 Ravikanth Vishnubhotla
Ravikanth Vishnubhotla Anand V. Kulkarni
Anand V. Kulkarni Mithun Sharma2
Mithun Sharma2 Duvvur Nageshwar Reddy
Duvvur Nageshwar Reddy
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Gastroenterol. , 30 November 2022
Sec. Hepatology
Volume 1 - 2022 | https://doi.org/10.3389/fgstr.2022.1030399
Worldwide, an estimated 2 billion individuals consume alcohol, which contributes to short-term or long-term consequences on health and social life. Alcohol is the cause of approximately 1.8 million deaths per year, representing 3.2% of all deaths worldwide. Of the 2 billion individuals who consume alcohol, more than 75 million are diagnosed with alcohol-use disorder (AUD) and are at an enhanced risk of developing alcoholic liver disease (ALD). However, not all individuals who consume alcohol develop liver disease suggesting the intricate interactions of host genetics with the environment in the precipitation of the phenotype. With advances in genomic technologies, it is now possible to sequence clinically relevant genomic loci associated with a phenotype with precision and faster turnaround times. Genomic data in the form of variants may be used to predict susceptibility to a phenotype in an unaffected individual or may assist the clinician in predicting the outcomes after the onset of the disease. Both of these are crucial as the former would aid in reducing the future burden of the disease, and the latter would help identify and treat individuals at risk of severe liver disease. In the current review, we summarize the pathogenic mechanisms of ALD and discuss the variants identified to date that may aid in predicting alcohol dependence and the development of cirrhosis in individuals with AUD.
Alcohol consumption historically has been practiced by many as a social engagement and for societal bonding. Though moderate alcohol consumption is associated with cardiovascular benefits, the term “moderate” may not be uniform to each individual (1). Furthermore, consumption of alcohol in excess leads to unpleasant and avoidable adverse outcomes, including an impact on social, occupational, interpersonal, and health. On average, the global consumption of alcohol was reported to be 6.18 liters per person (2). While alcohol use statistics in individuals from the United States of America suggest that 85.6% of people aged 18 or more reportedly drank alcohol at some point in their lifetime, 25.8% and 6.3% reported binge drinking and heavy alcohol use, respectively (3). According to the National Survey of Drug Use and Health (NSDUH) carried out in the year 2019, approximately 14.5 million people aged ≥12 years (9.0 million men and 5.5 million women) were reported to have alcohol use disorder (AUD) (3). The numbers of individuals with AUD are alarmingly high, even among the young aged between 12 and 17 years, where an estimated 4,14,000 (1,63,000 males and 2,51,000 females) are affected (3). However, only 7.2% of the people aged ≥12 years and 6.4% of adolescents who had AUD received treatment.
In 2016, 5.3% of all global deaths translating to 3 million, were attributed to the consumption of alcohol, and alcohol misuse was the seventh leading risk factor for premature death and disability (4, 5). While the World Health Organization (WHO) suggested that alcohol consumption contributed to >200 diseases that ranged from liver diseases, cardiovascular disease, cancer, tuberculosis, and acquired immunodeficiency syndrome (AIDS), the related deaths were 21.3% due to digestive diseases (primarily cirrhosis of the liver and pancreatitis), 19% due to cardiovascular diseases, 12.6% due to cancer and 12.9% due to infectious diseases (6). This puts the economic burden at 249 billion dollars in the United States due to alcohol misuse (7).
Alcohol is the leading cause of liver diseases worldwide (8). However, the number of people affected with liver diseases is disproportionately low compared to a higher number consuming alcohol points to the role of pathomechanics specific to a subset of individuals. Over the years, research points to the overwhelming role of the interactions between host genetics and environment, apart from other triggers for this progression. It is very pertinent to identify the role of genetics in the form of variants and their associated genes not only to understand the pathogenic mechanisms involved in the development of alcoholic liver disease (ALD) at the molecular level but also to explore the putative drug targets for developing advanced therapeutics. This review summarizes the genetics of ALD and variants associated with alcohol dependence.
Ethanol, a simple alcohol with two carbon atoms, is a small molecule known to diffuse quickly across cell membranes. The absorption of the molecule is influenced by many factors, including age, gender, metabolism, and body weight. Biotransformation of ethanol is predominantly carried out by its oxidation into acetaldehyde, which is the main pathway. The process utilizes nicotinamide adenine dinucleotide (NAD+), and is achieved primarily by alcohol dehydrogenase (ADH), which is mainly expressed by the cells of the liver and gastrointestinal tract. Other processes, including catalase and microsomal ethanol oxidation system based on CYP2E1, are also involved in ethanol metabolism. Irrespective of the oxidation pathway, ethanol is converted to acetate, a process catalyzed by aldehyde dehydrogenase (ALDH, expressed in cytoplasm and mitochondria). This oxidation and subsequent conversion of ethanol to acetate increase the amount of NADH in the liver. With the enzymes involved in this conversion process predominantly being expressed in the liver, most of the harmful effects of ethanol are, therefore, directly noted in the hepatocytes.
Accumulation of reactive oxygen species (ROS), predominantly hydrogen peroxide (H2O2) and superoxide anion (O2-) concomitant with hypoxia, translocation of bacterial species, and proinflammatory cytokine release is demonstrated to be a direct result of ethanol metabolism. The ROS bind to ethanol/iron atoms to form reactive metabolites (Ferrous oxide: FeO, hydroxyethyl radical: CH3CHOH) and are involved in lipid peroxidation of cell membranes. During the pathogenesis of alcohol-induced liver injury, iron is also involved in oxidative stress and is known to promote fibrosis by mediating the catalyzing ROS formation (9). The double-edged cytotoxic effects comprising ethanol metabolism and ROS formation reportedly contribute to apoptosis and necrosis, eventually leading to cell death. Continuous exposure to ethanol is also known to induce glutathione depletion, making the hepatocyte cells more sensitive to oxidative stress (10).
Maturation of proteins, including the folding process, usually occurs in the endoplasmic reticulum (ER). Incomplete maturation of the proteins in response to various stimuli leads to the accumulation of unfolded/misfolded proteins in the ER. This triggers multiple pathways leading to ER stress, which activates NF-kB and JNK, thereby initiating and promoting inflammation. In addition, disruption of calcium homeostasis and activation of pathways, including CHOP-GADD153, triggers apoptosis. Upregulation of aldose reductase (AR) concomitant with an elevation of its metabolites (uric acid, sorbitol, and fructose) in liver specimens was demonstrated in patients with alcohol-associated hepatitis. These events correlated with increased lipid peroxidation byproducts, ER stress, decreased protective ER chaperones, and greater cell death and liver injury (11).
Gender differences in the progression of ALD with females being more susceptible than males in the background of similar levels of consumption of alcohol, interethnic differences, and higher concordance for cirrhosis in monozygotic twins as compared to dizygotic twins, all point to the role of genetic modifiers for the phenotype. Studies have provided unequivocal evidence in favor of genetic predisposition to organ-specific complications of alcohol consumption. Twin studies in ALD cohorts identified a nearly three-fold higher prevalence in monozygotic compared to dizygotic twins, again pointing to the genetic predisposition to the phenotype (12). Most of the genetic susceptibility identified thus far is predominantly in genes concerning alcohol metabolism and/or those influencing the risk of hepatic damage in individuals with continued alcohol consumption.
The volume, the concentration of ethanol ingested, and the duration of exposure determine the extent of damage to the organs. Metabolism of ethanol involves ADH, ALDH, and CYP2E1 pathways. Multiple ADH and ALDH enzymes are reportedly identified and are encoded by different genes. The details of these genes, including the location, size, number of exons, and the total variants identified thus far, are summarized (Table 1). Depending on the type of variants in these genes, the ability to metabolize ethanol varies in individuals and is known to influence an individual’s drinking habits and subsequent risk of alcohol dependence (13). Conversely, certain variants in these genes predispose individuals to a significantly reduced risk of alcohol dependence. In addition, the prevalence of these variants differs among ethnicities confirming the higher risk of ALD associated with particular ethnicities.
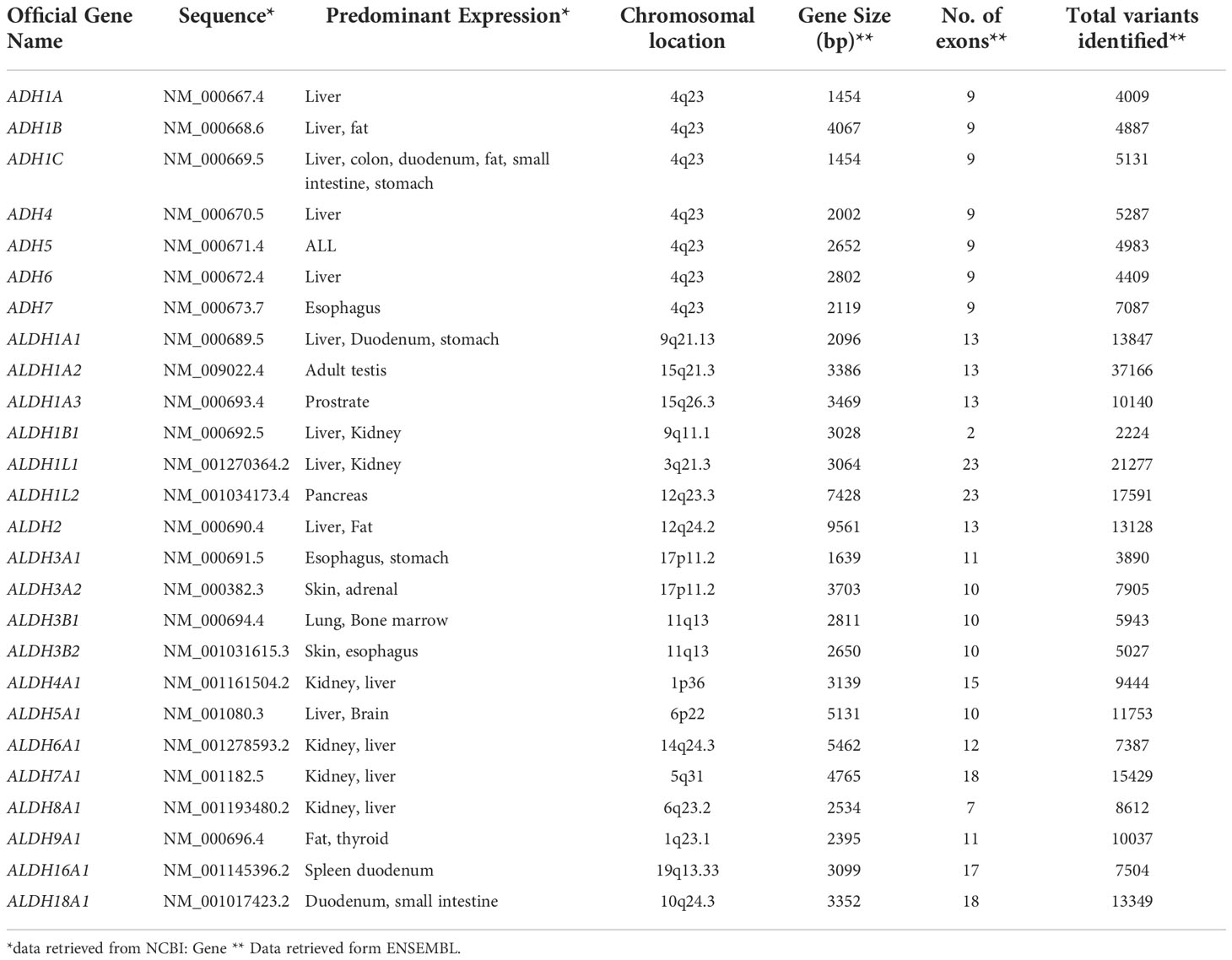
Table 1 Genes concerning Alcohol Dehydrogenase (ADH) and Aldehyde Dehydrogenase (ALDH).
Variants in ADH1B and ADH1C genes are known to lead to the production of enzymes with altered kinetic properties. Three different ADH1B alleles have altered amino acids in the sequence that encodes the β-subunit. The amino acid substitutions in both β2 and β3 subunits are known to occur at a position that makes contact with the (NAD+) coenzyme that is essential for the oxidation of ethanol. The variants and their associated effect on the metabolism of alcohol are summarized in Table 2.
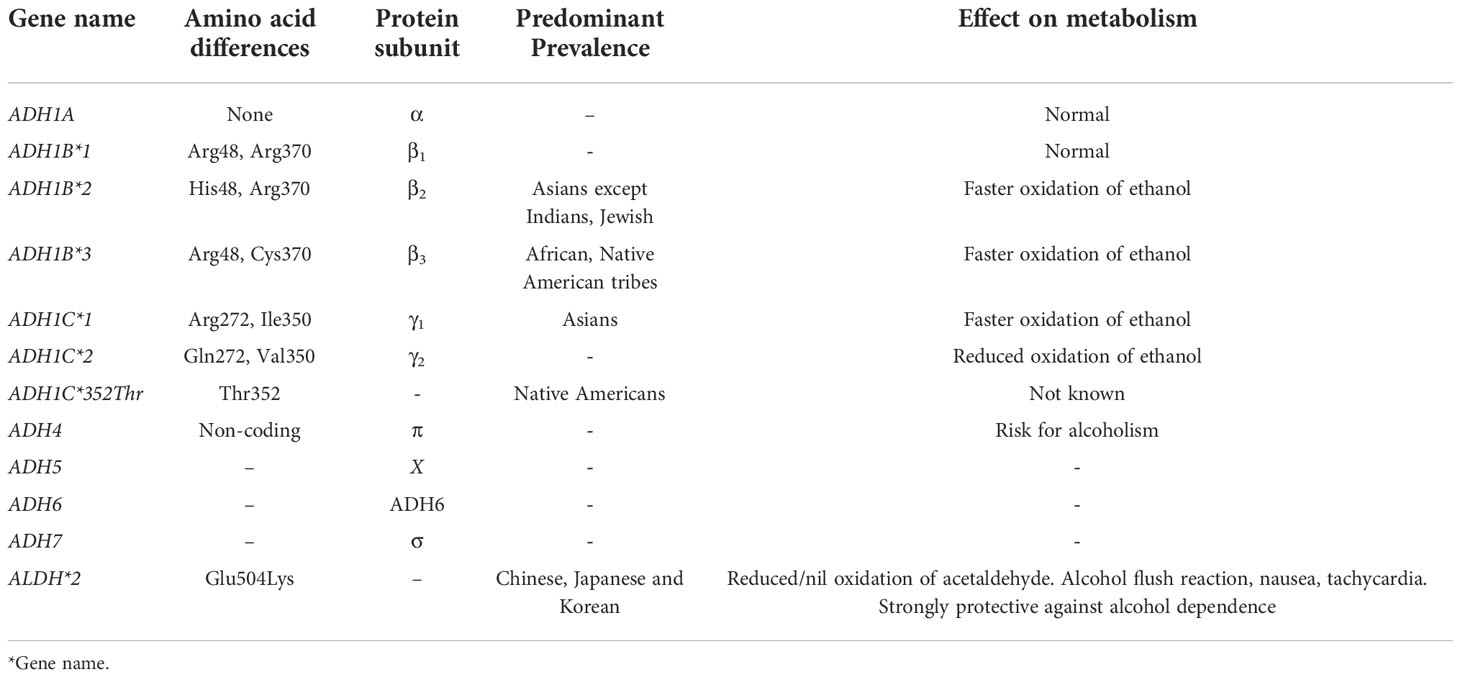
Table 2 Summary of variants in Alcohol Dehydrogenase (ADH) and Aldehyde dehydrogenase (ALDH) genes and their effect.
Two predominant enzymes, ALDH1 encoded by ALDH1A1 and ALDH2 encoded by ALDH2, are known to metabolize acetaldehyde produced during ethanol oxidation. The proteins encoded by these two genes are identical to a more significant extent (70%), including the sequence and structure of genes. The predominant variant in ALDH2 gene that is extensively worked out is ALDH*2 (Table 2). The substitution (Glu504Lys) results in the production of an inactive enzyme; therefore, individuals with this variant cannot oxidize acetaldehyde to acetate. Due to the dominant inheritance, the enzyme may be undetectable even in heterozygous individuals. Individuals with ALDH*2 allele show an alcohol flush reaction even with smaller amounts of alcohol consumption that triggers highly aversive reactions, including severe flushing, tachycardia, and nausea. Studies have reported strong protection against alcohol dependence even with a single copy of the ALDH*2 allele. This beneficial effect is because of the severe adverse effects demonstrated with even small amounts of alcohol consumption. However, environmental interactions are known to modulate the protective effects of this allele, and this was demonstrated by a study where the investigators found an increase of this allele (2.5% to 13% between 1979 and 1992) in Japanese individuals who consumed alcohol (14).
A meta-analysis of 50 association studies of variants reported in genes namely ADH2, ADH3, CYP2E1, and ADLH2 reported significant associations of ADH2*1, ADH3*2, and ALDH2*1 alleles with the risk of alcoholism (15). In addition, subgroup analyses showed an association for ADH2*1 and ADH3*2 only in East Asians and ADH2*1 in Caucasians. Further, the study revealed no significant associations for ADH2*1, ADH3*2, or ALDH2*1 in all subpopulations and no association for the CYP2E1 variant whatsoever. All the alleles were inherited in a dominant mode (15).
A study that genotyped for variants in ADH2, ADH3, CYP4502E1 (Pst-I and Dra-I), and ALDH2 in Spaniard alcoholic subjects (without liver disease, with non-cirrhotic liver disease, and with cirrhosis) and compared with non-alcoholic subjects (healthy controls with non-cirrhotic non-alcoholic liver disease and with cirrhosis unrelated to alcohol) reported that variants in the studied genes were not related to alcoholism or susceptibility to alcoholic liver disease in addition to ALDH2 locus being monomorphic (16).
Variants known to predict alcohol sensitivity are currently being investigated by firms that offer variant-based testing directly to consumers under nutrigenetics testing. These firms predominantly test under food sensitivity and include variants that predict alcohol dependence, alcohol consumption, and alcohol use disorder, generating problematic use scores (17). A list of variants that are currently being used is depicted in Figure 1.
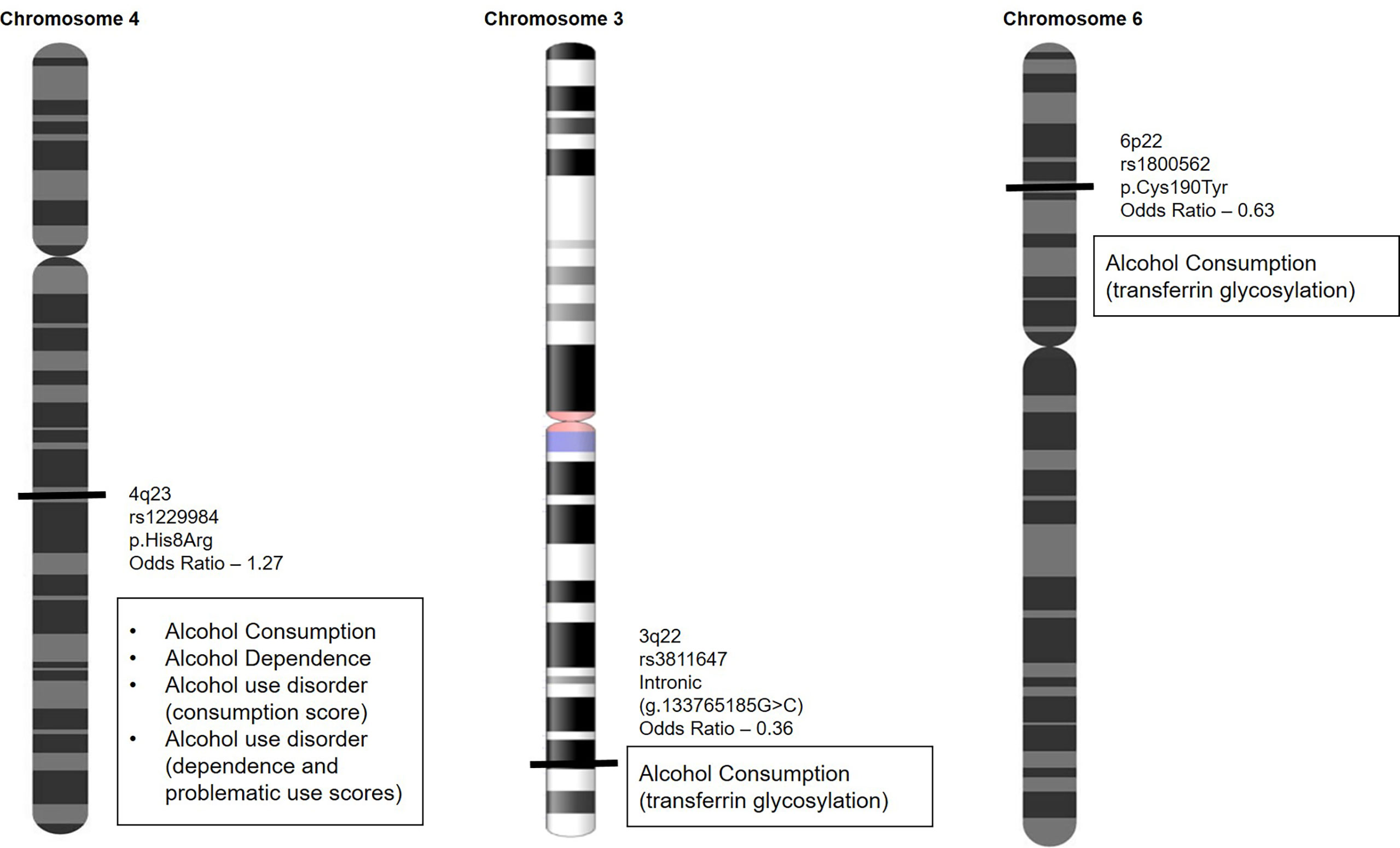
Figure 1 Variants that predict alcohol dependence, alcohol consumption, and alcohol use disorder are used to generate problematic use scores to identify individuals at higher risk of alcohol abuse.
Certain loci confer a higher risk of ALD (18). Most prominent among them are variants in genes of methylenetetrahydrofolate reductase (MTHFR), interleukin 1β (IL1B), NFE2L2, RASGRF2, CYP2E1, DRD2, TM6SF2, MBOAT7, and PNPLA3 that were reportedly associated with the phenotype. The summary of information on the genes (Table 3) and their mechanism (Figure 2) is given. A detailed description of the role of these genes in ALD is discussed below.
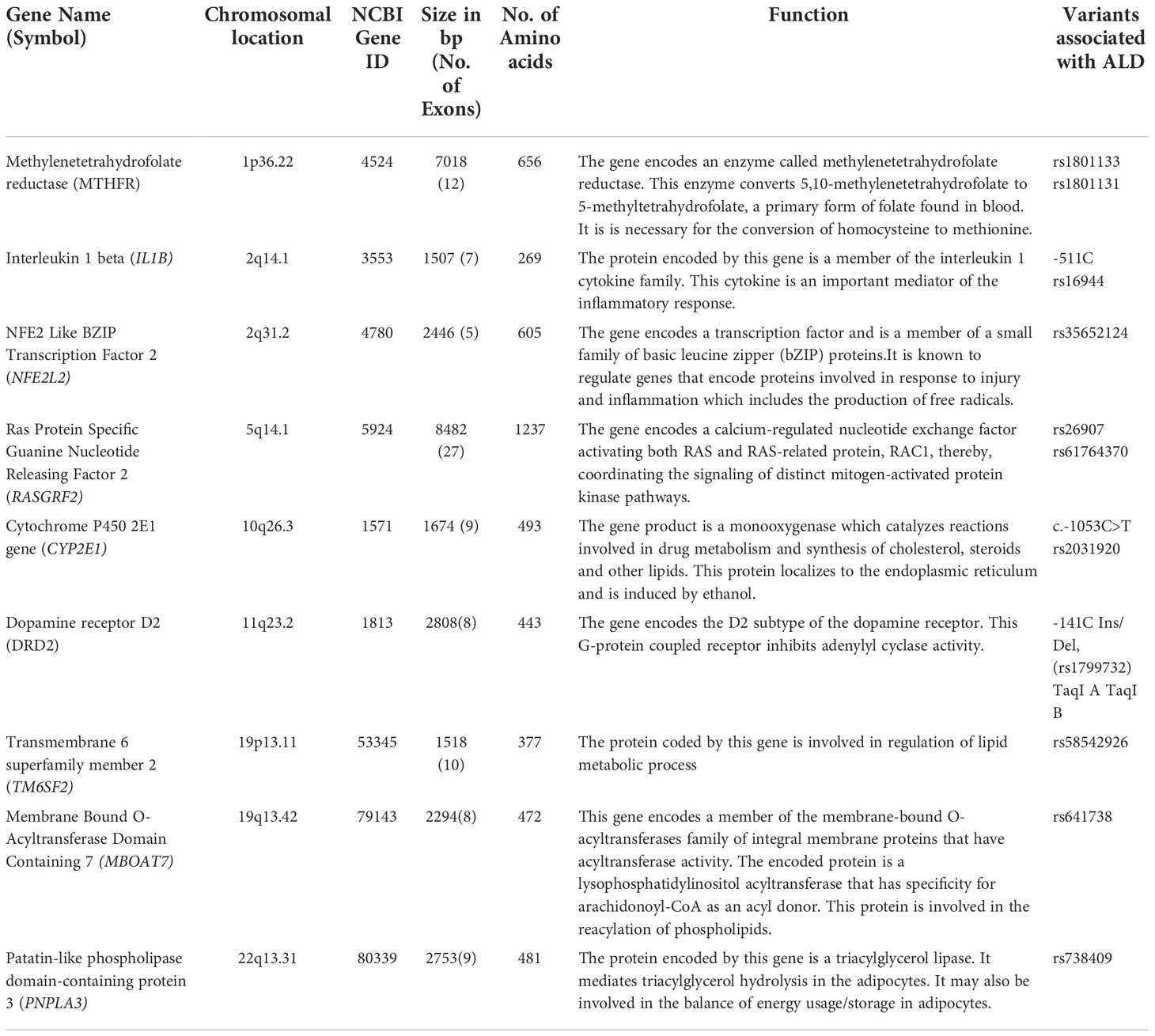
Table 3 Summary of information on genes associated with Alcoholic Liver Disease (ALD).
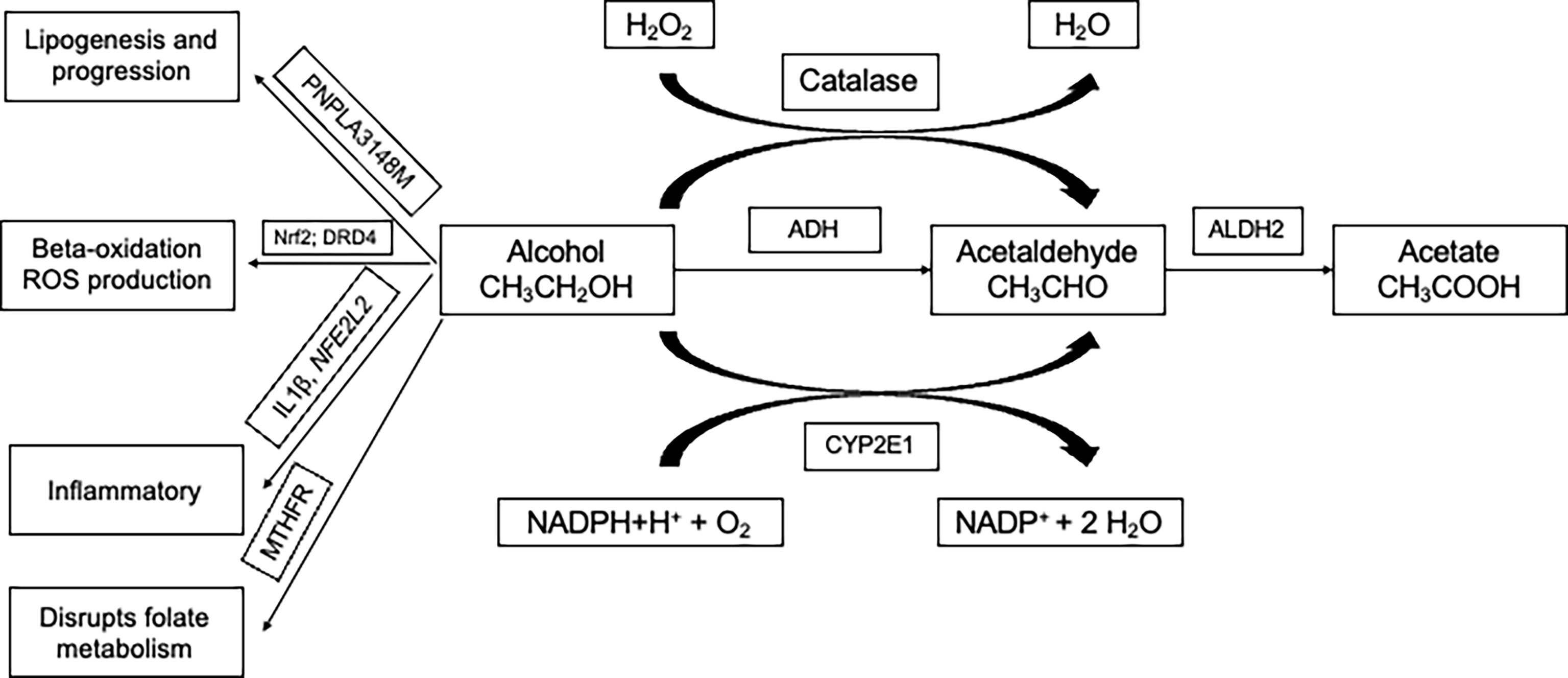
Figure 2 Three main pathways involved in the metabolism of alcohol and involves genes namely ADH, ALDH, and CYP2E1. Multiple ADH and ALDH enzymes are reportedly identified and are encoded by different genes. PNPLA3-148M variant accelerates lipogenesis and progression. Alcohol abuse in the presence of variants in Nrf2 and DRD4 are involved in beta-oxidation and production of ROS (reactive oxygen species). Variants in inflammatory genes are known to modulate alcohol-induced liver injury. Alcohol is known to disrupt the folate cycle involved in the metabolism of methionine, which is crucial in providing methyl groups required for the methylation of DNA. ADH, Alcohol dehydrogenase; CYP2E1, cytochrome P450 2E1 subfamily; ALDH, Aldehyde dehydrogenase; PNPLA3, Patatin-like phospholipase domain-containing protein 3; IL1B - Interleukin 1 beta; NFE2L2 - NFE2 Like BZIP Transcription Factor 2; DRD2, Dopamine receptor D2; MTHFR, Methylenetetrahydrofolate reductase; NADPH, Nicotinamide adenine dinucleotide phosphate; H2O2, Hydrogen peroxide; H2O, Water; O2, Oxygen.
The protein coded by the gene TM6SF2 is localized to the ER membrane and intermediate compartment membrane and is involved in regulating the lipid metabolic process. A variant in this gene (p.Glu167Lys; rs58542926) was initially identified by an exome-wide screening of variants for non-alcoholic fatty liver disease (NAFLD) (19). Subsequent to the identification, the variant was demonstrated to regulate liver fat metabolism by influencing triglyceride secretion and hepatic lipid droplet content (20). In addition, it has been shown to induce lipid biosynthesis and reduce the secretion of Apolipoprotein B (21). A genome-wide association study for alcohol-related cirrhosis in individuals of European descent identified this variant in TM6SF2 apart from MBOAT7 as new risk loci (22). A similar alcohol-related cirrhosis risk for the variant was reported in the British and Irish populations (23). However, few studies could not replicate the association. A study that explored the association of this variant in TM6SF2 gene apart from other variants with the risk of hepatic fibrosis or liver cirrhosis of different etiologies suggested that the variant was not associated with hepatic fibrosis, liver cirrhosis induced by alcohol in an Eastern European population (24).
This gene encodes a lysophosphatidylinositol acyltransferase protein and has an affinity for arachidonoyl-CoA as an acyl donor. It is reportedly involved in the reacylation of phospholipids as part of the phospholipid remodeling pathway, also known as the Land cycle. A genome-wide association study that followed up with fine mapping identified a variant (rs641738) in MBOAT7 gene to be associated with alcohol-related cirrhosis (22). Later, the variant was also reported to be associated with an increased risk of NAFLD in individuals of European descent. However, other studies have reported a negative association of the variant (rs641738) in MBOAT7 with hepatic fibrosis, alcohol, or hepatitis C-related liver cirrhosis in an Eastern European population (24, 25). Although there is unequivocal evidence for the association of the variant with liver fat, ALT, and fibrosis in NAFLD, a similar association with ALD is lacking (26). Further studies involving a larger multi-ethnic sample are warranted before arriving at a definite conclusion.
The protein encoded by the gene PNPLA3 is a lipid droplet-associated protein that is demonstrated to have triglyceride and retinyl esters hydrolase activity. In humans, this gene is highly expressed in the liver and moderately in the brain, kidney, adipose, and skin (27, 28). A genome-wide association study (GWAS) in a multiethnic population comprising Hispanic, African American, and European American individuals identified a variant in this gene associated with hepatic fat concentration for the first time. The variant (rs738409; C>G) is a cytosine to guanine change that causes methionine to isoleucine conversion at the 148-amino acid position (I148M) (29). Following this discovery, multiple studies have associated this variant with the risk of ALD and elevated liver enzymes (30, 31). A cross-sectional study on the US population explored the interaction between alcohol consumption and PNPLA3 variant (rs738409) (32). The results from this study suggested that the variant conferred a strong genetic susceptibility to liver disease and was modifiable by the level of alcohol consumption. They further reiterated that keeping the consumption of alcohol low may offset the genetic predisposition to liver disease in variant carriers (32). Another vital study demonstrated that although the variant (rs738409) in the PNPLA3 gene is associated with ALD, it did not correlate with serum IL6, IL10, IL8, and CCL2 levels in the Russian population (33).
A GWAS in patients with alcohol-related cirrhosis of European ethnicity found a genome-wide significant risk association of rs738409 in PNPLA3 (Odds ratio = 2.19; G allele) and rs4607179 near HSD17B13 (Odds ratio = 0.57; C allele. In addition, the study identified a protective association at rs374702773 in Fas-associated factor family member 2 (FAF2; Odds ratio = 0.61; del(T) allele) for alcohol-related cirrhosis when conditional analysis accounting for the PNPLA3 and HSD17B13 loci was carried out (34). A GWAS was carried out in the Korean Genome and Epidemiology Study Health Examination cohort data to assess the risk of developing ALD in nondrinkers, light drinkers, and heavy drinkers. The study reported an association of loci, namely gamma-glutamyltransferase 1 (GGT1; rs2006227), zinc protein finger 827 (ZNF827; rs1183910), and HNF1 homeobox A (HNF1A; rs1183910) with ALD risk, in addition to 5 variants on Chromosome 11 that showed protective effects against ALD (35).
Interleukin 1β apart from interleukin 1α is known to bind to the same receptor IL-1R type 1. They have similar functions and are involved in proinflammatory responses (36). The levels of IL-1β in the serum are high in patients with ALD, especially those with cirrhosis. A study carried out on East Indians suggested that variants in genes, including IL-1β, modulate the indices of alcohol-induced liver injury. The study identified that a variant (-511C) in the promoter region of the IL-1β gene was significantly overrepresented in ALD patients compared to individuals who misused alcohol without liver disease (controls). They also suggested that the risk genotype (-511CC) influenced the total levels of bilirubin, albumin, and alanine transaminase among alcohol users (37). Significantly higher occurrence of variants in IL-1 β gene in Japanese heavy drinkers who progressed to ALD-cirrhosis also strengthens the suspicion of its role in ALD (38). However, a meta-analysis reported no significant association between variants in IL-1β (− 511C > T and + 3953T > C) and ALD susceptibility. The meta-analysis included a total of 825 ALD patients and 743 controls that were pooled from 10 studies (39). These results point to the role of ethnicity-specific susceptibility and needs to be explored further. In addition, the role of variants in IL-1β both in conferring susceptibility and/or progression of ALD needs to be explored in a large multi-ethnic population.
NFE2 Like BZIP Transcription Factor 2 (NFE2L2) codes for transcription factor nuclear factor erythroid-related factor 2 (Nrf2). Nrf2 is known to regulate the expression of proteins that protect against inflammation and oxidative stress caused by excess consumption of alcohol and therefore is considered a potential candidate marker for ALD susceptibility. A study that explored the association of variants rs35652124, rs4893819, and rs6721961 in NFE2L2 gene identified that the variant rs35652124 predisposed individuals to ALD. Hematoxylin & eosin and immunohistochemistry of Nrf2 staining suggested that the variant was associated with a lower level of Nrf2 expression. In addition, liver tissues from ALD patients with this variant were associated with severe inflammatory activity (40).
Variants in the gene have been associated with various diseases, including alcohol-related disorders, predominantly through their involvement in neurotransmission and inflammation (41). A variant (rs26907) in the Ras-specific guanine nucleotide-releasing factor 2 gene (RASGRF2) was associated with higher alcohol consumption and also variability in alcohol-induced reward response (42). These findings added credence to the role of the gene in alcohol misuse susceptibility. This role is further reiterated by its regulation by RasGRF2 in synaptic transmission and its putative role in inflammation-related pathways via the activation of MAPKs (mitogen-activated protein kinases) and ERKs (extracellular signal-regulated kinases) (43–45). Other important variants also have been reported for their potential association with alcoholism. A variant (rs61764370) in KRAS gene is promising as it is located in the miRNA binding site for the member of the let-7 family. Individuals with the G allele at this position have impaired binding of let-7 and may be relevant in alcoholism, as a study showed the relevance of K-ras activity and alcohol craving in an animal model (46). Furthermore, let-7 is shown to be overexpressed in the brains of alcohol abusers and has been associated with an inflammatory response triggered by alcohol consumption (47). Also, the regulatory functions of let-7 are demonstrated to affect the activation of stellate cells in the liver and are involved in liver injury (48).
The central dopaminergic system is known to influence and regulate brain reward mechanisms and is considered to play a critical role in the development of dependence on a range of psychoactive substances, including ethanol (49–53). Alcohol reportedly stimulates dopamine receptors, promoting its release in the ventral striatum, activating the mechanism involving craving and incentive salience attributions paving the way for increased ethanol consumption (54). Therefore, studying the role of variants in genes concerning dopaminergic pathways in alcohol dependence is relevant. Various studies focused on association studies of the D1-D5 dopamine receptors and transporter protein (DAT) have identified the relevance of DRD2 gene in susceptibility to alcohol dependence. Although many studies focussed on exploring the role of DRD2 variants in alcohol dependence, the findings have primarily been controversial. Most of the significant associations of variants in the gene have been reported from the European/European American ethnicities, with studies in the Taiwanese population (Atayal natives) registering a negative association (55). The most commonly investigated variants in the DRD2 gene are -141C Ins/Del, TaqI B, and TaqI A, which are associated with alcohol dependence (56, 57). Insertion of the nucleotide “C” in the promoter region of the DRD2 gene at the -141 position (rs1799732) is reported to be associated with receptor density (58). As the TaqI B variant is in the 5`untranslated region (5` UTR), it is predicted to play an essential role in transcriptional regulation (59). The TaqI A variant is associated with low levels of the availability of D2 dopamine receptors in the striatum (60). Although this variant was initially reported to be in the 3`UTR region of the DRD2 gene, it is now identified that the variant is located in the gene ANKK1 (ankyrin repeat and kinase domain containing), where it is reportedly a missense variant. The variant in ANKK1 gene is associated with increased activity of the final enzyme named aromatic L-amino acid decarboxylase, which is involved in the biosynthesis of dopamine (61). A study from India that explored the association of these three variants in their association with alcohol dependence concluded that two variants (-141C Ins allele and TaqI A1) were associated with alcohol dependence (62).
This gene belongs to a member of the cytochrome P450 superfamily of enzymes that catalyze many reactions, including drug metabolism, synthesis of cholesterol, and lipids, and being induced by ethanol. The enzyme is involved in various processes, such as gluconeogenesis, and is implicated in liver cirrhosis (63). Functionally relevant variants in the CYP2E1 gene are therefore crucial in susceptibility to ALD. A study that explored the genetic polymorphisms in the 5`-flanking region of the CYP2E1 with the transcriptional regulation of the gene reported that polymorphisms affect the transcriptional regulation and may lead to inter-individual variation (64). A study that did not find any association between CYP2E1 c.-1053C>T variant with susceptibility to alcohol dependence reported that it might influence the expression of obsessive-compulsive and anxiety symptoms (65). A family-based genome-wide linkage analysis involving sibling pairs reported that linkage analysis detected significant linkage to CYP2E1 gene but was diminished. The authors suggested that variants in and around CYP2E1 affect the level of response to alcohol consumption, thus providing evidence for its role in alcohol addiction (66). A study reported no association of variants (CYP2E1*1D, *5B, *6, and *1B) with alcohol dependence in the Han Taiwanese population (67).
This gene is involved in the conversion of folate to methionine. Specifically, this enzyme is required to convert 5,10 methylenetetrahydrofolate to 5-methyltetrahydrofolate, the primary circulatory form. This circulatory form of 5-methyltetrahydrofolate is necessary for the subsequent multistep conversion of homocysteine to methionine. In animal models, folate deficiency was shown to disturb hepatic methionine metabolism, thereby promoting liver injury in ethanol-fed micropigs (68). A variant at the 677 position (rs1801133; C677T; A222V) in the human MTHFR gene is associated with reduced protein and increased homocysteine levels. The variant is associated with multiple diseases, including hepatic steatosis, cirrhosis, and hepatocellular carcinoma, predominantly due to higher homocysteine levels. Another variant, A1298C (rs1801131), in this gene, is reported to be in high linkage disequilibrium with the C677T and is also clinically relevant. However, a recent meta-analysis examining the association between the C677T variant in the MTHFR gene and the risk of alcohol dependence confirmed that there was no association (69). In addition, the study concluded that there was no association between the variant and alcohol dependence in Asians and Caucasians on sub-group analysis (69).
Women are at a higher risk of ALD and acute liver failure as compared to males (70, 71). It is known that for a given dose of alcohol, women are more susceptible to toxic effects as compared to males, although the latter is known to abuse alcohol more than the former. A population-based 13-year prospective study carried out in Denmark that followed 13,000 participants demonstrated that the risk of developing ALD in women was higher as compared to the males, even with higher consumption of alcohol in the males [7 to 13 beverages per week (84-156 g) in females Vs. 14 to 27 beverages per week (168-324 g) in males] (70). In addition, a higher relative risk of alcohol-induced cirrhosis was noted in women as compared to men [7.0 (95% CI, 3.8-12.8) Vs. 17.0 (95% CI, 6.8-40.8)], although it is less frequently diagnosed in women (72).Experiments in animal models demonstrate that this effect is due to relatively higher endotoxin levels and increased gut permeability, variability in the concentrations of estrogen receptors in the liver, and activation of Kupffer cells by estrogen. All these lead to increased inflammation and necrosis. In addition, alcohol induces differential expression of genes responsible for higher oxidative stress, inflammation, and injury in female rats (73). Therefore, females have an increased risk of developing ALD even with small amounts of alcohol consumed.
Substantial ethnicity-based differences in hospitalizations and mortality have been reported for patients with ALD. A study that assessed the liver and alcohol-related hospitalizations and deaths in ALD and specific alcohol-related diseases (ARD) reported ethnicity-based differences in the population. While Indians had a 75% higher risk for men, White British and Pakistani men had a relatively lower risk of ALD. A 2-fold higher risk for ARD was noted for White Irish men and any mixed background women, lower risk was noted for Pakistani and Chinese men and women (74). ARD included diseases attributed to alcohol: liver disease, chronic pancreatitis, poisoning, mental and behavioural disorders, nervous system disorders, cardiomyopathy, and gastritis. In addition, Hispanics present at 4-10 years younger age for ALD than White/Caucasian patients (75).
The most frequent complication of ALD is malnutrition. The reason for malnutrition is increased resting energy expenditure, limited ingestion or improper absorption, digestion, and utilization of nutrients due to the effect of alcohol metabolism (76, 77). The relationship between alcoholism and malnutrition is biderctional. Loss of muscle mass of the skeleton or sarcopenia and disturbances in energy metabolism are the two major known components of malnutrition in liver disease. Although it is known that alcohol is metabolized directly in the skeletal muscle contributing to the loss of muscle mass, however, its contribution to other organ injuries is currently unknown. Duration and the use/abuse of alcohol, quantity consumed, and other underlying causes/severity of liver disease are few of the factors that affect the severity of malnutrition in ALD (78). Furthermore, patients with ALD frequently are known to have deficiencies in proteins and vitamins that contribute to the severity. The highly reactive toxic metabolites generated by alcohol metabolism, including acetaldehyde and/or oxygen-containing molecules, interfere with the metabolism of other nutrients, primarily lipids, thereby contributing to the damage of the hepatic cell. Although a number of clinical trials and meta-analyses have been reported in the literature on nutritional support in both cirrhosis and ALD, they have not been conclusive for various reasons (79, 80). Therefore, a balanced diet can compensate for the general existence of malnutrition. In general, a daily intake of about 2000kcal comprising ~1.5g/kg/d protein may generally benefit patients with ALD (81). The intake may be distributed over the day giving short intervals between meals with an appropriate route of administration (78).
It is well recognized that the presence of comorbidities contributes to the progression of ALD. The most common comorbidities at diagnosis of cirrhosis were predominantly arterial hypertension, type 2 diabetes, and obesity (82). The presence of diabetes increases the risk for cirrhosis and hepatocellular carcinoma among patients with both ALD and non-alcoholic fatty liver disease (82, 83). A large population-based study reported that age, male sex, obesity, and components of metabolic syndrome (MetS) were independent predictors of liver-related mortality in ALD patients (84). Diabetes is a significant predictor for severe liver disease and development of cirrhosis among patients who misuse alcohol (85, 86).
Advances in genomics over the years has enabled us to probe the genome to understand its role in the pathogenesis of the disease. Alterations in the genome are majorly being used to predict the susceptibility to a phenotype/clinical condition or the risk of progression after the onset of the disease. Alterations in the genome may be at the single nucleotide level (variants) or large microscopically visible chromosomal aberrations. The variants can occur in the coding and non-coding regions of the genome and may be classified as Single nucleotide variants (SNV), multi-nucleotide variants (MNV), or indels (Insertions/deletions) depending on the number of nucleotides that are changed. Copy number variants (CNVs) that include inversions, translocations, deletions, duplications, and insertions are large, typically greater than 50 base pair rearrangements in the genome. Karyotype analysis by GTG banding is the preferred technique for detecting chromosomal aberrations (>5 Mb). Other advanced molecular techniques, including fluorescent in-situ hybridization (FISH) and microarray-based comparative genomic hybridization (aCGH) are routinely applied for a more accurate analysis. However, these platforms do not detect mosaicism, balanced translocations, and inversions. Other relevant molecular biology techniques including multiplex ligation-dependent probe amplification (MLPA) may also be used. Restriction Fragment Length Polymorphism (RFLP), Allele-specific PCR (Amplification-refractory mutation system (ARMS)-PCR), Probe based detection (Taqman probes) on Real time PCR (qRT-PCR) are the choice of methods for detecting single nucleotide variants that have prior susceptibility information. DNA microarray, DNA sequencing (Sangers sequencing and Next generation sequencing) are powerful techniques to map variants at the genome level with the disease and are predominantly employed by the Genome-wide association studies that are considered an unbiased way of identifying genetic susceptibility.
Progression to hepatocellular Carcinoma (HCC) is seen in patients with underlying infections (hepatitis B and C) and metabolic disease (non-alcoholic steatohepatitis [NASH]) apart from ALD. While the incidence of HBV/HCV-related HCC has declined with the introduction of newer therapies, a rise in cases of HCC associated with NASH and ALD is seen (87–89). Although non-cirrhotic HCC is common in NAFLD and viral hepatitis, the progression to HCC in cases with underlying NASH/ALD is typically after steatohepatitis, fibrosis, and cirrhosis (90). A study that characterized the role of IL-17A (a tumour-promoting cytokine) and its receptor IL-17RA signaling in the pathogenesis of HCC in an animal model and human specimens demonstrated that global deletion of IL17-RA suppressed HCC in an alcohol-fed animal model. They suggested that IL-17A may be a potential therapeutic target for patients with alcohol-induced HCC (91). Although it was known that individuals who consume alcohol and are heterozygous for ALDH2*1/*2 or homozygous for ALDH2*2/*2 variants have ALDH2 deficiency and are at a higher risk of developing digestive tract cancers, the mechanism for the same was unknown (92). An in vivo and in vitro mechanistic study indicated that Aldh2-deficient hepatocytes produced a large amount of harmful oxidized mitochondrial DNA via the extracellular vesicles after CCL4 plus ethanol exposure. These were then transferred into the neighboring HCC cells and, along with acetaldehyde, activated multiple oncogenic pathways, including the JNK, STAT3, BCL-2, and TAZ, thereby promoting HCC (93).
Risk stratification scores predominantly based on susceptibility conferring genetic loci were developed primarily to identify individuals with alcohol consumption who may progress to develop cirrhosis/HCC. A study compared three variants, PNPLA3:rs738409, SUGP1-TM6SF2:rs10401969, and HSD17B13:rs6834314, between cases with alcohol-related cirrhosis and controls with a history of alcohol consumption but no evidence of liver disease. The study concluded that stratification of heavy drinkers is possible based on the three variant risk scores and diabetes status, and early preventative interventions might benefit the patient (86).
Understanding the genetic susceptibility both in asymptomatic individuals who are at a higher risk to develop ALD as well as patients with ALD with a risk of progression would be beneficial to reduce the burden of the disease on the healthcare system. Genotyping for variants with an ability to predict alcohol dependence in an individual and assessing the risk will aid in counselling the individual to curtail alcohol consumption. Variants may be routinely genotyped in ALD patients to assess the genotype and the risk of progression with an intention of treating them aggressively. Identifying additional variants by employing whole exome sequencing at the genome level that is associated with susceptibility and risk of progression would aid in developing personalized programs for prevention and/or treatment. Additionally, novel gene targets from the data may be identified and explored as potential therapeutic targets. Currently genotping of variant in PNPLA3 gene is routinely being used by the clinicians to assess the risk. With access to Next generation sequening and routine use of whole exome and clinical exome sequencing in clinical practice, in furture, polygenic risk scores and algorithms that integrate environmental risks with genetic data would aid the clinician in accurate prediction of onset in high-risk individuals and the risk of progression.
Although higher numbers of individuals reportedly consume alcohol, a fraction of individuals progresses to develop ALD and cirrhosis. ALD is a complex, multi-factorial disease where interactions between genetic susceptibility and the environment precipitate the phenotype. While a compelling role for a few variants across ethnicities is reported (PNPLA3), conflicting results are reported for other genes. Identifying the comprehensive genetic susceptibility to the risk of developing ALD (Figure 3) and developing algorithms that aid in identifying individuals at-risk of ALD may be beneficial in reducing the burden of the disease. In addition, routing genotyping of risk variants predictive of developing the severe disease as part of diagnostic workup aids in identifying this subset of patients who may require aggressive treatment.
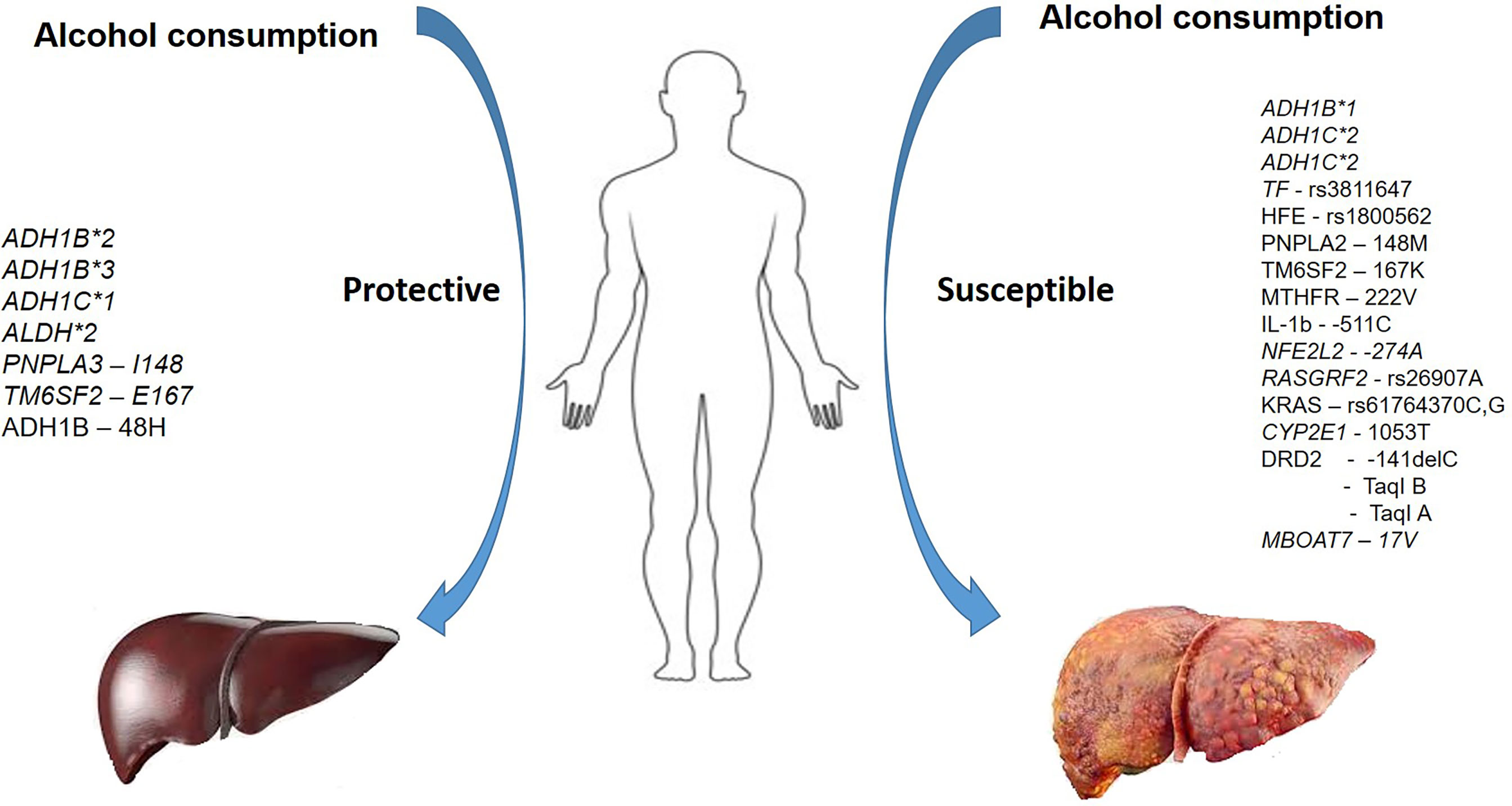
Figure 3 A summary of variants in various genes conferring protection and susceptibility in the presence of alcohol use.
AK and RV made the study concept and design. Figures by MS. RV prepared the initial draft. AK, PR, and DR critically edited and supervised the project. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Fernández-Solà J. Cardiovascular risks and benefits of moderate and heavy alcohol consumption. Nat Rev Cardiol (2015) 12(10):576–87. doi: 10.1038/nrcardio.2015.91
3. Abuse S. Key substance use and mental health indicators in the united states: results from the 2019 national survey on drug use and health. (USA) (2020).
4. Griswold MG, Fullman N, Hawley C, Arian N, Zimsen SRM, Tymeson HD, et al. Alcohol use and burden for 195 countries and territories, 1990–2016: a systematic analysis for the global burden of disease study 2016. Lancet (2018) 392(10152):1015–35. doi: 10.1016/S0140-6736(18)31310-2
5. Fuster D, Samet JH. Alcohol use in patients with chronic liver disease. N Engl J Med (2018) 379(13):1251–61. doi: 10.1056/NEJMra1715733
6. Poznyak V, Rekve D. Global status report on alcohol and health 2018. Geneva: World Health Organization (2018). p. 450.
7. Sacks JJ, Gonzales KR, Bouchery EE, Tomedi LE, Brewer RD. 2010 National and state costs of excessive alcohol consumption. Am J Prev Med (2015) 49(5):e73–e9. doi: 10.1016/j.amepre.2015.05.031
8. Idalsoaga F, Kulkarni AV, Mousa OY, Arrese M, Arab JP. Non-alcoholic fatty liver disease and alcohol-related liver disease: Two intertwined entities. Front Med (Lausanne) (2020) 7:448. doi: 10.3389/fmed.2020.00448
9. Liu CY, Wang M, Yu HM, Han FX, Wu QS, Cai XJ, et al. Ferroptosis is involved in alcohol-induced cell death in vivo and in vitro. Biosci Biotechnol Biochem (2020) 84(8):1621–8. doi: 10.1080/09168451.2020.1763155
10. Vogt BL, Richie JP Jr. Glutathione depletion and recovery after acute ethanol administration in the aging mouse. Biochem Pharmacol (2007) 73(10):1613–21. doi: 10.1016/j.bcp.2007.01.033
11. Wang M, Chen WY, Zhang J, Gobejishvili L, Barve SS, McClain CJ, et al. Elevated fructose and uric acid through aldose reductase contribute to experimental and human alcoholic liver disease. Hepatology (2020) 72(5):1617–37. doi: 10.1002/hep.31197
12. Stickel F, Hampe J. Genetic determinants of alcoholic liver disease. Gut (2012) 61(1):150–9. doi: 10.1136/gutjnl-2011-301239
13. Hurley T, Edenberg H, Li T. The pharmacogenomics of alcoholism. In: Pharmacogenomics: the search for individualized therapies. Weinheim: Pharmacogenomics: the search for individualized therapies (2002). p. 417–41.
14. Edenberg HJ. The genetics of alcohol metabolism: role of alcohol dehydrogenase and aldehyde dehydrogenase variants. Alcohol Res Health (2007) 30(1):5–13.
15. Zintzaras E, Stefanidis I, Santos M, Vidal F. Do alcohol-metabolizing enzyme gene polymorphisms increase the risk of alcoholism and alcoholic liver disease? Hepatology (2006) 43(2):352–61.
16. Vidal F, Lorenzo A, Auguet T, Olona M, Broch M, Gutiérrez C, et al. Genetic polymorphisms of ADH2, ADH3, CYP4502E1 dra-I and pst-I, and ALDH2 in Spanish men: lack of association with alcoholism and alcoholic liver disease. J Hepatol (2004) 41(5):744–50.
17. Floris M, Cano A, Porru L, Addis R, Cambedda A, Idda ML, et al. Direct-to-Consumer nutrigenetics testing: An overview. Nutrients (2020) 12(2).
18. Stickel F, Moreno C, Hampe J, Morgan MY. The genetics of alcohol dependence and alcohol-related liver disease. J Hepatol (2017) 66(1):195–211.
19. Kozlitina J, Smagris E, Stender S, Nordestgaard BG, Zhou HH, Tybjærg-Hansen A, et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet (2014) 46(4):352–6. doi: 10.1038/ng.2901
20. Mahdessian H, Taxiarchis A, Popov S, Silveira A, Franco-Cereceda A, Hamsten A, et al. TM6SF2 is a regulator of liver fat metabolism influencing triglyceride secretion and hepatic lipid droplet content. Proc Natl Acad Sci U S A. (2014) 111(24):8913–8. doi: 10.1073/pnas.1323785111
21. Prill S, Caddeo A, Baselli G, Jamialahmadi O, Dongiovanni P, Rametta R, et al. The TM6SF2 E167K genetic variant induces lipid biosynthesis and reduces apolipoprotein b secretion in human hepatic 3D spheroids. Sci Rep (2019) 9(1):11585. doi: 10.1038/s41598-019-47737-w
22. Buch S, Stickel F, Trépo E, Way M, Herrmann A, Nischalke HD, et al. A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat Genet (2015) 47(12):1443–8. doi: 10.1038/ng.3417
23. Goffredo M, Caprio S, Feldstein AE, D'Adamo E, Shaw MM, Pierpont B, et al. Role of TM6SF2 rs58542926 in the pathogenesis of nonalcoholic pediatric fatty liver disease: A multiethnic study. Hepatology (2016) 63(1):117–25. doi: 10.1002/hep.28283
24. Basyte-Bacevice V, Skieceviciene J, Valantiene I, Sumskiene J, Petrenkiene V, Kondrackiene J, et al. TM6SF2 and MBOAT7 gene variants in liver fibrosis and cirrhosis. Int J Mol Sci (2019) 20(6). doi: 10.3390/ijms20061277
25. Mancina RM, Dongiovanni P, Petta S, Pingitore P, Meroni M, Rametta R, et al. The MBOAT7-TMC4 variant rs641738 increases risk of nonalcoholic fatty liver disease in individuals of European descent. Gastroenterology (2016) 150(5):1219–30.e6. doi: 10.1053/j.gastro.2016.01.032
26. Teo K, Abeysekera KWM, Adams L, Aigner E, Anstee QM, Banales JM, et al. rs641738C>T near MBOAT7 is associated with liver fat, ALT and fibrosis in NAFLD: A meta-analysis. J Hepatol (2021) 74(1):20–30. doi: 10.1016/j.jhep.2020.08.027
27. Wilson PA, Gardner SD, Lambie NM, Commans SA, Crowther DJ. Characterization of the human patatin-like phospholipase family. J Lipid Res (2006) 47(9):1940–9. doi: 10.1194/jlr.M600185-JLR200
28. Huang Y, He S, Li JZ, Seo YK, Osborne TF, Cohen JC, et al. A feed-forward loop amplifies nutritional regulation of PNPLA3. Proc Natl Acad Sci U S A. (2010) 107(17):7892–7. doi: 10.1073/pnas.1003585107
29. Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet (2008) 40(12):1461–5.
30. Tian C, Stokowski RP, Kershenobich D, Ballinger DG, Hinds DA. Variant in PNPLA3 is associated with alcoholic liver disease. Nat Genet (2010) 42(1):21–3. doi: 10.1038/ng.488
31. Stickel F, Buch S, Lau K, Meyer zu Schwabedissen H, Berg T, Ridinger M, et al. Genetic variation in the PNPLA3 gene is associated with alcoholic liver injury in caucasians. Hepatology (2011) 53(1):86–95. doi: 10.1002/hep.24017
32. Lazo M, Bilal U, Mitchell MC, Potter J, Hernaez R, Clark JM. Interaction between alcohol consumption and PNPLA3 variant in the prevalence of hepatic steatosis in the US population. Clin Gastroenterol Hepatol (2021) 19(12):2606–14.e4. doi: 10.1016/j.cgh.2020.08.054
33. Peregud DI, Baronets VY, Lobacheva AS, Ivanov AS, Arisheva OS, Garmash IV, et al. PNPLA3 rs738409 associates with alcoholic liver cirrhosis but not with serum levels of IL6, IL10, IL8 or CCL2 in the Russian population. Ann Hepatol (2021) 20:100247. doi: 10.1016/j.aohep.2020.08.065
34. Schwantes-An TH, Darlay R, Mathurin P, Masson S, Liangpunsakul S, Mueller S, et al. Genome-wide association study and meta-analysis on alcohol-associated liver cirrhosis identifies genetic risk factors. Hepatology (2021) 73(5):1920–31. doi: 10.1002/hep.31535
35. Kim KY, Kim JO, Kim YS, Choi JE, Park JM, Han K, et al. Genome-wide association of individual vulnerability with alcohol-associated liver disease: A Korean genome and epidemiology study. Hepatology (2022) 75(2):391–402. doi: 10.1002/hep.32115
36. Tilg H, Moschen AR, Szabo G. Interleukin-1 and inflammasomes in alcoholic liver disease/acute alcoholic hepatitis and nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology (2016) 64(3):955–65. doi: 10.1002/hep.28456
37. Roy N, Mukhopadhyay I, Das K, Pandit P, Majumder PP, Santra A, et al. Genetic variants of TNFα, IL10, IL1β, CTLA4 and TGFβ1 modulate the indices of alcohol-induced liver injury in East Indian population. Gene (2012) 509(1):178–88. doi: 10.1016/j.gene.2012.07.077
38. Takamatsu M, Yamauchi M, Maezawa Y, Saito S, Maeyama S, Uchikoshi T. Genetic polymorphisms of interleukin-1beta in association with the development of alcoholic liver disease in Japanese patients. Am J Gastroenterol (2000) 95(5):1305–11. doi: 10.1111/j.1572-0241.2000.02030.x
39. Xiao M, Yang R, Zhao Y-Y, Zeng T. The common genetic variants in IL-1β and IL-1RN may have no predisposition to alcoholic liver disease: A meta-analysis. Meta Gene (2017) 13:26–31. doi: 10.1016/j.mgene.2017.04.005
40. Nunes Dos Santos K, Florentino RM, França A, Lima Filho ACM, Santos MLD, Missiaggia D, et al. Polymorphism in the promoter region of NFE2L2 gene is a genetic marker of susceptibility to cirrhosis associated with alcohol abuse. Int J Mol Sci (2019) 20(14). doi: 10.3390/ijms20143589
41. Barbacid M. Ras genes. Annu Rev Biochem (1987) 56(1):779–827. doi: 10.1146/annurev.bi.56.070187.004023
42. Schumann G, Coin LJ, Lourdusamy A, Charoen P, Berger KH, Stacey D, et al. Genome-wide association and genetic functional studies identify autism susceptibility candidate 2 gene (AUTS2) in the regulation of alcohol consumption. Proc Natl Acad Sci U S A. (2011) 108(17):7119–24. doi: 10.1073/pnas.1017288108
43. Schwechter B, Rosenmund C, Tolias KF. RasGRF2 rac-GEF activity couples NMDA receptor calcium flux to enhanced synaptic transmission. Proc Natl Acad Sci U S A. (2013) 110(35):14462–7. doi: 10.1073/pnas.1304340110
44. Tian X, Gotoh T, Tsuji K, Lo EH, Huang S, Feig LA. Developmentally regulated role for ras-GRFs in coupling NMDA glutamate receptors to ras, erk and CREB. EMBO J (2004) 23(7):1567–75. doi: 10.1038/sj.emboj.7600151
45. Wang Q, Siminovitch KA, Downey GP, McCulloch CA. Ras-guanine-nucleotide-releasing factors 1 and 2 interact with PLCγ at focal adhesions to enable IL-1-induced Ca(2+) signalling, ERK activation and MMP-3 expression. Biochem J (2013) 449(3):771–82. doi: 10.1042/BJ20121170
46. Repunte-Canonigo V, van der Stap LD, Chen J, Sabino V, Wagner U, Zorrilla EP, et al. Genome-wide gene expression analysis identifies K-ras as a regulator of alcohol intake. Brain Res (2010) 1339:1–10. doi: 10.1016/j.brainres.2010.03.063
47. Lewohl JM, Nunez YO, Dodd PR, Tiwari GR, Harris RA, Mayfield RD. Up-regulation of microRNAs in brain of human alcoholics. Alcohol Clin Exp Res (2011) 35(11):1928–37. doi: 10.1111/j.1530-0277.2011.01544.x
48. McDaniel K, Huang L, Sato K, Wu N, Annable T, Zhou T, et al. The let-7/Lin28 axis regulates activation of hepatic stellate cells in alcoholic liver injury. J Biol Chem (2017) 292(27):11336–47. doi: 10.1074/jbc.M116.773291
49. Tupala E, Tiihonen J. Dopamine and alcoholism: neurobiological basis of ethanol abuse. Prog Neuropsychopharmacol Biol Psychiatry (2004) 28(8):1221–47. doi: 10.1016/j.pnpbp.2004.06.022
50. Koob GF, Le Moal M. Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology (2001) 24(2):97–129. doi: 10.1016/S0893-133X(00)00195-0
51. Dick DM, Foroud T. Candidate genes for alcohol dependence: a review of genetic evidence from human studies. Alcohol Clin Exp Res (2003) 27(5):868–79. doi: 10.1097/01.ALC.0000065436.24221.63
52. Munafò M, Johnstone E, Murphy M, Walton R. New directions in the genetic mechanisms underlying nicotine addiction. Addict Biol (2001) 6(2):109–17. doi: 10.1080/13556210020040181
53. Lingford-Hughes A, Nutt D. Neurobiology of addiction and implications for treatment. Br J Psychiatry (2003) 182:97–100. doi: 10.1192/bjp.182.2.97
54. Kienast T, Heinz A. Dopamine and the diseased brain. CNS Neurol Disord Drug Targets (2006) 5(1):109–31. doi: 10.2174/187152706784111560
55. Gelernter J, Kranzler H. D2 dopamine receptor gene (DRD2) allele and haplotype frequencies in alcohol dependent and control subjects: no association with phenotype or severity of phenotype. Neuropsychopharmacology (1999) 20(6):640–9. doi: 10.1016/S0893-133X(98)00110-9
56. Ferguson RA, Goldberg DM. Genetic markers of alcohol abuse. Clin Chim Acta (1997) 257(2):199–250. doi: 10.1016/S0009-8981(96)06444-3
58. Arinami T, Gao M, Hamaguchi H, Toru M. A functional polymorphism in the promoter region of the dopamine D2 receptor gene is associated with schizophrenia. Hum Mol Genet (1997) 6(4):577–82. doi: 10.1093/hmg/6.4.577
59. Hauge XY, Grandy DK, Eubanks JH, Evans GA, Civelli O, Litt M. Detection and characterization of additional DNA polymorphisms in the dopamine D2 receptor gene. Genomics (1991) 10(3):527–30. doi: 10.1016/0888-7543(91)90431-D
60. Pohjalainen T, Rinne JO, Någren K, Lehikoinen P, Anttila K, Syvälahti EK, et al. The A1 allele of the human D2 dopamine receptor gene predicts low D2 receptor availability in healthy volunteers. Mol Psychiatry (1998) 3(3):256–60. doi: 10.1038/sj.mp.4000350
61. Laakso A, Pohjalainen T, Bergman J, Kajander J, Haaparanta M, Solin O, et al. The A1 allele of the human D2 dopamine receptor gene is associated with increased activity of striatal l-amino acid decarboxylase in healthy subjects. Pharmacogenet Genomics (2005) 15(6):387–91. doi: 10.1097/01213011-200506000-00003
62. Prasad P, Ambekar A, Vaswani M. Dopamine D2 receptor polymorphisms and susceptibility to alcohol dependence in Indian males: a preliminary study. BMC Med Genet (2010) 11:24. doi: 10.1186/1471-2350-11-24
63. Fisher CD, Lickteig AJ, Augustine LM, Ranger-Moore J, Jackson JP, Ferguson SS, et al. Hepatic cytochrome P450 enzyme alterations in humans with progressive stages of nonalcoholic fatty liver disease. Drug Metab Dispos (2009) 37(10):2087–94. doi: 10.1124/dmd.109.027466
64. Hayashi S, Watanabe J, Kawajiri K. Genetic polymorphisms in the 5'-flanking region change transcriptional regulation of the human cytochrome P450IIE1 gene. J Biochem (1991) 110(4):559–65. doi: 10.1093/oxfordjournals.jbchem.a123619
65. Plemenitas A, Kastelic M, Porcelli S, Serretti A, Rus Makovec M, Kores Plesnicar B, et al. Genetic variability in CYP2E1 and catalase gene among currently and formerly alcohol-dependent male subjects. Alcohol Alcohol (2015) 50(2):140–5. doi: 10.1093/alcalc/agu088
66. Webb A, Lind PA, Kalmijn J, Feiler HS, Smith TL, Schuckit MA, et al. The investigation into CYP2E1 in relation to the level of response to alcohol through a combination of linkage and association analysis. Alcohol Clin Exp Res (2011) 35(1):10–8. doi: 10.1111/j.1530-0277.2010.01317.x
67. Huang CY, Kao CF, Chen CC, Kuo PH, Huang MC. No association of CYP2E1 genetic polymorphisms with alcohol dependence in han Taiwanese population. J Formos Med Assoc (2018) 117(7):646–9. doi: 10.1016/j.jfma.2018.03.007
68. Halsted CH, Villanueva JA, Devlin AM, Niemelä O, Parkkila S, Garrow TA, et al. Folate deficiency disturbs hepatic methionine metabolism and promotes liver injury in the ethanol-fed micropig. Proc Natl Acad Sci U S A. (2002) 99(15):10072–7. doi: 10.1073/pnas.112336399
69. Rai V, Kumar P. Methylenetetrahydrofolate reductase (MTHFR) gene C677T (rs1801133) polymorphism and risk of alcohol dependence: a meta-analysis. AIMS Neurosci (2021) 8(2):212–25. doi: 10.3934/Neuroscience.2021011
70. Becker U, Deis A, Sørensen TI, Grønbaek M, Borch-Johnsen K, Müller CF, et al. Prediction of risk of liver disease by alcohol intake, sex, and age: a prospective population study. Hepatology (1996) 23(5):1025–9. doi: 10.1002/hep.510230513
71. Reuben A, Koch DG, Lee WM. Drug-induced acute liver failure: results of a U.S. multicenter, prospective study. Hepatology (2010) 52(6):2065–76. doi: 10.1002/hep.23937
72. Jepsen P, Ott P, Andersen PK, Sørensen HT, Vilstrup H. Risk for hepatocellular carcinoma in patients with alcoholic cirrhosis: a Danish nationwide cohort study. Ann Intern Med (2012) 156(12):841–7, w295. doi: 10.7326/0003-4819-156-12-201206190-00004
73. Guy J, Peters MG. Liver disease in women: the influence of gender on epidemiology, natural history, and patient outcomes. Gastroenterol Hepatol (N Y). (2013) 9(10):633–9.
74. Bhala N, Cézard G, Ward HJ, Bansal N, Bhopal R. Ethnic variations in liver- and alcohol-related disease hospitalisations and mortality: The Scottish health and ethnicity linkage study. Alcohol Alcohol (2016) 51(5):593–601. doi: 10.1093/alcalc/agw018
75. Levy R, Catana AM, Durbin-Johnson B, Halsted CH, Medici V. Ethnic differences in presentation and severity of alcoholic liver disease. Alcohol Clin Exp Res (2015) 39(3):566–74. doi: 10.1111/acer.12660
76. Lieber CS. Relationships between nutrition, alcohol use, and liver disease. Alcohol Res Health (2003) 27(3):220–31.
77. Mehtani R, Premkumar M, Kulkarni AV. Nutrition in critical care hepatology. Curr Hepatol Rep (2022). doi: 10.1007/s11901-022-00586-0
78. Dasarathy S. Nutrition and alcoholic liver disease: Effects of alcoholism on nutrition, effects of nutrition on alcoholic liver disease, and nutritional therapies for alcoholic liver disease. Clin Liver Dis (2016) 20(3):535–50. doi: 10.1016/j.cld.2016.02.010
79. Antar R, Wong P, Ghali P. A meta-analysis of nutritional supplementation for management of hospitalized alcoholic hepatitis. Can J Gastroenterol (2012) 26(7):463–7. doi: 10.1155/2012/945707
80. Nasrallah SM, Galambos JT. Aminoacid therapy of alcoholic hepatitis. Lancet (1980) 2(8207):1276–7. doi: 10.1016/S0140-6736(80)92338-7
81. Bischoff SC, Bernal W, Dasarathy S, Merli M, Plank LD, Schütz T, et al. ESPEN practical guideline: Clinical nutrition in liver disease. Clin Nutr (2020) 39(12):3533–62. doi: 10.1016/j.clnu.2020.09.001
82. Vaz J, Eriksson B, Strömberg U, Buchebner D, Midlöv P. Incidence, aetiology and related comorbidities of cirrhosis: a Swedish population-based cohort study. BMC Gastroenterol (2020) 20(1):84. doi: 10.1186/s12876-020-01239-6
83. Mantovani A, Targher G. Type 2 diabetes mellitus and risk of hepatocellular carcinoma: spotlight on nonalcoholic fatty liver disease. Ann Transl Med (2017) 5(13):270. doi: 10.21037/atm.2017.04.41
84. Stepanova M, Rafiq N, Younossi ZM. Components of metabolic syndrome are independent predictors of mortality in patients with chronic liver disease: a population-based study. Gut (2010) 59(10):1410–5. doi: 10.1136/gut.2010.213553
85. Åberg F, Helenius-Hietala J, Puukka P, Färkkilä M, Jula A. Interaction between alcohol consumption and metabolic syndrome in predicting severe liver disease in the general population. Hepatology (2018) 67(6):2141–9. doi: 10.1002/hep.29631
86. Whitfield JB, Schwantes-An TH, Darlay R, Aithal GP, Atkinson SR, Bataller R, et al. A genetic risk score and diabetes predict development of alcohol-related cirrhosis in drinkers. J Hepatol (2022) 76(2):275–82. doi: 10.1016/j.jhep.2021.10.005
87. Morgan RL, Baack B, Smith BD, Yartel A, Pitasi M, Falck-Ytter Y. Eradication of hepatitis c virus infection and the development of hepatocellular carcinoma: a meta-analysis of observational studies. Ann Intern Med (2013) 158(5 Pt 1):329–37. doi: 10.7326/0003-4819-158-5-201303050-00005
88. Kulkarni AV, Duvvuru NR. Management of hepatitis b and c in special population. World J Gastroenterol (2021) 27(40):6861–73. doi: 10.3748/wjg.v27.i40.6861
89. Kulkarni AV, Fatima S, Sharma M, Kumar P, Gupta R, Padaki NR, et al. Lenvatinib for unresectable hepatocellular carcinoma: the first Indian experience. GastroHep (2021) 3(6):407–8. doi: 10.1002/ygh2.477
90. Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology (2011) 141(5):1572–85. doi: 10.1053/j.gastro.2011.09.002
91. Ma HY, Yamamoto G, Xu J, Liu X, Karin D, Kim JY, et al. IL-17 signaling in steatotic hepatocytes and macrophages promotes hepatocellular carcinoma in alcohol-related liver disease. J Hepatol (2020) 72(5):946–59. doi: 10.1016/j.jhep.2019.12.016
92. Li H, Borinskaya S, Yoshimura K, Kal'ina N, Marusin A, Stepanov VA, et al. Refined geographic distribution of the oriental ALDH2*504Lys (nee 487Lys) variant. Ann Hum Genet (2009) 73(Pt 3):335–45. doi: 10.1111/j.1469-1809.2009.00517.x
Keywords: Alcohol-related liver disease, susceptibility, genetics, variants, PNPLA3, TM6SF2
Citation: Vishnubhotla R, Kulkarni AV, Sharma M, Rao PN and Reddy DN (2022) An update on the genetics of alcoholic liver disease. Front. Gastroenterol. 1:1030399. doi: 10.3389/fgstr.2022.1030399
Received: 28 August 2022; Accepted: 04 November 2022;
Published: 30 November 2022.
Edited by:
Luis Antonio Díaz Piga, Pontificia Universidad Católica de Chile, ChileReviewed by:
Debanjali Dasgupta, Mayo Clinic, United StatesCopyright © 2022 Vishnubhotla, Kulkarni, Sharma, Rao and Reddy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anand V. Kulkarni, YW5hbmR2azkwQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.