
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 11 July 2012
Sec. Epigenomics and Epigenetics
Volume 3 - 2012 | https://doi.org/10.3389/fgene.2012.00129
This article is part of the Research Topic Reproductive genetics and epigenetics View all 9 articles
 Michelle M. Denomme1,2,3
Michelle M. Denomme1,2,3 Carlee R. White1,2,3
Carlee R. White1,2,3 Carolina Gillio-Meina1,4
Carolina Gillio-Meina1,4 William A. MacDonald1,2 Bonnie J. Deroo1,2,3,5
William A. MacDonald1,2 Bonnie J. Deroo1,2,3,5 Gerald M. Kidder1,4
Gerald M. Kidder1,4 Mellissa R. W. Mann1,2,3 *
Mellissa R. W. Mann1,2,3 *Growth and maturation of healthy oocytes within follicles requires bidirectional signaling and intercellular gap junctional communication. Aberrant endocrine signaling and loss of gap junctional communication between the oocyte and granulosa cells leads to compromised folliculogenesis, oocyte maturation, and oocyte competency, consequently impairing fertility. Given that oocyte-specific DNA methylation establishment at imprinted genes occurs during this growth phase, we determined whether compromised endocrine signaling and gap junctional communication would disrupt de novo methylation acquisition using ERβ and connexin37 genetic models. To compare mutant oocytes to control oocytes, DNA methylation acquisition was first examined in individual, 20–80 μm control oocytes at three imprinted genes, Snrpn, Peg3, and Peg1. We observed that each gene has its own size-dependent acquisition kinetics, similar to previous studies. To determine whether compromised endocrine signaling and gap junctional communication disrupted de novo methylation acquisition, individual oocytes from Esr2- and Gja4-deficient mice were also assessed for DNA methylation establishment. We observed no aberrant or delayed acquisition of DNA methylation at Snrpn, Peg3, or Peg1 in oocytes from Esr2-deficient females, and no perturbation in Snrpn or Peg3 de novo methylation in oocytes from Gja4-null females. However, Gja4 deficiency resulted in a loss or delay in methylation acquisition at Peg1. One explanation for this difference between the three loci analyzed is the late establishment of DNA methylation at the Peg1 gene. These results indicate that compromised fertility though impaired intercellular communication can lead to imprinting acquisition errors. Further studies are required to determine the effects of subfertility/infertility originating from impaired signaling and intercellular communication during oogenesis on imprint maintenance during preimplantation development.
The tight regulation of monoallelic gene expression based on gametic origin is termed genomic imprinting (Bartolomei and Ferguson-Smith, 2011). This dynamic process relies on epigenetic modifications such as DNA methylation to mark, or “imprint”, one of the two parental alleles, resulting in differential gene expression in progeny (Verona et al., 2003). Gametogenesis encompasses the critical period of heritable epigenetic reprogramming for imprinted genes. Imprinted DNA methylation is first erased in primordial germ cells, subsequently allowing for de novo differential methylation at imprinted loci in oocytes and sperm (Li and Sasaki, 2011). In males, de novo DNA methylation acquisition occurs during the prenatal stages of spermatogenesis, beginning in prospermatogonia and is completed by birth (Kafri et al., 1992; Davis et al., 1999,2000; Ueda et al., 2000). In females, de novo DNA methylation is acquired after oocytes enter the growth phase of follicular development, from the primary to antral follicle stage (Lucifero et al., 2004; Hiura et al., 2006; Sato et al., 2007; Song et al., 2009). Importantly for oocytes, imprinted methylation acquisition is dependent on oocyte size and not oocyte age, with methylation levels increasing as oocyte diameter increases.
The correct establishment of germline imprints is significant as disruptions to this process can result in the development of imprinting disorders such as Beckwith–Wiedemann syndrome (BWS), Silver–Russell Syndrome (SRS), and Angelman syndrome (AS). BWS is an overgrowth disorder that is caused by imprinting defects that result in a gain of maternal methylation at the H19 imprinting control region (ICR) or a loss of maternal-specific methylation at the KCNQ1OT1 (KCNQ1 overlapping transcript 1) ICR (Weksberg et al., 2010). SRS is an intrauterine growth restricted imprinting disorder with imprinting defects at the H19 and possibly at the paternally expressed gene 1 (Peg1) imprinted domains (Eggermann, 2010). AS is a neurological disorder that is caused by loss of maternal-specific methylation at the small nuclear ribonucleoprotein N (SNRPN) ICR (Mabb et al., 2011). Sporadic epigenetic errors resulting in these disorders are reported to occur more frequently in the assisted reproductive technologies (ARTs) population (Cox et al., 2002; DeBaun et al., 2003; Gicquel et al., 2003; Maher et al., 2003; Orstavik et al., 2003; Halliday et al., 2004; Chang et al., 2005; Ludwig et al., 2005; Rossignol et al., 2006; Azzi et al., 2009; Bliek et al., 2009; Lim et al., 2009; Lennerz et al., 2010; Turner et al., 2010). For AS, patients at the highest risk for an imprinting defect have parents with prolonged infertility undergoing infertility treatment (Ludwig et al., 2005; Doornbos et al., 2007). This raises the question as to whether imprinting errors in ART patients are associated with parental infertility/subfertility. While studies have been conducted to determine the effects of ARTs on genomic imprinting, investigations of how impaired fertility may contribute to imprinting errors are lacking. In this study, we queried whether impaired fertility arising during oogenesis could lead to imprinting defects.
Development of healthy oocytes is dependent on interactions between the growing oocyte and surrounding follicular cells (Kidder and Vanderhyden, 2010). Oocytes play an important role in regulating granulosa cell development, proliferation, and differentiation, as well as steroid hormone production. In turn, follicular cells play a critical role in oocyte growth, meiotic progression, and transcriptional activity and chromatin remodeling of the oocyte genome. This synergistic partnership is facilitated by endocrine and paracrine signaling, and intercellular gap junctional communication, ensuring meiotic and developmental competence of the oocyte. In this study, we specifically examined the effects of aberrant signaling and communication on imprint acquisition.
A complex endocrine signaling pathway is active in the ovary that regulates follicle and oocyte development. 17β-estradiol acting through nuclear estrogen receptor beta (ERβ) augments the actions of follicle stimulating hormone (FSH). In the ovary, ERβ is expressed primarily in granulosa cells and at low levels in the oocyte (Drummond and Fuller, 2011). Female mice bearing a targeted deletion of the ERβ (Esr2) gene are subfertile, producing fewer oocytes following superovulation, as well as litters with fewer pups (Krege et al., 1998; Couse et al., 2000,2003,2005; Dupont et al., 2000; Emmen et al., 2005). Attenuated differentiation of granulosa cells following gonadotropin stimulation in Esr2-null mice leads to decreased antrum formation, delayed follicle maturation, and reduced follicular rupture, producing greater numbers of atretic follicles and fewer preovulatory oocytes. In addition, vascularization of the thecal layer, which is required for follicular growth, is impaired (Inzunza et al., 2007). Mechanistically, ERβ is required for optimal cAMP production in mouse granulosa cells following gonadotropin stimulation (Deroo et al., 2009). ERβ deficiency causes disruption of cAMP second messenger signaling in granulosa cells in response to FSH, producing aberrant FSH-regulated gene expression, decreased response to luteinizing hormone, and impaired ovulation and fertility.
Gap junctions are specialized channels composed of six membrane proteins termed connexins (CX). These channels are essential for communication between neighboring cells (Harris, 2001). In the mouse, CX37 and CX43 are the only connexins known to be required in developing follicles (Kidder and Vanderhyden, 2010). CX43 localizes to gap junctions in the granulosa cell membranes, enabling granulosa cell to granulosa cell communication. By comparison, CX37 constitutes the gap junctions coupling the oocyte with surrounding granulosa cells and is specifically located at the interface between the oocyte and the first layer of granulosa cells (Simon et al., 1997). Gap junctions allow the transport of nutrients, metabolites and second messengers, such as cAMP, between the granulosa cells and the oocyte (Kidder and Vanderhyden, 2010). Targeted deletion of the CX37 (Gja4) gene causes arrested folliculogenesis at the early antral stage, impaired oocyte development and meiotic competency, and premature luteinization of the follicles (Simon et al., 1997; Carabatsos et al., 2000).
In this study, we employed the Esr2– / – and Gja4– / – genetic models to interfere specifically with endocrine signaling and gap junctional communication, compromising fertility. We hypothesized that inhibition of the ERβ pathway and/or oocyte–granulosa cell gap junctional communication would lead to perturbations in imprinted methylation acquisition. To compare mutant oocytes to control oocytes, DNA methylation acquisition was first examined in individual, 20–80 μm diameter control oocytes at three imprinted genes, Snrpn, Peg3, and Peg1 (also known as Mest). Similar to previous studies (Lucifero et al., 2004; Hiura et al., 2006; Sato et al., 2007; Song et al., 2009), we observed that each gene had its own size-dependent acquisition kinetics. To determine whether compromised endocrine signaling and gap junctional communication disrupted de novo methylation acquisition, preovulatory oocytes from Esr2– / – females, and early antral stage oocytes from Gja4– / – mice were assessed for DNA methylation establishment at Snrpn, Peg3, and Peg1. We observed no aberrant or delayed acquisition of DNA methylation at Snrpn, Peg3, and Peg1 in preovulatory oocytes from ERβ-deficient females. Similarly, we found no perturbation of Snrpn and Peg3 de novo methylation in oocytes from CX37-null follicles. However, Peg1 methylation acquisition was lost or delayed in Gja4-deficient oocytes compared to controls. We attribute this to the late establishment of DNA methylation at the Peg1 gene. These results indicate that compromised fertility though impaired intercellular communication can lead to imprinting acquisition errors. Further studies are required to determine the post-fertilization effects of subfertility/infertility originating from impaired signaling and intercellular communication during oogenesis.
Ovaries were obtained from C57BL/6 female mice (Charles River) at 10, 14, 21, and 28 days postpartum (dpp), and placed in Waymouth MB 752/1 medium (Invitrogen) supplemented with 10% fetal bovine serum (Li et al., 2007). For further follicle separation, ovaries were digested in the same medium containing 2 mg/ml type I collagenase (Sigma-Aldrich) at 37°C. Primary, secondary and early tertiary (antral) follicles were liberated by repeated aspiration and expulsion with a 1 ml pipette. Follicles were washed several times in culture medium without collagenase. For oocyte isolation, follicles were centrifuged for 5 min at 4,000 rpm, re-suspended and digested in 0.05% Trypsin/EDTA in a culture dish for 15 min at 37°C. Oocytes were dissociated from the granulosa cells by repeated aspiration and expulsion with a 1 ml pipette. Oocytes were retrieved through mouth pipetting and placed in 30 μl drops of M2 medium (Sigma) for further analysis.
Ovaries were removed from Gja4– / – female mice (C57BL/6 background) at 21 and 28 dpp, and placed in Waymouth MB 752/1 medium (Invitrogen) supplemented with 10% fetal bovine serum. Gja4-null oocytes were retrieved through the same collection method as control oocytes and placed in 30 μl M2 medium (Sigma) for further analysis.
Ovaries were removed from Esr2– / – females (C57BL/6 background) at 28 dpp and placed in a 100-mm cell culture dish containing 15 ml ice-cold M199 medium (Sigma) supplemented with 1 mg/ml BSA (Invitrogen) and 2.5 g/ml gentamicin (Invitrogen; Deroo et al., 2009). Follicles were released by manual puncture with 25-gauge needles and subsequent pressure applied with a sterile spatula. Oocytes were retrieved through mouth pipetting and transferred to 30 μl drops of M2 medium (Sigma) for further analysis.
Processing, embedding, and bisulfite mutagenesis of individual oocytes was performed as previously described (Denomme et al., 2011). Briefly, oocytes were treated with 0.3 mg/ml hyaluronidase (Sigma) to remove any surrounding cumulus cells (if present), washed three times in 30 μl drops of M2 medium (Sigma), and then imaged using the Olympus IX81 microscope. Oocyte diameter was measured using Macnification v.1.8 (Orbicule). Following treatment with acidic Tyrode’s solution (Sigma) to remove the zona pellucida (if present), oocytes were washed twice in M2 medium, then individual oocytes were embedded in 10 μl of 2:1 LMP agarose and lysis solution [100 mM Tris–HCl, pH 7.5 (Bioshop), 500 mM LiCl (Sigma), 10 mM EDTA, pH 8.0 (Sigma), 1% LiDS (Bioshop), and 5 mM DTT (Sigma), 1 μl 2 mg/ml proteinase K (Sigma), and 1 μl 10% Igepal (Sigma)] under 300 μl of mineral oil (Sigma), and placed on ice for 10 min for the agarose to harden. Mineral oil was replaced with 500 μl SDS lysis buffer [450 μl 1× Tris EDTA (TE), pH 7.5 [10 mM Tris (Bioshop), 1 mM EDTA], 50 μl 10% SDS (Bioshop), 1 μl 2 mg/ml proteinase K] and incubated at 50°C overnight. Following overnight incubation, lysis buffer was replaced with 300 μl mineral oil and oocytes were either immediately treated for bisulfite conversion or frozen at -20°C for up to 5 days. Firstly, samples were placed at 90°C for 2.5 min to heat inactivate the proteinase K, and then DNA was denatured using 0.1 M NaOH (Sigma) at 37°C for 15 min. Treatment with 2.5 M bisulfite solution (0.125 M hydroquinone (Sigma), 3.8 g sodium hydrogen sulfite (Sigma), 5.5 ml water, and 1 ml 3 M NaOH) at 50°C for 3.5 h was followed by desulfonation using 0.3 M NaOH at 37°C for 15 min. Samples were washed twice in 1× TE pH 7.5 and twice in water, and then added directly to a Ready-To-Go PCR bead (GE) consisting of 15 μl water, gene-specific primers and 1 μl of 240 ng/ml transfer RNA as a carrier, with 25 μl mineral oil overlay. Negative controls (no oocyte) were processed alongside each bisulfite reaction. PCR amplification of the Snrpn ICR, Peg3 DMR, and Peg1 DMR was performed as previously described (Market-Velker et al., 2010). Following ligation into the PGEM-T easy vector (Promega) and cloning, 30 μl of colony PCR product was sent to Bio-Basic Inc. (Markham, Ontario, Canada) for sequencing. For each sample, five clones were sequenced. As MI oocytes have not extruded the first polar body, both alleles were successfully amplified in some oocytes, and only one allele was detectable in other oocytes. However, oocytes with more than two clones having very different methylation patterns and different non-CpG conversion rates were excluded from analysis, as cumulus cell contamination could not be ruled out. Table 1 gives the number of oocytes included and excluded from analysis per gene.
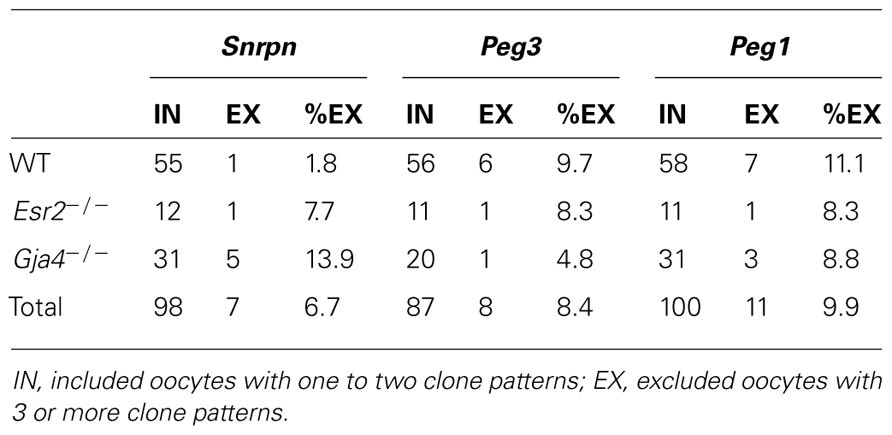
TABLE 1. Number of oocytes included and excluded from analysis
For each imprinted gene, significant difference of CpG methylation percentage was determined by a two-tailed Mann–Whitney U test between mutant oocytes and control oocytes matched for size. A diameter range of 65–80 μm was used to compare Esr2-deficient oocytes to control oocytes, while the 35–60 μm diameter range (including KO468 for Snrpn with a diameter of 60.5 μm) was used to compare the Gja4 deficient to control oocytes. A p-value of <0.05 was taken to be statistically significant.
In female mammals, imprinted DNA methylation has been shown to arise during follicle growth from the primary to the antral stage in correlation with oocyte diameter (Lucifero et al., 2004; Hiura et al., 2006), with gene-specific kinetics for imprint acquisition. However, these analyses were performed with pooled oocytes of different sizes. To compare individual mutant oocytes to control oocytes, we first needed to examine imprinted DNA methylation acquisition in individual control oocytes. C57BL/6 oocytes were collected at 10, 14, 21, and 28 dpp to obtain oocytes with a diameter range of 20–80 μm. Oocytes that were collected at 10 dpp displayed a diameter range of 20–70 μm, those at 14 dpp were 40–80 μm in diameter, at 21 dpp ranged from 50 to 70 μm, and at 28 dpp ranged from 60 to 80 μm in diameter.
Analysis of de novo methylation acquisition at the Snrpn ICR showed mean methylation levels of 8.7% in 20–40 μm, 12.6% in 40–45 μm, 9.3% in 45–50 μm, 39.3% in 50–55 μm, 82.7% in 55–60 μm, 97.0% in 60–65 μm, 82.8% in 65–70 μm, 93.8% in 70–75 μm, and 98.0% in 75–80 μm oocytes (Figures 1 and 2). Likewise, mean methylation levels at the Peg3 DMR were 1.6% in 20–40 μm, 11.2% in 40–45 μm, 16.1% in 45–50 μm, 22.9% in 50–55 μm, 47.5% in 55–60 μm, 51.7% in 60–65 μm, 82.6% in 65–70 μm, 85% in 70–75 μm, and 94.0% in 75–80 μm oocytes (Figures 3 and 4). For the Peg1 DMR, mean methylation levels were 4.3% in 20–40 μm, 4.7% in 40–45 μm, 12.2% in 45–50 μm, 15.9% in 50–55 μm, 45.5% in 55–60 μm, 51.6% in 60–65 μm, 91.0% in 65–70 μm, 92.0% in 70–75 μm, and 93.2% in 75–80 μm oocytes (Figures 5 and 6). Thus, we observed that each gene had its own acquisition kinetics. DNA methylation acquisition began first for Snrpn at ~50 μm and was near completion at >60 μm. Next was Peg3, where DNA methylation acquisition was initiated at ~45 μm and nearly complete at >65 μm, which was followed by Peg1, where DNA methylation acquisition began at ~55 μm and was near completion by >70 μm. Snrpn had the shortest acquisition interval while Peg3 had the longest.
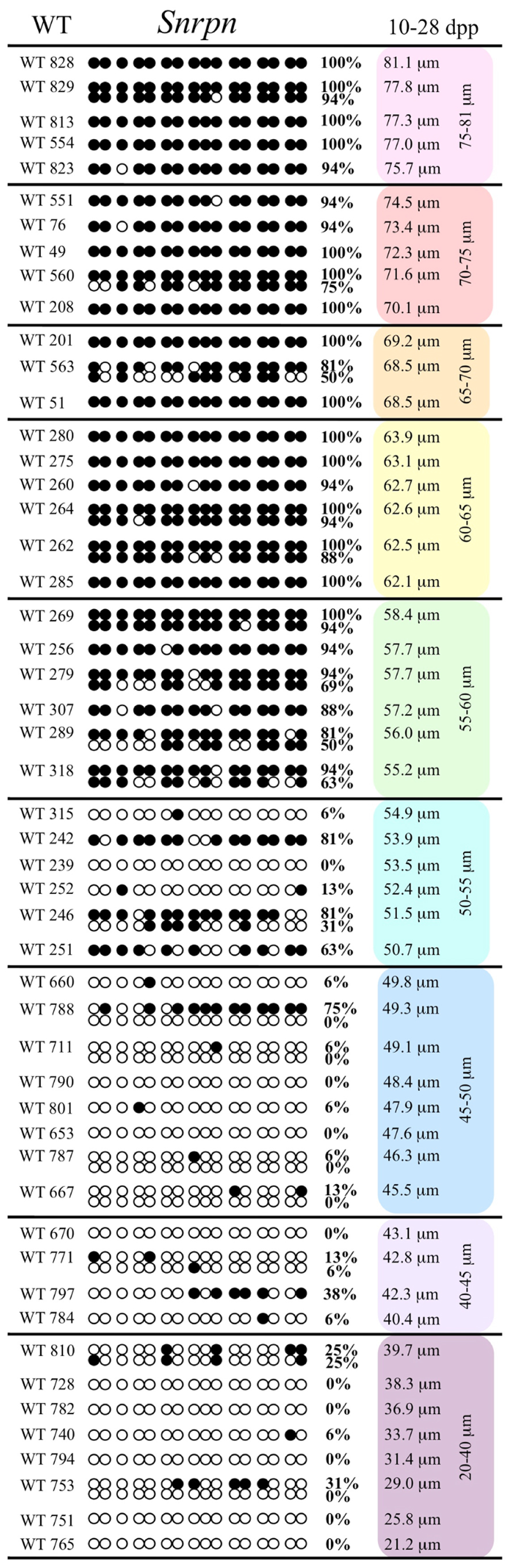
FIGURE 1. Methylation analysis of the Snrpn ICR in individual oocytes derived from control C57BL/6 female mice. The Snrpn ICR region analyzed contains 16 CpGs. Black circles indicate methylated CpGs while white circles indicate unmethylated CpGs. Each row represents an individual oocyte (designation indicated to the left). Methylation percentage and diameter for each oocyte is shown at the right. Oocytes are grouped into cohorts ranging from 20 to 80 μm diameters in 5 μm increments. Oocytes with one methylation pattern represent one of the two parental alleles detected. Oocytes with two methylation patterns represent detection of both parental alleles.
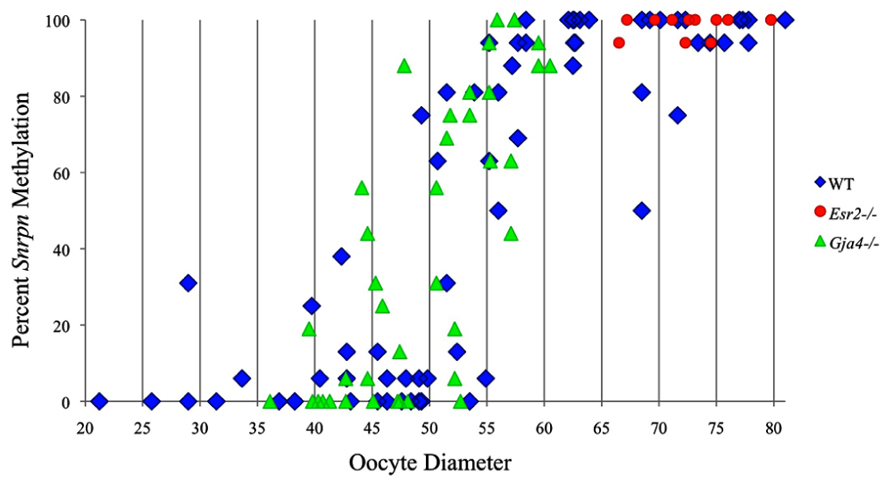
FIGURE 2. Methylation percentage of each parental allele at the Snrpn ICR in relation to oocyte diameter (μm). For oocytes with two parental alleles, each allele was graphed separately. Blue diamonds represent oocytes from control females, red circles represent oocytes from Esr2– / – females, and green triangles represent oocytes from Gja4– / – females.
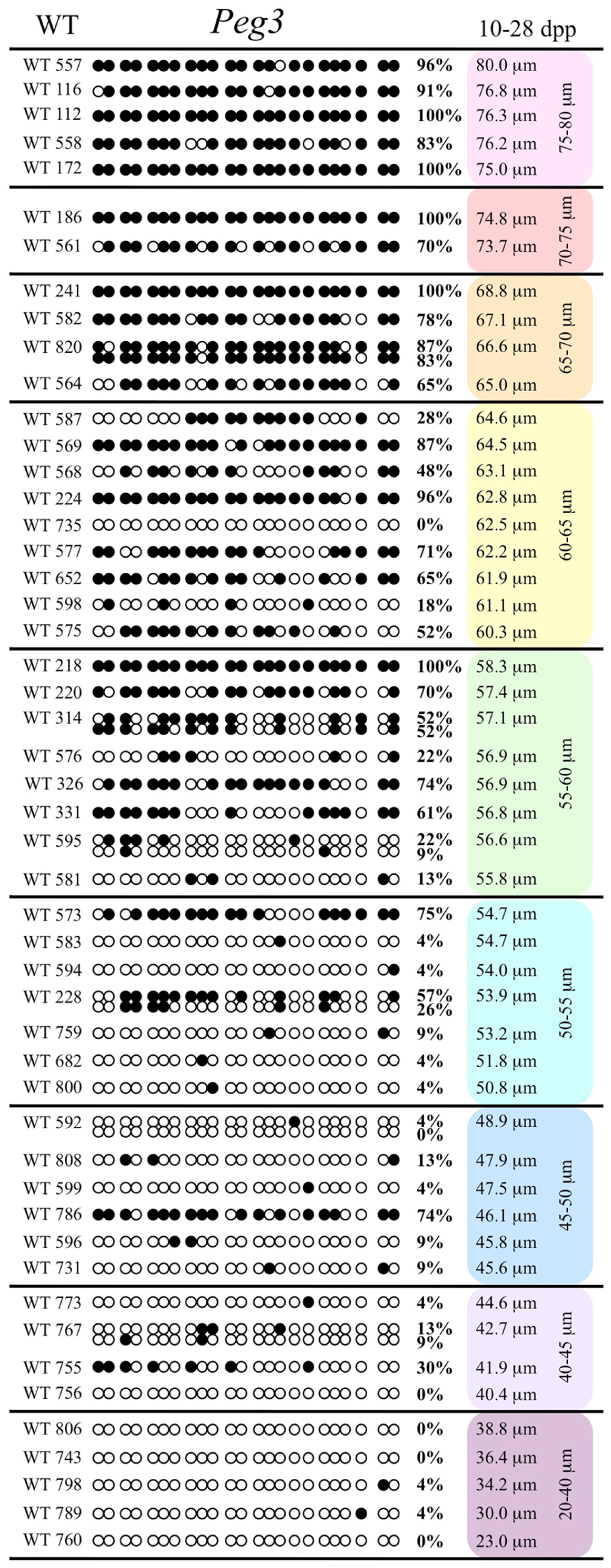
FIGURE 3. Methylation analysis of the Peg3 DMR in individual oocytes derived from control C57BL/6 females. The Peg3 DMR region analyzed contains 23 CpGs. Details are described in Figure 1.
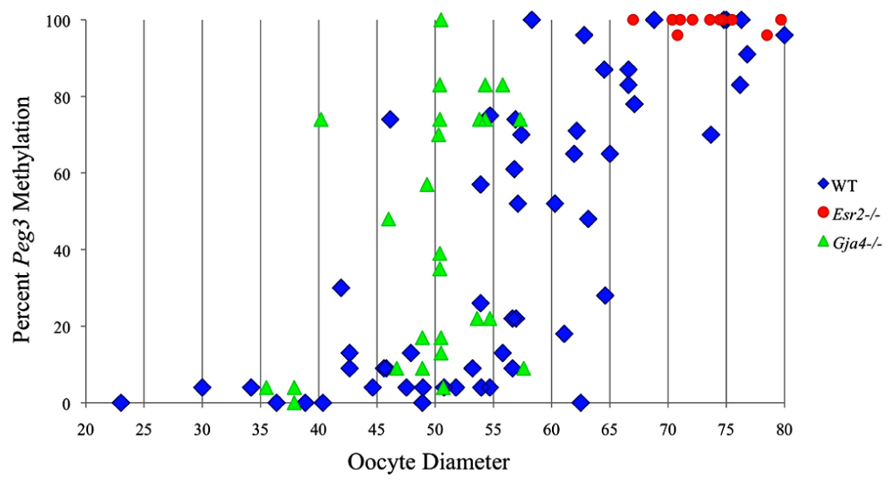
FIGURE 4. Methylation percentage of each parental allele at the Peg3 DMR in relation to oocyte diameter (μm). Oocytes from control females, Esr2– / – females and Gja4– / – females are represented by blue diamonds, red circles and green triangles, respectively.
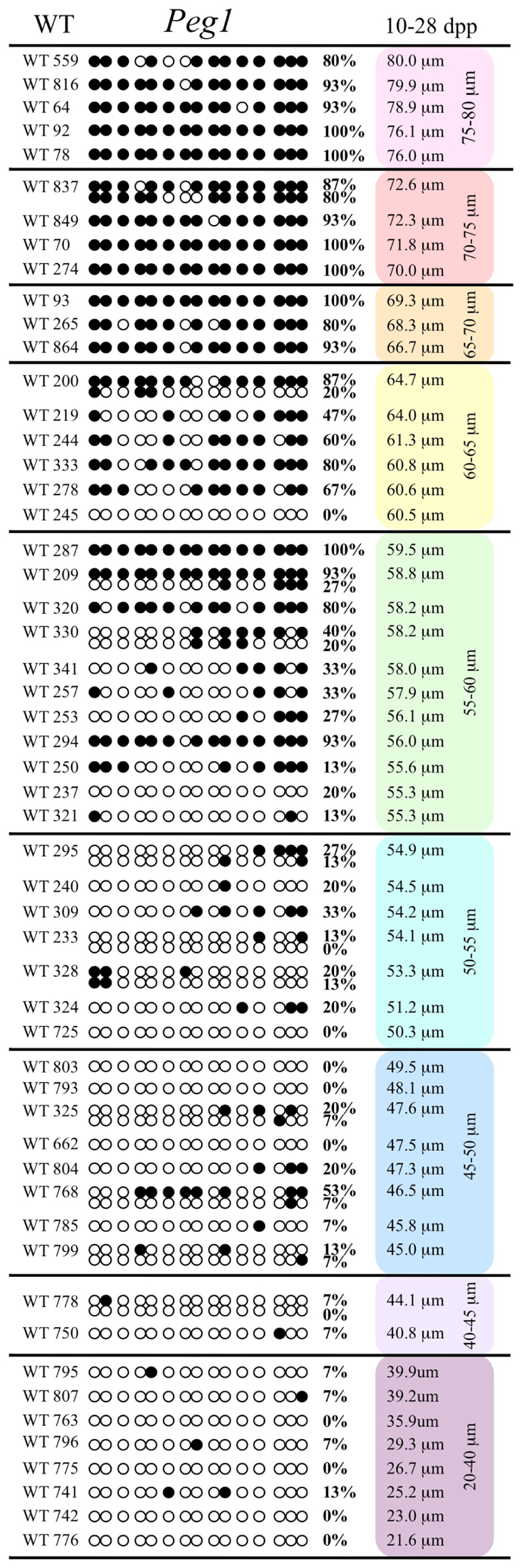
FIGURE 5. Methylation analysis of the Peg1 DMR in individual oocytes derived from the control C57BL/6 mice. The Peg1 DMR region analyzed contains 15 CpGs. Details are described in Figure 1.
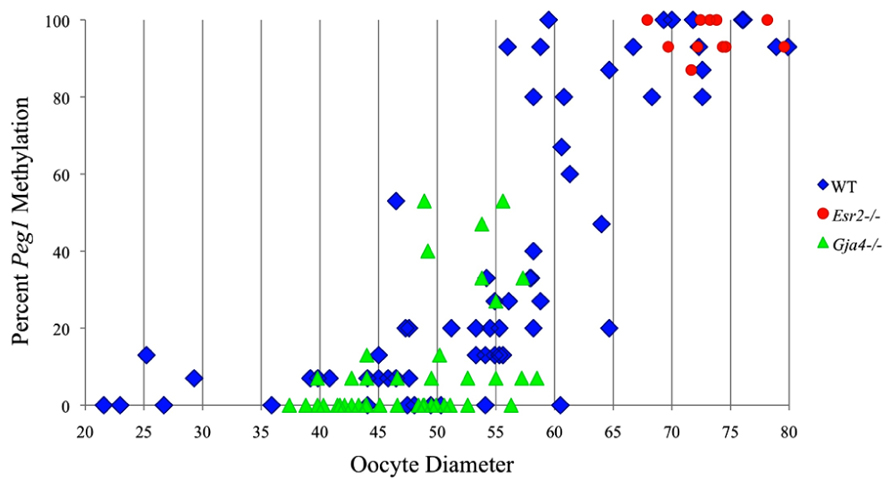
FIGURE 6. Methylation percentage of each parental allele at the Peg1 DMR in relation to oocyte diameter (μm). Oocytes from control females, Esr2– / – females and Gja4– / – females are represented by blue diamonds, red circles and green triangles, respectively.
Ovaries deficient in Esr2 produce a reduced number of maturing oocytes, but those that do mature appear to not be developmentally compromised (Krege et al., 1998). Consistent with this, we recovered a small number of oocytes from 28 dpp females, ranging in diameter size from 66 to 82 μm, corresponding to the preovulatory stage in oocyte growth. To investigate the role of reduced hormone signaling on imprint acquisition, we analyzed the progression of DNA methylation acquisition in developing oocytes from mice deficient in Esr2. For the Snrpn ICR, mean methylation levels were 98.0% for 65–70 μm, 97.0% for 70–75 μm, and 100.0% for 75–80 μm oocytes (Figures 2 and 7). For the Peg3 DMR, mean methylation was 100.0% in 65–70 μm, 99.4% in 70–75 μm, and 98.7% in 75–80 μm oocytes (Figures 4 and 8). For the Peg1 DMR, mean methylation levels were 96.5% for 65–70 μm, 95.1% for 70–75 μm, and 96.5% for 75–80 μm oocytes (Figures 6 and 9). Thus, oocytes from Esr2-null females had comparable DNA methylation levels to control oocytes, indicating that imprint DNA methylation acquisition was unaffected by Esr2 deficiency.
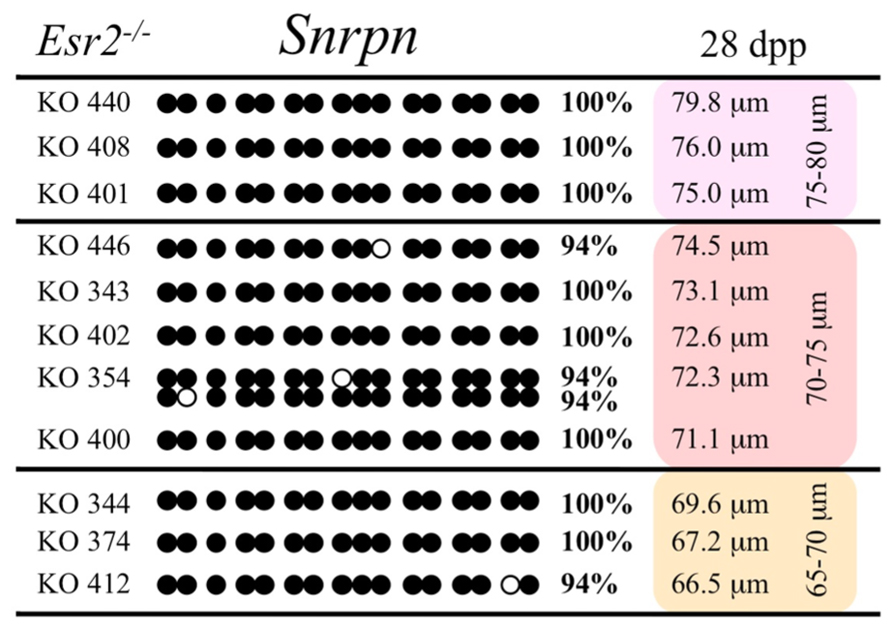
FIGURE 7. Methylation analysis of the Snrpn ICR in individual oocytes derived from Esr2– / – females. Details are described in Figure 1.
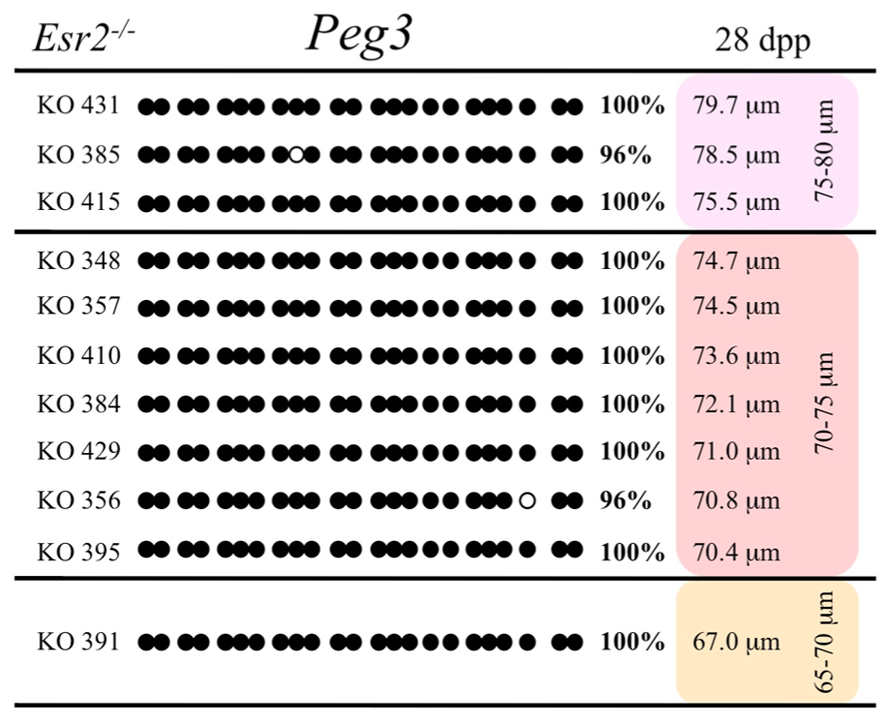
FIGURE 8. Methylation analysis of the Peg3 DMR in individual oocytes derived from Esr2– / – mice. Details are described in Figure 1.
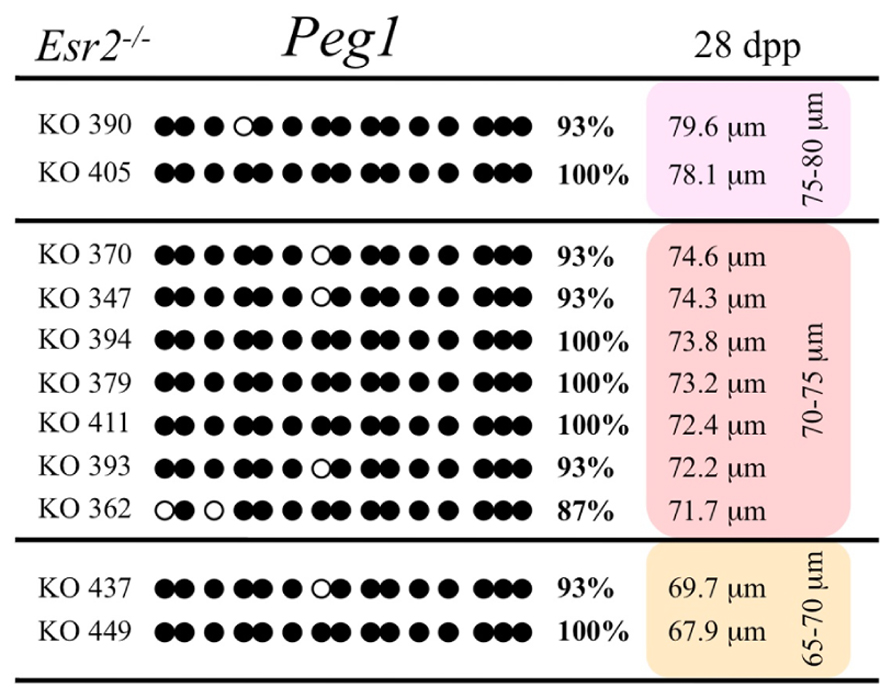
FIGURE 9. Methylation analysis of the Peg1 DMR individual oocytes derived from Esr2– / – female mice. Details are described in Figure 1.
Previous analyses have shown that oocytes in CX37-null ovaries arrest development before reaching meiotic competence, around the time the antrum begins to form (~21 dpp; Simon et al., 1997; Carabatsos et al., 2000; Li et al., 2007). We collected and analyzed oocytes from Gja4-null 21 dpp females, which ranged in diameter sizes from 35 to 55 μm and from 28 dpp females, which ranged in size from 50 to 60.5 μm. The maximum diameter obtained was 60.5 μm, consistent with previous studies (Simon et al., 1997; Carabatsos et al., 2000). To explore the relationship between gap junction loss and imprint acquisition, we analyzed the progression of DNA methylation establishment in developing oocytes from Gja4-deficient mice.
At the Snrpn ICR, mean methylation levels were 6.3% in 35–40 μm, 14.0% in 40–45 μm, 17.4% in 45–50 μm, 45.8% in 50–55 μm, 80.8% in 55–60 μm, and 88.0% in 60–65 μm oocytes (Figures 2 and 10). No significant difference was observed in methylation levels between Gja4-null and control oocytes. Analysis at the Peg3 DMR showed mean methylation levels of 2.7% for 35–40 μm, 74.0% in 40–45 μm, 28.0% for 45–50 μm, 50.7% for 50–55 μm, and 55.3% for 55–60 μm oocytes (Figures 4 and 11). No significant difference was observed in methylation levels between Gja4-null and control oocytes. For the Peg1 DMR, mean methylation levels were 1.8% in 35–40 μm, 2.8% in 40–45 μm, 9.7% in 45–50 μm, 14.3% in 50–55 μm, and 19.1% in 55–60 μm oocytes (Figures 6 and 12). Statistical analysis of Peg1 showed a significant difference in methylation acquisition between control and Gja4-deficient oocytes (P = 0.0006). Because Gja4-null oocytes stop growing and are eventually lost from the follicles, it could not be determined whether this is a delay or a disruption in Peg1 DNA methylation acquisition.
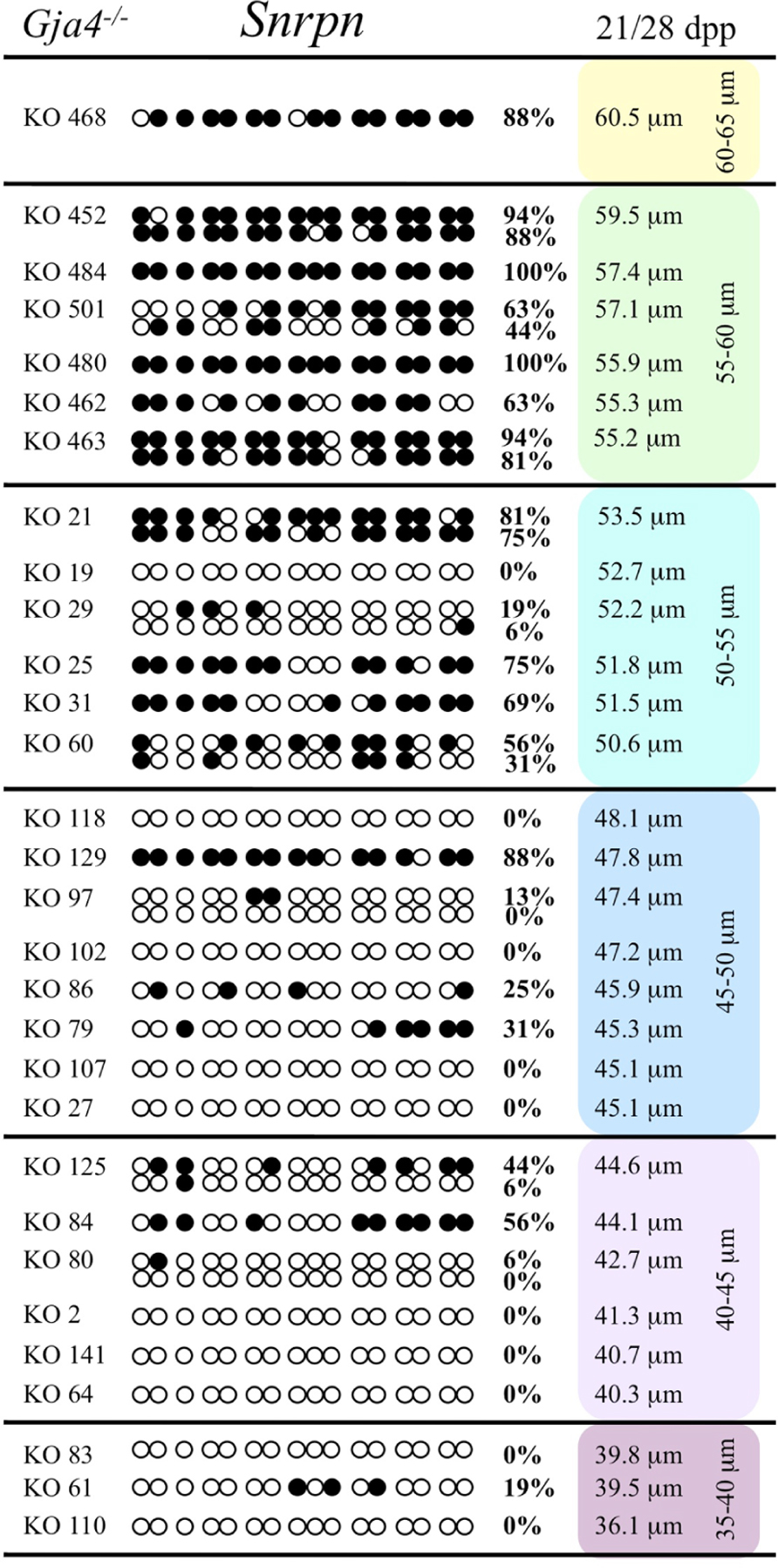
FIGURE 10. Methylation analysis of the Snrpn ICR in individual oocytes derived from Gja4– / – mice. Details are described in Figure 1.
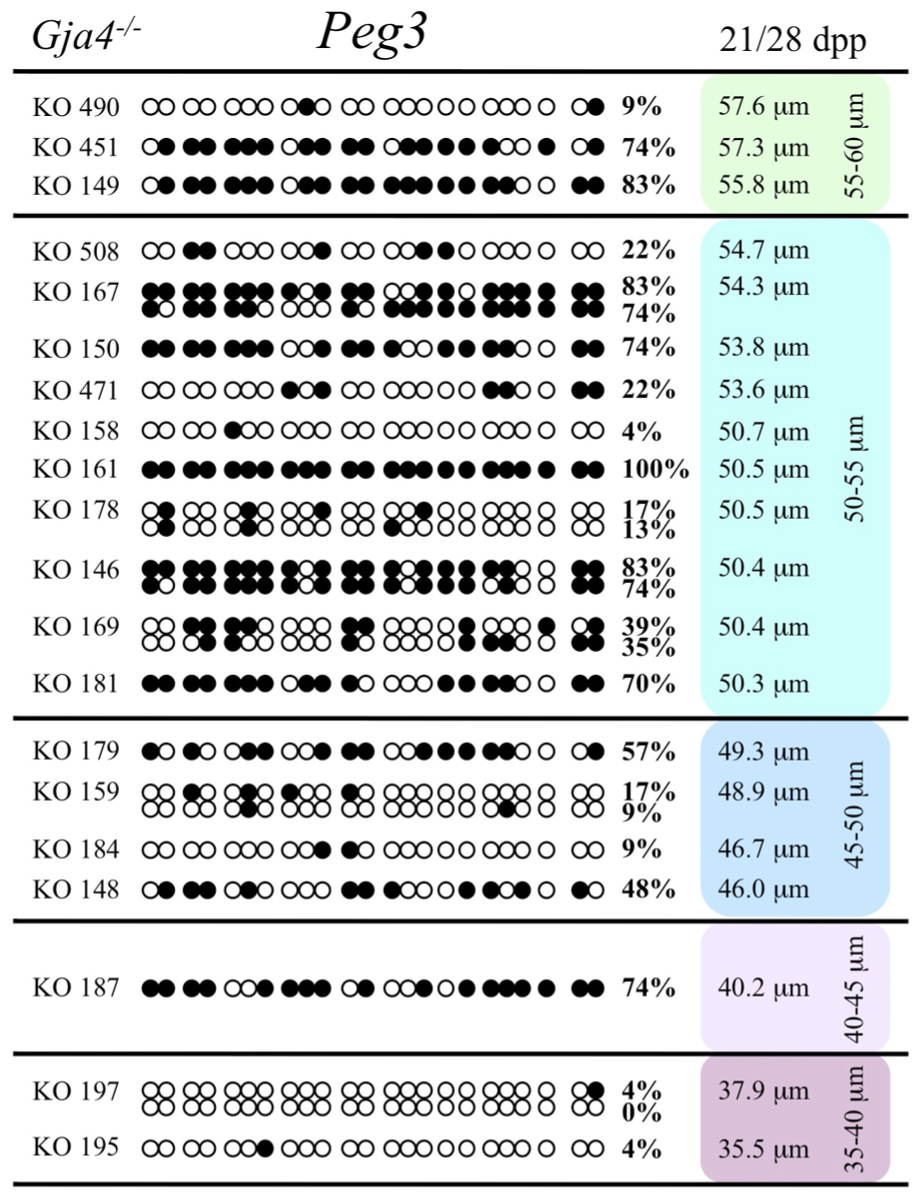
FIGURE 11. Methylation analysis of the Peg3 DMR individual GV oocytes derived from Gja4– / – female mice. Details are described in Figure 1.
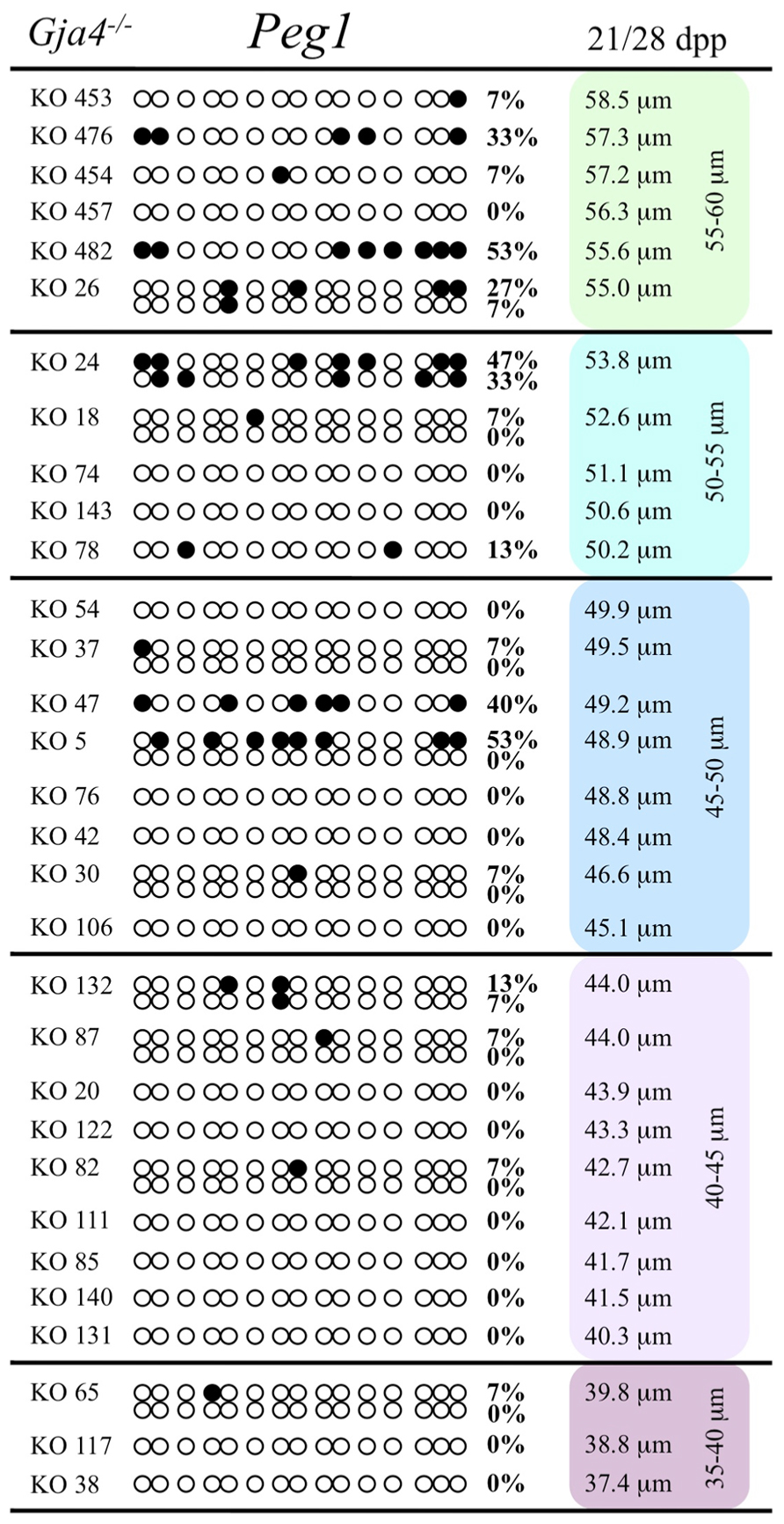
FIGURE 12. Methylation analysis of the Peg1 DMR individual GV oocytes derived from Gja4– / – females. Details are described in Figure 1.
Growth and maturation of oocytes within follicles requires bidirectional signaling and exchange of nutrients, metabolites, and second messengers through gap junctions between the oocyte and granulosa cells (Matzuk et al., 2002; Gilchrist et al., 2008; Su et al., 2009). Aberrant endocrine signaling and loss of gap junctional communication between the oocyte and granulosa cells leads to compromised folliculogenesis, oocyte maturation, and oocyte competency, consequently impairing fertility. Given that oocyte-specific DNA methylation establishment at imprinted genes occurs during this growth phase, we determined whether compromised endocrine signaling and gap junctional communication would disrupt de novo methylation acquisition. Individual oocytes from Esr2- and Gja4-deficient mice were assessed for DNA methylation establishment at Snrpn, Peg3, and Peg1. We observed no aberrant or delayed acquisition of DNA methylation at Snrpn, Peg3, or Peg1 in oocytes from Esr2-deficient females, and no perturbation in Snrpn or Peg3 de novo methylation in oocytes from Gja4-null females. However, Gja4 deficiency resulted in a loss or delay in methylation acquisition at Peg1. One possible explanation for this difference between the three loci analyzed is the late establishment of DNA methylation at the Peg1 gene. These results indicate that compromised fertility though impaired intercellular communication can lead to imprinting acquisition errors. Further studies are required to determine whether subfertility/infertility originating from impaired signaling and intercellular communication during oogenesis has an effect post-fertilization on imprint maintenance in the preimplantation embryo.
Our study is the first to investigate imprint methylation acquisition of Snrpn, Peg3, and Peg1 in individual oocytes. We observed that each gene has its own size-dependent acquisition kinetics. Snrpn had the shortest acquisition interval with de novo methylation beginning at ~50 μm and near completion at >60 μm. Peg3 had the earliest and longest acquisition interval. DNA methylation acquisition was initiated at ~45 μm and was nearly complete at >65 μm. Peg1 had the latest acquisition of de novo methylation, beginning at ~55 μm and near completion by >70 μm. Previous studies reported similar findings using pooled oocytes where methylation level increased with days postpartum, follicular stage or with oocyte diameter/size, and initiation of acquisition was gene-specific (Lucifero et al., 2004; Hiura et al., 2006; Sato et al., 2007; Song et al., 2009). Oocyte-specific de novo methylation was also found to occur differentially with the maternal allele acquiring methylation prior to the paternal allele for Snrpn, Zac1, and Peg1 (Lucifero et al., 2004; Hiura et al., 2006). Our data are consistent with this observation. Firstly, in oocytes for which two alleles were successfully amplified, one allele possessed higher and the other allele lower methylation levels, indicative of maternal and paternal contribution, respectively. For example, Snrpn WT563 oocyte had 81 and 50% methylation (Figure 1). Secondly, for oocytes within each diameter range (see Peg3 control oocytes between 60 and 65 μm; Figure 3), a subset of oocytes had high methylation percentages (68, 71, 87, and 96%, indicative of the maternal allele) while others had low methylation percentages (18, 28, 48, 52%, indicative of the paternal allele). Finally, scatter plots show two distinct cohorts within the same range of diameter measurements. For example, Peg1 control oocytes between 55 and 65 μm grouped into 0–40% methylation and 75–100% methylation (Figure 6).
While Gja4-deficient oocytes ceased development and did not achieve mature size, our analyses indicated that they were not compromised in their ability to catalyze DNA methylation as de novo DNA methylation was initiated for the Snrpn and Peg3 imprinted genes. The failure to initiate Peg1 methylation acquisition may simply be due to the fact that oocytes lacking CX37 never reach the size necessary for de novo methylation to commence at late-acquiring loci. However, control oocytes of comparable size (55–60 μm) displayed initiation of de novo Peg1 methylation. This suggests that Peg1 methylation acquisition was lost or delayed in mutant oocytes. Alternatively, CX37-null oocytes may have reduced stores of methyl donors or other metabolites required for DNA methylation that would normally be transported from granulosa cells to the oocyte via gap junctions. If this is the case, then there must have been sufficient availability of methyl donors in mutant oocytes for Snrpn and Peg3 de novo methylation, but oocytes lacking junctional coupling with the granulosa cells may have exhausted their methyl donors during oocyte growth, preventing de novo methylation at late-acquiring genes like Peg1. To investigate the requirement for methyl donors during follicle development, Anckaert et al. (2010) cultured preantral follicles in medium with low methyl donors. While this led to impaired antrum development and polar body formation, it did not impede the acquisition of DNA methylation at the Snrpn ICR and the Peg3 DMR. However, a reduced level of DNA methylation was found at the Peg1 DMR. This provides support for the argument that gap junctional communication provides important metabolites for DNA methylation acquisition. To better understand the mechanism leading to loss or delayed methylation acquisition, further studies are required to assess the level of methyl donors, amount of S-adenosylmethionine, and ability to carry out global and gene-specific methylation in 55–60 μm CX37-null or CX37-depleted oocytes. Furthermore, methylation studies should be carried out using F1 females. For Peg1 CX37 oocytes between 45 and 60 μm, oocytes possessed 0–53% methylation. DNA methylation acquisition was likely initiated on the maternal Peg1 allele in some oocytes, while other oocytes lacked methylation on both parental alleles. Thus, loss or delayed Peg1 methylation acquisition may preferentially lead to a failure of the paternal allele to become methylated. Further studies are required to investigate this potential grandpaternal effect.
Peg1 may also be more susceptible to perturbation by assisted reproductive technologies. Loss of Peg1/PEG1 methylation was observed in mouse oocytes following in vitro maturation (Kerjean et al., 2003), and human oocytes following ovarian stimulation (Sato et al., 2007). Further studies are required to determine whether the susceptibility of Peg1 to perturbation relates to its late acquisition of methylation or whether a different epigenetic regulatory mechanism(s) operates at this gene. Superovulation also caused imprinting errors in the mouse preimplantation embryo (Market-Velker et al., 2010), although imprinted methylation acquisition was not perturbed in mouse oocytes by exogenous hormone treatment (Anckaert et al., 2009; Denomme et al., 2011). We hypothesized that superovulation disrupts maternal-effect gene products required for imprint maintenance during embryo development. Thus, impaired fertility may not only disrupt Peg1 methylation acquisition but may also lead to inadequate stores of maternal products, including those from granulosa cells, that may disrupt imprint maintenance at Peg1 as well as at Snrpn and Peg3 during preimplantation development. Extending studies to preimplantation embryos generated from fertilized ERβ-deficient and CX37-depleted oocytes will be required to determine their effects on imprint maintenance. In addition, further studies are required to determine whether assisted reproductive technologies, such as in vitro oocyte maturation and superovulation, lead to aberrant endocrine and paracrine signaling as well as granulosa cell–oocyte gap junctional communication.
It is important to understand granulosa cell–oocyte communication as technological advances move forward. Procedures such as slow-freezing cryopreservation and ultra-fast vitrification of oocyte-enclosed follicles, which employ cryoprotectants and very low temperatures, may permanently or temporally disrupt actin- or tubulin-rich projections that extend from granulosa cells through the zona pellucida to the oocyte (Kidder and Mhawi, 2002). Slow-freezing of mouse, rhesus monkey, and human preantral follicles disrupted projections and uncoupled the oocyte and granulosa cells (Barrett and Albertini, 2010). Temporal disruption of oocyte–granulosa cell contacts was also observed following vitrification (Trapphoff et al., 2010). Thus, transfer of molecules between the two compartments may be temporarily disturbed. While low levels of imprinting errors were detected in a subset of oocyte pools following vitrification (Trapphoff et al., 2010), further studies are required to determine whether disruption of oocyte–granulosa coupling leads to errors in imprint acquisition and/or maintenance.
Continued studies in animal models and in humans are required to understand the molecular mechanisms regulating genomic imprinting acquisition and maintenance as well as how impaired fertility and assisted reproductive technologies induce epigenetic changes and disease.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank Dr. Greg Gloor for statistical advice, Dr. Chris Pin for control C57BL/6 female mice, Lucimar Teodoro, Linsay Drysdale, and Adrian Buensuceso for technical assistance, Kevin Barr for management of the CX37 deleted mouse colony, and staff at the University of Western Ontario and the London Health Science Centre for animal care. This work was supported by the University of Western Ontario, the Department of Obstetrics and Gynaecology to Mellissa R. W. Mann; and grants from the Ministry of Research and Innovation, Early Researcher Award to Mellissa R. W. Mann (ER06-02-188) and the Canadian Institutes of Health Research to Bonnie J. Deroo (MOP 93658) and to Gerald M. Kidder (MOP14150). Michelle M. Denomme was supported by a CIHR Training Program in Reproduction, Early Development and the Impact on Health (REDIH) Graduate Scholarship, Carlee R. White by an Ontario Graduate Scholarship, and William A. MacDonald by a Children’s Health Research Institute Postdoctoral Fellowship.
Anckaert, E., Adriaenssens, T., Romero, S., Dremier, S., and Smitz, J. (2009). Unaltered imprinting establishment of key imprinted genes in mouse oocytes after in vitro follicle culture under variable follicle-stimulating hormone exposure. Int. J. Dev. Biol. 53, 541–548.
Anckaert, E., Romero, S., Adriaenssens, T., and Smitz, J. (2010). Effects of low methyl donor levels in culture medium during mouse follicle culture on oocyte imprinting establishment. Biol. Reprod. 83, 377–386.
Azzi, S., Rossignol, S., Steunou, V., Sas, T., Thibaud, N., Danton, F., Le Jule, M., Heinrichs, C., Cabrol, S., Gicquel, C., Le Bouc, Y., and Netchine, I. (2009). Multilocus methylation analysis in a large cohort of 11p15-related foetal growth disorders (Russell–Silver and Beckwith–Wiedemann syndromes) reveals simultaneous loss of methylation at paternal and maternal imprinted loci. Hum. Mol. Genet. 18, 4724–4733.
Barrett, S. L., and Albertini, D. F. (2010). Cumulus cell contact during oocyte maturation in mice regulates meiotic spindle positioning and enhances developmental competence. J. Assist. Reprod. Genet. 27, 29–39.
Bartolomei, M. S., and Ferguson-Smith, A. C. (2011). Mammalian genomic imprinting. Cold Spring Harb. Perspect. Biol. 3, pii: a002592.
Bliek, J., Verde, G., Callaway, J., Maas, S. M., De Crescenzo, A., Sparago, A., Cerrato, F., Russo, S., Ferraiuolo, S., Rinaldi, M. M., Fischetto, R., Lalatta, F., Giordano, L., Ferrari, P., Cubellis, M. V., Larizza, L., Temple, I. K., Mannens, M. M., Mackay, D. J., and Riccio, A. (2009). Hypomethylation at multiple maternally methylated imprinted regions including PLAGL1 and GNAS loci in Beckwith–Wiedemann syndrome. Eur. J. Hum. Genet. 17, 611–619.
Carabatsos, M. J., Sellitto, C., Goodenough, D. A., and Albertini, D. F. (2000). Oocyte–granulosa cell heterologous gap junctions are required for the coordination of nuclear and cytoplasmic meiotic competence. Dev. Biol. 226, 167–179.
Chang, A. S., Moley, K. H., Wangler, M., Feinberg, A. P., and DeBaun, M. R. (2005). Association between Beckwith–Wiedemann syndrome and assisted reproductive technology: a case series of 19 patients. Fertil. Steril. 83, 349–354.
Couse, J. F., Curtis Hewitt, S., and Korach, K. S. (2000). Receptor null mice reveal contrasting roles for estrogen receptor alpha and beta in reproductive tissues. J. Steroid Biochem. Mol. Biol. 74, 287–296.
Couse, J. F., Yates, M. M., Deroo, B. J., and Korach, K. S. (2005). Estrogen receptor-beta is critical to granulosa cell differentiation and the ovulatory response to gonadotropins. Endocrinology 146, 3247–3262.
Couse, J. F., Yates, M. M., Walker, V. R., and Korach, K. S. (2003). Characterization of the hypothalamic–pituitary–gonadal axis in estrogen receptor (ER) Null mice reveals hypergonadism and endocrine sex reversal in females lacking ERalpha but not ERbeta. Mol. Endocrinol. 17, 1039–1053.
Cox, G. F., Burger, J., Lip, V., Mau, U. A., Sperling, K., Wu, B. L., and Horsthemke, B. (2002). Intracytoplasmic sperm injection may increase the risk of imprinting defects. Am. J. Hum. Genet. 71, 162–164.
Davis, T. L., Trasler, J. M., Moss, S. B., Yang, G. J., and Bartolomei, M. S. (1999). Acquisition of the H19 methylation imprint occurs differentially on the parental alleles during spermatogenesis. Genomics 58, 18–28.
Davis, T. L., Yang, G. J., McCarrey, J. R., and Bartolomei, M. S. (2000). The H19 methylation imprint is erased and re-established differentially on the parental alleles during male germ cell development. Hum. Mol. Genet. 9, 2885–2894.
DeBaun, M. R., Niemitz, E. L., and Feinberg, A. P. (2003). Association of in vitro fertilization with Beckwith–Wiedemann syndrome and epigenetic alterations of LIT1 and H19. Am. J. Hum. Genet. 72, 156–160.
Denomme, M. M., Zhang, L., and Mann, M. R. (2011). Embryonic imprinting perturbations do not originate from superovulation-induced defects in DNA methylation acquisition. Fertil. Steril. 96, 734–738.e732.
Deroo, B. J., Rodriguez, K. F., Couse, J. F., Hamilton, K. J., Collins, J. B., Grissom, S. F., and Korach, K. S. (2009). Estrogen receptor beta is required for optimal cAMP production in mouse granulosa cells. Mol. Endocrinol. 23, 955–965.
Doornbos, M. E., Maas, S. M., Mcdonnell, J., Vermeiden, J. P., and Hennekam, R. C. (2007). Infertility, assisted reproduction technologies and imprinting disturbances: a Dutch study. Hum. Reprod. 22, 2476–2480.
Drummond, A. E., and Fuller, P. J. (2011). Activin and inhibin, estrogens and NFκB, play roles in ovarian tumourigenesis is there crosstalk? Mol. Cell. Endocrinol. 359, 85–91.
Dupont, S., Krust, A., Gansmuller, A., Dierich, A., Chambon, P., and Mark, M. (2000). Effect of single and compound knockouts of estrogen receptors alpha (ERalpha) and beta (ERbeta) on mouse reproductive phenotypes. Development 127, 4277–4291.
Eggermann, T. (2010). Russell–Silver syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 154C, 355–364.
Emmen, J. M., Couse, J. F., Elmore, S. A., Yates, M. M., Kissling, G. E., and Korach, K. S. (2005). In vitro growth and ovulation of follicles from ovaries of estrogen receptor (ER){alpha} and ER{beta} null mice indicate a role for ER{beta} in follicular maturation. Endocrinology 146, 2817–2826.
Gicquel, C., Gaston, V., Mandelbaum, J., Siffroi, J. P., Flahault, A., and Le Bouc, Y. (2003). In vitro fertilization may increase the risk of Beckwith–Wiedemann syndrome related to the abnormal imprinting of the KCN1OT gene. Am. J. Hum. Genet. 72, 1338–1341.
Gilchrist, R. B., Lane, M., and Thompson, J. G. (2008). Oocyte-secreted factors: regulators of cumulus cell function and oocyte quality. Hum. Reprod. Update 14, 159–177.
Halliday, J., Oke, K., Breheny, S., Algar, E., and Amor, D. J. (2004). Beckwith–Wiedemann syndrome and IVF: a case-control study. Am. J. Hum. Genet. 75, 526–528.
Harris, A. L. (2001). Emerging issues of connexin channels: biophysics fills the gap. Q. Rev. Biophys. 34, 325–472.
Hiura, H., Obata, Y., Komiyama, J., Shirai, M., and Kono, T. (2006). Oocyte growth-dependent progression of maternal imprinting in mice. Genes Cells 11, 353–361.
Inzunza, J., Morani, A., Cheng, G., Warner, M., Hreinsson, J., Gustafsson, J. A., and Hovatta, O. (2007). Ovarian wedge resection restores fertility in estrogen receptor beta knockout (ERbeta-/-) mice. Proc. Natl. Acad. Sci. U.S.A. 104, 600–605.
Kafri, T., Ariel, M., Brandeis, M., Shemer, R., Urven, L., Mccarrey, J., Cedar, H., and Razin, A. (1992). Developmental pattern of gene-specific DNA methylation in the mouse embryo and germ line. Genes Dev. 6, 705–714.
Kerjean, A., Couvert, P., Heams, T., Chalas, C., Poirier, K., Chelly, J., Jouannet, P., Paldi, A., and Poirot, C. (2003). In vitro follicular growth affects oocyte imprinting establishment in mice. Eur. J. Hum. Genet. 11, 493–496.
Kidder, G. M., and Mhawi, A. A. (2002). Gap junctions and ovarian folliculogenesis. Reproduction 123, 613–620.
Kidder, G. M., and Vanderhyden, B. C. (2010). Bidirectional communication between oocytes and follicle cells: ensuring oocyte developmental competence. Can. J. Physiol. Pharmacol. 88, 399–413.
Krege, J. H., Hodgin, J. B., Couse, J. F., Enmark, E., Warner, M., Mahler, J. F., Sar, M., Korach, K. S., Gustafsson, J. A., and Smithies, O. (1998). Generation and reproductive phenotypes of mice lacking estrogen receptor beta. Proc. Natl. Acad. Sci. U.S.A. 95, 15677–15682.
Lennerz, J. K., Timmerman, R. J., Grange, D. K., DeBaun, M. R., Feinberg, A. P., and Zehnbauer, B. A. (2010). Addition of H19 ‘loss of methylation testing’ for Beckwith–Wiedemann syndrome (BWS) increases the diagnostic yield. J. Mol. Diagn. 12, 576–588.
Li, T. Y., Colley, D., Barr, K. J., Yee, S. P., and Kidder, G. M. (2007). Rescue of oogenesis in Cx37-null mutant mice by oocyte-specific replacement with Cx43. J. Cell Sci. 120, 4117–4125.
Li, Y., and Sasaki, H. (2011). Genomic imprinting in mammals: its life cycle, molecular mechanisms and reprogramming. Cell Res. 21, 466–473.
Lim, D., Bowdin, S. C., Tee, L., Kirby, G. A., Blair, E., Fryer, A., Lam, W., Oley, C., Cole, T., Brueton, L. A., Reik, W., Macdonald, F., and Maher, E. R. (2009). Clinical and molecular genetic features of Beckwith–Wiedemann syndrome associated with assisted reproductive technologies. Hum. Reprod. 24, 741–747.
Lucifero, D., Mann, M. R., Bartolomei, M. S., and Trasler, J. M. (2004). Gene-specific timing and epigenetic memory in oocyte imprinting. Hum. Mol. Genet. 13, 839–849.
Ludwig, M., Katalinic, A., Gross, S., Sutcliffe, A., Varon, R., and Horsthemke, B. (2005). Increased prevalence of imprinting defects in patients with Angelman syndrome born to subfertile couples. J. Med. Genet. 42, 289–291.
Mabb, A. M., Judson, M. C., Zylka, M. J., and Philpot, B. D. (2011). Angelman syndrome: insights into genomic imprinting and neurodevelopmental phenotypes. Trends Neurosci. 34, 293–303.
Maher, E. R., Brueton, L. A., Bowdin, S. C., Luharia, A., Cooper, W., Cole, T. R., Macdonald, F., Sampson, J. R., Barratt, C. L., Reik, W., and Hawkins, M. M. (2003). Beckwith–Wiedemann syndrome and assisted reproduction technology (ART). J. Med. Genet. 40, 62–64.
Market-Velker, B. A., Zhang, L., Magri, L. S., Bonvissuto, A. C., and Mann, M. R. (2010). Dual effects of superovulation: loss of maternal and paternal imprinted methylation in a dose-dependent manner. Hum. Mol. Genet. 19, 36–51.
Matzuk, M. M., Burns, K. H., Viveiros, M. M., and Eppig, J. J. (2002). Intercellular communication in the mammalian ovary: oocytes carry the conversation. Science 296, 2178–2180.
Orstavik, K. H., Eiklid, K., Van Der Hagen, C. B., Spetalen, S., Kierulf, K., Skjeldal, O., and Buiting, K. (2003). Another case of imprinting defect in a girl with Angelman syndrome who was conceived by intracytoplasmic semen injection. Am. J. Hum. Genet. 72, 218–219.
Rossignol, S., Steunou, V., Chalas, C., Kerjean, A., Rigolet, M., Viegas-Pequignot, E., Jouannet, P., Le Bouc, Y., and Gicquel, C. (2006). The epigenetic imprinting defect of patients with Beckwith–Wiedemann syndrome born after assisted reproductive technology is not restricted to the 11p15 region. J. Med. Genet. 43, 902–907.
Sato, A., Otsu, E., Negishi, H., Utsunomiya, T., and Arima, T. (2007). Aberrant DNA methylation of imprinted loci in superovulated oocytes. Hum. Reprod. 22, 26–35.
Simon, A. M., Goodenough, D. A., Li, E., and Paul, D. L. (1997). Female infertility in mice lacking connexin 37. Nature 385, 525–529.
Song, Z., Min, L., Pan, Q., Shi, Q., and Shen, W. (2009). Maternal imprinting during mouse oocyte growth in vivo and in vitro. Biochem. Biophys. Res. Commun. 387, 800–805.
Su, Y. Q., Sugiura, K., and Eppig, J. J. (2009). Mouse oocyte control of granulosa cell development and function: paracrine regulation of cumulus cell metabolism. Semin. Reprod. Med. 27, 32–42.
Trapphoff, T., El Hajj, N., Zechner, U., Haaf, T., and Eichenlaub-Ritter, U. (2010). DNA integrity, growth pattern, spindle formation, chromosomal constitution and imprinting patterns of mouse oocytes from vitrified pre-antral follicles. Hum. Reprod. 25, 3025–3042.
Turner, C. L., Mackay, D. M., Callaway, J. L., Docherty, L. E., Poole, R. L., Bullman, H., Lever, M., Castle, B. M., Kivuva, E. C., Turnpenny, P. D., Mehta, S. G., Mansour, S., Wakeling, E. L., Mathew, V., Madden, J., Davies, J. H., and Temple, I. K. (2010). Methylation analysis of 79 patients with growth restriction reveals novel patterns of methylation change at imprinted loci. Eur. J. Hum. Genet. 18, 648–655.
Ueda, T., Abe, K., Miura, A., Yuzuriha, M., Zubair, M., Noguchi, M., Niwa, K., Kawase, Y., Kono, T., Matsuda, Y., Fujimoto, H., Shibata, H., Hayashizaki, Y., and Sasaki, H. (2000). The paternal methylation imprint of the mouse H19 locus is acquired in the gonocyte stage during foetal testis development. Genes Cells 5, 649–659.
Verona, R. I., Mann, M. R., and Bartolomei, M. S. (2003). Genomic imprinting: intricacies of epigenetic regulation in clusters. Annu. Rev. Cell Dev. Biol. 19, 237–259.
Keywords: genomic imprinting, DNA methylation, imprint acquisition, infertility, oocyte, connexin37, estrogen receptor beta, oocyte diameter
Citation: Denomme MM, White CR, Gillio-Meina C, MacDonald WA, Deroo BJ, Kidder GM and Mann MRW (2012) Compromised fertility disrupts Peg1 but not Snrpn and Peg3 imprinted methylation acquisition in mouse oocytes. Front. Gene. 3:129. doi: 10.3389/fgene.2012.00129
Received: 31 March 2012; Accepted: 22 June 2012;
Published online: 11 July 2012.
Edited by:
Wendy Robinson, University of British Columbia, CanadaReviewed by:
Sushma S. Iyengar, University of Southern California, USACopyright: © 2012 Denomme, White, Gillio-Meina, MacDonald, Deroo, Kidder and Mann. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Mellissa R. W. Mann, Children’s Health Research Institute, 4th Floor, Victoria Research Laboratories, A4-130a, 800 Commissioners Road East, London, ON, Canada N6C 2V5. e-mail:bW1hbm4yMkB1d28uY2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.