 Leilani S. Dacones
Leilani S. Dacones Robert C. Kemerait Jr.2
Robert C. Kemerait Jr.2 Marin T. Brewer
Marin T. Brewer
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Fungal Biol. , 22 July 2022
Sec. Fungi-Plant Interactions
Volume 3 - 2022 | https://doi.org/10.3389/ffunb.2022.910232
Numerous plant-pathogenic fungi secrete necrotrophic effectors (syn. host-selective toxins) that are important determinants of pathogenicity and virulence in species that have a necrotrophic lifestyle. Corynespora cassiicola is a necrotrophic fungus causing emerging target spot epidemics in the southeastern United States (US). Previous studies revealed that populations of C. cassiicola from cotton, soybean, and tomato are clonal, host specialized and genetically distinct. Additionally, cassiicolin – the necrotrophic effector identified in some C. cassiicola isolates – is an important toxin for virulence on rubber. It is encoded by seven Cas gene variants. Our goal was to conduct comparative genomic analyses to identify variation among putative necrotrophic effector genes and to determine if lack of one of the mating-types explained clonal populations in C. cassiicola causing outbreaks in the southeastern US and the apparent absence of sexual reproduction worldwide. A total of 12 C. cassiicola genomes, with four each from isolates from tomato, soybean, and cotton, were sequenced using an Illumina Next Seq platform. Each genome was assembled de novo, compared with the reference genome from rubber, and searched for known Cas, and other gene clusters with homologs of secondary metabolites. Cas2 and/or Cas6 were present in isolates from soybean in the southeastern US, whereas Cas1 and Cas2 were present in isolates from cotton in the southeastern US. In addition, several toxin genes, including the T-toxin biosynthetic genes were present in all C. cassiicola from cotton, soybean, and tomato. The mating-type locus was identified in all of the sequenced genomes, with the MAT1-1 idiomorph present in all cotton isolates and the rubber isolate, whereas the MAT1-2 idiomorph was present in all soybean isolates. We developed a PCR-based marker for mating-type in C. cassiicola. Both mating types were present in isolates from tomato. Thus, C. cassiicola has both mating-types necessary for sexual reproduction, but the absence of both mating-types within soybean and cotton populations could explain clonality in these populations. Variation in necrotrophic effectors may underlie host specialization and disease emergence of target spot on cotton, soybean, and tomato in the southeastern US.
Evolutionary processes play a vital role in shaping plant disease epidemics and populations of their causal pathogens over time and space (Milgroom and Peever, 2003). In agricultural environments extensive monoculture combined with disease management practices that result in strong selective pressures, especially single-site fungicides and host plant resistance mediated by a single gene, can drive evolutionary arms races over relatively short timescales. These selective pressures contribute to changes in virulence and major shifts in the predominant pathogen populations interacting with the hosts and causing disease outbreaks. An example of a major shift is the rapid emergence in the 1970’s of Bipolaris maydis (syn. Cochliobolus heterostrophus) race T that caused severe epidemics of Southern Corn Leaf Blight (SCLB). Race T produces T-toxin (Kono et al., 1980), which was lethal to the widely-planted Texas cytoplasmic male sterile maize (T-cms) (Dewey et al., 1988; Levings, 1990). T-toxin is a host-selective toxin (syn. necrotrophic effector) produced by B. maydis race T (Kono and Daly, 1979) that is absent in B. maydis race O, which was not very virulent and had been the predominant race prior to the SCLB epidemics. The maize mitochondrial protein, Urf13 (Levings, 1993), conferring male sterility also confers sensitivity to T-toxin, thereby resulting in elevated virulence of race T and widespread epidemics of SCLB in the 1970’s. In addition to increasing virulence, host selective toxins also contribute to the evolution of host specificity.
Plant pathogens are often adapted to different host species, or even more specifically to different host cultivars (Burdon and Silk, 1997). The initial cultivar or host jump may lead to the breakdown of host resistance and emergence of a new disease. Over time population genetic subdivision resulting from host specialization results in genetic differences and allows pathogens to maintain population diversity and subsequently help to evade extinction (Fournier and Giraud, 2007). An understanding genetic differentiation by host aids in implementing proper disease management. Host specialization is particularly common for fungal pathogens with biotrophic lifestyles, such as rusts and powdery mildews, that have intimate associations with their host (Duplessis et al., 2011; Ohm et al., 2012). For necrotrophic plant pathogens, host specialization often involves mechanisms such as the production of host-selective toxins (Friesen et al., 2008; Friesen and Faris, 2012), or acquisition of conditionally dispensable chromosomes (also known as supernumerary, accessory, or lineage-specific chromosomes) (Hatta et al., 2002; Ma et al., 2010; Vlaardingerbroek et al., 2016) that carry genes for pathogenicity or virulence that can be moved via horizontal gene transfer (HGT) (Soanes and Richards, 2014). Pyrenophora tritici-repentis is a necrotroph that causes tan spot of wheat. It is virulent on wheat due to the production of the necrotrophic effector ToxA that mediates host-specific interactions (Ciuffetti et al., 1997). The increase in virulence was attributed to the acquisition of ToxA via HGT (Friesen et al., 2006) from another wheat pathogen, Stagonospora nodorum, which also produces ToxA. Another example where necrotrophic effectors are involved in virulence and host specificity is in Alternaria alternata, which has a wide host range (Gilchrist and Grogan, 1976); however, host specificity of pathotypes conferred by diverse necrotrophic effectors results in at least 11 different diseases of unique host plants (Tsuge et al., 2013).
Corynespora cassiicola is a ubiquitous saprotrophic and necrotrophic fungus commonly found in tropical and subtropical areas (Dixon et al., 2009). It has a wide host range across plant species (Farr and Rossman, 2017), but may be best known for causing Corynespora leaf fall of rubber (Hevea brasiliensis), a devastating disease resulting in severe economic losses in Asia and Africa (Chee, 1990). Recently, C. cassiicola has been causing emerging target spot epidemics in the southeastern United States (US) on cotton (Gossypium hirsutum) (Campbell et al., 2012; Edmisten, 2012; Fulmer et al., 2012; Mehl and Phipps, 2013; Price et al., 2015; Butler et al., 2016), soybean (Glycine max) (Koenning et al., 2006; Bennett, 2016; Faske, 2016; Edwards Molina et al., 2022), and tomato (Solanum lycopersicon) (Schlub et al., 2009). Phylogenetic and population genetic analyses of C. cassiicola from cotton, soybean, and tomato in the southeastern US showed three genetically distinct populations that clustered based on the host of origin (Sumabat et al., 2018a; Sumabat et al., 2018b). In addition, isolates were shown to be most aggressive when inoculated on the same host as the host of origin providing evidence for host specialization. The underlying genetic basis for host specialization in these populations is unknown.
As a necrotroph C. cassiicola kills plant tissue, which is most often the foliage and sometimes fruits. Characteristic symptoms include necrotic lesions forming a target-like appearance with concentric rings surrounded by a yellow margin. Often, these symptoms are followed by leaf drop or premature defoliation of both mature and immature leaves (Fulmer et al., 2012). Severe infections can lead to massive defoliation and subsequent death of infected plants (de Lamotte et al., 2007). The symptoms of target spot are characteristic of toxin involvement. Cassiicolin – a phytotoxic protein – is a necrotrophic effector in C. cassiicola isolates from rubber and determined to play a role in pathogenicity and virulence (Breton et al., 2000; de Lamotte et al., 2007). The precursor of cassiicolin is encoded by the gene Cas1, which is expressed by C. cassiicola in the early stages of infection of rubber (Déon et al., 2012). Seven variants of cassiicolin-encoding genes (Cas1 to Cas7) have been identified among isolates from different hosts and diverse geographic regions (Déon et al., 2014; Lopez et al., 2018). The Cas gene variants encode different isoforms of the cassiicolin toxin. The variants are located in different regions of C. cassiicola genomes and when more than one variant was detected in a single genome they were not clustered (Lopez et al., 2018). Only 47% of the characterized isolates had Cas genes encoding for cassiicolin; however, isolates without Cas genes were still virulent, including some to rubber, showing that factors other than cassiicolin are involved in virulence and host specialization (Déon et al., 2014). A secreted toxin in culture filtrate was determined to play a role in pathogenicity of C. cassiicola to tomato but was not further characterized (Onesirosan et al., 1975). The role of Cas variants in pathogenicity and virulence of C. cassiicola populations causing epidemics in the southeastern US is unknown.
In fungal populations causing emerging plant diseases sexual reproduction is often absent and reproduction is strictly clonal. Sexual reproductive structures have not been observed in C. cassiicola and it is considered strictly clonal and assumed to only reproduce asexually (Schoch et al., 2009). All phylogenetic and population genetic studies to date, including those on the host-specialized populations of C. cassiicola causing emerging target spot epidemics in the southeastern US, indicate that C. cassiicola is clonal and lacks sexual reproduction (Silva et al., 2002; Dixon et al., 2009; Déon et al., 2014; Sumabat et al., 2018a; Sumabat et al., 2018b). Sumabat et al. (2018a) showed evidence of recombination among the C. cassiicola lineages from different hosts in the southeastern US. However, these recombination events did not likely occur recently (Sumabat et al., 2018b) or could be due to homoplasy since the reticulations were detected deep among the lineages. Aside from recombination, another indirect molecular method to infer sexual reproduction in fungi involves detection of compatible mating-types within the species or population of interest (Fraser and Heitman, 2003). The mating-type locus, MAT1, is present in both sexually and asexually reproducing members of Ascomycota (Debuchy and Turgeon, 2006). In heterothallic Ascomycota, MAT1 has two variants, or idiomorphs, which together contain the genes necessary for sexual reproduction to occur; individuals must be different idiomorphs at MAT1 to be sexually compatible. In homothallic Ascomycota, all of the genes for mating are present in a single individual. Therefore, the structure of MAT1 in a species and composition of idiomorphs in a population provide valuable information on the mating system of that fungal species or population. Until now the MAT1 locus in C. cassiicola had not been described, so it was unknown if the species is homothallic or heterothallic, or if populations are not able to reproduce sexually since they are lacking one of the mating-type idiomorphs.
The objectives of this study were to conduct comparative genomic analysis of host-specialized populations of C. cassiicola to: 1) identify variation among putative necrotrophic effector genes, including Cas variants and secondary metabolites, and 2) to identify and characterize MAT1 in C. cassiicola and variation in mating-types among populations causing outbreaks in the southeastern US. Knowledge on the diversity of necrotrophic effector genes and the reproductive biology is critical in understanding the genetic basis of host specialization and disease emergence of target spot of cotton, soybean, and tomato in the southeastern US.
Twelve C. cassiicola isolates from the three host-specialized populations in the southeastern US – cotton, soybean, and tomato – were selected for whole genome sequencing (Table 1). High quality genomic DNA was extracted using the cetyl trimethylammonium bromide (CTAB) method (Fulton et al., 1995) adapted from the 1000 Fungal Genome Project of Joint Genome Institute – Department of Energy (JGI-DOE, 1000.fungalgenomes.org). Briefly, isolates were grown on quarter-strength potato dextrose agar (qPDA) overlaid with sterile cellophane and incubated at 25°C for 7 days in the dark. Mycelium of each isolate was scraped from the cellophane with a spatula and ground in liquid nitrogen. Approximately 500 mg finely-ground mycelium was mixed with 17.5 ml CTAB lysis buffer, which consisted of: 6.5 ml Buffer A (0.35 M sorbitol; 0.1 M Tris-HCl, pH 9; and 5 mM EDTA, pH 8), 6.5 ml Buffer B (0.2 M Tris-HCl, pH 9; 50 mM EDTA, pH 8; 2 M NaCl; and 2% CTAB), 2.6 ml of Buffer C (5% Sarkosyl), 1.75 ml PVP (0.1%), and 1.25 μl Proteinase K. The mixture was shaken with two 5-mm glass beads (VWR Soda Lime, Radnor, PA, USA) at 1750 RPM for 2 min followed by 1 min using a 2010 Geno/Grinder (SPEX SamplePrep, Metuchen, NJ, USA). Next, 5.75 ml of 5 M potassium acetate was added to the tube then it was inverted 10 times. The mixture was incubated on ice for 30 min then centrifuged for 20 min at 14, 000 g. The supernatant was added to one volume of chloroform:isoamylalcohol (v/v 24:1) and subsequently centrifuged at 14, 000 g for 10 min. The resulting supernatant was mixed with 100 μl Rnase A (10 mg/ml) and incubated at 37°C for 2 hr. Isopropanol at equal volume and sodium acetate at 1/10 volume were then added, incubated at 25°C for 5 min, and centrifuged at 14, 000 g for 30 min. The supernatant was discarded, and the resulting pellet was rinsed twice with 70% ethanol and air-dried overnight. The DNA pellet was eluted with 500 μl deionized H2O. Genomic DNA was submitted to the Georgia Genomics and Bioinformatics Core (Athens, GA, USA) for library preparation of each isolate and Illumina sequencing using NextSeq platform based on a paired-end 150-bp (PE150) protocol. The genome of C. cassiicola isolate CCP from rubber (Lopez et al., 2018) was downloaded from NCBI (assembly ID: GCA_003016335.1, BioProject accession: PRJNA234811) for further analyses.
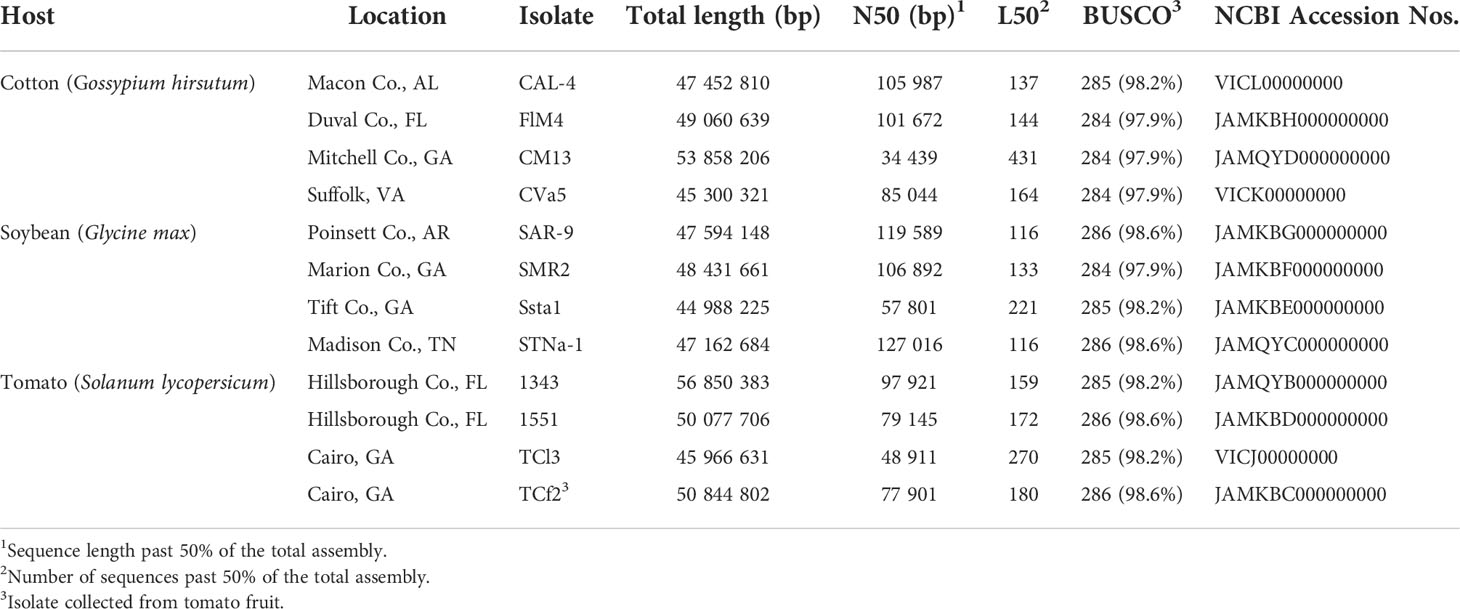
Table 1 Origin and basic metrics of the assembled Corynespora cassiicola genomes.
Illumina short reads from the replicated runs were concatenated to their corresponding read direction (all forward reads together; all reverse reads together) for each of the sequenced isolates. Quality assessment of the raw reads was conducted using FastQC v.0.11.4 (Andrews, 2010) on the Georgia Advanced Computing Resource Center (GACRC) Linux cluster. Trimming was performed using TrimGalore v.0.3.7 (Krueger, 2015) to remove adapter, and other short and low-quality sequences. Filtering parameters were set based on the initial evaluation of the raw reads with FastQC. Trimmed paired-end read files for each isolate were assembled de novo using SPAdes v.3.11.0 (Bankevich et al., 2012). Optimal k-mer values were based on the iterative feature of SPAdes that selects those that exhibit the best quality metrics from multiple k-mer values. The resulting genome assemblies were assessed using QUAST v.4.5 (Gurevich et al., 2013) and BUSCO v.3.0.2 (Kriventseva et al., 2015) with the conserved fungal dataset “Fungi odb9” that contains 290 genes. The 12 draft genomes (Table 1) and raw reads were deposited at NCBI under BioProject number PRJNA549429
Each of the 12 C. cassiicola assembled draft genomes, as well as the reference genome from the rubber isolate CCP, was searched for homologs of the Cas genes (Déon et al., 2014) using NCBI BLAST+ (Altschul et al., 1990) with the following nucleotide sequences of C. cassiicola: Cas1 from isolate CCP from rubber (JF915148), Cas2 from isolate ATI17 from cotton (JF915159), Cas3 from isolate E70 from rubber (JF915169), Cas4 from isolate E79 from rubber (JF915171), Cas5 from isolate SS1 from rubber (JF915173), Cas6 from isolate ATI17 from cotton (JF915182), and Cas7 from isolate IA from cucumber (MF564202).
Nucleotide sequences of Cas genes identified by BLAST+ in the 12 C. cassiicola draft genomes and the reference genome, as well as sequences representative of the 6 Cas variants, were visually edited and aligned in Geneious v.7 (Biomatters) using ClustalW (Thompson et al., 2002). Phylogenetic analyses were performed using Maximum Likelihood (ML) in MEGA5 (Tamura et al., 2011) and Bayesian Inferences (BI) in MrBayes (Ronquist and Huelsenbeck, 2003). An evolutionary model of nucleotide substitution was determined based on goodness-of-fit in MEGA5. The Tamura three-parameter model of evolution assuming a γ distribution with invariant sites was identified as the most appropriate. Support for each node was determined by 500 bootstrap replicates for ML whereas four incrementally heated Markov chains were run, and samples were taken every 100 generations for 5,000,000 generations for BI. To compare differences in coding regions among the variants, the predicted amino acid sequences of the identified cassiicolin-encoding genes were aligned in Geneious v.7 using ClustalW.
To identify putative necrotrophic effector genes, each of the C. cassiicola draft genomes and the reference genome were scanned for biosynthetic loci of known secondary metabolite compound classes in fungi using the webserver tool of the antiSMASH pipeline (Blin et al., 2011). Gene prediction was performed through GlimmerHMM (Pertea et al., 2004) with the FASTA file of the input eukaryotic data. The amino acid sequence translations of all protein-encoding genes were mined with profile Hidden Markov Models (pHMM) using the HMMer3 tool (Eddy, 2009). The models are based on multiple sequence alignments of previously described protein signatures or protein domains. Subsequent BLAST searches were performed on the 12 C. cassiicola draft genomes and the reference genome to query individual biosynthetic genes of the secondary metabolite initially identified by the antiSMASH pipeline.
To identify the mating-type locus in C. cassiicola each of the draft genomes and the reference genome were scanned for MAT1 by BLAST searches. The mating-type genes MAT1-1-1 (AAB82945) and MAT1-2-1 (AAB84004) as well as the flanking genes GTPase activating protein (gap1, AAB82943), unknown open reading frame (orf1, AAB82944), and ß-glucosidase (bgl1, AAB82946) from B. maydis were used as queries.
PCR primers (Table 2) were designed to amplify sequences of the MAT1-1 and MAT1-2 idiomorphs of C. cassiicola isolates in a multiplex reaction. The expected fragment size for MAT1-1 was 1494 bp, whereas for MAT1-2 was 998 bp. The PCR was initially tested on the 12 C. cassiicola isolates sequenced by Illumina since their mating-type idiomorphs were known based on BLAST searches. Multiplex PCR amplification was performed in a 12.5 μl reaction containing the following: 1.25 μl of 10× PCR buffer (Takara Bio Inc., San Jose, CA, USA), 1.25 μl dNTPs (2.5 mM each), 0.56 μl each of the 10 μM primers CCMAT1-1F2, CCMAT1-1R2, CCMAT1-2F4, and CCMAT1-2R4, 0.75 U ExTaq (Takara Bio Inc.), and 1 μl (20-300 ng) DNA template. Thermal cycling conditions had an initial denaturation for 5 min at 94°C followed by; 28 cycles of 30 s at 94°C, 30 s at 52°C, and 30 s at 72°C; and a final elongation of 2 min at 72°C. Confirmation of PCR products was performed by electrophoresis on a 1% (w/v) agarose gel with 1×TBE buffer. PCR products were cleaned with ExoSAP-IT PCR product cleanup reagent (Affymetrix, Santa Clara, CA, USA). Representative DNA fragments were sent for Sanger sequencing (Eurofins Scientific, Louisville, KY, USA). All PCR products were sequenced in both directions using the same primers used for PCR.

Table 2 Primers for multiplex PCR-based mating-type (MAT1) marker in C. cassiicola.
The multiplex PCR-based assay for mating type was conducted on 58 additional C. cassiicola isolates causing recent epidemics in the southeastern U.S., including 25 from cotton, 20 from soybean, and 13 from tomato to identify their mating type. These isolates came from different fields across states (Georgia, Florida, Alabama, Mississippi, Tennesee, Louisiana, Virginia) where epidemics were severe, and were sampled over multiple years (Sumabat et al., 2018a; Sumabat et al., 2018b).
Twelve C. cassiicola isolates were sequenced and draft genomes were assembled (Table 1). The optimal k-mer value determined in SPAdes was k = 77 for all genomes. Assembled genome size ranged from 45.0 to 56.9 Mbp. The N50 ranged from 34 to 127 Kbp with a mean of 87 Kbp. The L50 ranged from 116 to 431 with a mean of 187 contiguous sequences. Genome completeness values measured in BUSCO ranged from 97.9% to 98.6% complete for the 290 conserved reference genes in the dataset “Fungi odb9”.
BLAST analysis using nucleotide sequence queries revealed homologs of cassiicolin-encoding genes in the assembled genomes of C. cassiicola from cotton and soybean. The genes were identified to Cas variant based on how they clustered with known Cas variants in the gene tree (Figure 1) and their amino acid identity with known Cas variants (Figure 2). We found more than one Cas variant in some of the C. cassiicola isolates (Supplemental Table S1). All four sequenced C. cassiicola isolates from cotton have both Cas1 and Cas2. Although the nucleotide sequences of the Cas1 precursor genes from cotton isolates from the southeastern US vary by more than 20 nucleotides from the reference Cas1 variants (Figure 1), the translated amino acid sequences are identical (Figure 2). One soybean isolate (Ssta1) has both Cas2 and Cas6. The other isolates from soybean, SAR-9 and STNa-1 have only Cas2, whereas isolate SMR2 has only Cas6. When more than one Cas variant was detected within the genome of a single isolate, the variants were found on different contigs (Supplemental Table S1). No Cas variants were detected in any of the C. cassiicola assembled genomes from tomato.
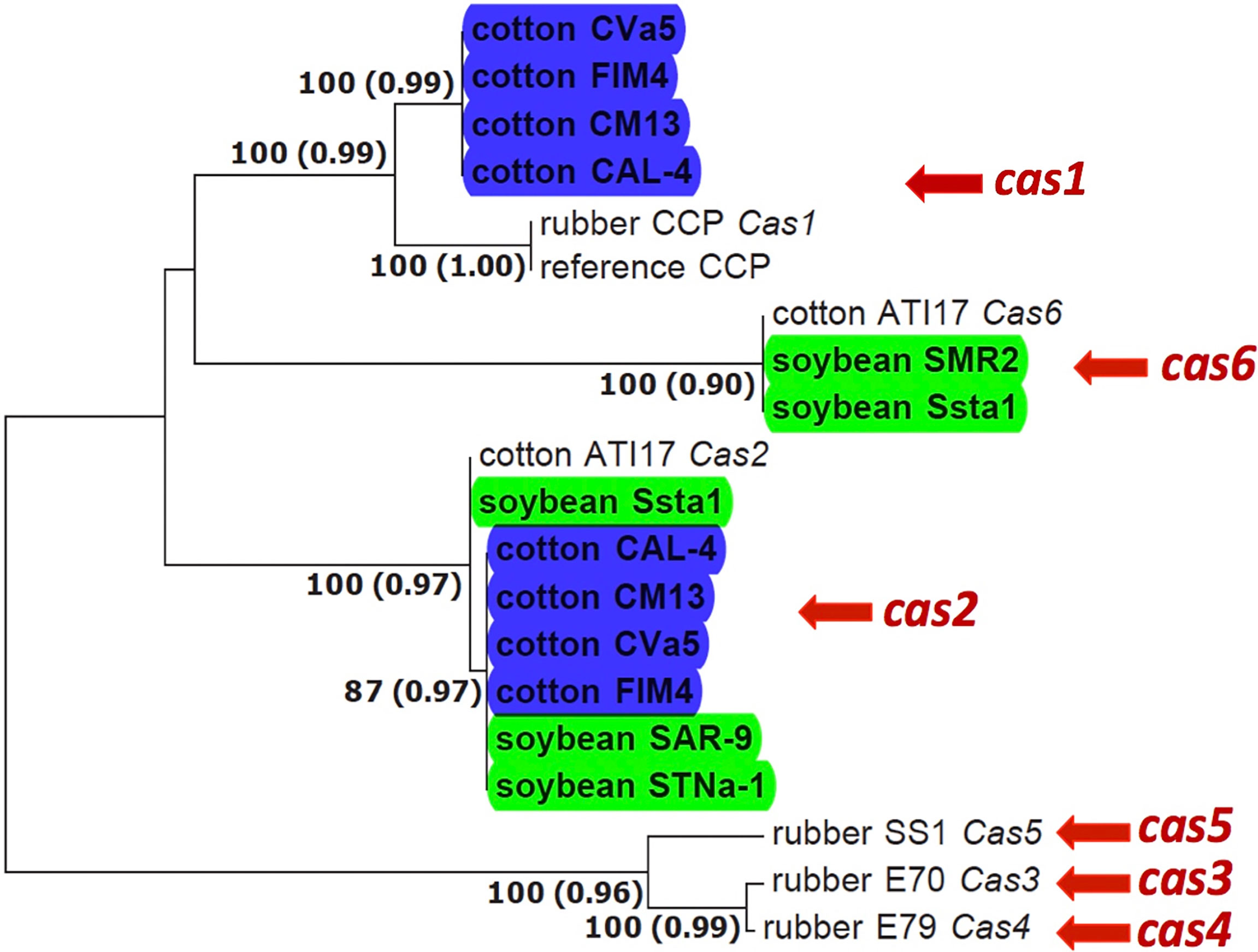
Figure 1 Phylogenetic tree of the cassiicolin-encoding genes in Corynespora cassiicola from different hosts and geographic regions based on Maximum Likelihood (ML) and Bayesian Inference (BI). C. cassiicola genomes sequenced and used in this study are in bold and highlighted blue (cotton) and green (soybean). ML bootstrap values > 70% are shown before the parenthesis for each of the supported branches. Posterior probabilities > 90% are shown in parenthesis. Original hosts and isolate names are indicated for all isolates. Cas7, which is divergent from the other Cas variants, especially for nucleotide sequences, and not found in any of the genomes from this study (Lopez et al., 2018), is not included.

Figure 2 Amino acid sequence alignment of the cassiicolin precursor proteins of the 7 known Cas variants in Corynespora cassiicola. Cas designation and isolate name are shown before each corresponding sequence. Isolates that we collected and sequenced for this study are indicated in bold. The sequences with names in red are from cotton isolates and the sequences with names in green are from soybean isolates.
A web-server tool of the antiSMASH pipeline was used to identify additional putative necrotrophic effectors or secondary metabolite gene clusters in C. cassiicola. Orthologs for genes needed for synthesis of T-toxin were detected in all of the assembled genomes of C. cassiicola from cotton, soybean, tomato, and rubber with high similarity to the biosynthetic genes of T-toxin in B. maydis (Figure 3). These gene clusters were located on a single contig in each of the C. cassiicola genomes. Other putative toxin gene clusters detected in silico in the C. cassiicola genomes showed similarity to gene clusters for brefeldin, ectoine, terrein, aspirochlorine, zearalenone, and acetylaranotin (Table 3). Some of these secondary metabolite gene clusters, including the one for zearalenone, had two similar gene clusters in all C. cassiicola genomes. A cluster with genes similar to the brefeldin cluster was found in all cotton and soybean isolates however, absent in all tomato isolates and the rubber isolate. A gene cluster with genes similar to those in the ectoine biosynthetic cluster was uniquely identified in one tomato isolate, whereas a cluster with genes similar to the terrein cluster was only found in two cotton isolates. A cluster similar to the aspirochlorine biosynthetic cluster was detected in all soybean and two tomato isolates, whereas on cotton isolates this cluster was lacking. A gene cluster with similarity to acetylaranotin was detected in all cotton isolates and two tomato isolates. Aside from the T-toxin cluster, none of these identified clusters contained all of the genes necessary for synthesis of the known secondary metabolite.
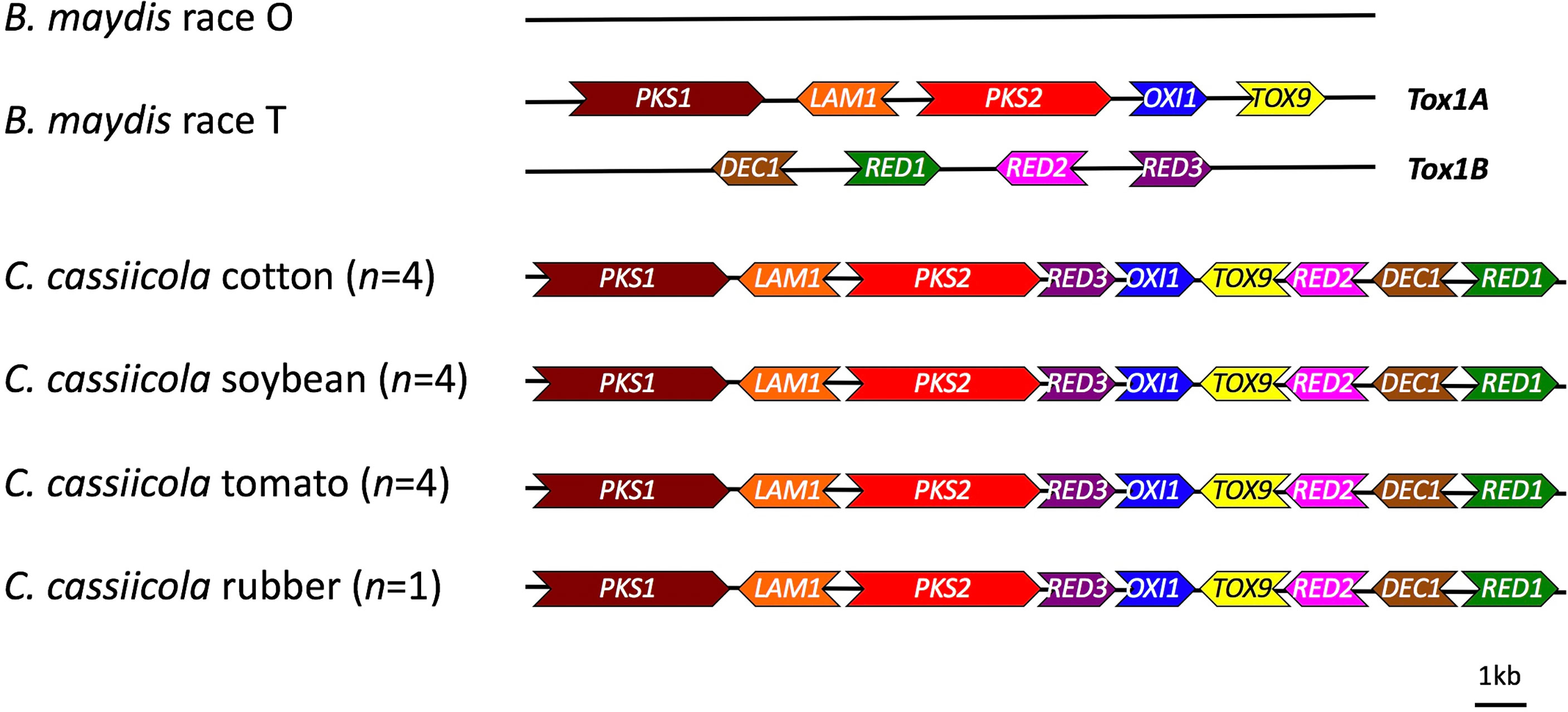
Figure 3 Schematic diagram of the structure of T-toxin biosynthetic genes identified in Corynespora cassiicola isolates sampled and sequenced for this study. The genes pks1 (AAB08104.3), lam1 (ACP43390.1), pks2 (ABB76806.1), oxi1 (ADB23430.1), tox9 (ADB23431.1), dec1 (AAM88291.1), red1 (AAM88292.1), red2 (ACP34152.1), and red3 (ACP34153.1) from Bipolaris maydis race T were used as references for comparison.
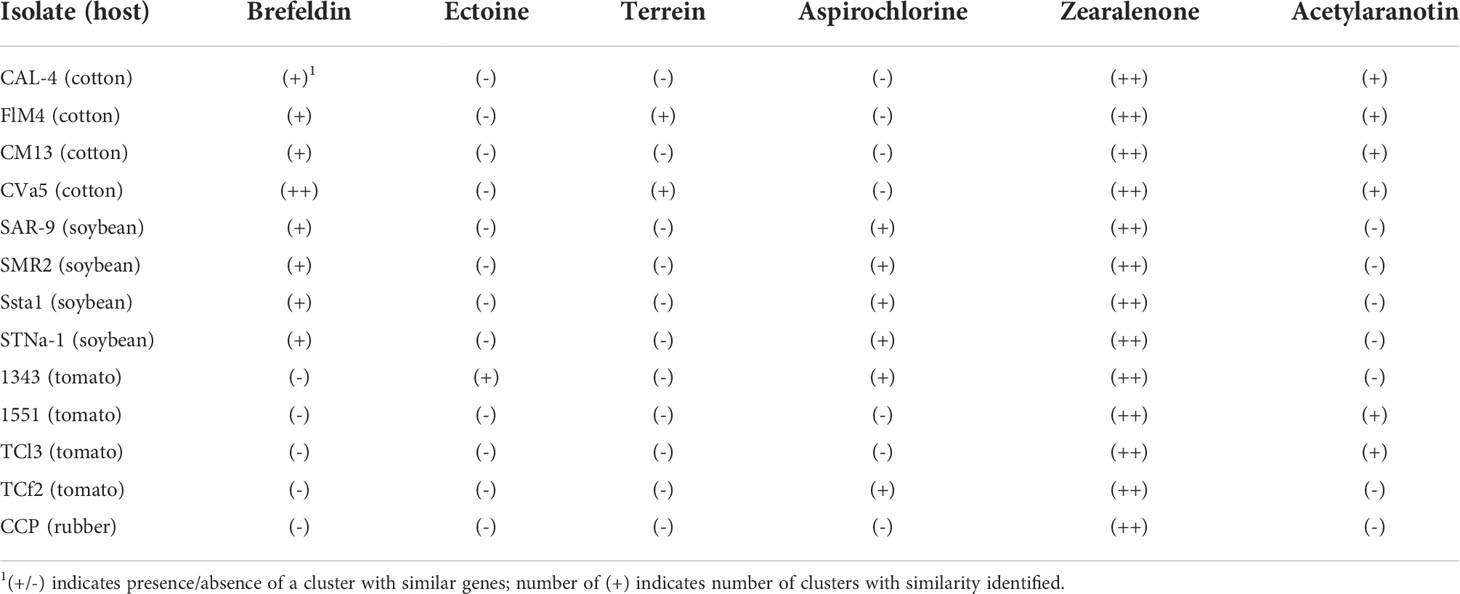
Table 3 Gene clusters identified in the Corynespora cassiicola genomes containing genes with similarity to toxin gene clusters.
The mating-type locus was identified in all of the draft C. cassiicola genomes, as well as the reference genome (Figure 4, Supplemental Table S1). The genes flanking the MAT1 locus in all of the assembled genomes were also identified. The structure of MAT1-1 in C. cassiicola is similar to B. maydis (Debuchy and Turgeon, 2006) with isolates having either the MAT1-1-1 or MAT1-2-1 gene flanked by ß-glucosidase (BGL1) downstream of MAT1 and an unknown open reading frame (ORF1) and GTPase activating protein (GAP1) upstream of MAT1. The 8 sequenced isolates of C. cassiicola from cotton, tomato, and the reference isolate from rubber have the MAT1-1 idiomorph, whereas the four from soybean have the MAT1-2 idiomorph.
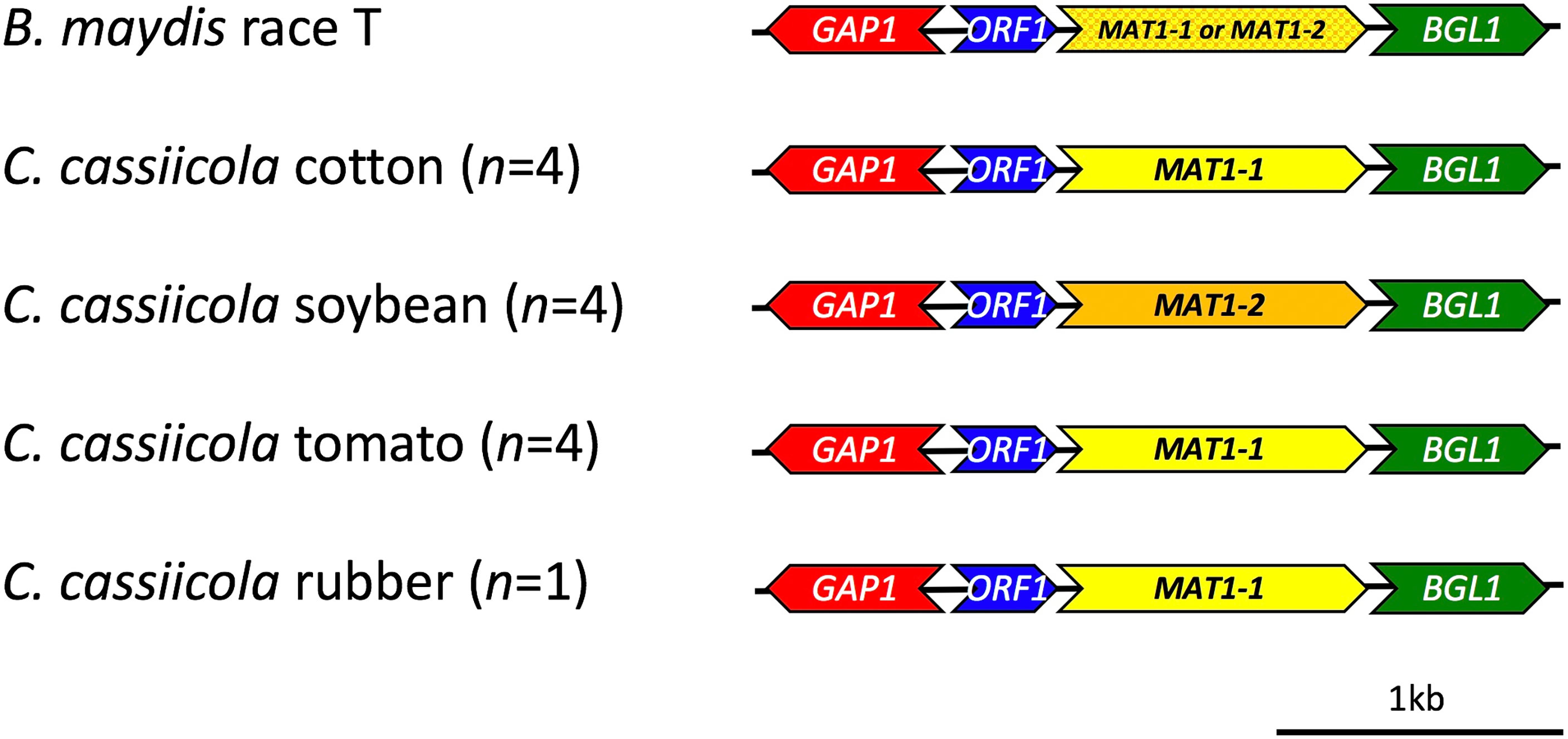
Figure 4 Schematic diagram of the mating-type loci (MAT1) in Corynespora cassiicola isolates sampled and sequenced for this study. The mating-type idiomorphs MAT1-1 (AAB82945.1) and MAT1-2 (AAB84004.1), as well as the flanking genes gap1 (AAB82943.1), unknown orf (AAB82944.1), and bgl1 (AAB82946.1) from Bipolaris maydis were used as references for comparison.
The primers CCMAT1-1F2 and CCMAT1-1R2 for MAT1-1 marker and then CCMAT1-2F4 and CCMAT1-2R4 for MAT1-2 marker (Table 2) consistently amplified the corresponding mating-type genes in C. cassiicola using a multiplexed reaction. PCR amplification confirmed on gel electrophoresis showed fragment sizes that are 1494 bp and 998 bp for MAT1-1 and MAT1-2, respectively (Supplemental Figure S1).
The 25 C. cassiicola isolates and the four whole genome sequenced isolates from cotton in the southeastern U.S. all have only MAT1-1, the 20 isolates and the four whole genome sequenced isolates from soybean in the southeastern U.S. all have only MAT1-2, whereas the 13 isolates from tomato in the southeastern U.S. have either MAT1-1 (n = 10) or MAT1-2 (n = 3) (Supplemental Table S2). The four genome sequenced isolates from tomato have MAT1-1.
In this study, twelve C. cassiicola isolates from three host plants (cotton, soybean and tomato) were sequenced with Illumina technology and assembled de novo. The assembled genomes of C. cassiicola ranged from 45 Mbp to 57 Mbp in genome length (Table 1). We hypothesize that the variation in the size can be attributed to intraspecies differences in the phylogenetic lineages (PL). Three of the larger genomes here are from tomato isolates. Previous studies have shown that this lineage (PL4) is quite distinct from the lineage that contains the cotton and soybean isolates (PL1) and could explain the observed differences (Dixon et al., 2009; Sumabat et al., 2018a). The annotated CCP C. cassiicola reference genome from rubber is 44.9 Mbp (Lopez et al., 2018) and an average genome size for members of Dothideomycetes is 44.6 Mbp (Mohanta and Bae, 2015), which indicates that the range of our assembled genomes is similar in length to the references. Four of the genomes – CM13, CVa5, Ssta1, and TCl3 – had lower mean sequencing read coverage depths (~50×) relative to the other genomes (~150×). This is evident by the low N50 value indicated by smaller length of the contigs as well as the high L50 value indicative of a higher number of contigs suggesting more fragmented sequence reads. However, the overall genome assemblies were similar, as reflected in the genome sizes as well as in the genome completeness assessment by BUSCO analysis.
Homologs of the cassiicolin-encoding genes were detected in all sequenced cotton and soybean isolates (Figure 1). All four sequenced cotton isolates have two Cas variants (Cas1 and Cas2); two of the sequenced soybean isolates have only Cas2, one has only Cas6, and one has both Cas2 and Cas6. Three soybean isolates from Brazil were shown to have only Cas2 (Déon et al., 2014). Two different Cas genes within the same genome has been previously documented (Déon et al., 2014). Two isolates from soybean from Brazil and one isolate from cotton from Brazil were all shown to have both Cas2 and Cas6. Interestingly, population genetic analyses have shown C. cassiicola isolates from cotton and soybean sampled from Brazil cluster with the soybean isolates from the southeastern US, whereas the cotton isolates from the southeastern US form a distinct population (Sumabat et al., 2018b). Although there was some variation in the nucleotide sequences of the Cas precursor genes, the predicted amino acid sequences of the Cas1, Cas2 and Cas6 variants identified in the C. cassiicola isolates from cotton and soybean from the southeastern US (Figure 2) were identical to those of the same previously described Cas variants (Déon et al., 2014). Although Cas variants were detected in silico, it is not clear if these genes are expressed, and if so, what role they have in pathogenicity, host specialization, and virulence of C. cassiicola from epidemics in the US. Functional characterization of Cas1 in an isolate from rubber showed that it is involved in virulence (Ribeiro et al., 2019). Additional functional analyses, such as construction of genetic knockouts, would need to be conducted to evaluate the roles of Cas variants in C. cassiicola isolates in the southeastern US, and their role(s) as virulence factors or necrotrophic effectors in C. cassiicola on cotton and soybean.
Interestingly, no Cas variants were detected in any of the assembled genomes of C. cassiicola isolates from tomato. Moreover, isolates from tomato were not shown to produce cassiicolin in previous studies (Déon et al., 2014), and the phytotoxin(s) produced by isolates on tomato has yet to be characterized (Onesirosan et al., 1975). In addition, the lineage of C. cassiicola to which isolates from tomato in the southeastern US belong is distantly related to the lineage to which the cotton and soybean isolates belong. This evolutionary distance may explain the absence of cassiicolin production in isolates from tomato (Sumabat et al., 2018a; Sumabat et al., 2018b).
A gene cluster containing the orthologs of the Tox1 genes in B. maydis race T has been found in C. cassiicola from tomato, cucumber, and rubber (Condon et al., 2018). T-toxin biosynthesis requires at least nine genes that encode for the following Tox1 genes: polyketide synthases (pks1 and pks2), decarboxylase (dec1), dehydrogenases (lam1, oxi1, red1, red2, and red3), and a hypothetical protein with unknown function (tox9) (Inderbitzin et al., 2010). While these genes in B. maydis are located on two unlinked loci, the Tox1 orthologs from C. cassiicola were located in a compact linear array (Condon et al., 2018). This complete set of nine genes was detected in all of our assembled genomes of C. cassiicola from cotton, soybean, and tomato, as well as from rubber (Figure 3). It is unlikely that C. cassiicola produces T-toxin since a microbial bioassay with Escherichia coli cells with the Urf13 protein from T-cms maize were not killed as would be expected if T-toxin were produced (Condon et al., 2018). However, all of the Tox1-like genes in C. cassiicola were expressed. Although a Tox1-like gene cluster was detected in C. cassiicola, it is possible that a different compound with a different biological function is synthesized in C. cassiicola, or the genes synthesize an entirely different metabolite with a different target that has yet to be characterized.
Additionally, gene clusters with genes showing similarity to other putative secondary metabolite or necrotrophic effector genes were detected in silico in the C. cassiicola genomes. These included genes involved in synthesis of brefeldin, ectoine, terrein, aspirochlorine, zearalenone, and acetylaranotin. This suggests the possible involvement of additional secondary metabolites in pathogenicity and virulence particularly those from tomato, where none of the Cas variants were detected. In silico prediction from other recent studies have identified putative effectors in C. cassiicola (Looi et al., 2017) as well as those previously detected in other fungal taxa with necrotrophic lifestyle and broad host range (Lopez et al., 2018)
To better understand the reproductive biology of C. cassiicola, the mating-type locus (MAT1) was identified in all 12 assembled genomes of C. cassiicola from cotton, soybean, tomato, and the reference genome CCP from rubber. This locus has not been previously identified despite the fact that C. cassiicola has long been reported as strictly asexual or clonal (Dixon et al., 2009; Déon et al., 2014; Sumabat et al., 2018a; Sumabat et al., 2018b). Only one mating-type idiomorph was detected within each of the C. cassiicola genomes; however, both mating types were detected among sequenced genomes indicating that C. cassiicola is heterothallic. The four isolates we sequenced from cotton and the reference isolate from rubber have only MAT1-1, whereas the four isolates we sequenced from soybean have only MAT1-2. The four isolates we sequenced from tomato have MAT1-1; however, when we used the PCR-based mating-type assay for C. cassiicola that we developed here, we detected additional tomato isolates with either MAT1-1 or MAT1-2. The additional isolates from cotton and soybean were all MAT1-1 or MAT1-2, respectively. Additional studies that identify the mating-type of more isolates from these host plants, as well as from other hosts, especially those that have evolved with a different ecological lifestyle, are needed to determine if host-specialized populations are reproductively isolated since each may contain isolates of only a single mating-type. Moreover, some of these populations and lineages may have diverged due to strictly clonal reproduction. However, the C. cassiicola populations from tomato in the southeastern US have both mating types and could potentially reproduce sexually. Determination of the mating-type ratio among a larger set of isolates from tomato or other hosts would help us to infer if cryptic sexual reproduction occurs within or among these populations or lineages (Debuchy and Turgeon, 2006; Brewer et al., 2011); a mating-type ratio not significantly different from 1:1 is expected in randomly mating populations.
The in silico identification of putative necrotrophic effectors in C. cassiicola does not provide direct evidence for host specialization but allows further investigation on the evolution and function of these different genes in host specialized populations. Further exploration of their evolutionary pathways via diverse mechanisms such as horizontal gene transfer, a mechanism for virulence and host specificity that has been documented for several fungal pathogens is now possible. Additionally, we can now target these genes for functional analyses related to host specificity.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA549429.
LD, RK and MB conceived and designed the research. LD conducted the experiments. LD and MB analyzed the data. LD, RK and MB wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding for this research project was provided by Cotton Incorporated contract 14-280 to MTB
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank T. Faske, A. Hagan, H. Kelly, H. Mehl, and P. Price for collecting and providing target spot samples; and H. Seitz, C. Chan and X. French for their technical assistance.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/ffunb.2022.910232/full#supplementary-material
Supplementary Figure 1 | Gel image of PCR products amplified by the multiplexed PCR-based mating-type assay for C. cassiicola. Fragment sizes are 1494 bp for isolates with MAT1-1 (lane indicated by “1”) and 998 bp for isolates with MAT1-2 (lane indicated by “2”). “M” indicates the lane with the size marker. Isolate names are listed at the top of each lane.
Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J. (1990). Basic local alignment search tool J. Mol. Biol 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Andrews S. (2010) FastQC: A quality control tool for high throughput sequence data. Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
Bankevich A., Nurk S., Antipov D., Gurevich A. A., Dvorkin M., Kulikov A. S., et al. (2012). SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bennett D. (2016). What’s known about target spot in soybeans? (Delta Farm Press). Available at: http://www.deltafarmpress.com/soybeans/what-s-known-about-target-spot-soybeans.
Blin K., Medema M. H., Fischbach M. A., Cimermancic P., Zakrzewski P., Breitling R., et al. (2011). antiSMASH: Rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 39, W339–W346. doi: 10.1093/nar/gkr466
Breton F., Sanier C., D'Auzac J. (2000). Role of cassiicolin, a host-selective toxin, in pathogenicity of Corynespora cassiicola, causal agent of a leaf fall disease of hevea. J. Rubber Res. 3, 115–128.
Brewer M. T., Cadle-Davidson L., Cortesi P., Spanu P. D., Milgroom M. G. (2011). Identification and structure of the mating-type locus and development of PCR-based markers for mating type in powdery mildew fungi. Fungal Genet. Biol. 48, 704–713. doi: 10.1016/j.fgb.2011.04.004
Burdon J. J., Silk J. (1997). Sources patterns Diversity plant-pathogenic fungi. Phytopathology. 87, 664–669. doi: 10.1094/PHYTO.1997.87.7.664
Butler S., Young-Kelly H., Raper T., Cochran A., Jordan J., Shrestha S. (2016). Sources and patterns of diversity in plant pathogenic fungi. Plant Dis. 100, 535. doi: 10.1094/PDIS-07-15-0785-PDN
Campbell H. L., Hagan A., Bowen K., Nightengale S. P. (2012). Corynespora leaf spot: A new disease in Alabama cotton. Am. Phytopathological Society St. Paul MN 18–19.
Chee K. H. (1990). Present status of rubber diseases and their control. Rev. Plant Pathol. 69, 423–430.
Ciuffetti L. M., Tuori R. P., Gaventa J. M. (1997). A single gene encodes a selective toxin causal to the development of tan spot of wheat. Plant Cell. 9, 135–144. doi: 10.1105/tpc.9.2.135.
Condon B. J., Elliott C., González J. B., Yun S. H., Akagi Y., Wiesner-Hanks T., et al. (2018). Clues to an evolutionary mystery: The genes for T-toxin, enabler of the devastating 1970 southern corn leaf blight epidemic, are present in ancestral species, suggesting an ancient origin. Mol. Plant Microbe Interact. 31, 1154–1165. doi: 10.1094/MPMI-03-18-0070-R
Debuchy R., Turgeon B. G. (2006). “Mating-type structure, evolution, and function in euascomycetes,” in Growth, differentiation and sexuality. Eds. Kües U., Fischer R. (Berlin, Heidelberg: Springer Berlin Heidelberg), 293–323.
de Lamotte F., Duviau M. P., Sanier C., Thai R., Poncet J., Bieysse D., et al. (2007). Purification and characterization of cassiicolin, the toxin produced by Corynespora cassiicola, causal agent of the leaf fall disease of rubber tree. J. Chromatogr. B. 849, 357–362. doi: 10.1016/j.jchromb.2006.10.051
Déon M., Fumanal B., Gimenez S., Bieysse D., Oliveira R. R., Shuib S. S., et al. (2014). Diversity of the cassiicolin gene in Corynespora cassiicola and relation with the pathogenicity in Hevea brasiliensis. Fungal Biol. 118, 32–47. doi: 10.1016/j.funbio.2013.10.011
Déon M., Scomparin A., Tixier A., Mattos C. R. R., Leroy T., Seguin M., et al. (2012). First characterization of endophytic Corynespora cassiicola isolates with variant cassiicolin genes recovered from rubber trees in Brazil. Fungal Divers. 54, 87–99. doi: 10.1007/s13225-012-0169-6
Dewey R. E., Siedow J. N., Timothy D. H., Levings C. S. (1988). A 13-kilodalton maize mitochondrial protein in E. coli confers sensitivity to Bipolaris maydis toxin. Science. 239, 293–295. doi: 10.1126/science.3276005
Dixon L. J., Schlub R. L., Pernezny K., Datnoff L. E. (2009). Host specialization and phylogenetic diversity of Corynespora cassiicola. Phytopathology. 99, 1015–1027. doi: 10.1094/PHYTO-99-9-1015
Duplessis S., Cuomo C. A., Lin Y. C., Aerts A., Tisserant E., Veneault-Fourrey C., et al. (2011). Obligate biotrophy features unraveled by the genomic analysis of rust fungi. Proc. Natl. Acad. Sci. 108, 9166–9171. doi: 10.1073/pnas.1019315108
Eddy S. R. (2009). A new generation of homology search tools based on probabilistic inference the genome inform. Genome Informatics 23, 205–211. doi: 10.1142/9781848165632_0019
Edmisten K. (2012). Target leaf spot found in north Carolina cotton. online publication (Southeast Farm Press). Available at: http://www.southeastfarmpress.com/cotton/target-leaf-spot-found-north-carolina-cotton.
Edwards Molina J. P., Navarro B. L., Allen T. W., Godoy C. V. (2022). Soybean target spot caused by Corynespora cassiicola: a resurgent disease in the americas. Trop. Plant Pathol 47, 315–331. doi: 10.1007/s40858-022-00495-z
Farr D. F., Rossman A. Y. (2017). Fungal databases, U.S. national fungus collections. online publication (United States Department of Agriculture – Agricultural Research Services). Available at: https://nt.ars-grin.gov/fungaldatabases/.
Faske T. (2016). Arkansas Soybeans: Target spot – what do we know? (Agfax). Available at: https://agfax.com/2016/11/02/arkansas-soybeans-target-spot-what-do-we-know/.
Fournier E., Giraud T. (2007). Sympatric genetic differentiation of a generalist pathogenic fungus, Botrytis cinerea, on two different host plants, grapevine and bramble. J. Evol. Biol. 21, 122–132. doi: 10.1111/j.1420-9101.2007.01462.x
Fraser J. A., Heitman J. (2003). Fungal mating-type loci. Curr. Biol. 13, R792–R795. doi: 10.1016/j.cub.2003.09.046
Friesen T. L., Faris J. D. (2012). “Characterization of plant-fungal interactions involving necrotrophic effector-producing plant pathogens,” in Plant fungal pathogens: Methods and protocols. Eds. Bolton M. D., Thomma B. P. (Totowa, NJ: Humana Press), 191–207.
Friesen T. L., Faris Justin D., Solomon Peter S., Oliver Richard P. (2008). Host-specific toxins: Effectors of necrotrophic pathogenicity. Cell Microbiol. 10, 1421–1428. doi: 10.1111/j.1462-5822.2008.01153.x
Friesen T. L., Stukenbrock E. H., Liu Z., Meinhardt S., Ling H., Faris J. D., et al. (2006). Emergence of a new disease as a result of interspecific virulence gene transfer. Nat. Genet. 38, 953–956. doi: 10.1038/ng1839
Fulmer A. M., Walls J. T., Dutta B., Parkunan V., Brock J., Kemerait R. C. (2012). First report of target spot caused by Corynespora cassiicola on cotton in Georgia. Plant Dis. 96, 1066–1066. doi: 10.1094/PDIS-01-12-0035-PDN
Fulton T. M., Chunwongse J., Tanksley S. D. (1995). Microprep protocol for extraction of DNA from tomato and other herbaceous plants. Plant Mol. Biol. Rep. 13, 207–209. doi: 10.1007/BF02670897
Gilchrist D., Grogan R. (1976). Production and nature of a host-specific toxin from Alternaria alternata f. sp. lycopersici. Phytopathology. 66, 165–171. doi: 10.1094/Phyto-66-165
Gurevich A., Saveliev V., Vyahhi N., Tesler G. (2013). QUAST: Quality assessment tool for genome assemblies. Bioinformatics. 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Hatta R., Ito K., Hosaki Y., Tanaka T., Tanaka A., Yamamoto M., et al. (2002). A conditionally dispensable chromosome controls host-specific pathogenicity in the fungal plant pathogen Alternaria alternata. Genetics. 161, 59–70. doi: 10.1093/genetics/161.1.59
Inderbitzin P., Asvarak T., Turgeon B. G. (2010). Six new genes required for production of T-toxin, a polyketide determinant of high virulence of Cochliobolus heterostrophus to maize. Mol. Plant Microbe Interact. 23, 458–472. doi: 10.1094/MPMI-23-4-0458
Koenning S. R., Creswell T. C., Dunphy E. J., Sikora E. J., Mueller J. D. (2006). Increased occurrence of target spot of soybean caused by Corynespora cassiicola in the southeastern united states. Plant Dis. 90, 974. doi: 10.1094/PD-90-0974C
Kono Y., Daly J. M. (1979). Characterization of the host-specific pathotoxin produced by Helminthosporium maydis, race T, affecting corn with Texas male sterile cytoplasm. Bioorg. Chem. 8, 391–397. doi: 10.1016/0045-2068(79)90064-6
Kono Y., Takeuchi S., Kawarada A., Daly J. M., Knoche H. W. (1980). Structure of the host-specific pathotoxins produced by Helminthosporium maydis, race T. Tetrahedron Lett. 21, 1537–1540. doi: 10.1016/S0040-4039(00)92768-0
Kriventseva E. V., Zdobnov E. M., Simão F. A., Ioannidis P., Waterhouse R. M. (2015). BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Krueger F. (2015) TrimGalore!: a wrapper tool around cutadapt and FastQC to consistently apply quality and adapter trimming to FastQ files. Available at: http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/.
Levings C. S. (1990). The texas cytoplasm of maize: Cytoplasmic male sterility and disease susceptibility. Science. 250, 942–947. doi: 10.1126/science.250.4983.942
Levings C. S. (1993). Thoughts on cytoplasmic male sterility in cms-T maize. Plant Cell. 5, 1285–1290. doi: 10.2307/3869781
Looi H. K., Toh Y. F., Yew S. M., Na S. L., Tan Y.-C., Chong P.-S., et al. (2017). Genomic insight into pathogenicity of dematiaceous fungus Corynespora cassiicola. PeerJ 5, e2841. doi: 10.7717/peerj.2841
Lopez D., Ribeiro S., Label P., Fumanal B., Venisse J.-S., Kohler A., et al. (2018). Genome-wide analysis of Corynespora cassiicola leaf fall disease putative effectors. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.00276
Ma L. J., van der Does H. C., Borkovich K. A., Coleman J. J., Daboussi M. J., Di Pietro A., et al. (2010). Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature. 464, 367–373. doi: 10.1038/nature08850
Mehl H. L., Phipps P. M. (2013). Applied research on field crop disease control (Tidewater Agricultural Research and Extension Center, Suffolk: Virginia Polytechnic Institute and State University, College of Agriculture and Life Sciences).
Milgroom M. G., Peever T. L. (2003). Population biology of plant pathogens: The synthesis of plant disease epidemiology and population genetics. Plant Dis. 87, 608–617. doi: 10.1094/PDIS.2003.87.6.608
Mohanta T. K., Bae H. (2015). The diversity of fungal genome. Biol. Proced. Online. 17, 8. doi: 10.1186/s12575-015-0020-z
Ohm R. A., Feau N., Henrissat B., Schoch C. L., Horwitz B. A., Barry K. W., et al. (2012). Diverse lifestyles and strategies of plant pathogenesis encoded in the genomes of eighteen dothideomycetes fungi. PLsoS Pathog. 8, 12. doi: 10.1371/journal.ppat.1003037
Onesirosan P., Mabuni C. T., Durbin R. D., Morin R. B., Rich D. H., Arny D. C. (1975). Toxin production by Corynespora cassiicola. Physiol. Plant Pathol. 5, 289–295. doi: 10.1016/0048-4059(75)90095-8
Pertea M., Salzberg S. L., Majoros W. H. (2004). TigrScan and GlimmerHMM: Two open source ab initio eukaryotic gene-finders. Bioinformatics. 20, 2878–2879. doi: 10.1093/bioinformatics/bth315
Price T., Singh R., Fromme D. (2015). First report of target spot caused by Corynespora cassiicola in Louisiana cotton. Plant Health Prog. 15, 223–224. doi: 10.1094/PHP-BR-15-0036
Ribeiro S., Tran D. M., Déon M., Clément-Demange A., Garcia D., Soumahoro M., et al. (2019). Gene deletion of corynespora cassiicola cassiicolin Cas1 suppresses virulence in the rubber tree. Fungal Genet. Biol. 129, 101–114. doi: 10.1016/j.fgb.2019.05.004
Ronquist F., Huelsenbeck J. P. (2003). MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 19, 1572–1574. doi: 10.1093/bioinformatics/btg180
Schlub R. L., Smith L. J., Datnoff L. E., Pernezny K. (2009). An overview of target spot of tomato caused by Corynespora cassiicola. Int. Symp. Tomato Dis. 808, 25–28. doi: 10.17660/ActaHortic.2009.808.1
Schoch C. L., Crous P. W., Groenewald J. Z., Boehm E. W. A., Burgess T. I., de Gruyter J., et al. (2009). A class-wide phylogenetic assessment of dothideomycetes. Stud. Mycol. 64, 1–15, S10. doi: 10.3114/sim.2009.64.01
Silva W. P. K., Deverall B. J., Lyon B. R. (2002). Molecular, physiological and pathological characterization of corynespora leaf spot fungi from rubber plantations in Sri Lanka. Plant Pathol. 47, 267–277. doi: 10.1046/j.1365-3059.1998.00245.x
Soanes D., Richards T. A. (2014). Horizontal gene transfer in eukaryotic plant pathogens. Annu. Rev Phytopathol. 52, 583–614. doi: 10.1146/annurev-phyto-102313-050127
Sumabat L. G., Kemerait R. C., Brewer M. T. (2018a). Phylogenetic diversity and host specialization of Corynespora cassiicola responsible for emerging target spot disease of cotton and other crops in the southeastern united states. Phytopathology. 108, 892–901. doi: 10.1094/PHYTO-12-17-0407-R
Sumabat L. G., Kemerait R. C. Jr., Kim D. K., Mehta Y. R., Brewer M. T. (2018b). Clonality and geographic structure of host-specialized populations of corynespora cassiicola causing emerging target spot epidemics in the southeastern united states. PLoS One 13, 10. doi: 10.1371/journal.pone.0205849
Tamura K., Peterson D., Peterson N., Stecher G., Nei M., Kumar S. (2011). MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. doi: 10.1093/molbev/msr121
Thompson J. D., Gibson T., Higgins D. G. (2002). Multiple sequence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinf. 00, 2.3.1–2.3.22. doi: 10.1002/0471250953.bi0203s00.
Tsuge T., Harimoto Y., Akimitsu K., Ohtani K., Kodama M., Akagi Y., et al. (2013). Host-selective toxins produced by the plant pathogenic fungus Alternaria alternata. FEMS Microbiol. Rev. 37, 44–66. doi: 10.1111/j.1574-6976.2012.00350.x
Keywords: mating-type, MAT1, necrotrophic effector, cassiicolin, T-toxin
Citation: Dacones LS, Kemerait RC Jr. and Brewer MT (2022) Comparative genomics of host-specialized populations of Corynespora cassiicola causing target spot epidemics in the southeastern United States. Front. Fungal Biol. 3:910232. doi: 10.3389/ffunb.2022.910232
Received: 01 April 2022; Accepted: 27 June 2022;
Published: 22 July 2022.
Edited by:
Fabiano Sillo, Istituto per la Protezione Sostenibile delle Piante, ItalyReviewed by:
Minou Nowrousian, Ruhr University Bochum, GermanyCopyright © 2022 Dacones, Kemerait and Brewer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marin T. Brewer, bXRicmV3ZXJAdWdhLmVkdQ==
†Present address: Leilani S. Dacones, Institute of Biology, University of the Philippines Diliman, Quezon City, Philippines
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.