 Adolfo Sequeira1†
Adolfo Sequeira1† Maureen V. Martin1† Brandi Rollins1†
Maureen V. Martin1† Brandi Rollins1† Emily A. Moon1 William E. Bunney2 Fabio Macciardi2,3 Sara Lupoli3
Emily A. Moon1 William E. Bunney2 Fabio Macciardi2,3 Sara Lupoli3 Erin N. Smith4
Erin N. Smith4 John Kelsoe5,6
John Kelsoe5,6 Christophe N. Magnan7
Christophe N. Magnan7 Mannis van Oven8 Pierre Baldi7 Douglas C. Wallace9
Mannis van Oven8 Pierre Baldi7 Douglas C. Wallace9 Marquis P. Vawter1*
Marquis P. Vawter1*- 1 Functional Genomics Laboratory, Department of Psychiatry and Human Behavior, University of California, Irvine, CA, USA
- 2 Department of Psychiatry and Human Behavior, University of California, Irvine, CA, USA
- 3 Department of Medicine, Surgery and Dentistry, University of Milan, Milan, Italy
- 4 Department of Pediatrics, School of Medicine, Rady’s Children’s Hospital, University of California, San Diego, CA, USA
- 5 Psychiatry Service, Veterans Affairs San Diego Healthcare System, San Diego, CA, USA
- 6 Department of Psychiatry, University of California, San Diego, CA, USA
- 7 School of Information and Computer Sciences, Institute for Genomics and Bioinformatics, University of California Irvine, Irvine, CA, USA
- 8 Department of Forensic Molecular Biology, Erasmus MC University Medical Center, Rotterdam, Netherlands
- 9 Department of Pathology and Laboratory Medicine, University of Pennsylvania, Philadelphia, PA, USA
Mitochondrial deficiencies with unknown causes have been observed in schizophrenia (SZ) and bipolar disorder (BD) in imaging and postmortem studies. Polymorphisms and somatic mutations in mitochondrial DNA (mtDNA) were investigated as potential causes with next generation sequencing of mtDNA (mtDNA-Seq) and genotyping arrays in subjects with SZ, BD, major depressive disorder (MDD), and controls. The common deletion of 4,977 bp in mtDNA was compared between SZ and controls in 11 different vulnerable brain regions and in blood samples, and in dorsolateral prefrontal cortex (DLPFC) of BD, SZ, and controls. In a separate analysis, association of mitochondria SNPs (mtSNPs) with SZ and BD in European ancestry individuals (n = 6,040) was tested using Genetic Association Information Network (GAIN) and Wellcome Trust Case Control Consortium 2 (WTCCC2) datasets. The common deletion levels were highly variable across brain regions, with a 40-fold increase in some regions (nucleus accumbens, caudate nucleus and amygdala), increased with age, and showed little change in blood samples from the same subjects. The common deletion levels were increased in the DLPFC for BD compared to controls, but not in SZ. Full mtDNA genome resequencing of 23 subjects, showed seven novel homoplasmic mutations, five were novel synonymous coding mutations. By logistic regression analysis there were no significant mtSNPs associated with BD or SZ after genome wide correction. However, nominal association of mtSNPs (p < 0.05) to SZ and BD were found in the hypervariable region of mtDNA to T195C and T16519C. The results confirm prior reports that certain brain regions accumulate somatic mutations at higher levels than blood. The study in mtDNA of common polymorphisms, somatic mutations, and rare mutations in larger populations may lead to a better understanding of the pathophysiology of psychiatric disorders.
Introduction
Mitochondria are subcellular organelles enriched in energetic tissues, such as muscle and brain, and located in the cytoplasm. Although there are over 1500 human mitochondrial genes (Wallace, 2005), only a small fraction of these are directly encoded by the mitochondrial DNA (mtDNA) genome. Human mtDNA, which is inherited in a matrilineal pattern (Giles et al., 1980), is a double stranded circular molecule of approximately 16,569 nucleotides (Wallace, 2005) containing 37 genes that encode two ribosomal RNAs, 22 transfer RNAs, and 13 polypeptides (Wallace, 2005). The current study is of mutations and single nucleotide polymorphisms (SNPs) found only in mtDNA in psychiatric conditions.
One possible indication of a mitochondrial disorder (Wallace et al., 1988; Wallace, 1994; McMahon et al., 1995) is matrilineal inheritance. For neuropsychiatric disorders, higher rates of disease are observed in offspring of maternal probands compared to offspring of paternal probands in SZ (Goldstein et al., 1992; Wolyniec et al., 1992; Verge et al., 2011) and bipolar disorder (BD; McMahon et al., 1995). Additionally, previous studies have also shown that mtDNA mutations and variations can play a role of susceptibility to both BD and SZ (Rollins et al., 2009; Bertolin et al., 2011; Kato et al., 2011; Verge et al., 2011), suggesting a direct role for the mitochondrial genome in neuropsychiatric disorders (Shao et al., 2008). Imaging studies also support the hypothesis that mtDNA alterations lead to neurotransmitter and metabolic changers in BD and SZ (Dager et al., 2004; Stork and Renshaw, 2005). Failure to find a pattern of maternal inheritance in SZ and BD (DeLisi et al., 2000), as well as evidence for the role of paternal inheritance have also been reported in family studies (Kornberg et al., 2000). Researchers have previously speculated that parent-of-origin effects might reflect subtle ascertainment biases, imprinting, X-linked genetic factors, vertical transmission of infectious agents, and in utero or other early maternal influences on offspring (McMahon et al., 2000).
Somatic mutations that arise during aging in brain and variation in length of mtDNA have been reported for select regions of the mtDNA genome (Cortopassi and Arnheim, 1990, 1992; Cortopassi et al., 1992; Soong et al., 1992; Cortopassi and Hutchin, 1994; Hutchin and Cortopassi, 1995). mtDNA is particularly susceptible to a higher rate of somatic deletions because mtDNA is not protected by histones and lacks the complete set of DNA repair machinery associated with nuclear DNA. Mutation rates are higher in mtDNA compared to nuclear DNA, and mutation rates are particularly high in non-coding regions of the mtDNA in human brain such as the hypervariable region of the mtDNA genome (Jazin et al., 1996; Cavelier et al., 2000). The accumulation of mtDNA mutations is tissue-specific, believed to be proportional to metabolic rate (Shoffner and Wallace, 1995) and could be a result of increased reactive oxygen species (ROS) in mitochondria. Oxidative stress and accumulation of ROS have also been observed in BD and SZ, suggesting a role for mitochondrial dysfunction in those disorders (Ben-Shachar, 2002; Rezin et al., 2009; Wood et al., 2009; Bitanihirwe and Woo, 2011; Pickrell et al., 2011; Verge et al., 2011).
Previously, our group and others have studied the mtDNA association with SZ and BD cases in a small number of subjects, and these case-control analyses of mtDNA SNPs have focused on rare mutations or haplogroup-defining SNPs and have not found compelling evidence for association (Shao et al., 2008; Rollins et al., 2009), although a recently large mitochondria SNP (mtSNP) study reported no excess prevalence of SZ in major European haplogroups (Mosquera-Miguel et al., 2012). In our previous study we used a re-sequencing array to study mtDNA in the dorsolateral prefrontal cortex (DLPFC) of SZ, BD, and major depressive disorder (MDD) patients and observed an increase of synonymous substitutions in SZ (Rollins et al., 2009). Recently, another group also used a re-sequencing array to study mtDNA in blood samples from patients with SZ and BD but did not find additional evidence that mtDNA contributes significantly to the maternal component of SZ and BD predisposition (Bertolin et al., 2011). To date, few studies have examined somatic mutations in multiple regions, and most studies have concentrated on DLPFC only (Shao et al., 2008). However, other regions have been shown to be particularly susceptible to mitochondrial alterations in oxidative phosphorylation such as the striatum and the substantia nigra (SN; Pickrell et al., 2011; Surmeier et al., 2011).
The relationships between a common somatic mtDNA variation, a 4977 bp deletion (Cortopassi et al., 1992; Soong et al., 1992), and age, sex, and diagnosis (Shao et al., 2008) was also tested in several brain regions and blood (cohorts 1 and 2). We hypothesized that psychiatric disorders (BD, MDD, and SZ) would have increased common deletion compared to controls after adjusting for the strong effect of age. The present study surveyed mtDNA SNPs in a total of 11 brain regions and in blood DNA samples by next generation sequencing to determine the influence of genetic variation in psychiatric disorders (cohort 3). Finally, mtDNA SNPs from the Affymetrix 6.0 SNP chip were tested in blood for association with SZ, BD, or both, using the Genetic Association Information Network (GAIN; SZ, BD, and controls) and Wellcome Trust Case Control Consortium 2 (WTCCC2) controls comprising a cohort of 6,040 EA cases and controls. The controls from GAIN and WTCCC2 were combined, and duplicate control subjects in GAIN were excluded in this analysis.
Materials and Methods
Post-Mortem Brain Samples
Informed consent was obtained from the next of kin for each subject and the study was reviewed and approved by the University of California, Irvine (UCI) Institutional Review Board. Three brain tissue cohorts were processed. Cohort 1 consisted of 11 subjects (6 controls, and 5 cases with schizophrenia) with 11 brains regions per subject (121 samples in total; Table 1), this cohort was used for the mitochondrial common deletion assay. Cohort 2 was comprised of 76 DLPFC samples from BD, MDD, SZ, and controls subjects (Table 1) that was used for the common deletion assay with no overlap between cohort 1 and 2 subjects. Cohort 3 was comprised of 23 DLPFC samples (four BD, five MDD, four SZ, and 10 controls) that were used for mitochondrial whole genome sequencing. Five of the 10 control subjects from cohort 3 were re-sequenced in 10 additional brain regions, and three were also re-sequenced in blood (74 samples in total; Table 1). Cohorts 1 and 3 shared five controls in common.
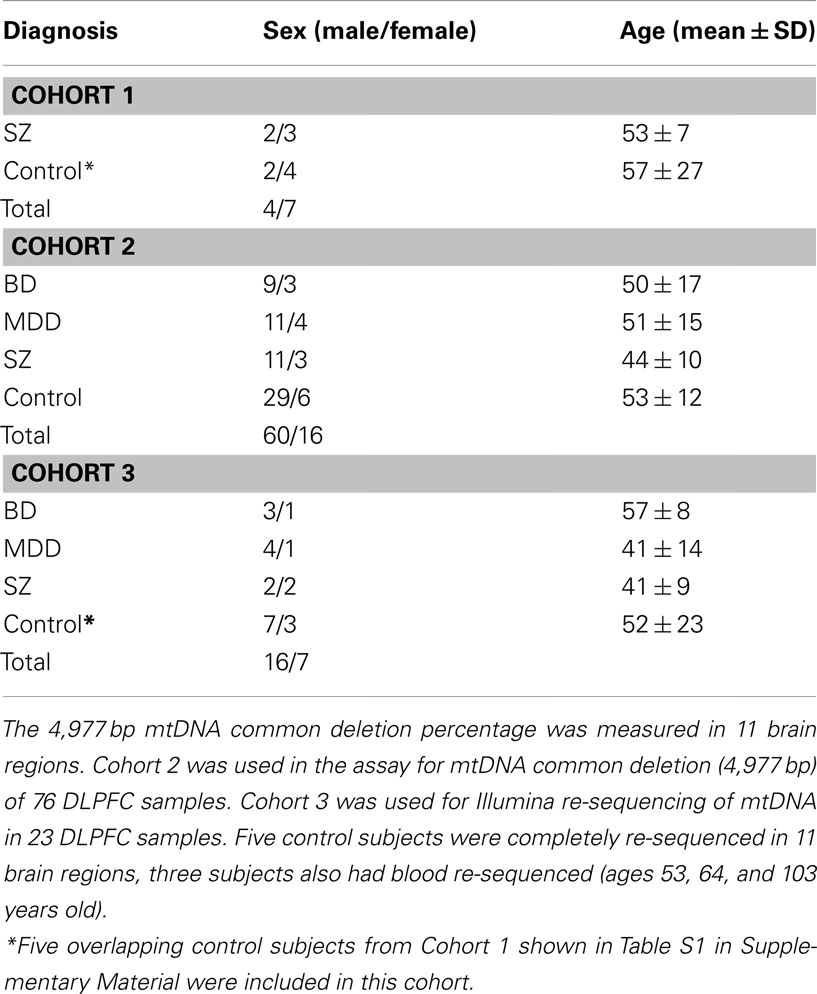
Table 1. Clinical and demographic characteristics for cohorts 1, 2, and 3/cohort 1 (n = 11) diagnosis and age.
Brain Dissections
For cohort 1, DNA was extracted from the following tissues: anterior cingulate cortex (ACC), amygdala (AMY), caudate nucleus (CAUN), cerebellum (CB), DLPFC, hippocampus (HIPP), nucleus accumbens (NACC), orbitofrontal cortex (OFC), putamen (PUT), SN, and thalamus (THAL) for 11 subjects and whole blood was obtained for three of those subjects. Briefly, prefrontal cortex samples were dissected from orbital gyrus (OFC) and the dorsal lateral prefrontal cortex at the level of the anterior end of the corpus callosum. The ACC was sampled just above the most anterior part of the corpus callosum. Three striatal structures (CAUN, putamen, and NACC) were dissected from the same coronal slice at a level where the three structures were visible anterior to the anterior commissure. The amygdala was sampled from the temporal lobe, ventral to the putamen just anterior of the hippocampus. The whole hippocampus and parahippocampal gyrus were dissected from the subsequent posterior coronal slices as a block and sampled together. The SN and the whole thalamus (from the lateral ventricle to the medial lemniscus and from the internal capsule to the midline of the brain), were individually dissected from the same coronal slice. Finally, a cortical sample of the lateral left cerebellum was dissected for processing and quality control. DNA was extracted from 25 mg of dissected brain tissue using a DNeasy Blood and Tissue Kit (Qiagen), according to the manufacturer’s protocol. For cohort 2 DNA was extracted from 76 DLPFC samples using the phenol phase of a Trizol protocol and precipitated with ethanol (Shao et al., 2008). Cohort 2 samples were assayed for the common deletion using copy number standards to quantitatively validate previous findings.
Mitochondrial Common Deletion Quantification
Primers were designed targeting the deleted or non-deleted region around the region of the deletion spanning mt8224–13501. As described previously (Shao et al., 2008), these primers allow the specific amplification of the deleted and non-deleted version of the mtDNA. A 300 bp amplicon was detected by quantitative polymerase chain reaction (qPCR) only in the presence of the common deletion, which occurs from mt8470 to 13447, by using a forward primer that binds to the flanking region. Primers for the 325 bp non-deleted amplicon spanned mt13176–13501 and the amplicon was detected only in the absence of the common deletion. Eight linearly diluted copy number standards were created from clones of the deletion or non-deletion amplicon and each clone was sequenced verified for presence of the correct insert.
Quantitative polymerase chain reaction was performed using the 7900 Sequence Detection System (Applied Biosystems) with cloned standards that ranged from 1,000,000 to 100 copies/μl, and the SYBR Green chemistry (Applied Biosystems). Two separate reactions were run for the deleted and non-deleted amplicon detection. The ABI Prism 7900 Sequence Detection System default thermal cycler program was used for each reaction: 10 min of pre-incubation at 95°C followed by 40 cycles of 15 s at 95°C and 1 min at 60°C. Individual real-time PCR reactions were performed in duplicate in a total volume of 12.5 μl in 384-well plates (Applied Biosystems) containing 6.25 μl SYBR Green (Applied Biosystems), 2.5 μl DNA (10 ng/μl), or 2.5 μl standards, 0.25 μl forward and reverse primers (10 pmol/μl), and 3.25 μl H2O. The cycle threshold (CT) was manually set at the level that reflected the best kinetic PCR parameters. Using the copy number standard curve, CT values were used to calculate copy number for the deleted and non-deleted amplicons: percent common deletion = (deletion copy number)/(deletion copy number + non-deletion copy number) × 100 (Shao et al., 2008). This equation estimates the percentage of common deletion normalized to the total number of mitochondrial copies present in a given sample. Common deletion levels showed a normal distribution across brain regions therefore we adopted parametric comparisons for region, age, and diagnosis effects. The common deletion amplicons were confirmed by gel electrophoresis to make sure only one fragment was amplified and by direct sequencing to assure specificity. The mitochondrial common deletion was assayed in two cohorts of subjects (cohorts 1 and 2). For cohort 1, the effect of age was estimated by linear regression and repeated measures ANOVA was used to analyze the effects of 11 brain regions (the within subjects measure), sex, and diagnosis on age-adjusted residuals. For cohort 2, the DLPFC data was analyzed by ANCOVA, with age as a covariate and sex and diagnosis as factors (Statistica 9.1, Tulsa, OK, USA).
Cloning Common Deletion and Wildtype Amplicons
Non-deleted and deletion-specific fragments were amplified by PCR and gel purified according to the QIAquick Gel Extraction Kit protocol (Qiagen) and TA cloned using the Original TA Cloning Kit (Invitrogen). The ligation reaction consisted of 1 μl PCR product (2.60 ng), 1 μl 10× ligation buffer, 2 μl PCR 2.1 vector (25ng/μl), 5 μl H2O, and 1 μl T4 DNA ligase run at 14°C overnight. Two μl of each ligation reaction was pipetted into a 50 μl vial of frozen One Shot competent cells (Invitrogen) per sample and heat shocked for 30 s at 42°C. After cooling the vials on ice, S.O.C. medium (Invitrogen) was added to each vial and 10–50 μl from each transformation vial was spread on LB agar plates containing 60 μg/ml X-gal and 50 μg/ml of kanamycin (Teknova). Plates were incubated overnight at 37°C and white colonies were selected and grown overnight at 37°C in a 2–5 ml LB broth tube containing 50 μg/ml kanamycin (Teknova). The plasmid containing the amplicon was extracted using a PureLink Quick Plasmid MiniPrep Kit (Qiagen). The plasmid was amplified and the samples were run on a 1% agarose gel and sequenced to confirm the presence of the correct amplicons. To further validate the qPCR amplicons, randomly selected qPCR amplicon reactions from the deletion and non-deletion assay plates were sequenced (Genewiz, Inc.; Ann Arbor, MI, USA). QPCR products for sequencing were run on a 1% agarose gel, purified using a QIAquick Gel Extraction Kit (Qiagen), and 10 ng DNA was mixed with 5 μl forward deletion or non-deletion specific forward primers (5 μM) in a total volume of 15 μl for capillary sequencing.
Mitochondrial Whole Genome Sequencing for Homoplasmic Mutations
Mutations were identified in 11 brain regions and blood (cohort 1) and in a separate cohort (cohort 3) of a single brain region (DLPFC) in subjects with SZ, BD, MDD, and controls by Illumina GAII sequencing. DNA (50 ng) was used to amplify the entire mitochondrial genome in two overlapping fragments of 9,289 bp (I) and 7,626 bp (II) in length. The primer sequences used to amplify fragment (I) are 5′-AACCAAACCCCAAAGACACC-3′ and 5′-GCCAATAATGACGTGAAGTCC-3′. The primer sequences for fragment (II) are 5′-TCCCACTCCTAAACACA TCC-3′ and 5′-TTTATGGGGTGATGTGAGCC-3′. PCR reactions, 50 μl using TaKaRa LA Taq polymerase, were performed with the following cycling conditions: 95°C 2 min; 35 cycles of 95°C 20 s, 59°C 30 s, 68°C 10 min; 68°C 20 min. PCR reactions were electrophoresed on 0.8% TAE agarose gels at 120 V. Fragments were gel extracted using the QIAquick gel extraction kit (Qiagen) and eluted in 30 μl elution buffer. Eluted samples were measured using the SpectraMax Plus 384 microplate spectrophotometer (Molecular Devices). The parallel DNA sequencing was performed using the Illumina Genome Analyzer II at Ambry Genetics Corporation (Aliso Viejo, CA, USA) according to the manufacturer’s protocol. Briefly, pooled, amplified mtDNA samples (fragments I and II) were sonicated and fragment ends were repaired and phosphorylated using Klenow T4 DNA polymerase and T4 polynucleotide kinase. An A base was added to the 3′ end of the blunted fragments and the resulting fragments were ligated to custom index adapters. The ligated products were size selected by gel purification and then PCR amplified. Using the Agilent Bioanalyzer (Santa Clara, CA, USA), each library size and concentration was determined so that samples were combined at equal molar ratios and multiplexed at 16 libraries per lane in a flow cell. This approach allows the purification of mtDNA from genomic DNA but has the disadvantage of excluding deleted mtDNA molecules.
Alignment to the Reference and Consensus Sequence
Illumina GAII single reads were aligned to the mitochondrial revised Cambridge Reference Sequence (rCRS; GenBank accession number NC_012920; Andrews et al., 1999) using Illumina’s Efficient Local Alignment of Nucleotide Data (ELAND) algorithm (Bentley et al., 2008). The consensus sequence was built using the reads aligned with ELAND using in-house software developed at the Institute for Genomics and Bioinformatics of the University of California, Irvine. A read was considered as successfully aligned to a given position of the reference sequence if the number of mismatches between the read sequence and the reference sequence was strictly lower than three. Reads with more than two mismatches with the reference sequence were rejected and considered as not aligned to the reference sequence. For each sample, around 90% of the reads were successfully aligned with the NC_012920 reference sequence for the entire human mtDNA genome. A phylogenetic tree was constructed from the consensus sequences according to PhyloTree1 Build 12 (van Oven and Kayser, 2009).
The Illumina sequencing data was screened for novel or rare homoplasmic variants using the following criteria: (1) coverage for a position was greater than 500×, the called allele was not the reference allele, (2) the% call at the variant position was >96%, and (3) the mutation had a reported incidence less than 1% in PhyloTree Build 12 (van Oven and Kayser, 2009), or in a total of 5,102 downloaded sequences from GenBank, or the mtDB-Human Mitochondrial Genome Database (Ingman and Gyllensten, 2006). The mutation data was further screened at MitoMap (Ruiz-Pesini et al., 2007) for submitted and unpublished mutations and disease associations. We further validated the observed variants by comparing for the same subjects different brain regions and also confirmed new or novel mutations by allele-specific PCR and by the dye-terminator sequencing method.
Association Study of mtSNPs
The GAIN datasets were obtained from the database of Genotypes and Phenotype (dbGaP). Data access was granted by the GAIN Data Access Committee for analysis of an approved project “Genetic Variation in Mitochondria in Schizophrenia and Bipolar Disorder.” The WTCCC2 cohort was obtained by Fabio Macciardi and Sara Lupoli. A certain number of control samples were shared between the GAIN cohorts; we made sure that there was no overlap between the cohorts and that only unique control subjects were included in the final analysis. Further, we used a common control group consisting of pooled WTCCC2 and GAIN samples to compare against BD and SZ subjects from GAIN. The subjects from GAIN chosen for inclusion in the analysis were controls (general research consent use, GRU), subjects with schizophrenia and related conditions (SARC), and bipolar subjects (bipolar disorder only, BDO; bipolar and related disorders, BARD), demographic details and distribution per cohort are showed in Table S1 in Supplementary Material. In order to minimize population specific mtDNA variants that would confound case-control analysis, African Americans were not included in the mtDNA analysis and individuals of European ancestry (EA) were analyzed. Affymetrix 6.0 SNP CEL files were extracted for 465 mtSNPs using Affymetrix Power Tools scripts (available upon request from author) and annotated at NetAffx2. In brief, median normalization for perfect match probes was used with a Birdseed (version 2) algorithm on 465 mtSNPs annotated from NetAffx (Korn et al., 2008). The output was forced for allele call only. After the mitochondria allele calls were determined for 465 SNPs and annotated with NetAffx, these 465 mtSNPs were pruned to 362 SNPs by removing SNPs with a no call rate >5% and individual subjects with <95% call rates. A genetic distance hierarchical clustering was performed to see whether site of origin (GAIN vs. WTCCC2) of control DNA potentially stratified the data, however this effect did not appear. Multidimensional scaling (MDS) analysis and logistic regression were calculated in PLINK with mtSNPs (Purcell et al., 2007) using four MDS covariates. The logistic regression results were compared with the Ingman database of mtSNP frequencies to remove outlier nominally significant associations due to obvious miscalls with the Affymetrix genotyping platform. There has not been a previous association study of mtDNA SNPs in psychiatry that has the same power as the present study; we calculated a 99% power to detect a 5% allelic difference.
Results
Common Deletion in Human Brain Increases with Age
We measured the levels of the common 4977 bp deletion across all brain regions in cohort 1. We observed, in a multivariate analysis, that age was significantly correlated with common deletions levels, R2 for age and common deletion calculated for 11 brain regions can be seen in Table S2 in Supplementary Material (p = 0.024). The most significant individual brain regional correlations with age were observed in the NACC, CAUN, and AMY (Figures 1A–C) for cohort 1; the additional figures for the other brain regions can be seen in Figure S1 in Supplementary Material. Common deletion percentages for individual regions were significantly correlated with age in seven regions, while four regions did not show significant Pearson correlations (Table S3 in Supplementary Material). The regional correlation matrix of the common deletion shows nominally significant (p < 0.05) relationships of the common deletion between brain regions for 31 pair wise correlations of 55 pairs. Thus, slightly over half of these paired brain regions show significant correlations for accumulation of the common deletion with age.
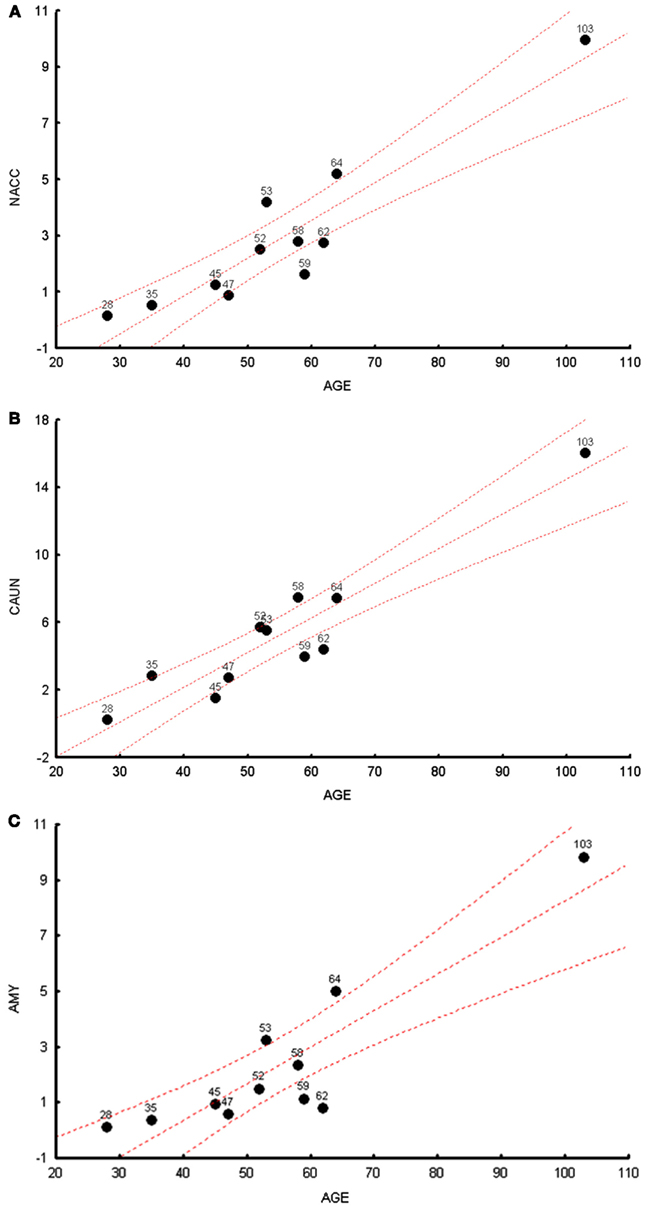
Figure 1. The highest correlations between the common deletion and age are shown for cohort 1 subjects. (A) The correlation between age (x-axis) and mtDNA common deletion (y-axis) showing the nucleus accumbens (NACC), r = 0.97. (B) The correlation between age (x-axis) and mtDNA common deletion (y-axis) showing caudate nucleus (CAUN), r = 0.98. (C) The correlation between age (x-axis) and mtDNA common deletion (y-axis) showing amygdala (AMY), r = 0.96.
The highest levels of the common deletion were observed in regions that have origins in the mesencephalon (SN) and telencephalon (CAUN). Overall, common deletion levels were greatest in mesencephalon (SN) > telencephalon (CAUN, putamen, NACC, cortical regions) > diencephalon (thalamus) ( rhomboencephalon (cerebellum). An ongoing study that includes over 500 samples will be used to independently retest these correlations of the common deletion across all brain regions. The OFC showed the highest number of significant correlations to all other brain regions of common deletion, in fact was significantly correlated to all other 10 regions (Table S3 in Supplementary Material).
A repeated measures ANCOVA was also performed for the common deletion with age as a covariate, region as a repeated measure factor, and sex and diagnosis as between-subjects factors for cohort 1. There was a significant overall effect of region [F(9, 63) = 6.49; p = 1.7 × 10−06], while sex [F(1, 6) = 0.39; p = 0.55], and diagnosis [F(1, 6) = 3.73; p = 0.10] factors were not significant, and interaction terms were not statistically significant (Figure 2). The significant region effect was further examined by post hoc comparisons of the mean percentage of common deletion per brain region and p-values were corrected for multiple comparisons using Tukey’s Honest Significant Difference test for each regional-comparison (AMY, ACC, CAUN, CB, DLPFC, HIPP, NACC, OFC, PUT, SN, and THAL). The two regions with the highest levels of common deletion were the CAUN and SN, with a 39- to 40-fold increase compared to the CB, which had the lowest common deletion level (Figure S2 in Supplementary Material) which is consistent with earlier reports (Corral-Debrinski et al., 1992; Soong et al., 1992; Meissner et al., 2008). These three regional differences account for most of the significant post hoc differences observed, while the remaining regions were not significantly different when corrected for multiple comparisons (Table S4 in Supplementary Material). The mtDNA common deletion percentage was 0.5 ± 0.4 SEM for blood (n = 3), but was greatly increased in the brain for the same subjects, consistent with prior observation of low common deletion levels in blood and higher levels in post mitotic tissue.
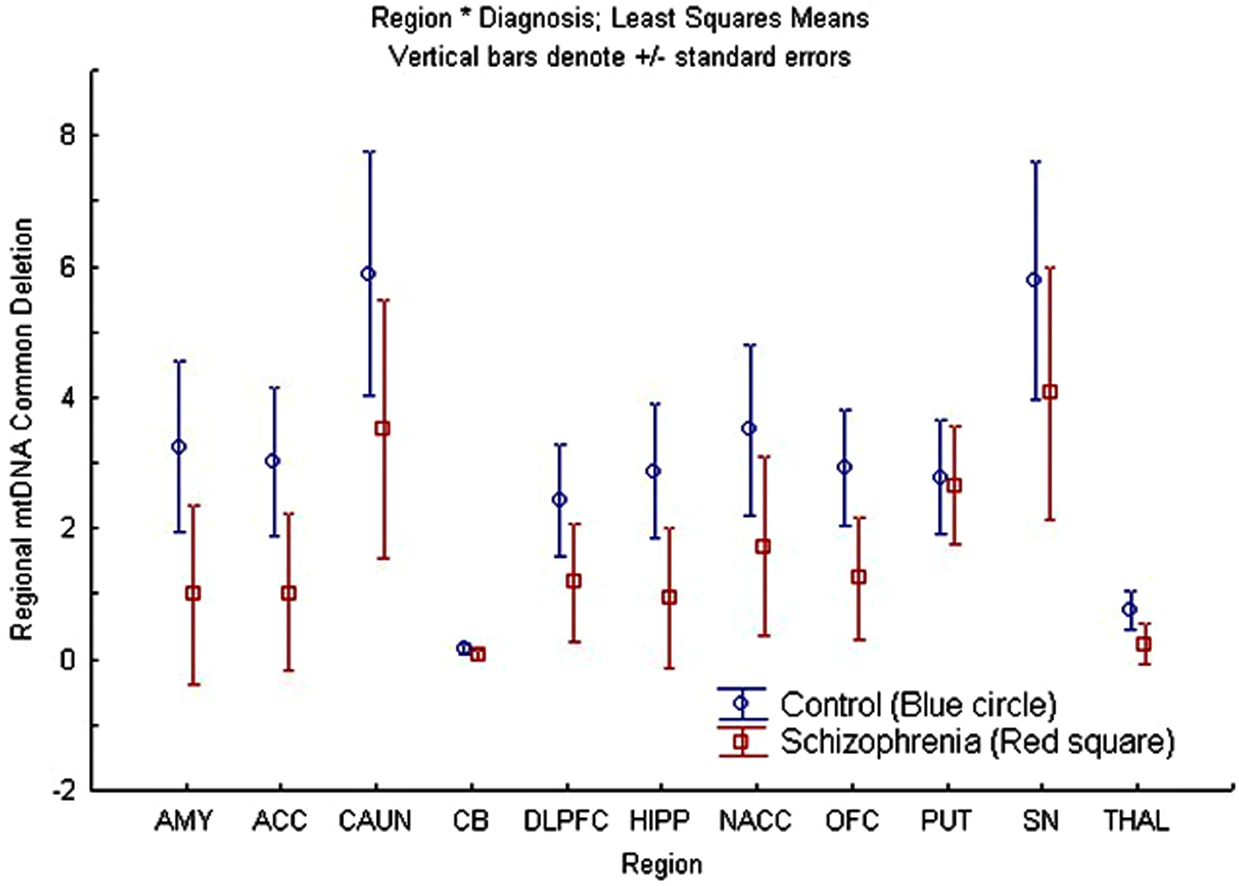
Figure 2. There was no overall region × diagnosis (schizophrenia compared to control) interaction effect on age-adjusted mtDNA common deletion percentages but regional differences were observed. This figure shows the least squares mean and standard errors of non-age-adjusted common deletion percentages per region, plotted by diagnosis in cohort 1 (schizophrenia compared to controls).
Results of the common deletion study in DLPFC only (cohort 2) showed also a significant relationship between age and common deletion levels (F = 6.7, p = 0.01), (Figure S3 in Supplementary Material) as well as sex and common deletion (F = 5.5, p = 0.02; Figure 3A). Our hypothesis that psychiatric disorders (BD, MDD, and SZ) have increased common deletion was tested in a planned comparison of the age-adjusted data in DLPFC (cohort 2). The BD group showed a significant increase in the common deletion compared to controls (p = 0.022, Figure 3B) by a fold change of 2.4 using age-adjusted least square means, MDD showed a 2.0 fold increase compared to controls (p = 0.058), while SZ was not significantly different compared to controls (p = 0.59). These findings validated previous data for BD and MDD for the same subjects in cohort 2 (Shao et al., 2008), however in the present study we used a more accurate method using a standard curve. Thus, we show increased mtDNA common deletion in BD, no change in SZ or MDD, compared to controls in DLPFC.
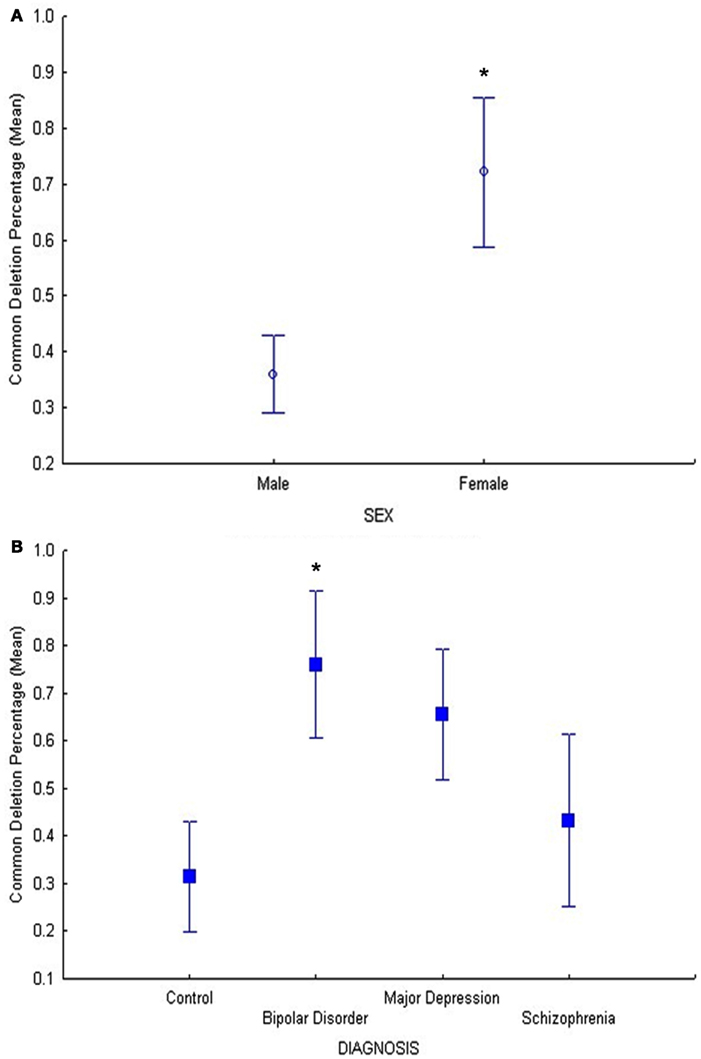
Figure 3. (A) In cohort 2, the main effect of sex was significant (F = 5.5, p = 0.02) on common deletion percentage in DLPFC, with females exhibiting significantly higher percentages of the mitochondrial common deletion compared to males. The age-adjusted means and standard error bars are displayed.(B) In cohort 2, the BD group showed a significant increase in the common deletion compared to controls (p = 0.022) and MDD showed a trend compared to controls (p = 0.058) while schizophrenia compared to controls was not different (p = 0.59).
Homoplasmic Novel and Rare Variants in mtDNA
There were 149 homoplasmic variants that were either novel or rare found by Illumina re-sequencing (Table S5 in Supplementary Material): 88% (131/149) were transitions, 11% (17/149) were transversions (Table S6 in Supplementary Material). Of these 149 homoplasmic variants, seven were novel and not reported in four databases: dbSNP (Sherry et al., 2001), mtDB (Ingman and Gyllensten, 2006), PhyloTree (van Oven and Kayser, 2009), and MITOMAP3. Six of the novel variants were in coding genes, five were synonymous, one was in a transfer RNA; one was in the non-coding D-Loop region (Table 2) and these were not found in an analysis of 5140 human mtDNA genomes (Pereira et al., 2009). We selected and successfully confirmed by dye-terminator direct sequencing of the following five variants: C4796T, G4924C, A6578G, T8945C, and A11026G.
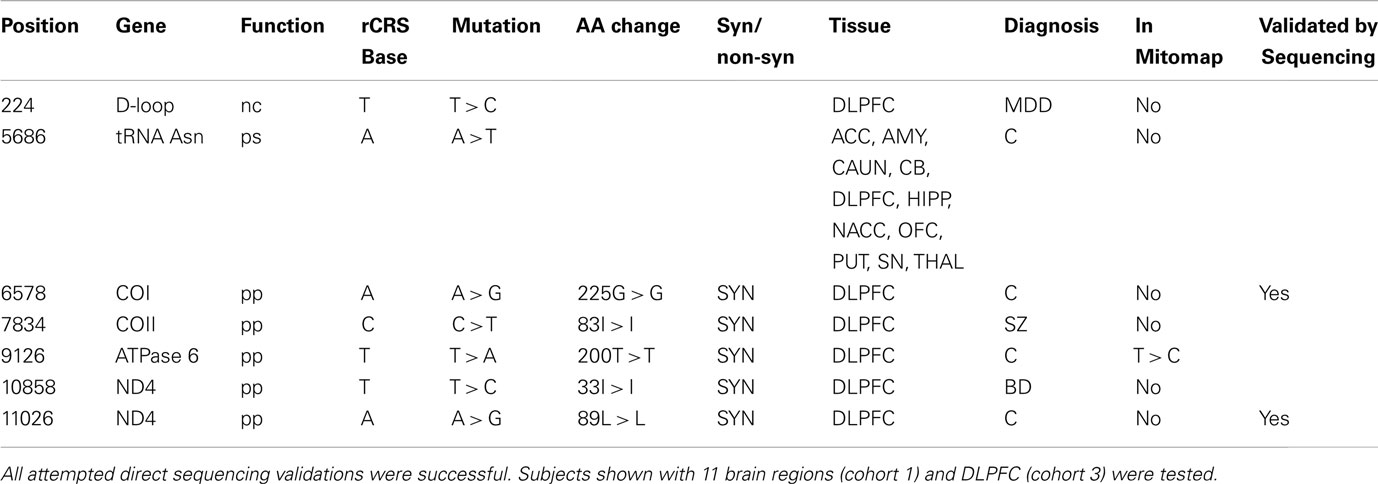
Table 2. Novel mitochondrial homoplasmic mutations (7) found in brain of neuropsychiatric patients and controls detected with Illumina sequencing.
Only one homoplasmic deletion was detected, an AC deletion at mt523-524, present in 11 brain regions sampled from two control subjects that have distant haplogroups (L1cb2 and H5, Figure 4). This mt523-524 AC deletion was previously reported (Han et al., 2003; Wanrooij et al., 2004). Two homoplasmic insertions were detected. One control subject had a T insertion at mt455. A second common insertion was also observed in the mononucleotide repeat (poly-C) region known as D310 for Displacement Loop (D-Loop), which consists of a series of C nucleotides (between 7 and 9) intercalated with a single T at position 310 followed by another series of C nucleotides (Sanchez-Cespedes et al., 2001), a hot spot for mutations possibly involved in Alzheimer’s (Wang et al., 2009), and cancer (Tang et al., 2004). Six subjects (three controls, two BP, and one SZ) had 309.1C or 309.1CC insertions in this poly-C tract. Due to the small number of subjects in this re-sequencing arm of the study, the rates of de novo mutations were not compared among groups. In summary, there were seven novel mutations in Cohort 3: four novel mutations in the 10 control subjects, one novel mutation in the four SZ subjects, one novel mutation in the five MDD subjects, and one novel mutation in the four BD subjects.
Considering further the rate of novel coding and non-coding mutations in cohort 3 (23 DLPFC subjects), there was one novel mutation in the non-coding region of the mitochondrial genome, resulting in a novel mutation frequency of 3.87 × 10−5 (23 genomes × 1121 bp non-coding = 25,783 total non-coding bp, one mutation/25,783 bp = 3.87 × 10−5 or 1:25,783; Table S7 in Supplementary Material). There were six novel mutations in the coding region for these subjects leading to a 1.69 × 10−5 novel mutation frequency (23 genomes × 15,447 bp total coding sequence = six novel mutations per 35,5281 bp or 1:59,214). These calculations show that novel mutations occurred more frequently in the non-coding region of the mitochondria compared to the coding region.
A total of 142 of the 149 variants were rare (Table S5 in Supplementary Material) with minor allele frequency <1% based upon mtDB (Ingman and Gyllensten, 2006) and PhyloTree (van Oven and Kayser, 2009). We observed a higher number of both transitions and transversions per genome for the MDD group compared to controls and to the other two diagnoses particularly in the non-coding region, yet had fewer transversions in the coding region, resulting in a higher ratio of transitions over transversions in the coding region (Table S8 in Supplementary Material).
Phylogenetic Analysis of mtDNA from Brain
We conducted phylogenetic analysis using the consensus sequence of the whole mitochondrial genome which allowed us to determine the specific haplogroup for each sample (van Oven and Kayser, 2009). All brain regions for each individual subject were correctly grouped together showing the accuracy and consistency of our sequencing algorithms (Figure 4).The distribution of diagnoses for each haplogroup built using the entire mtDNA consensus sequences can be seen in Table S9 in Supplementary Material. We detected back mutations in the haplogroups shown in Figure 4. Notably, the mtDNA sequence of individual 58 allowed us to refine the mutation motif of haplogroup L1c3b2, which thus far was based on a single mtDNA sequence only (Behar et al., 2008), viz. sequence 597 from (Howell et al., 2004). When we detected a homoplasmic mutation or rare variant in one brain region, it was inevitably found in the remaining 10 brain regions (Figure 4).
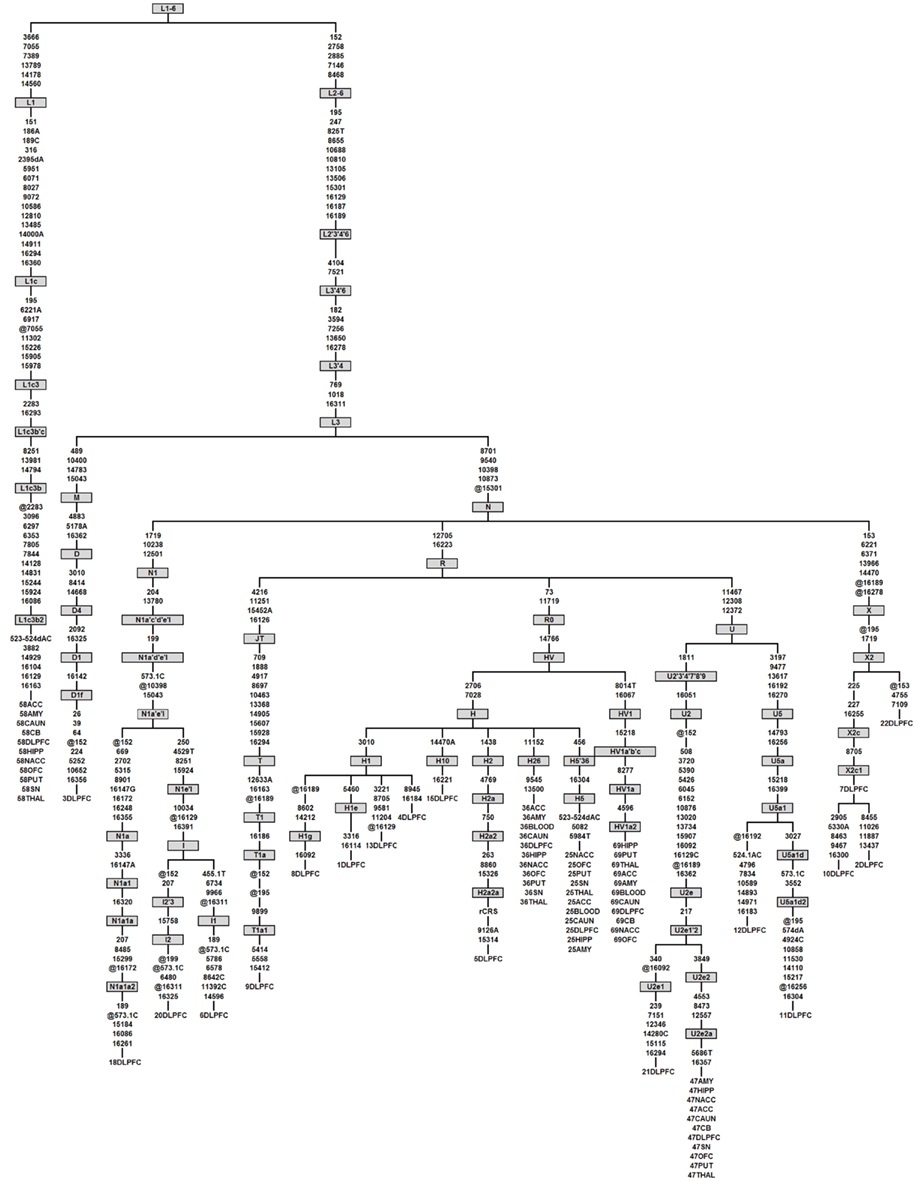
Figure 4. Phylogenetic tree of the 23 complete mtDNA sequences generated in this study. Haplogroups are labeled in boxes and follow PhyloTree Build 12 nomenclature (van Oven and Kayser, 2009). Mutations are denoted as nucleotide position numbers corresponding to the rCRS. Mutations are transitions unless an exact base change is specified. Deletions and insertions are indicated by “d” and “0.1,” respectively, followed by the base(s) involved. Back mutations (from the perspective of the ancestral root) are preceded by “@.” Variants 309.1C, 315.1C, 16182C, 16183C, and 16519 were not considered for tree construction and are therefore not shown. The samples that have appeared in the study are given numerical labels followed by brain region abbreviations. As an example, in the first column individual 58, belonging to haplogroup L1c3b2, was sequenced in 11 brain regions which all showed the same phylogenetic relationship, indicating a lack of novel mutations between brain regions from the same individual.
mtSNP Association in Psychiatric Disorders
PLINK logistic regression was run with genotypes for 362 mtSNPs from 6,040 subjects using MDS covariates. In all analyses, the overlap of SZ and BD controls from the GAIN study were removed before undertaking association or normalization.
The GAIN and WTCCC2 controls were pooled into one common control group for comparison to BD and SZ subjects from GAIN. There were no significant mtSNP associations with psychiatric disorders after genome-wide correction. However, the logistic regression analysis showed nominally significant association with SZ, BD, and pooled analysis for nine different mtSNP alleles that passed nominal threshold of p < 0.05 for SZ or BD association (Table 3) without correction for multiple testing. Some of these nominally associated mtSNPs were located in mutation hotspots in the hypervariable I (between positions 16001 and 16568) and II (between positions 1 and 574) regions (Table 4). The remaining associated coding mtSNPs were primarily synonymous. MtSNPs with relatively low no call rates compared to MAF were included in Table 4. The “no calls” are from the Affymetrix 6.0 SNP chip analysis, for an individual SNP when the algorithm is unable to assign an allele call. The listing of additional SNPs that had nominal p-value for association, but had high no call rates is shown (Table S10 in Supplementary Material). Interestingly, the minor population allele showed an equal number of risk and protective associations for psychiatric disorders (Table 4).
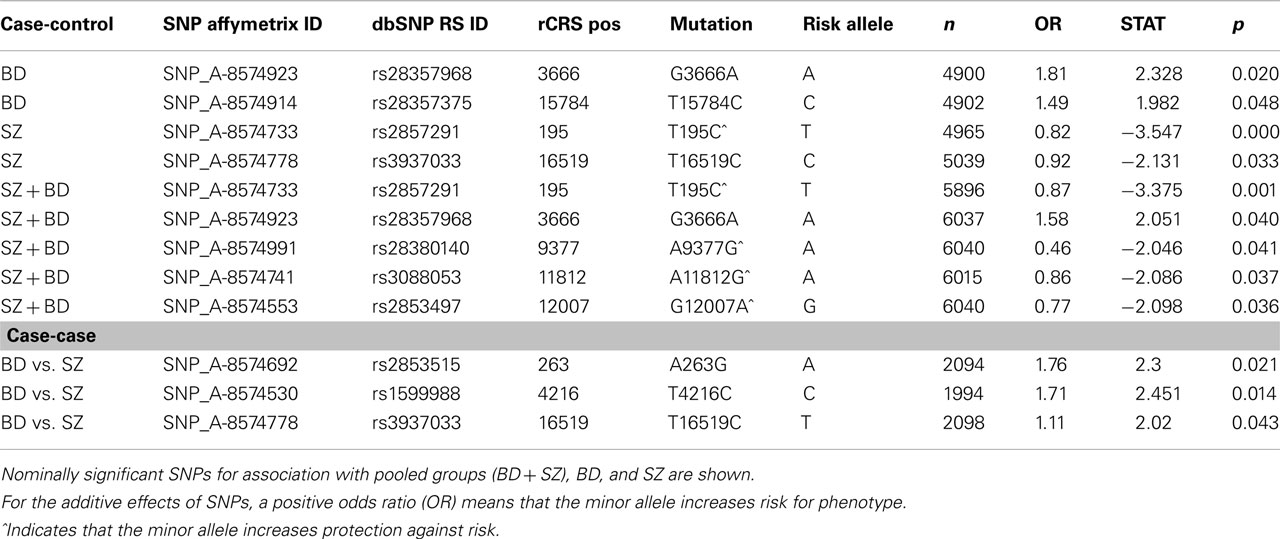
Table 3. mtDNA SNPs from Affymetrix cel files were used to calculate association with MDS covariates.
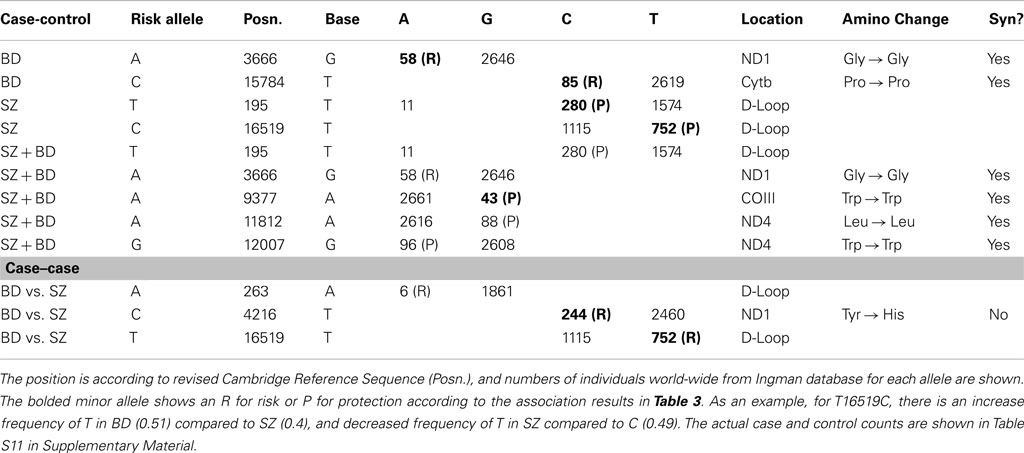
Table 4. Annotation of nominally significant SNPs from association with pooled groups (BD + SZ), BD, and SZ are shown.
The haplogroup-defining SNP (A12308G) and the T195C allele in D-Loop region were combined in an exploratory post hoc analysis (Table 5) by using a Cochran-Mantel-Haenszel test for repeated 2 × 2 tests of independence. The SZ group showed less 195C alleles than controls (CMH = 10.6, df = 2, p = 0.0011), while similar test conducted for BD was not significant (CMH = 2.6, df = 2, p = 0.10); however, the combined SZ + BD CMH was significant (p = 0.001). These results confirm the under-representation of the 195C allele in SZ, independent of a major ethnic stratification of SNP A12308G. The 195C allele was associated with a reduced relative risk in controls by 0.67–0.80 depending on the A12308G allele call.
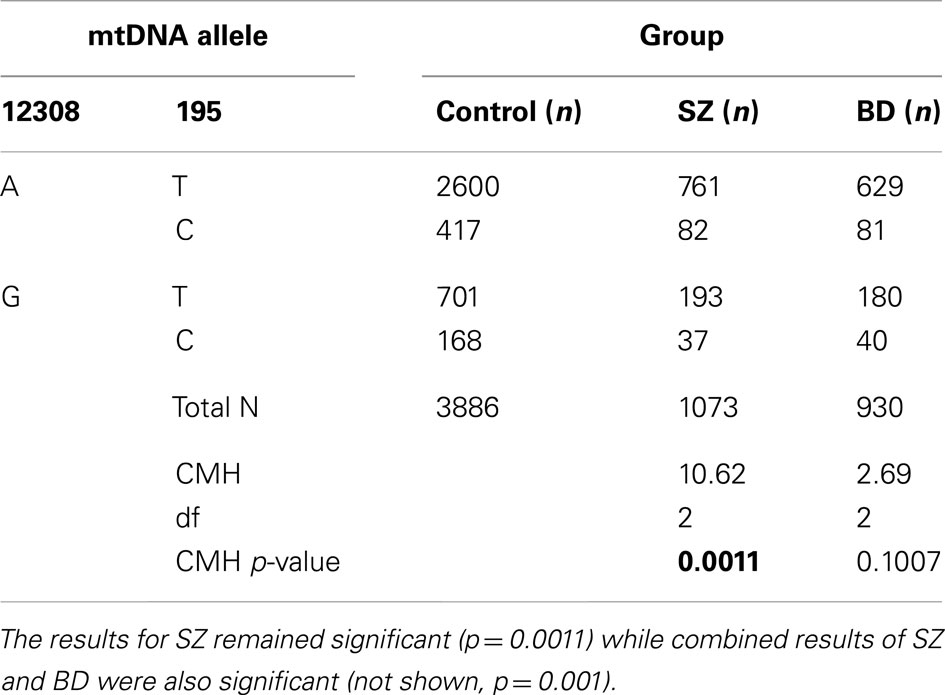
Table 5. The haplogroup defining SNP (A12308G) and the T195C allele in D-Loop region were tested in a post hoc analysis using a Cochran–Mantel–Haenszel (CMH) test for repeated 2 × 2 tests of independence.
In summary, when SZ and BD were compared to combined controls from GAIN and WTCCC2 datasets, there was weak evidence of association after correction for MDS as well as following the removal of stratification effect of a major ethnic defining haplogroup SNP. The present findings suggest that similar frequencies of SZ or BD were found at major haplogroup specific EA mtSNPs, while more sporadically occurring mtSNPs, T195C and T16519C, in the hypervariable region showed preliminary associations to SZ, and pooled SZ and BD.
Discussion
Common Deletion
We confirmed an increase in brain levels of the 4,977 bp mtDNA common deletion previously reported in BD, aging, and sex (Kato et al., 1997; Fuke et al., 2008). There was no difference in the common deletion in SZ compared to controls which is also consistent with prior reports (Kato et al., 1997; Fuke et al., 2008). The 4,977 bp common deletion accumulated with age in brain compared to blood as previously reported (Corral-Debrinski et al., 1992; Bender et al., 2006; Kraytsberg et al., 2006). We also showed that in advanced aging, the levels can remain low in blood while reaching high levels in brain, particularly in regions such as the SN, CAUN, and putamen. The mtDNA common deletion is an example of a somatic mutation that can reach fairly high levels in some brain regions while remaining relatively absent in other brain regions (CB and thalamus) and in blood.
The levels of common deletion vary significantly across brain regions, and are particularly high in mesostriatal and mesolimbic regions such as the SN, putamen, NACC, CAUN, and amygdala (Figure 1; Table S2 in Supplementary Material) as shown before in post-mortem brains (Corral-Debrinski et al., 1992; Soong et al., 1992; Meissner et al., 2008). Previous reports have also shown high levels of common deletion in the SN of both aged controls and Parkinson’s patients (Bender et al., 2006; Kraytsberg et al., 2006). Similarly, we also observed high levels of common deletion in some striatal-nigral structures, such as the SN, CAUN, and putamen and to a lesser extent the NACC, innervated and populated by dopaminergic neurons. Other regions such as the cerebellum, with unknown dopamine innervations, had a significantly lower accumulation of the common deletion in the same subjects (Figure 2). The regional susceptibility to accumulation of the common deletion might be in both dopamine and glutamatergic innervated brain regions, a result of excessive ROS due to toxic dopamine metabolites, as well as excess calcium ion flow (Ben-Shachar, 2002; Lin and Beal, 2006; Trushina and McMurray, 2007; Keating, 2008; Pivovarova and Andrews, 2010; Surmeier et al., 2011). Both dopaminergic and glutamatergic neurotransmitter systems have been consistently shown to be involved in psychiatric disorders and our findings of higher accumulation of common deletion levels in heavily dopaminergic and glutamatergic regions in BD and MDD supports the involvement of mitochondrial dysfunction in those disorders as previously suggested (Ben-Shachar, 2002; Rezin et al., 2009; Kato et al., 2011; Verge et al., 2011). Further studies of the mechanism of protection and susceptibility to the mtDNA common deletion is required to understand this diverse regional pattern and the functional significance in the aging human brain.
A caveat to measurements of homogenized dissected post-mortem brain regions is the lack of specific cellular phenotypes for the mtDNA common deletion, although it is unlikely that cells with deleterious mutations survived with huge burdens of heteroplasmy. Another issue we could not address in the current study is sub-regional or layer specific differences mtDNA common deletion, or the cell specific differences. While it is possible to use laser capture microdissection to look at separate cells, in post-mortem brain, for clonal expansion possibility, we did not initially perform this analysis, as we were interested in the brain regional distribution of somatic mutations. In the future, with certain regions that show very high common deletion rates, this approach could be very fruitful to pursue, although initial studies capturing cell nuclei are not promising as very few mitochondria are captured from cell bodies (Mamdani, F., personal communication). Important details about patients are not specifically known, for example the exact age of onset, there is only partial information regarding the decade of life when psychotic symptoms began for SZ or mood symptoms for BD. The lifetime medication histories are lengthy, most patients receive at least 5–10 different medications, and we are not aware of what the cumulative effects of those medications are on common deletion levels in brain, although we have proposed to study this relationship. These potential factors could contribute to our results as false negatives or false positives in this study.
mtDNA Mutation in the DLPFC
This study found 7 novel and 142 rare homoplasmic mutations that appeared in 23 mtDNA genomes derived from DLPFC (Table S5 in Supplementary Material) and in blood samples also analyzed. Thus, we did not find somatic homoplasmic mutations that differed in brain compared to blood. In this study we focused on homoplasmic mutations with high coverage. Although heteroplasmy was observed, we are currently in the process of validating and analyzing our heteroplasmy findings, to be reported in a future manuscript. Five homoplasmic mutations selected for validation by direct sequencing were confirmed. However, we did observe some false positives with this sequencing approach at some heteroplasmic loci, such as the previously reported A > C at mtDNA 3492 position, a known sequencing error hotspot (Li et al., 2010) that we were unable to confirm by Sanger sequencing or by allele-specific amplification.
Our findings support prior reports that the non-coding hypervariable mtDNA regions have more novel mutations than coding regions (Jazin et al., 1998; Mishmar et al., 2003; Soares et al., 2009). This is of interest for the etiology of psychiatric disorders based upon consideration of the partial contribution from mtDNA variants. Since the rate of introduction of mtDNA variants varies from 2- to 20-fold depending upon coding and non-coding regions of mtDNA genome, the introduction of mtDNA variants has been particularly high in the hypervariable region of mtDNA through the world population (Cavelier et al., 2000). In fact, the T16519C is the most frequent variant that occurs world-wide, for which we have found a preliminary association (Soares et al., 2009). The overall ratio of transition to transversion mutations observed in the present study, 18, is similar to the ratio for human mtDNA of 13.75 previously reported (Brown et al., 1982; Tamura and Nei, 1993; Belle et al., 2005). This preliminary result will require large samples of fully sequenced mtDNA genomes, comparisons between the groups of subjects were not significant due to the small number of total mutations observed per subject.
Schizophrenia and Bipolar Disorder Risk Association mtDNA SNP Alleles
In this study we tested the largest sample to date for common mtSNP association with either SZ or BD (n = 6,040) using a pool of common controls from GAIN and WTCCC2 and did not observe any significant disease associated alleles following genome-wide correction. Prior reports with smaller sample sizes that have shown trends for association, perhaps did not adequately address population stratification (Kirk et al., 1999; Amar et al., 2007; Rollins et al., 2009). We previously showed in a small sample, that T195C was associated with BD risk (Rollins et al., 2009). However, in the current study of 6,040 subjects, we show that T195C is protective for SZ, and in the SZ plus BD pooled analysis. The major haplogroup-defining SNP (A12308G) stratification does not affect the outcome of the protective association of T195C, in this larger sample. These results require replication in an independent study to rule out the possibility of unknown biases accounting for these preliminary GWAS results of 6040 individuals and genotyping by another method to confirm that the presence of a small number of no calls on the Affymetrix 6 chip did not cause this false positive. Others have used a haplogroup analysis to determine differential prevalences of SZ (Mosquera-Miguel et al., 2012) or BD in major haplogroups which might be a more sensitive method to avoid effects of unbalanced cases and control ascertainments; however, we chose to look at selected haplogroups in a post hoc analysis following our case-control logistic regression analysis covaried with four MDS scores derived from mtSNPs.
While not directly affecting mitochondrial function by altering the protein sequence, several of the nine mtSNPs in the hypervariable region that showed preliminary association, such as 16519, have the highest world-wide mutation rates (Soares et al., 2009) and have been previously associated with diseases. T16519C has been associated with cancer (Navaglia et al., 2006; Bai et al., 2007; Peng et al., 2011), diabetes (Liao et al., 2008), exercise oxygen consumption (Murakami et al., 2002), and other common disorders such as migraine (Zaki et al., 2009) in studies of Asian and European cases and controls. This is the first association study of mtDNA 16519T and 16519C variants in psychiatric disorders of BD, while a recent report showed a positive trend for SZ (Mosquera-Miguel et al., 2012).
A recent paper did not find an association of T16519C with SZ in a Spanish EA haplogroup case-control analysis (Mosquera-Miguel et al., 2012). This study used primarily Spaniards of self-reported EA with SZ (n = 942), and a control group consisting of ethnicity-matched Spanish individuals (N = 1,231). Although the T16519C, which was studied in only one-half of the subjects was not significant, this SNP had the same magnitude and direction of change in odds-ratio that we report of 0.92 for SZ (Mosquera-Miguel et al., 2012). Pooling the T16519C data from Mosquera-Miguel et al. (2012) data with our SZ data shows a simple combined OR = 0.87 (95% CI 0.76–0.98, p = 0.027) with a total SZ cases (n = 1975) and controls (n = 4133) between both studies. The studies are also in agreement with other haplogroup markers such as A12308G, which were not more prevalent in SZ (Mosquera-Miguel et al., 2012). As mentioned, there have been reports of T16519C association with metabolic disorders, such as exercise oxygen consumption and diabetes mellitus.
Differences between the current study and Mosquera-Miguel et al. (2012) are that they examined 25 mtDNA SNPs; a smaller number of total subjects especially controls, and did not include a BD case-control analysis. Further, the present study used slightly different mtDNA analyses, correcting possible mtSNP stratification using MDS, preferring this method instead of a formal haplogroup stratification test. Taken together, however, both studies with different approaches, reinforce conceptually that with larger Ns, compared to all prior studies of mtDNA common SNPs, in Southern and Northern EA populations, there was not an overwhelmingly strong association of SZ or BD to common haplogroup-defining SNPs in the coding region of mtDNA. This makes the current T195C and T16519C findings more salient as associated alleles since they are both more sporadically occurring mutations. Importantly, the functionality of both mtSNPs are unknown, T195C is a back mutation to the original ancestral allele which was the 195C, which we find to be protective for both SZ and BD.
Conclusion
In conclusion, the large somatic common deletion was the most variable result in post-mortem brain, highly age-dependent, and replicates many prior reports (Corral-Debrinski et al., 1992; Soong et al., 1992; Meissner et al., 2008). We observed the higher levels of common deletion in mesostriatal and mesolimbic regions such as the SN, putamen, NACC, CAUN, and amygdala, suggesting that oxidative stress mechanisms related to dopamine metabolism to be involved in the accumulation of the common deletion. With multiple caveats stated above, there seems to be some evidence (that requires independent replication) of protective and risk alleles in the hypervariable region of mtDNA, which mutates at a fast rate in the human population. We continue to test the hypothesis that accumulation of large somatic mutations in the coding region is relevant to psychiatric disorders and coupled with sporadic mtSNPs in the hypervariable region these two parts of the molecule may independently exert functional impacts in brain and mitochondrial function.
We also tested whether single point mutations would be found in different brain regions from the same brain. We were unable in this initial study to find evidence for homoplasmic mutations occurring independently across brain. We found the NGS technique to work reliably with some notable exceptions, and have since improved our throughput, by multiplexing 24 samples per lane on a Hi-Seq Illumina instrument, maintaining excellent coverage with 100 bp single reads. We also found evidence that some mtDNA variants are associated with SZ and BD protection or susceptibility at the population level using a large sample of patients and controls and available genotype data.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Anja Kammesheidt and Wei Guo at Ambry Genetics for their efforts in mtDNA sequencing and alignment. We appreciate the contributions of the UC Irvine Davis Brain Bank personnel: Preston Cartagena, Psy.D, David Walsh, Psy.D, Richard Stein, Ph.D., Kathleen Burke, and Claudia Cervantes, as well as Jacque Berndt and the investigators and medical examiners at the Orange County Coroner’s Office. The collection of brain tissue was supported by funding from the NIMH Conte Center Grant P50 MH60398, Pritzker Family Philanthropic Fund, and NIMH Mitochondria Grant R01MH085801 (MPV). We thank Drs. Taosheng Huang and Sha Tang for discussion and early access to their data for heteroplasmy detection. M.v.O. received financial support by the Netherlands Forensic Institute (NFI) and Erasmus MC via the Department of Forensic Molecular Biology of Erasmus MC, and by a grant from the Netherlands Genomics Initiative (NGI)/Netherlands Organization for Scientific Research (NWO) within the framework of the Forensic Genomics Consortium Netherlands (FGCN). The research was further supported by the William Lion Penzner Foundation (Dept of Psychiatry), Della Martin Foundation (WEB, AS), and NIMH Grant R01MH085801 (MPV). This study makes use of data generated by the Wellcome Trust Case-Control Consortium. A full list of the investigators who contributed to the generation of the data is available from http://www.wtccc.org.uk. Funding for the project was provided by the Wellcome Trust under award 085475. Erin N. Smith is supported in part by NIH/NCRR Grant Number UL1 RR025774.
Whole Genome-Wide Association Study of Bipolar Disorder
Funding support for the whole genome association study of bipolar disorder was provided by the National Institute of Mental Health (NIMH) and the genotyping of samples was provided through the GAIN. The datasets used for the analyses described in this manuscript were obtained from the dbGaP found at http://www.ncbi.nlm.nih.gov/gap through dbGaP accession numbers (phs000017.v3.p1). Samples and associated phenotype data for the collaborative genomic study of bipolar disorder were provided by the The NIMH Genetics Initiative for Bipolar Disorder. Data and biomaterials were collected in four projects that participated in NIMH Bipolar Disorder Genetics Initiative. From 1991 to 1998, the Principal Investigators and Co-Investigators were: Indiana University, Indianapolis, IN, U01 MH46282, John Nurnberger, M.D., Ph.D., Marvin Miller, M.D., and Elizabeth Bowman, M.D.; Washington University, St. Louis, MO, U01 MH46280, Theodore Reich, M.D., Allison Goate, Ph.D., and John Rice, Ph.D.; Johns Hopkins University, Baltimore, MD U01 MH46274, J. Raymond DePaulo, Jr., M.D., Sylvia Simpson, M.D., MPH, and Colin Stine, Ph.D.; NIMH Intramural Research Program, Clinical Neurogenetics Branch, Bethesda, MD, Elliot Gershon, M.D., Diane Kazuba, B.A., and Elizabeth Maxwell, M.S.W. Data and biomaterials were collected as part of ten projects that participated in the NIMH Bipolar Disorder Genetics Initiative. From 1999 to 2003, the Principal Investigators and Co-Investigators were: Indiana University, Indianapolis, IN, R01 MH59545, John Nurnberger, M.D., Ph.D., Marvin J. Miller, M.D., Elizabeth S. Bowman, M.D., N. Leela Rau, M.D., P. Ryan Moe, M.D., Nalini Samavedy, M.D., Rif El-Mallakh, M.D. (at University of Louisville), Husseini Manji, M.D. (at Wayne State University), Debra A. Glitz, M.D. (at Wayne State University), Eric T. Meyer, M.S., Carrie Smiley, R.N., Tatiana Foroud, Ph.D., Leah Flury, M.S., Danielle M. Dick, Ph.D., Howard Edenberg, Ph.D.; Washington University, St. Louis, MO, R01 MH059534, John Rice, Ph.D., Theodore Reich, M.D., Allison Goate, Ph.D., Laura Bierut, M.D.; Johns Hopkins University, Baltimore, MD, R01 MH59533, Melvin McInnis M.D., J. Raymond DePaulo, Jr., M.D., Dean F. MacKinnon, M.D., Francis M. Mondimore, M.D., James B. Potash, M.D., Peter P. Zandi, Ph.D., Dimitrios Avramopoulos, and Jennifer Payne; University of Pennsylvania, PA, R01 MH59553, Wade Berrettini M.D., Ph.D.; University of California at Irvine, CA, R01 MH60068, William Byerley M.D., and Mark Vawter Ph.D., M.S. Ed,.; University of Iowa, IA, R01 MH059548, William Coryell M.D., and Raymond Crowe M.D.; University of Chicago, IL, R01 MH59535, Elliot Gershon, M.D., Judith Badner Ph.D., Francis McMahon M.D., Chunyu Liu Ph.D., Alan Sanders M.D., Maria Caserta, Steven Dinwiddie M.D., Tu Nguyen, Donna Harakal; University of California at San Diego, CA, R01 MH59567, John Kelsoe, M.D., Rebecca McKinney, B.A.; Rush University, IL, R01 MH059556, William Scheftner M.D., Howard M. Kravitz, D.O., M.P.H., Diana Marta, B.S., Annette Vaughn-Brown, MSN, RN, and Laurie Bederow, MA; NIMH Intramural Research Program, Bethesda, MD, 1Z01MH002810-01, Francis J. McMahon, M.D., Layla Kassem, PsyD, Sevilla Detera-Wadleigh, Ph.D., Lisa Austin, Ph.D., Dennis L. Murphy, M.D.
Genome-Wide Association Study of Achizophrenia
Funding support for the genome-wide association of schizophrenia study was provided by the National Institute of Mental Health (R01 MH67257, R01 MH59588, R01 MH59571, R01 MH59565, R01 MH59587, R01 MH60870, R01 MH59566, R01 MH59586, R01 MH61675, R01 MH60879, R01 MH81800, U01 MH46276, U01 MH46289 U01 MH46318, U01 MH79469, and U01 MH79470) and the genotyping of samples was provided through the Genetic Association Information Network (GAIN). The datasets used for the analyses described in this manuscript were obtained from the database of Genotypes and Phenotypes (dbGaP) found at http://www.ncbi.nlm.nih.gov/gap through dbGaP accession number (phs000021.v3.p2). Samples and associated phenotype data for the genome-wide association of schizophrenia study were provided by the Molecular Genetics of Schizophrenia Collaboration (PI: Pablo V. Gejman, Evanston Northwestern Healthcare (ENH) and Northwestern University, Evanston, IL, USA).
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Behavioral_and_Psychiatric_Genetics/10.3389/fgene.2012.00103/abstract
Footnotes
References
Amar, S., Shamir, A., Ovadia, O., Blanaru, M., Reshef, A., Kremer, I., Rietschel, M., Schulze, T. G., Maier, W., Belmaker, R. H., Ebstein, R. P., Agam, G., and Mishmar, D. (2007). Mitochondrial DNA HV lineage increases the susceptibility to schizophrenia among Israeli Arabs. Schizophr. Res. 94, 354–358.
Andrews, R. M., Kubacka, I., Chinnery, P. F., Lightowlers, R. N., Turnbull, D. M., and Howell, N. (1999). Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 23, 147.
Bai, R. K., Leal, S. M., Covarrubias, D., Liu, A., and Wong, L. J. (2007). Mitochondrial genetic background modifies breast cancer risk. Cancer Res. 67, 4687–4694.
Behar, D. M., Villems, R., Soodyall, H., Blue-Smith, J., Pereira, L., Metspalu, E., Scozzari, R., Makkan, H., Tzur, S., Comas, D., Bertranpetit, J., Quintana-Murci, L., Tyler-Smith, C., Wells, R. S., Rosset, S., and Genographic, C. (2008). The dawn of human matrilineal diversity. Am. J. Hum. Genet. 82, 1130–1140.
Belle, E. M., Piganeau, G., Gardner, M., and Eyre-Walker, A. (2005). An investigation of the variation in the transition bias among various animal mitochondrial DNA. Gene 355, 58–66.
Bender, A., Krishnan, K. J., Morris, C. M., Taylor, G. A., Reeve, A. K., Perry, R. H., Jaros, E., Hersheson, J. S., Betts, J., Klopstock, T., Taylor, R. W., and Turnbull, D. M. (2006). High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 38, 515–517.
Ben-Shachar, D. (2002). Mitochondrial dysfunction in schizophrenia: a possible linkage to dopamine. J. Neurochem. 83, 1241–1251.
Bentley, D. R., Balasubramanian, S., Swerdlow, H. P., Smith, G. P., Milton, J., Brown, C. G., Hall, K. P., Evers, D. J., Barnes, C. L., Bignell, H. R., Boutell, J. M., Bryant, J., Carter, R. J., Keira Cheetham, R., Cox, A. J., Ellis, D. J., Flatbush, M. R., Gormley, N. A., Humphray, S. J., Irving, L. J., Karbelashvili, M. S., Kirk, S. M., Li, H., Liu, X., Maisinger, K. S., Murray, L. J., Obradovic, B., Ost, T., Parkinson, M. L., Pratt, M. R., Rasolonjatovo, I. M., Reed, M. T., Rigatti, R., Rodighiero, C., Ross, M. T., Sabot, A., Sankar, S. V., Scally, A., Schroth, G. P., Smith, M. E., Smith, V. P., Spiridou, A., Torrance, P. E., Tzonev, S. S., Vermaas, E. H., Walter, K., Wu, X., Zhang, L., Alam, M. D., Anastasi, C., Aniebo, I. C., Bailey, D. M., Bancarz, I. R., Banerjee, S., Barbour, S. G., Baybayan, P. A., Benoit, V. A., Benson, K. F., Bevis, C., Black, P. J., Boodhun, A., Brennan, J. S., Bridgham, J. A., Brown, R. C., Brown, A. A., Buermann, D. H., Bundu, A. A., Burrows, J. C., Carter, N. P., Castillo, N., Chiara, E. C. M., Chang, S., Neil Cooley, R., Crake, N. R., Dada, O. O., Diakoumakos, K. D., Dominguez-Fernandez, B., Earnshaw, D. J., Egbujor, U. C., Elmore, D. W., Etchin, S. S., Ewan, M. R., Fedurco, M., Fraser, L. J., Fuentes Fajardo, K. V., Scott Furey, W., George, D., Gietzen, K. J., Goddard, C. P., Golda, G. S., Granieri, P. A., Green, D. E., Gustafson, D. L., Hansen, N. F., Harnish, K., Haudenschild, C. D., Heyer, N. I., Hims, M. M., Ho, J. T., Horgan, A. M., Hoschler, K., Hurwitz, S., Ivanov, D. V., Johnson, M. Q., James, T., Huw Jones, T. A., Kang, G. D., Kerelska, T. H., Kersey, A. D., Khrebtukova, I., Kindwall, A. P., Kingsbury, Z., Kokko-Gonzales, P. I., Kumar, A., Laurent, M. A., Lawley, C. T., Lee, S. E., Lee, X., Liao, A. K., Loch, J. A., Lok, M., Luo, S., Mammen, R. M., Martin, J. W., McCauley, P. G., McNitt, P., Mehta, P., Moon, K. W., Mullens, J. W., Newington, T., Ning, Z., Ling Ng, B., Novo, S. M., O’Neill, M. J., Osborne, M. A., Osnowski, A., Ostadan, O., Paraschos, L. L., Pickering, L., Pike, A. C., Pike, A. C., Chris Pinkard, D., Pliskin, D. P., Podhasky, J., Quijano, V. J., Raczy, C., Rae, V. H., Rawlings, S. R., Chiva Rodriguez, A., Roe, P. M., Rogers, J., Rogert Bacigalupo, M. C., Romanov, N., Romieu, A., Roth, R. K., Rourke, N. J., Ruediger, S. T., Rusman, E., Sanches-Kuiper, R. M., Schenker, M. R., Seoane, J. M., Shaw, R. J., Shiver, M. K., Short, S. W., Sizto, N. L., Sluis, J. P., Smith, M. A., Ernest Sohna Sohna, J., Spence, E. J., Stevens, K., Sutton, N., Szajkowski, L., Tregidgo, C. L., Turcatti, G., Vandevondele, S., Verhovsky, Y., Virk, S. M., Wakelin, S., Walcott, G. C., Wang, J., Worsley, G. J., Yan, J., Yau, L., Zuerlein, M., Rogers, J., Mullikin, J. C., Hurles, M. E., McCooke, N. J., West, J. S., Oaks, F. L., Lundberg, P. L., Klenerman, D., Durbin, R., and Smith, A. J. (2008). Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456, 53–59.
Bertolin, C., Magri, C., Barlati, S., Vettori, A., Perini, G. I., Peruzzi, P., Mostacciuolo, M. L., and Vazza, G. (2011). Analysis of complete mitochondrial genomes of patients with schizophrenia and bipolar disorder. J. Hum. Genet. 56, 869–872.
Bitanihirwe, B. K., and Woo, T. U. (2011). Oxidative stress in schizophrenia: an integrated approach. Neurosci. Biobehav. Rev. 35, 878–893.
Brown, W. M., Prager, E. M., Wang, A., and Wilson, A. C. (1982). Mitochondrial DNA sequences of primates: tempo and mode of evolution. J. Mol. Evol. 18, 225–239.
Cavelier, L., Jazin, E., Jalonen, P., and Gyllensten, U. (2000). mtDNA substitution rate and segregation of heteroplasmy in coding and noncoding regions. Hum. Genet. 107, 45–50.
Corral-Debrinski, M., Horton, T., Lott, M. T., Shoffner, J. M., Beal, M. F., and Wallace, D. C. (1992). Mitochondrial DNA deletions in human brain: regional variability and increase with advanced age. Nat. Genet. 2, 324–329.
Cortopassi, G. A., and Arnheim, N. (1990). Detection of a specific mitochondrial DNA deletion in tissues of older humans. Nucleic Acids Res. 18, 6927–6933.
Cortopassi, G. A., and Arnheim, N. (1992). Using the polymerase chain reaction to estimate mutation frequencies and rates in human cells. Mutat. Res. 277, 239–249.
Cortopassi, G. A., and Hutchin, T. P. (1994). Germline inheritance of a rare mtDNA variant leads to greatly increased risk for Alzheimer’s disease. (Abstract 857). Am. J. Hum. Genet. 55, A149.
Cortopassi, G. A., Shibata, D., Soong, N. W., and Arnheim, N. (1992). A pattern of accumulation of a somatic deletion of mitochondrial DNA in aging human tissues. Proc. Natl. Acad. Sci. U.S.A. 89, 7370–7374.
Dager, S. R., Friedman, S. D., Parow, A., Demopulos, C., Stoll, A. L., Lyoo, I. K., Dunner, D. L., and Renshaw, P. F. (2004). Brain metabolic alterations in medication-free patients with bipolar disorder. Arch. Gen. Psychiatry 61, 450–458.
DeLisi, L. E., Razi, K., Stewart, J., Relja, M., Shields, G., Smith, A. B., Wellman, N., Larach, V. W., Loftus, J., Vita, A., Comazzi, M., and Crow, T. J. (2000). No evidence for a parent-of-origin effect detected in the pattern of inheritance of schizophrenia. Biol. Psychiatry 48, 706–709.
Fuke, S., Kametani, M., and Kato, T. (2008). Quantitative analysis of the 4977-bp common deletion of mitochondrial DNA in postmortem frontal cortex from patients with bipolar disorder and schizophrenia. Neurosci. Lett. 439, 173–177.
Giles, R. E., Blanc, H., Cann, H. M., and Wallace, D. C. (1980). Maternal inheritance of human mitochondrial DNA. Proc. Natl. Acad. Sci. U.S.A. 77, 6715–6719.
Goldstein, J. M., Faraone, S. V., Chen, W. J., and Tsuang, M. T. (1992). Gender and the familial risk for schizophrenia. Disentangling confounding factors. Schizophr. Res. 7, 135–140.
Han, C. B., Li, F., Zhao, Y. J., Ma, J. M., Wu, D. Y., Zhang, Y. K., and Xin, Y. (2003). Variations of mitochondrial D-loop region plus downstream gene 1 2S rRNA-tRNA(phe) and gastric carcinomas. World J. Gastroenterol. 9, 1925–1929.
Howell, N., Elson, J. L., Turnbull, D. M., and Herrnstadt, C. (2004). African haplogroup L mtDNA sequences show violations of clock-like evolution. Mol. Biol. Evol. 21, 1843–1854.
Hutchin, T., and Cortopassi, G. (1995). A mitochondrial DNA clone is associated with increased risk for Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 92, 6892–6895.
Ingman, M., and Gyllensten, U. (2006). mtDB: Human Mitochondrial Genome Database, a resource for population genetics and medical sciences. Nucleic Acids Res. 34, D749-751.
Jazin, E., Soodyall, H., Jalonen, P., Lindholm, E., Stoneking, M., and Gyllensten, U. (1998). Mitochondrial mutation rate revisited: hot spots and polymorphism. Nat. Genet. 18, 109–110.
Jazin, E. E., Cavelier, L., Eriksson, I., Oreland, L., and Gyllensten, U. (1996). Human brain contains high levels of heteroplasmy in the noncoding regions of mitochondrial DNA. Proc. Natl. Acad. Sci. U.S.A. 93, 12382–12387.
Kato, M., Nakamura, M., Ichiba, M., Tomiyasu, A., Shimo, H., Higuchi, I., Ueno, S., and Sano, A. (2011). Mitochondrial DNA deletion mutations in patients with neuropsychiatric symptoms. Neurosci. Res. 69, 331–336.
Kato, T., Stine, O. C., Mcmahon, F. J., and Crowe, R. R. (1997). Increased levels of a mitochondrial DNA deletion in the brain of patients with bipolar disorder. Biol. Psychiatry 42, 871–875.
Keating, D. J. (2008). Mitochondrial dysfunction, oxidative stress, regulation of exocytosis and their relevance to neurodegenerative diseases. J. Neurochem. 104, 298–305.
Kirk, R., Furlong, R. A., Amos, W., Cooper, G., Rubinsztein, J. S., Walsh, C., Paykel, E. S., and Rubinsztein, D. C. (1999). Mitochondrial genetic analyses suggest selection against maternal lineages in bipolar affective disorder. Am. J. Hum. Genet. 65, 508–518.
Korn, J. M., Kuruvilla, F. G., Mccarroll, S. A., Wysoker, A., Nemesh, J., Cawley, S., Hubbell, E., Veitch, J., Collins, P. J., Darvishi, K., Lee, C., Nizzari, M. M., Gabriel, S. B., Purcell, S., Daly, M. J., and Altshuler, D. (2008). Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat. Genet. 40, 1253–1260.
Kornberg, J. R., Brown, J. L., Sadovnick, A. D., Remick, R. A., Keck, P. E. Jr., Mcelroy, S. L., Rapaport, M. H., Thompson, P. M., Kaul, J. B., Vrabel, C. M., Schommer, S. C., Wilson, T., Pizzuco, D., Jameson, S., Schibuk, L., and Kelsoe, J. R. (2000). Evaluating the parent-of-origin effect in bipolar affective disorder. Is a more penetrant subtype transmitted paternally? J. Affect. Disord. 59, 183–192.
Kraytsberg, Y., Kudryavtseva, E., Mckee, A. C., Geula, C., Kowall, N. W., and Khrapko, K. (2006). Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet. 38, 518–520.
Li, M., Schonberg, A., Schaefer, M., Schroeder, R., Nasidze, I., and Stoneking, M. (2010). Detecting heteroplasmy from high-throughput sequencing of complete human mitochondrial DNA genomes. Am. J. Hum. Genet. 87, 237–249.
Liao, W. Q., Pang, Y., Yu, C. A., Wen, J. Y., Zhang, Y. G., and Li, X. H. (2008). Novel mutations of mitochondrial DNA associated with type 2 diabetes in Chinese Han population. Tohoku J. Exp. Med. 215, 377–384.
Lin, M. T., and Beal, M. F. (2006). Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795.
McMahon, F. J., Chen, Y. S., Patel, S., Kokoszka, J., Brown, M. D., Torroni, A., Depaulo, J. R., and Wallace, D. C. (2000). Mitochondrial DNA sequence diversity in bipolar affective disorder. Am. J. Psychiatry 157, 1058–1064.
McMahon, F. J., Stine, O. C., Meyers, D. A., Simpson, S. G., and Depaulo, J. R. (1995). Patterns of maternal transmission in bipolar affective disorder. Am. J. Hum. Genet. 56, 1277–1286.
Meissner, C., Bruse, P., Mohamed, S. A., Schulz, A., Warnk, H., Storm, T., and Oehmichen, M. (2008). The 4977 bp deletion of mitochondrial DNA in human skeletal muscle, heart and different areas of the brain: a useful biomarker or more? Exp. Gerontol. 43, 645–652.
Mishmar, D., Ruiz-Pesini, E., Golik, P., Macaulay, V., Clark, A. G., Hosseini, S., Brandon, M., Easley, K., Chen, E., Brown, M. D., Sukernik, R. I., Olckers, A., and Wallace, D. C. (2003). Natural selection shaped regional mtDNA variation in humans. Proc. Natl. Acad. Sci. U.S.A. 100, 171–176.
Mosquera-Miguel, A., Torrell, H., Abasolo, N., Arrojo, M., Paz, E., Ramos-Rios, R., Agra, S., Paramo, M., Brenlla, J., Martinez, S., Vilella, E., Valero, J., Gutierrez-Zotes, A., Martorell, L., Costas, J., and Salas, A. (2012). No evidence that major mtDNA European haplogroups confer risk to schizophrenia. Am. J. Med. Genet. B, Neuropsychiatri. Genet. 159B, 414–421.
Murakami, H., Ota, A., Simojo, H., Okada, M., Ajisaka, R., and Kuno, S. (2002). Polymorphisms in control region of mtDNA relates to individual differences in endurance capacity or trainability. Jpn. J. Physiol. 52, 247–256.
Navaglia, F., Basso, D., Fogar, P., Sperti, C., Greco, E., Zambon, C. F., Stranges, A., Falda, A., Pizzi, S., Parenti, A., Pedrazzoli, S., and Plebani, M. (2006). Mitochondrial DNA D-loop in pancreatic cancer: somatic mutations are epiphenomena while the germline 16519 T variant worsens metabolism and outcome. Am. J. Clin. Pathol. 126, 593–601.
Peng, Z., Xie, C., Wan, Q., Zhang, L., Li, W., and Wu, S. (2011). Sequence variations of mitochondrial DNA D-loop region are associated with familial nasopharyngeal carcinoma. Mitochondrion 11, 327–333.
Pereira, L., Freitas, F., Fernandes, V., Pereira, J. B., Costa, M. D., Costa, S., Maximo, V., Macaulay, V., Rocha, R., and Samuels, D. C. (2009). The diversity present in 5140 human mitochondrial genomes. Am. J. Hum. Genet. 84, 628–640.
Pickrell, A. M., Fukui, H., Wang, X., Pinto, M., and Moraes, C. T. (2011). The striatum is highly susceptible to mitochondrial oxidative phosphorylation dysfunctions. J. Neurosci. 31, 9895–9904.
Pivovarova, N. B., and Andrews, S. B. (2010). Calcium-dependent mitochondrial function and dysfunction in neurons. FEBS J. 277, 3622–3636.
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A., Bender, D., Maller, J., Sklar, P., De Bakker, P. I., Daly, M. J., and Sham, P. C. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575.
Rezin, G. T., Amboni, G., Zugno, A. I., Quevedo, J., and Streck, E. L. (2009). Mitochondrial dysfunction and psychiatric disorders. Neurochem. Res. 34, 1021–1029.
Rollins, B., Martin, M. V., Sequeira, P. A., Moon, E. A., Morgan, L. Z., Watson, S. J., Schatzberg, A., Akil, H., Myers, R. M., Jones, E. G., Wallace, D. C., Bunney, W. E., and Vawter, M. P. (2009). Mitochondrial variants in schizophrenia, bipolar disorder, and major depressive disorder. PLoS ONE 4, e4913.
Ruiz-Pesini, E., Lott, M. T., Procaccio, V., Poole, J. C., Brandon, M. C., Mishmar, D., Yi, C., Kreuziger, J., Baldi, P., and Wallace, D. C. (2007). An enhanced MITOMAP with a global mtDNA mutational phylogeny. Nucleic Acids Res. 35, D823-828.
Sanchez-Cespedes, M., Parrella, P., Nomoto, S., Cohen, D., Xiao, Y., Esteller, M., Jeronimo, C., Jordan, R. C., Nicol, T., Koch, W. M., Schoenberg, M., Mazzarelli, P., Fazio, V. M., and Sidransky, D. (2001). Identification of a mononucleotide repeat as a major target for mitochondrial DNA alterations in human tumors. Cancer Res. 61, 7015–7019.
Shao, L., Martin, M. V., Watson, S. J., Schatzberg, A., Akil, H., Myers, R. M., Jones, E. G., Bunney, W. E., and Vawter, M. P. (2008). Mitochondrial involvement in psychiatric disorders. Ann. Med. 40, 281–295.
Sherry, S. T., Ward, M. H., Kholodov, M., Baker, J., Phan, L., Smigielski, E. M., and Sirotkin, K. (2001). dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29, 308–311.
Shoffner, J. M., and Wallace, D. (1995). The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill.
Soares, P., Ermini, L., Thomson, N., Mormina, M., Rito, T., Rohl, A., Salas, A., Oppenheimer, S., Macaulay, V., and Richards, M. B. (2009). Correcting for purifying selection: an improved human mitochondrial molecular clock. Am. J. Hum. Genet. 84, 740–759.
Soong, N. W., Hinton, D. R., Cortopassi, G., and Arnheim, N. (1992). Mosaicism for a specific somatic mitochondrial DNA mutation in adult human brain. Nat. Genet. 2, 318–323.
Stork, C., and Renshaw, P. F. (2005). Mitochondrial dysfunction in bipolar disorder: evidence from magnetic resonance spectroscopy research. Mol. Psychiatry 10, 900–919.
Surmeier, D. J., Guzman, J. N., Sanchez-Padilla, J., and Schumacker, P. T. (2011). The role of calcium and mitochondrial oxidant stress in the loss of substantia nigra pars compacta dopaminergic neurons in Parkinson’s disease. Neuroscience 198, 221–231.
Tamura, K., and Nei, M. (1993). Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 10, 512–526.
Tang, M., Baez, S., Pruyas, M., Diaz, A., Calvo, A., Riquelme, E., and Wistuba, I. I. (2004). Mitochondrial DNA mutation at the D310 (displacement loop) mononucleotide sequence in the pathogenesis of gallbladder carcinoma. Clin. Cancer Res. 10, 1041–1046.
Trushina, E., and McMurray, C. T. (2007). Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience 145, 1233–1248.
van Oven, M., and Kayser, M. (2009). Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum. Mutat. 30, E386–E394.
Verge, B., Alonso, Y., Valero, J., Miralles, C., Vilella, E., and Martorell, L. (2011). Mitochondrial DNA (mtDNA) and schizophrenia. Eur. Psychiatry 26, 45–56.
Wallace, D. C. (1994). Mitochondrial DNA mutations in diseases of energy metabolism. J. Bioenerg. Biomembr. 26, 241–250.
Wallace, D. C. (2005). The mitochondrial genome in human adaptive radiation and disease: on the road to therapeutics and performance enhancement. Gene 354, 169–180.
Wallace, D. C., Zheng, X. X., Lott, M. T., Shoffner, J. M., Hodge, J. A., Kelley, R. I., Epstein, C. M., and Hopkins, L. C. (1988). Familial mitochondrial encephalomyopathy (MERRF): genetic, pathophysiological, and biochemical characterization of a mitochondrial DNA disease. Cell 55, 601–610.
Wang, P. N., Lee, H. C., Wang, C. H., Ping, Y. H., Liu, T. Y., Chi, C. W., Lin, K. N., and Liu, H. C. (2009). Heteroplasmy of mitochondrial D310 mononucleotide repeat region in the blood of patients with Alzheimer’s disease. J. Alzheimers Dis. 18, 345–353.
Wanrooij, S., Luoma, P., Van Goethem, G., Van Broeckhoven, C., Suomalainen, A., and Spelbrink, J. N. (2004). Twinkle and POLG defects enhance age-dependent accumulation of mutations in the control region of mtDNA. Nucleic Acids Res. 32, 3053–3064.
Wolyniec, P. S., Pulver, A. E., Mcgrath, J. A., and Tam, D. (1992). Schizophrenia: gender and familial risk. J. Psychiatr. Res. 26, 17–27.
Wood, S. J., Yucel, M., Pantelis, C., and Berk, M. (2009). Neurobiology of schizophrenia spectrum disorders: the role of oxidative stress. Ann. Acad. Med. Singap. 38, 396–396.
Keywords: mitochondria, homoplasmy, common deletion, novel mutations, schizophrenia, bipolar disorder
Citation: Sequeira A, Martin MV, Rollins B, Moon EA, Bunney WE, Macciardi F, Lupoli S, Smith EN, Kelsoe J, Magnan CN, van Oven M, Baldi P, Wallace DC and Vawter MP (2012) Mitochondrial mutations and polymorphisms in psychiatric disorders. Front. Gene. 3:103. doi: 10.3389/fgene.2012.00103
Received: 18 January 2012; Paper pending published: 31 March 2012;
Accepted: 20 May 2012; Published online: 20 June 2012.
Edited by:
Berit Kerner, University of California Los Angeles, USAReviewed by:
Dawn Thiselton, Virginia Commonwealth University, USAMaria R. Dauvermann, University of Edinburgh, UK
Copyright: © 2012 Sequeira, Martin, Rollins, Moon, Bunney, Macciardi, Lupoli, Smith, Kelsoe, Magnan, van Oven, Baldi, Wallace and Vawter. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Marquis P. Vawter, Functional Genomics Laboratory, GNRF Room 2119, Department of Psychiatry and Human Behavior, School of Medicine, Irvine, CA 92697-4260, USA. e-mail:bXZhd3RlckB1Y2kuZWR1
†Adolfo Sequeira, Maureen V. Martin and Brandi Rollins have contributed equally to this work