 Valerie Hsiao
Valerie Hsiao Natalia G. Erazo
Natalia G. Erazo Ruth Reef
Ruth Reef Catherine Lovelock
Catherine Lovelock Jeff Bowman
Jeff Bowman
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. For. Glob. Change , 25 July 2024
Sec. Forest Growth
Volume 7 - 2024 | https://doi.org/10.3389/ffgc.2024.1336037
This article is part of the Research Topic Tree-Microbe Interactions in Changing and Adverse Environments View all articles
Mangroves offer many important ecosystem services including carbon sequestration, serving as nursery grounds to many organisms, and acting as barriers where land and sea converge. Mangroves exhibit environmental flexibility and resilience and frequently occur in nutrient-limited systems. Despite existing research on mangrove microbiomes, the effects of nutrient additions on microbial community structure, composition, and function in intertidal and landward zones of mangrove ecosystems remain unclear. We utilized a long-term nutrient amendment study in Exmouth Gulf, Western Australia conducted in two zones, the intertidal fringe and supralittoral scrub forests, dominated by Avicennia marina. Root samples were fractionated into rhizosphere, rhizoplane and endosphere compartments and analyzed by 16S rRNA gene amplicon sequencing to determine the effects of nutrient stress on community structure and function. Our data showed species richness and evenness were significantly higher in the scrub forest zone. PERMANOVA analysis revealed a significant effect of nutrient enrichment on beta diversity (p = 0.022, R2 = 0.012) in the fringe forest zone only. Cylindrospermopsis, which has been associated with harmful algal blooms, was found to be significantly enriched in fringe phosphate-fertilized plots and nitrogen-fixing Hyphomicrobiales were significantly depleted in the scrub nitrogen-fertilized plots. Meanwhile, root compartments and forest zone had a greater effect on beta diversity (p = 0.001, R2 = 0.186; p = 0.001, R2 = 0.055, respectively) than nutrient enrichment, with a significant interaction between forest zone and root compartment (p = 0.001, R2 = 0.025). This interaction was further observed in the distinct divergence identified in degradative processes of the rhizosphere compartment between the two forest zones. Degradation of aromatic compounds were significantly enriched in the fringe rhizosphere, in contrast to the scrub rhizosphere, where degradation of carbohydrates was most significant. Despite the highly significant effect of forest zone and root compartments, the long-term effect of nutrient enrichment impacted community structure and function, and potentially compromised overall mangrove health and ecosystem stability.
Mangroves inhabit tropical and subtropical coastlines around the world and are well known for their efficiency in sequestering carbon (Macreadie et al., 2021). On a global scale, mangroves demonstrate the capacity to sequester a disproportionately high amount of carbon relative to their spatial extent and are responsible for burying an estimated 9.6–15.8 Tg Corg per year in mangrove soils and sediments (Alongi, 2022). In addition to their substantive role in blue carbon storage, mangroves provide a range of other vital ecosystem services, from storm protection to serving as nurseries for various organisms (Barbier et al., 2011). Despite the high biomass accumulation associated with these habitats, mangroves frequently exist in oligotrophic systems (Reis et al., 2017), and are affected by biotic and abiotic factors that regulate nutrient availability (Reef et al., 2010).
Mangrove root-microbial assemblages are influenced by host taxonomy, nutrients, and environmental conditions (Reinhold-Hurek et al., 2015; Francioli et al., 2018). Diazotrophic microbes and other microorganisms involved in biogeochemical cycling are key contributors to the supply of new nutrients in these oligotrophic ecosystems (Romero et al., 2012; Srivastava et al., 2021). Microbially-mediated provisions include not only nutrients, but also plant growth-promoting metabolites, such as hormones and antibiotics (Chen et al., 2016). To this end, the mangrove root microbiome plays an important role in mangrove function (Allard et al., 2020). Root community members are recruited in part through plant exudates and metabolites that exist along a gradient from the exterior root compartments of the rhizosphere and rhizoplane, into the interior endosphere root compartment (Reinhold-Hurek et al., 2015; Francioli et al., 2018). Microbial assembly and distribution are additionally influenced by variations in sediment redox conditions (Alongi, 2015), which can differ depending on the physicochemical characteristics associated with the intertidal position of the forest zone (Thomson et al., 2022). Forest zones differ in nutrient availability, pH, and salinity, all of which contribute to microbial selection pressures and functional diversity of mangrove soils (De Souza Rocha et al., 2020).
Anthropogenic activities have led to eutrophication in coastal ecosystems around the world (Dai et al., 2023). It is important to understand the long-term impacts of nutrient pollution on mangrove root-microbe interactions by elucidating nutrient profiles that alter microbial communities. Nutrient-limited systems select for oligotrophic species, however, with the addition of nutrients, oligotrophic community members have been reported to give way to copiotrophic taxa, which are characterized by faster growth rates and preferential usage of labile carbon sources, as opposed to slow growing oligotrophic species that are able to metabolize complex carbon sources (Bledsoe et al., 2020). Changes in community structure related to increased nutrient loads may negatively affect and diminish forest function and subsequent valuable ecosystem services provided by mangrove habitats. Past terrestrial plant studies have found that nutrient enrichment can alter bacteria community structure, from which reduced soil function leads to the emission of greenhouse gases (Cusack et al., 2011; Chen M. et al., 2020). Likewise, shifts in community structure are often indicators of soil function in mangrove ecosystems (Thomson et al., 2022).
Numerous studies comparing natural and polluted ecosystems have demonstrated that nutrient pollution can lead to a reduction in microbial diversity (Wang et al., 2018; Craig et al., 2021; Erazo and Bowman, 2021). However, contrasting research suggests microbial communities do not consistently exhibit distinct differences and microbial functional changes are proposed to be better predictors of nutrient stress (Graves et al., 2016; Liao et al., 2020). Despite the existing body of research, the effects of nutrient additions on microbial community structure in mangrove ecosystems remains unclear (Zhou et al., 2014; Liu et al., 2015; Yuan et al., 2020).
There are few studies that explore microbial communities and metabolic pathways to describe root compartment function in the context of forest type and nutrient stress, such as nutrient limitation and long-term enrichment (Zhuang et al., 2020; Thomson et al., 2022). Our study included nitrogen (N), phosphorus (P), and iron (Fe) amendments across two forest zones. In an effort to identify differences in microbial community structure among these fertilization treatments, we sought to answer three research questions: (i) Are there differences in the microbial community structure and composition as a result of long-term nutrient amendments compared to untreated controls, and are these differences distinct between N, P or Fe enriched plots? (ii) Are there significant differences in the microbial community structure and function within rhizosphere, rhizoplane and endosphere root compartments compared to the surrounding bulk soil? (iii) Do microbial metabolic pathways differ among the various treatments?
We hypothesized that the long-term effect of nutrient enriched soils would be nutrient dependent and detectable by the divergence of community composition. Specifically, we expected to observe the enrichment of denitrifiers or nitrate reducers and depletion of nitrogen fixers in nitrogen-amended soils, the depletion of phosphate solubilizers in phosphorous-amended soils, and the depletion of microbes that mediate iron redox cycles. Lastly, we hypothesized that the long-term effect of nutrient fertilization would be detected by differences in bacterial signatures, such as maximal growth rate predictions, genome size and ribosomal RNA gene copy number (RRN), which would represent a shift from oligotrophic-copiotrophic community members. We anticipated the presence of oligotrophic taxa characteristic of oligotrophic mangrove ecosystems would give way to copiotrophic taxa with nutrient amendments. As root community members are recruited from bulk soil and community refinement begins in the rhizosphere, we expected to observe divergent community structure among the root compartments of treated soils compared to the untreated control soils (Edwards et al., 2015; Reinhold-Hurek et al., 2015; Zhuang et al., 2020; Moroenyane et al., 2021). To test these hypotheses, we leveraged a longstanding nutrient enrichment experiment of N, P, and Fe enriched mangrove trees located at Exmouth Gulf, Western Australia, dominated by the gray mangrove Avicennia marina (Reef et al., 2010). This study offers insight to the long-term effect of nutrient enrichment, root compartments and forest type on microbial community composition and diversity to identify divergent community members and inferred metabolic pathways in this mangrove ecosystem.
The coastal land at Exmouth Gulf spans 2,600 km2, of which 161 km2 is dominated by the gray mangrove Avicennia marina (Reef et al., 2010). A. marina mangroves are a frequent pioneer species resilient to harsh environmental conditions such as hypersalinity, aridity, high water temperatures and frost frequency (Alzubaidy et al., 2016). Due to the low population density in Exmouth Gulf, the mangroves at this location are largely unaffected by human influences and offer a unique opportunity to study the effects of nutrient enrichment in an input-controlled environment. Many studies have evaluated the effects of N and P amended mangrove soils, but few have included the effects of Fe enrichment.
Nutrient amended experimental plots were established in 2004, at 21°44’S, 144°35’ E, in Exmouth Gulf, Western Australia. There is no rainy season in Exmouth, with an annual average rainfall of 233.68 mm (Climate Data, 2022). On April 28, 2022, nutrient amendment was conducted on a total of 9 replicate trees per fertilizer treatment, in each of the two forest zones: seaward fringe and landward scrub forests (Figure 1A). Each forest zone contained 5 experimental lots, three of which consisted of triplicate N, P, and control (C) treatments, and two of which were Fe-fertilized lots flanking the north and south ends of the experimental transect. Individual experimental trees were fertilized with 200–300 g of urea for N fertilization, and triple superphosphate was used for P fertilization (Lovelock et al., 2009). Fe fertilization was established in 2015 and was applied as iron EDTA chelate fertilizer (13% Fe, Manutec, Cavan, South Australia 5094, Australia) at a dose rate recommended by the manufacturers for fruit trees (50 g per tree delivered in two 25 g doses). All fertilizer treatments were inserted into 30 cm deep holes cored on either side of the main stem of the tree, according to a previously established fertilization procedure described in Feller et al. (1999). The fertilizers were inserted into cored holes on either side of the main stem of the tree rather than fertilizing the surface around the tree due to the tides, which can wash fertilizer away from the target trees, leading to diffusion of impacts. The holes were then sealed with a portion of the extracted sediment core. Control trees were cored but fertilizer was not inserted into the holes. There were 36 trees in each of the two forests, totaling 72 experimental mangrove trees (18 N, 18 P, 18 Fe and 18C). Fertilizers were added annually during the cool months of Exmouth Gulf, between April and August since 2004, however most of the nutrient additions occurred in the month of April. Fertilization was not applied in 2019 and 2020, due to COVID-19 travel restrictions, and was resumed in 2021.
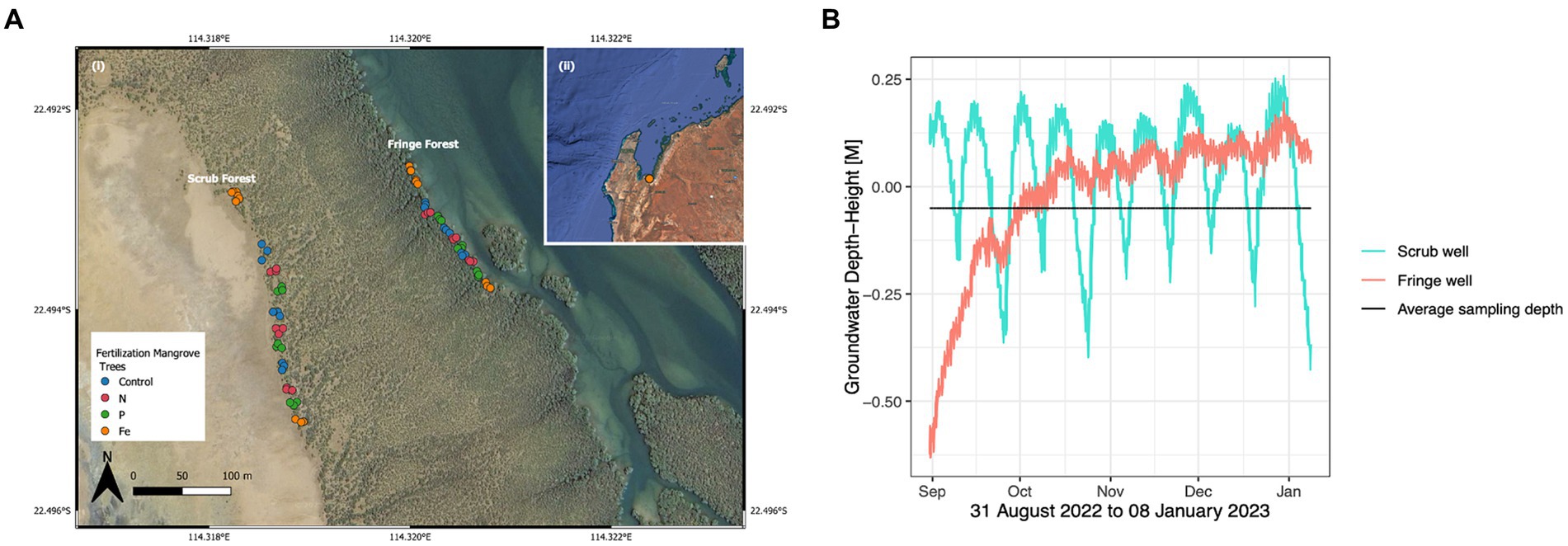
Figure 1. (A) Mangrove nutrient fertilization experimental site was established in 2004 in (i) Exmouth Gulf, Western Australia. Map was created utilizing QGIS, a free and open source software. (ii) Triplicates of fertilization treatments, nitrogen (N), phosphorous (P), and iron (Fe) were applied to triplicate mangrove tree lots in the seaward fringe and landward scrub forest zones, for a total of 9 replicates per nutrient amendment in each of the forest zones. (B) Soil hydrology in the fringe (salmon) and scrub (turquoise) forest zones during the period of 31 August 2022 to 08 January 2023 showing groundwater connectivity with average sampling field depth (black) over time (x-axis).
Conductivity, temperature, and depth (CTD) were measured at 5-min intervals in a series of three wells dug across the sampling area. The CTD loggers (LTC M10, Solinst, Canada) were placed within vented wells made of 50 mm diameter PVC pipes, sealed with a cap at both ends, with vents for pressure equalization cut into the top section. The bottom hole was at 1 m below the surface for all wells and a mesh cover prevented the entry of sediments into the well. The excavation of the well was carried out with a 50 mm hand auger. The wells were located within the sampling area, one in the fringe section of the mangrove adjacent to the fertilized trees and one near the fertilized trees of the scrub. Measurements began on the 25th of August 2022; however, the drawdown period has been removed for the purpose of this study, thus the hydrological statistics are described for the period of 01 September 2022 to 08 January 2023. Dry periods were defined as periods where the water level within the well exceeded the average sediment sampling depth of 5 cm. Salinity was calculated from conductivity and temperature data collected from the time period between 24 August 2022 to 01 September 2022 with R package wql, using the ec2pss function (Jassby and Cloern, 2022). Wilcoxon test method was used to analyze data for statistical differences in average temperature and salinity among the forest zones.
Root samples with fine root hairs were collected over a four-day period between the 20th and 24th days of August 2022 by digging near the tree base at a depth ranging from 2 to 8 cm below the surface. Cutting size ranged between 5 and 10 cm in length with 2 exceptions from a fringe iron-fertilized plot where appropriate roots were very difficult to collect without substantial breakage. In those cases, the average length of collected root segments were 3 cm. The roots were placed in sterile containers and placed on ice until transported back to the field laboratory for root compartment fractionation. We sterilized collection tools with 70% ethanol between treatment plots. One root (with corresponding compartments) was sampled per tree, for a total of 36 fringe forest and 36 scrub forest samples (Nroots = 72). Triplicate bulk soil samples were collected approximately 2–3 m from the base of each tree to avoid roots.
Root samples were separated into rhizosphere, rhizoplane and endosphere compartments following previous work (Edwards et al., 2015; Duran et al., 2018; Zhuang et al., 2020). To extract rhizosphere samples, the roots were first manually shaken to remove excess soil, leaving approximately 1 mm of soil adhered to the root surface. Each root was then placed into a sterile 50 mL conical tube, filled with 10–20 mL of phosphate buffered saline (PBS), and stirred vigorously with sterile forceps to remove the attached layer of soil from the root surface. The resulting PBS suspension was then decanted into bead-beating tubes and stored on dry ice at −80°C for transport back to our home laboratory.
Following rhizosphere isolation, the rhizoplane compartment from each root was extracted by transferring the root into a sterile 15 mL conical tube with PBS to isolate the remaining tightly adhering layer of soil with sonication for 30 s at 50–60 Hz (output frequency 42 kHz, power 90 W). The resulting suspension was filtered with 25 mm 0.22 μm size Supor PES membranes (Pall Corporation), to extract microbial biomass from the rhizosphere compartment. Filters containing biomass were then placed into bead-beating tubes and stored on dry ice at −80°C for transport.
The endosphere microbiome was collected by first conducting surface sterilization according to Duran et al. (2018) (also implemented by Richter-Heitmann et al., 2016; Zhuang et al., 2020). Each root was rinsed with 80% ethanol for a duration of 1 min, followed by additional sterilization with 0.25% NaClO (bleach) for a duration of 1 min, followed by rinsing with DI water for 1 min, 3 times (Zhuang et al., 2020). Surface sterilized root samples were then stored at −80°C on dry ice for transport to our home laboratory. At the home laboratory individual root samples were homogenized to extract microbial biomass of the endosphere compartment by first flash freezing in liquid nitrogen for 30 s then pulverizing with a mortar and pestle sterilized with 70% ethanol between treatment-grouped samples. For each endosphere and bulk soil sample, 250 mg of homogenized material or soil was placed into bead beating tubes for DNA extraction. Root compartment fractions and bulk soil samples resulted in a total of 224 samples.
DNA extractions for root compartments were performed on a King Fisher™ Flex Purification system using the MagMax Microbiome Ultra Nucleic Acid Extraction Kit (Thermo Fisher Scientific) and were quantified using the Qubit HS DNA quantification kit (Invitrogen). Particularly low concentrations (<0.100 ng μl−1) were quality checked by gel electrophoresis and PCR amplification of the 16S rRNA gene using primers 515F and 806R (Walters et al., 2015) for bacteria and archaea. Bulk soil samples were handled with 50 mM sodium pyrophosphate-amended lysis buffer to improve extracted DNA concentrations (Liu et al., 2010). DNA extractions were sent to Argonne National Laboratory Environmental Sample Processing Center for amplification and library preparation with the above-mentioned primer set, followed by 2 × 151 paired end sequencing on the Illumina Miseq platform. Sequences were submitted to NCBI Bioproject under accession number PRJNA1035549.
Following methods in Erazo and Bowman (2021) and Chamberlain et al. (2022) for sequence analysis, Illumina Miseq reads were demultiplexed using the ‘iu-demultiplex’ command in Illumina utils (Eren et al., 2013). Demultiplexed reads were quality controlled and denoised using the ‘FilterandTrim’ and ‘dada’ commands within the R package dada2 (Callahan et al., 2016), and assembled with the ‘mergePairs’ command. The non-redundant fasta files of the generated unique reads produced by dada2 were used as an input for the paprica pipeline v0.70 (Bowman and Ducklow, 2015), a phylogenetic placement approach for microbial community structure and metabolic inference.1 Paprica places sample reads for Bacteria and Archaea onto a reference tree created from the concatenated 16S and 23S rRNA genes from all completed genomes in the Genbank RefSeq database (Haft et al., 2018). Each ASV is placed to either an internal or terminal branch on the tree (Nawrocki and Eddy, 2013; Barbera et al., 2019; Czech et al., 2020). Based on phylogenetic assignments, the paprica pipeline provides predictions for taxonomic maximal growth rate potential, genome size, and RRN. The paprica model for maximal growth rate potential is based on the R package gRodon, developed by Weissman et al. (2021). The model was extended from the work of Vieira-Silva and Rocha that leveraged high codon usage bias (CUB) in genes coding for ribosomal proteins and highly expressed genes for maximal growth rate predictions with partial genomic data. Further improvements were made on predictive accuracy for applications to bulk community data by analyzing codon usage in additional dimensions based on species abundance from metagenomes (Weissman et al., 2021).
Microbial community structure and metabolic pathway sequencing data were analyzed in R v.4.3.1 (2023-06-16) and R Studio v. 2023.06.2 + 561, utilizing R package vegan. To test the hypothesis that beta diversity would be significant among samples, (dis)similarity as a function of treatment, root compartments, and/or forest type were analyzed. We visualized (dis)similarities between treatments, root compartments and forest type with nonmetric multi-dimensional scaling (NMDS) ordination, utilizing Bray–Curtis similarity distance matrix of multivariate data separated into two dimensions using a log10-transformed, relative abundance table. Quality control was performed on ASV and predicted metabolic pathway datasets for samples with low reads. The ASV dataset was prefiltered to exclude samples with less than 1,000 reads and the predicted metabolic pathways dataset was prefiltered to exclude samples with less than 3,600 reads. To test for statistical significance, the function adonis2 from the R vegan package (Oksanen et al., 2022) was used to conduct Permutational Analysis of Variance (PERMANOVA). Utilizing the Bray-Curtis method to calculate the distance matrix for PERMANOVA, the model design tested for the main effects of Site, Treatment, Compartment and their interaction terms, with 999 permutations. Due to an interaction between site and root compartment, additional PERMANOVA analyses was conducted on partitioned data of the two forest zones, fringe and scrub. The previously described procedure was used to test the effects of Treatment, Compartment, Lot and their interaction terms in each of the forest zones. To diminish potentially spurious results from the PERMANOVA analysis, the heterogeneity of dispersion between groups was tested. A one-way ANOVA of the distances to group centroids was performed to interpret the significance of the pseudo-F statistic (Anderson, 2006; Anderson et al., 2006). Multivariate dispersions were calculated with the betadisper function on the distance matrix previously created with each factor: Treatment, Compartment and Site. The resulting objects were then tested for significant variance with a one-way ANOVA.
To test the hypothesis that richness and evenness (alpha diversity) would be different among forest zones, treatments and root compartments, R package phyloseq (McMurdie and Holmes, 2013) was used to calculate alpha diversity via Shannon, Simpson, and Inverse Simpson indices (Supplementary Figures 1A–C). Alpha diversity with Inverse Simpson diversity index was analyzed for the significance of forest zone with the Wilcoxon test method, and the Kruskal-Wallis method was utilized to test root compartments and treatments, setting the alpha threshold to 0.05. Kruskal-Wallis is a nonparametric alternative to a one-way ANOVA and allows the comparison of three or more groups. Similarly, to test the hypothesis that the effect of treatments could be detected by a shift from oligotrophic to copiotrophic communities, bacterial signatures described as the means of maximal growth rate potential, genome size and RRN were analyzed.
Differential abundance (DA) analysis was conducted using R package DESeq2, which is based on the negative binomial/Gamma-Poisson distribution (Love et al., 2014). DA analysis was used to test the hypothesis that differences in community composition between treatments and compartments could be detected by divergently abundant ASVs and inferred metabolic pathways. When hypothesis testing for divergence in community structure for treatments and compartments, the log likelihood ratio test was used instead of the default Wald test due to the multiple levels present in the factors of our metadata. When analyzing the data for differences in compartments between forest zones, the Wald test was used because the factor of forest zone had only two levels. To minimize false discovery rates, the ashr log fold change shrinkage method was used to reduce noise and preserve large differences (Stephens, 2016; Zhu et al., 2018). DESeq2 adjusts p-values of significant ASVs for false discovery rate using the Benjamini-Hochberg adjustment (Love et al., 2014). The adjusted p-value threshold was set to 0.05 and the LFC threshold was set to 1. The ashr LFC shrinkage method was selected to allow usage of the contrast function in DESeq2 to compare levels within a factor to one another as well as to the set control.
For identifying differentially abundant ASVs between the treatments and the control, the DESeq matrix was calculated in which the model design tested for the main effect of Treatment, controlling for the effect of Compartment, with an interaction term of Treatment:Compartment. In the DA analysis for ASVs in compartments between forest zones, the data was partitioned by root compartment and a separate DESeq matrix was calculated for each compartment, setting the scrub forest as the control, testing for the main effect of Site, controlling for Treatments.
DA analysis was conducted on inferred metabolic pathways between compartments and the DESeq matrix was calculated for the scrub forest only, setting the bulk soil as the control. The model design tested for the main effect of Compartment, controlling for Treatment, with an interaction term of Compartment:Treatment. In the analysis for differentially abundant inferred metabolic pathways in compartments, contrasting forest types, data was partitioned by root compartment, setting the scrub forest as the control, utilizing a model design that tested the main effect of Site, controlling for the effect of Treatment.
Hydrological measurements indicated that the soil hydrology was markedly different at the two forest zones. Over a period of 8 months, sediments at the sampling depths were inundated at all times in the fringe zone, while scrub zone sediments experienced wet/dry cycles. The scrub zone sediments experienced drying episodes on a biweekly basis in conjunction with neap tide cycle. These dry episodes lasted on average 4.4 days. Overall, the sediments were dry (at or below field capacity) 26% of the time in the scrub forest zone and the fringe forest zone was dry 0% of the time (Figure 1B; Supplementary Datasheet 1). Porewater temperature and salinity were significantly higher in the scrub zone relative to the fringe (p < 2.2 × 10−16) (Figure 2A), averaging 62.36 and 50.35 ppt respectively, with an average temperature difference of 1.03°C (Figure 2B; Supplementary Dataheet 2).
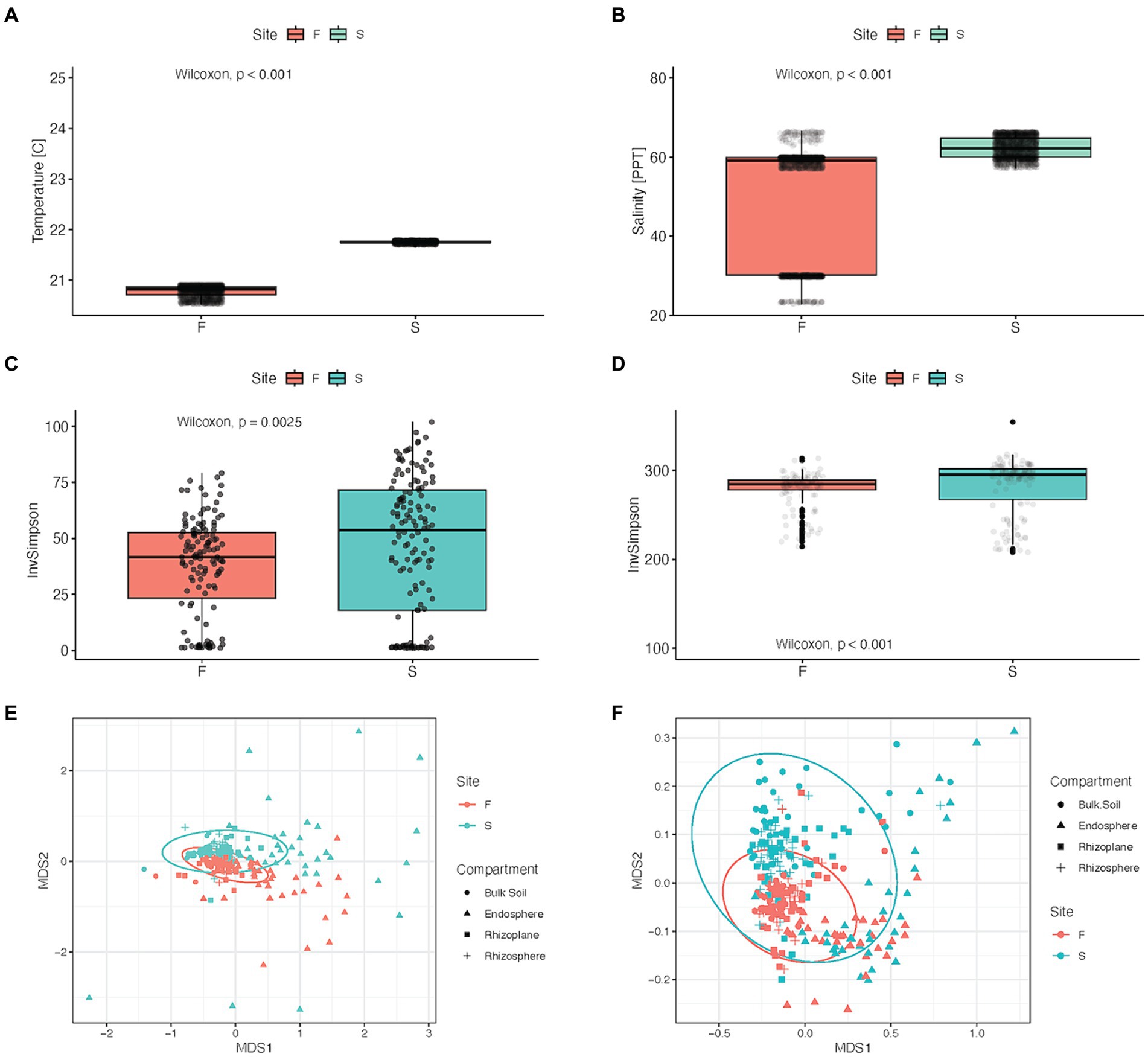
Figure 2. Physicochemical properties and microbial alpha and beta diversity between forest zones: Fringe (F) and Scrub (S). (A) Average temperature and (B) salinity from groundwater wells located near sampling field in each of the forest zones. (C) Microbial alpha diversity in Inverse Simpson diversity index between forest types, (Kruskal-Wallis, p = 0.002). (D) Alpha diversity of inferred metabolic pathways between forest zones, (Kruskal-Wallis, p < 0.001). (E) Nonmetric Multidimensional scaling (NMDS) of Bray-Curtis (dis)similarity of log10- transformed relative-abundance data, stress = 0.149. Ellipses were set to a confidence level of 0.95, used to visualize microbial beta diversity tested with PERMANOVA, p = 0.001. (F) NMDS (stress = 0.1065) visualization of metabolic beta diversity (PERMANOVA) p = 0.001.
Taxonomic and metabolic alpha diversity was significantly lower in the fringe forest zone compared to the scrub (p = 0.002, p < 0.001, Figures 2C,D, respectively). Separation of microbial community structure was observed in multidimensional scaling ordination (Figure 2E), which supported significant PERMANOVA results for beta diversity between forest zones (p = 0.001), explaining 5.53% of the variance (R2 = 0.055, Supplementary Table S1). There was an interaction between compartment and site explaining 3.06% (p = 0.001, R2 = 0.031) of the variance between samples (Supplementary Table S1) with significant dispersion in compartments and forest zone data (p = 0.000 and 0.039, respectively, Supplementary Table S2). Data partitioned by forest zone revealed a significant effect of Lot in fringe and scrub forests (p = 0.001, 0.008, respectively), explaining 5.7% of the variance in the scrub forest, and 6.7% in the fringe forest. Additionally, there were significant compositional differences in predicted metabolic pathways (p = 0.001, R2 = 0.035, Figure 2F) with a significant interaction observed between forest zone and root compartment (p = 0.002, R2 = 0.021, F = 2.538; values available in Supplementary Table S3).
Maximal growth rate predictions were significant among forest zones (p < 0.001, Supplementary Figure S2B). The fringe forest zone displayed higher maximum growth rate predictions, with an average value of 10.0, while the scrub forest exhibited a mean value of 8.5.
The effect of root compartments on beta diversity of ASVs and predicted metabolic pathways were highly significant (p = 0.001, Figures 3A,B), explaining 18.6% of the variance among ASVs and 19.5% among predicted metabolic pathways (Supplementary Tables S1, S3, respectively). Data partitioned by forest type revealed a significant interaction between Compartment and Lot (p = 0.022) explaining 1.2% of the variance. Significant dispersion was found among ASVs in root compartment samples (p ≤ 0.001). A post-hoc Dunn pairwise comparison of bacterial and metabolic alpha diversity revealed Kruskal-Wallis test scores of 140.89 (p ≤ 0.0001) and 127.87 (p ≤ 0.0001) respectively, which demonstrated the endosphere root compartment with the lowest diversity (Figures 3C,D).
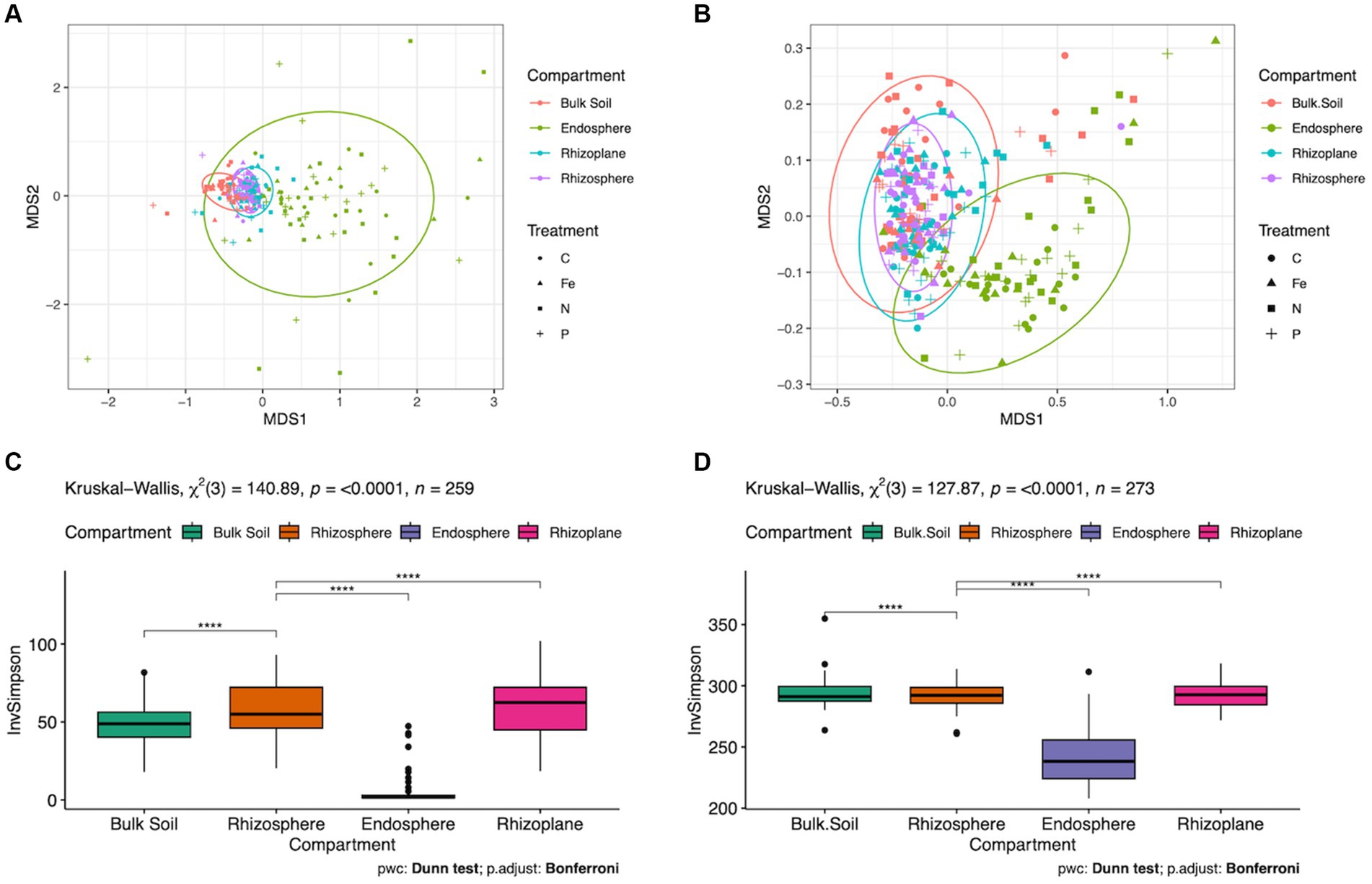
Figure 3. Microbial and metabolic diversity among root compartments. (A) NMDS ordination used to visualize of differences in microbial beta diversity (PERMANOVA p = 0.001) and (B) Metabolic beta diversity (p = 0.001). Bulk soil (red), endosphere (green), rhizosphere (blue) and rhizoplane (purple) are highlighted. (C) Microbial alpha diversity and (D) metabolic alpha diversity, utilizing Dunn pairwise test with Bonferroni p adjustment, p < 0.0001.
Means of maximal growth rate predictions among root compartments were found to be significant (p < 0.001; Figure 4A), with the endosphere microbiome exhibiting the lowest growth rate mean. Comparisons of mean genome sizes and RRN among the root compartment microbiomes also yielded significant results (p < 0.001). Notably, the endosphere community exhibited the highest mean genome size, exceeding 6 × 106 bp, with approximately 3.5 RRNs (Figures 4B,C).
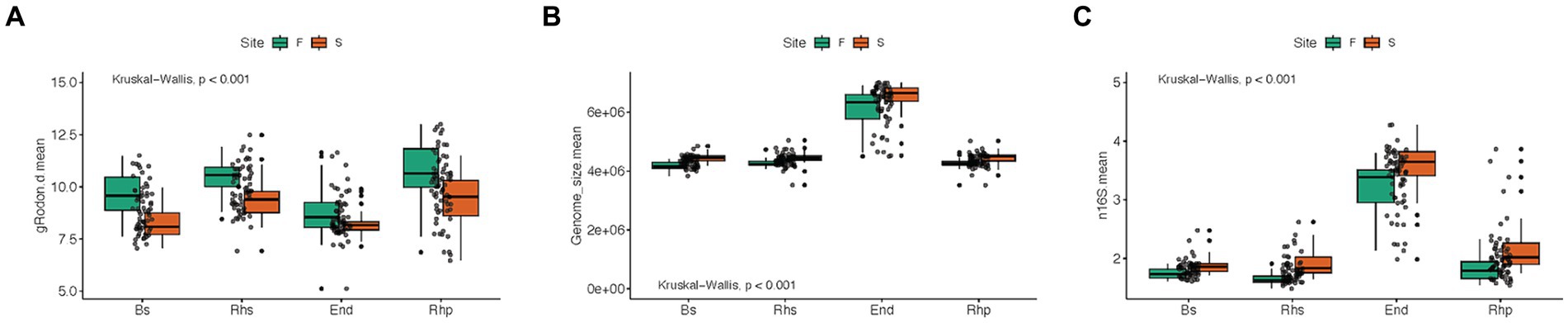
Figure 4. Boxplots of maximal growth rate potential, genome size and ribosomal RNA operon copy number (n16S) averages, between root compartments, and forest types, using Kruskal-Wallis test method. S = scrub and F = fringe forest zones. Bs = bulk soil samples, Rhs = rhizosphere samples, Rhp = rhizoplane samples and End = endosphere samples. (A) Maximal growth rate potential (p < 0.001), (B) Genome size (p < 0.001), (C) Ribosomal RNA operon copy number (p < 0.001).
Across both forest zones, 32% of the variance in differentially abundant microbiota between root compartments were explained on principle coordinate axis 1 (PC1), and 8% of the variance was explained along PC2 (Supplementary Figure 3A). In fringe forest samples, 36% of the variance in differential root microbiota was explained on PC1 and 8% was explained on PC2 (Supplementary Figure 3B). In the scrub forest samples, differentially abundant microbiota explained 35% variance on PC1, and 11% was explained on PC2 (Supplementary Figure 3C).
There was a total of 79 significantly enriched and depleted ASVs in rhizosphere samples. Forty-four taxa were significantly enriched in fringe forest rhizospheres while 35 taxa were significant in scrub forest rhizosphere samples. DA analysis of the rhizoplane identified a total of 59 significantly differential ASVs, 28 of which were enriched in the fringe and 31 in scrub forest samples. Pseudoanabaenales, and Halomonas sp. SS10-MC5 were most significant in the scrub rhizosphere and rhizoplane compartments while Sulfurovum and Sulfuriflexus mobilis were highly enriched in the fringe. The 10 most significant differential taxa of the rhizosphere and rhizoplane compartments (p < 0.001) are highlighted by their calculated z-scores (Figures 5A,B; all significant ASVs and specific p values are available in Supplementary Datasheets 3, 4). Additionally, the fringe and scrub endospheres revealed a total of 16 differentially enriched ASVs. Fourteen significant taxa were identified in fringe endosphere samples, with 2 significant taxa, Vibrio alginolyticus and Halomonas sp. SS10-MC5 in the corresponding scrub root compartment (Figure 5C; Supplementary Datasheet 5).
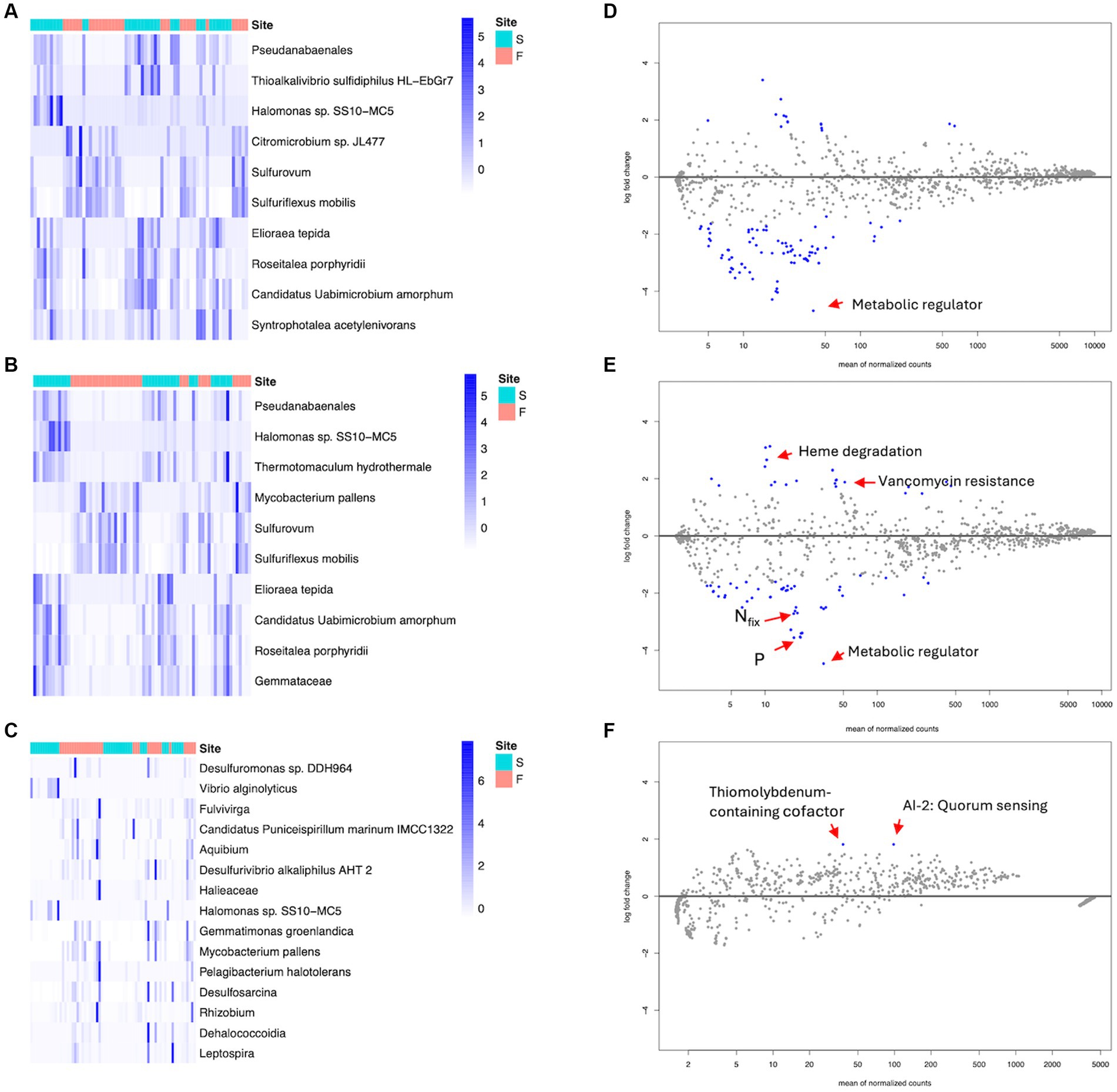
Figure 5. Heatmap of differentially abundant ASVs (DESeq2) and annotated for scrub (S) and fringe (F) forest types, among root compartments. Z scores were calculated from DESeq2 result matrix and the top 10–15 most significant differentially abundant ASVs between forest types. (A) Top 10 significant ASVs in rhizosphere root compartment. (B) Top 10 Significant ASVs in rhizoplane root compartment. (C) All significant ASVs in endosphere compartment. (D–F) MA plots from differential abundance analysis for divergent metabolic pathways (DESeq2), in root compartments, contrasting forest zones. The scrub forest was set as the control (Log2 fold change <0). Blue points represent significant differentially enriched pathways. (D) Rhizosphere, fringe vs. scrub. (E) Rhizoplane, fringe vs. scrub. (F) Endosphere, fringe vs. scrub.
Similarly, DA analysis was performed for potential metabolic pathways among root compartments, contrasting forest zones. A total of 70 significant differential metabolic pathways were observed between fringe and scrub rhizospheres, 11 of which were enriched in the fringe, while 59 significant processes were observed in the scrub. Enriched predicted degradative processes were identified in the rhizosphere for each of the forest zones (Table 1, see Supplementary Datasheet 6 for all significant metabolic pathway predictions). A total of 84 significant differential pathways were revealed in the rhizoplane (Supplementary Datasheet 7). Twenty-five were significantly enriched in the fringe and 59 were significant in the scrub forest. Among other processes, a highly significant pathway, ppGpp metabolism (p < 0.001), was enriched in the exterior scrub root compartments (Figures 5D,E).
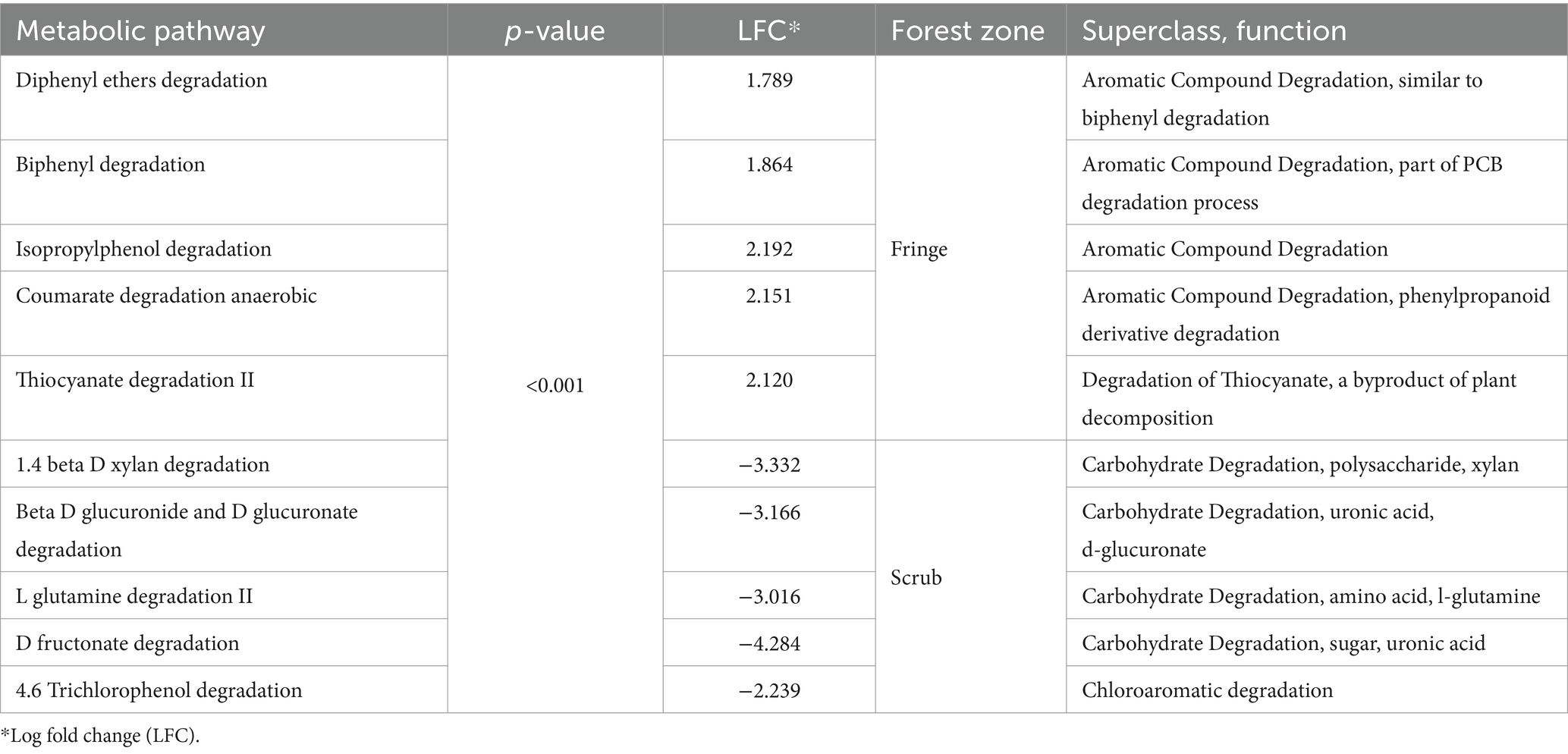
Table 1. Ten most significantly differential degradative metabolic pathways classified in the rhizosphere compartment, contrasting fringe and scrub forest zones (Meta-Cyc.org was utilized for classifications).
DA analysis of fringe and scrub endosphere revealed two significant differential pathways (both p < 0.001) observed in the fringe endosphere, degradation of a secondary metabolite, autoinducer 2, and the biosynthesis of molybdenum-containing cofactor biosynthesis (Figure 5F). There were no significant differentially abundant pathways observed in the scrub endosphere.
DA analysis contrasting root compartments to bulk soil samples in the scrub forest zone identified a total of 28 significantly differential predicted pathways associated with the rhizosphere root compartment. Out of the 28 significant pathways, 16 were enriched in the rhizosphere while 12 were depleted compared to bulk soil samples. In the rhizoplane, 128 significant differentially abundant metabolic pathways were uncovered, 40 of which were enriched and 80 were depleted. DA analysis results of the endosphere and bulk identified a total of 312 significantly differential predicted pathways. 126 pathways were significantly enriched while 186 were depleted in the endosphere, compared to the bulk soil. Significant nutrient metabolisms were identified within each root compartment of the rhizosphere, rhizoplane and endosphere (Figures 6A–C and Table 2, Supplementary Datasheets 8–10, respectively, for all significant metabolic processes in the root compartments).
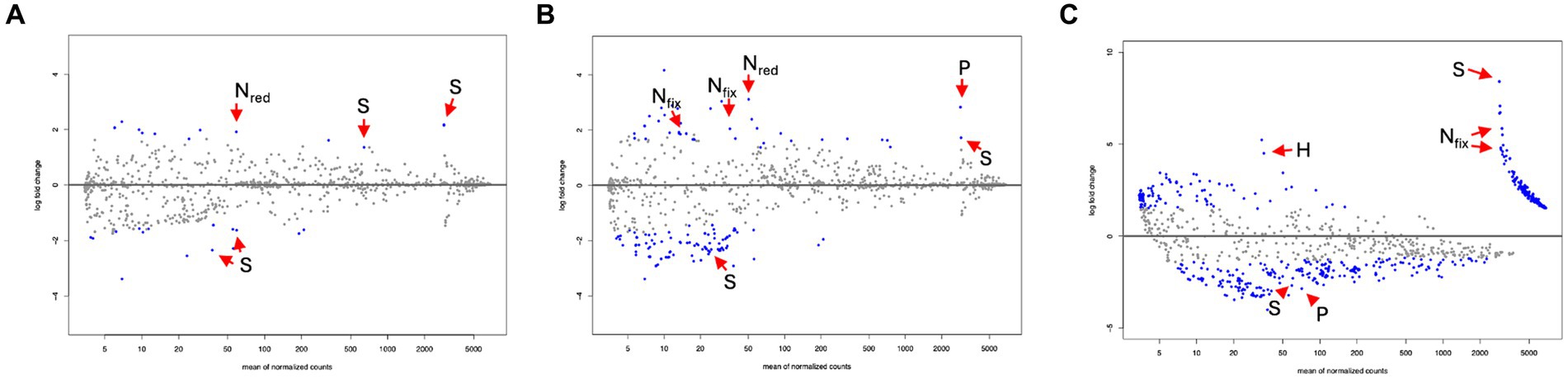
Figure 6. MA plots from differential abundance analysis for potential metabolic pathways, comparing root compartments of the scrub forest, setting the bulk soil as the control (Log2 fold change <0), (A) Rhizosphere vs. Bulk Soil. Nitrogen reduction (Nred), and sulfur (S) metabolisms are noted. (B) Rhizoplane vs. Bulk Soil. Nitrogen fixation (Nfix), Nred, phosphorus (P), and S metabolisms are noted. (C) Endosphere vs. Bulk Soil. Nfix, S, hydrogen (H) and P metabolisms are noted.
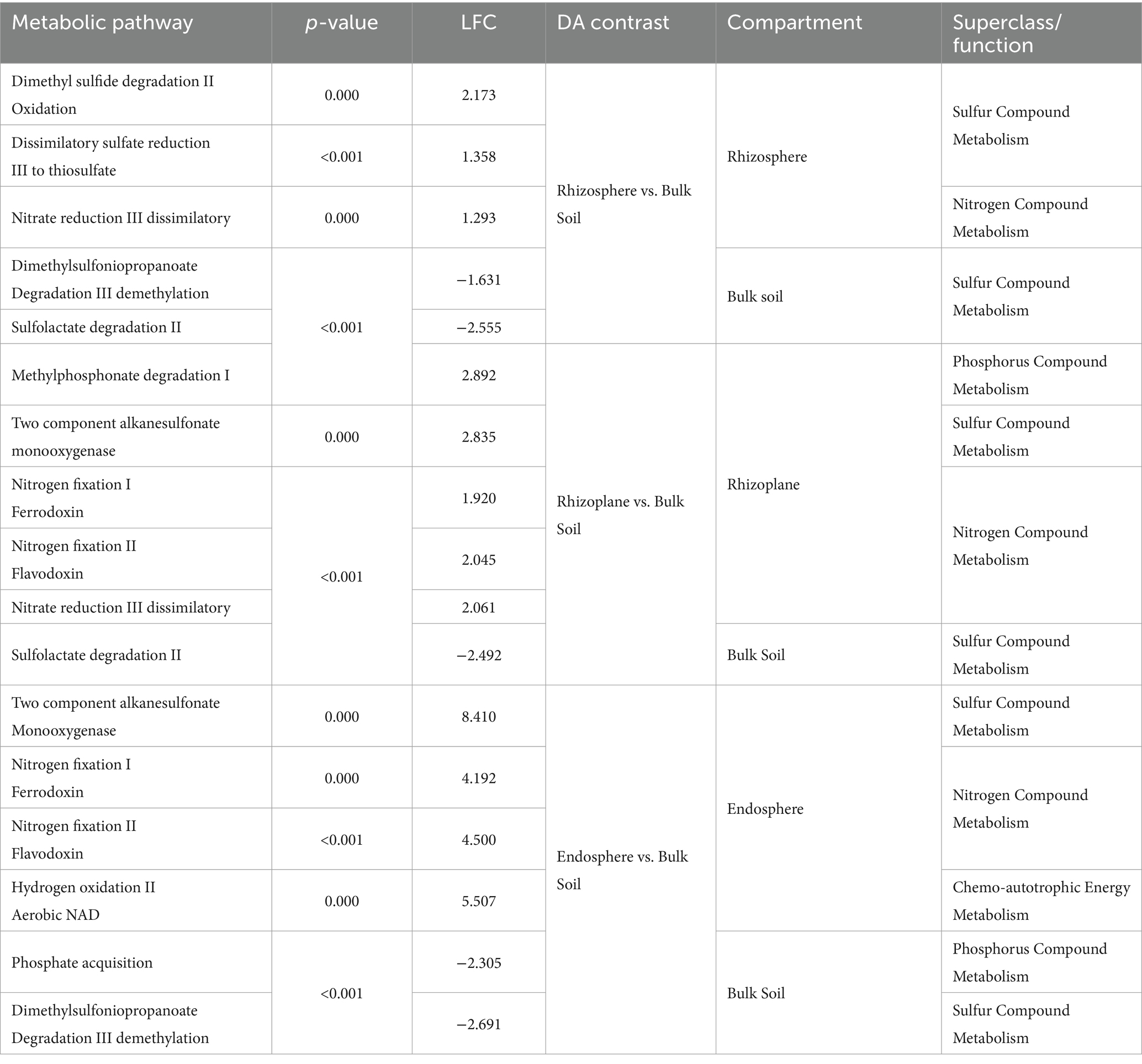
Table 2. Identification of inorganic nutrient metabolisms, contrasting root compartments to bulk soil samples in the scrub forest zone (Meta-Cyc.org was utilized for classifications).
Treatments did not have a significant effect on alpha diversity of ASVs in either of the forest zones (Supplementary Figures 1D,E). The effect of nutrient fertilization on microbial community composition (beta diversity) was significant (p = 0.022); however, the effect size was low, explaining only 1.2% of the variance among samples (R2 = 0.012) (Figure 7A; Supplementary Table S1). Nutrient enrichment (Treatment) in the scrub forest did not have a significant effect on microbial community structure (p = 0.094). In contrast, treatments in the fringe forest zone were found to have a significant effect on sample communities (p = 0.014, R2 = 0.026). Treatments had a significant effect on compositional differences in predicted metabolic pathways (Supplementary Table S3) from both forest zones combined, however was not detectable in either of the forest zones individually. Bacterial signatures were not significant among experimental treatments (Supplementary Figure S2A).

Figure 7. Bacteria community composition and divergence among treatments. (A) NMDS visualization (stress = 0.1478) of microbial beta diversity (PERMANOVA, p = 0.022). Nutrient treatments are highlighted: C (red) = untreated control plots, Fe (green) = iron-amended plots, N (sky blue) = nitrogen-amended plots, and p (purple) = phosphorous-amended plots. Differential abundance analysis was performed among treatments, within scrub forest (B,C) and fringe forest (D-F) zones. Significantly enriched ASVs in treatments are shown in Log2 Fold Change > 0, untreated control is shown in Log2 Fold Change < 0. (B) Nitrogen vs Control in the scrub forest, (C) Iron vs Control in the scrub forest, (D) Nitrogen vs Control in the fringe forest, (E) Phosphorous vs Control in the fringe forest, (F) Iron vs Control in the fringe forest.
DA analysis on the effect of nutrient enrichment revealed a total of 7 significantly differential ASVs in the scrub forest zone (Figures 7B,C). Six ASVs were found differentially significant from DA analysis of scrub forest N-fertilized plots compared to control plots (Figure 7C). Rhodobacterales, Ectothiorhodospiraceae (both p < 0.001), and Allocoleopsis franciscana (p = 0.000) were enriched in N-fertilized plots. Thalassococcus sp. S3, Salinisphaera, and Salaquimonas pukyongi (all p < 0.001) were enriched in the control plots. Hymenobacter (p < 0.001) was significant in iron-fertilized soils.
Significant ASVs observed in fringe control plots included Spiribacter curvatus, Desulfatibacillum aliphaticivorans, Muriicola soli, Rhodobacterales Aquirufa, Roseivivax marinus, Hyphomonas sp. Mor2 and Sulfurovum lithotrophicum (all p ≤ 0.001), and Thalassotalea crassostreae and Erythrobacter (both p = 0.000). There were no significant ASVs enriched in the N-fertilized plots (Figure 7D). A total of 9 significant ASVs were observed between the P-fertilized and control plots. Cylindrospermopsis (p < 0.001) was significantly enriched in the P-fertilized plots compared to 8 significant ASVs identified in the controls, which included Bulleidia sp. Zg-1006, Aureimonas sp. SA4125, and Thalassococcus sp. S3, (all p < 0.001) (Figure 7E). Thalassotalea sp. PS06, and Microbulbifer hydrolyticus (both p < 0.001) were significantly enriched in Fe-fertilized soils. Erythrobacter (p < 0.001) was significant in the controls compared to Fe-fertilized plots (Figure 7F).
Microbial community structure, metabolic potential, and diversity varied significantly among mangrove forest zones and root compartments. Physicochemical properties such as temperature and salinity of mangrove sediments and variable hydrologic connectivity across the intertidal landscape are contributing factors in microbial community structure and diversity (Alongi et al., 1993; Zhou et al., 2014; Langenheder and Lindstrom, 2019; Zhuang et al., 2020). At the field site in Exmouth Gulf, microbial community composition and diversity differed between fringe and scrub forest zones, likely influenced by the differential groundwater connectivity related to intertidal position. Intermittent hydrologic connectivity in the scrub forest zone facilitated desiccation and increased salinity, fostering environmental heterogeneity conducive to diverse taxa that exhibit survival adaptations to harsh conditions. In contrast, environmental conditions in the fringe forest zone of the field site were subjected to homogenization and dispersion by tidal inundation (Thrush et al., 2006; Hewitt et al., 2010; De Santana et al., 2021; Junkins et al., 2022) and continual groundwater connectivity.
Bacterial communities of the rhizosphere and rhizoplane compartments displayed overall similarity in their 15 most significantly enriched taxa within each of the forest zones; however, the scrub forest zone was distinct from the fringe with its regime of highly diverse taxa and ecological roles reflecting its environmental niches. These taxa included halophilic or halotolerant species, such as those belonging to the genus of Halomonas (Kim et al., 2013), which have been identified to promote plant growth through its production of exopolysaccharide production during abiotic stress such as increased salinity (Mukherjee et al., 2019). Other bacterial members included fermenters such as Roseilatalea porphyridii (Hyeon et al., 2017) and Thermotoamaculum hydrothermale, also capable of sulfur reduction (Izumi et al., 2012).
Diverse ecological roles were further indicated by the enriched presence of Candidatus Uabmicrobium amorpheum, which is known for its predation on community bacteria and microeukaryotes via phagocytosis (Shiratori et al., 2019). Reugeria was another significant community member contributing to the functional diversity observed in the exterior root compartments of the scrub forest. It has been described to possess a unique combination of amino acid metabolism and sulfur oxidation, along with the ability to upregulate gene expression when subjected to high carbon concentrations in culture, a quality indicative of copiotrophic growth strategies (Su et al., 2023). These overlapping bacterial community members reflect the functional diversity likely recruited to the rhizoplane from the rhizosphere.
The scrub rhizoplane compartment was distinct from the rhizosphere with the presence of Stella humosa and Gemmataceae, which belong to the Planctomycetes, Verruomicrobiae and Chlamydiae (PVC) superphylum. Members within the PVC group play important roles in the global carbon and nitrogen cycles (Rivas-Marín and Devos, 2018). S. humosa exhibits oligocarbophilic characteristics, thriving in environments with low nutrient concentrations while utilizing organic carbon and amino acids (Vasilyeva, 1985). These community members are collectively indicative of the low nutrient availability in the scrub mangrove forest zone and have roles as beneficial nutrient-cycling mangrove root microbiota.
The fringe rhizoplane and rhizosphere also shared significant overlap, again highlighting the ecological continuity between these two microenvironments. In addition to this overlap, we observed enrichment of Desulfobulbus oligotrophicus, a novel and strict anaerobic sulfate reducer and propionate oxidizer. This unique metabolic profile suggested the presence of sulfur and carbon cycling processes specific to the fringe forest ecosystem (El Houari et al., 2017). Additionally, the most enriched community members of the rhizosphere and rhizoplane within the fringe forest were predominantly characterized with sulfur metabolism, specifically sulfur oxidation, which is associated with low oxygen levels (Brune et al., 2000). Prominent taxa in this context included Sulfurovum, Sulfuriflexus mobilis, and Sulfurimonas. Mycobacterium pallens was a highly significant taxa found in our fringe forest samples, classified for its rapid growth, a copiotrophic bacterial signature, coupled with its ability for nitrate reduction. However, it has been previously described as having limited tolerance to salinity (Hennessee et al., 2009), and its enrichment could be attributed to the continuous groundwater connectivity observed in the fringe zone, where salinity was observed to be significantly lower than the scrub and also highly variable.
The exterior root compartments of the scrub forest zone were enriched with a higher number of ecologically diverse taxa compared to the corresponding compartments in the fringe zone. This distinction suggested that a broader range of microbial diversity was supported by the multifarious environment of the scrub forest zone and could ultimately influence nutrient cycling, ecosystem stability, and overall ecological dynamics (Langenheder and Lindstrom, 2019). However, the endosphere compartment did not follow this pattern, with a higher number of significant ASVs observed in the fringing mangroves. The scrub endosphere was significantly enriched with two halophilic taxa. Halomonas sp. SS10-MC5l possesses genes for denitrification, assimilation, and stress response, strategies necessary to survive the extreme environmental conditions. Vibrio alginolyticus is capable of proteolysis and considerable phosphate solubilization (Mamangkey et al., 2021). Although identified as a pathogen of humans and marine animals commonly found in estuaries and coastal ecosystems (Mustapha et al., 2013; Zhou et al., 2021, its enrichment in the scrub endosphere is likely dictated by the increased salinity and recruitment for nutrient metabolisms rather than a biproduct of reduced mangrove health or function.
We expected that the endosphere compartment would contain taxa with the smallest mean genome size and lowest RRN content, given streamlined genomic features typically associated with endosymbionts and insect symbionts (Pini et al., 2011). However, our findings revealed the contrary, with the endosphere exhibiting the largest predicted mean genome size in comparison to the exterior root compartments. Of note, the endosphere can select for a diverse range of function found in both obligate and facultative symbiotic bacteria (Mitter et al., 2013). In our data, significantly differential taxa partially making up the seaward fringe mangrove endosphere community had a range of bacteria capable of a free-living lifestyle, such as taxa belonging to Helleaceae (Li et al., 2023), Mycobacteria pallens (Hennessee et al., 2009), and Desulfourivibrio alkaliphilus AHT2 (Melton et al., 2016). Taxa known for nitrogen-fixation in plant-associated symbiosis, such as Rhizobium (Díez-Méndez and Menéndez, 2021) were also found significant in the fringe endosphere. Subsequently, higher maximal growth rate predictions were observed in the exterior root compartments, compared to the endosphere. This would be expected with higher concentrations of carbon sources in the immediate root exterior, to which copiotrophic taxa belonging to Alpha-, Beta- and Gamma- proteobacteria and Actinobacteria phyla would congregate (Fierer et al., 2007; Chen et al., 2016).
Significant differences in degradative pathways between fringe and scrub rhizospheres were identified. The fringe rhizosphere was highly enriched with degradative processes of aromatic compounds, in particular phenols, which are produced by plants as defense against pathogens (Liao et al., 2020) and more specifically, biphenyls have been reported to be produced when the plant is under pathogenic attack (Zhou et al., 2016; Li et al., 2017; Wang Y. et al., 2022). Rhizoplane compartments in the fringe forest zone were depleted of inorganic nutrient metabolisms such as phosphonate degradative processes and nitrogen fixation compared to the scrub zone. Instead, the fringe was highly enriched in biosynthesis of vancomycin resistance, and degradation of heme, which are present in many pathogens and mycobacteria (Chim et al., 2010). Significant degradative processes shifted from predominantly aromatic compounds in the fringe rhizosphere to more diverse processes such as degradation of sugars and alcohols in the rhizoplane, suggestive of stronger plant root influence on microbial community structure. Two pathways enriched in the fringe endosphere included degradation of a secondary metabolite autoinducer, which is responsible for cell–cell communication, and quorum sensing (Pereira et al., 2013). Together, these two pathways are likely indicators of high cell density in the fringe endosphere compartment (Compant et al., 2021).
In contrast to the fringe zone, carbohydrate degradative processes were most significant in the scrub forest. However, the single most significant pathway overall was a metabolic regulating pathway associated with ppGpp nucleotides (guanosine 3′-diphophate 5′-diphosphate). The ppGpp pathway upregulates gene expression with bacterial stringent response, and is a global regulator activated during nutrient or energy starvation and environmental stress (Traxler et al., 2008). Additionally, biosynthesis of glutathione, which plays an important role in the detoxification of ROS stress (Wongsaroj et al., 2018), was significantly enriched in the scrub forest zone. These inferred metabolic pathways are indicators for divergent environmental stressors characterizing physicochemical differences between forest zones, particularly that higher temperatures and hypersaline conditions that are associated with oxidative stress (Lee and Park, 2012).
Since there are not many existing studies describing root metabolic function of a scrub mangrove forest zone, we sought to contribute to this knowledge. Significant divergence of predicted metabolic function was observed among the root compartments with some overlap of nutrient metabolisms observed. Compared to bulk soil samples, the rhizosphere and rhizoplane compartments were differentially enriched with sulfur and nitrate compound metabolisms for DMS degradation, dissimilatory sulfate reduction to thiosulfate, and nitrate reduction. Our results are comparable to other studies (Edwards et al., 2015; Liao et al., 2020; Zhuang et al., 2020), where sulfate reduction was found more enriched in the rhizosphere and bulk soil. These findings are reflective of an oxygen gradient characterized by mangrove soil conditions, in which redox potentials can facilitate sulfate reduction. Among other significant nutrient metabolisms, the rhizoplane was additionally enriched with two component alkanesulfonate monooxygenase, which has been associated with Beta- and Gammaproteobacteria and plays a protective role as controllers of the sulfur cycle against oxidative stress in sulfur-limited environments (Park et al., 2020). Our results differed from other earlier studies (Edwards et al., 2015; Liao et al., 2020; Zhuang et al., 2020), which showed enrichment of methanogens and methanotrophs in Kandelia obovata and Arabidopsis thaliana endospheres. However, decreased methanogen and methanotroph presence has been correlated to higher saline conditions (Chen S. et al., 2020), which is congruous with the physicochemical conditions in the scrub forest zone of the experimental site in Exmouth Gulf.
Bacterial signatures of maximal growth rate predictions, genome size and RRN were not significant among treatments. Even in nutrient-available systems and optimal growth conditions, such as absence of competition, variations in maximal potential growth rates are dependent upon cell machinery capabilities (Bledsoe et al., 2020). It has been thought that intermittent nutrient availability strongly selects for taxa capable of upregulating their growth rate to transfer the energy source into biomass (Westoby et al., 2021). Therefore, it is difficult to detect overall shifts in trophic strategies due to the episodic availability and usage of nutrients and are more likely to be represented by a spectrum (Westoby et al., 2021). In our study, nutrient treatments did not have a significant long-term effect on trophic strategies within each of the forest zones; however, bacterial beta diversity was significantly affected in the fringe forest zone. As ground porewater is another source of nutrients (Wang F. et al., 2022), the continual hydrologic connectivity in the fringe forest zone could be a contributing factor to these findings; however, it is not well understood and further research would be needed. Other contributing factors could include the homogenizing forces of tidal variation characteristic of the intertidal fringe forest zone, which selected for taxa with faster growth rates and metabolic flexibility (Junkins et al., 2022; Ngugi et al., 2023), while the lower growth rates observed in the scrub forest zone were selected for extreme and adverse environmental stressors.
Differentially enriched taxa capable of thriving in harsh, desiccated environments were observed in N-fertilized plots compared to untreated control plots in the scrub forest zone. Interestingly, cyanobacteria Allocoleopsis franciscana from the family Coleoasciculacea is not capable of nitrogen fixation, but rather, reported to be a pioneer of biological soil crust formation. In contrast, the control plots relative to N-fertilized plots, were enriched with nutrient cycling taxa characteristic of oligotrophic mangrove microbiomes, such as nitrogen-fixing plant symbiont belonging to Hyphomicrobiales and Thalassococcus, belonging to Rhodobacteraceae, a family entrenched with sulfur and carbon biogeochemical cycling species (Pujalte et al., 2014). The family of Phyllobacteriaceae of the order Hyphomicrobiales, characterized by nitrogen cycling and as beneficial plant root-associated taxa (Willems, 2014), were found significantly enriched in control soils. Significant differential enrichment of denitrifiers or nitrate-reducers were not observed when comparing N-amended to untreated mangrove soils and further conclusions could not be made on the long-term effects of nitrogen fertilization. In previous studies where N addition has impacted microbial community composition, higher relative abundance in taxa possessing nitrate-reducing and ammonium-oxidizing metabolic pathways were observed (Palacios et al., 2021).
Few taxa were significantly enriched in treatment plots compared to the control plots in the fringe forest zone, indicating depletion of nutrient-cycling taxa. Significant ASVs enriched in fringe forest control plots consisted of marine and mangrove-associated microbes included in genera from Alpha-, Delta-, Gamma- and Epsilon- Proteobacteria. Oligotrophic nitrate reducing Hyphomonas, a member of the Hyphomonadaceae family (Abraham and Rohde, 2014), were identified in addition to other oligotrophic taxa from the Rhodobacteraceae family (Simon et al., 2017) in fringe zone control plots. Additionally, nitrogen-fixing Aureimonas sp. SA4125 (Sakai et al., 2021) and several taxa involved in sulfur cycles were observed, including Spiribacter curvatus (León et al., 2015), Roseivivax marinus, a sulfur oxidizer (Simon et al., 2017), and sulfur-reducing Desulfobacticillus aliphaticivorans (Cravo-Laureau et al., 2004).
Thalassotalea SP06 was enriched in fringe Fe-fertilized plots and are known to contribute to the biodegradation of hydrocarbons. Additionally, Thalassotalea possess gene repertoires for assimilatory nitrate and sulfate reduction (Kim et al., 2020). It has been suggested that Thalassotalea SP06 is capable of adapting to habitat changes or evolutionary challenges and are predicted to be metabolically flexible, a characteristic that has been correlated with requisite bacterial lifestyle strategies of the intertidal fringe zone (Junkins et al., 2022; Ngugi et al., 2023).
The long-term effect of nutrient enrichment was clearly indicated in P-amended soils located in the fringe forest zone. The differentially abundant Cylindrospermopsis is a harmful algal bloom-forming cyanobacteria associated with eutrophication (Burford et al., 2016), placing it on the spectrum of copiotrophy. Studies conducted on Cylindrospermopsis revealed C. raciborskii has become widely distributed and globally invasive (Antunes et al., 2015), with toxin-producing strains isolated in Australia and Asia (Piccini et al., 2011).
DA analysis suggested long-term nutrient fertilization have altered community structure to some extent. The results of this study were partially consistent with our hypotheses that community divergence would be driven by the enrichment of particular taxa associated with nutrient pollution such as copiotrophs, nitrate reducers or ammonia oxidizers, resulting in reduced diversity (Erazo and Bowman, 2021). While there was not a clear signal of the aforementioned variety of taxa observed across all treatments, beta diversity in the fringe forest zone was slightly affected by nutrient enrichment. Coupled with reduced alpha diversity and the presence of metabolic pathways associated with pathogenicity, these results could collectively suggest reduced health and stability.
Overall, our study indicated the effects of root compartment mechanisms and physicochemical properties of forest zones outweighed the long-term effects of nutrient enrichments. DA analysis provided insights into the microbial communities inhabiting these distinct ecosystems, shedding light on their diverse ecological roles and functional adaptations within their respective forest zone-related environmental characteristics. The observed differences in community structure and functional diversity between forest zones highlight the need for deeper understanding of ecosystem-specific microbial interactions. Our data indicated abiotic and biotic interactions are shaping microbial community composition. In the context of climate change and rising sea levels, environmental changes are implicated to drastically influence the mangrove’s microbiome. These adverse environmental factors and long-term nutrient stress could have a negative impact on beneficial root-microbial partnerships in mangrove ecosystems, as well as diminish mangrove ecosystem function.
The data that support the findings of this study and sequences were submitted to the NCBI sequence read archive (SRA) under BioProject ID: PRJNA1035549.
VH: Data curation, Formal analysis, Investigation, Visualization, and Writing - original draft. NGE: Investigation, Methodology, Visualization, Supervision, Writing - review & editing. RR: Conceptualization, Data curation, Investigation, Writing - review & editing. CL: Investigation, Writing-review & editing. JB: Conceptualization, Data curation, Funding acquisition, Investigation, Supervision, Writing - review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was funded by the University of California San Diego – Monash University Seed Fund. NGE was supported by SENESCYT fellowship.
We would like to express gratitude and appreciation to the members of Bowman Lab for their support. Special thanks to Riley Hale and Benjamin Kemplay for helpful input and guidance on analysis methods, and Prudence for field work assistance.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/ffgc.2024.1336037/full#supplementary-material
Abraham, W. R., and Rohde, M. (2014). “The family Hyphomonadaceae” in The prokaryotes: alphaproteobacteria and betaproteobacteria. eds. E. Rosenberg, E. F. DeLong, S. Lory, E. Stackebrandt, and F. Thompson (Berlin Heidelberg: Springer), 283–299.
Allard, S. M., Costa, M. T., Bulseco, A. N., Helfer, V., Wilkins, L. G. E., Hassenrück, C., et al. (2020). Introducing the mangrove microbiome initiative: identifying microbial research priorities and approaches to better understand, protect, and rehabilitate mangrove ecosystems. mSystems 5:e00658-20. doi: 10.1128/mSystems.00658-20
Alongi, D. M. (2015). The impact of climate change on mangrove forests. Curr. Clim. Chang. Rep. 1, 30–39. doi: 10.1007/s40641-015-0002-x
Alongi, D. M. (2022). Impacts of climate change on blue carbon stocks and fluxes in mangrove forests. Forests 13:149. doi: 10.3390/f13020149
Alongi, D. M., Christoffersen, P., and Tirendi, F. (1993). The influence of forest type on microbial-nutrient relationships in tropical mangrove sediments. J. Exp. Mar. Biol. Ecol. 171, 201–223. doi: 10.1016/0022-0981(93)90004-8
Alzubaidy, H., Essack, M., Malas, T. B., Bokhari, A., Motwalli, O., Kamanu, F. K., et al. (2016). Rhizosphere microbiome metagenomics of gray mangroves (Avicennia marina) in the Red Sea. Gene 576, 626–636. doi: 10.1016/j.gene.2015.10.032
Anderson, M. J. (2006). Distance-based tests for homogeneity of multivariate dispersions. Biometrics 62, 245–253. doi: 10.1111/j.1541-0420.2005.00440.x
Anderson, M. J., Ellingsen, K. E., and McArdle, B. H. (2006). Multivariate dispersion as a measure of beta diversity. Ecol. Lett. 9, 683–693. doi: 10.1111/j.1461-0248.2006.00926.x
Antunes, J. T., Leao, P. N., and Vasconcelos, V. M. (2015). Cylindrospermopsis raciborskii: review of the distribution, phylogeography, and ecophysiology of a global invasive species. Front. Microbiol. 6:473. doi: 10.3389/fmicb.2015.00473
Barbera, P., Kozlov, A. M., Czech, L., Morel, B., Darriba, D., Flouri, T., et al. (2019). EPA-ng: massively parallel evolutionary placement of genetic sequences. Syst. Biol. 68, 365–369. doi: 10.1093/sysbio/syy054
Barbier, E. B., Hacker, S. D., Kennedy, C., Koch, E. W., Stier, A. C., and Silliman, B. R. (2011). The value of estuarine and coastal ecosystem services. Ecol. Soc. Am. 81, 169–193. doi: 10.1890/10-1510.1
Bledsoe, R. B., Goodwillie, C., and Peralta, A. L. (2020). Long-term nutrient enrichment of an Oligotroph-dominated wetland increases bacterial diversity in bulk soils and plant rhizospheres. mSphere 5:e00035-20. doi: 10.1128/mSphere.00035-20
Bowman, J. S., and Ducklow, H. W. (2015). Microbial communities can be described by metabolic structure: a general framework and application to a seasonally variable, depth-stratified microbial community from the coastal West Antarctic peninsula. PLoS One 10:e0135868. doi: 10.1371/journal.pone.0135868
Brune, A., Frenzel, P., and Cypionka, H. (2000). Life at the oxic–anoxic interface: microbial activities and adaptations. FEMS Microbiol. Rev. 24, 691–710. doi: 10.1016/S0168-6445(00)00054-1
Burford, M., Beardall, J., Willis, A., Orr, P. T., Magalhaes, V. F., Rnagel, L. M., et al. (2016). Understanding the winning strategies used by the bloom-forming cyanobacterium Cylindrospermopsis raciborskii. Harmful Algae 54, 44–53. doi: 10.1016/j.hal.2015.10.012
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Chen, L., Brookes, P. C., Xu, J., Zhang, J., Zhang, C., Zhou, X., et al. (2016). Structural and functional differentiation of the root-associated bacterial microbiomes of perennial ryegrass. Soil Biol. Biochem. 98, 1–10. doi: 10.1016/j.soilbio.2016.04.004
Chamberlain, E. J., Balmonte, J. P., Torstensson, A., Fong, A. A., Snoejis-Leijonmalm, P., and Bowman, J. S. (2022). Impacts of sea ice melting procedures on measurements of microbial community structure. Elementa Science of the Anthropocene, 10: 00017. doi: 10.1525/elementa.2022.00017
Chen, M., Chang, L., Zhang, J., Guo, F., Vymazal, J., He, Q., et al. (2020). Global nitrogen input on wetland ecosystem: the driving mechanism of soil labile carbon and nitrogen on greenhouse gas emissions. Environ. Sci. Technol. 4:100063. doi: 10.1016/j.ese.2020.100063
Chen, S., Wang, P., Liu, H., Xie, W., Wan, X. S., Kao, S.-J., et al. (2020). Population dynamics of methanogens and methanotrophs along the salinity gradient in Pearl River estuary: implications for methane metabolism. Appl. Microbiol. Biotechnol. 104, 1331–1346. doi: 10.1007/s00253-019-10221-6
Chim, N., Iniguez, A., Nguyen, T. Q., and Goulding, C. W. (2010). Unusual Diheme conformation of the Heme-degrading protein from Mycobacterium tuberculosis. J. Mol. Biol. 395, 595–608. doi: 10.1016/j.jmb.2009.11.025
Climate Data (2022). Exmouth Climate (Australia). Creative Commons license Canonical URL: https://creativecommons.org/licenses/by-nc/4.0. Data was retrieved from https://en.climate-data.org/oceania/australia/western-australia/exmouth-30413/ (Accessed April 14, 2024).
Compant, S., Cambon, M. C., Vacher, C., Mitter, B., Samad, A., and Sessitsch, A. (2021). The plant endosphere world – bacterial life within plants. Environ. Microbiol. 23, 1812–1829. doi: 10.1111/1462-2920.15240
Craig, H., Antwis, R. E., Cordero, I., Ashworth, D., Robinson, C. H., Osborne, T. Z., et al. (2021). Nitrogen addition alters composition, diversity, and functioning of microbial communities in mangrove soils: an incubation experiment. Soil Biol. Biochem. 153:108076. doi: 10.1016/j.soilbio.2020.108076
Cravo-Laureau, C., Matheron, R., Cayol, J. L., Joulian, C., and Hirschler-Réa, A. (2004). Desulfatibacillum aliphaticivorans gen. nov., sp. nov., an n-alkane- and n-alkene-degrading, sulfate-reducing bacterium. Int. J. Syst. Evol. Microbiol. 54, 77–83. doi: 10.1099/ijs.0.02717-0
Cusack, D. F., Silver, W. L., Torn, M. S., Burton, S. D., and Firestone, M. K. (2011). Changes in microbial community characteristics and soil organic matter with nitrogen additions in two tropical forests. Ecology, 92, 621–632. doi: 10.1890/10-0459.1
Czech, L., Barbera, P., and Stamatakis, A. (2020). Genesis and Gappa: processing, analyzing and visualizing phylogenetic (placement) data. Bioinformatics 36, 3263–3265. doi: 10.1093/bioinformatics/btaa070
Dai, M., Zhao, Y., Chai, F., Chen, M., Chen, N., Chen, Y., et al. (2023). Persistent eutrophication and hypoxia in the coastal ocean. Coastal Futures 1:E19. doi: 10.1017/CFT.2023.7
De Santana, C. O., Spealman, P., Melo, V. M. M., Gresham, D., De Jesus, T. B., and Chinalia, F. A. (2021). Effects of tidal influence on the structure and function of prokaryotic communities in the sediments of a pristine Brazilian mangrove. Biogeosciences 18, 2259–2273. doi: 10.5194/bg-18-2259-2021
De Souza Rocha, A. F., Vitorino, L. C., Bessa, L. A., Costa, R. R. G. F., Da Silva Brasil, M., and Souchie, E. L. (2020). Soil parameters affect the functional diversity of the symbiotic microbiota of Hymenaea courbaril L., a Neotropical fruit tree. Rhizosphere 16:100237. doi: 10.1016/j.rhisph.2020.100237
Díez-Méndez, A., and Menéndez, E. (2021). “Rhizobium presence and functions in microbiomes of non-leguminous plants” in Symbiotic soil microorganisms. soil biology. eds. N. Shrivastava, S. Mahajan, and A. Varma, vol. 60 (Cham: Springer).
Duran, P., Thiergart, T., Garrdio-Oter, R., Kemen, E., Schulze-Lefert, P., and Hacquard, S. (2018). Microbial interkingdom interactions in roots promote Arabidopsis survival. Cell 175:973. doi: 10.1016/j.cell.2018.10.020
Edwards, J., Johnson, C., Santos-Medellín, C., Lurie, E., Podishetty, N. K., Bhatnagar, S., et al. (2015). Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. 112, E911–E920. doi: 10.1073/pnas.1414592112
El Houari, A., Ranchou-Peyruse, M., Ranchou-Peyruse, A., Dakdaki, A., Guignard, M., Idouhammou, L., et al. (2017). Desulfobulbus oligotrophicus sp. nov., a sulfate-reducing and propionate-oxidizing bacterium isolated from a municipal anaerobic sewage sludge digester. Int. J. Syst. Evol. Microbiol. 67, 275–281. doi: 10.1099/ijsem.0.001615
Erazo, N. G., and Bowman, J. S. (2021). Sensitivity of the mangrove-estuarine microbial community to aquaculture effluent. iScience 24:102204. doi: 10.1016/j.isci.2021.102204
Eren, A. M., Vineis, J. H., Morrison, H. G., and Sogin, M. L. (2013). A filtering method to generate high quality short reads using Illumina paired-end technology. PLoS One 8:e66643. doi: 10.1371/journal.pone.0066643
Feller, I. C., Whigham, D. F., O’Neill, J. P., and McKee, K. L. (1999). Effects of nutrient enrichment on within-stand cycling in a mangrove forest. Ecology 80, 2193–2205. doi: 10.1890/0012-9658(1999)080[2193,EONEOW]2.0.CO;2
Fierer, N., Bradford, M. A., and Jackson, R. B. (2007). Toward an ecological classification of soil bacteria. Ecology, 88, 1354–1364.
Francioli, D., Schulz, E., Buscot, F., and Reitz, T. (2018). Dynamics of Soil Bacterial Communities Over a Vegetation Season Relate to Both Soil Nutrient Status and Plant Growth Phenology. Microbial Ecology, 75, 216–227. doi: 10.1007/s00248-017-1012-0
Graves, C. J., Makrides, E. J., Schmidt, V. T., Giblin, A. E., Cardon, Z. G., and Rand, D. M. (2016). Functional responses of salt marsh microbial communities to long-term nutrient enrichment. Appl. Environ. Microbiol. 82, 2862–2871. doi: 10.1128/AEM.03990-15
Haft, D. H., DiCuccio, M., Badretdin, A., Brover, V., Chetvernin, V., O’Neill, K., et al. (2018). RefSeq: an update on prokaryotic genome annotation and curation. Nucleic Acids Res. 46, D851–D860. doi: 10.1093/nar/gkx1068
Hennessee, C. T., Seo, J.-S., Alvarez, A. M., and Li, Q. X. (2009). Polycyclic aromatic hydrocarbon-degrading species isolated from Hawaiian soils: Mycobacterium crocinum sp. nov., Mycobacterium pallens sp. nov., Mycobacterium rutilum sp. nov., Mycobacterium rufum sp. nov. and Mycobacterium aromaticivorans sp. nov. Int. J. Syst. Evol. Microbiol. 59, 378–387. doi: 10.1099/ijs.0.65827-0
Hewitt, J., Thrush, S., Lohrer, A., and Townsend, M. (2010). A latent threat to biodiversity: consequences of small-scale heterogeneity loss. Biodivers. Conserv. 19, 1315–1323. doi: 10.1007/s10531-009-9763-7
Hyeon, J. W., Jeong, S. E., Baek, K., and Jeon, C. O. (2017). Roseitalea porphyridii gen. nov., sp. nov., isolated from a red alga, and reclassification of Hoeflea suaedae Chung et al. 2013 as Pseudohoeflea suaedae gen. nov. comb. nov. Int. J. Syst. Evol. Microbiol. 67, 362–368. doi: 10.1099/ijsem.0.001633
Izumi, H., Nunoura, T., Miyazaki, M., Mino, S., Toki, T., Takai, K., et al. (2012). Thermotomaculum hydrothermale gen. nov., sp. nov., a novel heterotrophic thermophile within the phylum Acidobacteria from a deep-sea hydrothermal vent chimney in the Southern Okinawa trough. Extremophiles 16, 245–253. doi: 10.1007/s00792-011-0425-9
Jassby, A. D., and Cloern, J. E. (2022). Wq: exploring water quality monitoring data. R package version 1.0.0. Available at: https://CRAN.R-project.org/package=wq
Junkins, E. N., McWhirter, J. B., McCall, L. I., and Stevenson, B. S. (2022). Environmental structure impacts microbial composition and secondary metabolism. ISME Commun. 2:15. doi: 10.1038/s43705-022-00097-5
Kim, M., Cha, I., Lee, K., Lee, E., and Park, S. (2020). Genomics reveal the metabolic potential and functions in the redistribution of dissoved organic matter in marine environments of the genus thalassotalea. Microorganisms 8:1412. doi: 10.3390/microorganisms8091412
Kim, K. K., Jung-Sook, L., and Stevens, D. A. (2013). Microbiology and epidemiology of Halomonas species. Future Microbiol. 8, 1559–1573. doi: 10.2217/fmb.13.108
Langenheder, S., and Lindstrom, E. S. (2019). Factors influencing aquatic and terrestrial bacterial community assembly. Environ. Microbiol. Rep. 11, 306–315. doi: 10.1111/1758-2229.12731
Lee, S., and Park, C. M. (2012). Regulation of reactive oxygen species generation under drought conditions in Arabidopsis. Plant Signal. Behav. 7, 599–601. doi: 10.4161/psb.19940
León, M. J., Rodríguez-Olmos, Á., Sánchez-Porro, C., López-Pérez, M., Rodríguez-Valera, F., Soliveri, J., et al. (2015). Spiribacter curvatus sp. Nov., a moderately halophilic bacterium isolated from a saltern. Int. J. Syst. Evol. Microbiol. 65, 4638–4643. doi: 10.1099/ijsem.0.000621
Li, X.-B., Chen, G.-Y., Liu, R.-J., Zheng, C.-J., Song, X.-M., and Han, C.-R. (2017). A new biphenyl derivative from the mangrove endophytic fungus Phomopsis longicolla HL-2232. Nat. Prod. Res. 31, 2264–2267. doi: 10.1080/14786419.2017.1300799
Li, S.-H., Kang, I., and Cho, J.-C. (2023). Metabolic versatility of the family Halieaceae revealed by the genomics of novel cultured isolates. Microbiol. Spectr. 11:e03879-22. doi: 10.1128/spectrum.03879-22
Liao, S., Wang, Y., Liu, H., Fan, G., Sahu, S. K., Jin, T., et al. (2020). Deciphering the microbial taxonomy and functionality of two diverse mangrove ecosystems and their potential abilities to produce bioactive compounds. mSystems 5:e00851-19. doi: 10.1128/mSystems.00851-19
Liu, L., Gundersen, P., Zhang, W., Zhang, T., Chen, H., and Mo, J. (2015). Effects of nitrogen and phosphorus additions on soil microbial biomass and community structure in two reforested tropical forests. Sci. Rep. 5:14378. doi: 10.1038/srep14378
Liu, J., Li, J., Feng, L., Cao, H., and Cui, Z. (2010). An improved method for extracting bacteria from soil for high molecular weight DNA recovery and BAC library construction. J. Microbiol. 48, 728–733. doi: 10.1007/s12275-010-0139-1
Love, M. I., Huber, W., and Anders, S. (2014). Modrated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Lovelock, C. E., Ball, M. C., Martin, K. C., and Feller, I. (2009). Nutrient enrichment increases mortality of mangroves. PLoS One 4:e5600. doi: 10.1371/journal.pone.0005600
Macreadie, P. I., Costa, M. D. P., Atwood, T. B., Friess, D. A., Kelleway, J. J., Kennedy, H., et al. (2021). Blue carbon as a natural climate solution. Nat. Rev. Earth Environ. 2, 826–839. doi: 10.1038/s43017-021-00224-1
Mamangkey, J., Suryanto, D., Munir, E., Mustopa, A. Z., Sibero, M. T., Mendes, L. W., et al. (2021). Isolation and enzyme bioprospetion of bacteria associated to Brugueira cylindrica, a mangrove plant of North Sumatra, Indonesia. Biotechnol. Rep. 30:300617. doi: 10.1016/j.btre.2021.e00617
McMurdie, P. J., and Holmes, S. (2013). Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217
Melton, E. D., Sorokin, D. Y., Overmars, L., Chertkov, O., Clum, A., Pillay, M., et al. (2016). Complete genome sequence of Desulfurivibrio alkaliphilus strain AHT2T, a haloalkaliphilic sulfidogen from Egyptian hypersaline alkaline lakes. Stand. Genomic Sci. 11:67. doi: 10.1186/s40793-016-0184-4
Mitter, B., Petric, A., Shin, M. W., Chain, P. S. G., Hauberg-Lotte, L., Reinhold-Hurek, B., et al. (2013). Comparative genome analysis of Burkholderia phytofirmans PsJN reveals a wide spectrum of endophytic lifestyles based on interaction strategies with host plants. Front. Plant Sci. 4:120. doi: 10.3389/fpls.2013.00120
Moroenyane, I., Mendes, L., Tremblay, J., Tripathi, B., and Yergeau, É. (2021). Plant compartments and developmental stages modulate the balance between niche-based and neutral processes in soybean microbiome. Microb. Ecol. 82, 416–428. doi: 10.1007/s00248-021-01688-w
Mukherjee, P., Mitra, A., and Roy, M. (2019). Halomonas Rhizobacteria of Avicennia marina of Indian Sundarbans promote Rice growth under saline and heavy metal stresses through exopolysaccharide production. Front. Microbiol. 10:1207. doi: 10.3389/fmicb.2019.01207
Mustapha, S., Mustapha, E. M., and Nozha, C. (2013). Vibrio alginolyticus: an emerging pathogen of foodborne diseases. International Journal of Science and Technology, 2, 302–309.
Nawrocki, E. P., and Eddy, S. R. (2013). Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935. doi: 10.1093/bioinformatics/btt509
Ngugi, D. K., Acinas, S. G., Sanchez, P., Gasol, J. M., Agusti, S., Karl, D. M., et al. (2023). Abiotic selection of microbial genome size in the global ocean. Nat. Commun. 14:1384. doi: 10.1038/s41467-023-36988-x
Oksanen, J., Simpson, G., Blanchet, F., Kindt, R., Legendre, P., Minchin, P. R., et al. (2022). Vegan: community ecology package. R package version 2.4-3. Available at: https://CRAN.R-project.org/package=vegan
Palacios, M. M., Trevathan-Tackett, S. M., Malerba, M. E., and Macreadie, P. I. (2021). Effects of a nutrient enrichment pulse on blue carbon ecosystems. Mar. Pollut. Bull. 165:112024. doi: 10.1016/j.marpolbul.2021.112024
Park, C., Shin, B., and Park, W. (2020). Protective role of bacterial alkanesulfonate monooxygenase under oxidative stress. Appl. Environ. Microbiol. 86:e00692-20. doi: 10.1128/AEM.00692-20
Pereira, C. S., Thompson, J. A., and Xavier, K. B. (2013). AI-2-mediated signalling in bacteria. FEMS Microbiol. Rev. 37, 156–181. doi: 10.1111/j.1574-6976.2012.00345.x
Piccini, C., Aubriot, L., Fabre, A., Amaral, V., González-Piana, M., Giani, A., et al. (2011). Genetic and eco-physiological differences of South American Cylindrospermopsis raciborskii isolates support the hypothesis of multiple ecotypes. Harmful Algae 10, 644–653. doi: 10.1016/j.hal.2011.04.016
Pini, F., Galardini, M., Bazzicalupo, M., and Mengoni, A. (2011). Plant-Bacteria association and Symbiosis: are there common genomic traits in Alphaproteobacteria? Genes 2, 1017–1032. doi: 10.3390/genes2041017
Pujalte, M. J., Lucena, T., Ruvira, M. A., Arahal, D. R., and Macián, M. C. (2014). “The family Rhodobacteraceae” in The prokaryotes: alphaproteobacteria and betaproteobacteria. eds. E. Rosenberg, E. F. DeLong, S. Lory, E. Stackebrandt, and F. Thompson (Berlin Heidelberg: Springer), 439–512.
Reef, R., Feller, I. C., and Lovelock, C. E. (2010). Nutrition of mangroves. Tree Physiol. 30, 1148–1160. doi: 10.1093/treephys/tpq048
Reinhold-Hurek, B., Bünger, W., Burbano, C. S., Sabale, M., and Hurek, T. (2015). Roots shaping their microbiome: global hotspots for microbial activity. Annu. Rev. Phytopathol. 53, 403–424. doi: 10.1146/annurev-phyto-082712-102342
Reis, C. R. G., Nardoto, G. B., and Oliveira, R. S. (2017). Global overview on nitrogen dynamics in mangroves and consequences of increasing nitrogen availability for these systems. Plant Soil 410, 1–19. doi: 10.1007/s11104-016-3123-7
Richter-Heitmann, T., Eickhorst, T., Knauth, S., Friedrich, M. W., and Schmidt, H. (2016). Evaluation of strategies to separate root-associated microbial communities: a crucial choice in Rhizobiome research. Front. Microbiol. 7:773. doi: 10.3389/fmicb.2016.00773
Rivas-Marín, E., and Devos, D. P. (2018). The paradigms they are a changin’: past, present and future of PVC bacteria research. Antoine van Leeuwenhoek 111, 785–799. doi: 10.1007/s10482-017-0962-z
Romero, I. S., Jacobson, M., Fuhrman, J. A., Fogel, M., and Capone, D. G. (2012). Long-term nitrogen and phosphorous fertilization effects on N2 fixation rates and nifH gene community patterns in mangrove sediments. Mar. Ecol. 33, 117–127. doi: 10.1111/j.1439-0485.2011.00465.x
Sakai, H. D., Matsuda, R., Imura, S., and Kurosawa, N. (2021). Complete genome and plasmid sequences of the Psychrotolerant Aureimonas strain SA4125, isolated from Antarctic Moss vegetation. Microbiol. Resour. Announc. 10:e00878-21. doi: 10.1128/MRA.00878-21
Shiratori, T., Suzuki, S., Kakizawa, Y., and Ishida, K. (2019). Phagocytosis-like cell engulfment by a planctomycete bacterium. Nat. Commun. 10:5529. doi: 10.1038/s41467-019-13499-2
Simon, M., Scheuner, C., Meier-Kolthoff, J. P., Brinkhoff, T., Wagner-Döbler, I., et al. (2017). Phylogenomics of Rhodobacteraceae reveals evolutionary adaptation to marine and non-marine habitats. The ISME Journal, 11, 1483–1499. doi: 10.1038/ismej.2016.198
Srivastava, R., Kanda, T., Yadav, S., Mishra, R., and Atri, N. (2021). “Cyanobacteria in rhizosphere: dynamics, diversity and symbiosis” in Plant, soil and microbes in tropical ecosystems. eds. S. K. Dubey and S. K. Verma, vol. 4 (Singapore: Springer), 51–69.
Stephens, M. (2016). False discovery rates: a new deal. Biostatistics 18, kxw041–kxw294. doi: 10.1093/biostatistics/kxw041
Su, X., Cui, H., and Zhang, W. (2023). Copiotrophy in a marine-biofilm-derived roseobacteraceae bacterium can be supported by amino acid metabolism and thiosulfate oxidation. Int. J. Mol. Sci. 24:8617. doi: 10.3390/ijms24108617
Thomson, T., Fusi, M., Bennett-Smith, M. F., Prinz, N., Aylagas, E., Carvalho, S., et al. (2022). Contrasting effects of local environmental and biogeographic factors on the composition and structure of bacterial communities in arid monospecific mangrove soils. Microbiol. Spectr. 10:e00903-21. doi: 10.1128/spectrum.00903-21
Thrush, S. F., Gray, J. S., Hewitt, J. E., and Ugland, K. I. (2006). Predicting the effects of habitat homogenization on marine biodiversity. Ecol. Appl. 16, 1636–1642. doi: 10.1890/1051-0761(2006)016[1636:PTEOHH]2.0.CO;2
Traxler, M. F., Summers, S. M., Nguyen, H., Zacharia, V. M., Hightower, G. A., Smith, J. T., et al. (2008). The global, ppGpp-mediated stringent response to amino acid starvation in Escherichia coli. Mol. Microbiol. 68, 1128–1148. doi: 10.1111/j.1365-2958.2008.06229.x
Vasilyeva, L. V. (1985). Stella, a new genus of soil Prosthecobacteria, with proposals for Stella humosa sp. Nov. and Stella vacuolata sp. nov. Int. J. Syst. Evol. Microbiol. 35, 518–521. doi: 10.1099/02207713-35-4-518
Wang, Y., Huang, Q., Zhang, L., Zheng, C., and Xu, H. (2022). Biphenyls in Clusiaceae: isolation, structure diversity, synthesis and bioactivity. Front. Chem. 10:987009. doi: 10.3389/fchem.2022.987009
Wang, C., Liu, D., and Bai, E. (2018). Decreasing soil microbial diversity is associated with decreasing microbial biomass under nitrogen addition. Soil Biol. Biochem. 120, 126–133. doi: 10.1016/j.soilbio.2018.02.003
Wang, F., Xiao, K., Santos, I. R., Lu, Z., Tamborski, J., Wang, Y., et al. (2022). Porewater exchange drives nutrient cycling and export in a mangrove-salt marsh ecotone. J. Hydrol. 606:127401. doi: 10.1016/j.jhydrol.2021.127401
Walters, W, Embriette, H., Donna, Berg-Lyons, Gail, Ackermann, Greg, Humphrey, Alma, Parada, et al. (2015). Improved Bacterial 16S rRNA Gene (V4 and V4-5) and Fungal Internal Transcribed Spacer Marker Gene Primers for Microbial Community Surveys. mSystems, 1. doi: 10.1128/msystems.00009-15
Weissman, J. L., Hou, S., and Fuhrman, J. A. (2021). Estimating maximal microbial growth rates from cultures, metagenomes, and single cells via codon usage patterns. Proceedings of the National Academy of Sciences, 118, e2016810118. doi: 10.1073/pnas.2016810118
Westoby, M., Nielsen, D. A., Gillings, M. R., Litchman, E., Madin, J. S., Paulsen, I. T., et al. (2021). Cell size, genome size, and maximum growth rate are near-independent dimensions of ecological variation across bacteria and archaea. Ecol. Evol. 11, 3956–3976. doi: 10.1002/ece3.7290
Willems, A. (2014). “The family Phyllobacteriaceae” in The prokaryotes: Alphaproteobacteria and Betaproteobacteria. eds. E. Rosenberg, E. F. DeLong, S. Lory, E. Stackebrandt, and F. Thompson (Berlin Heidelberg: Springer), 355–418.
Wongsaroj, L., Saninjuk, K., Romsang, A., Duang-nkern, J., Trinachartvanit, W., Vattanaviboon, P., et al. (2018). Pseudomonas aeruginosa glutathione biosynthesis genes play multiple roles in stress protection, bacterial virulence and biofilm formation. PLoS One 13:e0205815. doi: 10.1371/journal.pone.0205815
Yuan, X., Decao, N., Weber-Grullon, L., and Fu, H. (2020). Nitrogen deposition enhances plant-microbe interactions in a semiarid grassland: the role of soil physicochemical properties. Geoderma 373:114446. doi: 10.1016/j.geoderma.2020.114446
Zhou, J., Deng, Y., Zhang, P., Xue, K., Liang, Y., Van Nostrand, J. D., et al. (2014). Stochasticity, succession, and environmental perturbations in a fluidic ecosystem. Proc. Natl. Acad. Sci. 111, E836–E845. doi: 10.1073/pnas.1324044111
Zhou, K., Tian, K., Liu, X., Liu, W., Zhang, X., Liu, J., et al. (2021). Characteristic and Otopathogenic analysis of a Vibrio alginolyticus strain responsible for chronic otitis externa in China. Front. Microbiol. 12:750642. doi: 10.3389/fmicb.2021.750642
Zhou, L., Yang, J., Yang, G., Kang, C., Xiao, W., Lv, C., et al. (2016). Biphenyl Phytoalexin in Sorbus pohuashanensis Suspension Cell Induced by Yeast Extract. Molecules, 21. doi: 10.3390/molecules21091180
Zhu, A., Ibrahim, J. G., and Love, M. I. (2018). Heavy-tailed prior distributions for sequence count data: removing the noise and preserving large differences. Bioinformatics 35, 2084–2092. doi: 10.1093/bioinformatics/bty895
Keywords: mangrove, root microbiomes, nutrient enrichment, root compartment, Western Australia, forest zone, metabolic function, community structure
Citation: Hsiao V, Erazo NG, Reef R, Lovelock C and Bowman J (2024) Forest zone and root compartments outweigh long-term nutrient enrichment in structuring arid mangrove root microbiomes. Front. For. Glob. Change. 7:1336037. doi: 10.3389/ffgc.2024.1336037
Edited by:
Jinping Wang, Nanchang Institute of Technology, ChinaReviewed by:
Hang Wang, Southwest Forestry University, ChinaCopyright © 2024 Hsiao, Erazo, Reef, Lovelock and Bowman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Valerie Hsiao, dmhzaWFvQHVjc2QuZWR1
†ORCID: Natalia G. Erazo, https://orcid.org/0000-0002-7498-0860
Ruth Reef, https://orcid.org/0000-0001-6639-520X
Catherine Lovelock, https://orcid.org/0000-0002-2219-6855
Jeff Bowman, https://orcid.org/0000-0002-8811-6280
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.