 Abbeah Navasca
Abbeah Navasca Jatinder Singh
Jatinder Singh Viviana Rivera-Varas
Viviana Rivera-Varas Upinder Gill
Upinder Gill Gary Secor
Gary Secor Thomas Baldwin
Thomas Baldwin- Department of Plant Pathology, North Dakota State University, Fargo, ND, United States
Fusarium solani is a species complex encompassing a large phylogenetic clade with diverse members occupying varied habitats. We recently reported a unique opportunistic F. solani associated with unusual dark galls in sugarbeet. We assembled the chromosome-level genome of the F. solani sugarbeet isolate strain SB1 using Oxford Nanopore and Hi-C sequencing. The average size of F. solani genomes is 54 Mb, whereas SB1 has a larger genome of 59.38 Mb, organized into 15 chromosomes. The genome expansion of strain SB1 is due to the high repeats and segmental duplications within its three potentially accessory chromosomes. These chromosomes are absent in the closest reference genome with chromosome-level assembly, F. vanettenii 77-13-4. Segmental duplications were found in three chromosomes but are most extensive between two specific SB1 chromosomes, suggesting that this isolate may have doubled its accessory genes. Further comparison of the F. solani strain SB1 genome demonstrates inversions and syntenic regions to an accessory chromosome of F. vanettenii 77-13-4. The pan-genome of 12 publicly available F. solani isolates nearly reached gene saturation, with few new genes discovered after the addition of the last genome. Based on orthogroups and average nucleotide identity, F. solani is not grouped by lifestyle or origin. The pan-genome analysis further revealed the enrichment of several enzymes-coding genes within the dispensable (accessory + unique genes) genome, such as hydrolases, transferases, oxidoreductases, lyases, ligases, isomerase, and dehydrogenase. The evidence presented here suggests that genome plasticity, genetic diversity, and adaptive traits in Fusarium solani are driven by the dispensable genome with significant contributions from segmental duplications.
1 Introduction
The Fusarium solani species complex (FSSC) comprises at least 77 phylogenetically distinct species known to thrive in diverse environments covering ecological, agricultural, and clinical settings (O’Donnell et al., 2020; Geiser et al., 2021). The FSSC has three distinct clades, denoted as Clades 1 through 3. Clade 1 comprises F. plagianthi and F. illudens, while Clade 2 contains several Fusarium species, including those that caused sudden death syndrome and bean root rot. Clade 3 is the largest, boasting over 60 distinct species. This clade houses clinically and agriculturally important species, including Fusarium solani (O’Donnell et al., 2008, 2020; Geiser et al., 2021).
The highly adaptive F. solani is infects humans, animals, and plants making it a trans-kingdom fungus. In humans, F. solani affects immunocompromised patients causing skin and nail infections (Gupta et al., 2000; Godoy et al., 2004; Zhang et al., 2006; Kuruvilla and Diaz, 2012), mycotic keratitis (Ahearn et al., 2008; Boral et al., 2018; Trovato et al., 2021) even in a healthy individual (Ortega-Rosales et al., 2019), and chronic diabetic ulcers (Pai et al., 2010). A recent case involving F. solani is the clinical meningitis outbreak in Mexico affecting Mexican and US residents who traveled for medical purposes (Smith et al., 2024; García-Rodríguez et al., 2024; Strong et al., 2024). F. solani is also a threat to animals. This fungus caused keratitis in rabbits (Kiryu et al., 1991), cutaneous hyalohyphomycosis and mass mortalities in loggerhead sea turtles (Cabanes et al., 1997; Sarmiento-Ramírez et al., 2010), tissue destruction and inflammation in shrimps and prawns (Hose et al., 1984; Bian and Egusa, 1981; Le et al., 2005), dermatitis in false killer whales (Tanaka et al., 2012), and submucosal nodules on the face, tongue, and waist of a Doberman dog (Kano et al., 2001). Undoubtedly, F. solani infects crops across various species. Several reports highlight this pathogen’s ability to cause multiple rot diseases such as dry rot in potato stems (Goss, 1940), fruit rot in pumpkin (Rampersad, 2009), sweet pepper (Ramdial and Rampersad, 2010), and strawberry (Mehmood et al., 2017); crown rot in cucumber (Li et al., 2010) and strawberry (Pastrana et al., 2014; Villarino et al., 2019); root rot in peas (VanEtten, 1978; Gibert et al., 2022), sweet potato (Wang et al., 2014), strawberry (Pastrana et al., 2014; Villarino et al., 2019), okra (Li et al., 2016), eggplant (Li et al., 2017), tobacco (Yang et al., 2020), olive (Perez et al., 2011), and many more. Apart from fruit and vegetables, F. solani also infects ornamental plants such as bulb rot in tulips (Nisa et al., 2021), soft rot (Han et al., 2017) and wilt (Xie et al., 2024) in orchids. Other symptoms caused by F. solani are cankers in sweet potato (Wang et al., 2014) and English Walnut (Chen and Swart, 2000; Mulero-Aparicio et al., 2019; Tuerdi et al., 2023), gummosis in rubber trees (Huang et al., 2016), wilt in cotton (Zhu et al., 2019), leaf-sheath rot in bush lily (Sun et al., 2022), and leaf spot in pineapple (Zhang et al., 2024).
Aside from being an important pathogen of humans, animals, and plants, F. solani also have other lifestyles. It thrives as an endophyte in mulberry (Kim et al., 2017) and arabidopsis (Mesny et al., 2021) and also as a saprophyte, increasing the insecticidal efficacy of the entomopathogenic nematode Steinernema diaprepesi (Wu et al., 2018). Recently, we reported F. solani as an opportunistic pathogen of sugarbeet (Navasca et al., 2023). This isolate, F. solani strain SB1, was recovered from dark galls of sugarbeets, with symptoms that differ from other Fusarium diseases in sugarbeets (Hanson, 2006; Rivera et al., 2008; Secor et al., 2014; Khan et al., 2021). Electron microscopy and sequencing work confirmed that the galls contain sugarbeet material. However, greenhouse tests show that SB1 can only cause mild vascular discoloration without developing any galls or gall-like structures, adding a layer of complexity to the disease.
The broad host range and adaptive lifestyle of Fusarium species can be attributed to their ability to take in genomic regions, making them very adaptive to specific environments (Coleman et al., 2009; Ma et al., 2010, 2013). Knowing this ability, we sequenced the F. solani strain SB1 to determine its genetic elements and how it compares to F. solani genomes, which could provide insight into this unique species and disease complex. The concept of supernumerary chromosomes, more commonly called accessory chromosomes (ACs), is well-established in Fusarium, particularly Fusarium solani and Fusarium oxysporum (Coleman et al., 2009; Ma et al., 2010, 2013; Zhang et al., 2020). These ACs are not essential for growth but offer added features such as pathogenicity or increased virulence. We hypothesize that F. solani SB1 contains accessory regions that enable it to be opportunistic to sugarbeet and are unique from other F. solani species. Here, we describe its complete genomic characteristics and how it compares with the genome of the pea pathogen, Fusarium vanettenii MPVI isolate 77-13-4 (previously reported as N. haematococca; Coleman et al., 2009). Given the ability of F. solani to thrive in various habitats, relying on reference genomes may overlook crucial adaptive genes present in unrepresented isolates. In this study, we performed a pan-genome analysis of publicly available twelve F. solani genomes encompassing pathogens of plants and animals, saprophytes, and endophytes, including the opportunistic pathogen of sugarbeet. We determined the enrichment of core and dispensable genes. Our findings underscore the remarkable genome plasticity, genetic diversity, and inherent adaptive ability of Fusarium solani.
2 Methodology
2.1 Isolation, DNA extraction, and draft genome sequencing
Procedures for isolation, DNA extraction, library preparation, and draft genome sequencing are available in Navasca et al., 2023. In brief, we isolated Fusarium solani from galled sugarbeet by excising and sterilizing small tissues with 0.5% v/v sodium hypochlorite for 10 minutes and washing with sterile distilled water. We extracted high-molecular-weight DNA from the mycelia of pure culture F. solani sugarbeet isolate strain SB1 grown in PDB for five days following the instructions of Liu et al. (2022). Agilent TapeStation and Qubit 4.0 determined the DNA quality and quantity, respectively. We utilized the Nanopore Protocol Lambda Control Experiment (SQK-LSK109) for library preparation and performed sequencing using the R10.3 version flowcell in MinIOn. Guppy version 6.0.1 (Oxford Nanopore Technologies, UK) base called the reads, followed by adapter removal using Porechop version 0.2.4 (Wick et al., 2017) and data quality control using LongQC version 1.2.0 (Fukasawa et al., 2020). Finally, we assembled the sequences using NECAT version 0.0.1 (Chen et al., 2021) and checked the quality using Quast 5.0.2 (Mikheenko et al., 2018).
2.2 Hi-C sequencing and chromosome-level genome assembly
Mycelia of F. solani SB1 grown in PDA broth for five days were collected and resuspended in 1% formaldehyde in molecular-grade water, followed by a 20-minute incubation with periodic mixing. Glycine (125 mM final concentration) was then added to the sample and incubated for 15 min with occasional mixing. A final spin down (1000 g) for 5 min separated the sample from the mixture, and the supernatant was removed before sending the sample to Phase Genomics (Seattle, Washington, USA) for high-throughput chromosome conformation capture, or Hi-C, library preparation using a Proximo v4.0 Fungal kit. Intact cells were crosslinked using a formaldehyde solution, then simultaneously digested using the DPNII, DDE1, HINF, MSEI restriction enzymes prior to proximity ligation with biotinylated nucleotides. Chimeric molecules composed of fragments from different regions of genomes that were physically proximal in vivo were pulled down with streptavidin beads, then processed into an Illumina-compatible sequencing library. Sequencing was performed on an Illumina NovaSeq system, generating PE150 read pairs.
Reads were aligned to the draft assembly using BWA-MEM (Li and Durbin, 2010) with the -5SP and -t 8 options specified and all other options default. SAMBLASTER (Faust and Hall, 2014) was used to flag PCR duplicates, which were later excluded from analysis. Alignments were then filtered with samtools (Li et al., 2009) using the -F 2304 filtering flag to remove non-primary and secondary alignments. Juicebox (Durand et al., 2016; Rao et al., 2014) was used to produce chromosome-scale scaffolds.
Genome completeness was assessed separately by Fungal Genome Mapping Project (FGMP) version 1.0.2 (Cissé and Stajich, 2019) and Benchmarking Universal Single-Copy Orthologs (BUSCO) version 5.2.2 (Simaão et al., 2015; Manni et al., 2021). In BUSCO analysis, F. graminearum gene model from Augustus version 3.4.0 (Stanke and Waack, 2003) was used for evaluating the genome completeness. Telomeric regions were identified by the ‘TTAGGG’ sequence specific for most ascomycetes (https://telomerase.asu.edu.
2.3 Genome annotation
We masked the genome and identified transposable elements via RepeatMasker version 4.0.9 (Smit et al., 2003) using a custom library produced from RepeatModeler2 version 2.0.5 (Flynn et al., 2020) with built-in three de-novo repeat finding programs RECON, RepeatScout, and LtrHarvest/Ltr_retriever. Evidence-based annotation (Batut et al., 2019) was performed using MAKER2 version 2.3.11 (Holt and Yandell, 2011) pipeline with built-in ab-initio gene annotation programs SNAP version 2013-11-29 (Korf, 2004) and Augustus version 3.4.0 (Stanke and Waack, 2003). We did three rounds of annotations utilizing ESTs and proteins of the well-studied reference genome F. vanettenii strain 77-13-4, formerly reported as Nectria haematococca mating population MPVI (Coleman et al., 2009), from EnsemblFungi (accessed November 12, 2023) and the reference genome F. solani strain FSSC 5 MPI-SDFR-AT-0091 (accessed November 28, 2023), reviewed proteins of F. solani from Uniprot (accessed December 14, 2023), and the custom library of repeats from RepeatModeler2 to produce a high-quality annotation for downstream analysis (Batut et al., 2019; Bretaudeau, 2023). We implemented the same annotation method to eight other Fusarium solani genomes from NCBI accessed on November 28, 2023 with only published raw data (Accessions: GCA_027574645.1, GCA_002215905.1, GCA_019320015.1, GCA_013168735.1, GCA_024220475.1, GCA_033085375.1, GCA_030014125.1, and GCA_029603225.1). Annotations of one genome from the submitter in NCBI (Accession: GCA_027945525.1) and F. vanettenii 77-13-4 v2.0, and F. solani FSSC 5 MPI-SDFR-AT-0091 (Accession: GCA_020744495.1) both from JGI, were used as is for further analysis. A total of 12 strains were used in this study. Following annotation, we determined genome completeness using proteins in BUSCO version 5.3.2 (Stanke and Waack, 2003; Manni et al., 2021) using ‘fungi_odb10’ database aided by F. graminearum protein libraries. We estimated gene density over 100kb region of each chromosome of F. solani strain SB1.
2.4 Prediction of pathogenicity-related genes
Genomic annotations were made for F. solani sugarbeet isolate strain SB1. Secondary metabolism potential was determined using the antiSMASH 7.0 fungal version (Blin et al., 2021) with the ‘relaxed’ option. SignalP 6.0 for eukaryotes (Teufel et al., 2022) and TargetP 2.0 with non-plant option (Armenteros et al., 2019) predicted the secretory signal peptides and mitochondrial proteins, respectively. After signal peptide prediction and removal of mitochondrial proteins, sequences were then subjected to DeepTMHMM 1.0.24 (Hallgren et al., 2022) and Phobius (Käll et al., 2004) to remove proteins with the transmembrane domain. The remaining protein sets were checked for glycosylphosphatidylinositol (GPI) anchors using NetGPI 1.1 (Gíslason et al., 2021). Ultimately, only proteins without GPI anchors were considered secreted proteins. The refined secreted proteins were further used for the analysis of pathogenesis-related proteins. The dbCAN3 (Zheng et al., 2023) meta server, which combines HMMER, DIAMOND, and eCAMI database, annotated the carbohydrate-active enzymes (CAZymes) while EffectorP 3.0 for fungi (Sperschneider and Dodds, 2022) predicted the effector proteins. We performed a BLAST search in the Pathogen-Host Interaction Database or PHI-base version 4.0 with protein sequences 4.14 database (Urban et al., 2022) to find homologs of refined secreted proteins functionally characterized on other organisms. We used e-value <1e-05 and at least 50% identity to screen the hits. We selected the gene with the highest bit score for genes with more than one hit. Bit scores consider both the alignment of the sequences and the gaps within the alignment. A high bit score indicates a better alignment (Madden, 2013). Finally, we determined the gene function intersection between CAZymes, effectors, and PHIs.
2.5 Genome comparisons and gene collinearity
We applied Mauve (Darling et al., 2004) plug-in software in Geneious Prime version 2023.1.2 to align the SB1 genome to itself to determine the global rearrangement structure within the SB1 genome. We utilized the alignment file to generate a plot using Circos version 0.69.8 (Krzywinski et al., 2009; Rasche and Hiltemann, 2020). We used Chromeister version 1.5a (Pérez-Wohlfeil et al., 2019) to fast-align DNA sequences and determine the sequence similarity of F. solani SB1 and F. vanettenii 77-13-4 v2.0. For synteny analysis, we performed all vs all protein alignment between SB1 and F. vanettenii using the BLASTP service of BLAST and prepared the bed files according to the requirements of McScanX (Wang et al., 2012). We also used the same procedure to identify gene collinearity within the SB1 genome with the duplicate_gene_classifier option in McScanX to determine the classification of gene duplications. Syntenic blocks were visualized in SynVisio (Bandi and Gutwin, 2020).
2.6 Pan-genome analysis and GO enrichment
OrthoFinder version 2.5.5 (Emms and Kelly, 2019) identified clusters of orthologous proteins between 12 strains. The pan-genome of F. solani was analyzed using core and dispensable proteins and was visualized in a curve in PanGP (Zhao et al., 2014; Li et al., 2022) in ‘totally random’ algorithm at 1000 combinations, replicated 50 times. Core proteins are those consistently found across all strains, whereas dispensable proteins cover accessory proteins (present in two or more strains) and unique proteins (exclusive to a single strain). Functional annotation of core and dispensable (accessory and unique) proteins was performed using InterProScan version 5.59-91.0 (Jones et al., 2014). We utilized ShinyGo version 0.80 (Ge et al., 2020) with false discovery rate (FDR) correction at p < 0.05 to evaluate the enrichment of core and dispensable genomes against the F. vanetteni genome.
2.7 Phylogenomic analysis
Protein sequences of F. solani strains in this study and those of the two outgroups Fusarium graminearum PH-1 NNRL 31084 (Accession: GCA_000240135.3) and Fusarium oxysporum f. sp. lycopersici 4287 strain (Accession: GCA_000149955.2) were utilized for phylogenomic analysis using OrthoFinder version 2.5.5 (Emms and Kelly, 2017, 2019). We used the software’s default parameters to produce the species tree using the information of the entire set of orthologous groups present in all F. solani strains. The tree was visualized using iTol version 6.8.1 (Letunic and Bork, 2021). We also computed for the average nucleotide identity of F. solani strains using FastANI version 1.3 (Jain et al., 2018).
3 Results
3.1 Genome features of Fusarium solani strain SB1
The draft genome assembly of F. solani SB1 was assembled previously in 19 contigs (Navasca et al., 2023) using Oxford Nanopore sequencing. High-throughput chromatin conformation capture (Hi-C) sequencing resolved the genome assembly in 15 chromosomes with a total size of 59.4 Mb and N50 of 4,122,546 bp with the largest contig at 6,537,432 bp. The chromosome size ranges from 6.54 to 2.11 Mb (Figure 1A). We achieved 96.1% genome completeness in Fungal Genome Mapping Project (FGMP) version 1.0.2 (Cissé and Stajich, 2019) and 98.5% in BUSCO version 5.2.2 (Simão et al., 2015; Manni et al., 2021). We found 13 chromosomes containing telomeric repeats at one end (Figure 1B). GC content of chromosomes ranged from 47 to 53% (Figure 1C). We also assembled the mitochondrial genome of F. solani SB1 isolate. To validate the number of chromosomes, we performed karyotyping on the SB1 isolate and independently confirmed that the observed chromosome count aligns closely with the Hi-C genomic assembly (Supplementary Figure 1). The SB1 genome contains 17,981 protein-coding genes predicted by MAKER2 version 2.3.11 (Holt and Yandell, 2011) and 41 secondary metabolites identified by antiSMASH 7.0 fungal version (Blin et al., 2021) and 10.71% repetitive elements. Screening of protein-coding genes by SignalP 6.0 for eukaryotes (Teufel et al., 2022) and TargetP 2.0 (Armenteros et al., 2019), DeepTMHMM version 1.0.24 (Hallgren et al., 2022), Phobius (Käll et al., 2004), and NetGPI (Gíslason et al., 2021) predicted 1,177 secreted proteins. EffectorP 3.0 for fungi (Sperschneider and Dodds, 2022) recognized a total of 440 putative effector proteins (37% of secreted proteins) classified as either cytoplasmic (194) or apoplastic (246) where at least 135 are potentially dual-localized effectors (cytoplasmic/apoplastic or apoplastic/cytoplasmic). A total of 308 carbohydrate-active enzymes (26% of secreted proteins) were predicted by the dbCAN3 meta server (Zheng et al., 2023). We found 141 proteins (12% of secreted proteins) having potential roles in pathogenesis from the BLAST results of PHI-base version 4 (Urban et al., 2022) based on the parameters we set (homology ≥ 50%, e-value <1e-05). All data are available in Supplementary Table 1.
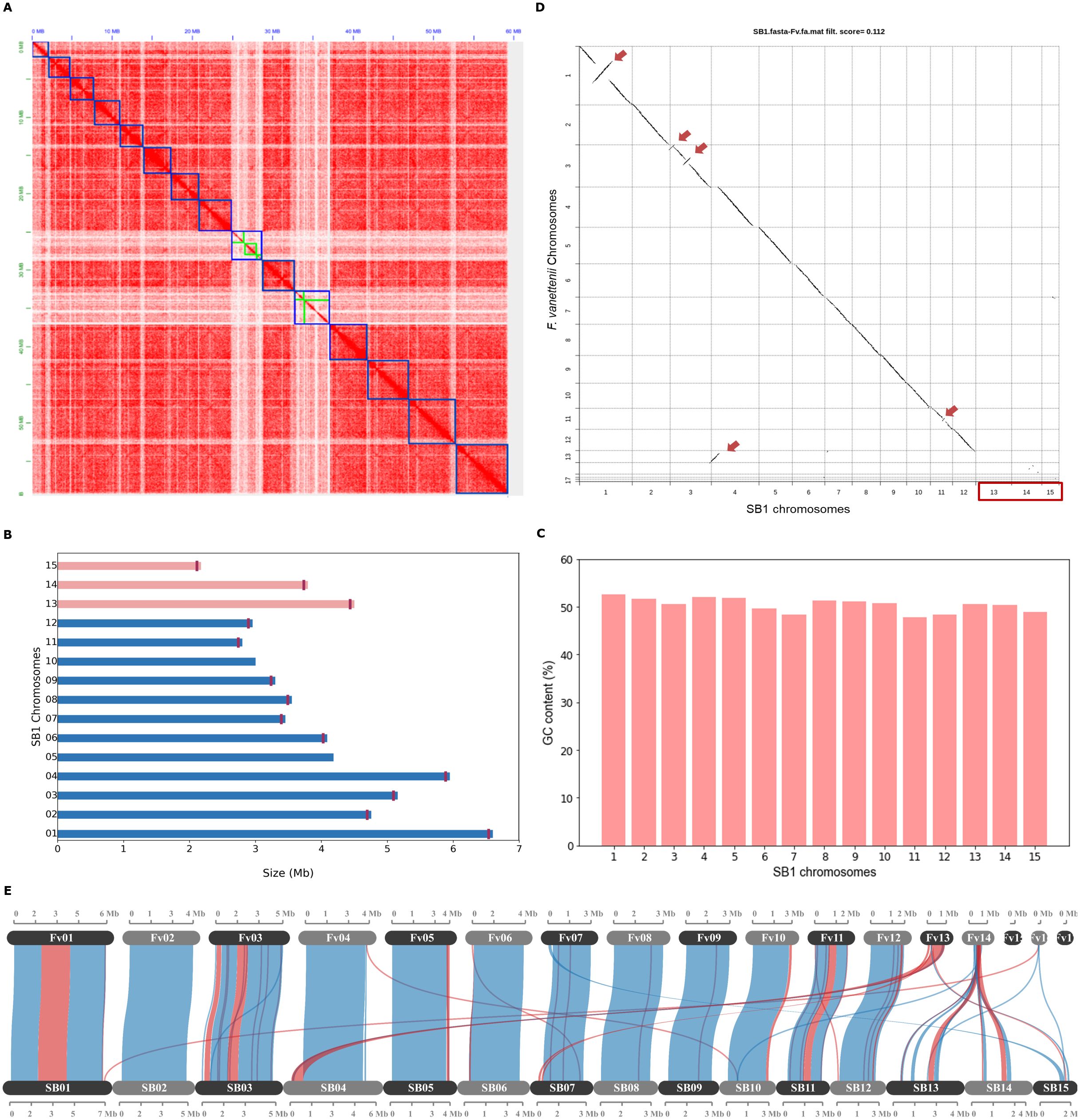
Figure 1. The Fusarium solani SB1 genome assembly and comparison with F. vanettenii strain 77-13-4. (A) Hi-C contact map of SB1 scaffolds, outlined with blue squares, correspond to the 15 chromosomes sorted according to their length. The color intensity represents the frequency of contact between two chromosomes. The green boxes on the heatmap represent contig boundaries. These contigs are long enough to span a significant portion of their respective scaffolds, making them noticeable at this resolution; (B) The Fusarium solani SB1 chromosomes arranged according to similarity to the reference genome F. vanettenii strain 77-13-4. Three chromosomes, Chr13, Chr14, and Chr15 (pink bars) are not found in the reference genome. Note the vertical lines marking the telomeres of each chromosome except Chr05 and Chr10; (C) GC content is similar across SB1 chromosomes; (D) Genome comparison of Fusarium solani SB1 and F. vanettenii shows high sequence similarity with inverted regions indicated by red arrows. Three chromosomes (Chr13, Chr14, and Chr15) are not found in F. vanettenii, indicated by the red box; (E) Gene collinearity F. vanettenii 77-13-4 and Fusarium solani SB1 shows syntenic (blue) and inverted regions (red). SB1 Chr13 and Chr14 have collinear genes with the accessory chromosome Chr14 of F. vanettenii. Fv - F. vanetenii 77-13-4, SB - Fusarium solani SB1.
3.2 F. solani SB1 harbors chromosomes not found in F. vanettenii strain 77-13-4
Fusarium vanettenii strain 77-13-4 was previously named Nectria haematococca (Coleman et al., 2009) and is the most studied strain in Fusarium solani group (Miao et al., 1991; Kistler et al., 1996; Wasmann and VanEtten, 1996; Han et al., 2001; Liu et al., 2003; Coleman et al., 2009). For this reason, we chose to compare the F. solani strain SB1 to this reference genome. We reversed-complemented five scaffolds (2, 3, 7, 10, 11) of the F. solani SB1 isolate and named them according to their matches with the chromosomes of the F. vanettenii strain 77-13-4 (Figures 1A−D). Direct pairwise genome comparison of F. solani SB1 and F. vanettenii 77-13-4 in Chromeister version 1.5a (Pérez-Wohlfeil et al., 2019) obtained a 0.112 score (‘zero’ as the perfect score for similarity), which indicates genomes are very similar but they also contain inversions indicated by red arrows in Figure 1D. Moreover, three chromosomes, Chr13, Chr14, and Chr15, are predominantly absent from F. vanettenii, highlighted by the red box in Figure 1D. These chromosomes will be highlighted in the succeeding sections. Further analysis using protein-coding genes in McScanX (Wang et al., 2012) visualized in SynVisio (Bandi and Gutwin, 2020) shows that F. solani SB1 and F. vanettenii 77-13-4 share 79.16% collinear genes but also harbor inversions indicated by red links in Figure 1E. Using our raw reads, we confirmed the inversions in our assembly by manually checking and ensuring that contigs on those areas overlap and do not have gaps. F. vanettenii harbor three ACs, Chr14, Chr15, and Chr17 (Coleman et al., 2009). We found collinear regions in Chr13 and Chr14 of F. solani strain SB1 with the AC Chr14 of F. vanettenii 77-13-4 (Figure 1E), suggesting these SB1 chromosomes could also be accessory. We checked these collinear genes and found that most are uncharacterized proteins of F. vanettenii (data not shown).
3.3 Genomic attributes of Chr13, Chr14, and Chr15
Among SB1 chromosomes, Chr13 had the highest repetitive elements, which make up a quarter of its genome at 26.34%, followed by Chr14 at 21.00% and Chr15 at 9.66% (Figure 2A). Class II DNA transposons mainly occupy these chromosomes, with 11% for Chr13 and Chr14 and about 4% for Chr15. Chr06 also contains Class II DNA transposons, while Chr06, Chr07, and Chr11 all contain Class I Retrotransposons. The average gene density of F. solani SB1 genome is 303 genes per Mb, but Chr13, Chr14, and Chr15 only have 206, 193, and 243 genes per Mb, respectively (Supplementary Table 1; Figure 2B). This is likely due to the highly repetitive elements in these chromosomes. Each chromosome has at least one predicted biosynthetic gene cluster (BCG) except Chr09, Chr13, and Chr14. These BCGs are classified as terpenes (3), phosphonate (1), isocyanide (1), fungal-RiPP-like (2), non-ribosomal peptide synthase, NRPS (18), and polyketide synthase, PKS (13), and NRPS-PKS hybrid (3). Three of the BCGs have 100% similarity to sansalvamide, choline, and ochratoxin A; three with 84-85% similarity to fusarubin, lucilactaean, and cyclosporin, one with 62% similarity with matachelin, while four have 28-40% similarity to squalestatin S1, gibepyrone-A, oxyjavanicin, and duclauxin. The remaining BCGs do not have similarities with known clusters. Among these known BCGs, sansalvamide, a cytotoxic cyclic depsipeptide, and fusarubin, a polyketide, are secondary metabolites produced by marine Fusarium and F. fujikoroi, respectively (Belofsky et al., 1999; Studt et al., 2012). Sansalvamide exhibits in vitro cytotoxicity against cancer cell lines (Belofsky et al., 1999; Romans-Fuertes et al., 2016), while fusarubin is responsible for perithecial pigmentation of F. fujikoroi (Studt et al., 2012). Most SB1 chromosomes had at least 66 secreted proteins, but Chr13, Chr14, and Chr15 only had 21, 20, and 40 secreted proteins, respectively (Supplementary Table 1; Figure 2B). For CAZymes, Chr13, Chr14, and Chr15 had three, seven, and six, respectively, compared to other chromosomes with at least 17 (Supplementary Table 1; Figure 2B). The CAZymes identified in these three chromosomes include chitinase 1 (Accession No.: RSL48174.1), chitinase 4 (Accession No.: KAJ4176553.1), glycoside hydrolases (Accession Nos.: XP_046123512.1, XP_046127030.1, XP_046127088.12, KIM92707.1, KAH7017800.1, KAH7009017.1), glycosyl hydrolases (Accession nos.: XP_046125026, XP_046127088.1, XP_046124973.1), glucanase (Accession No.: XP_053015740.1), and pectin lyase (Accession No.: XP_046125011.1). There are at least 21 effectors present in each chromosome except for Chr13 and Chr14 with six (Supplementary Table 1; Figure 2B). Most effectors found in Chr13, Chr14, and Chr15 were uncharacterized or hypothetical except for three out of six from Chr13 and seven out of 21 from Chr15. These effector proteins mainly code for enzymes such as lipase (Accession No.: KAI8649777.1), killer toxin Kp4/SMK (Accession No.: XP_046128691.1), lysophospholipase (Accession No.: XP_018242567.1), Chloroperoxidase (Accession No.: XP_046127231.1), Alpha/Beta hydrolase protein (Accession No.: XP_046127211.1), glycosyl hydrolase family 61-domain-containing protein (Accession No.: XP_046127088.1), pectin lyase fold/virulence factor (Accession No.: XP_046125011.1), tannase and feruloyl esterase-domain-containing protein (Accession No.: XP_046124962.1), and peptidase A4 family-domain-containing protein (Accession No.: XP_046123576.1). We highlight that even with its smallest size and high repeat sequences, Chr15 still has the highest number of genes and more secreted proteins compared to Chr13 and Chr14.
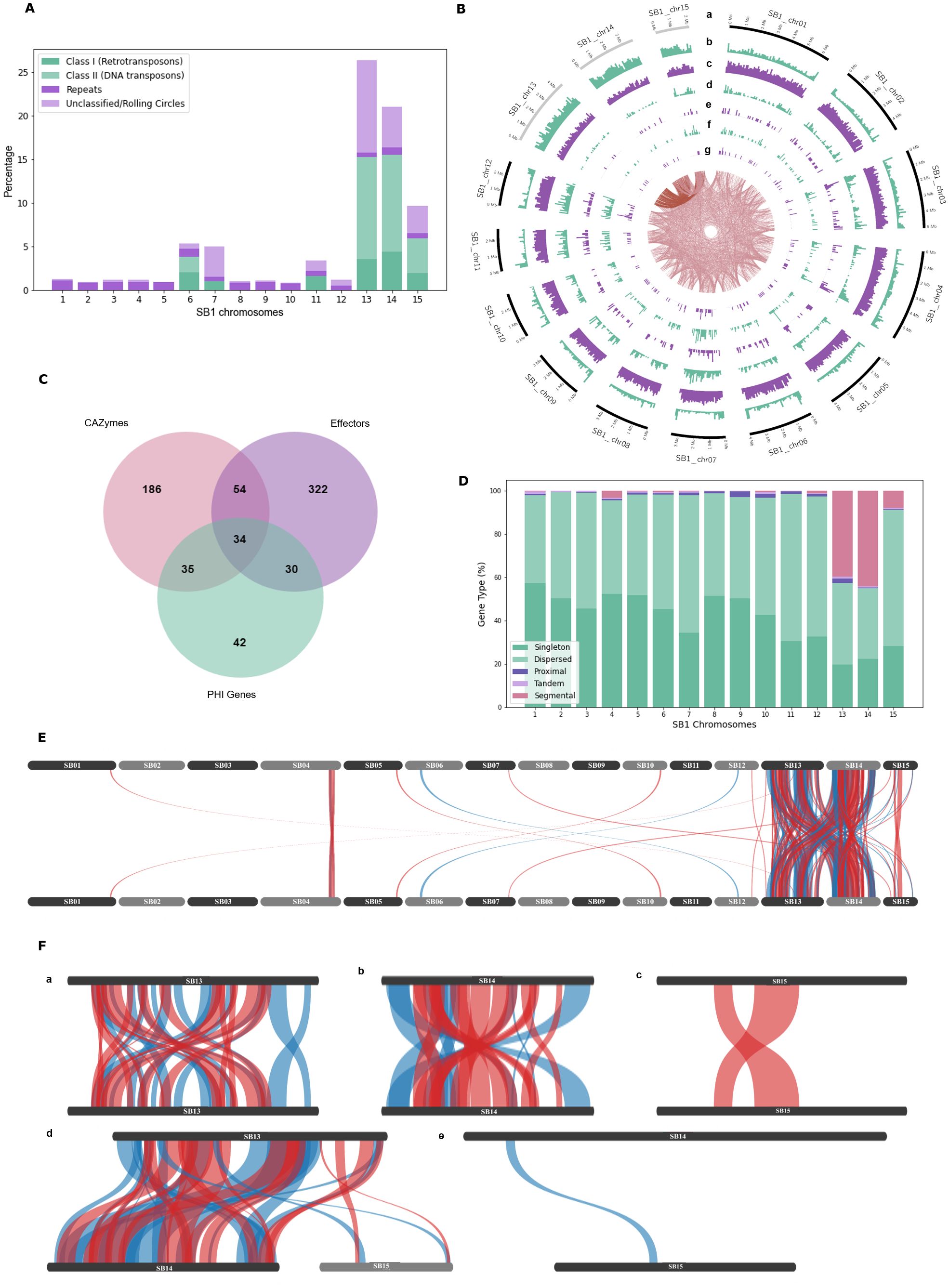
Figure 2. Genomic features of the Fusarium solani SB1. (A) Repeats and transposable elements of SB1 chromosomes showing Chr13, Chr14, and Chr15 with the highest content among chromosomes; (B) Circos plot showing F. solani SB1 15 chromosomes at different sizes, gray bands are potential accessory chromosomes (a), repeat element density (b), gene density (c), secreted proteins (d), carbohydrate-active enzymes (e), effectors (f), pathogenicity-host interaction genes (g), and links between SB1 chromosomes. Note the high repeats, fewer genes, CAZymes, effectors, and phi genes in Chr13, Chr14, and Chr15; (C) Venn diagram showing the number of genes playing various roles as CAZymes, effectors, and/or PHI genes; (D) Type of genes in each chromosome of SB1 strain. Note the high percentage of dispersed duplications all over the SB1 genome and segmental duplications within and between Chr13 and Chr14, followed by Chr15 and then Chr04. (E) Collinearity of F. solani SB1 genome showing duplications, particularly between Chr13, Chr14, and Chr15; (F) Close-up view of duplications within and between Chr13, Chr14, and Chr15; E, F only show duplicated regions where blue lines are synthetic while red lines are inverted.
Using the PHI-base, around 69 genes were associated with reduced virulence, five with effector functions, one with loss of pathogenicity, 47 with no effect on pathogenicity, and 19 had mixed functions. Further, around 44% (62) of these proteins are homologous to Fusarium species, mainly to F. graminearum (36), F. oxysporum (12), F. solani (12), F. verticillioides (1), and F. virguliforme (1). The remaining 56% (79) are homologous to proteins found in Magnaporthe oryzae (22), Botrytis sp. (10), Colletotrichum sp. (9), Trichoderma virens (9), Penicillium digitatum (6), and other organisms (23). A complete list is available in Supplementary Table 2. Focusing on F. solani protein homologs, four genes namely PELA (PHI: 179), PELD (PHI: 180), CutA (PHI: 2849), and CSN1 (PHI: 2403), comprise all 12 hits. PELA (PHI:179) and PELD (PHI:180) are essential genes coding for pectate lyase associated with root rots in Pisum sativum (Rogers et al., 2000). CutA (PHI:2849) coding for cutinase and CSN1 (PHI: 2403) coding for chitosanase were considered non-essential genes associated with storage rot in Maxima cucurbita and Maxima moscato (Crowhurst et al., 1997) and root, seedling, and pod rot in Pisum sativum (Liu et al., 2010), respectively. No PHIs from Chr13, Chr14, and Chr15 belong to F. solani. The number of PHI genes varies across chromosomes, as low as three for Chr15 and as high as 19 for Chr07. Four of 19 PHIs from Chr07 have similarities to Fusarium solani essential genes PELA (1) and PELD (3). One particular protein (Chr07_2628) has 99.07% identity with PELD coding for family 3 polysaccharide lyase in F. solani (Accession no.: XP_046134888.1). There were no hits of PEP genes in the SB1 secreted proteins.
Considering secreted proteins can be CAZymes, effectors, or PHIs, we determined how many genes have intersecting function. We found 34 genes with all three functions (Figure 2C). Around 80% (27 of 34) of genes code for enzymes such as glycoside hydrolase, pectin/pectate lyase, polygalacturonase, endoglucanase, glucanase, lyases, and cutinase. The remaining genes code for hypothetical and starch-binding domain-containing proteins. Even though most genes are coding for similar functions, they do not have the exact accession numbers in BLAST results and are showing 95-100% query cover and similarity. This displays the arsenal of enzymes encoded in the genome of F. solani SB1.
3.4 Segmental duplications between and within Chr13 and Chr14
We performed self-alignment of the F. solani SB1 DNA sequence and uncovered intriguing inter-chromosomal connections indicative of sequence similarity (Figure 2B, links in the innermost circle). Furthermore, our analysis highlights Chr13’s predominant alignment with Chr14 and Chr15. We further conducted syntenic gene analysis within the F. solani SB1 genome, revealing a total of 10,406 dispersed, 195 proximal, 123 tandem, and 963 segmental duplications, alongside 9,101 singletons (Figure 2D). The dispersed duplications explain the links indicating sequence similarity all over the SB1 genome. Chr13 accounts for almost half of the segmental duplications at 44%, followed by Chr14 at 38.5% (Figure 2E). Chr04 also contains segmental duplications at 7%, while Chr15 obtained 5%. The rest of the chromosomes have no segmental duplications. Moreover, our investigation revealed that duplications were predominantly concentrated within and between Chr13 and Chr14 (Figures 2F-a, c, d), with fewer occurrences observed within Chr15 and between Chr15 and either Chr13 or Chr14 (Figures 2F-c–e).
3.5 Comparative genomics of 12 Fusarium solani genomes
3.5.1 Genomic characteristics of F. solani group
The genome size of Fusarium solani isolates averaged 53.91 Mb, with the smallest at 45.81 Mb from JS-169 and the biggest at 66.64 Mb from CR12 (Figure 3A and Supplementary Table 3). The GC content (%) ranged from 49.5 to 51.5. Genomes achieved 95.6 to 99.7% completeness by BUSCO version 5.2.2 (Simão et al., 2015; Manni et al., 2021). Repeat contents contributed to large sizes of SB1 and CR12 with 11.50 and 17.45%, respectively (Figure 3A). Gene models ranged from 14,663 to 18,410, with an average gene density of 319 ranging from 276 to 334 per Mb where the two largest genomes and highest repeats, CR12 and SB1, also had the lowest gene densities with 276 and 303 per Mb, respectively. Average protein length per genome ranges from 452 to 496 and averages 477 amino acids (AA) (Figure 3B). The protein length distribution warrants good-quality annotation supplementing BUSCO analysis (Nevers et al., 2023).
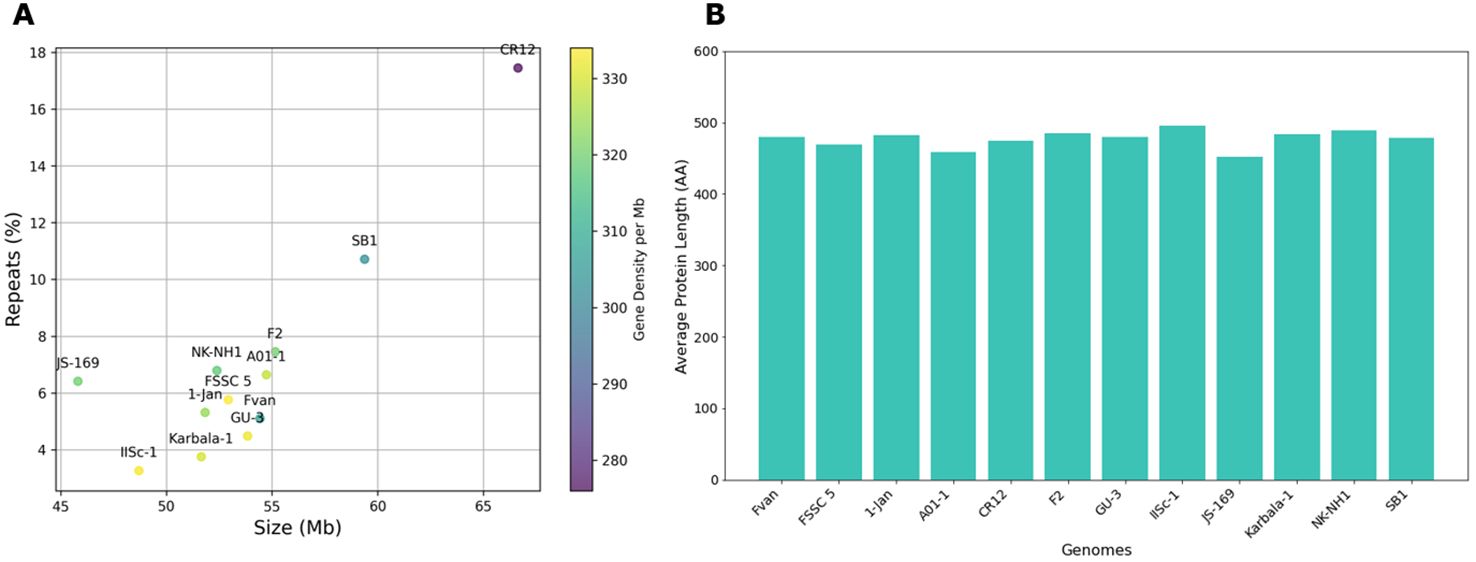
Figure 3. Genomic features of Fusarium solani genomes. (A) Size (Mb), repeats (%), and gene density (color gradient). The largest genomes, CR12 and SB1, also have the highest repeats but the lowest gene densities; (B) Protein length of Fusarium solani genomes, averages 477 AA.
3.5.2 Nearly saturated F. solani pan-genome
We constructed the pan-genome curve by clustering 204,225 predicted proteins derived from 12 accessions showing decreasing core proteins as new genomes are added. In contrast, new clusters increase with new genomes, indicating an open pan-genome of Fusarium solani (Figure 4A). However, we also observed a steady decline of new protein clusters with only under 300 as the 12th genome is added (Figure 4B). This suggests discovering fewer new genes when sequencing additional genomes of F. solani. The number of core proteins (present in all strains) is almost the same for all genomes at 11,400 on average, but this varies in percentage relative to the size of the genomes. For example, JS-169 has the lowest number of core proteins (n=11,141) but is equivalent to 76%, the highest among genomes (Figure 4C; Supplementary Table 3). Accessory proteins (present in two or more strains) ranged from 3,217 for JS-169 to 6,417 for SB1, accounting for 21% and 36% of their respective genomes. Unique proteins (present in a single strain) are as low as 97 or 0.6% of F2’s genome and as high as 582 or 3.2% of AO1-1 genomes. The ratio between the number of genes in orthogroups and the number of orthogroups present in a genome is >1 for all strains, indicating that each orthogroup contains more than one gene from each genome (Supplementary File 5). This is shown as paralogs primarily observed in AO1-1 and CR12 at 20% (Figure 4D).
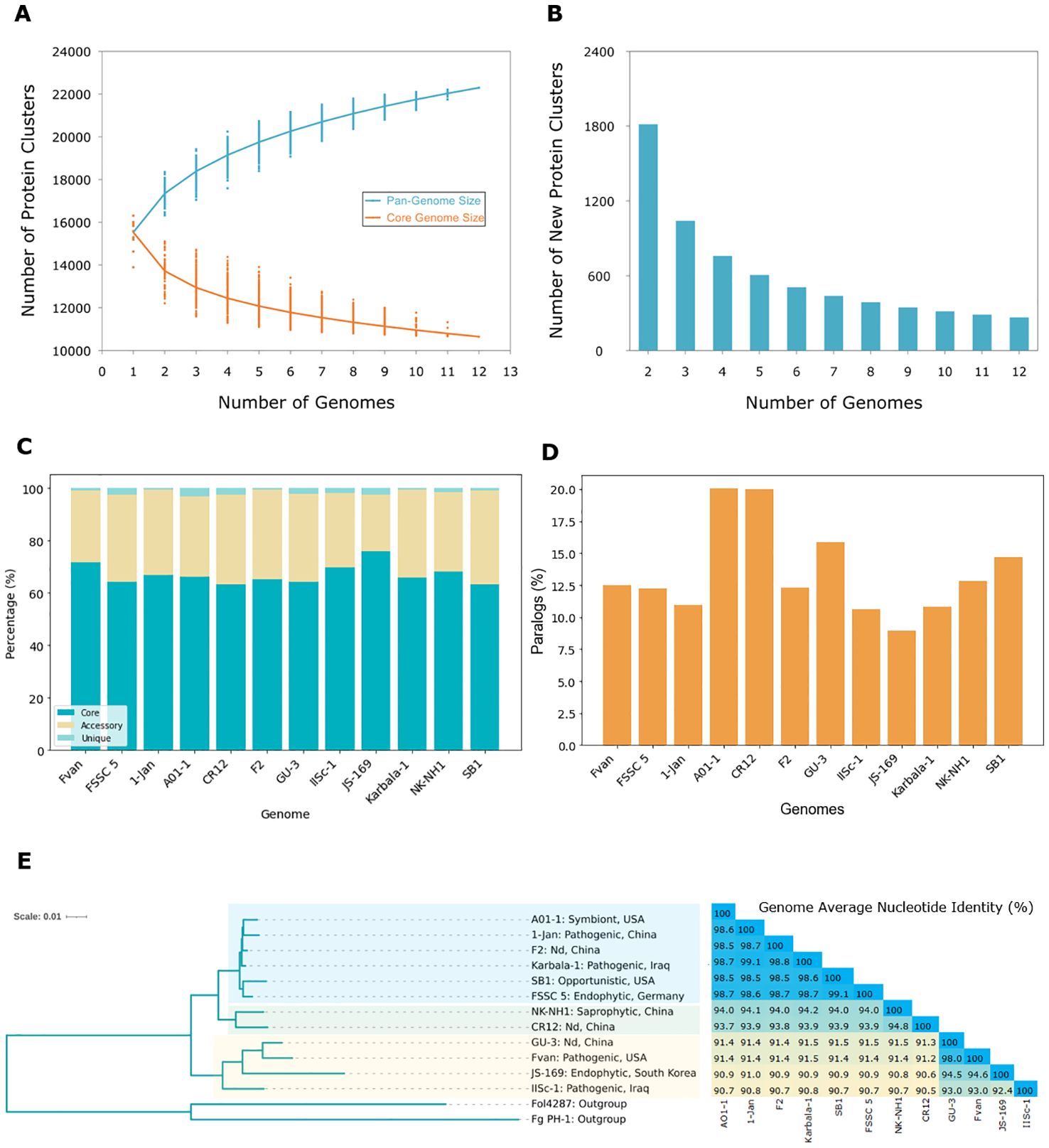
Figure 4. The pan-genome and phylogenetic relationship of Fusarium solani. (A) The pan-genome and core genome of 12 F. solani strains show decreasing core proteins as new genomes are added; (B) Number of new protein clusters of F. solani in the pan-genome with <300 new protein clusters after the 12th genome; (C) Core (present in all strains), accessory (present in two or more strains), and unique (present in one strain) proteins of 12 F. solani genomes accounting for an average of 66.7%, 31.6%, and 1.7% of the total pan-genome; (D) Percent paralogs (proteins present in more than one copy in an orthogroup) in each genome are primarily found in AO1-1 and CR12; (E) Phylogenetic relationships of F. solani based on orthogroups (n=10,650) and genome average nucleotide identity (ANI) show this species is not grouped according to lifestyle or origin. Nd, not determined.
3.5.3 Phylogenomic relationships between F. solani isolates
OrthoFinder version 2.5.5 (Emms and Kelly, 2017, 2019) generated the species tree by utilizing 10,650 orthogroups containing 136,545 genes of F. solani strains (Figure 4E). All F. solani strains are rooted in the two outgroups Fol4287 and Fg PH1. F. solani strains are divided into different clades but it is evident that the strains are not grouped based on their lifestyle or origin. For example, in one clade, the strains AO1-1, 1-Jan, F2, and Karbala-1 are closely related but have different lifestyles and origins. AO1-1 is a symbiont from sweet oranges in the USA, 1-Jan and Karbala-1 are pathogenic isolates from prawns in China and the cockscomb plant in Iraq (Shehan et al., 2023), respectively. F2 was isolated from the rhizospheric soil of Chinese ginseng with a lifestyle that needs to be clearly defined. These four strains share the same ancestor as SB1, an opportunistic isolate from sugarbeet in the USA, and FSSC 5, an endophytic isolate from Arabidopsis in Germany. The SB1 isolate was placed in the same clade as FSSC 5, confirming our previous result using the RPB2 gene (Navasca et al., 2023). Another clade sharing the same ancestor as the six previously mentioned strains is two isolates from China, the saprophyte NK-NH1 and CR12, with undetermined lifestyles. The remaining four strains, GU-3, Fvan, JS-169, and IISc-1, with various lifestyles and origins, comprise another clade. The GU3 strain from China and the pea pathogen Fvan from the USA are closely related and share a common ancestor with the mulberry endophytic JS-169 from South Korea. These three strains are related to the evergreen pathogen ISSc-1 from Iraq. Based on the branch lengths, JS-169 had the most significant genetic variation among F. solani genomes. The average nucleotide identity (ANI, %) analysis between F. solani genomes aligns with the outcomes of the species tree (Figure 4E). The closely related strains AO1-1, 1-Jan, F2, Karbala-1, SB1, and FSSC 5 exhibited a notable similarity, sharing more than 98.5% identity. CR12 and NK-NH1 displayed 93.7 to 94.8% identity with other strains and with each other. GU-3 and Fvan demonstrated a 91.2 to 91.5% similarity with other strains but a higher percentage at 98.0% with each other. JS-169 had comparatively lower ANI with other strains ranging from 90.6 to 91.0% except for its nearest neighbors GU-3 and Fvan, with 94.5% and 94.6%, respectively. IISc-1 exhibited the lowest ANI among strains with 90.5%, except when compared to strains, it shared a common ancestor with – GU-3, Fvan, and JS-169 – with identities ranging from 92.4 to 93.0%.
3.5.4 Enzymatic enrichment of F. solani dispensable genome
The top significant pathways enriched by core and dispensable (accessory and unique) genomes are shown in Figure 5. Over 100 core proteins (bars in dark blue in Figures 5A−C) belong to each of the several pathways in the core genome under biological processes (10/10), cellular components (6/10), and molecular function (4/20) while more than 50 proteins mostly occupy the rest. This contrasts with the dispensable genome, where most pathways are occupied by over 25 but less than 50 proteins (bars in lightest blue in Figures 5D−F). While this is the case, fold-enrichment of several pathways is higher in the dispensable genome than in the core genome. The role played by core proteins is crucial in the growth and development of fungi. This is evident in the enrichment of biological processes such as translation and several biosynthetic pathways, as well as activities occurring in the mitochondrion, ribosome, endoplasmic reticulum, and cellular envelopes. On the contrary, pathways enriched in the dispensable genome are more on metabolic pathways and vitamin biosynthetic processes, including cytoplasmic and ribosomal activities. These are additional measures that the fungi would undertake when necessary. We showed the top 10 pathways for biological processes (Figures 5A, D) and cellular components (Figures 5B, E) and the top 20 for molecular functions (Figures 5C, F) to emphasize the enrichment of enzymes in this category, particularly for dispensable proteins. Apart from structural and electron transport activities, six pathways of the core genome are enriched in enzymatic functions. Apart from transporters, 11 pathways are enriched with enzymatic functions in the dispensable genome, including hydrolases, transferases, oxidoreductases, lyases, ligase, isomerase, and dehydrogenase, with the highest fold enrichment at 9.65. All associations between core and dispensable proteins and GO terms are significant, with all FDR values below the threshold (p-value: 0.05).

Figure 5. The top gene ontology pathways of core (A−C) and dispensable (D−F) genomes are categorized under biological processes (A, D), cellular components (B, E), and molecular functions (C, F). The numbers after the bars represent statistical significance against the FDR threshold (p-value: 0.05). The dispensable genome has relatively higher enrichment in several pathways than the core genome. The highest enrichment was observed from shikimate 3-dehydrogenase (9.65) under the molecular function category of the dispensable genome.
4 Discussion
Fusarium species are known to acquire genomic regions or accessory chromosomes, allowing them to infect a wide range of hosts (van Dam et al., 2017; Yang et al., 2020). For instance, the nonpathogenic strain F. oxysporum 47 was able to infect the tomato when transformed with the pathogenicity chromosome of F. oxysporum f. sp. lycopersici 4287 (Ma et al., 2010). Members of the Fusarium solani species complex (FSSC) are pathogens, saprophytes, and opportunists associated with more than 100 hosts, including plants, animals, and even humans (Coleman et al., 2009). The ability of this group to adapt to different environments reflects their genetic diversity, making them notorious fungal pathogens. Coleman et al. (2009) reported that the expanded genome of Nectria haematococca MPVI, now called F. vanettenii strain 77-14-3, is due to the specific genes not found in other fungi and single-copy genes occurring in multiple copies. In this study, we identified repeat contents, transposable elements (TEs), and segmental duplications as contributing factors to the expanded genome of the F. solani SB1 isolate, particularly by the three chromosomes not found in the reference genome F. vanettenii strain 77-13-4. In addition to small size and low GC content, the high TEs and segmental duplications are characteristics of accessory chromosomes (Han et al., 2001; Hatta et al., 2002; Garmaroodi and Taga, 2007; Mehrabi et al., 2011; Coleman et al., 2009; Ma et al., 2010; Yang et al., 2020). A recent study established that segmental duplications shape accessory regions in F. oxysporum and F. solani (van Westerhoven et al., 2024). With these characteristics, we hypothesize that these three chromosomes, Chr13, Chr14, and Chr15 of the SB1 genome, are accessory chromosomes.
Accessory chromosomes (ACs) are linked to pathogenicity in F. oxysporum (Ma et al., 2010; Ma et al., 2013; Yang et al., 2020), but this has not been proven yet in Nectria haematococca (syn. F. solani). The pea pathogenicity (PEP) and pisatin demethylating ability (PDA) genes are present in the ACs of F. vanettenii 77-14-3 (Miao et al., 1991; Kistler et al., 1996; Wasmann and VanEtten, 1996; Han et al., 2001; Liu et al., 2003) and are known to enhance virulence. However, their absence does not significantly impact pathogenicity (Wasmann and VanEtten, 1996; Temporini and VanEtten, 2002). Because we did not find these genes in the SB1 genome, we suspect some other genes harbored by Chr13, Chr14, and Chr15 are responsible for the opportunistic habit of the SB1 isolate. Most genes carried by these chromosomes encode for chitinases 1 and 4, glycoside and glycosyl hydrolases, glucanase, lyase, tannase and esterase, and peptidase. These enzymes are involved in the assembly and degradation of carbohydrates (Lombard et al., 2014) and play a crucial role in pathogenesis by degrading the first line of plant defense, the cell walls (Kubicek et al., 2014). Glycoside hydrolases, an essential enzyme in cell wall degradation, are particularly enhanced in the SB1 genome, potentially aiding the entry of F. solani SB1 into sugarbeet.
Segmental duplications are long DNA segments (> 1 Kbp) with high sequence similarity (~ 90%) along multiple locations in chromosomes (Bailey et al., 2001; Hartmann, 2022). They are major sources of evolution and are found in genomes of primates and humans (Bailey et al., 2001; Vollger et al., 2022), Saccharomyces species (Dujon et al., 2004), and Candida albicans (Rustchenko et al., 1997). In the SB1 genome, massive segmental duplications are found in Chr13 and Chr14. This explains why the collinearity of F. vanettenii 77-14-3 Chr14 genes was found on both chromosomes. Because of this large duplication, we deliberated whether Chr13 and Chr14 are identical chromosomes. In the Hi-C contact map, Chr13 as scaffold 5 has a lighter red color, indicating minimal interaction with other chromosomes. In comparison, Chr14 as scaffold 8 is darker red, meaning its interaction with other chromosomes is more frequent. Apart from this difference in interaction, the genomic characteristics (size, GC content, repeat families, and number of genes) of these two chromosomes vary. These differences suggest that Chr13 and Chr14 are distinct chromosomes. However, telomeres were only assembled at one end of the chromosome, leaving the possibility that Chr13, Chr14, and Chr15 are arms of the same chromosomes or they are attached to one of the other chromosomes. Advancements in telomere-to-telomere assembly will resolve the genome structure and confirm karyotyping to confirm the chromosomes physically. We outsourced the independent and unbiased karyotyping services of www.lifeasible.com, where they estimated approximately 16 chromosomes of this strain (Supplementary Method and Supplementary Figure 1), which aligns closely with our findings. We acknowledge the inherent challenges in accurately determining the chromosome for this strain, as our attempts using the germ tube burst method were unsuccessful. However, this added confidence to the number of chromosomes in the final assembly. There are limited explanations for this case of Chr13 and Chr14. In Cryptococcus neoformans, Fraser et al. (2005) reported a meiotic event where two chromosomes fused, eventually splitting to form two new chromosomes sharing large segmental duplications. Another possible explanation is the involvement of transposable element activity in segmental duplications, as exhibited in the Fusarium banana pathogen Tropical Race 4 strain II (van Westerhoven et al., 2024). This is probable since Chr13 and Chr14 have high transposable elements, although we did not determine their proximity to the segmental duplications.
Fusarium solani has an open pan-genome but is nearing saturation, with only a few genes uncovered in 12 genomes. We speculate that despite the variation in size observed among F. solani genomes, the pan-genome is approaching saturation because the genome expansion is due to the duplication of genes, particularly those coding for enzymes, rather than the emergence of novel genes. This finding is also supported by the enrichment of enzymes in the dispensable genome rather than the core genome. Evident with the strains used in this study, F. solani has a wide host range and a diverse lifestyle - pathogens, saprophytes, and opportunistic. The enrichment of several enzymes, such as hydrolases, transferases, oxidoreductases, lyases, ligase, isomerase, and dehydrogenase, apparent by the GO terms in the dispensable genome, supports the ability of Fusarium solani to adapt to its varying environment. These enzymes carry out processes essential for adaptations and possibly the reason for the highly adaptive nature of this group.
Most evolutionary relationships are generated using single-copy orthologs, but recent papers argue that some information is lost by not including paralogs (Smith and Hahn, 2021; Smith et al., 2022; Ufimov et al., 2022). This is particularly important when inferring relationships in the concept of adaptation, which is one of the objectives of our study. Moreover, using single-copy orthologs in the phylogenomic analysis might bias the wide range of F. solani genomes from 45.81 to 66.64 Mb where paralogs occupy 8-10%. OrthoFinder version 2.5.5 (Emms and Kelly, 2017, 2019) generated the species tree using orthogroups (n=10,650) present in all strains of F. solani. Around 136,545 genes from these core orthogroups support that F. solani is not classified according to lifestyle or origin. The ANI values between F. solani genomes corroborate this idea. Our findings supplement those of Hoh et al. (2022), where members of the Fusarium solani species complex are not grouped by its animal or plant hosts.
Horizontal gene or chromosome transfer and hybridization between plant pathogenic fungi, especially Fusarium species, are ways for pathogens to broaden their host range (Ma et al., 2010, 2013; Mehrabi et al., 2011; Yang et al., 2022). Genome comparisons of other Fusarium species, particularly F. solani, residing in the soil where other crops, such as potato, dry bean, and soybean, are grown might shed more light on gene and chromosomal transfers within this group. Here, we identified Chr13, Chr14, and Chr15 of the F. solani SB1 isolate as potentially accessory chromosomes. Further investigation is needed to discern whether individual or all three chromosomes are necessary for the opportunistic habit of this isolate. The results we presented in this study provide additional evidence of the genome plasticity of the highly adaptive Fusarium solani.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
AN: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft, Writing – review & editing. JS: Methodology, Validation, Writing – review & editing. VR-v: Methodology, Writing – review & editing. UG: Methodology, Validation, Writing – review & editing. GS: Supervision, Writing – review & editing. TB: Conceptualization, Supervision, Validation, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
This work used resources of the Center for Computationally Assisted Science and Technology (CCAST) at North Dakota State University, which were made possible in part by NSF MRI Award No. 2019077. We would like to thank Lifeasible for performing the karyotyping analysis.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/ffunb.2025.1432339/full#supplementary-material
References
Ahearn D. G., Zhang S., Doyle Stulting R., Schwam B. L., Simmons R. B., Ward M. A., et al. (2008). Fusarium keratitis and contact lens wear: facts and speculations. Med. Myco. 46, 397–410. doi: 10.1080/13693780801961352
Armenteros J. J. A., Salvatore M., Emanuelsson O., Winther O., Von Heijne G., Elofsson, et al. (2019). Detecting sequence signals in targeting peptides using deep learning. Life Sci. Alliance. 2, e201900429. doi: 10.26508/lsa.201900429
Bailey J. A., Yavor A. M., Massa H. F., Trask B. J., Eichler E. E. (2001). Segmental duplications: organization and impact within the current human genome project assembly. Genome Res. 11, 1005–1017. doi: 10.1101/gr.187101
Bandi V., Gutwin C. (2020). “Interactive exploration of genomic conservation,” in Proceedings of the 46th Graphics Interface Conference 2020 (GI’20). Available online at: https://synvisio.github.io.
Batut B., Hiltemann S., Bagnacani A., Baker D., Bhardwaj V., Blank C., et al. (2019). Community-driven data analysis training for biology. Cell Syst. 6, 752–758. doi: 10.1016/j.cels.2018.05.012
Belofsky G. N., Jensen P. R., Fenical W. (1999). Sansalvamide: A new cytotoxic cyclic depsipeptide produced by a marine fungus of the genus Fusarium. Tetrahedron Lett. 40, 2913–2916. doi: 10.1016/S0040-4039(99)00393-7
Bian B. Z., Egusa S. (1981). Histopathology of black gill disease caused by Fusarium solani (Martius) infection in the Kuruma prawn, Penaeus japonicus Bate. J. Fish Dis. 4, 195–201. doi: 10.1111/j.1365-2761.1981.tb01126.x
Blin K., Shaw S., Kloosterman A. M., Charlop-Powers Z., Van Wezel G. P., Medema M. H., et al. (2021). antiSMASH 6.0: improving cluster detection and comparison capabilities. Nucleic Acids Res. 49, W29–W35. doi: 10.1093/nar/gkab335
Boral H., Van Diepeningen A., Erdem E., Yağmur M., De Hoog G. S., Ilkit M., et al. (2018). Mycotic keratitis caused by Fusarium solani sensu stricto (FSSC5): a case series. Mycopathologia 183, 835–840. doi: 10.1007/s11046-018-0280-7
Bretaudeau A. (2023). Genome annotation with maker (Galaxy training materials) (Accessed Feb 02, 2023).
Cabanes F. J., Alonso J. M., Castellá G., Alegre F., Domingo M., Pont S. (1997). Cutaneous hyalohyphomycosis caused by Fusarium solani in a loggerhead sea turtle (Caretta caretta L.). J. Clinic. Microbiol. 35, 3343–3345. doi: 10.1128/jcm.35.12.3343-3345.1997
Chen Y., Nie F., Xie S. Q., Zheng Y. F., Dai Q., Bray T., et al. (2021). Efficient assembly of nanopore reads via highly accurate and intact error correction. Nat. Commun. 12, 60. doi: 10.1038/s41467-020-20236-7
Chen W., Swart W. J. (2000). First report of stem canker of English walnut caused by Fusarium solani in South Africa. Plant Dis. 84, 592–592. doi: 10.1094/PDIS.2000.84.5.592A
Cissé O. H., Stajich J. E. (2019). FGMP: assessing fungal genome completeness. BMC Bioinf. 20, 1–9. doi: 10.1186/s12859-019-2782-9
Coleman J. J., Rounsley S. D., Rodriguez-Carres M., Kuo A., Wasmann C. C., Grimwood J., et al. (2009). The genome of Nectria haematococca: contribution of supernumerary chromosomes to gene expansion. PloS Genet. 5, e1000618. doi: 10.1371/journal.pgen.1000618
Crowhurst R. N., Binnie S. J., Bowen J. K., Hawthorne B. T., Plummer K. M., Rees-George J., et al. (1997). Effect of disruption of a cutinase gene (cutA) on virulence and tissue specificity of Fusarium solani f. sp. cucurbitae race 2 toward Cucurbita maxima and C. moschata. Mol. Plant-Microbe Interact. 10, 355–368. doi: 10.1094/MPMI.1997.10.3.355
Darling A. C., Mau B., Blattner F. R., Perna N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Dujon B., Sherman D., Fischer G., Durrens P., Casaregola S., Lafontaine I., et al. (2004). Genome evolution in yeasts. Nature 430, 35–44. doi: 10.1038/nature02579
Durand N. C., Robinson J. T., Shamim M. S., Machol I., Mesirov J. P., Lander E. S., et al. (2016). Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101. doi: 10.1016/j.cels.2015.07.012
Emms D. M., Kelly S. (2017). STRIDE: species tree root inference from gene duplication events. Mol. Bio. Evol. 34, 3267–3278. doi: 10.1093/molbev/msx259
Emms D. M., Kelly S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 1–14. doi: 10.1186/s13059-019-1832-y
Faust G. G., Hall I. M. (2014). SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics 30, 2503–2505. doi: 10.1093/bioinformatics/btu314
Flynn J. M., Hubley R., Goubert C., Rosen J., Clark A. G., Feschotte C., et al. (2020). RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. 117, 9451–9457. doi: 10.1073/pnas.1921046117
Fraser J. A., Huang J. C., Pukkila-Worley R., Alspaugh J. A., Mitchell T. G., Heitman J. (2005). Chromosomal translocation and segmental duplication in Cryptococcus neoformans. Eukaryot. Cell. 4, 401–406. doi: 10.1128/ec.4.2.401-406.2005
Fukasawa Y., Ermini L., Wang H., Carty K., Cheung M. S. (2020). LongQC: a quality control tool for third generation sequencing long read data. G3: Genes Genomes Genet. 10, 1193–1196. doi: 10.1534/g3.119.400864
García-Rodríguez G., Duque-Molina C., Kondo-Padilla I., Zaragoza-Jiménez C. A., González-Cortés V. B., Flores-Antonio R., et al. (2024). Outbreak of fusarium solani meningitis in immunocompetent persons associated with neuraxial blockade in durango, Mexico 2022–2023. Open Forum Infect. Dis. 11, ofad690. doi: 10.1093/ofid/ofad690
Garmaroodi H. S., Taga M. (2007). Duplication of a conditionally dispensable chromosome carrying pea pathogenicity (PEP) gene clusters in Nectria haematococca. Mol. Plant-Microbe Interact. 20, 1495–1504. doi: 10.1094/MPMI-20-12-1495
Ge S. X., Jung D., Yao R. (2020). ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics 36, 2628–2629. doi: 10.1093/bioinformatics/btz931
Geiser D. M., Al-Hatmi A. M., Aoki T., Arie T., Balmas V., Barnes I., et al. (2021). Phylogenomic analysis of a 55.1-kb 19-gene dataset resolves a monophyletic Fusarium that includes the Fusarium solani species complex. Phytopathol 111, 1064–1079. doi: 10.1094/PHYTO-08-20-0330-LE
Gibert S., Edel-Hermann V., Gautheron E., Gautheron N., Sol J. M., Capelle G., et al. (2022). First report of Fusarium avenaceum, Fusarium oxysporum, Fusarium redolens, and Fusarium solani causing root rot in pea in France. Plant Dis. 106, 1297. doi: 10.1094/PDIS-04-21-0833-PDN
Gíslason M. H., Nielsen H., Armenteros J. J. A., Johansen A. R. (2021). Prediction of GPI-anchored proteins with pointer neural networks. Curr. Biotechnol. 3, 6–13. doi: 10.1016/j.crbiot.2021.01.001
Godoy P., Nunes F., Silva V., Tomimori-Yamashita J., Zaror L., Fischman O. (2004). Onychomycosis caused by Fusarium solani and Fusarium oxysporum in Sao Paulo, Brazil. Mycopathol 157, 287–290. doi: 10.1023/B:MYCO.0000024186.32367.d4
Gupta A. K., Baran R., Summerbell R. C. (2000). Fusarium infections of the skin. Curr. Opin. Infect. Dis. 13, 121–128. doi: 10.1097/00001432-200004000-00005
Hallgren J., Tsirigos K. D., Pedersen M. D., Almagro Armenteros J. J., Marcatili P., Nielsen H., et al. (2022). DeepTMHMM predicts alpha and beta transmembrane proteins using deep neural networks. BioRxiv, 2022–2004. doi: 10.1101/2022.04.08.487609
Han M., Lee H. R., Choi M. N., Lee H. S., Lee S. W., Park E. J. (2017). First report of Fusarium solani causing soft rot on the tuber of Gastrodia elata in Korea. Plant Dis. 101, 1323–1323. doi: 10.1094/PDIS-03-17-0319-PDN
Han Y., Liu X., Benny U., Kistler H. C., VanEtten H. D. (2001). Genes determining pathogenicity to pea are clustered on a supernumerary chromosome in the fungal plant pathogen Nectria haematococca. Plant J. 25, 305–314. doi: 10.1046/j.1365-313x.2001.00969.x
Hanson L. E. (2006). First report of Fusarium yellows of sugar beet caused by Fusarium oxysporum in Michigan. Plant Dis. 90, 1554–1554. doi: 10.1094/PD-90-1554B
Hartmann F. E. (2022). Using structural variants to understand the ecological and evolutionary dynamics of fungal plant pathogens. New Phytol. 234, 43–49. doi: 10.1111/nph.17907
Hatta R., Ito K., Hosaki Y., Tanaka T., Tanaka A., Yamamoto M., et al. (2002). A conditionally dispensable chromosome controls host-specific pathogenicity in the fungal plant pathogen Alternaria alternata. Genet 161, 59–70. doi: 10.1093/genetics/161.1.59
Hoh D. Z., Lee H. H., Wada N., Liu W. A., Lu M. R., Lai C. K., et al. (2022). Comparative genomic and transcriptomic analyses of trans-kingdom pathogen Fusarium solani species complex reveal degrees of compartmentalization. BMC Biol. 20, 236. doi: 10.1186/s12915-022-01436-7
Holt C., Yandell M. (2011). MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinf. 12, 1–14. doi: 10.1186/1471-2105-12-491
Hose J. E., Lightner D. V., Redman R. M., Danald D. A. (1984). Observations on the pathogenesis of the imperfect fungus, Fusarium solani, in the California brown shrimp, Penaeus californiensis. J. Invert. Pathol. 44, 292–303. doi: 10.1016/0022-2011(84)90027-2
Huang G. X., Zhou X. M., Liu X. B., Cai J. M., Li B. X. (2016). First report of rubber tree gummosis disease caused by Fusarium solani in China. Plant Dis. 100, 1788–1788. doi: 10.1094/PDIS-10-15-1147-PDN
Jain C., Rodriguez-R L. M., Phillippy A. M., Konstantinidis K. T., Aluru S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9, 5114. doi: 10.1038/s41467-018-07641-9
Jones P., Binns D., Chang H. Y., Fraser M., Li W., McAnulla C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Käll L., Krogh A., Sonnhammer E. L. (2004). A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol. 338, 1027–1036. doi: 10.1016/j.jmb.2004.03.016
Kano R., Okayama T., Hamamoto M., Nagata T., Ohno K., Tsujimoto H., et al. (2001). Isolation of Fusarium solani from a dog: identification by molecular analysis. Med. Mycol. 40, 435–437. doi: 10.1080/mmy.40.4.435.437
Khan M. F. R., Liu Y., Bhuiyan M. Z. R., Lakshman D., Liu Z., Zhong S. (2021). First report of Fusarium equiseti causing seedling death on sugar beet in Minnesota, USA. Plant Dis. 105, 2017. doi: 10.1094/PDIS-10-20-2102-PDN
Kim J. A., Jeon J., Park S. Y., Kim K. T., Choi G., Lee H. J., et al. (2017). Genome sequence of an endophytic fungus, Fusarium solani JS-169, which has antifungal activity. Genome Announce. 5, 10–1128. doi: 10.1128/genomea.01071-17
Kiryu H., Yoshida S., Suenaga Y., Asahi M. (1991). Invasion and survival of Fusarium solani in the dexamethasone-treated cornea of rabbits. J. Med. Vet. Mycol. 29, 395–406. doi: 10.1080/02681219180000631
Kistler H. C., Meinhardt L. W., Benny U. (1996). Mutants of Nectria haematococca created by a site-directed chromosome breakage are greatly reduced in virulence toward pea. Mol. Plant-Microbe Interact. 9, 804–809. doi: 10.1094/MPMI-9-0804
Krzywinski M., Schein J., Birol I., Connors J., Gascoyne R., Horsman D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Kubicek C. P., Starr T. L., Glass N. L. (2014). Plant cell wall–degrading enzymes and their secretion in plant-pathogenic fungi. Annu. Rev. Phytopathol. 52, 427–451. doi: 10.1146/annurev-phyto-102313-045831
Kuruvilla T. S., Dias M. (2012). Fusarium solani: a causative agent of skin and nail infections. Indian J. Dermatol. 57, 308–309. doi: 10.4103/0019-5154.97680
Le V. K., Hatai K., Yuasa A., Sawada K. (2005). Morphology and molecular phylogeny of Fusarium solani isolated from kuruma prawn Penaeus japonicus with black gills. Fish Pathol. 40, 103–109. doi: 10.3147/jsfp.40.103
Letunic I., Bork P. (2021). Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Li H., Durbin R. (2010). Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Li B. J., Guo M. Y., Chai A. L. (2016). First report of Fusarium solani causing Fusarium root rot on okra (Abelmoschus esculentus) in China. Plant Dis. 100, 526. doi: 10.1094/PDIS-05-15-0588-PDN
Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li B. J., Li P. L., Li J., Chai A. L., Shi Y. X., Xie X. W. (2017). First report of fusarium root rot of Solanum melongena caused by Fusarium solani in China. Plant Dis. 101, 1956–1956. doi: 10.1094/PDIS-11-16-1559-PDN
Li B. J., Liu Y., Shi Y. X., Xie X. W., Guo Y. L. (2010). First report of crown rot of grafted cucumber caused by Fusarium solani in China. Plant Dis. 94, 1377–1377. doi: 10.1094/PDIS-03-10-0217
Li H., Wang S., Chai S., Yang Z., Zhang Q., Xin H., et al. (2022). Graph-based pan-genome reveals structural and sequence variations related to agronomic traits and domestication in cucumber. Nat. Commun. 13, 682. doi: 10.1038/s41467-022-28362-0
Liu X., Inlow M., VanEtten H. D. (2003). Expression profiles of pea pathogenicity (PEP) genes in vivo and in vitro, characterization of the flanking regions of the PEP cluster and evidence that the PEP cluster region resulted from horizontal gene transfer in the fungal pathogen Nectria haematococca. Curr. Genet. 44, 95–103. doi: 10.1007/s00294-003-0428-x
Liu Z., No E. G., Clement C. D., He P., Shan L. (2022). “Isolation of high-molecular-weight (HMW) DNA from fusarium oxysporum for long-read sequencing,” in Fusarium wilt. Methods in molecular biology, vol. 2391 . Ed. Coleman J. (Humana, New York, NY). doi: 10.1007/978-1-0716-1795-3_2
Liu H., Zhang B., Li C., Bao X. (2010). Knock down of chitosanase expression in phytopathogenic fungus Fusarium solani and its effect on pathogenicity. Curr. Genet. 56, 275–281. doi: 10.1007/s00294-010-0299-x
Lombard V., Golaconda Ramulu H., Drula E., Coutinho P. M., Henrissat B. (2014). The carbohydrate-active enzymes database (CAZy). Nucleic Acids Res. 42, D490–D495. doi: 10.1093/nar/gkt1178
Ma L. J., Geiser D. M., Proctor R. H., Rooney A. P., O’Donnell K., Trail F., et al. (2013). Fusarium pathogenomics. Annu. Rev. Microbiol. 67, 399–416. doi: 10.1146/annurev-micro-092412-155650
Ma L. J., van der Does H., Borkovich K., Coleman J. J., Daboussi M.-J., Di Pietro A., et al. (2010). Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 464, 367–373. doi: 10.1038/nature08850
Madden T. (2013). “The BLAST sequence analysis tool,” in The NCBI handbook, 2nd edition (National Center for Biotechnology Information, US).
Manni M., Berkeley M. R., Seppey M., Simão F. A., Zdobnov E. M. (2021). BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evolution. 38, 4647–4654. doi: 10.1093/molbev/msab199
Mehmood N., Riaz A., Jabeen N., Anwaar S., Hussain S. Z., Abbas M. F., et al. (2017). First report of Fusarium solani causing fruit rot of strawberry in Pakistan. Plant Dis. 101, 1681–1681. doi: 10.1094/PDIS-12-16-1825-PDN
Mehrabi R., Bahkali A. H., Abd-Elsalam K. A., Moslem M., Ben M’Barek S., Gohari A. M., et al. (2011). Horizontal gene and chromosome transfer in plant pathogenic fungi affecting host range. FEMS Microbio. Rev. 35, 542–554. doi: 10.1111/j.1574-6976.2010.00263.x
Mesny F., Miyauchi S., Thiergart T., Pickel B., Atanasova L., Karlsson M., et al. (2021). Genetic determinants of endophytism in the Arabidopsis root mycobiome. Nat. Commun. 12, 7227. doi: 10.1038/s41467-021-27479-y
Miao V. P., Matthews D. E., VanEtten H. D. (1991). Identification and chromosomal locations of a family of cytochrome P-450 genes for pisatin detoxification in the fungus Nectria haematococca. Mol. Gen. Gen. 226, 214–223. doi: 10.1007/BF00273606
Mikheenko A., Prjibelski A., Saveliev V., Antipov D., Gurevich A. (2018). Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 34, i142–i150. doi: 10.1093/bioinformatics/bty266
Mulero-Aparicio A., Agustí-Brisach C., Raya M. D. C., Lovera M., Arquero O., Trapero A. (2019). First report of fusarium solani causing stem canker in english walnut in Spain. Plant Dis. 103, 3281–3281. doi: 10.1094/PDIS-06-19-1163-PDN
Navasca A., Singh J., Rivera-Varas V., Geddes B., Secor G., Gill U., et al. (2023). First report and draft genome resource of a unique opportunistic Fusarium solani pathogen associated with unknown dark galls in sugarbeet. PhytoFront 3, 704–707. doi: 10.1094/PHYTOFR-01-23-0006-A
Nevers Y., Glover N. M., Dessimoz C., Lecompte O. (2023). Protein length distribution is remarkably uniform across the tree of life. Genome Biol. 24, 135. doi: 10.1186/s13059-023-02973-2
Nisa Q., Shahnaz E., Banday S., Anwar A., Masoodi K. Z., Hamid A., et al. (2021). First Report of Fusarium solani Associated with the Bulb Rot of Tulip (Tulipa spp.) in India. Plant Dis. 106, 759. doi: 10.1094/PDIS-05-21-0916-PDN
O’Donnell K., Al-Hatmi A. M., Aoki T., Brankovics B., Cano-Lira J. F., Coleman J. J., et al. (2020). No to Neocosmospora: phylogenomic and practical reasons for continued inclusion of the Fusarium solani species complex in the genus Fusarium. Msphere 5, 10–1128. doi: 10.1128/mSphere.00810-20
O’Donnell K., Sutton D. A., Fothergill A., McCarthy D., Rinaldi M. G., Brandt M. E., et al. (2008). Molecular phylogenetic diversity, multilocus haplotype nomenclature, and in vitro antifungal resistance within the Fusarium solani species complex. J. Clin. Microbiol. 46, 2477–2490. doi: 10.1128/JCM.02371-07
Ortega-Rosales A., Quizhpe-Ocampo Y., Montalvo-Flores M., Burneo-Rosales C., Romero-Ulloa G. (2019). A case of fungal keratitis due to Fusarium solani after an indigenous healing practice. IDCases 18, e00618. doi: 10.1016/j.idcr.2019.e00618
Pai R., Boloor R., Shreevidya K., Shenoy D. (2010). Fusarium solani: an emerging fungus in chronic diabetic ulcer. J. Lab. Phys. 2, 037–039. doi: 10.4103/0974-2727.66710
Pastrana A. M., Capote N., De los Santos B., Romero F., Basallote-Ureba M. J. (2014). First report of Fusarium solani causing crown and root rot on strawberry crops in southwestern Spain. Plant Dis. 98, 161–161. doi: 10.1094/PDIS-07-13-0682-PDN
Perez B. A., Farinon O. M., Berretta M. F. (2011). First report of Fusarium solani causing root rot of olive in southeastern Argentina. Plant Dis. 95, 1476–1476. doi: 10.1094/PDIS-02-11-0095
Pérez-Wohlfeil E., Diaz-del-Pino S., Trelles O. (2019). Ultra-fast genome comparison for large-scale genomic experiments. Sci. Rep. 9, 10274. doi: 10.1038/s41598-019-46773-w
Ramdial H. A., Rampersad S. N. (2010). First report of Fusarium solani causing fruit rot of sweet pepper in Trinidad. Plant Dis. 94, 1375–1375. doi: 10.1094/PDIS-06-10-0433
Rampersad S. N. (2009). First report of Fusarium solani fruit rot of pumpkin (Cucurbita pepo) in Trinidad. Plant Dis. 93, 547–547. doi: 10.1094/PDIS-93-5-0547B
Rao S. S., Huntley M. H., Durand N. C., Stamenova E. K., Bochkov I. D., Robinson J. T., et al. (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. doi: 10.1016/j.cell.2014.11.021
Rasche H., Hiltemann S. (2020). Galactic Circos: User-friendly Circos plots within the Galaxy platform. Gigascience 9, giaa065. doi: 10.1093/gigascience/giaa065
Rivera V., Rengifo J., Khan M., Geiser D. M., Mansfield M., Secor G. (2008). First report of a novel Fusarium species causing yellowing decline of sugar beet in Minnesota. Plant Dis. 92, 1589–1589. doi: 10.1094/PDIS-92-11-1589B
Rogers L. M., Kim Y. K., Guo W., González-Candelas L., Li D., Kolattukudy P. E. (2000). Requirement for either a host-or pectin-induced pectate lyase for infection of Pisum sativum by Nectria hematococca. Proc. Natl. Acad. Sci. 97, 9813–9818. doi: 10.1073/pnas.160271497
Romans-Fuertes P., Sondergaard T. E., Sandmann M. I. H., Wollenberg R. D., Nielsen K. F., Hansen F. T., et al. (2016). Identification of the non-ribosomal peptide synthetase responsible for biosynthesis of the potential anti-cancer drug sansalvamide in Fusarium solani. Curr. Genet. 62, 799–807. doi: 10.1007/s00294-016-0584-4
Rustchenko E. P., Howard D. H., Sherman F. (1997). Variation in assimilating functions occurs in spontaneous Candida albicans mutants having chromosomal alterations. Microbiol 143, 1765–1778. doi: 10.1099/00221287-143-5-1765
Sarmiento-Ramírez J. M., Abella E., Martín M. P., Tellería M. T., Lopez-Jurado L. F., Marco A., et al. (2010). Fusarium solani is responsible for mass mortalities in nests of loggerhead sea turtle, Caretta caretta, in Boavista, Cape Verde. FEMS Microbiol. Let. 312, 192–200. doi: 10.1111/j.1574-6968.2010.02116.x
Secor G. A., Rivera-Varas V., Christ D. S., Mathew F. M., Khan M. F., Varrelmann M., et al. (2014). Characterization of Fusarium secorum, a new species causing Fusarium yellowing decline of sugar beet in north central USA. Fungal Biol. 118, 764–775. doi: 10.1016/j.funbio.2014.06.001
Shehan S. J., Abdalmoohsin R. G., Jeddoa Z. M., Lahuf A. A. (2023). First report of cockscomb root rot caused by fusarium solani in Iraq. PHP 24, 525–526. doi: 10.1094/PHP-05-23-0048-BR
Simão F. A., Waterhouse R. M., Ioannidis P., Kriventseva E. V., Zdobnov E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Smit A., Hubley R., Green P. (2003). RepeatMasker open-4.0. Available online at: http://www.repeatmasker.org (Accessed 12 February 2023).
Smith D. J., Gold J. A., Chiller T., Bustamante N. D., Marinissen M. J., Rodriquez G. G., et al. (2024). Update on outbreak of fungal meningitis among US residents who received epidural anesthesia at two clinics in Matamoros, Mexico. Clin. Infect. Dis. 78 (6), 1554–1558. doi: 10.1093/cid/ciad570
Smith M. L., Hahn M. W. (2021). New approaches for inferring phylogenies in the presence of paralogs. Trend. Genet. 37, 174–187. doi: 10.1016/j.tig.2020.08.012
Smith M. L., Vanderpool D., Hahn M. W. (2022). Using all gene families vastly expands data available for phylogenomic inference. Mol. Bio. Evol. 39, msac112. doi: 10.1093/molbev/msac112
Sperschneider J., Dodds P. N. (2022). EffectorP 3.0: prediction of apoplastic and cytoplasmic effectors in fungi and oomycetes. Mol. Plant-Microbe Interact. 35, 146–156. doi: 10.1094/MPMI-08-21-0201-R
Stanke M., Waack S. (2003). Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19, 215–225. doi: 10.1093/bioinformatics/btg1080
Strong N., Meeks G., Sheth S. A., McCullough L., Villalba J. A., Tan C., et al. (2024). Neurovascular complications of iatrogenic fusarium solani meningitis. NEJM 390, 522–529. doi: 10.1056/NEJMoa2308192
Studt L., Wiemann P., Kleigrewe K., Humpf H. U., Tudzynski B. (2012). Biosynthesis of fusarubins accounts for pigmentation of Fusarium fujikuroi perithecia. Appl. Environ. Microbio. 78, 4468–4480. doi: 10.1128/AEM.00823-12
Sun Y., Wang R., Qiao K., Pan H., Wang F., Wang Y., et al. (2022). First report of Fusarium solani causing leaf sheath rot of Bush Lily in China. Plant Dis. 106, 1992. doi: 10.1094/PDIS-02-21-0414-PDN
Tanaka M., Izawa T., Kuwamura M., Nakao T., Maezono Y., Ito S., et al. (2012). Deep granulomatous dermatitis of the fin caused by Fusarium solani in a false killer whale (Pseudorca crassidens). J. Vet. Med. Sci. 74, 779–782. doi: 10.1292/jvms.11-0421
Temporini E. D., VanEtten H. D. (2002). Distribution of the pea pathogenicity (PEP) genes in the fungus Nectria haematococca mating population VI. Curr. Genet. 41, 107–114. doi: 10.1007/s00294-002-0279-x
Teufel F., Almagro Armenteros J. J., Johansen A. R., Gíslason M. H., Pihl S. I., Tsirigos K. D., et al. (2022). SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 40, 1023–1025. doi: 10.1038/s41587-021-01156-3
Trovato L., Marino A., Pizzo G., Oliveri S. (2021). Case report: molecular diagnosis of fungal keratitis associated with contact lenses caused by Fusarium solani. Front. Med. 8. doi: 10.3389/fmed.2021.579516
Tuerdi M., Lv C., Dan H., Shan L., Zhang R., Chen X. (2023). First Report of Fusarium solani Associated with Twig Canker of English Walnut (Juglans regia) in Xinjiang, China. Plant Dis. 107, 2858. doi: 10.1094/PDIS-03-23-0430-PDN
Ufimov R., Gorospe J. M., Fér T., Kandziora M., Salomon L., van Loo M., et al. (2022). Utilizing paralogues for phylogenetic reconstruction has the potential to increase species tree support and reduce gene tree discordance in target enrichment data. Mol. Ecol. Resour. 22, 3018–3034. doi: 10.1111/1755-0998.13684
Urban M., Cuzick A., Seager J., Wood V., Rutherford K., Venkatesh S. Y., et al. (2022). PHI-base in 2022: a multi-species phenotype database for Pathogen–Host Interactions. Nucleic Acids Res. 50, D837–D847. doi: 10.1093/nar/gkab1037
van Dam P., Fokkens L., Ayukawa Y., van der Gragt M., Ter Horst A., Brankovics B., et al. (2017). A mobile pathogenicity chromosome in Fusarium oxysporum for infection of multiple cucurbit species. Sci. Rep. 7, 9042. doi: 10.1038/s41598-017-07995-y
VanEtten H. D. (1978). Identification of additional habitats of Nectria haematococca mating population VI. Phytopathol 68, 1552–1556. doi: 10.1094/Phyto-68-1552
van Westerhoven A. C., Aguilera-Galvez C., Nakasato-Tagami G., Shi-Kunne X., Martinez de la Parte E., Chavarro-Carrero E., et al. (2024). Segmental duplications drive the evolution of accessory regions in a major crop pathogen. New Phytol. 242, 610:625. doi: 10.1111/nph.19604
Villarino M., de la Lastra E., Basallote-Ureba M. J., Capote N., Larena I., Melgarejo P., et al. (2019). Characterization of Fusarium solani populations associated with Spanish strawberry crops. Plant Dis. 103, 1974–1982. doi: 10.1094/PDIS-02-19-0342-RE
Vollger M. R., Guitart X., Dishuck P. C., Mercuri L., Harvey W. T., Gershman A., et al. (2022). Segmental duplications and their variation in a complete human genome. Science 376, eabj6965. doi: 10.1126/science.abj6965
Wang R. Y., Gao B., Li X. H., Ma J., Chen S. L. (2014). First report of Fusarium solani causing Fusarium root rot and stem canker on storage roots of sweet potato in China. Plant Dis. 98, 160–160. doi: 10.1094/PDIS-06-13-0651-PDN
Wang Y., Tang H., DeBarry J. D., Tan X., Li J., Wang X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49–e49. doi: 10.1093/nar/gkr1293
Wasmann C. C., VanEtten H. D. (1996). Transformation-mediated chromosome loss and disruption of a gene for pisatin demethylase decrease the virulence of Nectria haematococca on pea. Mol. Plant-Microbe Interact. 9, 793–803. doi: 10.1094/MPMI-9-0793
Wick R., Volkening J., Loman N. (2017). Porechop: adapter trimmer for Oxford Nanopore reads. Available online at: https://github.com/rrwick/Porechop (Accessed March 15, 2022).
Wu S. Y., El-Borai F. E., Graham J. H., Duncan L. W. (2018). The saprophytic fungus Fusarium solani increases the insecticidal efficacy of the entomopathogenic nematode Steinernema diaprepesi. J. Invertebr. Pathol. 159, 87–94. doi: 10.1016/j.jip.2018.10.004
Xie Y., Ying J., Fang L., Wu J., Wang L., Mao X., et al. (2024). First report of Fusarium solani causing Fusarium wilt of Bletilla striata (hyacinth orchid) in Zhejiang province of China. Plant Dis. 108, 204. doi: 10.1094/PDIS-01-23-0049-PDN
Yang M., Cao J., Zheng Y., Wang J., Zhou H., He M., et al. (2020). First report of Fusarium root rot of tobacco caused by Fusarium solani in Lincang, China. Plant Dis. 104, 1541. doi: 10.1094/PDIS-08-19-1711-PDN
Yang H., Yu H., Ma L. J. (2020). Accessory chromosomes in Fusarium oxysporum. Phytopathology 110, 1488–1496. doi: 10.1094/PHYTO-03-20-0069-IA
Zhang Y., Ma H. Y., Liu S. H., Hu H. Q., Ren L., Wang Y. Y. (2024). First report of leaf spot disease on pineapple caused by fusarium solani in China. Plant Dis. 108, 528. doi: 10.1094/PDIS-09-23-1791-PDN
Zhang N., O’Donnell K., Sutton D. A., Nalim F. A., Summerbell R. C., Padhye A. A., et al. (2006). Members of the Fusarium solani species complex that cause infections in both humans and plants are common in the environment. J. Clin. Microbiol. 44, 2186–2190. doi: 10.1128/jcm.00120-06
Zhang Y., Yang H., Turra D., Zhou S., Ayhan D. H., DeIulio G. A., et al. (2020). The genome of opportunistic fungal pathogen Fusarium oxysporum carries a unique set of lineage-specific chromosomes. Commun. Biol. 3, 50. doi: 10.1038/s42003-020-0770-2
Zhao Y., Jia X., Yang J., Ling Y., Zhang Z., Yu J., et al. (2014). PanGP: a tool for quickly analyzing bacterial pan-genome profile. Bioinformatics 30, 1297–1299. doi: 10.1093/bioinformatics/btu017
Zheng J., Ge Q., Yan Y., Zhang X., Huang L., Yin Y. (2023). dbCAN3: automated carbohydrate-active enzyme and substrate annotation. Nucleic Acids Res. 51, W115–W121. doi: 10.1093/nar/gkad328
Keywords: Fusarium solani, pan-genome, accessory chromosomes, segmental duplications, genome plasticity
Citation: Navasca A, Singh J, Rivera-Varas V, Gill U, Secor G and Baldwin T (2025) Dispensable genome and segmental duplications drive the genome plasticity in Fusarium solani. Front. Fungal Biol. 6:1432339. doi: 10.3389/ffunb.2025.1432339
Received: 13 May 2024; Accepted: 20 January 2025;
Published: 05 February 2025.
Edited by:
Toni Gabaldón, Barcelona Supercomputing Center, SpainCopyright © 2025 Navasca, Singh, Rivera-Varas, Gill, Secor and Baldwin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas Baldwin, dGhvbWFzLnQuYmFsZHdpbkBuZHN1LmVkdQ==