 Carolena Trocchia
Carolena Trocchia Maua Mosha3
Maua Mosha3 Neil Goldenberg
Neil Goldenberg Racha Khalaf
Racha Khalaf- 1School of Medicine, Stanford University, Stanford, CA, United States
- 2Department of Pediatric Gastroenterology, Hepatology and Nutrition, Stanford Healthcare, Stanford, CA, United States
- 3Institute for Clinical and Translational Research, Johns Hopkins All Children’s Hospital, Saint Petersburg, FL, United States
- 4Department of Pediatric Gastroenterology, Hepatology and Nutrition, University of South Florida, Tampa, FL, United States
Background: Gastrointestinal (GI) disease in pediatric patients with cystic fibrosis (CF) is a growing concern in the era of improved lung disease; however, the true prevalence of GI diagnoses, medical therapies, and frequency of diagnostic and therapeutic interventions in this population have yet to be explored on a multisystem scale. The aim of the present study was to describe, among pediatric patients with CF (PwCF) compared to a large cohort of healthy controls, the prevalence and types of 1) GI disorders; 2) GI medication use; and 3) GI procedural interventions.
Methods: This was a multicenter case-control analysis using the TriNetX electronic medical record (EMR) global anonymized data platform. Patients were included if they had an ICD9/10 diagnosis code and were between the age of zero to ≤ 21 years between January 1st, 2010, and January 1st, 2020. Those with a history of solid organ transplants were excluded.
Results: The cohort was comprised of 7,649 patients with a diagnosis of CF (cases) and 22,516,240 patients without CF (controls). The prevalence of any GI disorder was greater in pediatric PwCF compared to those without CF (73% versus <1%). The prevalence of diseases of the biliary tract and pancreas (43% vs. <1%; p<.0001), hepatic disease (8.8% vs. <1%; p<.0001), disease of the esophagus to duodenum (34% vs. <1%; p<.0001), and intestinal disease (43% vs. <1%; p<.0001) were each significantly greater in PwCF (p<0.0001). Furthermore, the frequencies of esophagogastroduodenoscopy (EGD), colonoscopy and endoscopic retrograde cholangiopancreatography (ERCP) were significantly higher among PwCF than controls (9.5% vs. <1%; 2.8%% vs. <1%; and 0.3% vs <.001% respectively; p<0.0001 for all comparisons).
Conclusion: The prevalence of GI diagnoses, use of GI medications, and frequency of GI procedures are all higher among pediatric patients PwCF. Prospective multicenter studies are warranted to substantiate these findings, further investigate risk factors for GI disorders, and to describe potential changes in GI disorders with novel CF disease-modifying therapies.
Introduction
Cystic fibrosis (CF) is a chronic disease with a pediatric onset most notably characterized by sinopulmonary and pancreatic complications but also involves gastrointestinal (GI) disorders (1–4). GI manifestations extend beyond the lumen and include hepatopancreatobiliary disease such as pancreatic insufficiency, pancreatitis, cholelithiasis, and CF-related liver disease (CF-LD) (3, 5, 6). Prior studies have shown that there is a high GI symptom burden—particularly bloating, early satiety, and abdominal pain—within this population (4, 7–9). Despite this knowledge, the prevalence of GI disorders in pediatric patients with, versus without, CF is unknown, and GI disorder prevalence has been poorly characterized in children with CF (1, 2).
The prevalence of GI diagnoses and medical therapies and the frequency of diagnostic and therapeutic interventions in this population have yet to be explored on a multisystem scale. Current knowledge is largely focused on GI symptoms rather than diagnoses (7, 8). Therefore, the aim of the present study was to describe, among pediatric PwCF, the prevalence and types of 1) GI disorders, 2) GI medication use, and 3) GI procedural interventions, as compared to a cohort of age-matched children without CF.
Methods
This study was approved by the Johns Hopkins Medicine Institutional Review Board.
The TriNetX database
We performed a nested case–control analysis using the global health data platform TriNetX, a deidentified web-based tool for research population cohort and feasibility queries that provides access to EMR-based clinical data from 67 healthcare organizations (HCOs) worldwide. The TriNetX database provides real-time access to longitudinal EMR information, derived from both inpatient and outpatient encounters. TriNetX performs recurring refreshers and spot checks of the data on a monthly basis, with varying data from HCOs.
Cases and controls
We included all patients between the ages of 0 and 21 years (inclusive) between 1 January 2010 and 1 January 2020. Those with a history of solid-organ transplants were excluded due to the greater complexity of these patients and a greater likelihood of biasing results. Out of this cohort, cases were defined as patients with an ICD-9 or ICD-10 diagnosis code for CF (Supplement). Controls were defined as those patients without a diagnosis of CF.
Statistical methods
Comparisons between the CF and non-CF cohorts were done using a chi-square test or Fisher’s exact test where appropriate. Bonferroni correction was used due to multiple comparisons, and a p-value of <0.0001 was considered significant. Demographic and clinical characteristics were summarized using descriptive statistics. Means with standard deviations or medians with interquartile ranges were used for continuous variables. Counts and percentages were used for categorical variables. GI manifestations in pediatric PwCF were categorized as intestinal, hepatobiliary, or pancreatic disease. Analyses were conducted using the TriNetX platform and SAS version 9.4.
Results
Demographics
Query of the TriNetX Research Network dataset identified a total of 81,126 PwCF, including 19,979 pediatric patients ≤21 years of age. Of these, 7,649 met our inclusion criteria for cases, compared to 22,516,240 patients without CF (controls). There was a statistically significant difference in cases versus controls for age, race, and ethnicity as seen in Table 1.
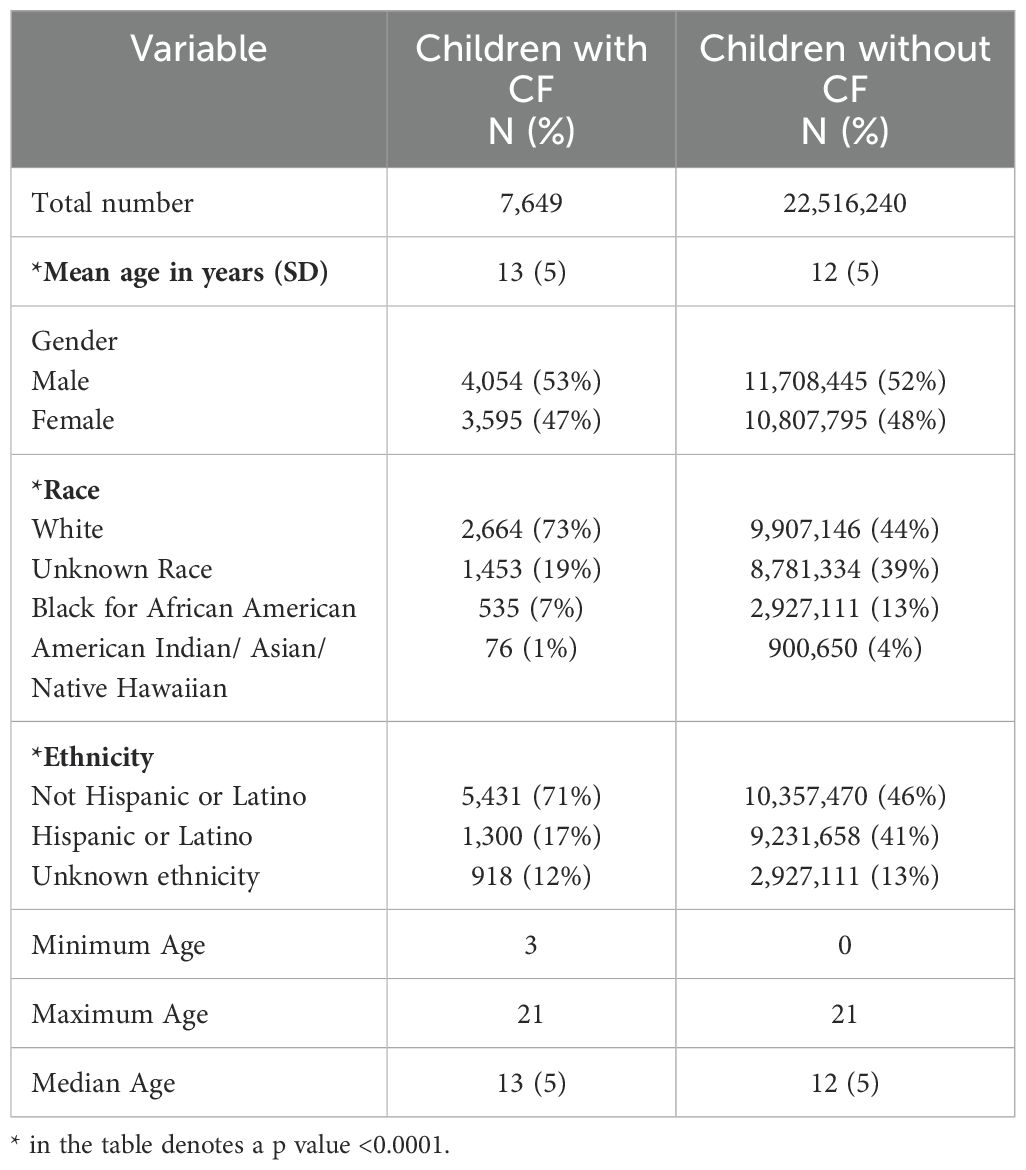
Table 1. Demographic data.
High burden of GI disorders in PwCF
The prevalence of GI disorders was significantly higher among children with CF (Table 2). GI disorders were diagnosed in 5,556 of pediatric PwCF (72%), compared to 120,804 controls (0.54%) (p < 0.0001). Similarly, the prevalence of each GI disorder subtype—pancreatic, hepatobiliary, and intestinal—was significantly higher among children with CF. Among pancreatic disorders in pediatric PwCF, exocrine pancreatic insufficiency was the most common (31%), followed by chronic pancreatitis (19%). Among hepatobiliary disorders, liver disease unspecified (4.7%) and other specified disease of the liver (3%) were the most common. For intestinal and luminal disorders, constipation was the most common at 38%, followed by gastroesophageal reflux disease (GERD) at 30%.
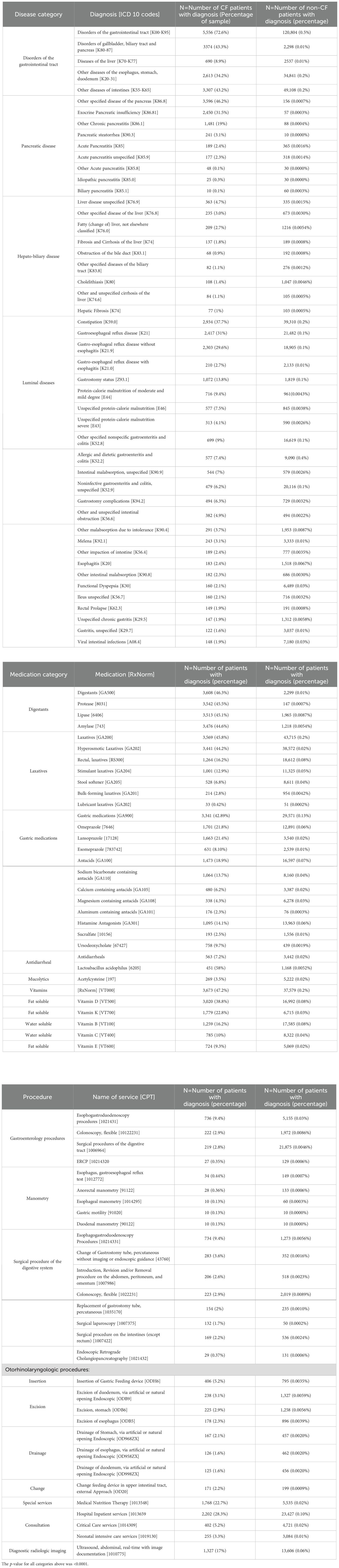
Table 2. Frequency of gastrointestinal manifestations, medications, and procedures within a sample of pediatric patients aged 0–21 diagnosed with and without cystic fibrosis from 2010 to 2020 obtained from the TriNetX database.
GI medication use including rectal therapies is higher in PwCF
Digestants were the most common GI medication class used in pediatric PwCF (Table 2). Compared to 0.01% of controls, 46% of PwCF were administered digestants. This is consistent with the higher prevalence of constipation seen in pediatric PwCF. Laxative medications and gastric medications (e.g., proton pump inhibitors, antacids) were the second and third most commonly administered GI medications among pediatric PwCF, consistent with the higher prevalence of constipation and GERD in PwCF.
GI procedures performed infrequently despite high disease prevalence in PwCF
The prevalence of GI procedures was less than 10% in both groups (Table 2). However, PwCF still had significantly higher rates of GI procedures, with the most common being esophagogastroduodenoscopy (EGD) (9.4%) and colonoscopy (2.8%) (p < 0.0001).
Discussion
Pulmonary and pancreatic diseases are typically the hallmarks of CF. In recent years, it has become increasingly clear that PwCF with GI disorders, including but not limited to pancreatic involvement, experience a reduced quality of life (3, 4, 7, 10–12). This has become of greater interest to the CF community as treatment outcomes for lung diseases have been transformed by the advent of highly effective modulator therapy (HEMT) (13). However, multicenter data are sparse on the prevalence and types of GI disorders in PwCF, especially beyond pancreatic disease. In this nested case–control analysis, we found a significantly higher prevalence of GI disorders—and accordingly, GI medication use—among pediatric PwCF when compared to those without CF, further exemplifying why research on CF GI disease is timely and can be transformative in this population. In addition, we determined that while the burden of GI disease is much greater in PwCF, and while GI procedural frequency is also significantly greater in this population, the total proportion of those undergoing procedures remains relatively low (<10%).
Prevalence of the disease and medication use
Our study highlights the broad and diverse diagnostic codes practitioners use to describe chronic pancreatic and liver diseases, none of which are specific for CF. Our findings of a high prevalence of chronic pancreatic disease (at 53.6%, including exocrine pancreatic insufficiency, chronic pancreatitis, and pancreatic steatorrhea, while 46.2% of patients had received a diagnosis of other specified disease of the pancreas) in pediatric PwCF are concordant with prior knowledge regarding pancreatic disease, indicating a prevalence of up to 85% and its underlying mechanisms in CF (1, 2, 14–18). This database further provides greater specificity to the different liver manifestations in CF while prior studies have previously assumed a broad prevalence of <5%–68% (19), and it also highlights the evolving landscape of CFLD as most recent clinical guidelines recommend stratifying disease as advanced CFLD (aCFLD) versus CF hepatobiliary involvement (CFHBI) (20). The variety of codes used especially in liver disease is reflective of the heterogeneity of this disease and the likely need for a CF-specific ICD-10 code which will be instrumental in future clinical trials (15, 16).
Our findings further demonstrate a high prevalence of constipation among pediatric PwCF and as such more frequent use of digestants and laxative medications. While most laxatives were oral, a surprisingly high proportion of patients needed rectal therapies to help alleviate constipation due to likely a higher severity of disease and due to the risk of distal intestinal obstruction syndrome (DIOS) in CF. Whether or not rectal therapies were more frequently used in the setting of DIOS or routinely for constipation management could not be appreciated through this study and could be a future area of investigation.
Despite GERD being the second most common intestinal diagnosis, correlating with the high use of gastric medications, there was a lower prevalence of esophageal manometry and impedance testing. There is a lack of diagnostic guidelines within the pediatric CF population for GERD, but typically, pH impedance, esophageal manometry, or endoscopy is not indicated unless the patient is resistant to therapy or has alarming symptoms (21). The use of HEMT has improved lung disease, its impact on GERD has not yet been examine. Defining the true prevalence of GERD in this population along with the prevalence of a healthy control group prior to the broad adoption of HEMT use is critical as it allows us to determine in the future if HEMT therapy allows PwCF to have a similar prevalence of GI manifestations with adequate and early therapy (13). Therefore, considering the role HEMT has on improving lung disease, we may better delineate the prevalence of a GERD diagnosis for either 1) true reflux or 2) to aid in the management of malnutrition in PwCF by administering acid blockade for presumed improved enzyme absorption.
The overall prevalence of GI procedures, although thought to be low, was comparably significantly greater in the PwCF compared to the non-CF control sample even with the Bonferroni adjustment due to such a large control sample. Despite the greater risk of sedation in PwCF, procedures were more likely to be performed on these patients again likely due to the significantly higher burden of disease. Endoscopies being more common in patients with CF over controls but still proportionally less common than controls may be secondary to multiple factors including: 1) historically, gastroenterologists were scarcely involved in the care of patients with CF as this was primarily managed by pulmonologists and pediatricians; 2) some of the GI CF manifestations especially reflux and dysmotility are widely accepted in the CF community as part of the disease; and 3) secondary to underlying lung disease and subsequently greater risk of sedation. Thus, it is likely that despite a high burden of disease, the use of procedures is less commonly felt to be needed for diagnosis.
Prior studies have shown a high GI symptom burden for these patients; thus, defining the prevalence of GI-related manifestations is important to develop measures that improve quality of life and define changes in disease state after using highly effective modulators.
Dataset limitations
We elected to use the TriNetX database despite its limitations for the following reasons. In other known registries such as the CF Foundation Registry, patients must opt-in to be recorded within the database, whereas TriNetX includes all recorded patients in a healthcare system. Furthermore, the TriNetX database collects data not yet available in the CF Foundation Registry and allows for comparison against an age-matched cohort. For our description, we relied on the search for GI diagnoses by diagnostic codes. Thus, patients may be diagnosed inaccurately, non-specifically, or by physicians of differing specialties, introducing further variability. To prevent this, diagnoses were reviewed by a pediatric CF gastroenterologist during analysis. Additionally, the TriNetX system includes an ICD-9 to ICD-10 mapping based on the General Equivalence Mappings (GEMs). GEMs use custom algorithms and curation to transform data from ICD-9-CM to ICD-10-CM. This allows diagnostic terms from historic data and HCOs who provide ICD-9-CM terms to be treated according to query. The time period—1 January 2010 to 1 January 2020—was chosen to include a full decade of data while balancing the exclusion of potential influence from COVID-19 and HEMT use. This may contribute to the findings of exocrine pancreatic insufficiency having a prevalence of 31% in the CF population when it is known to exist in greater than 85% of pediatric CF (22). Furthermore, while we anticipated detecting diagnoses such as small bacterial overgrowth in the dataset, this was not reported. There is also no dedicated ICD-10 code for cystic fibrosis-associated liver disease, which is a heterogeneous disease. Thus, the involvement of the liver in CF may be unknown with subclinical features until the patient presents with diffuse non-specific liver damage. Comparing patients with versus without CF was chosen to reduce complexity; however, other chronic diseases may contribute to the results. Lastly, this is a descriptive study and, thus, does not demonstrate causation of GI manifestations in CF.
Notwithstanding these limitations, our findings demonstrate that the prevalence of GI disorders, medication use, and procedure performance is greater in pediatric PwCF compared to those without CF.
Conclusion
This study uniquely describes the strikingly high prevalence of GI manifestations and associated GI medication use among pediatric PwCF internationally as compared to a large healthy control. Interestingly, we saw a comparatively lower prevalence of GI procedures in this population despite the high burden of disease, though still substantially greater when compared to controls. These results highlight the broad array of GI manifestations associated with CF, suggesting a need for further focus on these disorders in the new era of HEMT. This study better defines the complexities of GI disease including the prevalence of GI disease, medication use, and interventions prior to broad and disseminated use of HEMT, which will be critical in understanding the effects of highly effective modulator therapy on the full scope of CF manifestations.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by the Johns Hopskins Medicine Institutional Review Board. The studies were conducted in accordance with the local legislation and institutional requirements.
Author contributions
CT: Conceptualization, Funding acquisition, Investigation, Software, Writing – original draft, Writing – review & editing. MM: Formal analysis, Methodology, Resources, Writing – review & editing. BH: Writing – review & editing. NG: Conceptualization, Funding acquisition, Investigation, Methodology, Resources, Supervision, Writing – review & editing. RK: Conceptualization, Formal analysis, Funding acquisition, Investigation, Methodology, Resources, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The Cystic Fibrosis Foundation funding to RK (#005206Q123, #005583Q123) and the Johns Hopkins All Children’s Hospital Foundation grant to CT are acknowledged.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Dickinson KM, Collaco JM. Cystic fibrosis. Pediatr Rev. (2021) 42:55–67. doi: 10.1542/pir.2019-0212
2. Shteinberg M, Haq IJ, Polineni D, Davies JC. Cystic fibrosis. Lancet. (2021) 397:2195–211. doi: 10.1016/S0140-6736(20)32542-3
3. Sathe MN, Freeman AJ. Gastrointestinal, pancreatic, and hepatobiliary manifestations of cystic fibrosis. Pediatr Clin North Am. (2016) 63:679–98. doi: 10.1016/j.pcl.2016.04.008
4. Moshiree B, Freeman AJ, Vu PT, Khan U, Ufret-Vincenty C, Heltshe S, et al. GALAXY Study Group. Multicenter prospective study showing a high gastrointestinal symptom burden in cystic fibrosis. J Cyst Fibros;. (2023) 22:266–74. doi: 10.1016/j.jcf.2022.10.006
5. Kamal N, Surana P, Koh C. Liver disease in patients with cystic fibrosis. Curr Opin Gastroenterol. (2018) 34:146–51. doi: 10.1097/MOG.0000000000000432.PMID:29438119
6. Sasame A, Stokes D, Bourke B, Connolly L, Fitzpatrick E, Rowland M. The impact of liver disease on mortality in cystic fibrosis–a systematic review. J Cystic Fibrosis. (2022) 21:202–11. doi: 10.1016/j.jcf.2021.07.014
7. Freeman AJ, Sathe M, Aliaj E, Borowitz D, Fogarty B, Goss CH, et al. Designing the GALAXY study: partnering with the cystic fibrosis community to optimize assessment of gastrointestinal symptoms. J Cyst Fibros. (2021) 20:598–604. doi: 10.1016/j.jcf.2020.12.021
8. Sathe M, Moshiree B, Vu PT, Khan U, Heltshe SL, Romasco M, et al. Utilization of electronic patient-reported outcome measures in cystic fibrosis research: Application to the GALAXY study. J Cyst Fibros. (2021) 20:605–11. doi: 10.1016/j.jcf.2021.07.002
9. Nichols DP, Paynter AC, Heltshe SL, Donaldson SH, Frederick CA, Freedman SD, et al. PROMISE Study group. Clinical Effectiveness of Elexacaftor/Tezacaftor/Ivacaftor in People with Cystic Fibrosis: A Clinical Trial. Am J Respir Crit Care Med. (2022) 205:529–39. doi: 10.1164/rccm.202108-1986OC
10. Birimberg-Schwartz L, Wilschanski M. Cystic fibrosis related gastrointestinal manifestations - moving forward. J Cyst Fibros. (2021) 20:562–3. doi: 10.1016/j.jcf.2021.07.011
11. Zuloaga N, Vivallos N, Faúndez R, González M, Navarro E, Chávez E, et al. Manifestaciones gastrointestinales en fibrosis quística en una población pediátrica [Gastrointestinal manifestations of cystic fibrosis in children. Andes Pediatr. (2021) 92:526–33. doi: 10.32641/andespediatr.v92i4.2693
12. Thomas L, Kumar M, Lionel BAP, Varkki S, Rebekah G. Pancreatic, hepatobiliary, and gastrointestinal manifestations of children with cystic fibrosis: A 10-year experience from a tertiary care center in southern India. Indian J Gastroenterol. (2022) 41:266–72. doi: 10.1007/s12664-021-01225-0
13. Middleton PG, Mall MA, Dřevínek P, Lands LC, McKone EF, Polineni D, et al. Elexacaftor–tezacaftor–ivacaftor for cystic fibrosis with a single phe508del allele. NEJM. (2019) 381:1809–19. doi: 10.1056/NEJMoa1908639
14. Ley D, Turck D. Digestive outcomes in cystic fibrosis. Best Pract Res Clin Gastroenterol. (2022) 56:56–57, 101788. doi: 10.1016/j.bpg.2022.101788
15. Zolin A, Bossi A, Cirilli N, Kashirskaya N, Padoan R. Cystic fibrosis mortality in childhood. Data from european cystic fibrosis society patient registry. Int J Environ Res Public Health. (2018) 15(9):2020. doi: 10.3390/ijerph15092020
16. Assis DN, Freedman SD. Gastrointestinal disorders in cystic fibrosis. Clin Chest Med. (2016) 37:109–18. doi: 10.1016/j.ccm.2015.11.004
17. Cystic fibrosis foundation patient registry 2021 annual data report. Bethesda, Maryland: Cystic Fibrosis Foundation (2022). Available at: https://www.cff.org/media/2021-Patient-Registry-Annual-Data-Report.
18. O’Sullivan BP, Freedman SD. Cystic fibrosis. Lancet. (2009) 373:1891–904. doi: 10.1016/S0140-6736(09)60327-5
19. Valamparampil JJ. Gupte Gl. Cystic fibrosis associated liver disease in children. World J Hepatol. (2021) 13:1727–42. doi: 10.4254/wjh.v13.i11.1727
20. Sellers ZM, Assis DN, Paranjape SM, Sathe M, Bodewes F, Bowen M, et al. Cystic fibrosis screening, evaluation, and management of hepatobiliary disease consensus recommendations. Hepatology. (2024) 79:1220–38. doi: 10.1097/HEP.0000000000000646
21. Bongiovanni A, Manti S, Parisi GF, Papale M, Mulè E, Rotolo N, et al. Focus on gastroesophageal reflux disease in patients with cystic fibrosis. World J Gastroenterol. (2020) 26:6322–34. doi: 10.3748/wjg.v26.i41.6322
Keywords: cystic fibrosis, exocrine pancreatic insufficiency, elexacaftor/tezacaftor/ivacaftor (ETI), modulator therapy, gastrointestinal disease
Citation: Trocchia C, Mosha M, Hamner B, Goldenberg N and Khalaf R (2025) Prevalence and characteristics of gastrointestinal disorders, medication use, and diagnostic interventions in pediatric patients with cystic fibrosis: a nested case–control analysis from the TriNetX EMR-derived global research network real-world dataset. Front. Gastroenterol. 3:1489876. doi: 10.3389/fgstr.2024.1489876
Received: 04 September 2024; Accepted: 05 December 2024;
Published: 24 January 2025.
Edited by:
Vito Terlizzi, University of Florence, ItalyReviewed by:
Ariel Benson, Shaare Zedek Medical Center, IsraelAnuj Sharma, PeaceHealth St. John Medical Center Foundation, United States
Copyright © 2025 Trocchia, Mosha, Hamner, Goldenberg and Khalaf. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Racha Khalaf, a2hhbGFmckB1c2YuZWR1