 Jaysen Gregory Knezovich and Michèle Ramsay*
Jaysen Gregory Knezovich and Michèle Ramsay*- Molecular Genetics Laboratory, Division of Human Genetics, University of the Witwatersrand, Johannesburg, South Africa
Imprinted loci play a critical role in fetal development. Their expression is often regulated by CCCTC-binding factor (CTCF) protein binding at imprinting control regions (ICRs). Prenatal alcohol exposure has been shown to reduce global DNA methylation in the developing mouse fetus. This study explored the effect of preconception paternal alcohol exposure on DNA methylation at two paternally methylated ICRs (H19 and Rasgrf1) in the sperm of exposed males and somatic DNA of sired offspring. Significant reductions at the H19 CTCF 1 (p = 0.0027) and CTCF 2 (p = 0.0009) binding sites were observed in the offspring of ethanol-treated sires, which was significantly correlated with reduced weight at postnatal days 35–42 (p < 0.05). As birth weight was unaffected and growth was only delayed during the postnatal weaning period, with subsequent re-convergence, we hypothesize that this may be the result of a mental deficit causing delayed establishment of independent feeding following weaning and would explain why this effect is transient. No difference in DNA methylation was observed in the sperm of alcohol-exposed males, indicating that the transmission of the epigenetic signal at conception is not due to altered methylation, but may be the result of an RNA-mediated mechanism or altered chromatin remodeling.
Introduction
Excessive maternal alcohol exposure during pregnancy has been implicated in a range of unfavorable fetal outcomes, collectively known as fetal alcohol spectrum disorders (FASD; Burd et al., 2003). The most severe of these outcomes, fetal alcohol syndrome (FAS), is characterized by pre- and postnatal growth retardation, mental and cognitive disability, distinct craniofacial abnormalities, and severe mental retardation that persist throughout life.
Fetal alcohol syndrome has been described as the leading cause of preventable mental retardation in the world, and presents a considerable health concern, globally (Abel and Sokol, 1987; Guerri et al., 2009). The global prevalence of FAS has been reported to be 0.5–2.0/1000 live births (May and Gossage, 2001), and poses a significant financial burden on health care services (Klug and Burd, 2003). These prevalence rates are vastly increased in some communities (May, 1991; Burd and Moffatt, 1994; Sampson et al., 1997). In particular, the Western and Northern Cape regions of South Africa have been reported to have the highest rates of FAS in the world, with over 70/1000 school-going children affected (Viljoen et al., 2005; May et al., 2007; Urban et al., 2008).
The etiology of FASD is poorly understood despite its high prevalence and detailed clinical characterization. There is evidence for a genetic contribution as part of the underlying cause – which may affect alcohol susceptibility in the developing fetus (as reviewed in Ramsay, 2010). The globally escalating prevalence rates of FAS, in conjunction with our limited understanding of its etiology and the considerable cost to health services, highlight the importance of alcohol-related research.
Although FAS research has focused primarily on maternal alcohol consumption during pregnancy, paternal contributions have been reported where infants were born with characteristic FASD symptoms to mothers who had not consumed alcohol during pregnancy, but whose fathers were alcoholics (Lemoine et al., 1968). One study reported that 75% of children with FAS had biological fathers who were heavy drinkers or alcoholics (Abel et al., 1983); and several rodent and human studies have reported that preconception paternal alcohol intake has been associated with congenital malformations, low birth weights, growth retardation, and neonatal mortality (Friedler, 1996; Passaro et al., 1998). These effects are not consistent between inbred rodent strains, suggesting a genetic contribution to the phenotype outcome. On balance, however, there is evidence that FASD attributed to gestational alcohol exposure, may be the result of, or partly due to, excessive paternal preconception alcohol consumption (Abel, 2004). The etiology of paternal contributions to FASD is however, largely unexplored to date.
Epigenetic modifications are independent of the DNA sequence and regulate gene expression (as reviewed in Holliday, 2006). Epigenetic mechanisms are vital components in regulating cell cycle and developmental processes in a spatial and temporal manner. These mechanisms include DNA methylation of cytosine bases and modifications to the core histone proteins, as well as RNA interference. Imprinted genes rely on DNA methylation to silence and activate alleles in a parent-of-origin specific manner, ensuring the reciprocal expression of specific genes. In some instances, expression is regulated through the methylation-sensitive binding of the insulator protein, CCCTC-binding factor (CTCF), at differentially methylated imprinting control regions (ICRs), through its enhancer-blocking activity. Initial Igf2 knockout studies demonstrated that the Igf2/H19 imprinted locus is involved in mammalian embryonic development and placentation (Barlow et al., 1991; DeChiara et al., 1991). According to the mechanism by which the Igf2/H19 locus is regulated, the hypomethylated state of the maternal ICR allows for the binding of the CTCF protein, insulating the downstream common enhancer from accessing the upstream paternally expressed H19 gene promoter, while the downstream maternally expressed Igf2 gene promoter is selectively acted upon. Conversely, the hypermethylated ICR on the paternal allele prevents CTCF binding and allows downstream enhancers to act on Igf2, while H19 expression is suppressed – effectively achieving reciprocal, monoallelic, parent-of-origin gene expression.
Imprinted genes are vital for fetal growth and development (Fowden et al., 2006), and their dysregulation leads to profound congenital abnormalities (Piedrahita, 2011). The dynamic nature of the mechanisms governing epigenetic regulation and their sensitivity to environmental influences (Bernal and Jirtle, 2010; Chmurzynska, 2010; Skinner et al., 2010; Ferguson-Smith and Patti, 2011), make them an attractive target to study the effect of environmental contributions to disease etiology.
Alcohol is a well-studied teratogen and its metabolites have been shown to directly interfere with epigenetic mechanisms through its influence on one carbon metabolism and the methyl donor pathway. Alcohol enhances the catabolism and loss of methyl groups, which in turn disrupts subsequent SAM-dependent transmethylation reactions of the folate pathway, which are required for DNA methylation (Schalinske and Nieman, 2005).
Investigations using mouse models demonstrated that alcohol exposure during gestation can result in global hypomethylation of fetal DNA (Garro et al., 1991), locus specific hypermethylation (Kaminen-Ahola et al., 2010) and selected hypomethylation of the paternally imprinted H19 ICR in the mouse placenta (Haycock and Ramsay, 2009; although these authors did not observe a difference in fetal DNA). In addition, site-specific hypomethylation of male gametes have been noted in both mice (Stouder et al., 2011) and humans (Ouko et al., 2009) chronically exposed to alcohol. Together, these findings provide a link between altered DNA methylation and FAS.
Given that imprinted genes are essentially haploid in nature – due to their reliance on DNA methylation to silence one of the parental alleles and achieve monoallelic expression – and that perturbations to such regulation results in abnormal fetal phenotypes, makes this group of genes particularly vulnerable to epimutations. Considering the impact of alcohol on DNA methylation, this study aimed to investigate the effect of paternal alcohol exposure on the methylation status of two paternally imprinted loci [Insulin-like growth factor 2 (Igf2)/H19 and Ras protein-specific guanine nucleotide-releasing factor 1 (Rasgrf1)] in both the sperm of exposed males, and their sired offspring. H19 is highly expressed in the developing embryo (Poirier et al., 1991), where it works in conjunction with Igf2, a potent mitogen, to regulate fetal growth and development (Kim and Kim, 2005; Chao and D’Amore, 2008), while Rasgrf1 encodes a guanine-nucleotide exchange factor involved in fetal growth and memory (Brambilla et al., 1997; Itier et al., 1998). Expression of these genes is tightly regulated by the methylation-sensitive binding of CTCF at the ICRs contained within both of these loci (Schoenherr et al., 2003; Yoon et al., 2005).
Contrary to expectation, this study did not observe differences in DNA methylation at the H19 and Rasgrf1 ICRs in the sperm of alcohol-exposed male mice. We did however observe significantly decreased DNA methylation profiles at the H19 ICR CTCF binding sites in the somatic tissue of offspring sired by ethanol-exposed male mice, which was correlated with postnatal growth restriction.
Materials and Methods
Alcohol Exposure in the Mouse Model
Eight C57BL/6 male mice were treated with 7.5 μl/g of a 50% solution of ethanol (5.917 g/kg), simulating a pharmacologically significant effect (Rhodes et al., 2005), which resulted in a blood ethanol concentration of > 1.0 mg/ml. Seven control males received an isocaloric solution of a 0.704 g/ml sucrose solution at 7.5 μl/g. Food and water was given ad libitum. Oral gavaging was carried out over a five week period. During the first week, mice were gavaged every second day; during week two, daily for five days; and daily thereafter. In week six, each male mouse was mated with two C57BL/6 females. Successful mating was determined by the presence of a vaginal plug. A total of 46 offspring (17 sired by ethanol-treated males, 29 sired by sucrose-treated males) were obtained. Offspring were weighed every second day for 30 days, and weekly thereafter, until postnatal day 63. Tail biopsies were taken at four weeks of age. Animal ethics clearance was obtained from the Animal Ethics Screening Committee, University of the Witwatersrand, Johannesburg, South Africa (clearance certificate no. 2007/41/03).
DNA Extraction and Bisulfite Modification
Mature sperm was harvested within 24 h of the last day of mating by epididymal dissection and spermatozoa isolated using the Differex™ System (Promega, Madison, WI, USA). DNA from sperm and offspring tail biopsies were extracted using the QIAamp DNA Micro Kit (Qiagen, Hilden, North Rhine-Westphalia, Germany) and bisulfite modified using the EpiTect® Bisulfite Kit (Qiagen, Hilden, North Rhine-Westphalia, Germany).
ICR PCR Amplification
The approximately 2 kb H19 ICR (located on contig AC13548.13) lies 2 kb upstream of the H19 transcriptional unit. The 423 bp region amplified in the present study contains two of the four H19 ICR CTCF binding sites and a total of 16 CpG sites (approximately 40% of the CpGs contained within the ICR). Six CpGs of the first and five CpGs of the second CTCF binding sites (CTCF 1 and CTCF 2, respectively) were analyzed. The 284 bp Rasgrf1 ICR region amplified is located on contig AC102545.9 and contains 29 CpG sites, with the CTCF binding site spanning CpGs 20–27. A 420 bp region of the Snrpn ICR, containing 16 CpGs, was included as a control for the detection of somatic contamination in sperm DNA. ICRs were PCR amplified using a GeneAmp 2720 thermal cycler (Applied Biosystems, Kotoku, Tokyo, Japan) in a final volume of 50 μl containing 0.2 mM dNTPs, 1 × PCR Gold Buffer, 1.5 mM MgCl2, 1 U AmpliTaq Gold (Applied Biosystems, Kotoku, Tokyo, Japan), 2 μl DNA (100 ng/μl), 10 μM of each forward (F) and reverse (R) primer according to the following conditions: 95°C 5 min (95°C 30 s, Ta°C 1 min, 72°C 1 min) for 50 cycles, 72°C 5 min. The annealing temperatures and primer sequences for amplification of the ICRs from bisulfite modified DNA were as follows: H19 55°C (F 5′-GAGTATTTAGGAGGTATAAGAATT-3′, R 5′-ATCAAAAACTAACATAAACCCCT-3′); Rasgrf1 59°C (F 5′-TAGAGAGTTTATAAAGTTAG-3′, R 5′-ACTAAAACAAAAACAACA-3′); Snrpn 59°C (F 5′-TATGTAATATGATATAGTTTAGAAATTAG-3′, R 5′-AATAAACCCAAATCTAAAATATTTTAATC-3′). A second round of PCR was performed using 1 μl PCR product from the first round under the same reaction conditions. Forward and reverse primers were designed using the PSQ Assay Design Software (Biotage, Uppsala, Uppsala County, Sweden). A universal biotin-labeled primer (5′-biotin-GACGGGACACCGCTGATCGTTTA-3′) was used to generate labeled DNA fragments (Colella et al., 2003). A priming site for the universal primer was achieved by the addition of a complementary 5′ 23 bp tail onto each reverse primer. H19 (CTCF 1) 60°C (F 5′-AGGAGGTATAAGAATTTTGTAAGG-3′, R 5′-GACGGGACACCGCTGATCGTTTATTAAATAACCCACAACATTACCAT-3′); H19 (CTCF 2) 66°C (F 5′-TTGTGTGTAAAGATTAGGGTTGTT-3′, R 5′-GACGGGACACCGCTGATCGTTTAAACTAACATAAACCCCTAACCTCA-3′); Snrpn 66°C (F 5′-TGGTAGTTGTTTTTTGGTAGGATA-3′, R 5′-GACGGGACACCGCTGATCGTTTATCCACAAACCCAACTAACCTT-3′); Rasgrf1 50°C (F 5′-GTTATAGTTGAGTTATAATTATTA-3′, R 5′-ACTAAAACAAAAACAACACTACAAC-3′).
Quantification of DNA Methylation at Selected CpG Sites
DNA methylation was quantified by pyrosequencing in triplicate using the PSQ 96 MA System in conjunction with Pyro Q-CpG Software (Biotage, Uppsala, Uppsala County, Sweden). Sequencing (S) primers were designed using the PSQ Assay Design Software (Biotage, Uppsala, Uppsala County, Sweden) as follows: H19 (CTCF 1; S 5′-GGAATTAGTTGTGGGGTT-3′); H19 (CTCF 2; S 5′-GTGTAAAGATTAGGGTTGTT-3′); Snrpn (S 5′-GTGTAGTTATTGTTTGGGA-3′); Rasgrf1 (S 5′-CCAAATACCACAACAACAA-3′). Two non-CpG cytosine bases were included in all pyrosequencing assays as an internal control for successful bisulfite conversion, and samples containing > 5% unsuccessfully converted non-CpG cytosines were discarded.
Statistical Analyses
Mann–Whitney U tests were used to assess changes in methylation in sperm DNA and somatic DNA of ethanol-exposed males and sired offspring, respectively, between the two treatment groups. Average methylation values were obtained by averaging triplicate data values. A Kruskal–Wallis analysis of variance (ANOVA) and Spearman’s correlations tested for differences in offspring postnatal growth. All data were tested for normality using the Shapiro–Wilk’s W test, and appropriate non-parametric tests were applied. P-values ≤ 0.05 were considered significant.
Results
Methylation Analyses of Offspring Somatic DNA
Analysis of individual CpG sites (Table 1) of H19 CTCF 1 revealed that sites 2, 3, 5–7 (p < 0.01), and 4 (p < 0.05) were significantly less methylated in offspring whose sires were exposed to ethanol. Likewise, the H19 CTCF 2 demonstrated significant hypomethylation at CpG sites 11, 12 and 13 (p = 0.0040, 0.0018 and 0.0014, respectively) when compared to controls. Analysis of the Rasgrf1 CTCF binding site revealed no significant differences in DNA methylation. It was interesting to note evidence of site-specific alterations in DNA methylation at the Snrpn locus in offspring of ethanol-treated sires, with increases at CpG sites 8 and 11 (p < 0.05), and a decrease at CpG site 9 (p = 0.0924) within the ICR. Deviation from the expected average methylation of 50% at these loci (as would normally be expected at imprinted regions) in the control group, is not uncommon, as documented in other studies (Ollikainen et al., 2010; Turan et al., 2010; Choufani et al., 2011). Thus, when comparing the offspring group sired by ethanol-treated males with the control group sired by sucrose-treated males, significant differences observed in DNA methylation can be attributed to the ethanol treatment of sires. The variation in DNA methylation percentage within each of the H19 CTCF binding sites was relatively low from site to site, and was reproducible. The decreases in methylation over consecutive CpG sites were consistent between treatment groups.
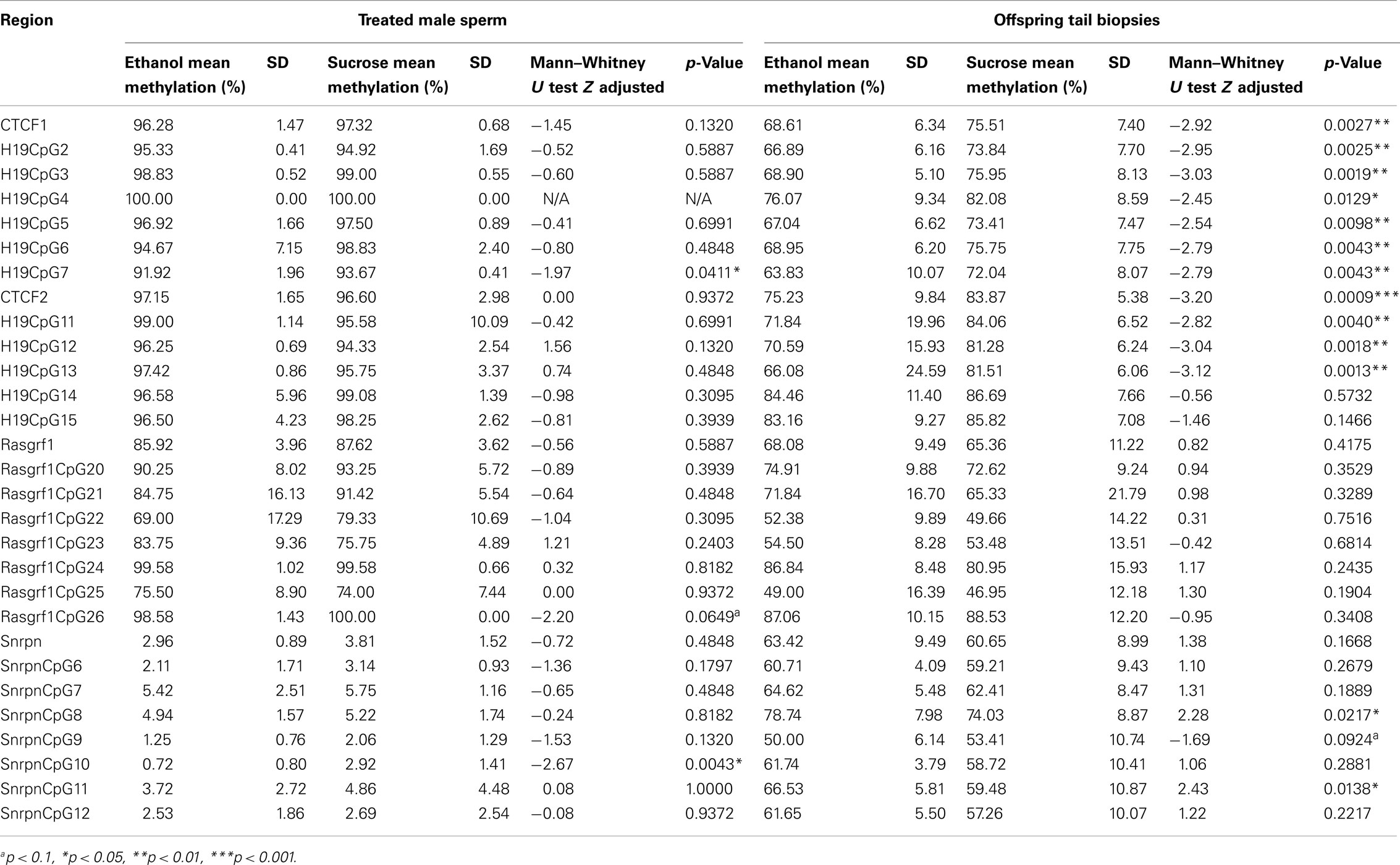
Table 1. Quantitative pyrosequencing data of sperm DNA from ethanol-treated males and tail biopsy DNA from sired offspring.
Offspring Postnatal Growth
The postnatal growth (weight) of offspring sired by ethanol-treated males (n = 17, “ethanol offspring”) and control males (n = 29, “sucrose offspring”) was recorded over a 63 day period. There was no observed difference in birth weight. When stratified by sex, there was a notable trend between the two treatment groups with both ethanol male and female offspring exhibiting a lower postnatal growth rate compared to their sucrose counterparts (Figure 1). This trend was most pronounced after day 30. A Kruskal–Wallis ANOVA (Supplementary Material) revealed that the divergence of sucrose male offspring from ethanol male offspring was significant at day 35 (p = 0.0389) with days 42, 49, and 56 approaching significance (p = 0.0640, 0.0640, and 0.0901, respectively). Similarly, female ethanol offspring showed lower weights than their sucrose counterparts at days 35 and 42 (p = 0.0353 and 0.0353, respectively).
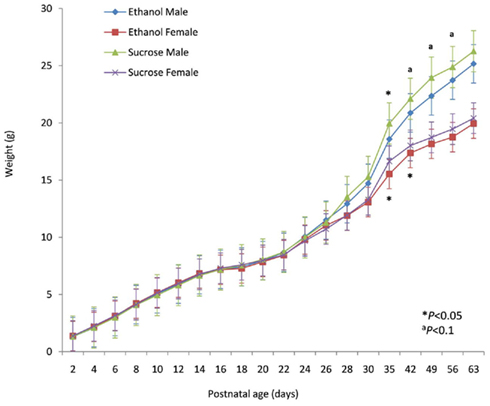
Figure 1. Postnatal growth rate of sired offspring. Postnatal weight of offspring sired by sucrose-exposed (control) males and ethanol-exposed males was measured over a 63 day period and stratified by sex (sucrose male/female and ethanol male/female, respectively). Measurements were taken every second day until postnatal day 30, and weekly thereafter.
Correlation of Offspring ICR DNA Methylation with Weight at Day 35
Analyses were conducted on offspring DNA methylation profiles at the level of individual CpG sites, and were correlated with offspring weight at day 35 – where a significant difference in weight between the treatment groups was observed (Table S1 in Supplementary Material). Significant, positive correlations suggested that decreased DNA methylation was associated with decreased offspring weight. Such correlations were observed for all CpG sites (2–7) comprising the H19 CTCF 1 region at day 35. Similarly, DNA methylation at the CpG sites contained within the H19 CTCF 2 region (11–14) were significantly correlated with offspring weight at day 35 (p < 0.05), with CpG 15 approaching significance (p = 0.0509).
Methylation Analyses of Sperm DNA
The Snrpn ICR was included as a control for the detection of contaminating somatic DNA in sperm samples. The DNA methylation status at this ICR was consistently low in 12 samples – ranging between 0.72 and 5.75% – and were used for subsequent analyses. The sperm of one of the 13 male mice had an average level of over 10% DNA methylation and was excluded from the study. The Snrpn ICR revealed significantly decreased DNA methylation at CpG 10 (p = 0.0043; Table 1). Overall, there was no significant demethylation at the H19 and Rasgrf1 ICRs, with only one CpG site demonstrating a significant reduction in DNA methylation in the ethanol-treated male group (H19 CpG 7, p = 0.0411), and Rasgrf1 CpG 26 bordering on significance (p = 0.0649; Table 1). Fluctuations in the Snrpn methylation status suggests that these difference are unlikely to be of significance.
Discussion
The findings of this study are consistent with the prediction that preconception alcohol treatment of male mice has an adverse effect on sired offspring. This included age restricted growth retardation and a reduction in DNA methylation at the H19 paternally imprinted ICR.
There is conflicting evidence about the effect of paternal preconception alcohol exposure on offspring birth weight. While some studies have shown that alcohol orally administered to male mice for periods of between 24 h and 3 months prior to conception tends to result in decreased birth weights of sired offspring (Cicero et al., 1994; Ledig et al., 1998); others have not observed such an effect in both humans and rodent models (Anderson and Willis, 1981; Abel and Lee, 1988; Abel and Tan, 1988; Abel, 1989, 1993; Passaro et al., 1998; Livy et al., 2004). Although these latter findings are supported by our study (when offspring are stratified by sex; data not shown), evidence of significant age-dependent growth restriction of both male and female offspring sired by ethanol-exposed males was observed, from approximately postnatal day 35–42. This is supported by rodent studies which noted inferior growth rates of offspring sired by alcohol-exposed males when compared to control offspring (Stockard, 1913; Stockard and Papanicolaou, 1916a,b); and findings of significantly decreased postnatal growth in children of alcoholic men (Abel and Sokol, 1983; Little and Sing, 1987).
When the regulation of imprinted loci is dependent on the methylation-sensitive binding of CTCF within the ICR, changes in DNA methylation may have profound effects on CTCF binding and gene expression. In the present study, the average decrease in DNA methylation across the CpG sites of the H19 ICR in the offspring sired by ethanol-treated males when compared to their controls, ranged between 2.23 and 15.43% (Table 1). This decrease could potentially allow increased CTCF binding at the paternal ICR, essentially switching the allele to a maternal epigenotype, and significantly reducing paternally expressed Igf2, while overexpressing H19, contributing to a growth restricted phenotype (as highlighted in Bourque et al., 2010). It has been demonstrated that inhibition of DNA methyltransferases (Dnmts) results in decreased DNA methylation at the Igf2/H19 locus in mice, and subsequent activation of H19 from the paternal allele (Li et al., 1992). Furthermore, decreased placental methylation at the H19 ICR has been associated with significantly underexpressed IGF2 in placental villi, correlated with postnatal growth restriction in humans (Bartholdi et al., 2009; Bourque et al., 2010).
There is evidence to suggest that modest changes in DNA methylation at CTCF binding regions can elicit a significant biological effect. Induced loss of DNA methylation at the human telomerase reverse transcriptase (hTERT) promoter from between 10 and 39% (Renaud et al., 2007; Choi et al., 2010), resulted in significantly increased CTCF binding, with a concomitant reduction in hTERT expression. Furthermore, demethylation of the Interleukin 6 gene promoter (Nile et al., 2008) and a single CpG site within the Interleukin 2 promoter-enhancer region (Murayama et al., 2006), have been associated with elevated interleukin expression via increased protein interaction at these regions, and in the case of increased Interleukin 6 expression, has been shown to contribute to the risk of developing rheumatoid arthritis. The changes observed in the present study are similar to these findings and may suggest a functional impact on gene expression in the offspring of ethanol-treated sires.
Imprinted genes have collectively been associated with fetal growth and development (Fowden et al., 2006), postnatal adaptation to feeding (Plagge et al., 2004), and perturbations to both paternally and maternally expressed genes have been linked to behavioral phenotypes that include deficits in feeding, pup suckling and energy metabolism (Curley and Mashoodh, 2010). Sire treatment with ethanol was significantly correlated with decreased offspring somatic DNA methylation and weight restriction at day 35, but not at birth. This coincides with the post-weaning period, and may suggest difficulties in establishing independent feeding due to potential mental deficit and subsequent undernourishment during the transition period from maternal mammary milk to solid foods. It has been shown that rat pups prenatally exposed to alcohol demonstrated postnatal growth deficiencies which were attributed to suckling difficulties (Chen et al., 1982). Furthermore, children born with FAS frequently suffer from feeding difficulties – which are said to be an indication of central nervous dysfunction – and exacerbates postnatal growth deficiencies (Driscoll et al., 1990). The teratogenic effect of alcohol associated with FAS severely affects neural development (Kotch and Sulik, 1992a,b) which results in central nervous system dysfunction. The etiology of these effects is not fully understood, but alcohol exposure during early neurulation has been shown to ubiquitously decrease DNA methylation, which is associated with changes in gene expression in the developing mouse brain (Liu et al., 2009). This finding suggests that aberrant DNA methylation at several loci may contribute to the mental deficits associated with FAS. The fact that ethanol can exert its effect on DNA methylation throughout the genome, and that there is support for the hypothesis that environmental perturbations of the epigenotype occur most readily at loci where the epigenome is undergoing developmental change (Waterland et al., 2006), opens the possibility that several imprinted loci in the sperm may have been significantly affected by preconception alcohol exposure.
Aberrant H19 ICR methylation has been associated with the growth retardation syndrome, Silver–Russell syndrome (SRS), which is characterized by severe fetal growth restriction. Hypomethylation of the H19 ICR has been reported in 20–65% of SRS patients, whose degree of hypomethylation has been associated with phenotype severity (Bruce et al., 2009). Patients with moderate methylation (9–35%) did not appear to be significantly growth restricted at birth, but did exhibit postnatal growth deficiencies – where the degree of H19 ICR hypomethylation was associated with increasing feeding difficulties (Blissett et al., 2001). Recently, hypomethylation at the H19 ICR CTCF binding sites and the consequent aberrant binding of CTCF at this locus has been linked with SRS etiology (Nativio et al., 2011). Feeding difficulties associated with SRS have been attributed to cognitive defects that affect oral-motor dysfunction, including problems with chewing unfamiliar textures, and has been observed in 90% of children with SRS (Blissett et al., 2001). There are interesting parallels with the current study. Firstly, as noted by Bruce et al. (2009), we did not observe significant reductions in birth weight in offspring who demonstrated a moderate (approximately 15%), yet significant reduction in H19 ICR methylation, but did observe postnatal growth deficiencies. Secondly, a growth restricted phenotype similar to SRS, was correlated with hypomethylation of the H19 ICR CTCF binding sites that was associated with reduced offspring growth in the period following weaning (Marques et al., 2008; Nicholson et al., 2009). Lastly, weight recovery was observed during the post-weaning period, similar to findings of Blissett et al. (2001). Taken together, it is therefore plausible that the observed postnatal growth restricted phenotype during the post-weaning period could be attributed to feeding difficulties associated with altered DNA methylation of one or more ICRs that influence mental functioning and postnatal feeding behavior.
Although the exact mechanism that contributed to the age-dependent growth-restricted phenotype observed in the offspring in our study is only speculative, the fact that significantly reduced methylation profiles at the H19 ICR were evident in offspring of ethanol-treated sires, suggests that an epimutation was inherited via the paternal germline. It has been demonstrated that ethanol exposure causes epigenetic remodeling, affecting both histone proteins and DNA methylation. A recent study by Stouder et al. (2011), exposed pregnant mice to a low dose of ethanol (0.5g/kg) to determine aberrant effects on DNA methylation at imprinted loci in their offspring, including their gametes. These authors found that direct in utero exposure to ethanol resulted in significant decreases in sperm DNA methylation at the H19 ICR CTCF 2 binding site, but no difference in somatic tissues of offspring. This might suggest that sperm DNA methylation, specifically at the H19 ICR, is particularly sensitive to ethanol exposure. However, findings of reduced methylation in the sperm DNA of ethanol exposed males in our study were nuanced and significantly evident at only two CpG sites in the two loci investigated (H19 CpG 7 and Rasgrf1 CpG 26). Stouder et al. (2011) subsequently produced offspring from males with sperm H19 ICR DNA methylation aberrations, and contrary to their previous findings, did not observe methylation differences in the sperm DNA of these offspring, but rather, significantly decreased DNA methylation at the H19 ICR CTCF 2 binding site in whole brain tissue.
Despite the presence of significantly reduced DNA methylation at the H19 CTCF 1 and CTCF 2 binding sites in offspring sired by ethanol-treated males in our study, only a single CpG site (H19 CpG 7) correlated with a similar significant decrease in sire sperm DNA. The lack of evidence of reduced DNA methylation in the sperm of ethanol-exposed males, may also suggest that the methylation deficiency observed at the corresponding ICRs in their offspring occurs post conception, but may be "programmed" in sperm prior to conception via alternate mechanisms susceptible to ethanol. These could include RNA mediated effects and histone modifications. RNA molecules present in, and unique to, sperm (Ostermeier et al., 2002, 2005; Geijsen et al., 2004; Rassoulzadegan et al., 2006) have been reported to be successfully transmitted to the oocyte during fertilization (Ostermeier et al., 2004). These RNAs (which include miRNAs) play a vital role in gene regulation and fetal development (Geijsen et al., 2004), and perturbations to these molecules in sperm have been shown to elicit aberrant offspring phenotypes (Rassoulzadegan et al., 2006). Ethanol has been found to disrupt miRNA expression (Sathyan et al., 2007; Wang et al., 2008) and is correlated with significant decreases in offspring weight (Wang et al., 2009). Furthermore, evidence of RNA directed DNA methylation has been described in two studies that demonstrated the establishment of DNA methylation by small RNA intermediates (Kawasaki and Taira, 2004; Morris et al., 2004). Alcohol exposure is also known to result in folate deficiency which, causes significant biological effects by altering histone modifications at lysine residues (Park et al., 2003; Kim and Shukla, 2005, 2006; Esteller, 2008; Shukla et al., 2008) and are associated with altered gene expression (Pal-Bhadra et al., 2007). Although sperm DNA is mainly protamine-bound, approximately 15% of human sperm DNA is retained in histone-bound structures (Gatewood et al., 1987; Bench et al., 1996; Hammoud et al., 2009) which, through epigenetic modifications, acquire precise nucleosome organization that regulates gene expression. It has been demonstrated that nucleosomes retained in sperm are significantly enriched at developmentally significant loci, including imprinted regions (Wykes and Krawetz, 2003; Hammoud et al., 2009). It is interesting to note that the hypomethylated and hypermethylated state of the H19 ICR of the maternal and paternal alleles, respectively, are associated with allele-specific histone modifications, particularly at lysine residues (Kacem and Feil, 2009). Considering the effect of alcohol on these histone modifications, alterations at these residues could convert paternal allele histone modifications to those characteristic of the maternal allele – a type of epigenotype switching. Dobosy et al. (2008) demonstrated that although mice fed on choline-methionine deficient diets demonstrated significant changes in gene expression levels of Igf2 and H19, this occurred without depression in CpG methylation, but was associated with changes in lysine residue modifications at both Igf2 and H19 promoter regions. Taken together, the evidence that a methionine deficient diet can alter histone modifications without significant changes in DNA methylation, in conjunction with ethanol’s ability to induce changes in histone modifications – which are typically associated with hypomethylated regions (synonymous with the H19 ICR methylation status of the maternal allele), and the ability of such histone modifications to negatively influence the establishment of DNA methylation (Mutskov et al., 2002), supports the proposed mechanism for the transmission of epimutations without a loss of DNA methylation at imprinted loci in sperm, but which abrogates subsequent establishment of DNA methylation in sired offspring. These mechanisms do however require further investigation.
A caveat of the present study, is that bisulfite sequencing does not distinguish between 5-methylcytosine (5mC) and 5-hydroxymethylcytosine (5hmC) (Nestor et al., 2010). 5hmC occurs naturally via the hydroxylation of 5mC by the Tet dioxygenases and has been implicated in active DNA demethylation (Wossidlo et al., 2011). Its strong association with Purkinje cells, embryonic stem cells, and the paternal pronucleus may suggest an intermediate functional role in 5mC reprogramming by influencing the binding of 5mC-specific proteins (as described in Wossidlo et al., 2011). This may be of particular importance to the present study, as alcohol metabolism has been proposed to induce oxidation of 5mC to form 5hmC (Jenner et al., 1998; Wright et al., 1999). The apparent lack of methylation alterations in sperm DNA of ethanol-exposed males may be the result of the inadequacy of bisulfite sequencing to distinguish 5mC from 5hmC, but which may have manifested later as demethylated DNA in sired offspring, accounting for the observed reduction in somatic DNA methylation.
In conclusion, this study suggests that offspring sired by male mice exposed to ethanol for a five week period prior to conception inherit aberrant epigenetic signatures at a developmentally significant locus, the H19 ICR. These changes were correlated with an age-related growth retardation phenotype in the immediate post-weaning period, suggesting that it is a consequence of a postnatal feeding behavioral aberration, as opposed to a direct effect on growth regulation in utero. It is proposed that reduced levels of ICR DNA methylation are as a result of an ethanol-induced insult to the sperm DNA prior to conception. As ethanol exposure did not appear to reduce DNA methylation at the corresponding locus in sperm DNA, it is proposed that the inability of bisulfite sequencing to discriminate 5mC from 5hmC may have masked ethanol’s effect on sperm DNA, or that other epigenetic mechanisms susceptible to the effects of ethanol mediated this effect. Our data support an epigenetic mechanism for the transmission of altered epigenetic states through the male germline, caused by excessive paternal ethanol exposure prior to conception, with important implications for the etiology of FAS.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to acknowledge the March of Dimes (Grant # 6-FY04-70), the National Health Laboratory Service Research Trust and the Medical Faculty Research Endowment Fund (University of the Witwatersrand) for funding; and to thank all staff at the University of the Witwatersrand’s Central Animal Services for their support.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/epigenomics/10.3389/fgene.2012.00010/abstract
References
Abel, E. (2004). Paternal contribution to fetal alcohol syndrome. Addict. Biol. 9, 127–33; discussion 135–136.
Abel, E. L. (1989). Duration of paternal alcohol consumption does not influence offspring growth and development. Growth Dev. Aging 53, 195–199.
Abel, E. L., Jacobson, S., and Sherwin, B. T. (1983). In utero alcohol exposure: functional and structural brain damage. Neurobehav. Toxicol. Teratol. 5, 363–366.
Abel, E. L., and Lee, J. A. (1988). Paternal alcohol exposure affects offspring behavior but not body or organ weights in mice. Alcohol. Clin. Exp. Res. 12, 349–355.
Abel, E. L., and Sokol, R. J. (1987). Incidence of fetal alcohol syndrome and economic impact of FAS-related anomalies. Drug Alcohol Depend. 19, 51–70.
Abel, E. L., and Sokol, R. M. (1983). Fetal alcohol syndrome: how good is the criticism? Neurobehav. Toxicol. Teratol. 5, 491–492.
Abel, E. L., and Tan, S. E. (1988). Effects of paternal alcohol consumption on pregnancy outcome in rats. Neurotoxicol. Teratol. 10, 187–192.
Anderson, R. A., and Willis, B. R. (1981). Alcohol and male fertility. Br. J. Alcohol Alcohol. 16, 179–185.
Barlow, D. P., Stoger, R., Herrmann, B. G., Saito, K., and Schweifer, N. (1991). The mouse insulin-like growth factor type-2 receptor is imprinted and closely linked to the Tme locus. Nature 349, 84–87.
Bartholdi, D., Krajewska-Walasek, M., Ounap, K., Gaspar, H., Chrzanowska, K. H., Ilyana, H., Kayserili, H., Lurie, I. W., Schinzel, A., and Baumer, A. (2009). Epigenetic mutations of the imprinted IGF2-H19 domain in Silver-Russell syndrome (SRS): results from a large cohort of patients with SRS and SRS-like phenotypes. J. Med. Genet. 46, 192–197.
Bench, G. S., Friz, A. M., Corzett, M. H., Morse, D. H., and Balhorn, R. (1996). DNA and total protamine masses in individual sperm from fertile mammalian subjects. Cytometry 23, 263–271.
Bernal, A. J., and Jirtle, R. L. (2010). Epigenomic disruption: the effects of early developmental exposures. Birth Defects Res. Part A Clin. Mol. Teratol. 88, 938–944.
Blissett, J., Harris, G., and Kirk, J. (2001). Feeding problems in Silver-Russell syndrome. Dev. Med. Child Neurol. 43, 39–44.
Bourque, D. K., Avila, L., Penaherrera, M., von Dadelszen, P., and Robinson, W. P. (2010). Decreased placental methylation at the H19/IGF2 imprinting control region is associated with normotensive intrauterine growth restriction but not preeclampsia. Placenta 31, 197–202.
Brambilla, R., Gnesutta, N., Minichiello, L., White, G., Roylance, A. J., Herron, C. E., Ramsey, M., Wolfer, D. P., Cestari, V., Rossi-Arnaud, C., Grant, S. G., Chapman, P. F., Lipp, H. P., Sturani, E., and Klein, R. (1997). A role for the Ras signalling pathway in synaptic transmission and long-term memory. Nature 390, 281–286.
Bruce, S., Hannula-Jouppi, K., Peltonen, J., Kere, J., and Lipsanen-Nyman, M. (2009). Clinically distinct epigenetic subgroups in Silver-Russell syndrome: the degree of H19 hypomethylation associates with phenotype severity and genital and skeletal anomalies. J. Clin. Endocrinol. Metab. 94, 579–587.
Burd, L., and Moffatt, M. E. (1994). Epidemiology of fetal alcohol syndrome in American Indians, Alaskan natives, and Canadian aboriginal peoples: a review of the literature. Public Health Rep. 109, 688–693.
Burd, L., Selfridge, R. H., Klug, M. G., and Juelson, T. (2003). Fetal alcohol syndrome in the Canadian corrections system. J. FAS Int. 1, 1–7.
Chao, W., and D’Amore, P. A. (2008). IGF2: epigenetic regulation and role in development and disease. Cytokine Growth Factor Rev. 19, 111–120.
Chen, J. S., Driscoll, C. D., and Riley, E. P. (1982). Ontogeny of suckling behavior in rats prenatally exposed to alcohol. Teratology 26, 145–153.
Chmurzynska, A. (2010). Fetal programming: link between early nutrition, DNA methylation, and complex diseases. Nutr. Rev. 68, 87–98.
Choi, J.-H., Min, N. Y., Park, J., Kim, J. H., Park, S. H., Ko, Y. J., Kang, Y., Moon, Y. J., Rhee, S., and Ham, S. W. (2010). TSA-induced DNMT1 down-regulation represses hTERT expression via recruiting CTCF into demethylated core promoter region of hTERT in HCT116. Biochem. Biophys. Res. Commun. 391, 449–454.
Choufani, S., Shapiro, J. S., Susiarjo, M., Butcher, D. T., Grafodatskaya, D., Lou, Y., Ferreira, J. C., Pinto, D., Scherer, S. W., Shaffer, L. G., Coullin, P., Caniggia, I., Beyene, J., Slim, R., Bartolomei, M. S., and Weksberg, R. (2011). A novel approach identifies new differentially methylated regions (DMRs) associated with imprinted genes. Genome Res. 21, 465–476.
Cicero, T. J., Nock, B., O’Connor, L. H., Sewing, B. N., Adams, M. L., and Meyer, E. R. (1994). Acute paternal alcohol exposure impairs fertility and fetal outcome. Life Sci. 55, PL33–PL36.
Colella, S., Shen, L., Baggerly, K. A., Issa, J. P., and Krahe, R. (2003). Sensitive and quantitative universal pyrosequencing methylation analysis of CpG sites. Biotechniques 35, 146–150.
Curley, J. P., and Mashoodh, R. (2010). Parent-of-origin and trans-generational germline influences on behavioral development: the interacting roles of mothers, fathers, and grandparents. Dev. Psychobiol. 52, 312–330.
DeChiara, T. M., Robertson, E. J., and Efstratiadis, A. (1991). Parental imprinting of the mouse insulin-like growth factor II gene. Cell 64, 849–859.
Dobosy, J. R., Fu, V. X., Desotelle, J. A., Srinivasan, R., Kenowski, M. L., Almassi, N., Weindruch, R., Svaren, J., and Jarrard, D. F. (2008). A methyl-deficient diet modifies histone methylation and alters Igf2 and H19 repression in the prostate. Prostate 68, 1187–1195.
Driscoll, C. D., Streissguth, A. P., and Riley, E. P. (1990). Prenatal alcohol exposure: comparability of effects in humans and animal models. Neurotoxicol. Teratol. 12, 231–237.
Fowden, A. L., Sibley, C., Reik, W., and Constancia, M. (2006). Imprinted genes, placental development and fetal growth. Horm. Res. 65(Suppl. 3), 50–58.
Friedler, G. (1996). Paternal exposures: impact on reproductive and developmental outcome. An overview. Pharmacol. Biochem. Behav. 55, 691–700.
Garro, A. J., McBeth, D. L., Lima, V., and Lieber, C. S. (1991). Ethanol consumption inhibits fetal DNA methylation in mice: implications for the fetal alcohol syndrome. Alcohol. Clin. Exp. Res. 15, 395–398.
Gatewood, J. M., Cook, G. R., Balhorn, R., Bradbury, E. M., and Schmid, C. W. (1987). Sequence-specific packaging of DNA in human sperm chromatin. Science 236, 962–964.
Geijsen, N., Horoschak, M., Kim, K., Gribnau, J., Eggan, K., and Daley, G. Q. (2004). Derivation of embryonic germ cells and male gametes from embryonic stem cells. Nature 427, 148–154.
Guerri, C., Bazinet, A., and Riley, E. P. (2009). Foetal alcohol spectrum disorders and alterations in brain and behaviour. Alcohol Alcohol. 44, 108–114.
Hammoud, S., Liu, L., and Carrell, D. T. (2009). Protamine ratio and the level of histone retention in sperm selected from a density gradient preparation. Andrologia 41, 88–94.
Haycock, P. C., and Ramsay, M. (2009). Exposure of mouse embryos to ethanol during preimplantation development: effect on DNA methylation in the H19 imprinting control region. Biol. Reprod. 81, 618–627.
Itier, J. M., Tremp, G. L., Leonard, J. F., Multon, M. C., Ret, G., Schweighoffer, F., Tocque, B., Bluet-Pajot, M. T., Cormier, V., and Dautry, F. (1998). Imprinted gene in postnatal growth role. Nature 393, 125–126.
Jenner, A., England, T. G., Aruoma, O. I., and Halliwell, B. (1998). Measurement of oxidative DNA damage by gas chromatography-mass spectrometry: ethanethiol prevents artifactual generation of oxidized DNA bases. Biochem. J. 331(Pt 2), 365–369.
Kacem, S., and Feil, R. (2009). Chromatin mechanisms in genomic imprinting. Mamm. Genome 20, 544–556.
Kaminen-Ahola, N., Ahola, A., Maga, M., Mallitt, K. A., Fahey, P., Cox, T. C., Whitelaw, E., and Chong, S. (2010). Maternal ethanol consumption alters the epigenotype and the phenotype of offspring in a mouse model. PLoS Genet. 6, e1000811. doi:10.1371/journal.pgen.1000811
Kawasaki, H., and Taira, K. (2004). Induction of DNA methylation and gene silencing by short interfering RNAs in human cells. Nature 431, 211–217.
Kim, H. J., and Kim, T. Y. (2005). Regulation of vascular endothelial growth factor expression by insulin-like growth factor-II in human keratinocytes, differential involvement of mitogen-activated protein kinases and feedback inhibition of protein kinase C. Br. J. Dermatol. 152, 418–425.
Kim, J. S., and Shukla, S. D. (2005). Histone H3 modifications in rat hepatic stellate cells by ethanol. Alcohol Alcohol. 40, 367–372.
Kim, J. S., and Shukla, S. D. (2006). Acute in vivo effect of ethanol (binge drinking) on histone H3 modifications in rat tissues. Alcohol Alcohol. 41, 126–132.
Klug, M. G., and Burd, L. (2003). Fetal alcohol syndrome prevention: annual and cumulative cost savings. Neurotoxicol. Teratol. 25, 763–765.
Kotch, L. E., and Sulik, K. K. (1992a). Experimental fetal alcohol syndrome: proposed pathogenic basis for a variety of associated facial and brain anomalies. Am. J. Med. Genet. 44, 168–176.
Kotch, L. E., and Sulik, K. K. (1992b). Patterns of ethanol-induced cell death in the developing nervous system of mice; neural fold states through the time of anterior neural tube closure. Int. J. Dev. Neurosci. 10, 273–279.
Ledig, M., Misslin, R., Vogel, E., Holownia, A., Copin, J. C., and Tholey, G. (1998). Paternal alcohol exposure: developmental and behavioral effects on the offspring of rats. Neuropharmacology 37, 57–66.
Lemoine, P., Harousseau, H., Borteyru, J. P., and Menuet, J. C. (1968). Les enfants des parents alcoholiques: anomolies observees a propos de 127 cas (The children of alcoholic parents: anomalies observed in 127 cases). Quest Med. 25, 476–482.
Li, E., Bestor, T. H., and Jaenisch, R. (1992). Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 69, 915–926.
Little, R. E., and Sing, C. F. (1987). Father’s drinking and infant birth weight: report of an association. Teratology 36, 59–65.
Liu, Y., Balaraman, Y., Wang, G., Nephew, K. P., and Zhou, F. C. (2009). Alcohol exposure alters DNA methylation profiles in mouse embryos at early neurulation. Epigenetics 4, 500–511.
Livy, D. J., Maier, S. E., and West, J. R. (2004). Long-term alcohol exposure prior to conception results in lower fetal body weights. Birth Defects Res. B Dev. Reprod. Toxicol. 71, 135–141.
Marques, J. M., Olsson, I. A., Ogren, S. O., and Dahlborn, K. (2008). Evaluation of exploration and risk assessment in pre-weaning mice using the novel cage test. Physiol. Behav. 93, 139–147.
May, P. A. (1991). Fetal alcohol effects among North American Indians. Alcohol Health Res. World 15, 239–248.
May, P. A., and Gossage, J. P. (2001). Estimating the prevalence of fetal alcohol syndrome. A summary. Alcohol Res. Health 25, 159–167.
May, P. A., Gossage, J. P., Marais, A.-S., Adnams, C. M., Hoyme, H. E., Jones, K. L., Robinson, L. K., Khaole, N. C. O., Snell, C., and Kalberg, W. O. (2007). The epidemiology of fetal alcohol syndrome and partial FAS in a South African community. Drug Alcohol Depend. 88, 259–271.
Morris, K. V., Chan, S. W., Jacobsen, S. E., and Looney, D. J. (2004). Small interfering RNA-induced transcriptional gene silencing in human cells. Science 305, 1289–1292.
Murayama, A., Sakura, K., Nakama, M., Yasuzawa-Tanaka, K., Fujita, E., Tateishi, Y., Wang, Y., Ushijima, T., Baba, T., Shibuya, K., Shibuya, A., Kawabe, Y., and Yanagisawa, J. (2006). A specific CpG site demethylation in the human interleukin 2 gene promoter is an epigenetic memory. EMBO J. 25, 1081–1092.
Mutskov, V. J., Farrell, C. M., Wade, P. A., Wolffe, A. P., and Felsenfeld, G. (2002). The barrier function of an insulator couples high histone acetylation levels with specific protection of promoter DNA from methylation. Genes Dev. 16, 1540–1554.
Nativio, R., Sparago, A., Ito, Y., Weksberg, R., Riccio, A., and Murrell, A. (2011). Disruption of genomic neighbourhood at the imprinted IGF2-H19 locus in Beckwith-Wiedemann syndrome and Silver-Russell syndrome. Hum. Mol. Genet. 20, 1363–1374.
Nestor, C., Ruzov, A., Meehan, R., and Dunican, D. (2010). Enzymatic approaches and bisulfite sequencing cannot distinguish between 5-methylcytosine and 5-hydroxymethylcytosine in DNA. Biotechniques 48, 317–319.
Nicholson, A., Malcolm, R. D., Russ, P. L., Cough, K., Touma, C., Palme, R., and Wiles, M. V. (2009). The response of C57BL/6J and BALB/cJ mice to increased housing density. J. Am. Assoc. Lab. Anim. Sci. 48, 740–753.
Nile, C. J., Read, R. C., Akil, M., Duff, G. W., and Wilson, A. G. (2008). Methylation status of a single CpG site in the IL6 promoter is related to IL6 messenger RNA levels and rheumatoid arthritis. Arthritis Rheum. 58, 2686–2693.
Ollikainen, M., Smith, K. R., Joo, E. J., Ng, H. K., Andronikos, R., Novakovic, B., Abdul Aziz, N. K., Carlin, J. B., Morley, R., Saffery, R., and Craig, J. M. (2010). DNA methylation analysis of multiple tissues from newborn twins reveals both genetic and intrauterine components to variation in the human neonatal epigenome. Hum. Mol. Genet. 19, 4176–4188.
Ostermeier, G. C., Dix, D. J., Miller, D., Khatri, P., and Krawetz, S. A. (2002). Spermatozoal RNA profiles of normal fertile men. Lancet 360, 772–777.
Ostermeier, G. C., Goodrich, R. J., Moldenhauer, J. S., Diamond, M. P., and Krawetz, S. A. (2005). A suite of novel human spermatozoal RNAs. J. Androl. 26, 70–74.
Ostermeier, G. C., Miller, D., Huntriss, J. D., Diamond, M. P., and Krawetz, S. A. (2004). Reproductive biology: delivering spermatozoan RNA to the oocyte. Nature 429, 154.
Ouko, L. A., Shantikumar, K., Knezovich, J., Haycock, P., Schnugh, D. J., and Ramsay, M. (2009). Effect of alcohol consumption on CpG methylation in the differentially methylated regions of H19 and IG-DMR in male gametes: implications for fetal alcohol spectrum disorders. Alcohol. Clin. Exp. Res. 33, 1615–1627.
Pal-Bhadra, M., Bhadra, U., Jackson, D. E., Mamatha, L., Park, P. H., and Shukla, S. D. (2007). Distinct methylation patterns in histone H3 at Lys-4 and Lys-9 correlate with up- & down-regulation of genes by ethanol in hepatocytes. Life Sci. 81, 979–987.
Park, P. H., Miller, R., and Shukla, S. D. (2003). Acetylation of histone H3 at lysine 9 by ethanol in rat hepatocytes. Biochem. Biophys. Res. Commun. 306, 501–504.
Passaro, K. T., Little, R. E., Savitz, D. A., and Noss, J. (1998). Effect of paternal alcohol consumption before conception on infant birth weight. ALSPAC Study Team. Avon Longitudinal Study of Pregnancy and Childhood. Teratology 57, 294–301.
Piedrahita, J. A. (2011). The role of imprinted genes in fetal growth abnormalities. Birth Defects Res. Part A Clin. Mol. Teratol. 91, 682–692.
Plagge, A., Gordon, E., Dean, W., Boiani, R., Cinti, S., Peters, J., and Kelsey, G. (2004). The imprinted signaling protein XL alpha s is required for postnatal adaptation to feeding. Nat. Genet. 36, 818–826.
Poirier, F., Chan, C. T., Timmons, P. M., Robertson, E. J., Evans, M. J., and Rigby, P. W. (1991). The murine H19 gene is activated during embryonic stem cell differentiation in vitro and at the time of implantation in the developing embryo. Development 113, 1105–1114.
Ramsay, M. (2010). Genetic and epigenetic insights into fetal alcohol spectrum disorders. Genome Med. 2, 27.
Rassoulzadegan, M., Grandjean, V., Gounon, P., Vincent, S., Gillot, I., and Cuzin, F. (2006). RNA-mediated non-mendelian inheritance of an epigenetic change in the mouse. Nature 441, 469–474.
Renaud, S., Loukinov, D., Abdullaev, Z., Guilleret, I., Bosman, F. T., Lobanenkov, V., and Benhattar, J. (2007). Dual role of DNA methylation inside and outside of CTCF-binding regions in the transcriptional regulation of the telomerase hTERT gene. Nucleic Acids Res. 35, 1245–1256.
Rhodes, J. S., Best, K., Belknap, J. K., Finn, D. A., and Crabbe, J. C. (2005). Evaluation of a simple model of ethanol drinking to intoxication in C57BL/6J mice. Physiol. Behav. 84, 53–63.
Sampson, P. D., Streissguth, A. P., Bookstein, F. L., Little, R. E., Clarren, S. K., Dehaene, P., Hanson, J. W., and Graham, J. M. Jr. (1997). Incidence of fetal alcohol syndrome and prevalence of alcohol-related neurodevelopmental disorder. Teratology 56, 317–326.
Sathyan, P., Golden, H. B., and Miranda, R. C. (2007). Competing interactions between micro-RNAs determine neural progenitor survival and proliferation after ethanol exposure: evidence from an ex vivo model of the fetal cerebral cortical neuroepithelium. J. Neurosci. 27, 8546–8557.
Schalinske, K. L., and Nieman, K. M. (2005). Disruption of methyl group metabolism by ethanol. Nutr. Rev. 63, 387–391.
Schoenherr, C. J., Levorse, J. M., and Tilghman, S. M. (2003). CTCF maintains differential methylation at the Igf2/H19 locus. Nat. Genet. 33, 66–69.
Shukla, S. D., Velazquez, J., French, S. W., Lu, S. C., Ticku, M. K., and Zakhari, S. (2008). Emerging role of epigenetics in the actions of alcohol. Alcohol. Clin. Exp. Res. 32, 1525–1534.
Skinner, M. K., Manikkam, M., and Guerrero-Bosagna, C. (2010). Epigenetic transgenerational actions of environmental factors in disease etiology. Trends Endocrinol. Metab. 21, 214–222.
Stockard, C. R. (1913). The effect on the offspring of intoxicating the male parent and the transmission of the defects to subsequent generations. Am. Nat. 47, 641–682.
Stockard, C. R., and Papanicolaou, G. (1916a). A further analysis of the hereditary transmission of degeneracy and deformities by the descendants of alcoholized mammals. Am. Nat. 50, 65–88.
Stockard, C. R., and Papanicolaou, G. (1916b). A further analysis of the hereditary transmission of degeneracy and deformities by the descendants of alcoholized mammals II. Am. Nat. 50, 144–177.
Stouder, C., Somm, E., and Paoloni-Giacobino, A. (2011). Prenatal exposure to ethanol: a specific effect on the H19 gene in sperm. Reprod. Toxicol. 31, 507–512.
Turan, N., Katari, S., Gerson, L. F., Chalian, R., Foster, M. W., Gaughan, J. P., Coutifaris, C., and Sapienza, C. (2010). Inter- and intra-individual variation in allele-specific DNA methylation and gene expression in children conceived using assisted reproductive technology. PLoS Genet. 6, e1001033. doi:10.1371/journal.pgen.1001033
Urban, M., Chersich, M. F., Fourie, L. A., Chetty, C., Olivier, L., and Viljoen, D. (2008). Fetal alcohol syndrome among grade 1 schoolchildren in Northern Cape Province: prevalence and risk factors. S. Afr. Med. J. 98, 877–882.
Viljoen, D. L., Gossage, J. P., Brooke, L., Adnams, C. M., Jones, K. L., Robinson, L. K., Hoyme, H. E., Snell, C., Khaole, N. C., Kodituwakku, P., Asante, K. O., Findlay, R., Quinton, B., Marais, A. S., Kalberg, W. O., and May, P. A. (2005). Fetal alcohol syndrome epidemiology in a South African community: a second study of a very high prevalence area. J. Stud. Alcohol 66, 593–604.
Wang, G., Wang, X., Wang, Y., Yang, J. Y., Li, L., Nephew, K. P., Edenberg, H. J., Zhou, F. C., and Liu, Y. (2008). Identification of transcription factor and microRNA binding sites in responsible to fetal alcohol syndrome. BMC Genomics 9(Suppl. 1), :S19. doi:10.1186/1471-2164-9-S1-S19
Wang, L. L., Zhang, Z., Li, Q., Yang, R., Pei, X., Xu, Y., Wang, J., Zhou, S. F., and Li, Y. (2009). Ethanol exposure induces differential microRNA and target gene expression and teratogenic effects which can be suppressed by folic acid supplementation. Hum. Reprod. 24, 562–579.
Waterland, R. A., Lin, J. R., Smith, C. A., and Jirtle, R. L. (2006). Post-weaning diet affects genomic imprinting at the insulin-like growth factor 2 (Igf2) locus. Hum. Mol. Genet. 15, 705–716.
Wossidlo, M., Nakamura, T., Lepikhov, K., Marques, C. J., Zakhartchenko, V., Boiani, M., Arand, J., Nakano, T., Reik, W., and Walter, J. (2011). 5-Hydroxymethylcytosine in the mammalian zygote is linked with epigenetic reprogramming. Nat. Commun. 2, 241.
Wright, R. M., McManaman, J. L., and Repine, J. E. (1999). Alcohol-induced breast cancer: a proposed mechanism. Free Radic. Biol. Med. 26, 348–354.
Wykes, S. M., and Krawetz, S. A. (2003). The structural organization of sperm chromatin. J. Biol. Chem. 278, 29471–29477.
Yoon, B., Herman, H., Hu, B., Park, Y. J., Lindroth, A., Bell, A., West, A. G., Chang, Y., Stablewski, A., Piel, J. C., Loukinov, D. I., Lobanenkov, V. V., and Soloway, P. D. (2005). Rasgrf1 imprinting is regulated by a CTCF-dependent methylation-sensitive enhancer blocker. Mol. Cell. Biol. 25, 11184–11190.
Keywords: epigenetics, imprinting control region, preconception paternal alcohol exposure, H19, Rasgrf1, CTCF binding site
Citation: Knezovich JG and Ramsay M (2012) The effect of preconception paternal alcohol exposure on epigenetic remodeling of the H19 and Rasgrf1 imprinting control regions in mouse offspring. Front. Gene. 3:10. doi: 10.3389/fgene.2012.00010
Received: 02 November 2011; Accepted: 18 January 2012;
Published online: 22 February 2012.
Edited by:
Wendy Robinson, University of British Columbia, CanadaCopyright: © 2012 Knezovich and Ramsay. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Michèle Ramsay, Molecular Genetics Laboratory, National Health Laboratory Service, Division of Human Genetics, Faculty of Health Sciences, University of the Witwatersrand, Corner of Hospital and de Korte Street, Braamfontein, 2000 Johannesburg, South Africa. e-mail:bWljaGVsZS5yYW1zYXlAbmhscy5hYy56YQ==