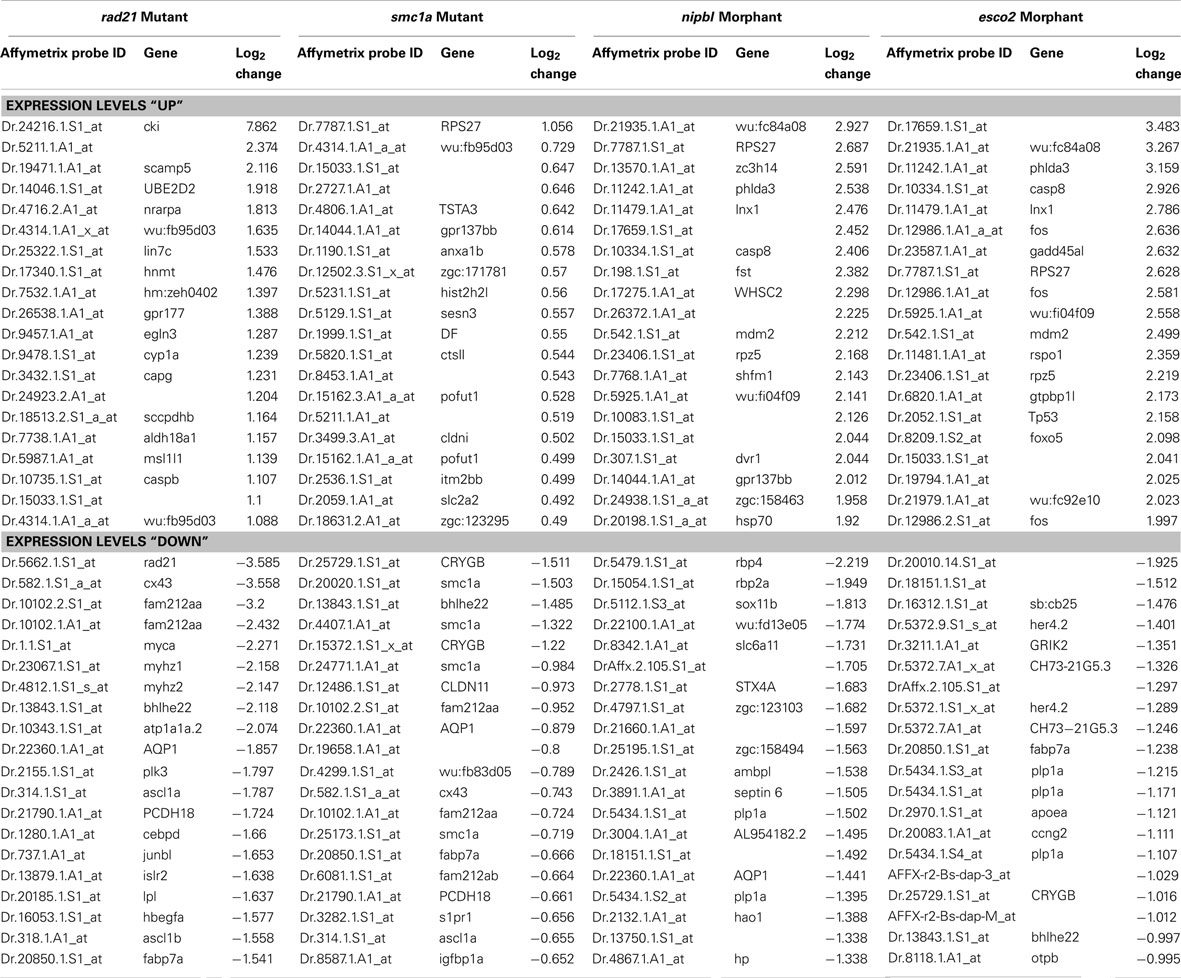
Table 2. Top 20 probe sets affected in zebrafish cohesinopathy microarrays at 24 h post-fertilization (p < 0.05).
We also found that certain Notch signaling targets of the hairy/enhancer of split family (such as her4, hey1) were consistently affected in our zebrafish cohesinopathy models (Rhodes et al., 2010), in agreement with cohesin/Nipbl regulation of the (E(spl)-C) in Drosophila (Schaaf et al., 2009). In combination with previous gene expression studies from Drosophila (Dorsett, 2009), strong evidence supports a link between cohesin-mediated transcription and cell fate in neuronal linages.
In addition to neuronal cell fate, it appears that cohesin together with CTCF could contribute to maintaining neuronal identity. Several studies show that cohesin and CTCF regulate expression of protocadherin genes (Kawauchi et al., 2009; Monahan et al., 2012; Remeseiro et al., 2012b). Cohesin-SA1 binds to the promoter of protocadherin genes and positively regulates their expression (Remeseiro et al., 2012b). Interestingly, CTCF and cohesin were recently found to modulate isoform expression of Pcdhα in a mouse neuroblastoma cell line (Monahan et al., 2012), by a mechanism assumed to involve enhancer-promoter communication. Cohesinopathy mutations could therefore have significant consequences for neuronal recognition of “self,” and the capacity to make functional synaptic connections (Dekker, 2012), since protocadherins are key players in these processes (Frank and Kemler, 2002; Esumi et al., 2005).
Evidence suggests that the widespread disruption of neuronal gene expression found in cohesinopathy mutants results in abnormal behavior and function of neurons. As discussed previously, localized disruption of cohesin subunits causes failure of axon pruning in the Drosophila mushroom body (Pauli et al., 2008; Schuldiner et al., 2008). Other model systems have highlighted a role for cohesinopathy proteins in axon pathfinding and/or migration. For example, Mau2, the Scc4 homolog that binds to Nipbl, is necessary for proper axon guidance and migration in C. elegans (Seitan et al., 2006). Consistent with a requirement of cohesin for migration, enteric neurons derived from neural crest cells failed to migrate in mice mutant for cohesin subunit Pds5b (Zhang et al., 2007). Furthermore, in esco2 morphant zebrafish, we observed defects that were consistent with abnormal neural crest cell migration (Monnich et al., 2011). In zebrafish mutant for rad21, we observed that while the trigeminal ganglia of the brain are specified, the axons clump together rather than extending forward (Figure 3). It is very likely that more subtle defects that are not so easily observed (for example, problems with neuronal connectivity) take place in the central nervous system of cohesinopathy patients and animal models.
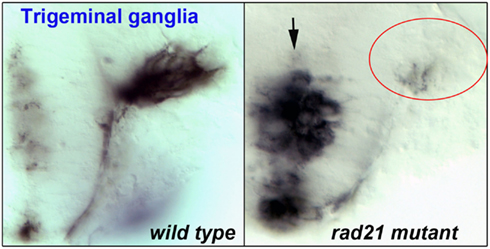
Figure 3. Flat-mount staining (anti-HNK-1) of trigeminal ganglia in wild type (left) and rad21 mutant (right) zebrafish embryos. In rad21 mutants, central neuronal clumping occurred (arrow), and axons failed to migrate and populate anterior regions (red oval).
Growth, metabolism, and development of the central nervous system appear to be processes that are universally sensitive to cohesinopathy mutations. Much of this pathology is likely to be caused by cohesin’s role in the regulation of gene expression. However, cohesin has another important role in the repair of DNA damage, and its loss is likely to trigger DNA damage checkpoints (Jessberger, 2009; Watrin and Peters, 2009). Activation of cell cycle checkpoints by cohesin depletion may represent additional biological processes contributing to cohesinopathies.
When damaged DNA is detected, cells respond by coordinating cell cycle arrest, DNA repair, and programmed cell death (Ciccia and Elledge, 2010). The crucial roles of cohesin and its regulators in DNA damage repair have been recently and comprehensively reviewed elsewhere (Dorsett and Strom, 2012; Wu and Yu, 2012). Inability to repair DNA and proceed through the cell cycle is accompanied by activation of cell cycle checkpoints, followed by cell death in the absence of repair.
Interestingly, mutations in other genes responsible for the DNA damage response underlie human syndromes with phenotypes that overlap the cohesinopathies (Ciccia and Elledge, 2010). Overlapping phenotypes include microcephaly, growth defects, neurological disorders, and facial/skeletal dysmorphology. These features are among the most conserved between the cohesinopathies, and raise the possibility that defects in the DNA damage response pathway might contribute to the etiology of cohesinopathy syndromes. In support of this, a patient with a mutation in a gene encoding the DNA helicase DDX11/ChlR1 had microcephaly, premature sister chromatid separation, and genome instability. This patient had features of both Fanconi Anemia (associated with other DNA helicases involved in DNA damage repair, XPD, and FANCJ) and RBS, in which ESCO2 is mutated. The syndrome, known as Warsaw Breakage Syndrome, is considered to reside at an interface between DNA damage repair and sister chromatid cohesion (van der Lelij et al., 2010).
It is possible that the CoAT ESCO2 has a particularly crucial role in DNA damage repair, since mutations in ESCO2 appear to resemble mutations in DNA damage repair pathways more than the other cohesinopathies do. Indeed, ESCO2-depleted cells are hypersensitive to DNA damaging agents such as Mitomycin C (van der Lelij et al., 2009; Whelan et al., 2012a). Acetylation of SMC3 is necessary for S phase checkpoint activation and cell survival (Kim et al., 2010a), which might explain the absolute requirement for ESCO2 at this stage of the cell cycle.
Other cohesinopathy mutations also have potential to compromise DNA damage repair. Mice heterozygous for a Rad21 null mutation are hypersensitive to IR, and exhibit problems with integrity and maintenance of the gastrointestinal tract and hematopoietic system post-irradiation (Xu et al., 2010). In humans, patients with RAD21 mutations also have impaired DNA damage repair (Deardorff et al., 2012b), and knock down of RAD21 sensitizes breast cancer cells to chemical agents that damage DNA (Atienza et al., 2005; Xu et al., 2011). Therefore, full dosage and function of the Rad21 gene is crucial for DNA damage repair. In addition, depletion of SMC1 sensitizes HeLa cells to DNA damage (Bauerschmidt et al., 2010). Interestingly, the cohesin regulator PDS5B (APRIN) and the cohesin subunits RAD21 and SMC3 were recently found to associate with the BRCA2 protein. PDS5B appears to have an essential function in both the DNA damage response and homologous recombination (Brough et al., 2012).
It is not clear to what extent DNA damage repair defects contribute the pathology of cohesinopathies (Dorsett and Strom, 2012), but evidence suggests that most cohesinopathy mutations are likely to impact on the cell cycle in intra-S and G2 phases, when DNA damage repair takes place. Insufficiency of DNA damage repair should lead to checkpoint activation and cell death, potentially resulting in a paucity of cells for adequate development. However, many cohesinopathy mutations give rise to altered transcription of developmental regulators rather than cell cycle phenotypes, raising the question of how distinct outcomes arise from mutations in proteins with a related function in the cell cycle.
We propose a model to explain the diverse phenotypes observed downstream of cohesinopathy genes, in which different phenotypes emerge according to the “phase” of the cohesin cycle that is most affected by a particular cohesinopathy mutation in a given population of cells (Figure 4). In this model, mutations affecting cohesin loading and its residency times on chromatin in interphase have a higher potential to influence the regulation of gene expression, since this function can be exquisitely sensitive to cohesin dose. Alternatively, mutations affecting the “cohesive” form of cohesin have more potential to impact on cell division, DNA damage repair, and cell cycle checkpoints. The consequences are that the latter mutations will affect sister chromatid cohesion, and initiate cell death pathways. Shared phenotypes such as microcephaly, craniofacial defects, and cognitive impairment are likely to lie at the interface between these two pathways.
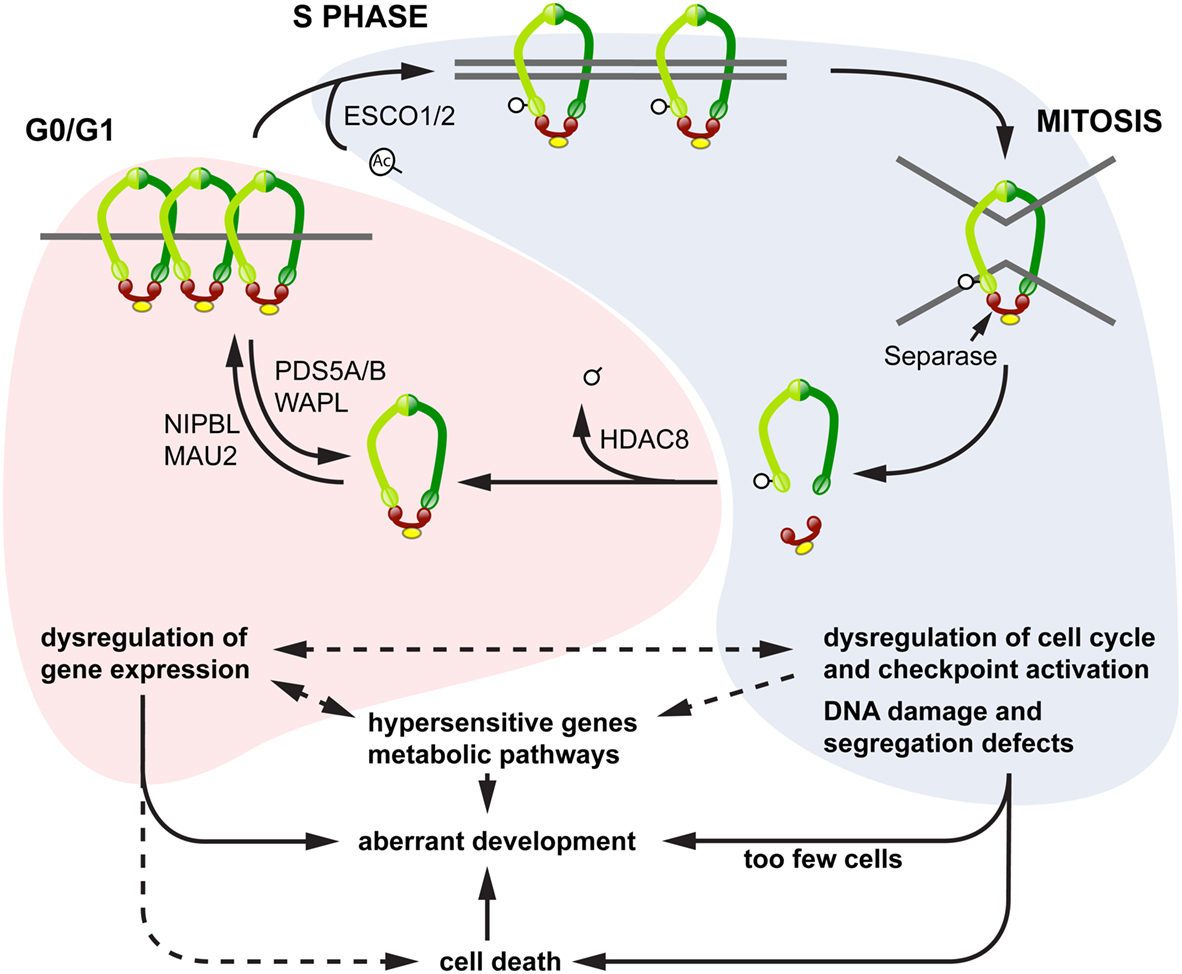
Figure 4. Model for diverse cohesinopathy phenotypes. In interphase (red shading), cohesin binding to chromatin is dynamic, with varying residency times. Interphase cohesin binding is likely to be cell type-specific and to contribute toward regulating developmental genes. Mutations in cohesin subunits and their key interphase regulators (e.g., Nipbl, Hdac8) primarily impact on the regulation of gene expression, including transcriptional regulation of growth pathways. This results in syndromic developmental defects that derive from dysregulated transcription, with the possibility of cell death as a contributing factor. From S phase to G2/M (blue shading), the overriding function of cohesin involves sister chromatid cohesion and DNA damage repair. Key regulators in this process include the CoAT ESCO2 and other DNA damage repair proteins. Mutations in these regulators result in chromosome segregation defects, genomic instability, and cell death. Increased cell death and reduced cell proliferation results in too few cells to make up body structures, leading to a different class of developmental defects and dysregulation of metabolic pathways. Transcription of a small subset of hypersensitive genes, including some in the Notch signaling pathway, appears to be sensitive to both interphase and S/G2 modes of cohesin binding.
It is important to note that cohesin subunits and the cohesin loading kollerin complex participate in all cohesin-related processes, including transcription regulation during interphase, chromatid cohesion during S phase, and DNA damage repair. Therefore mutations in genes encoding these proteins have potential to disrupt all the processes shown in the model (Figure 4). However, diverse outcomes from different cohesinopathy mutations could result if certain processes have differential sensitivity to loss of cohesin components and regulators, in distinct cell populations.
For example, zebrafish embryos zygotic null for rad21 contain heavy maternal loading of Rad21 protein and are able to develop for about 20 h before cell cycle deficiencies halt growth. However, well before cell cycle defects have any impact, rad21 null embryos fail to activate runx1 expression in the hematopoietic mesoderm (Horsfield et al., 2007). Thus, there is a threshold level of cohesin essential for runx1 expression that is below the level necessary to sustain cell division. The primary impact of suboptimal levels of Rad21 is that of altered gene expression in a subpopulation of cells, and the secondary impact of cell cycle arrest is not observed until Rad21 levels are further depleted. Radiation sensitivity observed in Rad21 heterozygous mice (Xu et al., 2010) and in cells of patients with compromised RAD21 function (Deardorff et al., 2012b) indicates other functions of Rad21 are also dose-sensitive.
In summary, the phenotypic outcome of cohesinopathy mutations may differ between cell populations and in any given cell population, depend upon the degree of sensitivity of gene expression to cohesin levels, the requirement for cell proliferation, and the presence of environmental stressors such as DNA damaging agents. There is likely to be significant overlap in these contributing factors to cohesin-related developmental disorders.
Although considerable progress has been made over the last 10 years in the understanding of cohesin function in the cell cycle, transcription, and human developmental disease, important questions remain. How is the transcriptional role of cohesin coordinated with its role in genome organization, cell division, and DNA repair? Why do some cohesinopathy mutations lead to developmental gene dysregulation, while others lead to chromosome segregation defects and cell death? Human syndromes and animal models have potential to lend important insight into the integration of cohesin functions in cell division and development. Continued research will be vital for understanding the pathology of cohesinopathy syndromes, and the development of future potential for clinical management or therapy.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors wish to thank Matt Deardorff for helpful discussions. Research on developmental roles for cohesin in the authors’ laboratory is supported by the Royal Society of NZ Marsden Fund, Lottery Health Research NZ, the KD Kirkby Trust, and Cure Kids.
Amsterdam, A., Nissen, R. M., Sun, Z., Swindell, E. C., Farrington, S., and Hopkins, N. (2004). Identification of 315 genes essential for early zebrafish development. Proc. Natl. Acad. Sci. U.S.A. 101, 12792–12797.
Atienza, J. M., Roth, R. B., Rosette, C., Smylie, K. J., Kammerer, S., Rehbock, J., Ekblom, J., and Denissenko, M. F. (2005). Suppression of RAD21 gene expression decreases cell growth and enhances cytotoxicity of etoposide and bleomycin in human breast cancer cells. Mol. Cancer Ther. 4, 361–368.
Ball, A. R. Jr., and Yokomori, K. (2008). Damage-induced reactivation of cohesin in postreplicative DNA repair. Bioessays 30, 5–9.
Bauerschmidt, C., Arrichiello, C., Burdak-Rothkamm, S., Woodcock, M., Hill, M. A., Stevens, D. L., and Rothkamm, K. (2010). Cohesin promotes the repair of ionizing radiation-induced DNA double-strand breaks in replicated chromatin. Nucleic Acids Res. 38, 477–487.
Beckouet, F., Hu, B., Roig, M. B., Sutani, T., Komata, M., Uluocak, P., Katis, V. L., Shirahige, K., and Nasmyth, K. (2010). An Smc3 acetylation cycle is essential for establishment of sister chromatid cohesion. Mol. Cell 39, 689–699.
Ben-Shahar, T. R., Heeger, S., Lehane, C., East, P., Flynn, H., Skehel, M., and Uhlmann, F. (2008). Eco1-dependent cohesin acetylation during establishment of sister chromatid cohesion. Science 321, 563–566.
Bermudez, V. P., Farina, A., Higashi, T.L., Du, F., Tappin, I., Takahashi, T. S., and Hurwitz, J. (2012). In vitro loading of human cohesin on DNA by the human Scc2-Scc4 loader complex. Proc. Natl. Acad. Sci. U.S.A. 109, 9366–9371.
Borges, V., Lehane, C., Lopez-Serra, L., Flynn, H., Skehel, M., Rolef Ben-Shahar, T., and Uhlmann, F. (2010). Hos1 deacetylates Smc3 to close the cohesin acetylation cycle. Mol. Cell 39, 677–688.
Bose, T., Lee, K. K., Lu, S., Xu, B., Harris, B., Slaughter, B., Unruh, J., Garrett, A., Mcdowell, W., Box, A., Li, H., Peak, A., Ramachandran, S., Seidel, C., and Gerton, J. L. (2012). Cohesin proteins promote ribosomal RNA production and protein translation in yeast and human cells. PLoS Genet. 8, e1002749. doi:10.1371/journal.pgen.1002749
Braunholz, D., Hullings, M., Gil-Rodriguez, M. C., Fincher, C. T., Mallozzi, M. B., Loy, E., Albrecht, M., Kaur, M., Limon, J., Rampuria, A., Clark, D., Kline, A., Dalski, A., Eckhold, J., Tzschach, A., Hennekam, R., Gillessen-Kaesbach, G., Wierzba, J., Krantz, I. D., Deardorff, M. A., and Kaiser, F. J. (2012). Isolated NIBPL missense mutations that cause Cornelia de Lange syndrome alter MAU2 interaction. Eur. J. Hum. Genet. 20, 271–276.
Brough, R., Bajrami, I., Vatcheva, R., Natrajan, R., Reis-Filho, J. S., Lord, C. J., and Ashworth, A. (2012). APRIN is a cell cycle specific BRCA2-interacting protein required for genome integrity and a predictor of outcome after chemotherapy in breast cancer. EMBO J. 31, 1160–1176.
Carretero, M., Remeseiro, S., and Losada, A. (2010). Cohesin ties up the genome. Curr. Opin. Cell Biol. 22, 781–787.
Chien, R., Zeng, W., Kawauchi, S., Bender, M. A., Santos, R., Gregson, H. C., Schmiesing, J. A., Newkirk, D., Kong, X., Ball, A. R. Jr., Calof, A. L., Lander, A. D., Groudine, M. T., and Yokomori, K. (2011). Cohesin mediates chromatin interactions that regulate mammalian {beta}-globin expression. J. Biol. Chem. 286, 17870–17878.
Ciccia, A., and Elledge, S. J. (2010). The DNA damage response: making it safe to play with knives. Mol. Cell 40, 179–204.
Craig, J. M., and Choo, K. H. (2005). Kiss and break up – a safe passage to anaphase in mitosis and meiosis. Chromosoma 114, 252–262.
Cuadrado, A., Remeseiro, S., Gomez-Lopez, G., Pisano, D. G., and Losada, A. (2012). The specific contributions of cohesin-SA1 to cohesion and gene expression: implications for cancer and development. Cell Cycle 11, 2233–2238.
de Lange, C. (1933). Sur un type nouveau de dégénération (typus Amstelodamensis). Archives de médecine des enfants 36, 713–719.
De Piccoli, G., Torres-Rosell, J., and Aragon, L. (2009). The unnamed complex: what do we know about Smc5-Smc6? Chromosome Res. 17, 251–263.
Deardorff, M., Bando, M., Nakato, R., Itoh, T., Minamino, M., Saitoh, K., Komata, M., Katou, Y., Clark, D., Cole, K., De Baere, E., Decroos, C., Ernst, S., Francey, L., Gyftodimou, Y., Hirashima, K., Hullings, M., Ishikawa, Y., Kaur, M., Kiyono, T., Lombardi, P., Mortier, G., Nozaki, N., Petersen, M., Seimiya, H., Siu, V., Suzuki, Y., Takagaki, K., Tyshchenko, N., Wilde, J., Willems, P., Gillessen-Kaesbach, G., Christianson, D., Kaiser, F., Jackson, L., Hirota, T., Krantz, I., and Shirahige, K. (2012a). HDAC8 mutations in Cornelia de Lange syndrome provide insight into the cohesin acetylation cycle. Nature. doi:10.1038/nature11316
Deardorff, M. A., Wilde, J. J., Albrecht, M., Dickinson, E., Tennstedt, S., Braunholz, D., Monnich, M., Yan, Y., Xu, W., Gil-Rodriguez, M. C., Clark, D., Hakonarson, H., Halbach, S., Michelis, L. D., Rampuria, A., Rossier, E., Spranger, S., Van Maldergem, L., Lynch, S. A., Gillessen-Kaesbach, G., Ludecke, H. J., Ramsay, R. G., Mckay, M. J., Krantz, I. D., Xu, H., Horsfield, J. A., and Kaiser, F. J. (2012b). RAD21 mutations cause a human cohesinopathy. Am. J. Hum. Genet. 90, 1014–1027.
Deardorff, M. A., Kaur, M., Yaeger, D., Rampuria, A., Korolev, S., Pie, J., Gil-Rodriguez, C., Arnedo, M., Loeys, B., Kline, A. D., Wilson, M., Lillquist, K., Siu, V., Ramos, F. J., Musio, A., Jackson, L. S., Dorsett, D., and Krantz, I. D. (2007). Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of Cornelia de Lange syndrome with predominant mental retardation. Am. J. Hum. Genet. 80, 485–494.
Dekker, J. (2012). CTCF and cohesin help neurons raise their self-awareness. Proc. Natl. Acad. Sci. U.S.A. 109, 8799–9599.
Denes, V., Pilichowska, M., Makarovskiy, A., Carpinito, G., and Geck, P. (2010). Loss of a cohesin-linked suppressor APRIN (Pds5b) disrupts stem cell programs in embryonal carcinoma: an emerging cohesin role in tumor suppression. Oncogene 29, 3446–3498.
Dorsett, D. (2009). Cohesin, gene expression and development: lessons from Drosophila. Chromosome Res. 17, 185–200.
Dorsett, D. (2010). Gene regulation: the cohesin ring connects developmental highways. Curr. Biol. 20, R886–R888.
Dorsett, D. (2011). Cohesin: genomic insights into controlling gene transcription and development. Curr. Opin. Genet. Dev. 21, 199–206.
Dorsett, D., and Strom, L. (2012). The ancient and evolving roles of cohesin in gene expression and DNA repair. Curr. Biol. 22, R240–R250.
Esumi, S., Kakazu, N., Taguchi, Y., Hirayama, T., Sasaki, A., Hirabayashi, T., Koide, T., Kitsukawa, T., Hamada, S., and Yagi, T. (2005). Monoallelic yet combinatorial expression of variable exons of the protocadherin-alpha gene cluster in single neurons. Nat. Genet. 37, 171–176.
Fay, A., Misulovin, Z., Li, J., Schaaf, C. A., Gause, M., Gilmour, D. S., and Dorsett, D. (2011). Cohesin selectively binds and regulates genes with paused RNA polymerase. Curr. Biol. 21, 1624–1634.
Gandhi, R., Gillespie, P. J., and Hirano, T. (2006). Human Wapl is a cohesin-binding protein that promotes sister-chromatid resolution in mitotic prophase. Curr. Biol. 16, 2406–2417.
Gause, M., Misulovin, Z., Bilyeu, A., and Dorsett, D. (2010). Dosage-sensitive regulation of cohesin chromosome binding and dynamics by Nipped-B, Pds5, and Wapl. Mol. Cell. Biol. 30, 4940–4951.
Gerlich, D., Koch, B., Dupeux, F., Peters, J.-M., and Ellenberg, J. (2006). Live-cell imaging reveals a stable cohesin-chromatin interaction after but not before DNA replication. Curr. Biol. 16, 1571–1578.
Gordillo, M., Vega, H., and Jabs, E. W. (1993). “Roberts syndrome,” in GeneReviews, eds R. A. Pagon, T. D. Bird, C. R. Dolan, K. Stephens, and M. P. Adam (Seattle: University of Washington). Available at: http://www.ncbi.nlm.nih.gov/books/NBK1153/
Gordillo, M., Vega, H., Trainer, A. H., Hou, F., Sakai, N., Luque, R., Kayserili, H., Basaran, S., Skovby, F., Hennekam, R. C., Uzielli, M. L., Schnur, R. E., Manouvrier, S., Chang, S., Blair, E., Hurst, J. A., Forzano, F., Meins, M., Simola, K. O., Raas-Rothschild, A., Schultz, R. A., McDaniel, L. D., Ozono, K., Inui, K., Zou, H., and Jabs, E. W. (2008). The molecular mechanism underlying Roberts syndrome involves loss of ESCO2 acetyltransferase activity. Hum. Mol. Genet. 17, 2172–2180.
Guacci, V., Koshland, D., and Strunnikov, A. (1997). A direct link between sister chromatid cohesion and chromosome condensation revealed through the analysis of MCD1 in S. cerevisiae. Cell 91, 47–57.
Hadjur, S., Williams, L. M., Ryan, N. K., Cobb, B. S., Sexton, T., Fraser, P., Fisher, A. G., and Merkenschlager, M. (2009). Cohesins form chromosomal cis-interactions at the developmentally regulated IFNG locus. Nature 460, 410–413.
Haering, C. H., Farcas, A. M., Arumugam, P., Metson, J., and Nasmyth, K. (2008). The cohesin ring concatenates sister DNA molecules. Nature 454, 297–301.
Hauf, S., Roitinger, E., Koch, B., Dittrich, C. M., Mechtler, K., and Peters, J. M. (2005). Dissociation of cohesin from chromosome arms and loss of arm cohesion during early mitosis depends on phosphorylation of SA2. PLoS Biol. 3, e69. doi:10.1371/journal.pbio.0030069
Heidinger-Pauli, J. M., Unal, E., Guacci, V., and Koshland, D. (2008). The kleisin subunit of cohesin dictates damage-induced cohesion. Mol. Cell 31, 47–56.
Heidinger-Pauli, J. M., Unal, E., and Koshland, D. (2009). Distinct targets of the Eco1 acetyltransferase modulate cohesion in S phase and in response to DNA damage. Mol. Cell 34, 311–321.
Higashi, T. L., Ikeda, M., Tanaka, H., Nakagawa, T., Bando, M., Shirahige, K., Kubota, Y., Takisawa, H., Masukata, H., and Takahashi, T. S. (2012). The prereplication complex recruits XEco2 to chromatin to promote cohesin acetylation in Xenopus egg extracts. Curr. Biol. 22, 977–988.
Hirano, T. (2006). At the heart of the chromosome: SMC proteins in action. Nat. Rev. Mol. Cell Biol. 7, 311–322.
Hornig, N. C., Knowles, P. P., Mcdonald, N. Q., and Uhlmann, F. (2002). The dual mechanism of separase regulation by securin. Curr. Biol. 12, 973–982.
Horsfield, J., Anagnostou, S., Hu, J. K. H., Cho, K. H.-Y., Geisler, R., Lieschke, G., Crosier, K., and Crosier, P. (2007). Cohesin-dependent regulation of runx genes. Development 134, 2639–2649.
Hou, C., Dale, R., and Dean, A. (2010). Cell type specificity of chromatin organization mediated by CTCF and cohesin. Proc. Natl. Acad. Sci. U.S.A. 107, 3651–3656.
Hou, F., and Zou, H. (2005). Two human orthologues of Eco1/Ctf7 acetyltransferases are both required for proper sister-chromatid cohesion. Mol. Biol. Cell 16, 3908–3918.
Hu, B., Itoh, T., Mishra, A., Katoh, Y., Chan, K. L., Upcher, W., Godlee, C., Roig, M. B., Shirahige, K., and Nasmyth, K. (2011). ATP hydrolysis is required for relocating cohesin from sites occupied by its Scc2/4 loading complex. Curr. Biol. 21, 12–24.
Huang, C. E., Milutinovich, M., and Koshland, D. (2005). Rings, bracelet or snaps: fashionable alternatives for Smc complexes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 360, 537–542.
Ireland, M., Donnai, D., and Burn, J. (1993). Brachmann-de Lange syndrome. Delineation of the clinical phenotype. Am. J. Med. Genet. 47, 959–964.
Ivanov, D., Schleiffer, A., Eisenhaber, F., Mechtler, K., Haering, C. H., and Nasmyth, K. (2002). Eco1 is a novel acetyltransferase that can acetylate proteins involved in cohesion. Curr. Biol. 12, 323–328.
Jackson, L., Kline, A. D., Barr, M. A., and Koch, S. (1993). de Lange syndrome: a clinical review of 310 individuals. Am. J. Med. Genet. 47, 940–946.
Jessberger, R. (2009). Cohesin’s dual role in the DNA damage response: repair and checkpoint activation. EMBO J. 28, 2491–2494.
Kagey, M. H., Newman, J. J., Bilodeau, S., Zhan, Y., Orlando, D. A., Van Berkum, N. L., Ebmeier, C. C., Goossens, J., Rahl, P. B., Levine, S. S., Taatjes, D. J., Dekker, J., and Young, R. A. (2010). Mediator and cohesin connect gene expression and chromatin architecture. Nature 467, 430–435.
Kawauchi, S., Calof, A. L., Santos, R., Lopez-Burks, M. E., Young, C. M., Hoang, M. P., Chua, A., Lao, T., Lechner, M. S., Daniel, J. A., Nussenzweig, A., Kitzes, L., Yokomori, K., Hallgrimsson, B., and Lander, A. D. (2009). Multiple organ system defects and transcriptional dysregulation in the Nipbl(+/-) mouse, a model of Cornelia de Lange syndrome. PLoS Genet. 5, e1000650. doi:10.1371/journal.pgen.1000650
Kenna, M. A., and Skibbens, R. V. (2003). Mechanical link between cohesion establishment and DNA replication: Ctf7p/Eco1p, a cohesion establishment factor, associates with three different replication factor C complexes. Mol. Cell. Biol. 23, 2999–3007.
Kim, B. J., Li, Y., Zhang, J., Xi, Y., Yang, T., Jung, S. Y., Pan, X., Chen, R., Li, W., Wang, Y., and Qin, J. (2010a). Genome-wide reinforcement of cohesin binding at pre-existing cohesin sites in response to ionizing radiation in human cells. J. Biol. Chem. 285, 22784–22792.
Kim, H. S., Baek, K. H., Ha, G. H., Lee, J. C., Kim, Y. N., Lee, J., Park, H. Y., Lee, N. R., Lee, H., Cho, Y., and Lee, C. W. (2010b). The hsSsu72 phosphatase is a cohesin-binding protein that regulates the resolution of sister chromatid arm cohesion. EMBO J. 29, 3544–3557.
Kim, Y. J., Cecchini, K. R., and Kim, T. H. (2011). Conserved, developmentally regulated mechanism couples chromosomal looping and heterochromatin barrier activity at the homeobox gene A locus. Proc. Natl. Acad. Sci. U.S.A. 108, 7391–7396.
Krantz, I. D., Mccallum, J., Descipio, C., Kaur, M., Gillis, L. A., Yaeger, D., Jukofsky, L., Wasserman, N., Bottani, A., Morris, C. A., Nowaczyk, M. J., Toriello, H., Bamshad, M. J., Carey, J. C., Rappaport, E., Kawauchi, S., Lander, A. D., Calof, A. L., Li, H. H., Devoto, M., and Jackson, L. G. (2004). Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nat. Genet. 36, 631–635.
Kueng, S., Hegemann, B., Peters, B. H., Lipp, J. J., Schleiffer, A., Mechtler, K., and Peters, J. M. (2006). Wapl controls the dynamic association of cohesin with chromatin. Cell 127, 955–967.
Lafont, A. L., Song, J., and Rankin, S. (2010). Sororin cooperates with the acetyltransferase Eco2 to ensure DNA replication-dependent sister chromatid cohesion. Proc. Natl. Acad. Sci. U.S.A. 107, 20364–20369.
Lara-Pezzi, E., Pezzi, N., Prieto, I., Barthelemy, I., Carreiro, C., Martinez, A., Maldonado-Rodriguez, A., Lopez-Cabrera, M., and Barbero, J. L. (2004). Evidence of a transcriptional co-activator function of cohesin STAG/SA/Scc3. J. Biol. Chem. 279, 6553–6559.
Lau, A., Blitzblau, H., and Bell, S. P. (2002). Cell-cycle control of the establishment of mating-type silencing in S. cerevisiae. Genes Dev. 16, 2935–2945.
Leem, Y. E., Choi, H. K., Jung, S. Y., Kim, B. J., Lee, K. Y., Yoon, K., Qin, J., Kang, J. S., and Kim, S. T. (2011). Esco2 promotes neuronal differentiation by repressing Notch signaling. Cell. Signal. 23, 1876–1884.
Lengronne, A., Katou, Y., Mori, S., Yokobayashi, S., Kelly, G. P., Itoh, T., Watanabe, Y., Shirahige, K., and Uhlmann, F. (2004). Cohesin relocation from sites of chromosomal loading to places of convergent transcription. Nature 430, 573–578.
Liu, J., and Krantz, I. D. (2008). Cohesin and human disease. Annu. Rev. Genomics Hum. Genet. 9, 303–320.
Liu, J., and Krantz, I. D. (2009). Cornelia de Lange syndrome, cohesin, and beyond. Clin. Genet. 76, 303–314.
Liu, J., Zhang, Z., Bando, M., Itoh, T., Deardorff, M. A., Clark, D., Kaur, M., Tandy, S., Kondoh, T., Rappaport, E., Spinner, N. B., Vega, H., Jackson, L. G., Shirahige, K., and Krantz, I. D. (2009). Transcriptional dysregulation in NIPBL and cohesin mutant human cells. PLoS Biol. 7, e1000119. doi:10.1371/journal.pbio.1000119
Losada, A., Hirano, M., and Hirano, T. (2002). Cohesin release is required for sister chromatid resolution, but not for condensin-mediated compaction, at the onset of mitosis. Genes Dev. 16, 3004–3016.
Mannini, L., Liu, J., Krantz, I. D., and Musio, A. (2010). Spectrum and consequences of SMC1A mutations: the unexpected involvement of a core component of cohesin in human disease. Hum. Mutat. 31, 5–10.
McNairn, A. J., and Gerton, J. L. (2008). Cohesinopathies: one ring, many obligations. Mutat. Res. 647, 103–111.
Mehta, G. D., Rizvi, S. M., and Ghosh, S. K. (2012). Cohesin: a guardian of genome integrity. Biochim. Biophys. Acta. 1823, 1324–1342.
Merkenschlager, M. (2010). Cohesin: a global player in chromosome biology with local ties to gene regulation. Curr. Opin. Genet. Dev. 20, 555–561.
Michaelis, C., Ciosk, R., and Nasmyth, K. (1997). Cohesins: chromosomal proteins that prevent premature separation of sister chromatids. Cell 91, 35–45.
Mishiro, T., Ishihara, K., Hino, S., Tsutsumi, S., Aburatani, H., Shirahige, K., Kinoshita, Y., and Nakao, M. (2009). Architectural roles of multiple chromatin insulators at the human apolipoprotein gene cluster. EMBO J. 28, 1234–1245.
Misulovin, Z., Schwartz, Y. B., Li, X. Y., Kahn, T. G., Gause, M., Macarthur, S., Fay, J. C., Eisen, M. B., Pirrotta, V., Biggin, M. D., and Dorsett, D. (2008). Association of cohesin and Nipped-B with transcriptionally active regions of the Drosophila melanogaster genome. Chromosoma 117, 89–102.
Moldovan, G. L., Pfander, B., and Jentsch, S. (2006). PCNA controls establishment of sister chromatid cohesion during S phase. Mol. Cell 23, 723–732.
Monahan, K., Rudnick, N., Kehayova, P., Pauli, F., Newberry, K., Myers, R., and Maniatis, T. (2012). Role of CCCTC binding factor (CTCF) and cohesin in the generation of single-cell diversity of Protocadherin-Œ± gene expression. Proc. Natl. Acad. Sci. U.S.A. 109, 9125–9155.
Monnich, M., Banks, S., Eccles, M., Dickinson, E., and Horsfield, J. (2009). Expression of cohesin and condensin genes during zebrafish development supports a non-proliferative role for cohesin. Gene Expr. Patterns 9, 586–594.
Monnich, M., Kuriger, Z., Print, C. G., and Horsfield, J. A. (2011). A zebrafish model of Roberts syndrome reveals that esco2 depletion interferes with development by disrupting the cell cycle. PLoS ONE 6, e20051. doi:10.1371/journal.pone.0020051
Musio, A., Selicorni, A., Focarelli, M. L., Gervasini, C., Milani, D., Russo, S., Vezzoni, P., and Larizza, L. (2006). X-linked Cornelia de Lange syndrome owing to SMC1L1 mutations. Nat. Genet. 38, 528–530.
Muto, A., Calof, A. L., Lander, A. D., and Schilling, T. F. (2011). Multifactorial origins of heart and gut defects in nipbl-deficient zebrafish, a model of Cornelia de Lange syndrome. PLoS Biol. 9, e1001181. doi:10.1371/journal.pbio.1001181
Nasmyth, K. (2011). Cohesin: a catenase with separate entry and exit gates? Nat. Cell Biol. 13, 1170–1177.
Nasmyth, K., and Haering, C. H. (2009). Cohesin: its roles and mechanisms. Annu. Rev. Genet. 43, 525–558.
Nativio, R., Wendt, K. S., Ito, Y., Huddleston, J. E., Uribe-Lewis, S., Woodfine, K., Krueger, C., Reik, W., Peters, J. M., and Murrell, A. (2009). Cohesin is required for higher-order chromatin conformation at the imprinted IGF2-H19 locus. PLoS Genet. 5, e1000739. doi:10.1371/journal.pgen.1000739
Nishiyama, T., Ladurner, R., Schmitz, J., Kreidl, E., Schleiffer, A., Bhaskara, V., Bando, M., Shirahige, K., Hyman, A. A., Mechtler, K., and Peters, J. M. (2010). Sororin mediates sister chromatid cohesion by antagonizing wapl. Cell 143, 737–749.
Nitzsche, A., Paszkowski-Rogacz, M., Matarese, F., Janssen-Megens, E. M., Hubner, N. C., Schulz, H., De Vries, I., Ding, L., Huebner, N., Mann, M., Stunnenberg, H. G., and Buchholz, F. (2011). RAD21 Cooperates with pluripotency transcription factors in the maintenance of embryonic stem cell identity. PLoS ONE 6, e19470. doi:10.1371/journal.pone.0019470
Parelho, V., Hadjur, S., Spivakov, M., Leleu, M., Sauer, S., Gregson, H. C., Jarmuz, A., Canzonetta, C., Webster, Z., Nesterova, T., Cobb, B. S., Yokomori, K., Dillon, N., Aragon, L., Fisher, A. G., and Merkenschlager, M. (2008). Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell 132, 422–433.
Pauli, A., Althoff, F., Oliveira, R. A., Heidmann, S., Schuldiner, O., Lehner, C. F., Dickson, B. J., and Nasmyth, K. (2008). Cell-type-specific TEV protease cleavage reveals cohesin functions in Drosophila neurons. Dev. Cell 14, 239–251.
Pauli, A., Van Bemmel, J. G., Oliveira, R. A., Itoh, T., Shirahige, K., Van Steensel, B., and Nasmyth, K. (2010). A direct role for cohesin in gene regulation and ecdysone response in Drosophila salivary glands. Curr. Biol. 20, 1787–1798.
Rankin, S., Ayad, N. G., and Kirschner, M. W. (2005). Sororin, a substrate of the anaphase-promoting complex, is required for sister chromatid cohesion in vertebrates. Mol. Cell 18, 185–200.
Remeseiro, S., Cuadrado, A., Carretero, M., Martinez, P., Drosopoulos, W. C., Canamero, M., Schildkraut, C. L., Blasco, M. A., and Losada, A. (2012a). Cohesin-SA1 deficiency drives aneuploidy and tumourigenesis in mice due to impaired replication of telomeres. EMBO J. 31, 2076–2089.
Remeseiro, S., Cuadrado, A., Gomez-Lopez, G., Pisano, D. G., and Losada, A. (2012b). A unique role of cohesin-SA1 in gene regulation and development. EMBO J. 31, 2090–2102.
Revenkova, E., Focarelli, M. L., Susani, L., Paulis, M., Bassi, M. T., Mannini, L., Frattini, A., Delia, D., Krantz, I., Vezzoni, P., Jessberger, R., and Musio, A. (2009). Cornelia de Lange syndrome mutations in SMC1A or SMC3 affect binding to DNA. Hum. Mol. Genet. 18, 418–427.
Rhodes, J. M., Bentley, F. K., Print, C. G., Dorsett, D., Misulovin, Z., Dickinson, E. J., Crosier, K. E., Crosier, P. S., and Horsfield, J. A. (2010). Positive regulation of c-Myc by cohesin is direct, and evolutionarily conserved. Dev. Biol. 344, 637–649.
Rhodes, J. M., Mcewan, M., and Horsfield, J. A. (2011). Gene regulation by cohesin in cancer: is the ring an unexpected party to proliferation? Mol. Cancer Res. 9, 1587–1607.
Rohatgi, S., Clark, D., Kline, A. D., Jackson, L. G., Pie, J., Siu, V., Ramos, F. J., Krantz, I. D., and Deardorff, M. A. (2010). Facial diagnosis of mild and variant CdLS: Insights from a dysmorphologist survey. Am. J. Med. Genet. A 152A, 1641–1653.
Rollins, R. A., Korom, M., Aulner, N., Martens, A., and Dorsett, D. (2004). Drosophila nipped-B protein supports sister chromatid cohesion and opposes the stromalin/Scc3 cohesion factor to facilitate long-range activation of the cut gene. Mol. Cell. Biol. 24, 3100–3111.
Rollins, R. A., Morcillo, P., and Dorsett, D. (1999). Nipped-B, a Drosophila homologue of chromosomal adherins, participates in activation by remote enhancers in the cut and Ultrabithorax genes. Genetics 152, 577–593.
Rowland, B. D., Roig, M. B., Nishino, T., Kurze, A., Uluocak, P., Mishra, A., Beckouet, F., Underwood, P., Metson, J., Imre, R., Mechtler, K., Katis, V. L., and Nasmyth, K. (2009). Building sister chromatid cohesion: smc3 acetylation counteracts an antiestablishment activity. Mol. Cell 33, 763–774.
Rubio, E. D., Reiss, D. J., Welcsh, P. L., Disteche, C. M., Filippova, G. N., Baliga, N. S., Aebersold, R., Ranish, J. A., and Krumm, A. (2008). CTCF physically links cohesin to chromatin. Proc. Natl. Acad. Sci. U.S.A. 105, 8309–8314.
Salah, S. M., and Nasmyth, K. (2000). Destruction of the securin Pds1p occurs at the onset of anaphase during both meiotic divisions in yeast. Chromosoma 109, 27–34.
Schaaf, C. A., Misulovin, Z., Sahota, G., Siddiqui, A. M., Schwartz, Y. B., Kahn, T. G., Pirrotta, V., Gause, M., and Dorsett, D. (2009). Regulation of the Drosophila enhancer of split and invected-engrailed gene complexes by sister chromatid cohesion proteins. PLoS ONE 4, e6202. doi:10.1371/journal.pone.0006202
Schmidt, D., Schwalie, P., Ross-Innes, C. S., Hurtado, A., Brown, G., Carroll, J., Flicek, P., and Odom, D. (2010). A CTCF-independent role for cohesin in tissue-specific transcription. Genome Res. 20, 578–588.
Schmitz, J., Watrin, E., Lenart, P., Mechtler, K., and Peters, J. M. (2007). Sororin is required for stable binding of cohesin to chromatin and for sister chromatid cohesion in interphase. Curr. Biol. 17, 630–636.
Schuldiner, O., Berdnik, D., Levy, J. M., Wu, J. S., Luginbuhl, D., Gontang, A. C., and Luo, L. (2008). piggyBac-based mosaic screen identifies a postmitotic function for cohesin in regulating developmental axon pruning. Dev. Cell 14, 227–238.
Schule, B., Oviedo, A., Johnston, K., Pai, S., and Francke, U. (2005). Inactivating mutations in ESCO2 cause SC phocomelia and Roberts syndrome: no phenotype-genotype correlation. Am. J. Hum. Genet. 77, 1117–1128.
Seitan, V. C., Banks, P., Laval, S., Majid, N. A., Dorsett, D., Rana, A., Smith, J., Bateman, A., Krpic, S., Hostert, A., Rollins, R. A., Erdjument-Bromage, H., Tempst, P., Benard, C. Y., Hekimi, S., Newbury, S. F., and Strachan, T. (2006). Metazoan Scc4 homologs link sister chromatid cohesion to cell and axon migration guidance. PLoS Biol. 4, e242. doi:10.1371/journal.pbio.0040242
Shintomi, K., and Hirano, T. (2009). Releasing cohesin from chromosome arms in early mitosis: opposing actions of Wapl-Pds5 and Sgo1. Genes Dev. 23, 2224–2236.
Skibbens, R. V. (2010). Buck the establishment: reinventing sister chromatid cohesion. Trends Cell Biol. 20, 507–513.
Skibbens, R. V., Corson, L. B., Koshland, D., and Hieter, P. (1999). Ctf7p is essential for sister chromatid cohesion and links mitotic chromosome structure to the DNA replication machinery. Genes Dev. 13, 307–319.
Strom, L., Karlsson, C., Lindroos, H. B., Wedahl, S., Katou, Y., Shirahige, K., and Sjogren, C. (2007). Postreplicative formation of cohesion is required for repair and induced by a single DNA break. Science 317, 242–245.
Strom, L., Lindroos, H. B., Shirahige, K., and Sjogren, C. (2004). Postreplicative recruitment of cohesin to double-strand breaks is required for DNA repair. Mol. Cell 16, 1003–1015.
Strom, L., and Sjogren, C. (2007). Chromosome segregation and double-strand break repair – a complex connection. Curr. Opin. Cell Biol. 19, 344–349.
Sutani, T., Kawaguchi, T., Kanno, R., Itoh, T., and Shirahige, K. (2009). Budding yeast Wpl1(Rad61)-Pds5 complex counteracts sister chromatid cohesion-establishing reaction. Curr. Biol. 19, 492–497.
Teo, A. K., Arnold, S. J., Trotter, M. W., Brown, S., Ang, L. T., Chng, Z., Robertson, E. J., Dunn, N. R., and Vallier, L. (2011). Pluripotency factors regulate definitive endoderm specification through eomesodermin. Genes Dev. 25, 238–250.
Tonkin, E. T., Wang, T. J., Lisgo, S., Bamshad, M. J., and Strachan, T. (2004). NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesion proteins and fly Nipped-B, is mutated in Cornelia de Lange syndrome. Nat. Genet. 36, 636–641.
Unal, E., Heidinger-Pauli, J. M., Kim, W., Guacci, V., Onn, I., Gygi, S. P., and Koshland, D. E. (2008). A molecular determinant for the establishment of sister chromatid cohesion. Science 321, 566–569.
van der Lelij, P., Chrzanowska, K. H., Godthelp, B. C., Rooimans, M. A., Oostra, A. B., Stumm, M., Zdzienicka, M. Z., Joenje, H., and De Winter, J. P. (2010). Warsaw breakage syndrome, a cohesinopathy associated with mutations in the XPD helicase family member DDX11/ChlR1. Am. J. Hum. Genet. 86, 262–266.
van der Lelij, P., Godthelp, B. C., van Zon, W., van Gosliga, D., Oostra, A. B., Steltenpool, J., de Groot, J., Scheper, R. J., Wolthuis, R. M., Waisfisz, Q., Darroudi, F., Joenje, H., and de Winter, J. P. (2009). The cellular phenotype of Roberts syndrome fibroblasts as revealed by ectopic expression of ESCO2. PLoS ONE 4, e6936. doi:10.1371/journal.pone.0006936
Vaur, S., Feytout, A., Vazquez, S., and Javerzat, J. P. (2012). Pds5 promotes cohesin acetylation and stable cohesin-chromosome interaction. EMBO Rep. 13, 645–652.
Vega, H., Trainer, A. H., Gordillo, M., Crosier, M., Kayserili, H., Skovby, F., Uzielli, M. L., Schnur, R. E., Manouvrier, S., Blair, E., Hurst, J. A., Forzano, F., Meins, M., Simola, K. O., Raas-Rothschild, A., Hennekam, R. C., and Jabs, E. W. (2010). Phenotypic variability in 49 cases of ESCO2 mutations, including novel missense and codon deletion in the acetyltransferase domain, correlates with ESCO2 expression and establishes the clinical criteria for Roberts syndrome. J. Med. Genet. 47, 30–37.
Vega, H., Waisfisz, Q., Gordillo, M., Sakai, N., Yanagihara, I., Yamada, M., Van Gosliga, D., Kayserili, H., Xu, C., Ozono, K., Jabs, E. W., Inui, K., and Joenje, H. (2005). Roberts syndrome is caused by mutations in ESCO2, a human homolog of yeast ECO1 that is essential for the establishment of sister chromatid cohesion. Nat. Genet. 37, 468–470.
Vierbuchen, T., Ostermeier, A., Pang, Z. P., Kokubu, Y., Sudhof, T. C., and Wernig, M. (2010). Direct conversion of fibroblasts to functional neurons by defined factors. Nature 463, 1035–1041.
Waizenegger, I., Gimenez-Abian, J. F., Wernic, D., and Peters, J. M. (2002). Regulation of human separase by securin binding and autocleavage. Curr. Biol. 12, 1368–1378.
Waizenegger, I. C., Hauf, S., Meinke, A., and Peters, J. M. (2000). Two distinct pathways remove mammalian cohesin from chromosome arms in prophase and from centromeres in anaphase. Cell 103, 399–410.
Wang, X., and Dai, W. (2005). Shugoshin, a guardian for sister chromatid segregation. Exp. Cell Res. 310, 1–9.
Watrin, E., and Peters, J. M. (2009). The cohesin complex is required for the DNA damage-induced G2/M checkpoint in mammalian cells. EMBO J. 28, 2625–2635.
Wendt, K. S., Yoshida, K., Itoh, T., Bando, M., Koch, B., Schirghuber, E., Tsutsumi, S., Nagae, G., Ishihara, K., Mishiro, T., Yahata, K., Imamoto, F., Aburatani, H., Nakao, M., Imamoto, N., Maeshima, K., Shirahige, K., and Peters, J. M. (2008). Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature 451, 796–801.
Whelan, G., Kreidl, E., Peters, J. M., and Eichele, G. (2012a). The non-redundant function of cohesin acetyltransferase Esco2: some answers and new questions. Nucleus 3. PMID: 22614755. [Epub ahead of print].
Whelan, G., Kreidl, E., Wutz, G., Egner, A., Peters, J. M., and Eichele, G. (2012b). Cohesin acetyltransferase Esco2 is a cell viability factor and is required for cohesion in pericentric heterochromatin. EMBO J. 31, 71–82.
Xiong, B., Lu, S., and Gerton, J. L. (2010). Hos1 is a lysine deacetylase for the Smc3 subunit of cohesin. Curr. Biol. 18, 1660–1665.
Xu, H., Balakrishnan, K., Malaterre, J., Beasley, M., Yan, Y., Essers, J., Appeldoorn, E., Thomaszewski, J. M., Vazquez, M., Verschoor, S., Lavin, M. F., Bertonchello, I., Ramsay, R. G., and Mckay, M. J. (2010). Rad21-cohesin haploinsufficiency impedes DNA repair and enhances gastrointestinal radiosensitivity in mice. PLoS ONE 5, e12112. doi:10.1371/journal.pone.0012112
Xu, H., Yan, M., Patra, J., Natrajan, R., Yan, Y., Swagemakers, S., Tomaszewski, J. M., Verschoor, S., Millar, E. K., Van der Spek, P., Reis-Filho, J. S., Ramsay, R. G., O’Toole, S. A., Mcneil, C. M., Sutherland, R. L., Mckay, M. J., and Fox, S. B. (2011). Enhanced RAD21 cohesin expression confers poor prognosis and resistance to chemotherapy in high grade luminal, basal and HER2 breast cancers. Breast Cancer Res. 13, R9.
Zhang, B., Chang, J., Fu, M., Huang, J., Kashyap, R., Salavaggione, E., Jain, S., Shashikant, K., Deardorff, M. A., Uzielli, M. L., Dorsett, D., Beebe, D. C., Jay, P. Y., Heuckeroth, R. O., Krantz, I., and Milbrandt, J. (2009). Dosage effects of cohesin regulatory factor PDS5 on mammalian development: implications for cohesinopathies. PLoS ONE 4, e5232. doi:10.1371/journal.pone.0005232
Zhang, B., Jain, S., Song, H., Fu, M., Heuckeroth, R. O., Erlich, J. M., Jay, P. Y., and Milbrandt, J. (2007). Mice lacking sister chromatid cohesion protein PDS5B exhibit developmental abnormalities reminiscent of Cornelia de Lange syndrome. Development 134, 3191–3201.
Zhang, J., Shi, X., Li, Y., Kim, B. J., Jia, J., Huang, Z., Yang, T., Fu, X., Jung, S. Y., Wang, Y., Zhang, P., Kim, S. T., Pan, X., and Qin, J. (2008a). Acetylation of Smc3 by Eco1 is required for S phase sister chromatid cohesion in both human and yeast. Mol. Cell 31, 143–151.
Keywords: cohesin, gene expression regulation, animal models, CdLS, RBS
Citation: Horsfield JA, Print CG and Mönnich M (2012) Diverse developmental disorders from The One Ring: distinct molecular pathways underlie the cohesinopathies. Front. Gene. 3:171. doi: 10.3389/fgene.2012.00171
Received: 08 July 2012; Paper pending published: 23 July 2012;
Accepted: 17 August 2012; Published online: 12 September 2012.
Edited by:
Michèle Amouyal, Centre National de la Recherche Scientifique, FranceReviewed by:
Jennifer Gerton, Stowers Institute for Medical Research, USACopyright: © 2012 Horsfield, Print and Mönnich. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Julia A. Horsfield, Department of Pathology, Dunedin School of Medicine, University of Otago, PO Box 913, Dunedin, New Zealand. e-mail:anVsaWEuaG9yc2ZpZWxkQG90YWdvLmFjLm56
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.