 Tianhua Sun
Tianhua Sun Yanan Zhao
Yanan Zhao Guona Zhou1
Guona Zhou1
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. For. Glob. Change , 14 June 2024
Sec. Pests, Pathogens and Invasions
Volume 7 - 2024 | https://doi.org/10.3389/ffgc.2024.1356511
This article is part of the Research Topic "Forentomics": forest pest and pathogen biology, ecology, and management using omics View all 10 articles
Pinus is a genus of great economic and ecological importance, and its members are dominant components of forests throughout the world. During the long evolutionary “arms race,” plants have developed complex and diverse systemic defense mechanisms to strategically and intelligently compete with herbivores. To study the alteration pattern and defensive response mechanism triggered by herbivorous feeding stimuli, we firstly built a biological model of the interrelationship between the Chinese pine (Pinus tabuliformis Carr.) and the Chinese pine caterpillar (Dendrolimus tabulaeformis Tsai et Liu). This model integrated proteomic and phosphoproteomic data, which were then normalized and combined with bioinformatics tools to evaluate and analyze changes in the phosphoproteomic profile in response to the caterpillar’s feeding stimulus on pine needles. Systematic identification of differentially significant phosphorylated proteins implicated in the pine’s defense mechanism against caterpillar stress was conducted. Furthermore, we predicted upstream kinases of phosphorylation sites and their activities. Through an analysis of Motif patterns of phosphorylated proteins, Mfuzz clustering of phosphorylation sites, and kinase regulatory networks, we explored the functional modules of phosphorylated protein interaction networks in response to stress within pine. In general, our study emphasized the significant role of kinase METK2, PTI12, PGK, as well as At3g59480 for the first time. The identification of these phosphorylated proteins was additionally confirmed through parallel reaction monitoring technology. Furthermore, genes associated with differentially expressed proteins were validated through real-time quantitative polymerase chain reaction detection. This investigation aids in understanding the mechanisms behind resistance formation and regulation of caterpillar feeding incentives in pine. Breeding more resistant pine varieties may benefit from a fuller understanding of these defense strategies in the future.
As a representative of coniferous trees, the Chinese pine (Pinus tabuliformis) is one of the ecological cornerstone species (Niu et al., 2022). A thorough analysis of the defense mechanisms employed by dominant phytophagous insects in response to biotic stress is crucial for understanding habitat regulation under forest tree species. In this study, our analysis specifically focuses on critical defense mechanisms and habitat regulation in forest tree species, particularly examining how these factors are influenced by herbivore feeding. Proteins are the direct carriers and implementers of functions in plant life activities. Exploring phenomena such as plant biotic stress response and laws utilizing proteins and modified proteins is an essential approach to studying forest ecological protection. The sequence tagging analysis of insect feeding trauma-induced defense genes in hybrid poplar identified two defense proteins: Kunitz trypsin inhibitor and chitinase. Additionally, the genes responsible for the feeding-induced emission of terpene volatiles from hybrid poplar by Trichoderma harzianum were cloned and analyzed for their functional characteristics and expression types (Arimura et al., 2009; Cheng et al., 2013; Song et al., 2014). Proteomics and other techniques were used to investigate the molecular characteristics of protein expression in maize in response to insect feeding damage. Additionally, the molecular characteristics of differential expression of resistance proteins in plant-insect co-evolution were examined (Baldwin et al., 2001; Conrath, 2011; Elsayed, 2011; Peng et al., 2011; Wei and Kang, 2011; Smith and Clement, 2012; Cheng et al., 2013). Tobacco lice prick the phloem of tobacco and inject Bt56 protein, which interacts with the transcription factor NTH202 in the phloem to activate the salicylic acid signaling pathway in the plant, which induces the accumulation of salicylic acid in the plant to inhibit jasmonic acid production in the plant and reduce the resistance (Xu et al., 2019). Although these explorations have greatly advanced the study of plant-induced insect resistance, previous studies have also shown that plant-induced defense responses are a complex process. The mechanism of defense response varies from plant to plant and from stimulus to stimulus. As a result, there are many new findings and confusions in the current studies. Here, we investigated for the first time the phosphoproteomics of Chinese pine in response to feeding stimuli of Chinese pine caterpillars under natural conditions. Changes in protein expression levels, protein modifications, identification of functional modules and pathways, and monitoring of resistance-related substances in response to feeding stimuli from Chinese pine caterpillars (Dendrolimus tabulaeformis) are involved in the defense process of Chinese pine.
Protein phosphorylation modification, first reported in 1926, has become the most well-known type of post-translational modification (PTM) (Sefton, 1998). The identification of protein phosphorylation modification sites holds significant value for vital biological regulatory processes such as protein phosphorylation, dephosphorylation, alterations in protein structure, changes in enzyme activity, modifications to substrate specificity, and more (Pawson and Scott, 2005). Modified proteomics is a commonly used method to investigate dynamic changes, including plant defense responses, in search of biomarkers, such as stably differentially expressed proteins. PTM is a small but diverse and central regulatory step in the protein mass transition process. PTMs of proteins have a profound impact on protein diversity and molecular function, as they represent highly regulated molecular patterns. Protein phosphorylation modificationomics is significant in plant adaptation mechanisms (Rampitsch, 2017; Fatma et al., 2019). The frequency of proteins involved in the activities of plant life is significant. High-throughput proteomics technology has expanded opportunities for thorough investigations of the protein modifier group (Meier et al., 2020; Demichev et al., 2022).
Phosphorylation modifications are the most prevalent and abundant type of PTMs in proteins. They have a significant impact on the various functions that proteins perform. Additionally, conducting thorough research on the PTM of proteins can provide insights into the mechanisms underlying plant life activities, facilitate the identification of key modification proteins that respond to biotic stress, and enable the identification of specific modification sites. Phosphorylation modifications activate substrates, modulate downstream pathways, and achieve biological significance through cascade amplification effects. Kinases trigger the phosphorylation modification of proteins by transferring phosphate groups to amino acids such as serine, threonine, and tyrosine. This process regulates various functions of target proteins, which in turn impact cellular metabolism, signaling, and other vital life processes. Phosphoproteomics is a crucial method for studying plant processes in response to biotic stresses in great detail. The utilization of phosphoproteomics provides valuable insights into the response mechanisms that trigger resistance in plants during adverse stress conditions (Perazzolli et al., 2016).
Phosphoproteomics unveils the phosphorylation modification between the plant’s target of rapamycin (TOR) kinase and the abscisic acid (ABA) receptor-coupled signaling pathways. This illustrates how plants use a conserved phosphorus-regulated feedback mechanism to balance growth, development, and stress responses. When external stressors impact plants, their typical growth activity is hindered by the hormones salicylic acid (SA), jasmonic acid (JA), and ethylene (ET). Instead, this leads to a heightened defense response (Waadt et al., 2022). Proteins undergo phosphorylation when a phosphate kinase transfers a phosphate group to a downstream protein, which may be a kinase or transcription factor. Simultaneously, the upstream kinase stimulates the protein’s phosphorylation, resulting in a multistage kinase pattern that facilitates amplification of the signal through cascading. Phosphorylated proteomes can quantify phosphorylated proteins that are significantly expressed in signaling pathways, thereby identifying phenotypically relevant activated signaling pathways and elucidating the associated mechanisms (Liu et al., 2018). The gene set enrichment analysis (GSEA) algorithm predicted the activity of phosphorylated kinases, and the activity did not correlate with their protein expression. Kinase regulation is a “many-to-many” relationship, with multiple phosphorylated kinases regulating one substrate and one phosphorylated kinase regulating multiple substrates. Site modification prediction performs site-by-site clustering analysis, which provides greater differentiation and accuracy than gene clustering analysis (Krug et al., 2019). The level of protein phosphorylation regulated by the protein kinase is correlated to the substrate expression, indicating its regulation state to some extent. Higher levels of protein phosphorylation are likely to indicate increased protein kinase activity (Hernandez-Armenta et al., 2017).
A proteome-based approach that focuses on phosphorylation was utilized to analyze and identify Arabidopsis modification profiles, and to screen conserved sites within a specific amount of phosphorylated peptides and their respective phosphorylation sites. Conservation of function is disrupted through point mutation mimicry, exposing the homeostatic mechanism of TOR kinase and ABA in mediating plant growth, development, and drought stress response (Wang et al., 2018). Phosphorylated proteomic techniques enable the investigation of plant regulation under low-temperature stress conditions (Zhao et al., 2017). Phytosulfokine (PSK) is a plant polypeptide hormone that promotes growth. When PSK signaling is absent, plant growth is inhibited, but disease resistance is increased (Förderer and Chai, 2023). PSK binds to phytosulfokine receptor 1 (PSKR1) and somatic embryogenesis receptor-like kinase (SERK) receptors during plant defense and growth periods, respectively, and induces activation of the calcium-dependent kinase 28 (CPK28) via the phosphorylation pathway. Glutamate synthase 2 (GS2) undergoes serine site phosphorylation at s334 and s360 induced by CPK28 kinase, promoting plant defense responses and regulating nitrogen availability for plant growth, respectively (Ding et al., 2023).
Plant cells utilize intricate signaling pathways in response to intricate biotic stress stimuli. Phosphorylated proteins play a essential role in researching the mechanisms of defense signaling induced by plants (Mukherji, 2005; Rampitsch, 2017). High-throughput phosphoproteomics provides a technological basis for the study of signaling cascades during plant-induced resistance responses, and specific protein-associated signals generated by phosphorylated kinase catalysis. The study of plant protein kinases and phosphorylated proteins has led to the identification of numerous protein modification signaling network components with prominent roles (Bentem and Hirt, 2007; Xing and Laroche, 2011). This creates the possibility of constructing signaling networks such as plant-induced resistance. Reversible protein phosphorylation is important for signal transduction mechanisms and signaling pathways (Rossignol, 2006), advancing the establishment of predictable models of plant protein phosphorylation and the process of plant post-genome integration.
By integrating the proteomic and phosphoproteomic results of Chinese pine caterpillar feeding stimulus-induced defense responses in Chinese pine, we systematically clarify the phosphorylation-modified proteins, kinases, and their defense regulatory functions that are involved in pine’s defense response mechanism against caterpillars under natural conditions. To analyze the key phosphorylation modification proteins, core genes and major defense products in the process of Chinese pine resistance induced by caterpillar, and to reveal the expression regulation law and upstream and downstream correlation of the three. A network of interactions and functional modules for phosphorylation-modified proteins of Chinese pine defense response were built. Determining the accuracy of key proteins in the oleander defense response mechanism at the validation level and revealing the specificity of the feeding stimulus in the formation of the oleander defense mechanism.
The experiment utilized Chinese pine test samples obtained from Huangtuliangzi State-owned Forest Farm in Pingquan City, Chengde City, Hebei Province, and Chinese pine caterpillars collected from Yushulinzi Forest in Pingquan City, Hebei Province, and the border region between Pingquan City and Liaoning Province. Pure Chinese pine forests with identical elevation, slope, aspect, tree thickness, and management level, unaffected by herbivores and disease, were selected for the experiment. Prior to each treatment, a group of sixth–seventh instar mature caterpillar larvae was collected and screened to ensure consistent feeding levels and stress levels on Chinese pine needles. The Chinese pine caterpillars were stored in insect bags to remove any remaining inactive or damaged bodies, including those infested with bacteria, fungi, or other insects, to minimize interference with the experiment. The caterpillars were also deprived of food for at least 12 h prior to testing.
A random sampling method was utilized to select test plants in the forest. Within the test site, sturdy 10-year-old Chinese pine trees with uniform growth, and no herbivores or diseases, were selected and sequentially tagged and labeled. One branch in each of the four cardinal directions was selected, ensuring good size, thickness, and consistent growth from the middle layer of the Chinese pine tree canopy. Wear gloves and gently spread 10 Chinese pine caterpillars (quickly climbing from the bottom of the bag to the opening, representing strong vitality) evenly onto the Chinese pine needles and leaves for each branch. Each branch should have 10 caterpillars. Use identical-sized net covers to wrap around the feeding areas for the Chinese pine caterpillars on each branch, secure the net mouth with a string to prevent the test insects from escaping.
Three treatment groups were established to observe Chinese pine caterpillar feeding at different time points (0 h for control, 2 h, and 8 h). Each group comprised six Chinese pine replicates to ensure reliable results. At each time point, the netting was removed to collect the Chinese pine caterpillars, which were subsequently returned to the insect bags. Three control groups were formed to control leaf-cutting, and the treatment duration was arranged as previously mentioned. For the leaf-cutting procedure, we selected pine needles from a single robust branch positioned in the middle layer of the Chinese pine’s canopy, using scissors to make the cuts. Each group had six Chinese pines as biological duplicates.
Controlling each Chinese pine specimen experimentally with identical treatments of pine caterpillar feeding and leaf-cutting, we selected direct-bearing tissue that responded to stress for our sample collection. We ensured that the pine trees were all of equal numbers and vigor. At each of the three intervals in the experimental design, pine needles located 3 cm below the point of injury caused by caterpillar feeding or leaf-cutting were harvested. The specimens were wrapped in aluminum foil, formed into squares that are smaller than the tank’s opening, labeled, and then quickly placed in a liquid nitrogen tank. The specimens were then delivered to the lab and stowed in an ultra-low temperature freezer at −80°C until needed.
The complete experimental procedures for tandem mass tag (TMT) proteomics, including protein ex-traction, trypsin digestion, TMT labeling, high performance liquid chromatography (HPLC) fractionation, liquid chromatography-mass spectrometry/mass spectrometry (LC-MS/MS) analysis, database search, and bioinformatics methods, are elaborated in Supplementary material. To compare protein sequences and collect annotation information on homologous proteins, a search was conducted on the Chinese pine transcriptome database.
Motif analysis of protein phosphorylation modifications using Motif-x algorithm (MoMo) software V5.0.2. Phosphate kinase family prediction upstream of the phosphate site using Group-based Prediction System (GPS) V5.0 (Wang et al., 2020). Phosphokinase activity prediction using GSEA V4.0.3 (Subramanian et al., 2005; Guo et al., 2019). Phosphokinase activity bar graph using ggplot2 R package4 V3.2.1 (Hadley, 2016). Phosphokinase activity heatmap using heatmap R package V1.0.12. Phosphokinase regulatory network visualization using networkD3 R package V0.4 and Cytoscape5 (Shannon, 2003).
The materials used for parallel reaction monitoring (PRM) testing included pine needles at 0-, 2-, and 8-h intervals after caterpillar feeding treatments. Each sample was analyzed using three biological replicates for PRM analysis. The complete procedure is presented in Supplementary material.
Pine needles were collected at 0, 2, and 8 h after feeding and leaf cutting for use as materials in real-time quantitative polymerase chain reaction (RT-qPCR). Comparison of the two treatments with a view to highlighting the specific defense response of Chinese pine to herbivore feeding stimuli. Six replicates were used for each time point treatment, with corresponding sample bags selected randomly. RNA extraction from Chinese pine needles was performed using the Tengen Polysaccharide and Polyphenol Plant Total RNA Extraction Kit DP441 based on its instruction manual. A sample volume of 100 mg of Chinese pine needle tissue was utilized. Thermo Scientific NanoDrop 2000 was employed to determine RNA and cDNA concentrations.
Reverse transcription was performed using the R223-01 Novozymes HiScript II Q RT SuperMix for qRCR (+gDNA wiper) kit. Amplification was conducted with a Thermal Cycler Block 5020, using a 20 μl volume that was incubated at 50°C for 15 min, followed by 85°C for 5 s. Subsequently, we evaluated the concentration of cDNA which passed successfully for the next stage of the experiment.
Primer software was utilized for designing the primers. The NCBI BLAST tool was utilized to screen the primers with the highest specificity. Genewiz synthesized the primers. ACT, also known as actin, served as the reference gene with the primer sequences (5′-3′) F: ATTCTCCTCACCGAAGCACC and R: ATGGGAACTGTGTGGCTGAC. The product size was 188 and the accession number is MN172177. The specific primers for genes are listed in Supplementary Table 1.
The primer sequences for RT-qPCR were selected from the Novozymes ChamQ Universal SYBR RT-qPCR Master Mix kit Q711-02/03. The Roche LightCycler 96 instrument was employed for this study. A 20 μl system was used for the RT-qPCR, and mixture configuration can be found in Q711-02/03. In order to enhance the amplification efficiency, the “three-step method” in the kit was employed and the extension time was extended. The specific reaction conditions are as follows: pre-denaturation for 30 s at 95°C was followed by a cycling reaction (40 times) at 95°C for 10 s, 56°C for 30 s, and 72°C for 60 s. Subsequently, a melting curve was run at 95°C for 15 s, 60°C for 60 s, and 95°C for 15 s, with an additional 15 s at 95°C and 60 s at 95°C. The genes’ relative expression was analyzed utilizing the 2–ΔΔCt method, and the data underwent statistical analysis using the DPS data processing system (Tang and Zhang, 2013). The experiments were conducted with a minimum of three technical replicates. In addition, at least use three biological replicates along with three technical replicates for RT-qPCR analysis.
The study of Chinese pine phosphorylated proteins generated 143,668 secondary mass spectra, of which 26,056 were valid (18.1% utilization). The findings revealed 10,817 peptides, 9,421 of which were phosphorylated, and 12,848 sites with phosphorylation-modification located on 4,073 proteins. Phosphorylated proteins were detected in pine needles through a T assay with a P-value < 0.05. Table 1 summarizes the data on phosphorylated proteins and modification sites normalized to the proteomic data. The phosphorylated proteome experiment was performed to analyze the differential proteins and phosphorylation modification sites in the control group (0 h) and the test group (2 and 8 h). Each set of protein abundance values and modification sites were compared, selecting a twofold change, and normalized with the proteome data. From this, the number of differentially expressed proteins was quantitatively determined. Compared to the control group, there were 171 upregulated phosphorylated proteins and 218 phosphorylation-modification sites, as well as 117 downregulated proteins and 147 phosphorylation-modification sites, 2 h after the Chinese pine caterpillar’s feeding stimulus. A total of 192 proteins with phosphorylation and 246 sites with upregulated expression of phosphorylation modification were discovered, along with 104 proteins with phosphorylation and 138 sites with downregulated expression of phosphorylation modification, all 8 h following the feeding stimulus. Comparing the phosphorylated proteins and phosphorylation-modified sites at 2 and 8 h after the feeding stimulus revealed that 11 proteins and 12 sites had increased expression while 17 proteins and 19 sites had decreased expression.
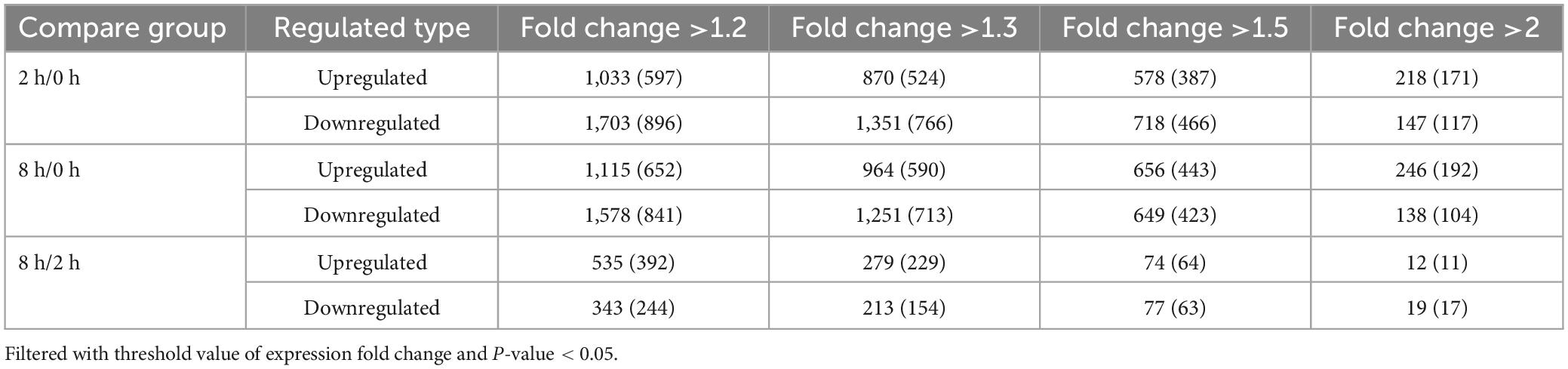
Table 1. Differentially phosphorylated modification sites (phosphorylated protein) summary after normalized.
The amino acid sequence pattern before and after phosphorylation modification sites, and their role in Chinese pine response to the caterpillar feeding stimulus were obtained and analyzed with statistical precision using MoMo software. The regularity pattern of the amino acid sequence trends in the region containing the phosphorylation modification site was calculated. Therefore, the characteristics of the sequence of phosphorylation modification sites can be determined, enabling the identification or hypothesization of the phosphorylase linked to the response of Chinese pine to the feeding stimulus of the caterpillar. The patterns at the serine and threonine phosphorylation modification sites and their enrichment details, along with the heatmap displaying the amino acid base sequence enrichment upstream and downstream of the phosphorylation modification sites, are presented in Figure 1.
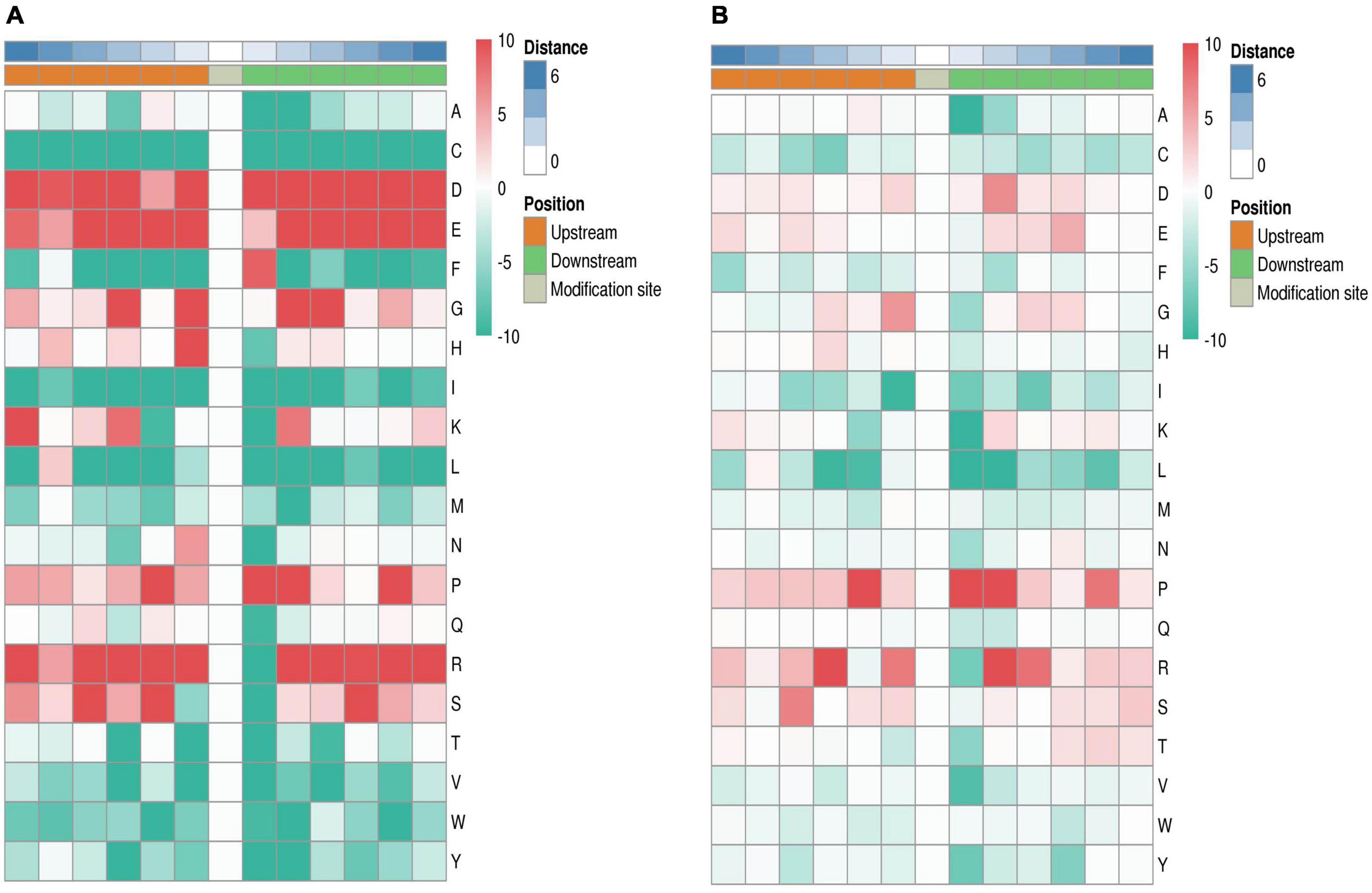
Figure 1. Heatmap of serine (A) and threonine (B) motif enrichment at upstream and downstream of phosphorylation modification sites. The red color in the graph denotes significant amino acid enrichment near the modification site, while the green color signifies a significant reduction in amino acids near the modification site.
Phosphorylated serine has 63 motifs, threonine has 10 motifs and tyrosine has only 5 motifs. Specific information can be found in the latest submitted Supplementary Table 2.
Cluster analysis was conducted to study changes in the abundance of phosphorylation modification sites among Chinese pine proteins after caterpillar feeding at different time points (2 h post-treatment, 8 h post-treatment, and 0 h control). A total of 9,629 modification sites were identified using a quantitative histological study of phosphorylation modifications. To eliminate modification sites with significant changes in abundance across 0, 2, and 8 h samples, we initially subjected the relative expression of modification sites to a Log2 logarithmic transformation. Next, we filtered out modification sites with SD (standard deviation) >0.5. The 1,684 modification sites that passed the initial screening were utilized for an expression pattern clustering analysis using the Mfuzz method. The columns in the cluster were labeled by corresponding expression pattern classes, and modification sites in the same cluster displayed similar trends in expression transformation (Figure 2). Mfuzz analysis parameters: the number of clusters k is set to six and the degree of cluster fuzzy m is set to two.
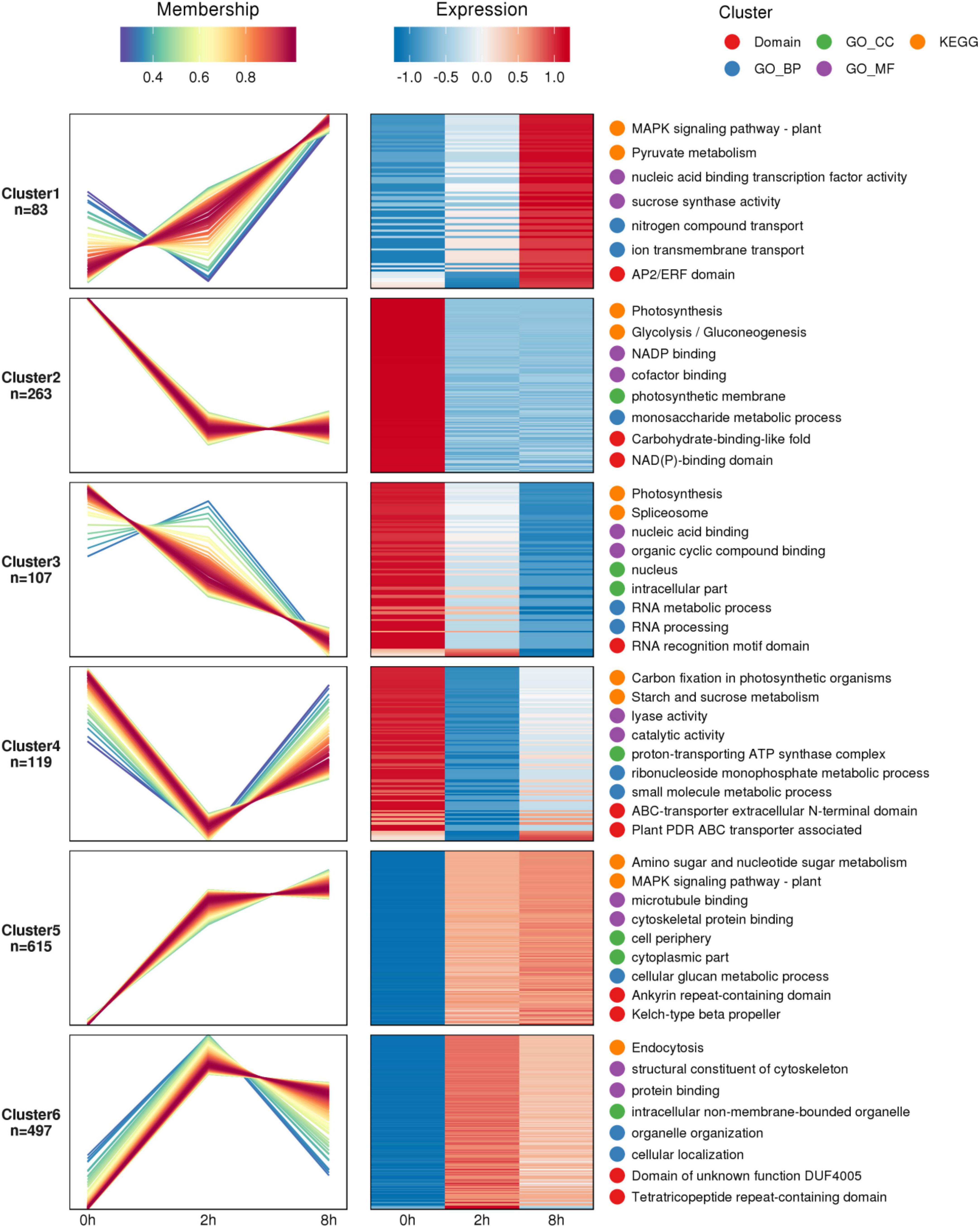
Figure 2. Summary graph of cluster analysis of differential phosphorylation site expression patterns. The figure depicts a line graph illustrating the expression of phosphorylation modification sites on the left side, and a heatmap representing the expression on the right side. Each cluster on the graph and map corresponds to a specific modification site. Line graph: the horizontal axis coordinate is the sample, the vertical coordinate is the relative expression of the phosphorylation modification site, a line represents a phosphorylation modification site, and the line color indicates the affiliation of the phosphorylation modification site in the current class. Heatmap: the horizontal axis represents the samples, while the vertical axis displays the different phosphorylation modification sites. The heatmap colors correspond to the relative expression levels of phosphorylation modification sites detected in the samples.
To facilitate presentation of all analysis results, the two most significantly enriched entries of GO molecular function.enrich.out (Supplementary Table 3), GO cellular component.enrich.out (Supplementary Table 4), KEGG.enrich.out (Supplementary Table 5), GO biological process.enrich.out (Supplementary Table 6), and Domain.enrich.out (Supplementary Table 7) enrichment analysis results are displayed on the right side of the corresponding cluster expression pattern clustering graph. Of the 83 phosphorylation sites in cluster 1, the MAPK signaling pathway-plant and pyruvate metabolism showed the highest levels of enrichment in terms of KEGG pathway. Additionally, both nucleic acid binding transcription factor activity and sucrose synthase activity displayed the most significant levels of enrichment for GO molecular function, while GO biological processes showed significant levels of enrichment for nitrogen compound transport and ion transmembrane transport. Among the 263 phosphorylation sites in cluster 2, the KEGG pathway showed the most significant enrichment in photosynthesis and glycolysis/gluconeogenesis. Additionally, the GO cellular composition demonstrated that the photosynthetic membrane was most significantly enriched, while the GO biological processes indicated that monosaccharide metabolic processes were significantly enriched. Of the 107 phosphorylation sites found in cluster 3, the photosynthesis pathway displays the greatest enrichment within the KEGG pathway. Additionally, the GO molecule function shows that nucleic acid binding and organic cyclic compound binding are the most significantly enriched. Similarly, the GO cellular composition demonstrates that intracellular part is the most significantly enriched. As for the GO biological process, RNA metabolic process and RNA processing were significantly enriched. Lastly, the structural domain shows that RNA recognition motif domain is the most significantly enriched. Among the 119 phosphorylation sites in cluster 4, carbon fixation in photosynthetic organisms and starch and sucrose metabolism were found to be significantly enriched KEGG pathways. Meanwhile, lyase activity and catalytic activity were the most significant parts of GO molecule functional enrichment. In terms of structural domains, the ABC-transporter extracellular N-terminal domain and plant PDR ABC transporter associated were the most significantly enriched. The KEGG pathway exhibited the highest level of enrichment for the amino sugar and nucleotide sugar metabolism and MAPK signaling pathway-plant pathways among the 615 phosphorylation sites within cluster 5. Concerning GO molecular functions, microtubule binding and cytoskeleton (protein binding) exhibited the most significant enrichment, while the most enriched GO cellular compositions were cell periphery and cytoplasmic part. Additionally, protein binding showed a significant enrichment as a GO molecular function. The 497 sites phosphorylated in cluster 6 showed significant enrichment for endocytosis in KEGG pathway analysis, structural composition of cytoskeleton and protein domain in GO molecular function, and organelle organization and cellular localization in GO biological processes.
Phosphokinase prediction upstream of the phosphorylation site was conducted utilizing the GPS 5.0 software. Based on the assumption that analogous short peptide segments may exert comparable biological functions and that akin protein kinases may regulate comparable short linear motifs, it is anticipated that the Chinese pine tree may govern a group of kinase proteins at particular phosphorylation sites prompted by feeding stimuli in caterpillars. The kinase proteins, pertaining to the kinase protein family in this species, were acquired through a comparison with the iEKPD database. Subsequently, protein–protein interaction information (PPI) was used to filter out potential false-positive interactions. A total of 1,736 regulatory connections were foreseen among 413 protein kinases and 780 sites where phosphorylation modification occurred on 485 identified proteins.
The study employed a gene set consisting of the regulatory correlation between phosphokinase and phosphorylation sites. The expression level of phosphosites served as the rank file in the samples, while the relative quantitative ratio value of differentially phosphorylated modification sites served as the rank file in the comparison groups. The kinase activities of the samples and the comparison group were predicted separately using the GSEA method. Fifty-six phosphokinases appeared to be inhibited, and one phosphokinase appeared to be activated in the Chinese pine needles 2 h after being stimulated by the caterpillars’ feeding. In the sample collected 8 h after exposure to stress, there was evidence of inhibition in 47 phosphokinases, while only 1 phosphokinase appeared to be activated. Compared to the treatment group, pine needle samples from the control group (0 h) showed inhibition of 15 phosphokinases and activation of 56 phosphokinases (Figure 3A). Phosphorylation of substrate sites can reflect the level of kinase regulation to a certain extent; the higher the protein phosphorylation level influenced by phosphokinase, the greater the kinase activity may be, and vice versa. The trends among the three comparison groups were consistent. In the 2 h vs. 0 h and 8 h vs. 0 h groups, respectively, the number of phosphokinases tending to inhibit was 59 and 57, while the number of phosphokinases tending to activate was 11 and 12. The comparison group at 8 h vs. 2 h had 8 phosphokinases that displayed tendencies toward inhibition and 12 phosphokinases displaying tendencies toward activation. The number of phosphokinases displaying tendencies toward activation was found to be essentially the same throughout all three comparison groups (Figure 3B).
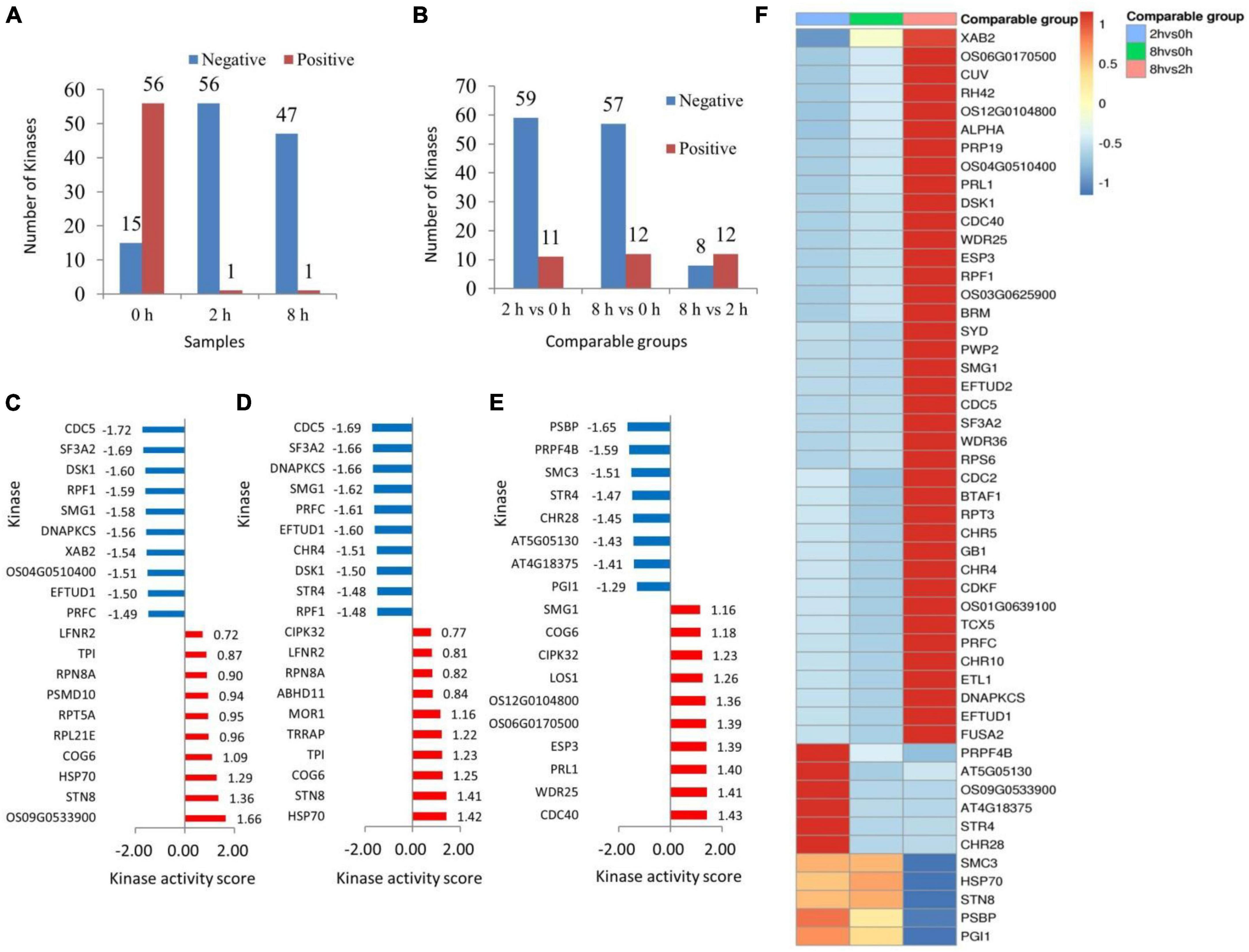
Figure 3. Statistical graph of sample (A) and comparable groups (B) phosphokinase activity. The x-axis denotes the sample name while the y-axis represents the number of kinases. Red color signifies activation tendency and blue color indicates inhibition tendency. Bar graph of phosphokinase activity scores in 2 h vs. 0 h (C), 8 h vs. 0 h (D), and 8 h vs. 2 h (E) comparison group. The enrichment analysis method provided the NES value which served as the kinase activity score. A kinase with a score >0 is typically activated, while a score <0 indicates inhibition. The x-axis is the phosphokinase activity score, and the y-axis is the top 10 phosphokinases in terms of activity score for the activated or inhibited state, with red representing the activated state and blue representing the inhibited state. Heatmap of phosphokinase activity matrix for the comparison group (F). Columns are sample names, rows are phosphokinases (clustering and Z-score normalization transformations on rows).
Activity prediction of kinases based on protein phosphorylation expression using GSEA.ES (enrichment score) responds to the extent to which members of the kinase-regulated locus are enriched at either end of the sorted list. Normalization of the enrichment score yields the normalized enrichment score (NES). In the comparison group 2 h vs. 0 h, the phosphokinases OS09G0533900 (endoglucanase 24), STN8 (serine/threonine-protein kinase STN8, chloroplastic) and HSP70 (heat shock 70 kDa protein 17), among others, were in the activated state. Phosphokinase CDC5 (cell division cycle 5-like protein), SF3A2 (splicing factor 3A subunit 2), and DSK1 (protein kinase dsk1), on the other hand, were in an inhibited state (Figure 3C). In the comparison group 8 h vs. 0 h, phosphokinases in activated state included HSP70, STN8, and COG6 (conserved oligomeric Golgi complex subunit 6), among others. Phosphokinases in the inhibited state were CDC5, SF3A2, and DNAPKCS (DNA-dependent protein kinase catalytic subunit) (Figure 3D). The phosphokinase CDC40 (pre-mRNA-processing factor), WDR25 (WD repeat-containing protein25), and PRL1 (protein pleiotropic regulatory locus) were in an activated state in the comparison group at 2 h vs. 8 h. Phosphate kinase PSBP (oxygen-evolving enhancer protein 2, chloroplastic), PRPF4B (serine/threonine-protein kinase PRP4 homolog), and SMC3 (structural maintenance of chromosomes protein 3) were in the inhibited state (Figure 3E).
The Kinase Activity Clustering Heat Map enables observation of kinase activity commonality within the same phenotype or specificity in different phenotypes. This aids exploration of potential relationships between kinase activity and phenotype for further screening of key kinases in Chinese pine in response to caterpillar feeding stimuli. A side-by-side comparison of the significantly enriched phosphokinases in the 2 h vs. 0 h, 8 h vs. 0 h, and 8 h vs. 2 h comparison groups showed that the phosphokinase XAB2 (pre-mRNA-splicing factor SYF1) was in a markedly inhibited state in the 2 h vs. 0 h comparison group, and in a markedly activated state in the 8 h vs. 2 h comparison group. Nineteen phosphokinases including OS06G0170500 (zinc finger CCCH domain-containing protein 40), CUV (pre-mRNA-splicing factor ATP-dependent RNA helicase DEAH7), and RH42 (DEAD-box ATP-dependent RNA helicase 42), demonstrated inhibition in the 2 h vs. 0 h comparator group, and activation in the 8 h vs. 2 h comparator group. Nineteen phosphokinases, such as SYD (chromatin structure-remodeling complex protein SYD), PWP2 (periodic tryptophan protein 2 homolog), and SMG1 (serine/threonine-protein kinase SMG1), were inhibited further in the comparison group of 8 h vs. 0 h, and were significantly activated in the comparison group of 8 h vs. 2 h. Phosphokinase PRPF4B and AT5G05130 (putative SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily A member 3-like) were activated in both 2 h vs. 0 h comparison groups. PRPF4B was inhibited in the 8 h vs. 0 h comparison group and also in the 8 h vs. 2 h comparison group. On the other hand, AT5G05130 was inhibited in both the 8 h vs. 0 h comparison group and the 8 h vs. 2 h comparison group. Four phosphokinases, including Phosphokinase OS09G0533900, AT4G18375 (KH domain-containing protein), STR4 (rhodanese-like domain-containing protein 4, chloroplastic) and CHR28 (helicase-like transcription factor CHR28), showed activation in the 2 h comparison group when compared to the 0 h group. Additionally, phosphokinase SMC3 was significantly inhibited in the 8 h vs. 2 h comparison group. Both HSP70 and STN8 phosphokinases tended to be activated in the 2 h vs. 0 h comparison group, whereas both phosphokinases PSBP and PGI1 (glucose-6-phosphate isomerase 1, chloroplastic) tended to be activated in the 8 h vs. 0 h comparison group, and were significantly inhibited in the 8 h vs. 2 h comparison group (Figure 3F).
For each comparative group of pine needle samples, we screened for phosphokinases that were significantly activated or inhibited and phosphorylation sites with significantly different expression levels (ratio threshold for fold change in differential expression is 2 and P-value threshold for significance of difference is 0.05) in response to insect feeding stress. Construct regulatory networks of kinases, observe regulatory relationships between kinases and substrates, and identify key regulatory kinases within complex networks. The kinases involved in the regulatory network in the control 0 h vs. 2 h post-feeding stimulation comparison groups were RPL21E (60S ribosomal protein L21-2), LOS1 (elongation factor 2), CDC5, SF3A2, and CHR4 (protein CHROMATIN REMODELING 4), and SMG1, DNAPKCS, OS09G0533900, and BTAF1 (TATA-binding protein-associated factor) (Figure 4A). The following kinases were involved in the regulatory network for the control 0 h vs. 8 h post-feeding stimulation comparison groups: AT4G18375, DSK1, SMG1, DNAPKCS, and TPI (triosephosphate isomerase, chloroplastic), CHR4, RPL21E, LOS1, BTAF1, and MOR1 (protein MOR1), TRRAP (transformation/transcription domain-associated protein), and STR4 (Figure 4B). The 2 and 8 h post-feeding stimulus comparison groups were examined for any inter-kinase associations. However, no such associations were found. Additionally, none of the MCODE score values reached the level of significance, indicating the absence of any clusters.
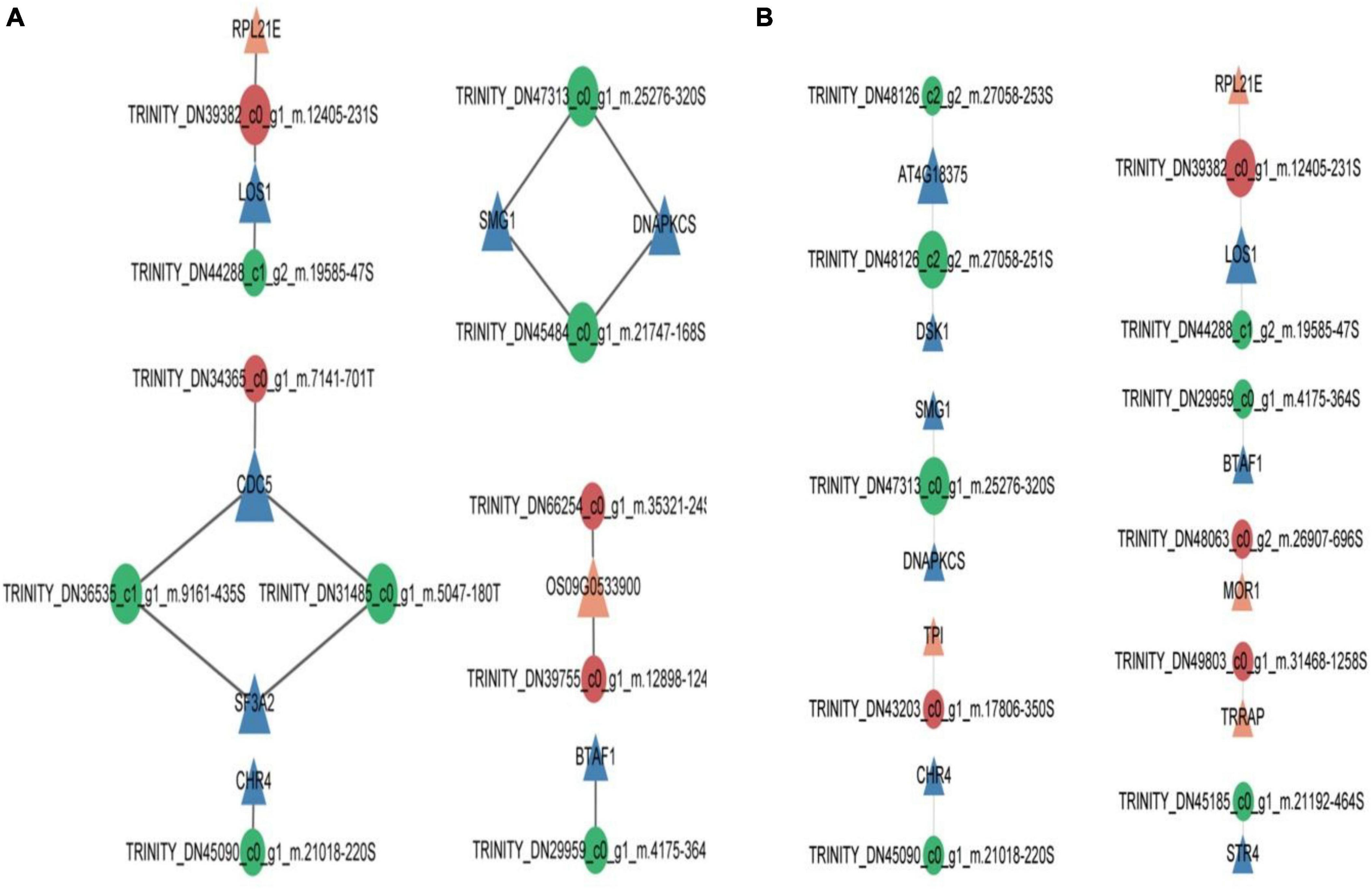
Figure 4. Comparison group of 0 h vs. 2 h (A) and 0 h vs. 8 h (B) kinase regulatory networks. Orange triangles indicate activated kinases while blue triangles indicate inhibited kinases. Red circles denote phosphorylation sites that are upregulated differentially, and green circles suggest phosphorylation sites that are downregulated differentially.
The MCODE add-on in the STRING application was utilized to extract high-connectivity modules in the protein interaction network. Following this, the module proteins underwent analysis for functional enrichment in a bid to identify significant biological processes in which they are involved. The differentially phosphorylated proteome was screened for 14 functional modules, focusing on PGK1 (phosphoglycerate kinase 1)-OS11g0171300 (fructose-bisphosphate aldolase, chloroplastic), and SHM4 (serine hydroxymethyltransferase 4), comparing the 0 and 2 h post-feeding stimulus groups (Figure 5A). Comparing the post-feeding stimulus comparison groups at 0 and 8 h, the differentially phosphorylated proteome was analyzed for 12 functional modules. Notably, At2g20050/At2g20040 (protein phosphatase 2C and cyclic nucleotide-binding/kinase domain-containing protein) stood out, followed by MOR1 (Figure 5B). No associations were found between differentially phosphorylated proteins in the comparison groups of 2 and 8 h post-feeding stimulus after screening, nor were there any associations of differentially phosphorylated proteins. Furthermore, no MCODE score values reached statistical significance, and consequently, no clusters were labeled.
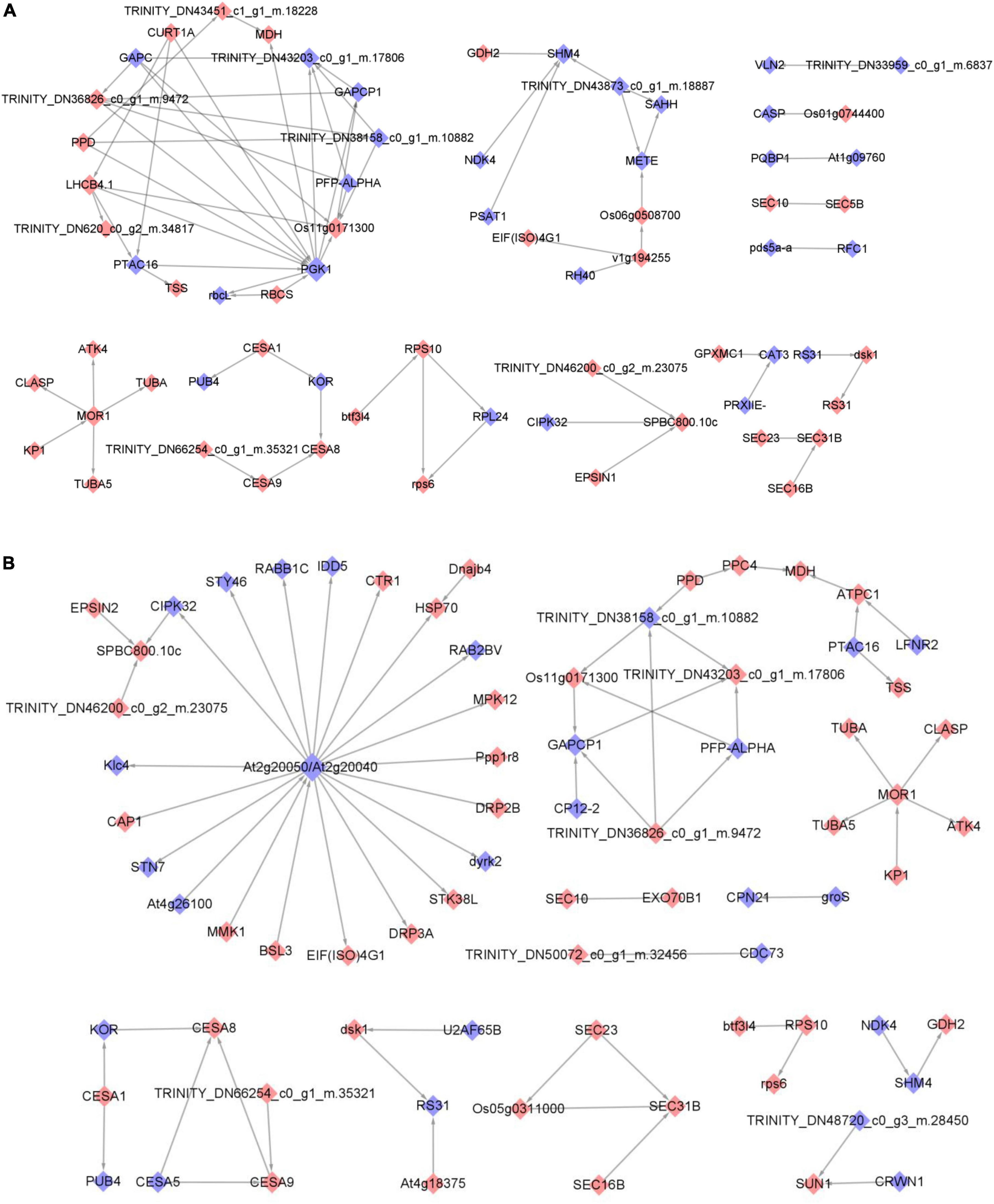
Figure 5. Protein Interaction Network Functional Module of 0 h vs. 2 h (A) and 0 h vs. 8 h (B) significantly different phosphorylated proteins. Phosphorylated protein database numbers or sequences from the comparison group based on a fold difference of 2.0 were compared with the STRING (v.11.0) protein interaction network database. The cytoscape tool was employed to visualize the differential phosphorylated protein interactions. The color scheme distinguished between upregulated proteins (red) and downregulated proteins (blue), with darker colors indicating greater differential folds in expression.
Parallel reaction monitoring quantification was conducted on four selected target proteins across nine samples (0 h-1, 0 h-2, 0 h-3, 2 h-1, 2 h-2, 2 h-3, 8 h-1, 8 h-2, and 8 h-3). For quantification, our experimental design used more than two distinct peptides per protein, although some proteins were identified with only one peptide due to sensitivity and other factors (Table 2). The precision of crucial proteins involved in the defense response mechanism of the Chinese pine was evaluated through protein-level validation. The validated PRM-assayed proteins consist of METK2 (S-adenosylmethionine synthase 2), PTI12 (PTI1-like tyrosine-protein kinase 2), pgk, and At3g59480 (probable fructokinase-4). METK2 plays a vital role in various biotic stresses in plants, and it is involved in the metabolism of cysteine and methionine. The function of PTI12 in plant stress response is focused on cytoskeletal regulation as a tyrosine kinase. pgk, a very conserved family of phosphorylated proteins, regulates senescence death in plant cells. Fructose kinase At3g59480 plays an important defense role in plant resistance pathways.
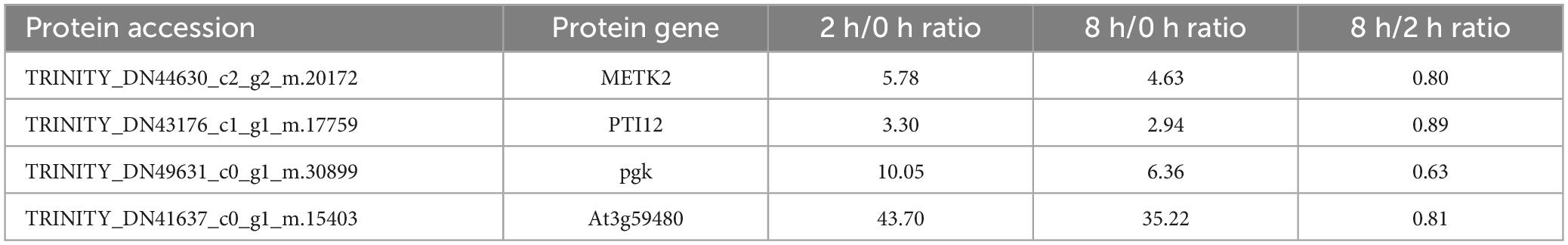
Table 2. Parallel reaction monitoring phosphorylation-related protein quantification results.
The four genes that correspond to the key phosphorylated proteins, verified through PRM, underwent RT-qPCR analysis. The study examined five groups: 0 h after the treatment (control), 2 h after pine caterpillar feeding stimulation, 2 h after leaf-cutting stimulation, 8 h after feeding stimulation, and 8 h after leaf-cutting stimulation. The trends in the relative expression of these four genes after feeding and leaf-cutting stimulation were as follows:
The expression levels of the METK2 gene (Figure 6A) initially decreased before increasing and remained lower than those of the control at two particular time points post-treatment. The expression of the METK2 gene showed a statistically significant (P < 0.01) decrease 2 h after feeding stimulation. There was also a significant (P < 0.05) difference in METK2 expression 2 h after treatment with leaf-cutting stimulation. METK2 expression was higher at 8 h compared to 2 h post-feeding, but this difference was not statistically significant. After cutting the leaves, the expression of METK2 increased after 8 h. The relative expression of METK2 was lower at 8 h after leaf-cutting compared to 2 h after leaf-cutting. Additionally, the differential relative expression of METK2 was lower at 8 h after leaf-cutting with stimulation compared to 8 h after leaf-cutting, but the difference was not significant.
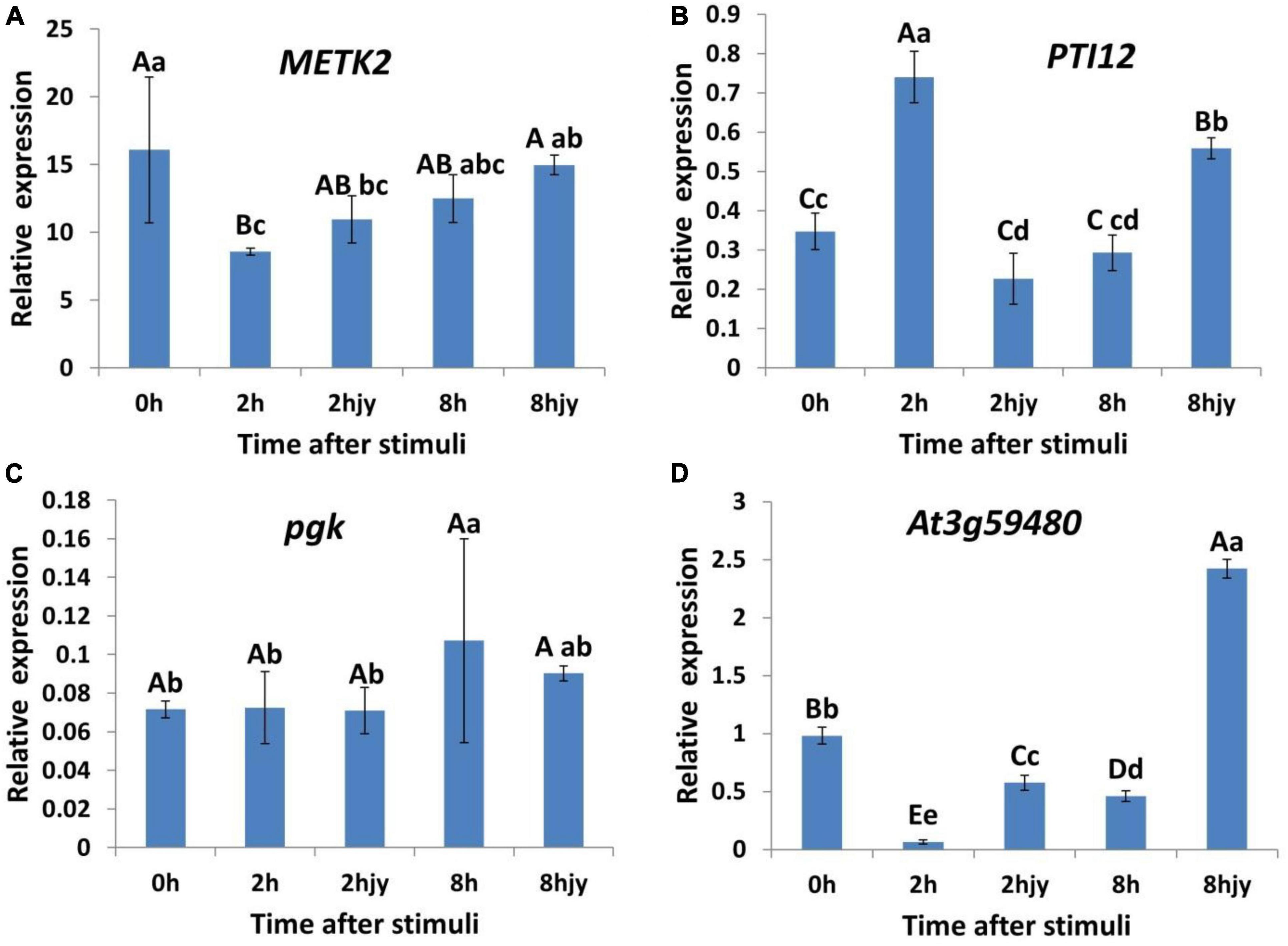
Figure 6. Real-time RT-PCR analysis of key phosphorylation-related protein upstream genes. Different letters represent significant differences, capital letters represent extremely significant differences (P < 0.01), lowercase letters represent significant differences (P < 0.05). (A) Relative expression of METK2 gene at 0 h in the control, 2 and 8 h after feeding stimulation and leaf cutting stimulation (2hjy and 8hjy). (B) Relative expression of PTI12 gene. (C) Relative expression of pgk gene. (D) Relative expression of At3g594800 gene.
PTI12 (Figure 6B) demonstrated a pattern of increasing and subsequently decreasing relative expression following feeding. The expression of PTI12 showed a significant increase (P < 0.01) 2 h after feeding, followed by a highly significant decrease (P < 0.01) 8 h after feeding. The expression level of PTI12 decreased 2 h after leaf-cutting stimulation. However, it was highly significantly elevated (P < 0.01) at 8 h after leaf-cutting in comparison to the expression level observed at 2 h after leaf-cutting. The expression level of PTI12 was significantly higher (P < 0.01) 2 h post-feeding stimulation compared to 2 h post-leaf-cutting stimulation. It is worth mentioning that the relative expression of PTI12 was lower than that of leaf-cutting stimulation at 8 h after feeding stimulation and reached the level of a highly significant difference (P < 0.01).
The relative expression of pgk at 2 h after feeding and leaf-cutting was barely different from that of the control at 0 h and was not significant. From 2 to 8 h after treatment, the relative expression of genes increased with both feeding and leaf-cutting stimuli. However, only the difference in the relative expression of pgk under feeding stimuli reached a significant (P < 0.05) level. Gene expression was higher 8 h after feeding compared to 8 h after leaf-cutting, but there was no significant difference between the two (Figure 6C).
The relative expression of At3g59480 (Figure 6D) was lower than that of the control and leaf-cutting treatments at both 2 and 8 h under feeding stimuli. This was shown by a notable reduction (P < 0.01) in the relative expression of At3g59480, 2 h following the feeding stimulus. The expression level of At3g59480 significantly increased (P < 0.01) at 8 h after feeding in contrast to its expression level at 2 h after feeding. Likewise, the gene’s expression level also significantly decreased (P < 0.01) 2 h after leaf-cutting stimulation. The expression level of At3g59480 was significantly higher (P < 0.01) at 8 h post leaf cutting than at 2 h after leaf cutting. It is worth noting that the relative expression of At3g59480 showed significant differences when comparing 2 h after feeding to 2 h post leaf-cutting, and again when comparing 8 h after feeding to 8 h post leaf-cutting (P < 0.01).
Exposing plants to external stressors triggers a cascade of resistant defensive processes. Extensive data indicates that stress responses in plants, induced by various stress factors and extracellular signals, are closely linked to changes in the phosphorylation status of proteins. This phosphorylation status is considered an important aspect of plant immunity. Stress response reactions, including mitotic initiation, isoprene synthesis, cytoplasmic streaming, and sucrose phosphate synthase activity, are regulated by intracellular processes of protein kinase and protein phosphatase. Tyrosine kinases play a crucial role in regulating plants’ response to stress, particularly in relation to the cytoskeleton (Samofalova et al., 2020). This study found that the expression of PTI12, a tyrosine-like protein kinase in Chinese pine, increased significantly at 2 and 8 h after caterpillar feeding. However, the expression level at 8 h was lower than at 2 h. Our results confirm that phosphorylated proteins play a critical role in regulating plant defense mechanisms against phytophagous insect feeding in Chinese pine. This is achieved through protein phosphorylation modifications in response to caterpillar feeding, demonstrating an involvement of phosphorylated proteins in regulating plant resistance. The expression level of tyrosine kinase experienced a significant decrease, which regulates a notable increase in the expression level of the heat shock protein Heat Shock 70 kDa Protein 6 (HSP70B). Its molecular chaperone function helps minimize protein aggregation, while also aiding in the repair and protection of cellular proteins from stress damage (Lyu et al., 2019). Tyrosine protein kinase is implicated in the upregulation of 12-oxophytodienoate reductase 3 (OPR3), a vital enzyme in the JA synthesis pathway, which may lead to enhanced resistance of rice against phytophagous insects (Guo et al., 2015).
METK2 participated in the metabolic process of cysteine and methionine following feeding stimulation by the Chinese pine caterpillar. METK2 expression displayed a significant increase at 2 and 8 h compared to 0 h but exhibited higher expression at the onset of stress. These findings suggest that METK2 plays a continuous role in resistance-related processes. METK2 is responsible for catalyzing the conversion of methionine into S-adenosyl-L-methionine. Adenosine homocysteine lyase is an enzyme that utilizes S-adenosyl-L-methionine as a substrate to produce homocysteine (HCY). Bioinformatics analysis of differentially expressed proteins in this study shows that there is an interaction between these two enzymes. METK2 is an upstream gene of S-adenosylmethionine synthase (SAM), SAM is an essential element in the induced defense of Arabidopsis and plays a direct role in the defense response (Zhang et al., 2010). ET regulates defense transduction signals, while SAM is responsible for the involvement of ET biosynthesis (Reymond et al., 2004). The defense gene SAM is activated by the two-spotted leaf mite infestation in lima bean leaves, which catalyzes the biosynthesis of ET and induces a resistance response (Arimura et al., 2002). The SAM gene improves plants’ defense against peach aphids. Technical term abbreviations will be explained upon their first use. Cloning and introducing the maize S-adenosylmethionine synthase (BtSAMS) gene into Arabidopsis thaliana reduced the number of peach aphids feeding on BtSAMS plants by over 50% compared to the control. Additionally, the number of neonate wakame on BtSAMS plants was less than 50% of the control (Yang et al., 2023). The expression of GmSAMS1, linked to defense response, underwent upregulation following phosphorylation modification in the Genetically Modified Calcium-dependent Protein Kinase 38 (GmCDPK38) soybean mutant. This modification ultimately intensified soybean resistance to Spodoptera litura (Li et al., 2022). Our results suggest a possible link between the expression of the METK2 gene and the phosphorylation modification of resistance proteins in response to the ingestion of Chinese pine needles by the Chinese pine caterpillar. This activation or enhancement of resistance function in Chinese pine against caterpillar feeding is likely to occur.
At3g59480 (probable fructokinase-4) participates in the defense response of coconut trees to Rhynchophorus ferrugineus stress. Specifically, fructokinase plays a defensive role in the resistance pathway linked to sugar metabolism after 5 and 20 days of R. ferrugineus’ feeding (Liu et al., 2023). This could explain why the expression of At3g59480 significantly increased in Chinese pine 2 and 8 h after feeding stimulation. It is hypothesized that At3g59480 plays a role in sugar metabolism related to Chinese pine’s resistance response to caterpillar feeding stimulation.
Key phosphorylated proteins that are upregulated in response to caterpillar feeding stimuli in Chinese pine include MOR1, MDH, RPS6A, and the chloroplast antibody AT4G01150. The phosphorylated protein MOR1 in pine plays a central role in functional protein modules related to plant microtubule structure, both 2 and 8 h after being exposed to caterpillar stress. MOR1 encodes a protein associated with the microtubule cytoskeleton structure in A. thaliana. Microtubule-binding proteins regulate transduction signals inside and outside of plant cells to facilitate the plant’s regulatory mechanisms in response to environmental changes. MOR1 is an essential component in the dynamic regulation of microtubules, as highlighted by Chen et al. (2022). The findings of this study suggest that the expression of MOR1 is closely associated with the defense response against resistance in Chinese pine.
Malate dehydrogenase (MDH) had a highly connected position in the interaction network of differentially significant phosphorylated proteins in oil pine, both at 2 and 8 h after insect infestation. This indicates that MDH is involved in the response process of Chinese pine against herbivore stress induced by caterpillar infestation. Previous research indicates that MDH plays a role in abiotic stress processes in plants. Specifically, the expression of genes in the MDH gene family of rice, such as OsMDH8.1, has been observed to have a significant correlation with the response of rice to salt stress (Zhang et al., 2022).
RPS6A is a downstream target gene within the Arabidopsis TOR signaling network (Kim et al., 2014). RPS6A’s primary functions include the regulation of protein synthesis and total RNA production (Jansen, 2017). Plant lifespan is strongly linked to nutrition and photosynthesis. According to Ren et al. (2012), Arabidopsis plants with RPS6A mutations have a longer lifespan, while plants overexpressing TOR ribosomal proteins have a shortened lifespan and accelerated life cycle. In this study, RPS6A was primarily present in the interaction network of phosphorylated Chinese pine resistance proteins 2 h after caterpillar feeding and was mildly less associated with herbivore stress after 8 h. This indicates that Chinese pine caterpillar stress activates the protective mechanism of RPS6A, and further research is required to investigate the specific nutritional and photosynthetic pathways involved, along with their regulatory mechanisms.
Novel chloroplast proteins have been identified in plant genomic and proteomic studies. At4g01150 is the only one of the 33 plastid goblet (PGs) proteins with a predicted transmembrane structural domain. It is localized in the chloroplast-like vesicle membrane (Friso et al., 2004; Peltier et al., 2004). Intense light stress caused a threefold rise in the relative accumulation of At4g01150 in plastid microspheres (Ytterberg et al., 2006). At4g01150 was more abundant in de-yellowed Arabidopsis cotyledons than the other three CURT1 family genes (Liang et al., 2021). Genes with unknown function in tobacco, which are homologous to Arabidopsis At4g01150, display higher expression levels in plants with high nitrogen application and topping for leaf retention (Lei et al., 2022). Arabidopsis chloroplast protein At4g01150 is significantly expressed during the heat stress response (Paul et al., 2020). During the analysis of the interaction network of phosphorylated proteins that are differentially and significantly upregulated in Chinese pine after a feeding boost from caterpillars, it was discovered that At4g01150 holds a prominent position and is linked to multiple proteins. It suggests that At4g01150 may be involved in the core mechanism of the Chinese pine’s response toward the feeding stimulus of caterpillars. This result holds significant importance in enhancing the functionality of novel chloroplast proteins.
Key phosphorylated proteins whose expression is reduced in response to insect-feeding stimuli in Chinese pine comprise PGK1, chloroplast antibody AT2G20050, GAPC1, RPL24, and PTAC16. Phosphoglycerate kinase PGK is a highly conserved protein family involved in carbon fixation and metabolic processes in the cytoplasm in vivo (Li et al., 2019). PGK1 is localized in the cytoplasm, while its homolog PGK2 is found in chloroplasts. PGK2 regulates programmed cell death in plant cells and performs functions that are comparable to those of the hypersensitive response, such as leaf yellowing, activation of PR genes, and accumulation of hydrogen peroxide (Huang and Zhao, 2017). PGK plays a critical role in regulating the conversion of ATP and ADP in plants. It is involved in various cellular activities such as glycolysis, gluconeogenesis, and the Calvin cycle. The study suggests that PGK is located within the cytoplasm and functions as an ATP generator while also acting as a transferase by transferring phosphate groups to related reactions. PGK participates in the first step of cellular respiration in the cytoplasm, and increased PGK expression suggests an improvement in plant respiration. Troncoso-Ponce et al. (2012) have extensively studied the biochemical profile of PGK isozymes during sunflower development. Pine needle PGK kinase exhibited the most significant downregulation of phosphorylation among proteins after caterpillar feeding for 2 h. The protein interactions that were differentially significant were centered around it. The decrease in Chinese pine PGK kinase activity, caused by caterpillar feeding, is believed to be a result of pine needle defense regulation and harmful substances in the insect’s saliva. Further investigation is required to determine the specifics.
The AT2G20050 chloroplast antibody is central to the phosphorylated protein interaction network of the oil pine caterpillar 8 h after feeding. During the 2–8-h feeding period, the Chinese pine’s damage focus shifted from PGK to AT2G20050, as evidenced by changes in pine phosphokinase activity. Many signaling kinases, receptor kinases, and phosphatases have close relationships with AT2G20050 (Mattei et al., 2016). These connections suggest potential functional roles for AT2G20050 in cellular signaling pathways. AT2G20050 negatively regulates protein kinase pathways involved in plant stress response, growth, and development (Kuhn et al., 2006). Chinese pine suppresses the expression of AT2G20050 kinase, triggering a complete defense response mechanism.
The plant’s cytoplasmic glyceraldehyde triphosphate dehydrogenase (GAPC) is believed to play a role in the mechanism of drought tolerance in wheat (Zhai, 2017), as well as in responding to iron deficiency stress (Li, 2000). GAPC1 is also recognized as a redox switch overexpressed in plants to improve Arabidopsis’ response to heat stress (Kim et al., 2020). GAPC1 also contributes to the defense response against reactive oxygen species (ROS) (Schneider et al., 2018). GAPC1 plays a crucial role in initiating resistance through a network of distinct, phosphorylated protein interactions. This process occurs 2 h after exposure to herbivore stimuli in Chinese pine. It is hypothesized that GAPC1 may be involved in the redox reaction process, including the clearance of reactive oxygen stress in Chinese pine. Further investigation is required to identify the specific resistance mechanism. The Arabidopsis transcription factor RPL acts as a resistance mechanism by regulating the conversion of energy needed for plant growth and development into a response against pathogen infestation. Its function is similar to that of a “switch” that turns on the defense system. This process involves the inhibition of the expression and transportation of pathogenic bacterial effector proteins, which play a role in the accumulation of flavonoids (Xu et al., 2022).
The expression of RPL24 in Chinese pine needles decreased 2 h after caterpillar feeding stimulation. It is hypothesized that the defense mechanism regulated by RPL24 may be gradually inhibited by insect pest stress, which could involve the regulation of flavonoids and related resistance products. The mechanism of action requires further and in-depth study. Eggermont et al. (2018) proposed that PTAC16 could be linked to the defense response against salt stress in Arabidopsis. Additionally, the PTAC16 protein is responsible for regulating the anchoring of the nucleoid membrane (Ingelsson and Vener, 2012). The plastid transcriptionally active kinase, PTAC16, exhibited high connectivity in the interaction network of differentially significant phosphorylated proteins in Chinese pine after 2 and 8 h of insect infestation, indicating a potential role of PTAC16 in the pine response to herbivore stress induced by caterpillars. Pest stress is believed to cause a notable decrease in the expression of PTAC16 kinase, leading to disruptions in signal recognition and plastid transcription in Chinese pine. Further investigation into the underlying mechanism is necessary to understand the phenomenon fully.
Overall, our study first established the interrelationship between wild Chinese pine and Chinese pine caterpillar under natural conditions. Additionally, our study also analyzed motif patterns of phosphorylated proteins and constructed a modification map for phosphorylated proteomics (Supplementary Table 2). Furthermore, the study emphasizes the significant role of kinase PTI12 At3g59480, as well as METK2. Additionally, MOR1, MDH, RPS6A, AT4G01150 PGK1, AT2G20050, GAPC1, RPL24, and PTAC16 are involved in Mfuzz clustering, kinase regulatory networks, and functional modules of phosphorylated proteins in the pine response to caterpillar stimuli. Our results advance our understanding of the mechanisms underlying Chinese pine resistance to Chinese pine caterpillar feeding and also PTM in plant immunity. The specific mechanisms by which phosphorylation regulate Chinese pine resistance to caterpillar remain to be investigated in the future.
The original contributions presented in this study are included in this article/Supplementary material, further inquiries can be directed to the corresponding author.
TS: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. YZ: Writing – original draft. GZ: Writing – original draft. SG: Writing – original draft. JL: Writing – review & editing. BG: Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research received funding from the Science and Technology Development Program at Hebei Agricultural University for Research on Molecular Mechanisms of Forest Plant-Insect Interactions. It also received support from Research on Molecular Mechanisms of Population Differentiation and Adaptation of Forest Pests and Insects under Environmental Stress (No. 30771739) and Forest Pests and Diseases (No. 1528003).
We express our gratitude to the Huangtuliangzi State forest farm for providing a test plot of natural, pure Chinese Pine forest.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/ffgc.2024.1356511/full#supplementary-material
Arimura, G., Matsui, K., and Takabayashi, J. (2009). Chemical and molecular ecology of herbivore-induced plant volatiles: Proximate factors and their ultimate functions. Plant Cell Physiol. 50, 911–923. doi: 10.1093/pcp/pcp030
Arimura, G., Ozawa, R., Nishioka, T., Boland, W., Koch, T., Kühnemann, F., et al. (2002). Herbivore-induced volatiles induce the emission of ethylene in neighboring lima bean plants. Plant J. 29, 87–98. doi: 10.1046/j.1365-313x.2002.01198.x
Baldwin, I. T., Halitschke, R., Kessler, A., and Schittko, U. (2001). Merging molecular and ecological approaches in plant–insect interactions. Curr. Opin. Plant Biol. 4, 351–358. doi: 10.1016/s1369-5266(00)00184-9
Bentem, S. D., and Hirt, H. (2007). Using phosphoproteomics to reveal signalling dynamics in plants. Trends Plant Sci. 12:411.
Chen, Y., Liu, X., Zhang, W., Li, J., Liu, H., Yang, L., et al. (2022). MOR1/MAP215 acts synergistically with katanin to control cell division and anisotropic cell elongation in Arabidopsis. Plant Cell 34, 3006–3027.
Cheng, X., Zhu, L., and He, G. (2013). Towards understanding of molecular interactions between rice and the brown planthopper. Mol. Plant 6, 621–634.
Demichev, V., Szyrwiel, L., Yu, F., Teo, G. C., Rosenberger, G., Niewienda, A., et al. (2022). dia-PASEF data analysis using FragPipe and DIA-NN for deep proteomics of low sample amounts. Nat. Commun. 13:3944. doi: 10.1038/s41467-022-31492-0
Ding, S., Lv, J., Hu, Z., Wang, J., Wang, P., Yu, J., et al. (2023). Phytosulfokine peptide optimizes plant growth and defense via glutamine synthetase GS2 phosphorylation in tomato. EMBO J. 42:e111858. doi: 10.15252/embj.2022111858
Eggermont, L., Stefanowicz, K., and Van Damme, E. J. (2018). Nictaba homologs from Arabidopsis thaliana are involved in plant stress responses. Front. Plant Sci. 8:2218. doi: 10.3389/fpls.2017.02218
Elsayed, G. (2011). Plant secondary substances and insects behaviour. Arch. Phytopathol. Plant Protect. 44, 1534–1549.
Fatma, B., Katrin, F. O., and Wilfried, S. (2019). Phosphorylation-dependent ribonuclease activity of Fra a 1 proteins. J. Plant Physiol. 233, 1–11. doi: 10.1016/j.jplph.2018.12.002
Förderer, A., and Chai, J. (2023). Die another day: Phytosulfokine at the molecular trade-off between growth and defense in plants. EMBO J. 42:113540. doi: 10.15252/embj.2023113540
Friso, G., Giacomelli, L., Ytterberg, A., Peltier, J.-B., Rudella, A., Sun, Q., et al. (2004). In-depth analysis of the thylakoid membrane proteome of Arabidopsis thaliana chloroplasts: New proteins, new functions, and a plastid proteome database. Plant Cell 16, 478–499. doi: 10.1105/tpc.017814
Guo, H. M., Sun, S. C., and Zhang, F. M. (2015). Identification of genes potentially related to herbivore resistance in OPR3 overexpression rice by microarray analysis. Physiol. Mol. Plant Pathol. 92, 166–174.
Guo, Y., Peng, D., Zhou, J., Lin, S., Wang, C., Ning, W., et al. (2019). iEKPD 2.0: An update with rich annotations for eukaryotic protein kinases, protein phosphatases and proteins containing phosphoprotein-binding domains. Nucleic Acids Res. 47, D344–D350. doi: 10.1093/nar/gky1063
Hernandez-Armenta, C., Ochoa, D., Gonçalves, E., Saez-Rodriguez, J., and Beltrao, P. (2017). Benchmarking substrate-based kinase activity inference using phosphoproteomic data. Bioinformatics 33, 1845–1851. doi: 10.1093/bioinformatics/btx082
Huang, X. Z., and Zhao, Y. C. (2017). Functional analysis of PGK gene family in Arabidopsis. J. Mountain Agric. Biol. 36:7.
Ingelsson, B., and Vener, A. V. (2012). Phosphoproteomics of Arabidopsis chloroplasts reveals involvement of the STN7 kinase in phosphorylation of nucleoid protein pTAC16. FEBS Lett. 586, 1265–1271. doi: 10.1016/j.febslet.2012.03.061
Jansen, W. M. (2017). Regulatory circuits linking energy status to growth. Utrecht: Utrecht University.
Kim, S. C., Guo, L., and Wang, X. (2020). Nuclear moonlighting of cytosolic glyceraldehyde-3-phosphate dehydrogenase regulates Arabidopsis response to heat stress. Nat. Commun. 11:3439. doi: 10.1038/s41467-020-17311-4
Kim, Y.-K., Kim, S., Shin, Y., Hur, Y. S., Kim, W.-Y., Lee, M.-S., et al. (2014). Ribosomal protein S6, a target of rapamycin, is involved in the regulation of rRNA genes by possible epigenetic changes in Arabidopsis. J. Biol. Chem. 289, 3901–3912. doi: 10.1074/jbc.M113.515015
Krug, K., Mertins, P., Zhang, B., Hornbeck, P., Raju, R., Ahmad, R., et al. (2019). A Curated Resource for Phosphosite-specific Signature Analysis. Mol. Cell. Proteomics 18, 576–593. doi: 10.1074/mcp.TIR118.000943
Kuhn, J. M., Boisson-Dernier, A., Dizon, M. B., Maktabi, M. H., and Schroeder, J. I. (2006). The protein phosphatase AtPP2CA negatively regulates abscisic acid signal transduction in Arabidopsis, and effects of abh1 on AtPP2CA mRNA. Plant Physiol. 140, 127–139. doi: 10.1104/pp.105.070318
Lei, B., Chang, W., Zhao, H., Zhang, K., Yu, J., Yu, S., et al. (2022). Nitrogen application and differences in leaf number retained after topping affect the tobacco (Nicotiana tabacum) transcriptome and metabolome. BMC Plant Biol. 22:38. doi: 10.1186/s12870-022-03426-x
Li, J. (2000). The cloning of the cytosolic glyceradehyde-3-phosphate dehydrogenase (GADPH) gene (TaGapC1) and its expression and sequence analysis in iron-deficient stress in wheat. Beijing: Capital Normal University.
Li, R., Qiu, Z., Wang, X., Gong, P., Xu, Q., Yu, Q.-B., et al. (2019). Pooled CRISPR/Cas9 reveals redundant roles of plastidial phosphoglycerate kinases in carbon fixation and metabolism. Plant J. 98, 1078–1089. doi: 10.1111/tpj.14303
Li, X., Hu, D., Cai, L., Wang, H., Liu, X., Du, H., et al. (2022). CALCIUM-DEPENDENT PROTEIN KINASE38 regulates flowering time and common cutworm resistance in soybean. Plant Physiol. 190, 480–499. doi: 10.1093/plphys/kiac260
Liang, Z., Yeung, W. T., and Ki Mai, K. K. (2021). CURT1A and CURT1C mediate distinct stages of plastid conversion in Arabidopsis. bioRxiv [Preprint]. bioRxiv 2021.12.01.470752.
Liu, J. J., Sharma, Ki, Zangrandi, L., Chen, C., Humphrey, S. J., Chiu, Y.-T., et al. (2018). In vivo brain GPCR signaling elucidated by phosphoproteomics. Science 360:eaao4927. doi: 10.1126/science.aao4927
Liu, L., Yan, W., and Liu, B. (2023). Transcriptome sequencing of Cocos nucifera leaves in response to Rhynchophorus ferrugineus infestation. Front. Genet. 14:1115392. doi: 10.3389/fgene.2023.1115392
Lyu, K., Gu, L., Wang, H., Zhu, X. X., Zhang, L., Sun, Y. F., et al. (2019). Transcriptomic analysis dissects the mechanistic insight into the Daphnia clonal variation in tolerance to toxic microcystis. Limnol. Oceanogr. 64, 272–283.
Mattei, B., Spinelli, F., Pontiggia, D., and Lorenzo, G. (2016). Comprehensive analysis of the membrane phosphoproteome regulated by oligogalacturonides in Arabidopsis thaliana. Front. Plant Sci. 7:1107. doi: 10.3389/fpls.2016.01107
Meier, F., Brunner, A.-D., Frank, M., Ha, A., Bludau, I., Voytik, E., et al. (2020). diaPASEF: Parallel accumulation–serial fragmentation combined with data-independent acquisition. Nat. Methods 17, 1229–1236.
Mukherji, M. (2005). Phophosproteomics in analyzing signaling pathways. Expert Rev. Proteomics 2, 117–128.
Niu, S., Li, J., Bo, W., Yang, W., Zuccolo, A., Giacomello, S., et al. (2022). The Chinese pine genome and methylome unveil key features of conifer evolution. Cell 185, 204–217. doi: 10.1016/j.cell.2021.12.006
Paul, P., Mesihovic, A., Chaturvedi, P., Ghatak, A., Weckwerth, W., Böhmer, M., et al. (2020). Structural and functional heat stress responses of chloroplasts of Arabidopsis thaliana. Genes 11:650. doi: 10.3390/genes11060650
Pawson, T., and Scott, J. D. (2005). Protein phosphorylation in signaling–50 years and counting. Trends Biochem Sci. 30, 286–290. doi: 10.1016/j.tibs.2005.04.013
Peltier, J.-B., Ytterberg, A. J., Sun, Q., and Wijk, K. J. (2004). New functions of the thylakoid membrane proteome of Arabidopsis thaliana revealed by a simple, fast, and versatile fractionation strategy. J. Biol. Chem. 279, 49367–49383. doi: 10.1074/jbc.M406763200
Peng, J., Loon, J. J. A., Zheng, S., and Dicke, M. (2011). Herbivore-induced volatiles of cabbage (Brassica oleracea) prime defence responses in neighbouring intact plants. Plant Biol. 13, 276–284. doi: 10.1111/j.1438-8677.2010.00364.x
Perazzolli, M., Palmieri, M. C., Matafora, V., Bachi, A., and Pertot, I. (2016). Phosphoproteomic analysis of induced resistance reveals activation of signal transduction processes by beneficial and pathogenic interaction in grapevine. J. Plant Physiol. 195, 59–72. doi: 10.1016/j.jplph.2016.03.007
Rampitsch, C. (2017). Phosphoproteomics analysis for probing plant stress tolerance. Methods Mol. Biol. 2017, 181–193. doi: 10.1007/978-1-4939-7136-7_11
Ren, M., Venglat, P., Qiu, S., Feng, Li, Cao, Y., Wang, E., et al. (2012). Target of rapamycin signaling regulates metabolism, growth, and life span in Arabidopsis. Plant Cell 24, 4850–4874. doi: 10.1105/tpc.112.107144
Reymond, P., Bodenhausen, N., Poecke, R. M. P., Krishnamurthy, V., Dicke, M., and Farmer, E. E. (2004). A conserved transcript pattern in response to a specialist and a generalist herbivore. Plant Cell 16, 3132–3147. doi: 10.1105/tpc.104.026120
Rossignol, M. (2006). Proteomic analysis of phosphorylated proteins. Curr. Opin. Plant Biol. 9, 538–543.
Samofalova, D. O., Karpov, P. A., Raevsky, A. V., and Blume, Y. B. (2020). “Interplay of protein phosphatases with cytoskeleton signaling in response to stress factors in plants,” in Protein phosphatases and stress management in plants: Functional genomic perspective, ed. G. K. Pandey (Cham: Springer), 261–287.
Schneider, M., Knuesting, J., Birkholz, O., Heinisch, J. J., and Scheibe, R. (2018). Cytosolic GAPDH as a redox-dependent regulator of energy metabolism. BMC Plant Biol. 18, 1–14.
Shannon, P. (2003). Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Smith, C. M., and Clement, S. L. (2012). Molecular bases of plant resistance to arthropods. Annu. Rev. Entomol. 57, 309–328.
Song, S., Huang, H., Gao, H., Wang, J., Wu, D., Liu, X., et al. (2014). Interaction between MYC2 and ETHYLENE INSENSITIVE3 modulates antagonism between jasmonate and ethylene signaling in Arabidopsis. Plant Cell 26, 263–279. doi: 10.1105/tpc.113.120394
Subramanian, A., Tamayo, P., Mootha, V. K., Mukherjee, S., Ebert, B. L., Gillette, M. A., et al. (2005). Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550.
Tang, Q. Y., and Zhang, C. X. (2013). Data processing system (DPS) software with experimental design, statistical analysis and data mining developed for use in entomological research. Insect Sci. 20, 254–260. doi: 10.1111/j.1744-7917.2012.01519.x
Troncoso-Ponce, M. A., Rivoal, J., Venegas-Calerón, M., Dorion, S., Sánchez, R., Cejudo, F. J., et al. (2012). Molecular cloning and biochemical characterization of three phosphoglycerate kinase isoforms from developing sunflower (Helianthus annuus L.) seeds. Phytochemistry 79, 27–38. doi: 10.1016/j.phytochem.2012.04.001
Waadt, R., Seller, C. A., Hsu, P.-K., Takahashi, Y., Munemasa, S., and Schroeder, J. I. (2022). Plant hormone regulation of abiotic stress responses. Nat. Rev. Mol. Cell Biol. 23, 680–694.
Wang, C., Xu, H., Lin, S., Deng, W., Zhou, J., Zhang, Y., et al. (2020). GPS 5.0: An update on the prediction of kinase-specific phosphorylation sites in proteins. Genom. Proteomics Bioinform. 18, 72–80. doi: 10.1016/j.gpb.2020.01.001
Wang, P., Zhao, Y., Li, Z., Hsu, C. C., Liu, X., Fu, L., et al. (2018). Reciprocal regulation of the TOR kinase and ABA receptor balances plant growth and stress response. Mol. Cell 69:100–112.e6. doi: 10.1016/j.molcel.2017.12.002
Wei, J., and Kang, L. (2011). Roles of (Z)-3-hexenol in plant-insect interactions. Plant Signal. Behav. 6, 369–371.
Xing, T., and Laroche, A. (2011). Revealing plant defense signaling: Getting more sophisticated with phosphoproteomics. Plant Signal. Behav. 6, 1469–1474. doi: 10.4161/psb.6.10.17345
Xu, H.-X., Qian, L.-X., Wang, X.-W., Shao, R. X., Hong, Y., Liu, S., et al. (2019). A salivary effector enables whitefly to feed on host plants by eliciting salicylic acid-signaling pathway. Proc. Natl. Acad. Sci. U.S.A. 116, 490–495. doi: 10.1073/pnas.1714990116
Xu, M., Wang, X., Liu, J., Jia, A., Xu, C., Deng, X. W., et al. (2022). Natural variation in the transcription factor REPLUMLESS contributes to both disease resistance and plant growth in Arabidopsis. Plant Commun. 3:100351. doi: 10.1016/j.xplc.2022.100351
Yang, L., Yang, X., and Gao, Y. (2023). S-adenosylmethionine synthase (BtSAMS) from Balsas teosinte confers resistance to peach aphids in Arabidopsis. Ann. Appl. Biol. 183, 15–22.
Ytterberg, A. J., Peltier, J. B., and Van Wijk, K. J. (2006). Protein profiling of plastoglobules in chloroplasts and chromoplasts. A surprising site for differential accumulation of metabolic enzymes. Plant Physiol. 140, 984–997. doi: 10.1104/pp.105.076083
Zhai, Q. H. (2017). Screening proteins interacting with cytosolic TAGAPC1 from wheat by yeast two hybrid. Xianyang: Northwest A&F University.
Zhang, J. H., Sun, L. W., Liu, L. L., Lian, J., An, S. L., Wang, X., et al. (2010). Proteomic analysis of interactions between the generalist herbivore Spodoptera exigua (Lepidoptera: Noctuidae) and Arabidopsis thaliana. Plant Mol. Biol. Rep. 28, 324–333.
Zhang, Y., Wang, Y., Sun, X., Yuan, J., Zhao, Z., Gao, J., et al. (2022). Genome-wide identification of MDH family genes and their association with salt tolerance in rice. Plants 11:1498. doi: 10.3390/plants11111498
Keywords: Pinus tabuliformis Carr., Dendrolimus tabulaeformis Tsai et Liu, proteomic, phosphoproteomics, induced defensive responses
Citation: Sun T, Zhao Y, Zhou G, Gao S, Liu J and Gao B (2024) Defense response to caterpillar feeding stress in wild Pinus tabuliformis unveiled by quantitative integrated proteomic and phosphoproteomic analyses. Front. For. Glob. Change 7:1356511. doi: 10.3389/ffgc.2024.1356511
Received: 15 December 2023; Accepted: 31 May 2024;
Published: 14 June 2024.
Edited by:
Amit Roy, Czech University of Life Sciences Prague, CzechiaReviewed by:
Kanakachari Mogilicherla, Czech University of Life Sciences Prague, CzechiaCopyright © 2024 Sun, Zhao, Zhou, Gao, Liu and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Baojia Gao, YmFvamlhZ2FvQDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.