 Michael Doulberis1,2,3Apostolis Papaefthymiou4,5,6Stergios A. Polyzos5Marina Boziki7Evangelos Kazakos3,8Maria Tzitiridou-Chatzopoulou3,8Elisabeth Vardaka3,9Carola Hammrich10Hasan Kulaksiz1Daniele Riva11Christos Kiosses1Ioannis Linas12Maria Touloumtzi3Aggeliki Stogianni3,13Jannis Kountouras3*
Michael Doulberis1,2,3Apostolis Papaefthymiou4,5,6Stergios A. Polyzos5Marina Boziki7Evangelos Kazakos3,8Maria Tzitiridou-Chatzopoulou3,8Elisabeth Vardaka3,9Carola Hammrich10Hasan Kulaksiz1Daniele Riva11Christos Kiosses1Ioannis Linas12Maria Touloumtzi3Aggeliki Stogianni3,13Jannis Kountouras3*- 1Private Gastroenterological Practice, Gastroklinik, Gastroklinik, Horgen, Switzerland
- 2Division of Gastroenterology and Hepatology, Medical University Department, Kantonsspital Aarau, Aarau, Switzerland
- 3Second Medical Clinic, School of Medicine, Aristotle University of Thessaloniki, Ippokration Hospital, Thessaloniki, Greece
- 4Department of Gastroenterology, University Hospital of Larisa, Larissa, Greece
- 5First Laboratory of Pharmacology, Aristotle University of Thessaloniki, Thessaloniki, Greece
- 6Pancreaticobiliary Medicine Unit, University College London Hospitals (UCLH), London, United Kingdom
- 7Second Neurological Department, Aristotle University of Thessaloniki, AHEPA University General Hospital of Thessaloniki, Thessaloniki, Greece
- 8School of Healthcare Sciences, Midwifery Department, University of West Macedonia, Kozani, Greece
- 9Department of Nutritional Sciences and Dietetics, School of Health Sciences, International Hellenic University, Macedonia, Greece
- 10School of Dental Medicine Georg-August University, Göttingen, Germany
- 11Private Gastroenterological Practice, Gastrocentro Plus, Lugano, Switzerland
- 12Private Gastroenterology Practice, Praxis Gastroenterologische Gruppenpraxis (GGP), Bern, Switzerland
- 13Department of Radiology, Ippokration Hospital, Thessaloniki, Greece
Helicobacter pylori, a widely renowned bacterium, has recently gained attention owing to its potential impact on extragastric health. The emergence of research linking H. pylori infection with metabolic syndrome (MetS)-related cardiovascular diseases (CVDs) has raised intriguing questions about the pathogenic linkage and its translational implications for clinicians. MetS encompasses a collection of metabolic abnormalities that considerably elevate the risk of CVDs and cerebrovascular diseases. Emerging evidence supports a potential pathogenetic role of H. pylori for MetS-related disorders through mechanisms implicating chronic smoldering inflammation, insulin resistance (IR), and modulation of immune responses. One intriguing aspect of this possible connection is the role of mast cells (MCs), a subset of immune cells representing innate immune system effector cells. They play a fundamental role in innate immune responses and the modulation of adaptive immunity. Activated MCs are commonly found in patients with MetS-related CVD. Recent studies have also suggested that H. pylori infection may activate MCs, triggering the release of pro-inflammatory mediators that contribute to IR and atherosclerosis. Understanding these intricate interactions at the cellular level provides new insights into the development of therapeutic strategies targeting both H. pylori infection and MetS-related MCs activation. This review investigates the current state of research regarding the potential impact of H. pylori infection and MetS-related MCs activation on the pathophysiology of CVD, thereby opening up new avenues for related research and paving the way for innovative approaches to prevention and treatment in clinical practice
1 Introduction
Helicobacter pylori infection, representing a high worldwide burden (1), has recently garnered growing attention for its potential role in various systemic disorders including cardiovascular diseases (CVDs) (2–7). Although H. pylori has been primarily associated with gastric disorders, particularly peptic ulcers, gastric adenocarcinoma, and MALT (mucosa-associated lymphoid tissue) lymphoma (8, 9), mounting evidence suggests that this bacterium can exert a systemic influence, affecting diverse pathophysiological processes beyond the gastrointestinal tract (2, 6, 7). The intricate relationship of H. pylori with the immune system and its potential for inducing chronic low-grade (“smoldering”) inflammation in remote organs have raised intriguing questions about its role in extragastric diseases.
Metabolic syndrome (MetS) constitutes a cluster of diverse metabolic risk factors that lead to a clinical syndrome. Despite the plethora of different definitions and cutoffs, MetS prerequisites the presence of the following conditions: abdominal obesity, insulin resistance (IR), type 2 diabetes mellitus (T2DM), dyslipidemia, nonalcoholic fatty liver disease (NAFLD), arterial hypertension (AH), and CVD (10).
In this respect, an emerging body of evidence supports a connection between MetS and active H. pylori infection: both pathologies appear to promote the pathogenesis of each other (11). H. pylori infection is positively associated with MetS (12), and its eradication affects MetS parameters (13). Moreover, H. pylori serves as an independent risk factor for CVD, and its eradication is safe from a cardiovascular perspective (14). The results of a recent meta-analysis also indicated a positive correlation between H. pylori infection and MetS and IR (15). A further piece of scientific evidence supports the suggestion that H. pylori infection is an independent risk factor for MetS-related NAFLD and is correlated with an increased degree of steatosis (16). Moreover, active H. pylori infection has been found to be independently positively associated with the severity of MetS-related non-alcoholic steatohepatitis (NASH) and other MetS-related parameters including IR, dyslipidemia, and AH (17). This association also extends to the context of pregnancy (18). In this regard, active H. pylori infection is equally associated with MetS-related systemic pathologies, especially cardio-cerebrovascular diseases (C-CVDs), the end point of MetS (19–21).
Mast cells (MCs), a subgroup of immune cells first detected to mediate allergic and anaphylactic reactions, represent innate immune system effector cells playing an important role in innate immune reactions and in modulating adaptive immunity (22).
Apart from local effects, some more recent data regarding the activation of MCs in H. pylori infection and/or MetS also concentrated on systemic effects. For instance, several infections, mostly H. pylori infection, have been associated with chronic urticaria (23). Many autoimmune disorders such as systemic lupus erythematosus, polymyositis, dermatomyositis, and rheumatoid arthritis have been connected with chronic urticaria (23). In this regard, H. pylori infection and/or MetS are linked to such disorders as systemic lupus erythematosus and rheumatoid arthritis (24–27). MCs also contribute to the pathophysiology of these autoimmune disorders (26, 28). Moreover, recent data have indicated the involvement of MCs in the pathophysiology of additional H. pylori and MetS-associated disorders, such as NAFLD (16, 17, 22). NAFLD is strongly associated with a higher risk of systemic disorders including C-CVD and neuroautoimmune pathologies, the end points of MetS (22, 29–31). In the same way, for instance, MCs induce dyslipidemia, atherosclerosis, and/or AH (32–34). All of these disorders, also connected with H. pylori infection and MetS, are risk factors for C-CVD and neurogenerative pathologies (21, 35–37). As another final example, H. pylori-induced vacuolating cytotoxin A (VacA) exhibits chemotactic activities toward bone marrow-derived MCs (BMD-MCs) and induces them to produce pro-inflammatory cytokines involved in topical pathologies. BMD-MCs, beyond the gastrointestinal tract, also reside adjacent to blood and lymphatic channels, predominantly under epithelial surfaces including the blood–brain barrier. MCs can be activated by the corticotropin-releasing hormone, secreted under stress, to induce mediators including chymase or tryptase, interleukin 8 (IL-8), and vascular endothelial growth factor, which disrupt the blood–brain barrier, leading to neurodegenerative pathologies (38). MCs activation, by mediating blood–brain barrier damage, contributes to cognitive impairment (39). Likewise, H. pylori VacA promotes the intracellular survival of H. pylori, and activated monocytes (possibly infected with H. pylori owing to defective autophagy) could access the brain (Trojan horse theory) through blood–brain barrier damage, thereby leading to neurodegeneration (6).
Specifically, concerning MCs-related pathogen internalization, related data indicated that Staphylococcus aureus (S. Aureus) is able to internalize within BMD-MCs (40) in a subterfuge manner, where it evades detection and elimination. Live S. aureus can be found viable up to 5 days later within MCs (41). The interactions between MCs and S. aureus, including the internalization procedure, demonstrated a noticeable role of staphylococcal enterotoxin B in stimulating the uptake of the bacteria into MCs (42). In contrast to other bacterial pathogens, S. aureus is able to internalize into MCs and could induce a unique response. There is a crosstalk between the internalized S. aureus and human MCs, and a fraction of the internalized S. aureus undergoes a profound transcriptional reprogramming within human MCs to adapt to the nutrients and stress encountered in the intracellular environment and remain viable (43). Of note is that MCs can act as professional phagocytes in the pathophysiology of biofilm-related disorders (44).
Since there is currently no scientifically established evidence in the literature regarding the possible capability of H. pylori to internalize MCs and evade their elimination, further investigation is required to elucidate this field.
Recent research has unveiled their implication in non-allergic and/or non-anaphylactic inflammatory pathologies, including MetS-related disorders such as NAFLD (45), the hepatic component of MetS having been recently renamed as metabolic (dysfunction)-associated fatty liver disease (MAFLD). MAFLD exhibits a 10-year high risk of atherosclerotic CVD, and it is strongly associated with a higher risk of systemic conditions including C-CVD and neuroautoimmune pathologies, the end points of MetS-linked MAFLD. Innate immune cell activation including MCs could contribute to the pathophysiology of MAFLD and its complications; therefore, restricting the activation of MCs may represent a possible novel therapeutic option for MAFLD and its complications (22, 46). Likewise, recent evidence has indicated that trained innate immunity or trained immunity can also lead to chronic inflammatory pathologies and, thus, to miscellaneous MetS-related disorders, such as atherosclerotic CVD (46). The training of innate immune cells, such as monocytes, macrophages, natural killer cells, and MCs, is possibly involved in the pathophysiology of MAFLD and its complications (46–48), and MCs may be involved in systemic MetS-connected inflammatory pathologies through several mechanisms (38, 49–51).
Apart from the implication of MCs in MetS-related disorders, their direct activation by H. pylori infection may also be implicated in H. pylori-related pathologies (52, 53). In this regard, at the topical level, the interactions between MCs and H. pylori appear to contribute to the pathophysiology of gastritis (53), and interactions among H. pylori, gastric epithelium, and MCs through inflammatory cytokine induction contribute to H. pylori-related gastric pathologies. While H. pylori can be eradicated by oral antibiotic administration in most patients, it is worth noting that chronic gastritis may persist even after successful H. pylori eradication regiments, particularly in the presence of chronic gastritis-related dysplasia (54, 55). Thus, it is beneficial to clinical prognosis when treatments can manage the underlying inflammatory process through several possible therapeutic targets, including MCs (56). At the systemic level, H. pylori-related stimulation of the innate immune cells including MCs may be involved in the aforementioned systemic MetS-related inflammatory pathologies through several mechanisms (38). Similarly, H. pylori infection may also be connected with trained immunity and, by provoking gut dysbiosis (57), may play a role in MALFD and other MetS-related CVD through several mechanisms (46). Repeated inflammation owing to H. pylori infection appears to be sufficient to induce trained immunity (58).
Furthermore, MCs activation may induce an augmented gut permeability, allowing potentially harmful gut symbiotic molecules to spread into circulation and contribute to systemic pathologies (59, 60).
Thus, H. pylori infection and MetS-related MCs involvement in the pathophysiology of topical and systemic pathologies may offer benefits through the application of novel related strategies toward increasing the eradication of H. pylori.
In view of the aforementioned considerations, we aimed to investigate the hypothesis of the potential pathogenetic link between H. pylori infection and the activation of MCs in the context of MetS with special focus on CVDs. Despite this connection seeming intricate and multifaceted, elucidating it could yield significant clinical insights. If substantiated, this hypothesis could provide clinicians with a novel perspective on the etiology and management of CVDs associated with H. pylori-related MetS-linked MCs activation. We will explore the current knowledge of each component of this hypothesis, aiming at shedding light on the intricate web of interactions that may culminate in CVDs. We will also discuss the potential translational implications for medical practitioners and the avenues for future research.
2 H. pylori and MetS-related CVD
Both H. pylori infection and MetS are highly prevalent worldwide. The mean global prevalence of H. pylori infection is 58%, partly owing to immigrants coming from nations with a high prevalence of H. pylori infection (61). The pooled prevalence estimate for MetS is 24%, and the prevalence rate of its components including being overweight/obese is from 35.6% to 44.1% (62, 63). There is growing evidence (Table 1) for a potential association between H. pylori infection and IR syndrome or MetS and its related morbidities, including abdominal obesity, T2DM, dyslipidemia, AH, MAFLD, and CVD (3). In particular, H. pylori infection contributes to IR, the central feature of MetS and a crucial component in the pathogenesis of atherosclerosis and AH-induced target tissue injuries.
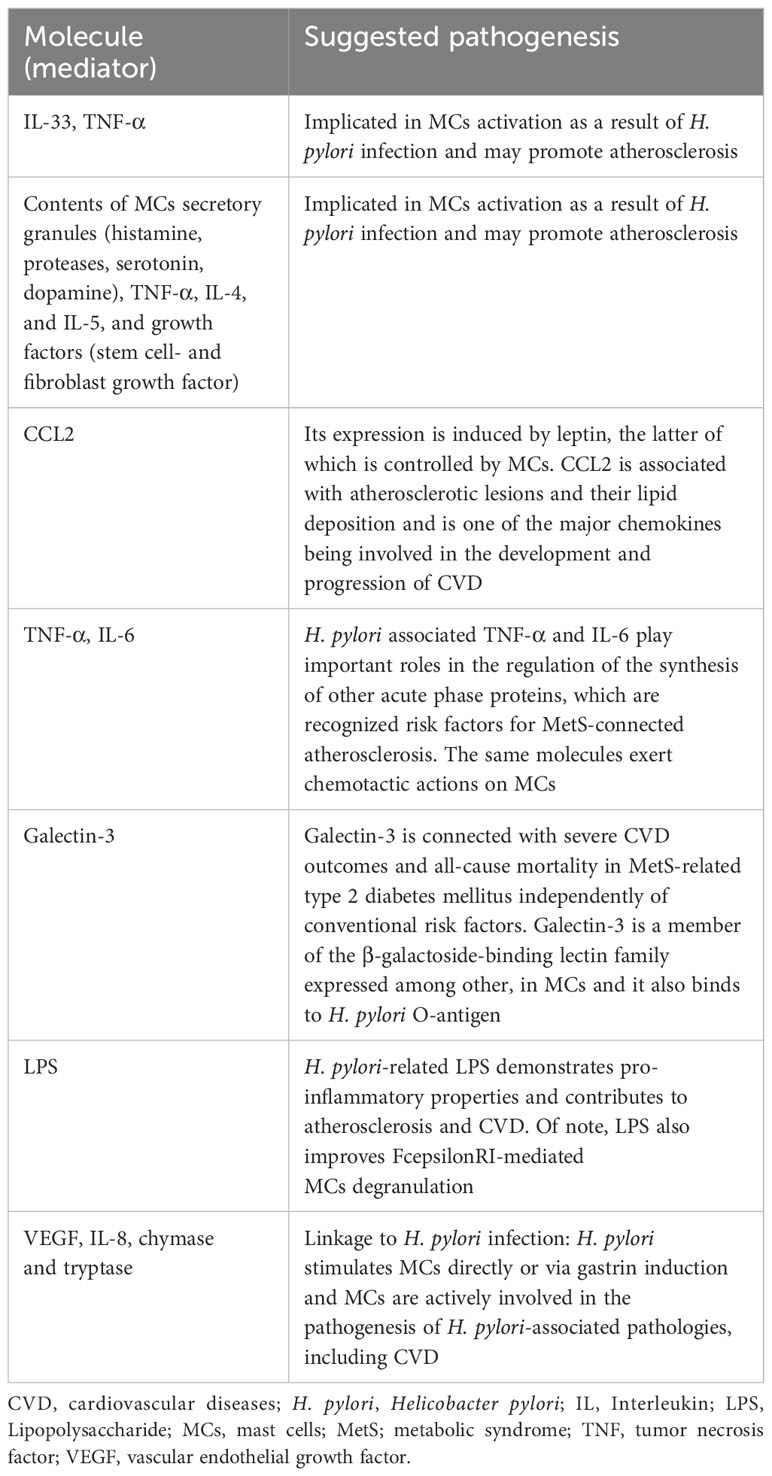
Table 1 Summary of the most important molecules (mediators) implicating in the possible interaction among Helicobacter pylori infection, mast cells, and cardiovascular disease.
To colonize the human gastric chamber, H. pylori first needs to overcome two essential physical barriers: stomach acid and the gastric mucous layer. The low-pH environment in the human stomach plays a fundamental role in preventing bacterial growth and colonization. H. pylori urease is thought to buffer the acid in the gastric lumen through the hydrolysis of urea to bicarbonate and ammonia, thereby inducing a neutral microenvironment around the bacterium (64, 65). The activity of urease is coupled with an acid-gated urea channel, which enables H. pylori to maintain a neutral cytoplasmic pH when exposed to acidic conditions (66). H. pylori bacteria that have survived the acid in the stomach are then able to negotiate the thick gastric mucous layer, courtesy of their spiral shape and multiple polar flagella (67).
After traversing the mucous layer and reaching the gastric epithelium, H. pylori avoids being flushed away during the continual replenishment of the gastric mucosa by using a range of adhesins to tightly attach to the cells of the gastric epithelium (68). This allows H. pylori to escape the acidic conditions in the lumen of the stomach and gain access to gastric epithelial cells, which could act as a source of essential nutrients following VacA-dependent cell permeabilization (69), thereby leading to its long-term persistency in the gastric mucosa and, thus, triggering chronic inflammation.
In addition, is well known that a direct interaction between H. pylori and MCs takes place in the gastric mucosa niche. Specifically, the bacterial ecosystem of the gastric niche is significantly affected by H. pylori infection (70), which may contribute to the broad spectrum of clinical manifestations in chronically infected patients. The natural niche for H. pylori is the human stomach, and H. pylori has developed a unique set of virulence factors that allow its survival in this unique ecological niche, the human stomach (71).
The interaction between H. pylori and MCs is associated with a significant increase in the number of activated MCs into the extracellular matrix of the gastric mucosa niche (53). The inflammatory process during H. pylori infection within the gastric mucosa niche involves the interactions between H. pylori, the gastric epithelium, and MCs via pro-inflammatory cytokines including IL-33 and tumor necrosis factor alpha (TNF-α) (56). Such cytokines, acting at a distance, are involved in the pathophysiology of atherosclerotic CVD (72–75). Similarly, in H. pylori-associated chronic gastritis, the density, and particularly the number of gastric niche MCs-related epithelial cells, is correlated with a high number of apoptotic epithelial cells (76).
Beyond the gastric mucosa niche, the oral cavity is another niche for H. pylori and could be the trigger of H. pylori infection, re-infection, and transmission into the stomach (77). Since MCs activation is also linked to various inflammatory conditions of the oral cavity (78), further research is warranted to elucidate the possible direct interaction between H. pylori and MCs taking place in the oral cavity niche, with a potential relation to CVDs.
In the context of CVDs, persistent H. pylori infection stimulates both local and systemic immune responses involved in the pathophysiology of atherosclerotic CVDs (79–81).
H. pylori infection is linked to arterial stiffness, an early indicator of atherosclerosis associated with MetS-related AH and an independent predictor of CVD complications and all-cause fatality (21). Interestingly, the occurrence of H. pylori DNA in atherosclerotic plaques and the H. pylori infection-related carotid plaque onset in adults without previous CVD suggest that this bacterium could play a role in the pathogenesis of atherosclerosis and that its eradication may decrease the burden of MetS-related atherosclerosis (82–84). H. pylori infection has been associated with fatal MetS-related ischemic heart disease and stroke outcomes than with nonfatal events ascertained by general practitioners. H. pylori infection is independently related to the future risk of CVD death; the assessed 10-year CVD risk increases as the grade of H. pylori-related gastritis increases. H. pylori-related galectin-3, which is positively associated with MetS, beyond MAFLD, is also connected to a higher risk of all-cause mortality and particularly with heart failure and C-CVD mortality (85). H. pylori-associated galectin-3 upregulation and MetS appear to be implicated in the persistent and progressive dysfunction of several organs including the liver, CVD, kidney, and the brain (86).
Focusing mainly on CVD, for instance, galectin-3 is associated with severe CVD outcomes and all-cause mortality in MetS-related T2DM independently of conventional risk factors (87). Acute myocardial infarction, a potentially fatal CVD complication, is closely linked to MetS (3), and H. pylori is a risk factor for potential lethal acute coronary syndrome including acute myocardial infarction. H. pylori infection also appears to provoke platelet activation/aggregation and an increment in various proatherogenic factors, such as MetS-related homocysteine, thereby contributing to H. pylori-associated CVD risk (88–90). Moreover, interactions between H. pylori infection and MetS-related hyperhomocysteinemia appear to contribute to atherosclerosis linked to systemic pathologies, including C-CVD (91).
Atrial fibrillation (AF) is an important contributor to CVD morbidity and mortality mainly due to AF-induced strokes. In this respect, H. pylori-related MetS could contribute to the development and/or progression of CVD, including AF, via several potential mechanisms including the pro-inflammatory cytokines TNF-α and IL-6 involved in atherosclerosis and AF. Similarly, IR is associated with AF and a twofold increase in CVD outcomes, including stroke (92). Active H. pylori infection-related MetS might also contribute to the pathophysiology of MAFLD and its related AF complications. Therefore, eradicating this bacterium may offer protection against the aforementioned pathologies; thus, further investigation is also needed (92, 93).
It is important to note that inflammatory pathways, including the nuclear factor kappa B (NF-κB) signaling cascade, are activated in response to the presence of H. pylori (37, 94). This inflammation can disrupt insulin signaling and contribute to the aforementioned IR (37, 95). Furthermore, H. pylori-related lipopolysaccharide (LPS) has demonstrated pro-inflammatory properties and contributes to atherosclerosis and CVD. The gut bacteria, including H. pylori, can initiate the inflammatory state of obesity and IR via the activity of LPS, and there is strong association between high LPS activity and the prevalence of MetS. For instance, the plasma LPS levels were reported to be 76% higher in patients with T2DM than in the control group (96). In this respect, H. pylori infection has been associated with alterations in the gut microbiota composition (5, 97, 98), and H. pylori-related MetS microbiota dysbiosis could play a role in the pathogenesis of MAFLD (17) and its systemic complications such as stroke. In contrast, fecal microbiota transplantation may prevent MAFLD and regulate the brain injury-induced dysbiosis, thereby improving stroke outcomes (99, 100).
Moreover, MetS is linked to dyslipidemia, and H. pylori infection can induce the host’s abnormal lipid metabolism, including lipoprotein (a), low-density lipoprotein cholesterol (LDL-C), high-density lipoprotein cholesterol (HDL-C), and total cholesterol (TC). H. pylori-related lower HDL-C can induce dyslipidemia, whereas H. pylori eradication significantly reduces TC, LDL-C, and fibrinogen, an independent risk factor for MetS-related CVD, but increases HDL-C. Furthermore, beyond dyslipidemia, H. pylori eradication improves the other MetS-related components, such as AH, IR, body mass index, and total oxidant status. Thus, H. pylori infection might serve as risk factor for MetS-related components, including dyslipidemia; thereby, its eradication possibly prevents the prevalence of MetS-related CVD (20).
Finally, H. pylori infection has been linked to alterations in appetite-regulating hormones. Ghrelin, an orexigenic hormone that stimulates hunger, is raised in individuals with H. pylori infection (101) and could contribute to increased food intake and obesity, both of which are central components of MetS. Although the linkage between H. pylori and MetS is evident, it is necessary to emphasize the role of confounding factors. Lifestyle factors, including diet and physical activity, play a pivotal role in the onset of MetS. These factors can also affect the rates of H. pylori infection and the course of disease (102). Therefore, unraveling the direct effects of H. pylori from lifestyle-related factors remains a challenge.
Apart from H. pylori, the rest of the gut microbiota is thought to be implicated in the pathobiology of CVD patients. Particularly, evidence remarkably supports an active crosstalk between CVDs and gut microbiota disturbances (dysbiosis) related to intestinal inflammation, while the stability of the gut microbiota exhibits a repressive effect on the development and progression of CVDs (103). Specifically, available evidence indicates a close connection between gut dysbiosis and the increased risk of cardiometabolic disorders and CVDs. Detrimental changes in the gut microbiota composition are associated with intestinal inflammatory reactions that lead to adverse remodeling of the host phenotype, predisposing to several pathological conditions such as obesity, IR, atherosclerosis, and associated disorders, ultimately leading to CVDs (104). Recent data indeed indicate that gut dysbiosis is related to chronic intestinal inflammation and MetS-related disorders inducing CVD, thereby appearing to be a promising target for the therapeutic management of these diseases (105). Gut dysbiosis may influence the cardiometabolic risk and CVD development either directly, via metabolite production, or indirectly, by interacting with the immune system (106). Gut dysbiosis triggers both the local intestinal inflammation involved in the pathophysiology of local disorders, including irritable bowel syndrome (IBS) and inflammatory bowel disease (IBD) (107), and systemic inflammation, contributing to the development and progression of MetS-related disorders including CVD (108).
In this respect, there is a relationship between H. pylori and the gut microbiota. Colonization of H. pylori in the gastric mucosa changes not only the local ecosystem but also the composition of the intestinal tract microbiota, consequently modulating the concentrations of microbial metabolites that can be introduced as disease markers (57). H. pylori infection induces profound alterations and increases the diversity in the human microbiota (109), and there is an interplay between H. pylori infection, gut dysbiosis, and extragastric disorders, particularly atherosclerotic CVD (110). Furthermore, MCs is associated with gut dysbiosis-related disorders such as IBD (111–113) and IBS (114).
Taken together, there is a potential crosstalk between CVD and gut dysbiosis related to H. pylori and MCs. Further research is warranted to elucidate in depth this critical topic offering significant clinical implications.
3 Role of MetS and H. pylori infection in the pathophysiology of MCs activation
MCs, originally renowned for their involvement in allergic and anaphylactic reactions, have emerged as pivotal players in the intricate web of MetS pathogenesis (115, 116). They were initially described by Waldeyer and later on by his student Ehrlich, who gave them their name (Mastzellen, namely, well fed, due to their high amount of cytoplasmatic granules) (51). Of note is that MCs are not found in the bloodstream as mature forms; instead, they end up in the epithelial and connective tissues of target organs and they reside there, where they undertake their final maturation, i.e., differentiation. MCs are multifunctional tissue-specific innate immune cells located in sites throughout the body, including the adipose tissue (117) and the heart (118). The topography of the adipose tissue places them at the forefront of the obesity-driven chronic inflammation that characterizes MetS (119). In this tissue, the activation of MCs chemotactically triggers the recruitment of immune cells, such as lipid-uptaking foamy macrophages, further amplifying the inflammatory milieu and angiogenesis (120, 121). This immune cell infiltration is a hallmark of IR adipose tissue and plays a crucial role in perpetuating the MetS phenotype (122, 123).
In the human heart, MCs are mainly distributed in the myocardial tissue, adjacent to capillaries and venules (124), and MCs degranulation, an early phase of their activation (125), occurs not only in the context of allergy but also in other pathologies including cardiac injury (126). Their secretory granules contain a plethora of vasoactive mediators including excessive specific substances (e.g., histamine), proteases (e.g., tryptase and chymase), non-MCs-specific proteases (cathepsin G), amines (serotonin and dopamine), cytokines (TNF-α, IL-4, and IL-5), and growth factors [stem cell factor (SCF) and fibroblast growth factor (FGF)] (127).
Regarding MetS-related MCs activation (51), the aforementioned mediators contribute to the onset of IR by compromising the insulin signaling pathways (51), which is associated with MetS-related atherosclerosis (128). The relationship between atherosclerosis and MetS is multifactorial, with obesity, dyslipidemia, and IR playing substantial roles in both its onset and progression (129).
In this regard, MCs have been associated with the dysregulation of MetS-related adipokines (130, 131), namely, bioactive molecules secreted directly by adipocytes. Leptin, an adipokine that orchestrates appetite and energy expenditure, is known to be increased in obesity (132). Higher concentrations of circulating leptin are linked to the augmented severity of MAFLD (130). In contrast, MetS-related adiponectin ameliorates liver histology in MAFLD, thereby exhibiting potential therapeutic benefits in MetS-related disorders including MAFLD (131). Specifically, related data have identified new roles for leptin and adiponectin in controlling responses from MCs (133). MCs from the rat peritoneal cavity were found to express the receptors for both leptin and adiponectin and to respond to these adipokines by producing cytokines and reactive oxygen species. As might be expected, the responses to leptin and adiponectin are rather different. While both induce the migration of MCs, leptin induces the secretion of histamine and cysteinyl leukotriene, as well as the expression of CCL2, while adiponectin induces the production of anti-inflammatory IL-10. CCL2 is associated with the amount of MetS-related atherosclerotic lesions and lipid deposition in atherosclerotic plaques (134) and is one of the major chemokines that are involved in the development and progression of CVD (135). In contrast, adiponectin appears to offer potential protection against the development of MetS (136). Thus, these molecules play quite different roles in controlling the MCs-driven inflammatory responses. The identification of the molecular mechanisms through which adipokines exert their effects on MCs may provide the potential to developing therapeutic strategies for controlling these adipokine-mediated effects.
Regarding H. pylori-related MCs stimulation, this bacterium, by activating MCs, induces cytokines such as IL-33 and, by altering the gut microbiota and metabolic hormone concentrations, such as leptin, may also be involved in MetS-related disorders at the topical and systemic levels (56, 137, 138). Patients with H. pylori infection are characterized by significantly higher counts of MCs in the gastric mucosa compared to those of normal subjects, a finding accompanied by a high number of apoptotic cells as well (76).
The infiltration of MCs in the gastric epithelium is significantly higher for the H. pylori group, but their numbers are diminished after a successful eradication (139). A further in vitro study deduced that H. pylori enhances the release of histamine by murine serosal MCs (140). Moreover, the H. pylori neutrophil activating protein, which is regarded as a major virulence factor of the bacterium (141), activates, among others, MCs that secondarily secrete pro-inflammatory molecules (142). The aforementioned IL-33 induced by H. pylori has been shown to orchestrate MCs responses, favor bacterial expansion, and synergize to gastritis development (56). In addition, H. pylori stimulates MCs directly or via gastrin induction, and they are actively involved in the pathogenesis of H. pylori-associated pathologies. Beyond activated MCs, vascular endothelial growth factor (VEGF), IL-8, chymase or tryptase, and MCs growth factor are linked to H. pylori infection (38).
The intestine appears to be another site for H. pylori infection that can contribute to the pathophysiology of intestinal inflammatory disorders (143, 144). Beyond the upper gastrointestinal tract, H. pylori can colonize the intestine (145), indicating that H. pylori strains could adapt to colonizing extragastric niches such as the intestine and can trigger intestinal inflammatory disorders such as IBS (144, 146, 147) and IBD, at least in certain populations (143, 148–151).
Similarly, MCs are residents of the whole gastrointestinal mucosa, and their dysregulation has been implicated in the pathophysiology of several inflammatory disorders such as IBS and IBD (152, 153). In this regard, increased numbers of MCs can be located in the intestine of patients with IBD, as well as an augmented amount of MCs products (i.e., histamine and tryptase), and there is abundant evidence of MCs activation in the intestinal mucosa of patients with IBD. Moreover, MCs activation also contributes to the pathophysiology of IBS (152, 154). Increased concentrations of MCs mediators in the gastrointestinal tract have been detected in these patients, signifying a direct interaction between MCs and nerves in IBD and IBS (152). MCs products including histamine, proteases, prostaglandins, and cytokines could also contribute to hypersensitivity and intestinal mucosal permeability defects in patients with IBS (155). Importantly, both IBS and IBD are associated with CVD (156, 157).
Therefore, a potential direct interaction between active H. pylori infection and activated MCs taking place in the intestinal niche might contribute to the pathophysiology of intestinal inflammatory disorders such as IBS and IBD associated with CVD. Thus, further studies are needed to shed light on this critical interaction, thereby offering related important clinical implications.
In addition, MCs themselves can be stimulated by the corticotropin-releasing hormone, secreted under stress, to release mediators including histamine, tryptase, IL-8, and VEGF, which are involved in systemic pathologies (38). Moreover, H. pylori-induced VacA displays chemotactic activities to BMD-MCs and induces these BMD-MCs to produce pro-inflammatory cytokines, including TNF-α (52). H. pylori infection is connected to the increased serum concentrations of TNF-α, a circulating cytokine able to exert its effects at a distance. TNF-α and IL-6 play important roles in the regulation of the synthesis of other acute-phase proteins, which are recognized risk factors for MetS-related atherosclerosis (38). These cytokines also exhibit profound actions on lipid metabolism directly at the site of the atherosclerotic lesion and could influence the atheroma process via blood circulating concentrations, distant production of cytokines, or via stimulating the circulating white blood cells to produce them, thus contributing to the pathogenesis of MetS-related CVD and C-CVD (158). Furthermore, MCs are implicated in vascular dysfunction, a prominent feature of MetS (159, 160), and the release of vasoactive substances by activated MCs can bring about endothelial dysfunction and impaired vasodilation, promoting AH and atherosclerosis (159, 160). MCs-derived mediators, such as tryptase, can directly damage the blood vessels and exacerbate vascular inflammation (159, 161).
More specifically, MCs acting in the first line of defense against pathogens have been observed to promote the development and progression of atherosclerosis. Chronic inflammation is regarded as a pivotal player in atherosclerosis, and both H. pylori and MCs-driven inflammation converge to amplify this process (51, 162, 163). In individuals with MetS, the occurrence of H. pylori infection can further deteriorate systemic inflammation, creating a microenvironment conducive to endothelial dysfunction and lipid accumulation within the arterial walls (102). The combined effects of H. pylori infection and MCs activation on endothelial function may accelerate the development of atherosclerotic lesions. H. pylori-induced inflammation, coupled with MCs-mediated cytokine release, fosters a pro-inflammatory microenvironment within the arterial wall, favoring the recruitment of immune cells and the formation of atherosclerotic plaques (162–164). MCs are between the crucial immune effectors in MetS-related atheroprogression because they release not only the MCs-specific neutral proteases tryptase and chymase upon activation but also a whole array of mediators including histamines, cathepsins, growth factors, cytokines, and chemokines (165).
Experimental evidence has generally illuminated the crucial role of MCs in atheroprogression and plaque vulnerability (166). These results are in line with related investigations where MCs abundance is associated with symptomatic carotid artery disease (167) and the prevalence of future adverse events (168).
Importantly, genetic and pharmacological interventions targeting MCs activation have demonstrated promising results in ameliorating MetS-related pathologies in experimental models (162). This highlights the therapeutic potential of modulating the activity of MCs in the context of MetS.
Beyond atherosclerosis, the impact of H. pylori infection and MCs activation extends to vasculitis, a manifold group of inflammatory conditions characterized by blood vessel inflammation (169). Vasculitis can influence various vascular beds, including small- and medium-sized arteries, and its clinical manifestations vary, ranging from skin rashes to multi-organ involvement. H. pylori has been reported to accompany certain forms of vasculitis, particularly with dermal and renal ones (170, 171). The ability of this bacterium to trigger immune responses and promote smoldering chronic inflammation may induce or deteriorate vasculitis processes (172). Similarly, in patients with vasculitis, the occurrence of MetS appears to be an important predictor of all-cause mortality (173).
Furthermore, the activation of MCs, richly populated around blood vessels, plays a crucial role in the pathophysiology of vasculitis (116). MCs contribute to vasculitis through the release of vasoactive substances, such as histamine, that can directly damage blood vessels (161). In medical disorders such as cutaneous leukocytoclastic vasculitis, MCs activation and the subsequent histamine release may be implicated in the formation of characteristic skin lesions (174). The interaction between H. pylori infection, MCs activation, and vasculitis demands further investigation to shed light to the precise mechanisms and clinical implications.
Whereas atherosclerosis and vasculitis constitute prominent vascular diseases and are the common denominator to both H. pylori infection and MCs activation, it is crucial to acknowledge that the impact of these factors extends beyond these conditions. Vascular diseases encompass a wide spectrum of pathologies, including aneurysms, peripheral arterial disease, and thromboembolic disorders. For instance, the role of H. pylori infection in the development of abdominal aortic aneurysms has garnered attention (175, 176). Chronic inflammation induced by H. pylori may contribute to the degradation of the arterial wall integrity, thereby predisposing individuals to aneurysm formation (177). Furthermore, MCs have been proposed as key players in the formation and progression of thromboembolic events (178). The release of MCs-derived mediators, such as tissue factor and plasminogen activator inhibitor-1, can promote a prothrombotic state, enhancing the risk of thrombosis and embolism (178, 179). In this respect, H. pylori appears to stimulate the expression of plasminogen activator inhibitor-1, an important player in the pathogenesis of CVD (88). Understanding the intricate interplay between H. pylori infection, MCs activation, and thromboembolic disorders is a fertile ground for future research.
In summary, MCs possess a central position in the complex landscape of H. pylori-related MetS. Their activation within the adipose tissue contributes to chronic inflammation, IR, and immune cell recruitment. Moreover, MCs are key players in vascular dysfunction and atherosclerosis, a hallmark of MetS and H. pylori infection. Comprehending the precise mechanisms underlying the involvement of MCs in H. pylori-related MetS pathophysiology holds promise for innovative therapeutic interventions and potential clinical applications.
4 Impact of H. pylori infection and MetS-related MCs activation on CVD
Activated MCs, which are associated with H. pylori infection and MetS, are frequently observed in the shoulder regions and particularly at the sites of erosion or rupture in coronary atherosclerotic lesions (Figure 1), i.e., in patients who died from myocardial infarction (180, 181). Moreover, human studies have shown an association of MCs with plaque neovascularization and hemorrhage that increase the risk of adverse CVD events (168). Furthermore, for instance, AH, which is prevalent in MetS and closely linked to both H. pylori infection and MCs activation, is associated with augmented premature morbidity and mortality due to CVD and stroke (182). Its high worldwide prevalence continues to increase and contributes to global morbidity and mortality, and as an important risk factor for H. pylori infection and MetS-related CVD, AH represents the principal cause of mortality related to H. pylori infection and MetS-associated ischemic heart disease and stroke (4). H. pylori-associated inflammation can lead to elevated blood pressure through multiple mechanisms, including compromised vascular function (4, 102). Similarly, MCs-derived mediators, such as histamine, can directly induce vasoconstriction, further exacerbating AH (183). In this respect, the MCs-revived tryptase induces cardiac fibrosis in the hypertensive heart (184). In addition, MCs may be implicated in hypertensive-related advance myocardial damage (185). Interestingly, activated MCs granules, beyond other mediators, also store and release the pro-inflammatory cytokine TNF-α, closely related to AH (186), which contributes to adverse myocardial remodeling by promoting cardiomyocytic apoptosis, hypertrophy, and inflammation. MCs degranulation is a significant contributor to inflammatory damaging processes (187), and in patients with MetS, TNF-α appears to be a CVD risk factor (188).
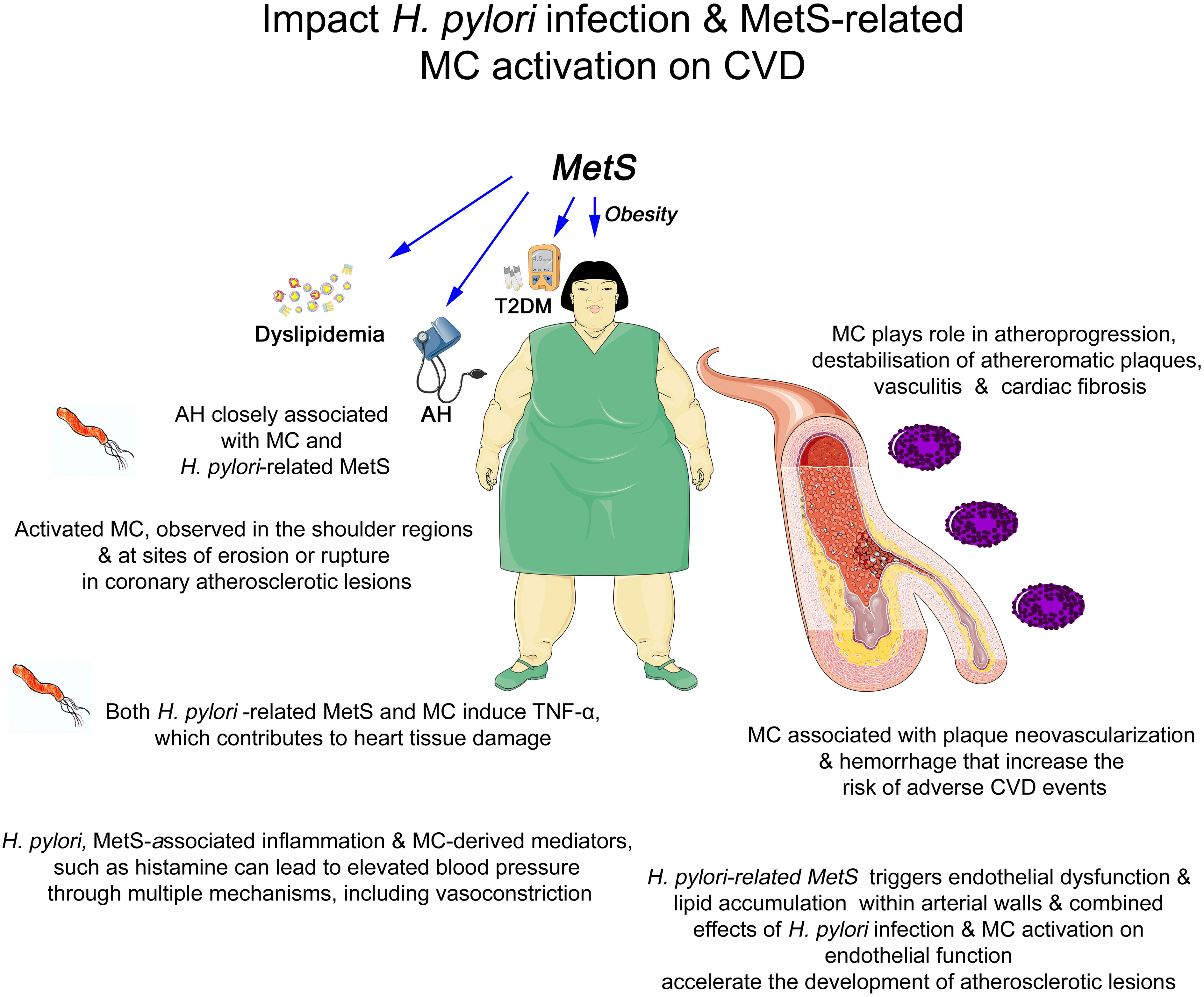
Figure 1 Graphical summary of the association between Helicobacter pylori infection, metabolic syndrome-related mast cell activation, and cardiovascular disorders. AH, arterial hypertension; CVD, cardiovascular disorders; MCs, mast cells; MetS, metabolic syndrome; T2DM, Type 2 Diabetes Mellitus; TNF-α, tumor necrosis factor alpha.
From a related clinical point of view, as the majority of hypertensive patients do not exhibit any symptoms, the disease is occasionally referred to as the “silent killer” and may be fatal due to cerebral strokes and ruptured arterial aneurysms. Heart disease, coronary artery disease, and stroke are just a few of the major issues caused by AH. Therefore, it is critical to understand high blood pressure risk factors and to take preventive measures (189).
As in the case of activated MCs-induced TNF-α that contributes to heart tissue damage, H. pylori-related MetS could also contribute to CVD by inducing pro-inflammatory cytokines, such as the aforementioned TNF-α (93). Beyond other mediators, H pylori-related increased circulating TNF-α, which is able to exert its effects at a distance, may also lead to heart tissue damage (158). Nevertheless, whether H. pylori and MetS-related MCs activation could also contribute to CVD progression by inducing pro-inflammatory mediators including TNF-α needs further investigation.
Existing related evidence indicates a central role for inflammatory cell-related processes, particularly MCs, in CVD (190). MCs accumulate excessively in the injured endothelium, which, by stimulating the formation of foam cells and atherosclerotic plaques and representing an important factor in plaque degradation and the initiation of coagulation due to the cytokines released from granules and proteolytic enzymes, plays a role in the pathogenesis of CVD (191). The MCs mediator, histamine, and its receptors profoundly impact the pathophysiology of the heart, leading to hypertension-induced cardiac hypertrophy and other cardiac disorders (190). Importantly, MCs degranulation occurs in cardiac injury (126). MCs degranulation increases rapidly and reaches to about 80% within a week after cardiac injury (192).
Especially, experimental studies in animals have clarified the critical role of MCs in atheroprogression and plaque vulnerability (166, 193). Chymase is linked to atherosclerotic plaque vulnerability (193). In humans, MCs have been shown to accumulate in advanced and ruptured coronary plaques (181). More recent data indicated that the connection of MCs with disease progression has been recognized in patients with CVD. Significantly augmented serum tryptase concentrations are detected in patients with acute coronary syndromes (168, 194), and the number of intraplaque MCs has been shown to increase upon atherosclerotic plaque destabilization (168). Equally, MCs in the human heart generate chymase and renin, which locally activate the angiotensin system and can cause arteriolar vasoconstriction, involved in the pathogenesis of CVD (195). Furthermore, an independent association of the number of intraplaque MCs in carotid plaques is connected to the prevalence of clinical CVD adverse events (168).
Finally, MCs are linked to cardiac fibrosis through several mechanisms (187). In this respect, for instance, MCs are able to produce various inflammatory cytokines including TNF-α, IL-6, amines (mostly histamine), and proteases, regulating the activity of matrix metalloproteases and collagen degradation, thus playing a role in the organization and resolution of inflammation and fibrosis in the myocardium (196). However, owing to the ability of MCs to induce both pro- and anti-fibrotic mediators, many studies have reported controversial results and defined MCs divergent functions, including negative, neutral, or protective activities of MCs in cardiac remodeling and fibrosis. Although preclinical and clinical data have identified MCs as important orchestrators of cardiac fibrosis, the exact mechanisms are yet to be clarified. Because MCs represent a valuable target for the therapeutic manipulation of fibrinogenesis, a better understanding of their involvement is required to stop and even reverse cardiac fibrosis.
In conclusion, the dual impact of H. pylori infection and MetS-related MCs activation extends to atherosclerosis, vasculitis, and a broader spectrum of vascular diseases. These interactions underscore the necessity for a comprehensive approach to understanding and managing vascular complications in individuals with MetS. Further research is essential to unraveling the intricate mechanisms driving these associations, potentially paving the way for innovative therapeutic strategies.
5 Translational insights for clinicians: reimagining diagnosis and treatment
The intricate interplay between H. pylori infection, MetS-related MCs activation, and their combined impact on CVDs presents clinicians with a unique opportunity to reimagine the diagnosis and treatment landscape. The multifaceted nature of these interactions proposes novel avenues for intervention, offering therefore a potential paradigm shift in managing patients at risk of CVD adverse events associated with H. pylori-related MetS.
One of the first translational insights lies in the identification of individuals at high risk of developing CVD within the context of both H. pylori infection and MetS. Recognizing that H. pylori infection may exacerbate the pro-inflammatory milieu in these patients, clinicians can consider routine testing for the presence of the bacterium. Although gastric histology for the detection of H. pylori represents the practical diagnostic gold standard for active H. pylori infection (197), noninvasive tests, such as the urea breath test or stool antigen test, can identify H. pylori infection with high sensitivity and specificity (197). In high-risk populations, such as those with MetS, routine screening may provide valuable insights into a patient’s overall risk profile.
Clinicians should consider not only the presence of H. pylori infection and MetS but also the immunological milieu, including markers of chronic inflammation and MCs activation accompanying these two disorders very frequent in routine clinical practice. Serum markers such as C-reactive protein, IL-6, IL-17, TNF-α, and/or IL-22 can serve as indicators of smoldering chronic inflammatory processes with concomitant MCs activation (198, 199). Increased levels of such markers, particularly in individuals with MetS and allergic and inflammatory disorders (199–201), may prompt further investigation into the potential involvement of H. pylori infection and MCs activation. In addition, the measurement of MCs-specific mediators, such as histamine, chymase, and tryptase, may offer insights into the extent of MCs activation (161, 201). While these tests are not routine in clinical practice, they represent a promising avenue for future research and development. Addressing these factors may complement the existing treatment approaches for MetS, thereby improving patient outcomes. In patients with both H. pylori infection and MetS, therapy to eradicate H. pylori could be considered as an adjunct to standard treatment (202, 203). Moreover, it may synergistically contribute to mitigate the inflammatory burden associated with H. pylori infection (6), potentially alleviating some of the systemic effects attributed to MetS and CVD (158, 204).
Regarding the impact of probiotics on the eradication of H. pylori and consequence of CVD, for more than 15 years, it has been well established that eradication regimens supplemented with probiotics increase the rates of eradication and reduces the side effects in H. pylori infection (205–208). The incorporation of probiotics into complex eradication therapy holds promise for optimizing the treatment of H. pylori infection. Probiotic strains, through their alteration of the gastric and gut microbiota, immunomodulatory properties, and their direct antagonistic effects against H. pylori (via bacteriocins and other factors such as bacterial synthesized acids and hydrogen peroxide), can increase the eradication rates, decrease the frequency and severity of side effects, and enhance patient compliance and treatment outcomes.
Apart from gastrointestinal pathologies, the impact of probiotic supplementation has been largely reported to reduce the risk of systemic pathologies, including cardiometabolic syndrome, and increase the H. pylori eradication rates (209). Supplementation of next-generation probiotics has been reported to reduce IR, plasma cholesterol, and the risk of developing liver dysfunction and inflammation, whereas no significant change is demonstrated in the gut microbiota (210). Probiotic supplementation can improve the clinical parameters of NAFLD, the hepatic component of MetS, and the eradication rates of H. pylori (211). Similarly, prebiotics and/or probiotics can exert a beneficial effect in the prevention of H. pylori-related neurodegenerative disorders (212).
Beyond MCs stabilization, MCs silencers constitute novel therapeutic approaches with translational potential (158, 162, 204, 213, 214). These agents could be evaluated as adjunctive therapies for individuals with H. pylori infection and MetS, particularly those with evidence of MCs activation-associated CVD adverse events.
Translational insights should also inform the education and lifestyle modification strategies that offer benefits to patients with MetS (215). Moreover, lifestyle interventions appear to decrease the risk of H. pylori-related gastric cancer prevalence and mortality (216). Of note is that H. pylori infection with MetS and/or MCs appears to be involved in gastrointestinal cancer pathophysiology (217–219) and metastases (217, 218, 220).
Emphasizing the potential impact of H. pylori infection and MetS-related MCs activation on CVDs can motivate patients to adhere to lifestyle interventions, i.e., dietary and exercise recommendations. Highlighting the potential role of infectious and immunological factors in the development of CVDs can enhance patient engagement and compliance with these interventions.
Translational insights are not static: they pave the way for future research directions and clinical advancements. Further investigations are warranted to elucidate the exact mechanisms linking H. pylori infection, MCs activation, and CVD within the context of MetS. Longitudinal studies should explore the impact of H. pylori eradication and MCs stabilization therapies and/or silencer novel therapeutic approaches on the clinical outcomes of individuals with MetS. These studies can help determine the effectiveness and safety of these interventions, providing valuable guidance for clinical practice. Biomarkers that reflect MCs activity and inflammation could enhance risk stratification and guide personalized treatment strategies. As mentioned earlier, very recent data have indicated that the oral cavity is an additional niche for H. pylori and could be the trigger of H. pylori infection, re-infection, and transmission into the stomach (77). Thus, further research is expected to elucidate the potential direct interaction between H. pylori and MCs taking place in the oral cavity, with a potential relation to CVD. In this respect, apart from H. pylori-related dysbiosis in the stomach, intestine, the ocular surface, and intraocular cavity, oral cavity H. pylori-related dysbiosis can also contribute to the pathophysiology of MetS component-associated glaucoma, CVD, and neurodegeneration (221, 222). Therefore, targeting such dysbiosis could be important to the management of glaucoma, CVD, and other neurodegenerative disorders (221, 222).
All in all, the translational insights derived from understanding the intricate crosstalk among H. pylori infection, MCs activation, MetS, and CVD offer a new era for clinicians. Identifying high-risk patients, adopting integrated diagnostic approaches, and tailoring therapeutic strategies represent tangible steps toward enhanced patient care. These insights not only improve our understanding of the pathophysiology of MetS but also permit innovative approaches that may ultimately lessen the impact of CVD in patients with MetS.
Author contributions
MD: Investigation, Visualization, Writing – original draft, Writing – review & editing. AP: Investigation, Validation, Writing – review & editing. SP: Investigation, Writing – review & editing. MB: Investigation, Writing – review & editing. EK: Investigation, Writing – review & editing. MT-C: Investigation, Writing – review & editing. EV: Investigation, Writing – review & editing. CH: Investigation, Writing – review & editing. HK: Investigation, Writing – review & editing. DR: Investigation, Writing – review & editing. CK: Investigation, Writing – review & editing. IL: Investigation, Writing – review & editing. MT: Investigation, Writing – review & editing. AS: Investigation, Writing – review & editing. JK: Conceptualization, Investigation, Project administration, Supervision, Validation, Writing – original draft, Writing – review & editing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Doulberis M, Kountouras J, Stadler T, Meerwein C, Polyzos SA, Kulaksiz H, et al. Association between helicobacter pylori infection and nasal polyps: A systematic review and meta-analysis. Microorganisms. (2023) 11. doi: 10.3390/microorganisms11061581
2. Doulberis M, Kotronis G, Thomann R, Polyzos SA, Boziki M, Gialamprinou D, et al. Review: impact of helicobacter pylori on alzheimer’s disease: what do we know so far? Helicobacter. (2018) 23. doi: 10.1111/hel.12454
3. Franceschi F, Gasbarrini A, Polyzos SA, Kountouras J. Extragastric diseases and helicobacter pylori. Helicobacter. (2015) 20 Suppl 1:40–6. doi: 10.1111/hel.12256
4. Kountouras J, Papaefthymiou A, Polyzos SA, Deretzi G, Vardaka E, Soteriades ES, et al. Impact of helicobacter pylori-related metabolic syndrome parameters on arterial hypertension. Microorganisms. (2021) 9. doi: 10.3390/microorganisms9112351
5. Malfertheiner P, Megraud F, O’Morain CA, Gisbert JP, Kuipers EJ, Axon AT, et al. Management of helicobacter pylori infection-the maastricht V/florence consensus report. Gut. (2017) 66:6–30. doi: 10.1136/gutjnl-2016-312288
6. Kountouras J, Doulberis M, Papaefthymiou A, Polyzos SA, Zavos C, Kazakos E, et al. Controlling the impact of helicobacter pylori-related hyperhomocysteinemia on neurodegeneration. Med (Kaunas). (2023) 59. doi: 10.3390/medicina59030504
7. Doulberis M, Papaefthymiou A, Kountouras J. Special issue “Extragastric disorders of helicobacter pylori infection: from diagnosis to treatment”: editorial. Microorganisms. (2023) 11. doi: 10.3390/microorganisms11030677
8. Parsonnet J, Friedman GD, Vandersteen DP, Chang Y, Vogelman JH, Orentreich N, et al. Helicobacter pylori infection and the risk of gastric carcinoma. N Engl J Med. (1991) 325:1127–31. doi: 10.1056/NEJM199110173251603
9. Doglioni C, Wotherspoon AC, Moschini A, de Boni M, Isaacson PG. High incidence of primary gastric lymphoma in northeastern Italy. Lancet. (1992) 339:834–5. doi: 10.1016/0140-6736(92)90280-g
10. Ahmed M, Kumari N, Mirgani Z, Saeed A, Ramadan A, Ahmed MH, et al. Metabolic syndrome; definition, pathogenesis, elements, and the effects of medicinal plants on it’s elements. J Diabetes Metab Disord. (2022) 21:1011–22. doi: 10.1007/s40200-021-00965-2
11. Xie Q, He Y, Zhou D, Jiang Y, Deng Y, Li R. Recent research progress on the correlation between metabolic syndrome and helicobacter pylori infection. PeerJ. (2023) 11:e15755. doi: 10.7717/peerj.15755
12. Liu Y, Shuai P, Chen W, Liu Y, Li D. Association between helicobacter pylori infection and metabolic syndrome and its components. Front Endocrinol (Lausanne). (2023) 14:1188487. doi: 10.3389/fendo.2023.1188487
13. Park J, Kim N, Kim WS, Lim SH, Choi Y, Jo HH, et al. Long-term effects of the eradication of helicobacter pylori on metabolic parameters, depending on sex, in South Korea. Gut Liver. (2023) 17:58–68. doi: 10.5009/gnl210588
14. Wernly S, Semmler G, Volkerer A, Flamm M, Aigner E, Niederseer D, et al. Helicobacter pylori and cardiovascular risk: only a dead helicobacter is a good helicobacter? Helicobacter. (2022) 27:e12928. doi: 10.1111/hel.12928
15. Azami M, Baradaran HR, Dehghanbanadaki H, Kohnepoushi P, Saed L, Moradkhani A, et al. Association of helicobacter pylori infection with the risk of metabolic syndrome and insulin resistance: an updated systematic review and meta-analysis. Diabetol Metab Syndr. (2021) 13:145. doi: 10.1186/s13098-021-00765-x
16. Abo-Amer YE, Sabal A, Ahmed R, Hasan NFE, Refaie R, Mostafa SM, et al. Relationship between helicobacter pylori infection and nonalcoholic fatty liver disease (Nafld) in a developing country: A cross-sectional study. Diabetes Metab Syndr Obes. (2020) 13:619–25. doi: 10.2147/DMSO.S237866
17. Doulberis M, Srivastava S, Polyzos SA, Kountouras J, Papaefthymiou A, Klukowska-Rotzler J, et al. Active helicobacter pylori infection is independently associated with nonalcoholic steatohepatitis in morbidly obese patients. J Clin Med. (2020) 9. doi: 10.3390/jcm9040933
18. Xia B, Wang W, Lu Y, Chen C. Helicobacter pylori infection increases the risk of metabolic syndrome in pregnancy: A cohort study. Ann Transl Med. (2020) 8:875. doi: 10.21037/atm-20-4863
19. Kountouras J, Papaefthymiou A, Polyzos SA, Liatsos C, Tzitiridou-Chatzopoulou M, Chatzopoulos D, et al. Potential impact of helicobacter pylori and metabolic syndrome-related non-alcoholic fatty liver disease on cardio-cerebrovascular disease. Metabolism. (2022) 135:155276. doi: 10.1016/j.metabol.2022.155276
20. Kountouras J, Papaefthymiou A, Polyzos SA, Kazakos E, Vardaka E, Touloumtzi M, et al. Impact of active helicobacter pylori infection-related metabolic syndrome on systemic arterial hypertension. Arq Bras Cardiol. (2022) 119:502–4. doi: 10.36660/abc.20210931
21. Kountouras J, Papaefthymiou A, Polyzos SA, Kyrailidi F, Doulberis M. Potential impact of helicobacter pylori-related metabolic syndrome on arterial hypertension outcomes. Am J Hypertens. (2022) 36. doi: 10.1093/ajh/hpac120
22. Kountouras J, Kazakos E, Kyrailidi F, Polyzos SA, Zavos C, Arapoglou S, et al. Innate immunity and nonalcoholic fatty liver disease. Ann Gastroenterol. (2023) 36:244–56. doi: 10.20524/aog.2023.0793
23. Hurtado-Aviles Hurtado-Aviles MG, Martinez-Reculez MGC, Vargas-Camano ME, Castrejon-Vazquez MI. [Autoimmunity in chronic urticaria. A historical and current perspective]. Rev Alerg Mex. (2022) 69 Suppl 1:s69–80. doi: 10.29262/ram.v69iSupl1.1037
24. Wu MCs, Leong PY, Chiou JY, Chen HH, Huang JY, Wei JC. Increased risk of systemic lupus erythematosus in patients with helicobacter pylori infection: A nationwide population-based cohort study. Front Med (Lausanne). (2019) 6:330. doi: 10.3389/fmed.2019.00330
25. Hamed AE, Moshrif A, Ryan M, Abdelmaguid MM, Azeem HA, Abdelkreem MI. The effect of helicobacter pylori seropositivity and activity on disease outcome in patients with rheumatoid arthritis, systemic lupus erythematosus and ankylosing spondylitis. Egypt J Immunol. (2021) 28:206–14. doi: 10.55133/eji.280421
26. Wang Y, Huang Z, Xiao Y, Wan W, Yang X. The shared biomarkers and pathways of systemic lupus erythematosus and metabolic syndrome analyzed by bioinformatics combining machine learning algorithm and single-cell sequencing analysis. Front Immunol. (2022) 13:1015882. doi: 10.3389/fimmu.2022.1015882
27. Grzechnik K, Targonska-Stepniak B. Metabolic syndrome and rheumatoid arthritis activity: an analysis of clinical, laboratory, and ultrasound parameters. Nutrients. (2023) 15. doi: 10.3390/nu15224756
28. Pisani F, Croia C, Petrelli F, Chericoni E, Migliorini P, Puxeddu I. The multifaceted role of mast cells in the pathogenesis of rheumatoid arthritis. Clin Exp Rheumatol. (2023). doi: 10.55563/clinexprheumatol/eontou
29. Nordestgaard LT, Christoffersen M, Frikke-Schmidt R. Shared risk factors between dementia and atherosclerotic cardiovascular disease. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms23179777
30. Yang S, Yang H, Zhang Y. Yao-shan of traditional chinese medicine: an old story for metabolic health. Front Pharmacol. (2023) 14:1194026. doi: 10.3389/fphar.2023.1194026
31. Weinstein G, O’Donnell A, Frenzel S, Xiao T, Yaqub A, Yilmaz P, et al. Nonalcoholic fatty liver disease, liver fibrosis, and structural brain imaging: the cross-cohort collaboration. Eur J Neurol. (2024) 31:e16048. doi: 10.1111/ene.16048
32. Cheng Y, Zhu Y, Zhang J, Duan X, Zhang Y. Large accumulation of collagen and increased activation of mast cells in hearts of mice with hyperlipidemia. Arq Bras Cardiol. (2017) 109:404–9. doi: 10.5935/abc.20170143
33. Ge W, Guo X, Song X, Pang J, Zou X, Liu Y, et al. The role of immunoglobulin E and mast cells in hypertension. Cardiovasc Res. (2022) 118:2985–99. doi: 10.1093/cvr/cvac010
34. Harper RL, Fang F, San H, Negro A, St Hilaire C, Yang D, et al. Mast cell activation and degranulation in acute artery injury: A target for post-operative therapy. FASEB J. (2023) 37:e23029. doi: 10.1096/fj.202201745RR
35. Jia G, Hill MA, Sowers JR. Vascular endothelial mineralocorticoid receptors and epithelial sodium channels in metabolic syndrome and related cardiovascular disease. J Mol Endocrinol. (2023) 71. doi: 10.1530/JME-23-0066
36. Duong MT, Nasrallah IM, Wolk DA, Chang CCY, Chang TY. Cholesterol, atherosclerosis, and apoe in vascular contributions to cognitive impairment and dementia (Vcid): potential mechanisms and therapy. Front Aging Neurosci. (2021) 13:647990. doi: 10.3389/fnagi.2021.647990
37. Riad M. Association of helicobacter pylori infection with coronary artery disease: is it an independent risk factor? Egypt Heart J. (2021) 73:61. doi: 10.1186/s43044-021-00185-2
38. Kountouras J, Boziki M, Polyzos SA, Katsinelos P, Gavalas E, Zeglinas C, et al. The emerging role of helicobacter pylori-induced metabolic gastrointestinal dysmotility and neurodegeneration. Curr Mol Med. (2017) 17:389–404. doi: 10.2174/1566524018666171219094837
39. Yue J, Tan Y, Huan R, Guo J, Yang S, Deng M, et al. Mast cell activation mediates blood-brain barrier impairment and cognitive dysfunction in septic mice in a histamine-dependent pathway. Front Immunol. (2023) 14:1090288. doi: 10.3389/fimmu.2023.1090288
40. Abel J, Goldmann O, Ziegler C, Holtje C, Smeltzer MS, Cheung AL, et al. Staphylococcus aureus evades the extracellular antimicrobial activity of mast cells by promoting its own uptake. J Innate Immun. (2011) 3:495–507. doi: 10.1159/000327714
41. Lowy FD. Staphylococcus aureus infections. N Engl J Med. (1998) 339:520–32. doi: 10.1056/NEJM199808203390806
42. Hayes SM, Biggs TC, Goldie SP, Harries PG, Walls AF, Allan RN, et al. Staphylococcus aureus internalization in mast cells in nasal polyps: characterization of interactions and potential mechanisms. J Allergy Clin Immunol. (2020) 145:147–59. doi: 10.1016/j.jaci.2019.06.013
43. Goldmann O, Sauerwein T, Molinari G, Rohde M, Forstner KU, Medina E. Cytosolic sensing of intracellular staphylococcus aureus by mast cells elicits a type I ifn response that enhances cell-autonomous immunity. J Immunol. (2022) 208:1675–85. doi: 10.4049/jimmunol.2100622
44. Lima HG, Pinke KH, Gardizani TP, Souza-Junior DA, Carlos D, Avila-Campos MJ, et al. Mast cells act as phagocytes against the periodontopathogen aggregatibacter actinomyceteMCsomitans. J Periodontol. (2013) 84:265–72. doi: 10.1902/jop.2012.120087
45. Kennedy L, Meadows V, Sybenga A, Demieville J, Chen L, Hargrove L, et al. Mast cells promote nonalcoholic fatty liver disease phenotypes and microvesicular steatosis in mice fed a western diet. Hepatology. (2021) 74:164–82. doi: 10.1002/hep.31713
46. Kountouras J, Kazakos E, Polyzos SA, Papaefthymiou A, Zavos C, Tzitiridou-Chatzopoulou M, et al. Potential impact of trained innate immunity on the pathophysiology of metabolic dysfunction-associated fatty liver disease. Clin Immunol. (2023) 256:109776. doi: 10.1016/j.clim.2023.109776
47. Lyons DO, Pullen NA. Beyond ige: alternative mast cell activation across different disease states. Int J Mol Sci. (2020) 21. doi: 10.3390/ijms21041498
48. De Zuani M, Dal Secco C, Tonon S, Arzese A, Pucillo CEM, Frossi B. Lps guides distinct patterns of training and tolerance in mast cells. Front Immunol. (2022) 13:835348. doi: 10.3389/fimmu.2022.835348
49. Sandhu JK, Kulka M. Decoding mast cell-microglia communication in neurodegenerative diseases. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms22031093
50. Gurung P, Moussa K, Adams-Huet B, Devaraj S, Jialal I. Increased mast cell abundance in adipose tissue of metabolic syndrome: relevance to the proinflammatory state and increased adipose tissue fibrosis. Am J Physiol Endocrinol Metab. (2019) 316:E504–E9. doi: 10.1152/ajpendo.00462.2018
51. Zhang J, Shi GP. Mast cells and metabolic syndrome. Biochim Biophys Acta. (2012) 1822:14–20. doi: 10.1016/j.bbadis.2010.12.012
52. Supajatura V, Ushio H, Wada A, Yahiro K, Okumura K, Ogawa H, et al. Cutting edge: vaca, a vacuolating cytotoxin of helicobacter pylori, directly activates mast cells for migration and production of proinflammatory cytokines. J Immunol. (2002) 168:2603–7. doi: 10.4049/jimmunol.168.6.2603
53. Shishkina VV, Klochkova SV, Alexeeva NT, Samodurova NY, Nikityuk DV. [Discussion of the immunomorphological role of interactions between mast cells and helicobacter pylori in the gastric mucosa]. Vopr Pitan. (2022) 91:98–108. doi: 10.33029/0042-8833-2022-91-1-98-108
54. Zhu F, Zhang X, Li P, Zhu Y. Effect of helicobacter pylori eradication on gastric precancerous lesions: A systematic review and meta-analysis. Helicobacter. (2023):e13013. doi: 10.1111/hel.13013
55. Veijola L, Oksanen A, Linnala A, Sipponen P, Rautelin H. Persisting chronic gastritis and elevated helicobacter pylori antibodies after successful eradication therapy. Helicobacter. (2007) 12:605–8. doi: 10.1111/j.1523-5378.2007.00549.x
56. Lv YP, Teng YS, Mao FY, Peng LS, Zhang JY, Cheng P, et al. Helicobacter pylori-induced il-33 modulates mast cell responses, benefits bacterial growth, and contributes to gastritis. Cell Death Dis. (2018) 9:457. doi: 10.1038/s41419-018-0493-1
57. Francisco AJ. Helicobacter pylori infection induces intestinal dysbiosis that could be related to the onset of atherosclerosis. BioMed Res Int. (2022) 2022:9943158. doi: 10.1155/2022/9943158
58. Soloski MJ, Poulain M, Pes GM. Does the trained immune system play an important role in the extreme longevity that is seen in the sardinian blue zone? Front Aging. (2022) 3:1069415. doi: 10.3389/fragi.2022.1069415
59. Girolamo F, Coppola C, Ribatti D. Immunoregulatory effect of mast cells influenced by microbes in neurodegenerative diseases. Brain Behav Immun. (2017) 65:68–89. doi: 10.1016/j.bbi.2017.06.017
60. Vanuytsel T, van Wanrooy S, Vanheel H, Vanormelingen C, Verschueren S, Houben E, et al. Psychological stress and corticotropin-releasing hormone increase intestinal permeability in humans by a mast cell-dependent mechanism. Gut. (2014) 63:1293–9. doi: 10.1136/gutjnl-2013-305690
61. Polyzos SA, Kountouras J, Mantzoros CS. Helicobacter pylori infection and nonalcoholic fatty liver disease: are the four meta-analyses favoring an intriguing association pointing to the right direction? Metabolism. (2019) 96:iii–v. doi: 10.1016/j.metabol.2019.05.006
62. Moyo-Chilufya M, Maluleke K, Kgarosi K, Muyoyeta M, Hongoro C, Musekiwa A. The burden of non-communicable diseases among people living with hiv in sub-saharan africa: A systematic review and meta-analysis. EClinicalMedicine. (2023) 65:102255. doi: 10.1016/j.eclinm.2023.102255
63. Beckett A, Scott JR, Chater AM, Ferrandino L, Aldous JWF. The prevalence of metabolic syndrome and its components in firefighters: A systematic review and meta-analysis. Int J Environ Res Public Health. (2023) 20. doi: 10.3390/ijerph20196814
64. Mobley HL, Hu LT, Foxal PA. Helicobacter pylori urease: properties and role in pathogenesis. Scand J Gastroenterol Suppl. (1991) 187:39–46.
65. Covacci A, Telford JL, Del Giudice G, Parsonnet J, Rappuoli R. Helicobacter pylori virulence and genetic geography. Science. (1999) 284:1328–33. doi: 10.1126/science.284.5418.1328
66. Weeks DL, Eskandari S, Scott DR, Sachs G. A H+-gated urea channel: the link between helicobacter pylori urease and gastric colonization. Science. (2000) 287:482–5. doi: 10.1126/science.287.5452.482
67. Wroblewski LE, Peek RM Jr., Wilson KT. Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clin Microbiol Rev. (2010) 23:713–39. doi: 10.1128/CMR.00011-10
68. Odenbreit S. Adherence properties of helicobacter pylori: impact on pathogenesis and adaptation to the host. Int J Med Microbiol. (2005) 295:317–24. doi: 10.1016/j.ijmm.2005.06.003
69. Szabo I, Brutsche S, Tombola F, Moschioni M, Satin B, Telford JL, et al. Formation of anion-selective channels in the cell plasma membrane by the toxin vaca of helicobacter pylori is required for its biological activity. EMBO J. (1999) 18:5517–27. doi: 10.1093/emboj/18.20.5517
70. Bik EM, Eckburg PB, Gill SR, Nelson KE, Purdom EA, Francois F, et al. Molecular analysis of the bacterial microbiota in the human stomach. Proc Natl Acad Sci U.S.A. (2006) 103:732–7. doi: 10.1073/pnas.0506655103
71. Plonka M, Targosz A, Jakubowska M, Reczynski W, Ptak-Belowska A, Szczyrk U, et al. The relationship between the presence of helicobacter pylori and the composition of ionic and organic microelements in drinking water from cracow. J Physiol Pharmacol. (2021) 72. doi: 10.26402/jpp.2021.3.10
72. Hernandez-Diaz Y, Tovilla-Zarate CA, Juarez-Rojop I, Banos-Gonzalez MA, Torres-Hernandez ME, Lopez-Narvaez ML, et al. The role of gene variants of the inflammatory markers crp and tnf-alpha in cardiovascular heart disease: systematic review and meta-analysis. Int J Clin Exp Med. (2015) 8:11958–84.
73. Gurgone D, MCshane L, MCsharry C, Guzik TJ, Maffia P. Cytokines at the interplay between asthma and atherosclerosis? Front Pharmacol. (2020) 11:166. doi: 10.3389/fphar.2020.00166
74. Ceolin J, de Borba EL, Mundstock E, de Oliveira JR, Mattiello R, Bodanese LC. Phase angle of bioimpedance as a marker of inflammation in cardiovascular diseases: A systematic review. Nutrition. (2023) 112:112064. doi: 10.1016/j.nut.2023.112064
75. Okada S, Yasudo H, Ohnishi Y, Matsuguma C, Fukano R, Motonaga T, et al. Interleukin-33/st2 axis as potential biomarker and therapeutic target in kawasaki disease. Inflammation. (2023) 46:480–90. doi: 10.1007/s10753-022-01753-7
76. Hofman V, Lassalle S, Selva E, Kalem K, Steff A, Hebuterne X, et al. Involvement of mast cells in gastritis caused by helicobacter pylori: A potential role in epithelial cell apoptosis. J Clin Pathol. (2007) 60:600–7. doi: 10.1136/jcp.2006.040741
77. Abdul NS, Khalid Alkhelaiwi A, Awadh Alenazi A, Fehaid Alrashidi R, Ghaleb Salma R. The association of helicobacter pylori in the oral cavity with dental caries in patients with and without gastric infection: A systematic review. Cureus. (2023) 15:e38398. doi: 10.7759/cureus.38398
78. Gaje PN, Amalia Ceausu R, Jitariu A, Stratul SI, Rusu LC, Popovici RA, et al. Mast cells: key players in the shadow in oral inflammation and in squamous cell carcinoma of the oral cavity. BioMed Res Int. (2016) 2016:9235080. doi: 10.1155/2016/9235080
79. Szwed P, Gasecka A, Zawadka M, Eyileten C, Postula M, Mazurek T, et al. Infections as novel risk factors of atherosclerotic cardiovascular diseases: pathophysiological links and therapeutic implications. J Clin Med. (2021) 10. doi: 10.3390/jcm10122539
80. Gravina AG, Zagari RM, De Musis C, Romano L, Loguercio C, Romano M. Helicobacter pylori and extragastric diseases: A review. World J Gastroenterol. (2018) 24:3204–21. doi: 10.3748/wjg.v24.i29.3204
81. Mladenova I. Helicobacter pylori and cardiovascular disease: update 2019. Minerva Cardioangiol. (2019) 67:425–32. doi: 10.23736/S0026-4725.19.04986-7
82. Du L, Liu J, Jin C, Ma Y, Yin L, Man S, et al. Association between helicobacter pylori infection and carotid atherosclerosis in chinese adults. Atheroscler Plus. (2021) 44:25–30. doi: 10.1016/j.athplu.2021.08.004
83. Farsak B, Yildirir A, Akyon Y, Pinar A, Oc M, Boke E, et al. Detection of chlamydia pneumoniae and helicobacter pylori DNA in human atherosclerotic plaques by pcr. J Clin Microbiol. (2000) 38:4408–11. doi: 10.1128/JCM.38.12.4408-4411.2000
84. Xie J, Xu L, Huang H, Chen Y, Wang J, Li Y, et al. A simpler definition of mafld precisely predicts incident metabolic diseases: A 7-year cohort study. Hepatol Int. (2023) 17:1182–91. doi: 10.1007/s12072-023-10558-1
85. Boziki M, Grigoriadis N, Doulberis M, Papaefthymiou A, Polyzos SA, Kountouras J. Potential impact of helicobacter pylori-related galectin-3 on chronic kidney, cardiovascular and brain disorders in decompensated cirrhosis. Dig Liver Dis. (2020) 52:121–3. doi: 10.1016/j.dld.2019.09.001
86. Boziki M, Polyzos SA, Papaefthymiou A, Doulberis M, Bakirtzis C, Sintila SA, et al. Potential impact of helicobacter pylori-related metabolic syndrome and galectin-3 on liver, chronic kidney and brain disorders. Metabolism. (2021) 118:154736. doi: 10.1016/j.metabol.2021.154736
87. Tan KCB, Cheung CL, Lee ACH, Lam JKY, Wong Y, Shiu SWM. Galectin-3 and risk of cardiovascular events and all-cause mortality in type 2 diabetes. Diabetes Metab Res Rev. (2019) 35:e3093. doi: 10.1002/dmrr.3093
88. Kountouras J, Polyzos SA, Zavos C, Deretzi G, Kountouras C, Vardaka E, et al. Helicobacter pylori might contribute to nonalcoholic fatty liver disease-related cardiovascular events by releasing prothrombotic and proinflammatory factors. Hepatology. (2014) 60:1450–1. doi: 10.1002/hep.27168
89. Zhang D, Zuo H, Yang H, Zhang M, Ge C, Song X. Comparison of clinical profiles and associated factors for acute myocardial infarction among young and very young patients with coronary artery disease. Coron Artery Dis. (2022) 33:655–60. doi: 10.1097/MCsA.0000000000001183
90. Ulloque-Badaracco JR, Hernandez-Bustamante EA, Alarcon-Braga EA, Al-Kassab-Cordova A, Cabrera-Guzman JC, Herrera-Anazco P, et al. Vitamin B12, folate, and homocysteine in metabolic syndrome: A systematic review and meta-analysis. Front Endocrinol (Lausanne). (2023) 14:1221259. doi: 10.3389/fendo.2023.1221259
91. Kountouras J, Doulberis M, Polyzos SA, Kazakos E, Vardaka E, Touloumtzi M, et al. Impact of helicobacter pylori-related metabolic syndrome with hyperhomocysteinemia on extragastric pathologies. J Gastroenterol Hepatol. (2022) 37:407–8. doi: 10.1111/jgh.15753
92. Kountouras C, Polyzos SA, Stergiopoulos C, Katsinelos P, Tzivras D, Zavos C, et al. A potential impact of helicobacter pylori infection on both obstructive sleep apnea and atrial fibrillation-related stroke. Sleep Med. (2017) 34:256. doi: 10.1016/j.sleep.2017.03.010
93. Kountouras J, Doulberis M, Papaefthymiou A, Polyzos SA. Impact of helicobacter pylori-linked metabolic syndrome on non-alcoholic fatty liver disease and its connected atrial fibrillation risk. Liver Int. (2020) 40:2036–7. doi: 10.1111/liv.14488
94. Soutto M, Bhat N, Khalafi S, Zhu S, Poveda J, Garcia-Buitrago M, et al. Nf-kb-dependent activation of stat3 by H. pylori is suppressed by tff1. Cancer Cell Int. (2021) 21:444. doi: 10.1186/s12935-021-02140-2
95. He C, Yang Z, Lu NH. Helicobacter pylori infection and diabetes: is it a myth or fact? World J Gastroenterol. (2014) 20:4607–17. doi: 10.3748/wjg.v20.i16.4607
96. Mohammad S, Thiemermann C. Role of metabolic endotoxemia in systemic inflammation and potential interventions. Front Immunol. (2020) 11:594150. doi: 10.3389/fimmu.2020.594150
97. Malfertheiner P, Megraud F, Rokkas T, Gisbert JP, Liou JM, Schulz C, et al. Management of helicobacter pylori infection: the maastricht vi/florence consensus report. Gut. (2022). doi: 10.1136/gutjnl-2022-327745
98. Doulberis M, Kotronis G, Gialamprinou D, Polyzos SA, Papaefthymiou A, Katsinelos P, et al. Alzheimer’s disease and gastrointestinal microbiota; impact of helicobacter pylori infection involvement. Int J Neurosci. (2021) 131:289–301. doi: 10.1080/00207454.2020.1738432
99. Wang J, Zhang H, He J, Xiong X. The role of the gut microbiota in the development of ischemic stroke. Front Immunol. (2022) 13:845243. doi: 10.3389/fimmu.2022.845243
100. Chen R, Xu Y, Wu P, Zhou H, Lasanajak Y, Fang Y, et al. Transplantation of fecal microbiota rich in short chain fatty acids and butyric acid treat cerebral ischemic stroke by regulating gut microbiota. Pharmacol Res. (2019) 148:104403. doi: 10.1016/j.phrs.2019.104403
101. Osawa H, Kita H, Ohnishi H, Nakazato M, Date Y, Bowlus CL, et al. Changes in plasma ghrelin levels, gastric ghrelin production, and body weight after helicobacter pylori cure. J Gastroenterol. (2006) 41:954–61. doi: 10.1007/s00535-006-1880-4
102. Konturek SJ, Bielanski W, Plonka M, Pawlik T, Pepera J, Konturek PC, et al. Helicobacter pylori, non-steroidal anti-inflammatory drugs and smoking in risk pattern of gastroduodenal ulcers. Scand J Gastroenterol. (2003) 38:923–30. doi: 10.1080/00365520310004696
103. Wang W, Zhu LJ, Leng YQ, Wang YW, Shi T, Wang WZ, et al. Inflammatory response: A crucial way for gut microbes to regulate cardiovascular diseases. Nutrients. (2023) 15. doi: 10.3390/nu15030607
104. Sanchez-Rodriguez E, Egea-Zorrilla A, Plaza-Diaz J, Aragon-Vela J, Munoz-Quezada S, Tercedor-Sanchez L, et al. The gut microbiota and its implication in the development of atherosclerosis and related cardiovascular diseases. Nutrients. (2020) 12. doi: 10.3390/nu12030605
105. Hamjane N, Mechita MB, Nourouti NG, Barakat A. Gut microbiota dysbiosis -associated obesity and its involvement in cardiovascular diseases and type 2 diabetes. A systematic review. Microvasc Res. (2024) 151:104601. doi: 10.1016/j.mvr.2023.104601
106. Kazemian N, Mahmoudi M, Halperin F, Wu JC, Pakpour S. Gut microbiota and cardiovascular disease: opportunities and challenges. Microbiome. (2020) 8:36. doi: 10.1186/s40168-020-00821-0
107. Vijay A, Valdes AM. Role of the gut microbiome in chronic diseases: A narrative review. Eur J Clin Nutr. (2022) 76:489–501. doi: 10.1038/s41430-021-00991-6
108. Tang WH, Kitai T, Hazen SL. Gut microbiota in cardiovascular health and disease. Circ Res. (2017) 120:1183–96. doi: 10.1161/CIRCRESAHA.117.309715
109. Frost F, Kacprowski T, Ruhlemann M, Bang C, Franke A, Zimmermann K, et al. Helicobacter pylori infection associates with fecal microbiota composition and diversity. Sci Rep. (2019) 9:20100. doi: 10.1038/s41598-019-56631-4
110. Candelli M, Franza L, Cianci R, Pignataro G, Merra G, Piccioni A, et al. The interplay between helicobacter pylori and gut microbiota in non-gastrointestinal disorders: A special focus on atherosclerosis. Int J Mol Sci. (2023) 24. doi: 10.3390/ijms242417520
111. De Zuani M, Dal Secco C, Frossi B. Mast cells at the crossroads of microbiota and ibd. Eur J Immunol. (2018) 48:1929–37. doi: 10.1002/eji.201847504
112. Traina G. The connection between gut and lung microbiota, mast cells, platelets and sars-cov-2 in the elderly patient. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms232314898
113. Hussain Z, Park H. Inflammation and impaired gut physiology in post-operative ileus: mechanisms and the treatment options. J Neurogastroenterol Motil. (2022) 28:517–30. doi: 10.5056/jnm22100
114. Valencia C, Fatima H, Nwankwo I, Anam M, Maharjan S, Amjad Z, et al. A correlation between the pathogenic processes of fibromyalgia and irritable bowel syndrome in the middle-aged population: A systematic review. Cureus. (2022) 14:e29923. doi: 10.7759/cureus.29923
115. Abraham SN, St John AL. Mast cell-orchestrated immunity to pathogens. Nat Rev Immunol. (2010) 10:440–52. doi: 10.1038/nri2782
116. Galli SJ, Borregaard N, Wynn TA. Phenotypic and functional plasticity of cells of innate immunity: macrophages, mast cells and neutrophils. Nat Immunol. (2011) 12:1035–44. doi: 10.1038/ni.2109
117. Winer DA, Winer S, Shen L, Wadia PP, Yantha J, Paltser G, et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic igg antibodies. Nat Med. (2011) 17:610–7. doi: 10.1038/nm.2353
118. Sperr WR, Bankl HC, Mundigler G, Klappacher G, Grossschmidt K, Agis H, et al. The human cardiac mast cell: localization, isolation, phenotype, and functional characterization. Blood. (1994) 84:3876–84. doi: 10.1182/blood.V84.11.3876.bloodjournal84113876
119. Liu J, Divoux A, Sun J, Zhang J, Clement K, Glickman JN, et al. Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nat Med. (2009) 15:940–5. doi: 10.1038/nm.1994
120. Michailidou Z, Gomez-Salazar M, Alexaki VI. Innate immune cells in the adipose tissue in health and metabolic disease. J Innate Immun. (2022) 14:4–30. doi: 10.1159/000515117
121. Lopez-Perez D, Redruello-Romero A, Garcia-Rubio J, Arana C, Garcia-Escudero LA, Tamayo F, et al. In patients with obesity, the number of adipose tissue mast cells is significantly lower in subjects with type 2 diabetes. Front Immunol. (2021) 12:664576. doi: 10.3389/fimmu.2021.664576
122. Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. (2007) 117:175–84. doi: 10.1172/JCI29881
123. Zatterale F, Longo M, Naderi J, Raciti GA, Desiderio A, Miele C, et al. Chronic adipose tissue inflammation linking obesity to insulin resistance and type 2 diabetes. Front Physiol. (2019) 10:1607. doi: 10.3389/fphys.2019.01607
124. Singh M, Saini HK. Resident cardiac mast cells and ischemia-reperfusion injury. J Cardiovasc Pharmacol Ther. (2003) 8:135–48. doi: 10.1177/107424840300800207
125. Mendoza RP, Fudge DH, Brown JM. Cellular energetics of mast cell development and activation. Cells. (2021) 10. doi: 10.3390/cells10030524
126. Zheng J, Wei CC, Hase N, Shi K, Killingsworth CR, Litovsky SH, et al. Chymase mediates injury and mitochondrial damage in cardiomyocytes during acute ischemia/reperfusion in the dog. PloS One. (2014) 9:e94732. doi: 10.1371/journal.pone.0094732
127. Wernersson S, Pejler G. Mast cell secretory granules: armed for battle. Nat Rev Immunol. (2014) 14:478–94. doi: 10.1038/nri3690
128. Perumalsamy S, Huri HZ, Abdullah BM, Mazlan O, Wan Ahmad WA, Vethakkan S. Genetic markers of insulin resistance and atherosclerosis in type 2 diabetes mellitus patients with coronary artery disease. Metabolites. (2023) 13. doi: 10.3390/metabo13030427
129. Razani B, Chakravarthy MV, Semenkovich CF. Insulin resistance and atherosclerosis. Endocrinol Metab Clin North Am. (2008) 37:603–21. doi: 10.1016/j.ecl.2008.05.001
130. Polyzos SA, Aronis KN, Kountouras J, Raptis DD, Vasiloglou MF, Mantzoros CS. Circulating leptin in non-alcoholic fatty liver disease: A systematic review and meta-analysis. Diabetologia. (2016) 59:30–43. doi: 10.1007/s00125-015-3769-3
131. Polyzos SA, Kountouras J, Zavos C, Tsiaousi E. The role of adiponectin in the pathogenesis and treatment of non-alcoholic fatty liver disease. Diabetes Obes Metab. (2010) 12:365–83. doi: 10.1111/j.1463-1326.2009.01176.x
132. Mechanick JI, Zhao S, Garvey WT. Leptin, an adipokine with central importance in the global obesity problem. Glob Heart. (2018) 13:113–27. doi: 10.1016/j.gheart.2017.10.003
133. Zelechowska P, Brzezinska-Blaszczyk E, Wiktorska M, Rozalska S, Wawrocki S, Kozlowska E, et al. Adipocytokines leptin and adiponectin function as mast cell activity modulators. Immunology. (2019) 158:3–18. doi: 10.1111/imm.13090
134. Tsilimigras DI, Thanopoulou K, Salagianni M, Siasos G, Oikonomou E, Perrea DD, et al. Rosuvastatin attenuates progression of atherosclerosis and reduces serum il6 and ccl2 levels in apolipoprotein-E-deficient mice. In Vivo. (2023) 37:994–1002. doi: 10.21873/invivo.13173
135. Zhang H, Yang K, Chen F, Liu Q, Ni J, Cao W, et al. Role of the ccl2-ccr2 axis in cardiovascular disease: pathogenesis and clinical implications. Front Immunol. (2022) 13:975367. doi: 10.3389/fimmu.2022.975367
136. Shabalala SC, Dludla PV, Mabasa L, Kappo AP, Basson AK, Pheiffer C, et al. The effect of adiponectin in the pathogenesis of non-alcoholic fatty liver disease (Nafld) and the potential role of polyphenols in the modulation of adiponectin signaling. BioMed Pharmacother. (2020) 131:110785. doi: 10.1016/j.biopha.2020.110785
137. Caruso RA, Parisi A, Crisafulli C, Bonanno A, Lucian R, Branca G, et al. Intraepithelial infiltration by mast cells in human helicobacter pylori active gastritis. Ultrastruct Pathol. (2011) 35:251–5. doi: 10.3109/01913123.2011.606964
138. Mohammadi SO, Yadegar A, Kargar M, Mirjalali H, Kafilzadeh F. The impact of helicobacter pylori infection on gut microbiota-endocrine system axis; modulation of metabolic hormone levels and energy homeostasis. J Diabetes Metab Disord. (2020) 19:1855–61. doi: 10.1007/s40200-020-00608-y
139. Nakajima S, Bamba N, Hattori T. Histological aspects and role of mast cells in helicobacter pylori-infected gastritis. Aliment Pharmacol Ther. (2004) 20 Suppl 1:165–70. doi: 10.1111/j.1365-2036.2004.01974.x
140. Bechi P, Dei R, Di Bello MG, Masini E. Helicobacter pylori potentiates histamine release from serosal rat mast cells in vitro. Dig Dis Sci. (1993) 38:944–9. doi: 10.1007/BF01295925
141. Kountouras J, Zavos C, Deretzi G, Gavalas E, Chatzopoulos D, Katsinelos P, et al. Potential implications of helicobacter pylori-related neutrophil-activating protein. World J Gastroenterol. (2012) 18:489–90. doi: 10.3748/wjg.v18.i5.489
142. Fu HW, Lai YC. The role of helicobacter pylori neutrophil-activating protein in the pathogenesis of H. pylori and beyond: from a virulence factor to therapeutic targets and therapeutic agents. Int J Mol Sci. (2022) 24. doi: 10.3390/ijms24010091
143. Kaakoush NO, Holmes J, Octavia S, Man SM, Zhang L, Castano-Rodriguez N, et al. Detection of helicobacteraceae in intestinal biopsies of children with crohn’s disease. Helicobacter. (2010) 15:549–57. doi: 10.1111/j.1523-5378.2010.00792.x
144. Shariati A, Fallah F, Pormohammad A, Taghipour A, Safari H, Chirani AS, et al. The possible role of bacteria, viruses, and parasites in initiation and exacerbation of irritable bowel syndrome. J Cell Physiol. (2019) 234:8550–69. doi: 10.1002/jcp.27828
145. Liu X, Ottemann KM. Methylation-independent chemotaxis systems are the norm for gastric-colonizing helicobacter species. J Bacteriol. (2022) 204:e0023122. doi: 10.1128/jb.00231-22
146. Wang C, Yin Y, Wang L, Guo X, Liu L, Qi X. Association between helicobacter pylori infection and irritable bowel syndrome: A systematic review and meta-analysis. Postgrad Med J. (2021) 99(1169):166–75. doi: 10.1136/postgradmedj-2021-141127
147. Hamad AJ, Alnaji EK, Almohana SJ, Kareem YS, Albdairi AJ. Association between H. pylori infection and irritable bowel syndrome. Wiad Lek. (2023) 76:406–14. doi: 10.36740/WLek202302123
148. Laharie D, Asencio C, Asselineau J, Bulois P, Bourreille A, Moreau J, et al. Association between entero-hepatic helicobacter species and crohn’s disease: A prospective cross-sectional study. Aliment Pharmacol Ther. (2009) 30:283–93. doi: 10.1111/j.1365-2036.2009.04034.x
149. Zhang L, Day A, MCsKenzie G, Mitchell H. Nongastric helicobacter species detected in the intestinal tract of children. J Clin Microbiol. (2006) 44:2276–9. doi: 10.1128/JCM.02017-05
150. Oliveira AG, das Gracas Pimenta Sanna M, Rocha GA, Rocha AM, Santos A, Dani R, et al. Helicobacter species in the intestinal mucosa of patients with ulcerative colitis. J Clin Microbiol. (2004) 42:384–6. doi: 10.1128/JCM.42.1.384-386.2004
151. Abd El-Wahab EW, Youssef EI, Hassouna E. Helicobacter pylori infection in patients with inflammatory bowel diseases: A single-centre, prospective, observational study in Egypt. BMJ Open. (2022) 12:e057214. doi: 10.1136/bmjopen-2021-057214
152. Rijnierse A, Nijkamp FP, Kraneveld AD. Mast cells and nerves tickle in the tummy: implications for inflammatory bowel disease and irritable bowel syndrome. Pharmacol Ther. (2007) 116:207–35. doi: 10.1016/j.pharmthera.2007.06.008
153. Shivji S, Conner JR, Kirsch R. Mast cell evaluation in gastrointestinal biopsies: should we be counting? A critical review and practical guide for the surgical pathologist. Histopathology. (2023) 82:960–73. doi: 10.1111/his.14897
154. Frieling T, Meis K, Kolck UW, Homann J, Hulsdonk A, Haars U, et al. Evidence for mast cell activation in patients with therapy-resistant irritable bowel syndrome. Z Gastroenterol. (2011) 49:191–4. doi: 10.1055/s-0029-1245707
155. Hasler WL, Grabauskas G, Singh P, Owyang C. Mast cell mediation of visceral sensation and permeability in irritable bowel syndrome. Neurogastroenterol Motil. (2022) 34:e14339. doi: 10.1111/nmo.14339
156. Hu WS, Lin CL. Association of irritable bowel syndrome in patients with atrial fibrillation: A nationwide population-based investigation. Perfusion. (2020) 35:847–52. doi: 10.1177/0267659120912363
157. Zaka A, Mridha N, Subhaharan D, Jones M, Niranjan S, Mohsen W, et al. Inflammatory bowel disease patients have an increased risk of acute coronary syndrome: A systematic review and meta-analysis. Open Heart. (2023) 10. doi: 10.1136/openhrt-2023-002483
158. Kountouras J, Polyzos SA, Katsinelos P, Zeglinas C, Artemaki F, Tzivras D, et al. Cardio-cerebrovascular disease and helicobacter pylori-related metabolic syndrome: we consider eradication therapy as a potential cardio-cerebrovascular prevention strategy. Int J Cardiol. (2017) 229:17–8. doi: 10.1016/j.ijcard.2016.11.265
159. Kunder CA, St John AL, Abraham SN. Mast cell modulation of the vascular and lymphatic endothelium. Blood. (2011) 118:5383–93. doi: 10.1182/blood-2011-07-358432
160. Stapleton PA, James ME, Goodwill AG, Frisbee JC. Obesity and vascular dysfunction. Pathophysiology. (2008) 15:79–89. doi: 10.1016/j.pathophys.2008.04.007
161. Theoharides TC, Kempuraj D, Tagen M, Conti P, Kalogeromitros D. Differential release of mast cell mediators and the pathogenesis of inflammation. Immunol Rev. (2007) 217:65–78. doi: 10.1111/j.1600-065X.2007.00519.x
162. Wang J, Shi GP. Mast cell stabilization: novel medication for obesity and diabetes. Diabetes Metab Res Rev. (2011) 27:919–24. doi: 10.1002/dmrr.1272
163. Vijayvergiya R, Vadivelu R. Role of helicobacter pylori infection in pathogenesis of atherosclerosis. World J Cardiol. (2015) 7:134–43. doi: 10.4330/wjc.v7.i3.134
164. Lindsberg PJ, Grau AJ. Inflammation and infections as risk factors for ischemic stroke. Stroke. (2003) 34:2518–32. doi: 10.1161/01.STR.0000089015.51603.CC
165. Kovanen PT. Mast cells as potential accelerators of human atherosclerosis-from early to late lesions. Int J Mol Sci. (2019) 20. doi: 10.3390/ijms20184479
166. Bot I, de Jager SC, Zernecke A, Lindstedt KA, van Berkel TJ, Weber C, et al. Perivascular mast cells promote atherogenesis and induce plaque destabilization in apolipoprotein E-deficient mice. Circulation. (2007) 115:2516–25. doi: 10.1161/CIRCULATIONAHA.106.660472
167. Lehtonen-Smeds EM, Mayranpaa M, Lindsberg PJ, Soinne L, Saimanen E, Jarvinen AA, et al. Carotid plaque mast cells associate with atherogenic serum lipids, high grade carotid stenosis and symptomatic carotid artery disease. Results from the helsinki carotid endarterectomy study. Cerebrovasc Dis. (2005) 19:291–301. doi: 10.1159/000084497
168. Willems S, Vink A, Bot I, Quax PH, de Borst GJ, de Vries JP, et al. Mast cells in human carotid atherosclerotic plaques are associated with intraplaque microvessel density and the occurrence of future cardiovascular events. Eur Heart J. (2013) 34:3699–706. doi: 10.1093/eurheartj/eht186
169. Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MCs, Ferrario F, et al. 2012 revised international chapel hill consensus conference nomenclature of vasculitides. Arthritis Rheum. (2013) 65:1–11. doi: 10.1002/art.37715
170. Aydi Z, Toujani S, Fatma D, Imen R, Baili L, Dhaou BB, et al. Cutaneous vasculitis associated with helicobacter pylori. J Dermatol Dermatol Surg. (2016) 20:132–4. doi: 10.1016/j.jdds.2015.12.001
171. Mohamed M, Shariff M, Al Hillan A, Haj RA, Kaunzinger C, Hossain M, et al. A rare case of helicobacter pylori infection complicated by henoch-schonlein purpura in an adult patient. J Med cases. (2020) 11:160–5. doi: 10.14740/jMCs3480
172. Kalia N, Bardhan KD. Of blood and guts: association between helicobacter pylori and the gastric microcirculation. J Gastroenterol Hepatol. (2003) 18:1010–7. doi: 10.1046/j.1440-1746.2003.03062.x
173. Park PG, Pyo JY, Ahn SS, Song JJ, Park YB, Huh JH, et al. Effect of numbers of metabolic syndrome components on mortality in patients with antineutrophil cytoplasmic antibody-associated vasculitis with metabolic syndrome. Clin Exp Rheumatol. (2022) 40:758–64. doi: 10.55563/clinexprheumatol/k4m3it
174. Theoharides TC, Singh LK, Boucher W, Pang X, Letourneau R, Webster E, et al. Corticotropin-releasing hormone induces skin mast cell degranulation and increased vascular permeability, a possible explanation for its proinflammatory effects. Endocrinology. (1998) 139:403–13. doi: 10.1210/endo.139.1.5660
175. Hirose H, Kugimiya T. Abdominal aortic aneurysm infected with helicobacter pylori–a case report. Angiology. (2000) 51:867–71. doi: 10.1177/000331970005101010
176. Ziver T, Yuksel P, Ipek G, Yekeler I, Bayramoglu Z, Tireli E, et al. Aneurysm and helicobacter pylori relationship: the seropositivity of caga, vaca and other antigens of helicobacter pylori in abdominal and ascending aortic aneurysms. New Microbiol. (2010) 33:233–42.
177. Golledge J, Norman PE, Murphy MP, Dalman RL. Challenges and opportunities in limiting abdominal aortic aneurysm growth. J Vasc Surg. (2017) 65:225–33. doi: 10.1016/j.jvs.2016.08.003
178. Huang AL, Bosco JJ, Peter K. Mast cell: an unexpected villain in venous thromboembolism? Circ Res. (2017) 121:899–901. doi: 10.1161/CIRCRESAHA.117.311777
179. Frischmuth T, Hindberg K, Aukrust P, Ueland T, Braekkan SK, Hansen JB, et al. Elevated plasma levels of plasminogen activator inhibitor-1 are associated with risk of future incident venous thromboembolism. J Thromb Haemost. (2022) 20:1618–26. doi: 10.1111/jth.15701
180. Kaartinen M, Penttila A, Kovanen PT. Accumulation of activated mast cells in the shoulder region of human coronary atheroma, the predilection site of atheromatous rupture. Circulation. (1994) 90:1669–78. doi: 10.1161/01.cir.90.4.1669
181. Laine P, Kaartinen M, Penttila A, Panula P, Paavonen T, Kovanen PT. Association between myocardial infarction and the mast cells in the adventitia of the infarct-related coronary artery. Circulation. (1999) 99:361–9. doi: 10.1161/01.cir.99.3.361
182. Soleimani M, Barone S, Luo H, Zahedi K. Pathogenesis of hypertension in metabolic syndrome: the role of fructose and salt. Int J Mol Sci. (2023) 24. doi: 10.3390/ijms24054294
183. Shepherd RK, Linden J, Duling BR. Adenosine-induced vasoconstriction in vivo. Role of the mast cell and A3 adenosine receptor. Circ Res. (1996) 78:627–34. doi: 10.1161/01.res.78.4.627
184. MCsLarty JL, Melendez GC, Brower GL, Janicki JS, Levick SP. Tryptase/protease-activated receptor 2 interactions induce selective mitogen-activated protein kinase signaling and collagen synthesis by cardiac fibroblasts. Hypertension. (2011) 58:264–70. doi: 10.1161/HYPERTENSIONAHA.111.169417
185. Kotov G, Landzhov B, Stamenov N, Stanchev S, Iliev A. Changes in the number of mast cells, expression of fibroblast growth factor-2 and extent of interstitial fibrosis in established and advanced hypertensive heart disease. Ann Anat. (2020) 232:151564. doi: 10.1016/j.aanat.2020.151564
186. Lamb FS, Choi H, Miller MR, Stark RJ. Tnfalpha and reactive oxygen signaling in vascular smooth muscle cells in hypertension and atherosclerosis. Am J Hypertens. (2020) 33:902–13. doi: 10.1093/ajh/hpaa089
187. Legere SA, Haidl ID, Legare JF, Marshall JS. Mast cells in cardiac fibrosis: new insights suggest opportunities for intervention. Front Immunol. (2019) 10:580. doi: 10.3389/fimmu.2019.00580
188. Chen L, Zhu H, Su S, Harshfield G, Sullivan J, Webb C, et al. High-mobility group box-1 is associated with obesity, inflammation, and subclinical cardiovascular risk among young adults: A longitudinal cohort study. Arterioscler Thromb Vasc Biol. (2020) 40:2776–84. doi: 10.1161/ATVBAHA.120.314599
189. Pokharel Y, Karmacharya BM, Neupane D. Hypertension-a silent killer without global bounds: what next? J Am Coll Cardiol. (2022) 80:818–20. doi: 10.1016/j.jacc.2022.05.043
190. Saheera S, Potnuri AG, Guha A, Palaniyandi SS, Thandavarayan RA. Histamine 2 receptors in cardiovascular biology: A friend for the heart. Drug Discovery Today. (2022) 27:234–45. doi: 10.1016/j.drudis.2021.08.008
191. Bergmann K, Sypniewska G. Is there an association of allergy and cardiovascular disease? Biochem Med (Zagreb). (2011) 21:210–8. doi: 10.11613/bm.2011.030
192. Luitel H, Sydykov A, Schymura Y, Mamazhakypov A, Janssen W, Pradhan K, et al. Pressure overload leads to an increased accumulation and activity of mast cells in the right ventricle. Physiol Rep. (2017) 5. doi: 10.14814/phy2.13146
193. Guo T, Chen WQ, Zhang C, Zhao YX, Zhang Y. Chymase activity is closely related with plaque vulnerability in a hamster model of atherosclerosis. Atherosclerosis. (2009) 207:59–67. doi: 10.1016/j.atherosclerosis.2009.04.014
194. Morici N, Farioli L, Losappio LM, Colombo G, Nichelatti M, Preziosi D, et al. Mast cells and acute coronary syndromes: relationship between serum tryptase, clinical outcome and severity of coronary artery disease. Open Heart. (2016) 3:e000472. doi: 10.1136/openhrt-2016-000472
195. Triggiani M, Patella V, Staiano RI, Granata F, Marone G. Allergy and the cardiovascular system. Clin Exp Immunol. (2008) 153 Suppl 1:7–11. doi: 10.1111/j.1365-2249.2008.03714.x
196. Janicki JS, Brower GL, Levick SP. The emerging prominence of the cardiac mast cell as a potent mediator of adverse myocardial remodeling. Methods Mol Biol. (2015) 1220:121–39. doi: 10.1007/978-1-4939-1568-2_8
197. Rupp S, Papaefthymiou A, Chatzimichael E, Polyzos SA, Spreitzer S, Doulberis M, et al. Diagnostic approach to helicobacter pylori-related gastric oncogenesis. Ann Gastroenterol. (2022) 35:333–44. doi: 10.20524/aog.2022.0725
198. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. (1993) 259:87–91. doi: 10.1126/science.7678183
199. Zhang Y, Shi Y, Lin J, Li X, Yang B, Zhou J. Immune cell infiltration analysis demonstrates excessive mast cell activation in psoriasis. Front Immunol. (2021) 12:773280. doi: 10.3389/fimmu.2021.773280
200. Davis DMR, Drucker AM, Alikhan A, Bercovitch L, Cohen DE, Darr JM, et al. American academy of dermatology guidelines: awareness of comorbidities associated with atopic dermatitis in adults. J Am Acad Dermatol. (2022) 86:1335–6 e18. doi: 10.1016/j.jaad.2022.01.009
201. Parente R, Giudice V, Cardamone C, Serio B, Selleri C, Triggiani M. Secretory and membrane-associated biomarkers of mast cell activation and proliferation. Int J Mol Sci. (2023) 24. doi: 10.3390/ijms24087071
202. Kountouras J, Doulberis M, Polyzos SA, Tzitiridou M, Chatzopoulos D, Gialamprinou D, et al. A potential impact of helicobacter pylori-related metabolic syndrome on early and long-term outcomes of bariatric surgery. Gastrointest Endosc. (2022) 96:1088–9. doi: 10.1016/j.gie.2022.07.003
203. Polyzos SA, Papaefthymiou A, Doulberis M, Mavridoglou G, Kountouras J. Helicobacter pylori infection and diabetes mellitus. Diabetes Metab Syndr. (2021) 15:845–6. doi: 10.1016/j.dsx.2021.03.034
204. Kountouras J, Polyzos SA, Katsinelos P, Zeglinas C, Artemaki F, Tzivras D, et al. Helicobacter pylori eradication to prevent cardio-cerebrovascular disease: are current data useful for clinical practice? Int J Cardiol. (2017) 233:92. doi: 10.1016/j.ijcard.2016.12.152
205. Moayyedi. Review: eradication therapy supplemented by probiotics increased eradication rates and reduced side effects in H. pylori infection. Evid Based Med. (2007) 12.
206. Liang B, Yuan Y, Peng XJ, Liu XL, Hu XK, Xing DM. Current and future perspectives for helicobacter pylori treatment and management: from antibiotics to probiotics. Front Cell Infect Microbiol. (2022) 12:1042070. doi: 10.3389/fcimb.2022.1042070
207. Wang Y, Wang X, Cao XY, Zhu HL, Miao L. Comparative effectiveness of different probiotics supplements for triple helicobacter pylori eradication: A network meta-analysis. Front Cell Infect Microbiol. (2023) 13:1120789. doi: 10.3389/fcimb.2023.1120789
208. Yao G, Fan X, Lu D. Efficacy and safety of probiotic-supplemented bismuth quadruple therapy for the treatment of helicobacter pylori infection: A systematic review and meta-analysis. J Int Med Res. (2023) 51:3000605231203841. doi: 10.1177/03000605231203841
209. Suez J, Zmora N, Segal E, Elinav E. The pros, cons, and many unknowns of probiotics. Nat Med. (2019) 25:716–29. doi: 10.1038/s41591-019-0439-x
210. Depommier C, Everard A, Druart C, Plovier H, Van Hul M, Vieira-Silva S, et al. Supplementation with akkermansia muciniphila in overweight and obese human volunteers: A proof-of-concept exploratory study. Nat Med. (2019) 25:1096–103. doi: 10.1038/s41591-019-0495-2
211. Buzas GM. Helicobacter pylori and non-alcoholic fatty liver disease. Minerva Gastroenterol Dietol. (2020) 66:267–79. doi: 10.23736/S1121-421X.20.02671-9
212. Franceschi F, Ojetti V, Candelli M, Covino M, Cardone S, Potenza A, et al. Microbes and alzheimer’ Disease: lessons from H. pylori and gut microbiota. Eur Rev Med Pharmacol Sci. (2019) 23:426–30. doi: 10.26355/eurrev_201901_16791
213. Costanzo G, Costanzo G, Del Moro L, Nappi E, Pelaia C, Puggioni F, et al. Mast cells in upper and lower airway diseases: sentinels in the front line. Int J Mol Sci. (2023) 24. doi: 10.3390/ijms24119771
214. Metz M, Kolkhir P, Altrichter S, Siebenhaar F, Levi-Schaffer F, Youngblood BA, et al. Mast cell silencing: A novel therapeutic approach for urticaria and other mast cell-mediated diseases. Allergy. (2024) 79(1):37–5. doi: 10.1111/all.15850
215. Kwon OY, Choi JY, Jang Y. The effectiveness of ehealth interventions on lifestyle modification in patients with nonalcoholic fatty liver disease: systematic review and meta-analysis. J Med Internet Res. (2023) 25:e37487. doi: 10.2196/37487
216. Guo Y, Li ZX, Zhang JY, Ma JL, Zhang L, Zhang Y, et al. Association between lifestyle factors, vitamin and garlic supplementation, and gastric cancer outcomes: A secondary analysis of a randomized clinical trial. JAMA Netw Open. (2020) 3:e206628. doi: 10.1001/jamanetworkopen.2020.6628
217. Kountouras J, Doulberis M, Papaefthymiou A, Polyzos SA, Vardaka E, Tzivras D, et al. A perspective on risk factors for esophageal adenocarcinoma: emphasis on helicobacter pylori infection. Ann N Y Acad Sci. (2019) 1452:12–7. doi: 10.1111/nyas.14168
218. Kountouras J, Polyzos SA, Doulberis M, Zeglinas C, Artemaki F, Vardaka E, et al. Potential impact of helicobacter pylori-related metabolic syndrome on upper and lower gastrointestinal tract oncogenesis. Metabolism. (2018) 87:18–24. doi: 10.1016/j.metabol.2018.06.008
219. Kountouras J, Doulberis M, Polyzos SA, Papaefthymiou A, Kapetanakis N, Arapoglou S, et al. Potential impact of active helicobacter pylori infection with or without concomitant metabolic syndrome on colorectal cancer invasion and mortality. Isr Med Assoc J. (2018) 20:725–6.
220. Kountouras J, Zavos C, Diamantidis MD, Deretzi G, Grigoriadis N, Tsapournas G, et al. A concept of helicobacterpylori and stress-secreted mast cells’ Potential involvement in brain metastases. J Neuroimmunol. (2009) 209:121–2. doi: 10.1016/j.jneuroim.2009.01.019
221. Huang L, Hong Y, Fu X, Tan H, Chen Y, Wang Y, et al. The role of the microbiota in glaucoma. Mol Aspects Med. (2023) 94:101221. doi: 10.1016/j.mam.2023.101221
Keywords: Helicobacter pylori, mast cells, vascular, metabolic syndrome, atherosclerosis
Citation: Doulberis M, Papaefthymiou A, Polyzos SA, Boziki M, Kazakos E, Tzitiridou-Chatzopoulou M, Vardaka E, Hammrich C, Kulaksiz H, Riva D, Kiosses C, Linas I, Touloumtzi M, Stogianni A and Kountouras J (2024) Impact of Helicobacter pylori and metabolic syndrome-related mast cell activation on cardiovascular diseases. Front. Gastroenterol. 3:1331330. doi: 10.3389/fgstr.2024.1331330
Received: 31 October 2023; Accepted: 31 January 2024;
Published: 04 March 2024.
Edited by:
Sahil Khanna, Mayo Clinic, United StatesReviewed by:
Yeonhee Cho, Amgen, United StatesPadhmanand Sudhakar, Kumaraguru College of Technology, India
Copyright © 2024 Doulberis, Papaefthymiou, Polyzos, Boziki, Kazakos, Tzitiridou-Chatzopoulou, Vardaka, Hammrich, Kulaksiz, Riva, Kiosses, Linas, Touloumtzi, Stogianni and Kountouras. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jannis Kountouras, jannis@auth.gr