 Amrita Chakraborty
Amrita Chakraborty Arnab Purohit
Arnab Purohit Arunabha Khara
Arunabha Khara Roman Modlinger
Roman Modlinger Amit Roy
Amit Roy- 1Forest Microbiome Team, EVA 4.0 Unit, Faculty of Forestry and Wood Sciences, Czech University of Life Sciences Prague, Prague, Czechia
- 2Forest Molecular Entomology Lab, Faculty of Forestry and Wood Sciences, Czech University of Life Sciences Prague, Prague, Czechia
- 3Excellent Team for Mitigation (ETM), Faculty of Forestry and Wood Sciences, Czech University of Life Sciences Prague, Prague, Czechia
Ips typographus L. (Coleoptera: Curculionidae) is one of the serious pests causing mass destruction of European spruce forests, with a substantial economic impact. Symbiotic microbes associated with bark beetles often play a definitive role in accomplishing their physiological and ecological functions by detoxifying chemicals, inhibiting pathogens, and offering nutrients. Although a few research works explored the microbes associated with I. typographus, much is yet to be studied to understand their adaptive ecology as holobionts comprehensively. The present study examined bacterial and fungal communities of larvae, adults, and feeding galleries from Austria and Czech Republic using high-throughput sequencing that elucidated the influence of geographic location, host, and life stage on the microbial assemblage in Eurasian spruce bark beetle, I. typographus. The most abundant bacterial genera in I. typographus included Pseudoxanthomonas, Spiroplasma, Pseudomonas, Cellulomonas, Tyzzerella, Bacillus, and Mycobacterium. Alternatively, Wickerhamomyces, Nakazawaea, Aspergillus, Ophiostoma, Cryptococcus, Rhexographium, Yamadazyma, Talaromyces, and Kuraishia were highly dominant fungal genera. Significant differences in bacterial and fungal community richness and diversity were detected among the tested samples. LEfSe analysis revealed species-specific bacterial and fungal biomarkers from different locations in the larvae, adults, and feeding gallery samples. PICRUSt2 and FUNGuild analysis documented putative roles of the bacterial and fungal communities in beetle holobiont and provided a foundation for downstream functional analyses. The current findings further enhanced our understanding of bark beetles as holobionts.
1. Introduction
Eurasian spruce bark beetle (ESBB) Ips typographus (L.) (Coleoptera: Curculionidae: Scolytinae), an essential species in the forest ecosystem, plays a crucial role in forest restoration by decomposition of dead or dying wood and nutrient recycling (Raffa et al., 2015; Hlásny et al., 2019; Powell et al., 2021). ESBB adults attack weakened, dead, or fallen spruce trees as secondary pests during suitable environmental conditions. Bark beetle-induced tree mortality begins with a loss of capacity to move water and nutrients (Mikkelson et al., 2013). Global climatic changes are reshaping forest ecosystems, where aggressive bark beetles like ESBB are causing massive destruction of healthy trees through pheromone co-ordinated mass attacks resulting in landscape-level mortality and enormous economic losses (Hlásny et al., 2021). In Europe, the average annual death of 2–14 million m3 of spruce by ESBB escalated massively to 118 million m3 in 2019 due to dry and hot summers (Seidl et al., 2014; Ebner, 2020).
Conifers are equipped with allelochemicals like terpenes and phenolic compounds to combat bark beetle attacks (Keeling and Bohlmann, 2006; Wang et al., 2023). Bark beetles successfully evade plant defense by metabolic degradation, modification or enhanced excretion of the toxic chemicals, and modified feeding behavior (Sauvard, 2007; Berasategui et al., 2017; Tanin et al., 2021). Bark beetles encounter another problem as they develop on the inner bark, which is nutritionally poor and recalcitrant. Bark and sapwood are rich in lignin, cellulose, and hemicellulose, but these biopolymers are not accessible to bark beetles as nutrients. Feeding on conifer tissues becomes more difficult due to low concentrations of nitrogen, phosphorus, vitamins, and other nutrients (Franceschi et al., 2005; Wang et al., 2023).
Symbiotic microbes associated with bark beetles play a definitive role in fulfilling their physiological and ecological functions to thrive in a hostile environment (Liu et al., 2022). For instance, several studies demonstrated the role of mountain pine beetle-associated bacteria Rahnella, Serratia, and Pseudomonas and ophiostomatoid fungi Grosmannia clavigera in monoterpene degradation (Wang et al., 2014; Zaman et al., 2023). Erwinia typographi sp. isolated from ESBB was resistant to acyclic monoterpene myrcene (Skrodenytė-Arbačiauskienė et al., 2012). Similarly, ESBB-associated fungi (Grosmannia penicillate, Grosmannia europhioides, and Endoconidiophora polonica syn. Ceratocystis polonica) are involved in the degradation of phenolic compounds stilbenes and flavonoids (Hammerbacher et al., 2013; Zhao et al., 2019b). Symbiotic microbes play a critical role in the nutrition of bark beetles by hydrolyzing complex biopolymers to simple sugars and providing assimilable nitrogen, phosphorus, amino acids, vitamins, and sterols (García-Fraile, 2018; Peral-Aranega et al., 2020). Bacteria and fungi associated with bark beetles synthesize necessary enzymes like cellulases, hemicellulases, xylanases, endoglucanases, peroxidases, and oxidases for hydrolysis complex starch- and non-starch plant polysaccharides to simple sugars (Valiev et al., 2009; Fabryová et al., 2018; Ceja-Navarro et al., 2019; Peral-Aranega et al., 2020). Aggregation and anti-aggregation pheromones produced by fungi and bacteria promote bark beetle colonization and reduce intraspecific competition (Xu et al., 2015; Zhao et al., 2015, 2019a; Netherer et al., 2021). Bark beetle symbionts also play a vital role in protecting bark beetle holobiont from antagonism. The role of different Pseudomonas isolates in entomopathogenic inhibition was demonstrated in various studies (Saati-Santamaría et al., 2018; Peral-Aranega et al., 2020). Leptographium abietinum, symbiotic fungi associated with spruce beetle, showed inhibition of entomopathogen Beauveria bassiana (Davis et al., 2019). Alternatively, symbiotic bacteria also benefit symbiotic fungi by promoting their growth, inhibiting antagonistic fungi, and reducing bark beetle-symbiotic fungi competition for nutrition (Adams et al., 2009; Zhou et al., 2016; Liu et al., 2020). The bacterial and fungal communities associated with the ESBB gut were recently identified using a metagenomic approach, and their potential role in bark beetle adaptation was predicted (Chakraborty et al., 2020a,b). Despite all those efforts, our current knowledge about the geographic location (including microhabitat, i.e., plant holobiont) and life stage influence on ESBB microbial assemblage is still limited and are yet to be conducted on broader geographic conditions (Moussa et al., 2023; Veselská et al., 2023).
It is already documented that diet, developmental stage, host phylogeny, and habitat environment influence insect gut microbial diversity (Yun et al., 2014). ESBB larval stage is the gregarious feeding stage, where the associated microbial communities primarily function in nutrient acquisition and detoxification of plant terpenes (Peral-Aranega et al., 2020). The adults have additional duties, including host finding, establishing new galleries, and reproduction. Such differing ecological roles can be associated with ESBB symbiont choice and maintenance throughout the developmental stages, including metamorphosis (Kaltenpoth, 2020). Notably, phloem consumption also differs between different life stages of beetles, and the nutritional quality of the host tissues varies after initial colonization, which could also influence microbial community structure. Moreover, different geographic locations can influence the bark beetle-microbial interactions due to the difference in host tree holobiont and environment (Adams et al., 2010; Liu et al., 2019; Moussa et al., 2023). We hypothesize that drivers such as bark beetle life stage and geo-location concurrently influence the ESBB microbial associations, and life stage, which undergoes metamorphosis, have a more predominant role. To test our hypothesis, we analyzed bacterial and fungal communities of ESBB larvae, adults, and galleries from Austria and Czech Republic to elucidate the impacts of geo-location and the beetle life stage on microbial assemblage. We successfully provided the first meticulous report on ESBB microbial associations under the influence of two crucial drivers (i.e., life stage and geo-location) together. Distinct microbial associations were observed in different life stages of ESBB, suggesting beetle life stage and physiology is the key determining factor for fine-tuning the microbial associations and their metabolic role in bark beetle holobiont. This study further expands our knowledge of ESBB holobiont, feeding-gallery microbiome and sheds light on the microbial acquisition and core microbiome during the complex life cycle of bark beetles under geographically isolated forest ecosystems.
2. Materials and methods
2.1. Sample collection
ESBB adults, larvae, their galleries, and control wood samples were obtained from two sites in Austria and Czech Republic, respectively. The samples were collected in June 2020. The samples from Czech Republic (CZ) include uninfested phloem wood as control (CZ.CW), gallery wood (CZ.GW), ESBB larvae (CZ.L), and whole-body ESBB adults (CZ.WB) collected from Rouchovany (49°04′08.0″ N 16°06′15.4″ E, 366 m a.s.l) a State Forest Enterprise, public forest, regular forest management, warm and drought zone. From Austria (AT), the samples were gallery wood (AT.GW), ESBB larvae (AT.L), and whole body of ESBB adults (AT.WB) collected from Stockerau (48° 22′ 59.99′′ N 16° 13′ 0.01′′ E, 176 m a.s.l). In both places, multiple bark beetle-infested logs (dbh ∼20 cm) from different spruce trees (6–8 trees) were collected and kept separately and brought back to the laboratory. The logs were kept overnight at 4°C until the beetles were harvested. Beetles and phloem wood samples were collected from the logs on the same or the next day of collection on sterile benches using sterile tools (i.e., sterilized tweezers) and placed into sterile 1.5 mL microcentrifuge tubes to minimize the chances of cross-contamination between different brood galleries. Unfortunately, we failed to obtain pure control wood from Austria. Live adults (sclerotized adults), larvae (3rd instar), and brood gallery wood (infested pigmented phloem) were carefully and separately sampled from 6 independent brood galleries from infested logs under sterile laboratory conditions, following the standard protocol comparable to the published one (Hulcr et al., 2022). The control wood (intact phloem) samples from Czech Republic were collected from the same spruce tree away from the beetle galleries, where there was no apparent bark beetle infestation. Parasitized galleries or beetles, including suspicious ones, were excluded from the current study. All the samples (beetles and wood) were shock frozen in liquid nitrogen for future nucleic acid extraction. Twelve individual beetle samples (larvae or adult) were randomly collected from infested logs obtained from a single infested tree to represent one biological replicate, and a total of six replications were used in the metagenomic analysis from 6 independent galleries belonging to different spruce trees per location. Similarly, each wood sample replicate comprised six phloem pieces (0.5 by 1 cm sections) collected and pooled from the same infested or control logs belonging to a single spruce tree. A total of six wood sample replicates were also taken from different spruce trees for metagenomics sequencing. The beetle adults and larvae were taxonomically identified based on published literature by Pfeffer (1955) and Nunberg (1981).
2.2. DNA extraction, amplification, and sequencing
Bark beetle samples were randomly selected and disinfected by first rinsing the beetles (adults and larvae) with 70% ethanol for 1 min and then washed in sterile water for 1 min. This step was repeated thrice to remove any surface contaminants. Each replicate of the adult whole body (2 beetles per extraction, 6 independent extractions per brood gallery), larvae (2 larvae per extraction, 6 independent extractions per brood gallery), gallery and control wood (∼ 120 mg per replicate) were homogenized and subjected to microbial DNA extraction using the Nucleospin soil DNA purification kit (Macherey Nagel, Germany) as outlined by the manufacturer. A total of six replicates for each sample were considered. It is worth mentioning that each beetle replicate contained an equimolar amount of pooled extracted DNA from six independent beetle DNA extractions from a single brood collection. The pooling of multiple extractions was adopted to provide a good representation of beetles in each replicate and minimize the higher sample sequencing requirements. Following DNA isolation, the samples were purified using a DNeasy Powerclean Pro cleanup kit (Qiagen, Germany) to remove PCR inhibitors, if any. The purified DNA quantity and quality were analyzed on Qubit Fluorometer (ThermoFisher Scientific, Germany) using Qubit High sensitivity dsDNA assay kit (ThermoFisher Scientific, Germany) and electrophoresed on 1% agarose gel, respectively. DNA was diluted (1 ng/μl) for amplicon sequencing in sterile water. The V3-V4 region of the bacterial 16S rRNA gene and fungal ITS2 region were amplified using specific primers [341F (5′-CCTAYGGGRBGCASCAG-3′)/806R (5′-GGACTACNNGGGTATCTAAT-3′) and ITS3 (5′-GCATCGATG AAGAACGCAGC-3′)/ITS4 (5′-TCCTCCGCTTATTGATATGC -3′), respectively] tagged with specific barcodes using Phusion High-Fidelity PCR Master Mix (New England Biolabs) for the PCR reactions (White et al., 1990; Klindworth et al., 2013). No template control was used to nullify the probability of contamination, similar to our previous studies (Chakraborty et al., 2020a,b). Equal amounts of amplicons were run on 2% agarose gel, and the bands were gel purified (Qiagen Gel Extraction Kit, Germany) before the library preparation. NEBNext Ultra II DNA Library Pre-Kit (Illumina) was used to construct the sequencing library. The constructed library was quantified on Qubit Fluorometer and qPCR. Agilent Bioanalyzer 2100 system was used to check the quality of the library. The Illumina NovaSeq 6000 sequencing platform generated 250bp paired-end reads from the library.
2.3. Sequencing data analysis
2.3.1. Data processing and species annotation
Bioinformatic analysis of the amplicon sequencing data was conducted using QIIME2 -2022.2 (Bolyen et al., 2019). Based on sample-specific barcodes, pair-end reads were assigned to the respective samples. Barcodes and primer sequences were removed, and pair-end reads were assembled using FLASH (V1.2.11)1 (Magoč and Salzberg, 2011). The “fastp” software was used to obtain high-quality clean tags after a quality check of the assembled raw tags. Chimera was detected and removed by VSEARCH software (Rognes et al., 2016), and effective tags were obtained for downstream bioinformatic analysis. Sequences with less than five read abundance were filtered to get the final amplicon sequence variants (ASVs) and the feature table using the DADA2 module in QIIME2 (Callahan et al., 2016). Taxa annotation of the ASVs was done with SILVA (Release v138.1)2 (Quast et al., 2012) and UNITE (Nilsson et al., 2019) databases for bacterial and fungal sequences, respectively, using Classify-sklearn module (Bokulich et al., 2018) in QIIME2 (Bolyen et al., 2019).
2.3.2. α-diversity
The microbial community richness and diversity within a sample were assessed using α-diversity indices. Sequence depth (Good’s coverage) (Good, 1953), observed species, microbial richness (Chao1), and Shannon and Simpson diversity indices (Good, 1953; Magurran, 1988) were estimated in QIIME2 and represented using R software ver 2.15.3 (R Core Team, 2013). The Kruskal-Wallis pairwise test evaluated the statistical differences between group comparisons for each α-diversity indices.
2.3.3. β-diversity
The unweighted UniFrac distance calculated by QIIME2 was used to estimate the variation in microbial diversity between the samples (Lozupone et al., 2011). Non-metric multidimensional scaling (NMDS) analysis was done based on unweighted UniFrac distances using vegan package in R software (Oksanen, 2011). Significant differences between communities of different samples were assessed using ADONIS and ANOSIM analyses (Clarke, 1993; Anderson, 2001) in the QIIME2 after 200 permutations. The ADONIS function is a non-parametric multivariate variance test based on the unweighted UniFrac distance matrix that uses permutation tests to determine the differences between sample groups and estimate their significance (Stat et al., 2013). Based on the unweighted UniFrac distance matrix, ANOSIM analysis determines whether the variance between groups is significantly greater than the variation within groups (Chapman and Underwood, 1999). Multiple hypothesis tests for sparsely sampled features and false discovery rate (FDR) were used in MetaStat analysis (Paulson et al., 2011) to assess the significant variations in observed species abundance among groups. Furthermore, T-test was used to evaluate whether there were significant differences in microbial species abundance groups (p-value < 0.05) (D’Argenio et al., 2014). Linear discriminant analysis Effect Size (LEfSe) analysis,3 where the linear discriminant analysis scores (LDA score [log10] > 4) were set as a threshold, was performed to determine the high-dimensional biomarkers to distinguish between tested samples (Segata et al., 2011). The putative functional spectrum of identified bacteriome was deduced using PICRUSt2 (Phylogenetic Investigation of Communities by Reconstruction of Unobserved Stats 2) software (Douglas et al., 2020). The metabolic function of the bacteria group was predicted by mapping the composition of the bacteria group obtained through sequencing to the database. The functional profiling based on different databases such as KEGG, METAcyc, COG, EC, PFAM, and TIGRFAM domain databases predicted different putative functions attributed to the bacterial communities in the samples. The abundance of functional annotation from different databases was used to generate heatmaps. Similarly, the putative ecological function of identified fungal communities was determined using the FUNGuild annotation tool4 that could taxonomically parse fungal ASVs by ecological or functional guild (Nguyen et al., 2016).
3. Results
3.1. Microbiome structure
3.1.1. Sequencing statistics
Illumina paired-end sequencing of larval and adult stages of ESBB collected from the Czech Republic and Austria (CZ.L, CZ.WB, AT.L, AT.WB) along with the control (CZ.CW) and gallery wood (CZ.GW, AT.GW) samples generated a total of 4686434 bacterial 16S rRNA reads, and 3363983 fungal ITS2 reads, respectively (Supplementary Material 1). From these, 3725513 bacterial and 3247655 fungal read counts were obtained after quality control with a Phred Quality score > 30 for further downstream bioinformatic analyses. Good’s coverage [>99%] and rarefaction analysis for all samples suggested sufficient sequencing depth (Table 1 and Supplementary Figure 1).
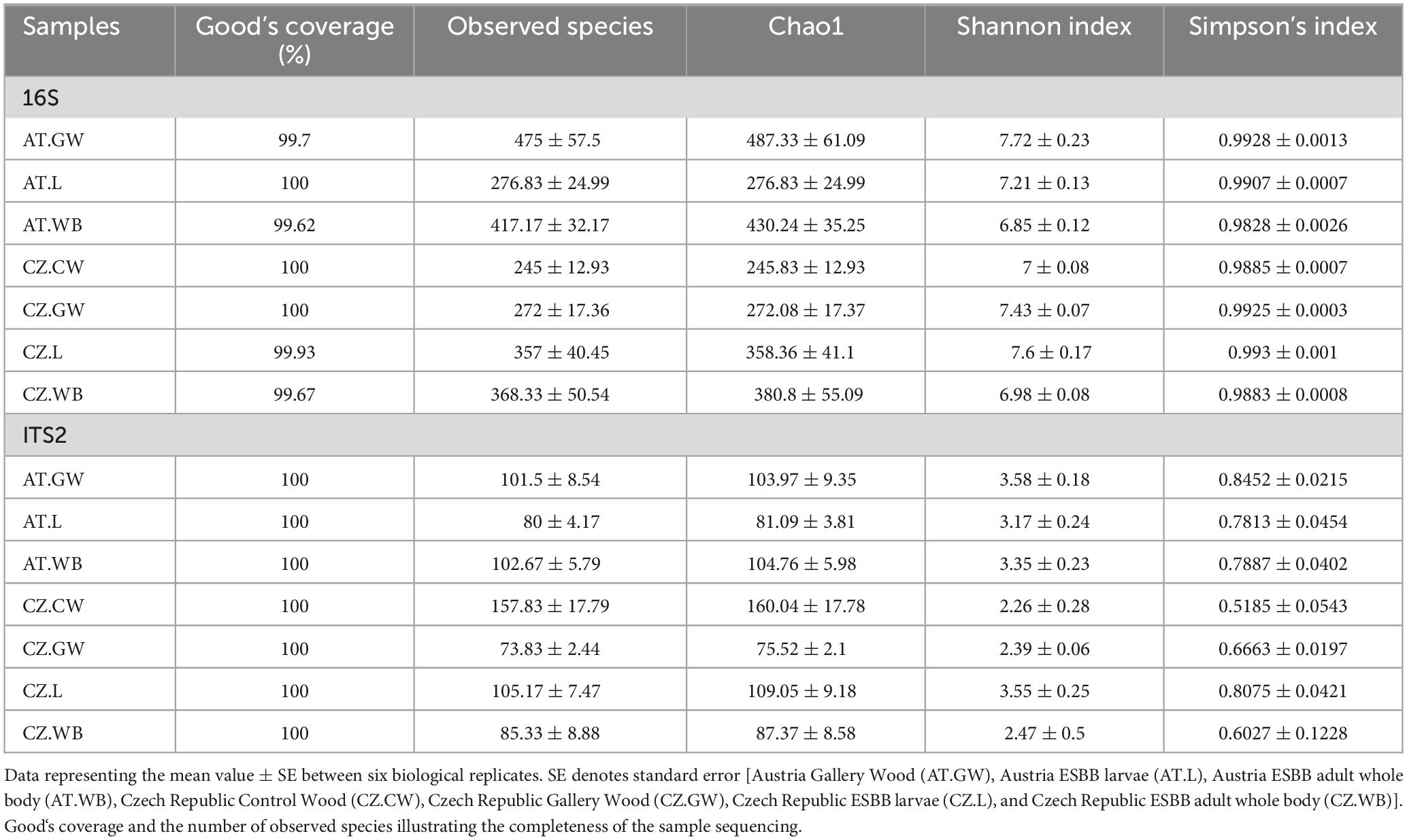
Table 1. Alpha diversity indices representing the bacterial and fungal community richness and diversity in ESBB life stages and their control and gallery wood.
3.1.2. Microbial community structure
3.1.2.1. Bacterial abundance
The bacterial 16S rRNA sequences were clustered into 12256 amplicon sequence varients (ASVs) belonging to 59 classes based on 97% sequence homology (Supplementary Material 2). Among these, Gammaproteobacteria was prevalent in all the samples [AT.WB (11.9%), AT.L (31%), CZ.L (24%), CZ.WB (20.2%), AT.GW (21.6%), CZ.GW (34.1%), CZ.CW (7.2%)] followed by Actinobacteria, Bacilli, Alphaproteobacteria, Clostridia, and Bacteroidia (Figure 1A and Supplementary Material 3). The heatmap depicting the 35 most abundant bacterial genera in ESBB and wood samples in both sites revealed the dominance of Roseomonas, Patulibacter, Novosphingomonas, Brevundimonas, and Microbacterium in CZ.L, while Acinetobacter, Chryseobacterium, Taibaiella, Desulfitobacterium, Tyzzerella, Bacillus, and Spiroplasma were prevalent in CZ.WB. On the contrary, AT.WB showed a high abundance of Streptomycetes, Ochrobactrum, Amycolatopsis, Saccharopolyspora, Candidatus_Lariskella, Mycobacterium, Cellulomonas (Figure 1B and Supplementary Material 4).
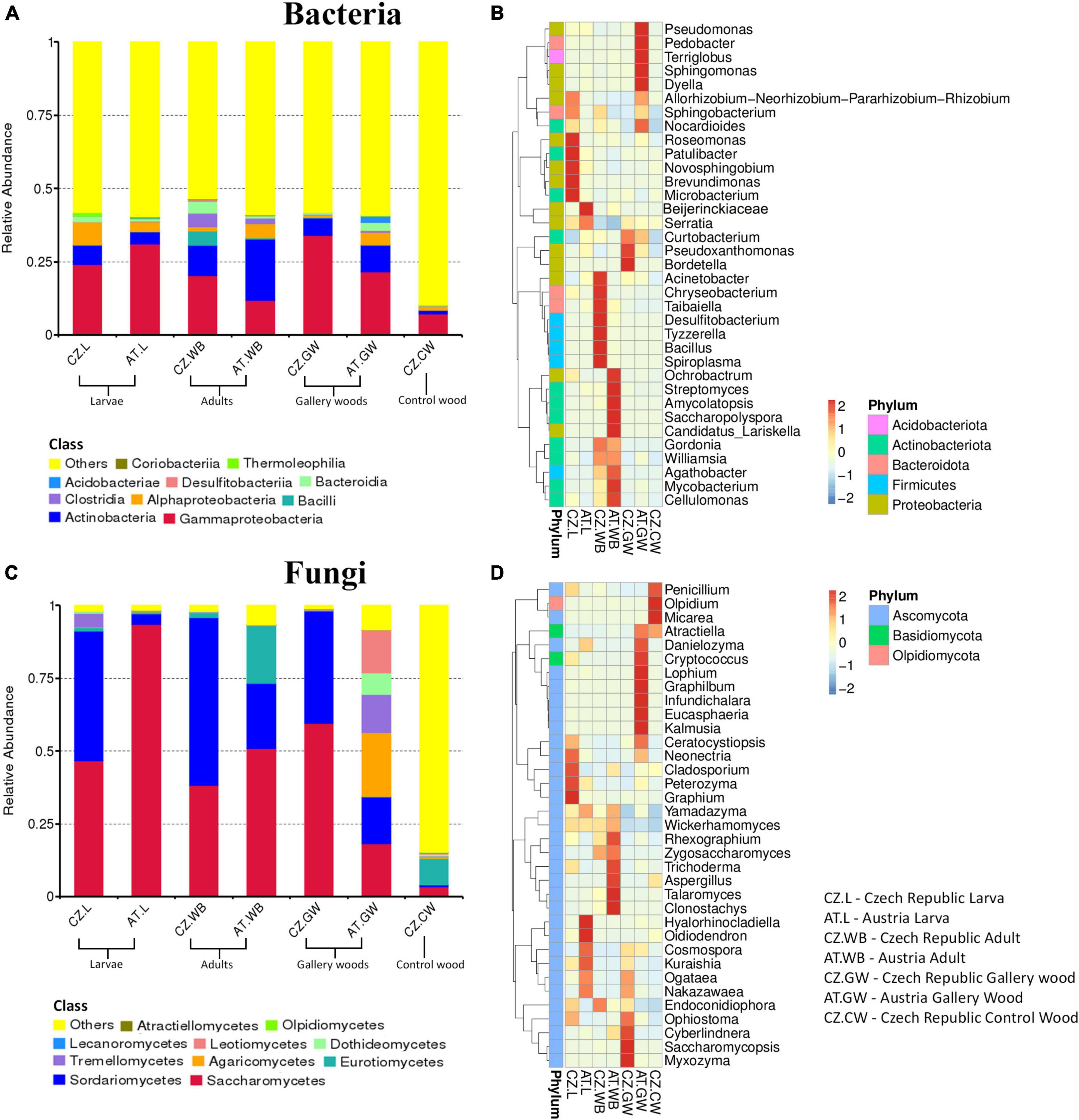
Figure 1. Microbial diversity in ESBB larvae, adults, gallery wood, and control wood samples. Representative bar graphs depict the relative abundance of the bacterial (A) and fungal (C) communities at the class level (top 10). “Others” represents the total relative abundance of the rest of the classes. Heatmaps illustrate the abundance of 35 abundant bacterial (B) and fungal (D) genera among the samples. The color gradient shows relative ASVs abundance for each sample, where darker and lighter colors denote higher and lower abundance, respectively [Austria Gallery Wood (AT.GW), Austria ESBB larvae (AT.L), Austria ESBB adult whole body (AT.WB), Czech Republic Control Wood (CZ.CW), Czech Republic Gallery Wood (CZ.GW), Czech Republic ESBB larvae (CZ.L), and Czech Republic ESBB adult whole body (CZ.WB)].
3.1.2.2. Fungal abundance
Similarly, the fungal ITS2 sequences were grouped into 1120 ASVs at a 97% similarity cut-off, belonging to 21 classes (Supplementary Material 2). Saccharomycetes and Sordariomycetes were the abundant fungal classes in all the ESBB samples and gallery wood, except for the larval samples from Austria (AT.L), where only Saccharomycetes (93.55%) was the most abundant class (Figure 1C and Supplementary Material 3). The relative abundance of Eurotiomycetes (19.9%) was higher in AT.WB compared to the rest of the beetle samples. The AT.GW showed a high relative abundance of Agaricomycetes (21.94%), Leotiomycetes (14.8%), Tremellomycetes (13.1%), and Dothideomycetes (7.3%) to rest of the samples (Figure 1C and Supplementary Material 3). The heatmap representing 35 most abundant fungal genera showed a prevalence of Neonectria, Cladosporium, Peterozyma, and Graphium in CZ.L, while AT.L showed an abundance of Hyalorhinocladiella, Cosmospora, Kuraishia, Ogataea, Nakazawaea (Figure 1D and Supplementary Material 4). Similarly, Rhexographium, Trichoderma, Aspergillus, Talaromyces, and Clonostachys were highly abundant in AT.WB (Figure 1D). Comparing the gallery woods, CZ.GW showed a high abundance of Ophiostoma, Cyberlindnera, Saccharomycopsis, and Myxozyma, while Danielozyma, Cryptococcus, Lophium, Graphilbum, and Kalmusia were abundant in AT.GW (Figure 1D and Supplementary Material 4).
3.2. α and β-diversity
3.2.1. Bacterial diversity
A significantly higher bacterial richness was observed in ESBB adults (AT.WB 430.24 ± 35.25) compared to the larval samples (AT.L 276.83 ± 24.99) from Austria (P < 0.01). However, no significant difference in the bacterial richness (P < 0.05) was detected between the two life stages of ESBB from Czechia (Chao1, CZ.L 358.36 ± 41, CZ.WB 380.8 ± 55.09) (Supplementary Figure 2A and Supplementary Material 5). Similarly, the same life stages of ESBB from two different locations did not show substantial variation in bacterial richness. Nevertheless, the gallery wood from two sites (Chao1, AT.GW 487.33 ± 61.09, CZ.GW 272.08 ± 17.37) revealed a significant variation in bacterial richness between them (Supplementary Figure 2A). The bacterial diversity differed significantly between the two life stages of ESBB, where the diversity in larvae was higher than in the adults (Supplementary Figures 2B, C). Furthermore, the bacterial diversity on the wood increased upon ESBB feeding (CZ.GW Shannon Index 7.43 ± 0.07, Simpson’s Index 0.9925 ± 0.0003; CZ.CW Shannon Index- 7 ± 0.08, Simpson’s Index- 0.9885 ± 0.0007). AT.GW (Shannon Index- 7.72 ± 0.23, Simpson’s Index- 0.9928 ± 0.0013) displayed a more diverse bacterial community than CZ.GW (Shannon Index- 7.43 ± 0.07, Simpson’s Index- 0.9925 ± 0.0003) (Supplementary Material 5). Furthermore, β- diversity represented by the NMDS plot (unweighted) clustered the respective samples from two locations in pairs. For instance, AT.L and CZ.L were grouped and separated from AT.WB and CZ.WB cluster. The gallery wood (CZ.GW) and the control wood (CZ.CW) from the Czech Republic were clustered separately (Figure 2A).
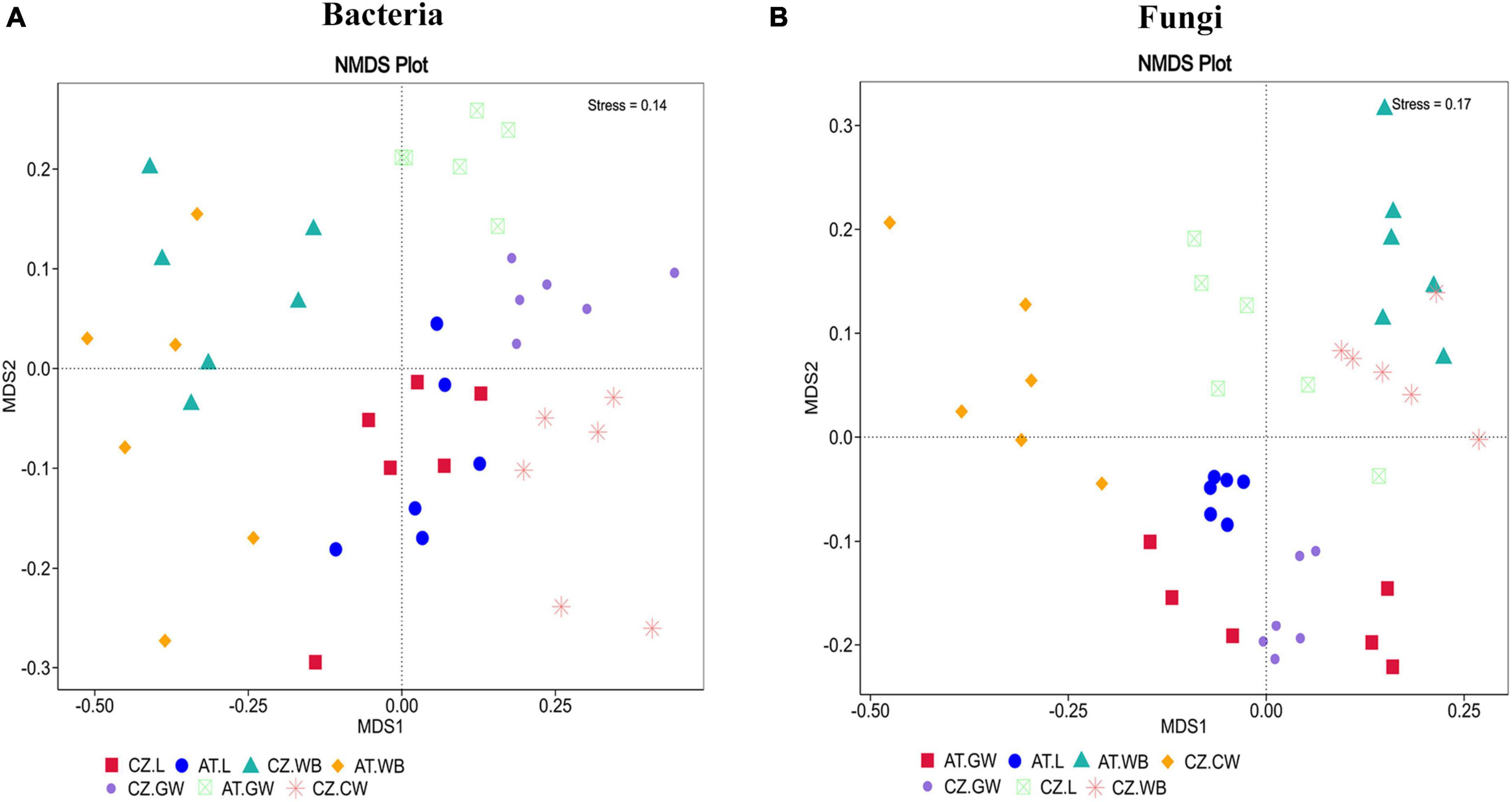
Figure 2. Non-metric multidimensional scaling analysis (NMDS) showing the variation in the bacterial (A) and fungal (B) communities in the ESBB larvae, adults, gallery wood, and control wood samples. The data points in the same color denote the same samples. Different symbols represent different samples. Larvae, adults, and gallery wood samples from two locations are clustered in pairs. In contrast, the control wood sample is clustered separately [Austria Gallery Wood (AT.GW), Austria Larvae (AT.L), Austria Adult Whole Body (AT.WB), Czech Republic Control Wood (CZ.CW), Czech Republic Gallery Wood (CZ.GW), Czech Republic Larvae (CZ.L), and Czech Republic Adult Whole Body (CZ.WB)].
3.2.2. Fungal diversity
The fungal richness in the gallery wood decreased (CZ.GW Chao1 75.52 ± 2.1) compared to the control wood (CZ.CW Chao1 160.04 ± 17.78) (Supplementary Figures 2D–F). Comparing the gallery wood from two different locations, AT.GW (Chao1- 103.97 ± 9.35, Shannon Index- 3.58 ± 0.18, Simpson’s Index-0.8452 ± 0.0215) showed higher fungal richness and diversity than CZ.GW (Chao1- 75.52 ± 2.1, Shannon Index- 2.39 ± 0.06, Simpson’s Index- 0.6663 ± 0.0197) (Supplementary Figure 2D and Supplementary Material 5). No significant difference in the fungal community richness and diversity was observed between the two life stages of ESBB collected from the two locations. Similar to the bacterial diversity, the adult beetle samples from the two locations were grouped together in the fungal NMDS plot (Figure 2B). However, the fungal communities in the larval samples varied between the two locations. Moreover, the gallery wood from the two locations (AT.GW and CZ.GW) was clustered together and distinctly separated from the control wood (CZ.CW) (Figure 2B).
Furthermore, significant differences in the bacterial and fungal communities due to different life stages (AT.L vs AT.WB, CZ.L vs CZ.WB), different geographic locations (AT.L vs CZ.L, AT.WB vs CZ.WB and AT.GW vs CZ.GW), and bark beetle feeding (CZ.CW vs CZ.GW) were demonstrated by ANOSIM analysis (Table 2). Similarly, ADONIS analysis showed significant variations in the overall microbiome (Supplementary Table 1).
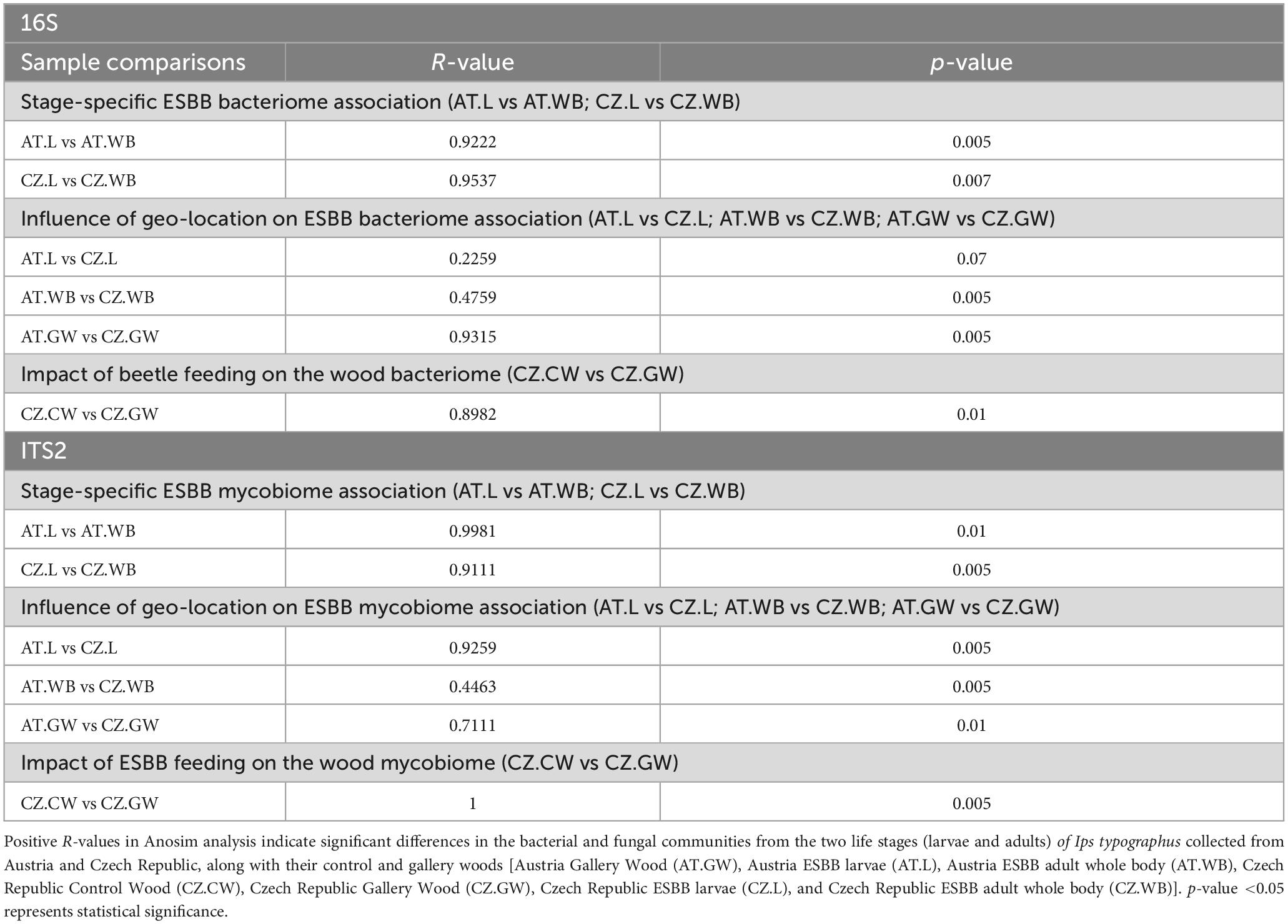
Table 2. ANOSIM analysis [Unweighted] representing the extent of variation in the bacterial and fungal communities present in different samples.
3.3. Beetle-specific associations and influence of geographic location
3.3.1. Life stage-specific microbial association
3.3.1.1. Bacterial assemblage in larvae and adult ESBB
ESBB larvae and adults collected from Austria shared 21 ASVs, while 1383 and 2440 unique ASVs were observed in AT.L and AT.WB respectively (Figure 3A). ASVs shared between AT.L and AT.WB includes Pseudoxanthomonas, Mycobacterium, and Nocardioides (Supplementary Material 6). MetaStat and t-test analyses revealed a significantly higher abundance (P < 0.05) of Mycobacterium, Gordonia, Streptomyces, Williamsia, and Amycolatopsis in AT.WB, while Serratia, Patulibacter, Sphingobacterium, and Novosphingobium were more abundant in AT.L (Table 3 and Supplementary Table 2). Furthermore, LEfSe analysis showed the presence of Mycobacteriace, Nocardiaceae, Streptomycetales, Cornybacteriales, Actinobacteria as biomarkers of AT.WB, while Erwiniaceae, Yersiniaceae, Enterobacterales, and Gammaproteobacteria were included as biomarkers of AT.L (Figure 3B and Supplementary Figure 3A).
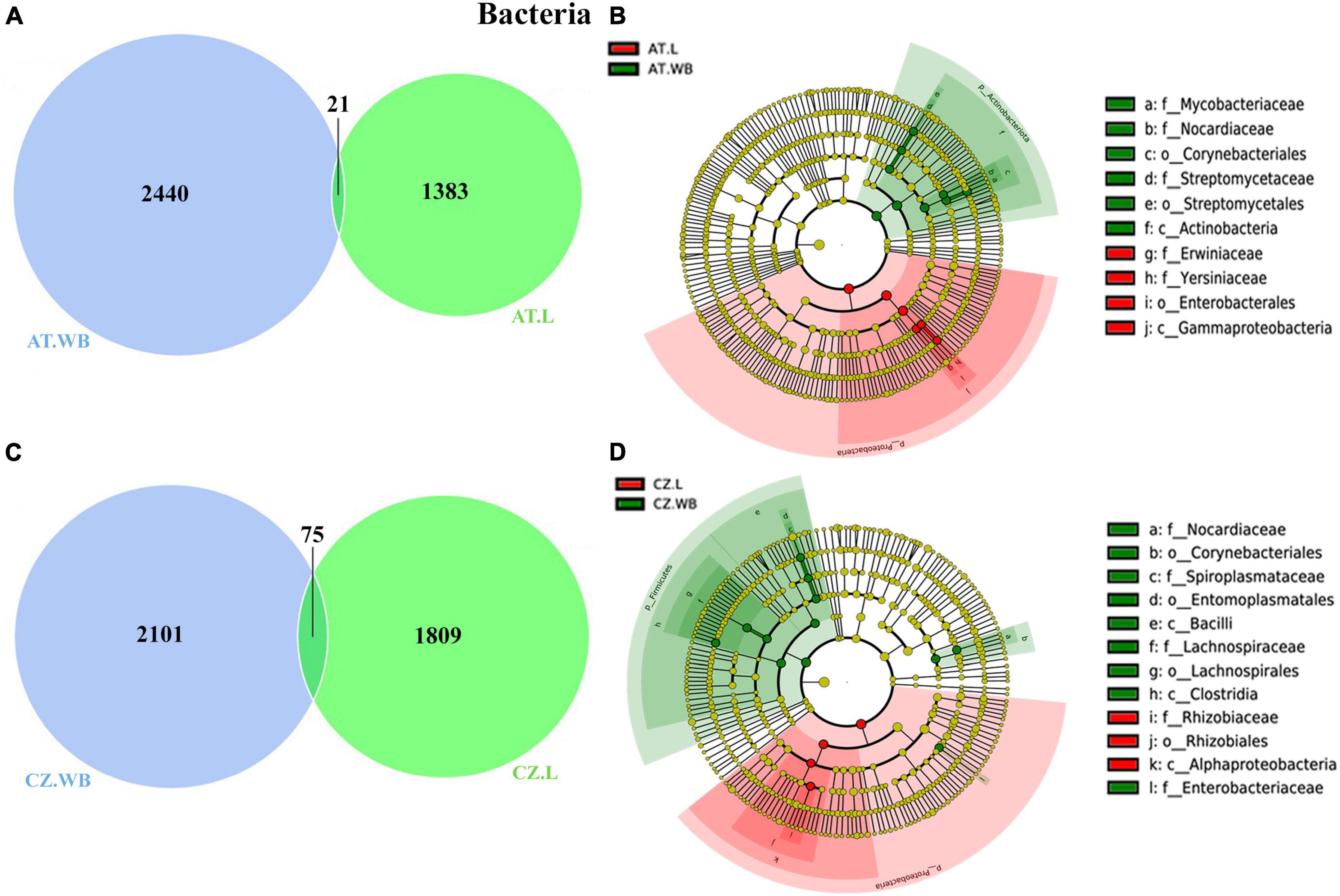
Figure 3. Life stage-specific bacterial association in ESBB. Venn diagrams illustrating bacterial ASVs distribution between adult and larval samples from Austria (A) and Czech Republic (C). The number of shared or unique OTUs is denoted. LEfSe analysis displaying the significantly different bacterial biomarkers between larvae and adults from Austria (B) and Czech Republic (D). Each circle represents a distinct taxon at the respective taxonomic level. The color of the bacterial biomarkers corresponds with the color of the life stages. Yellowish-green circles denote non-significant bacterial species. Red and green nodes resemble bacterial taxon, highly contributing to the group. Letters above the circles represent bacterial biomarkers [Austria Larva (AT.L), Austria Adult Whole Body (AT.WB), Czech Republic Larvae (CZ.L), and Czech Republic Adult Whole Body (CZ.WB)].
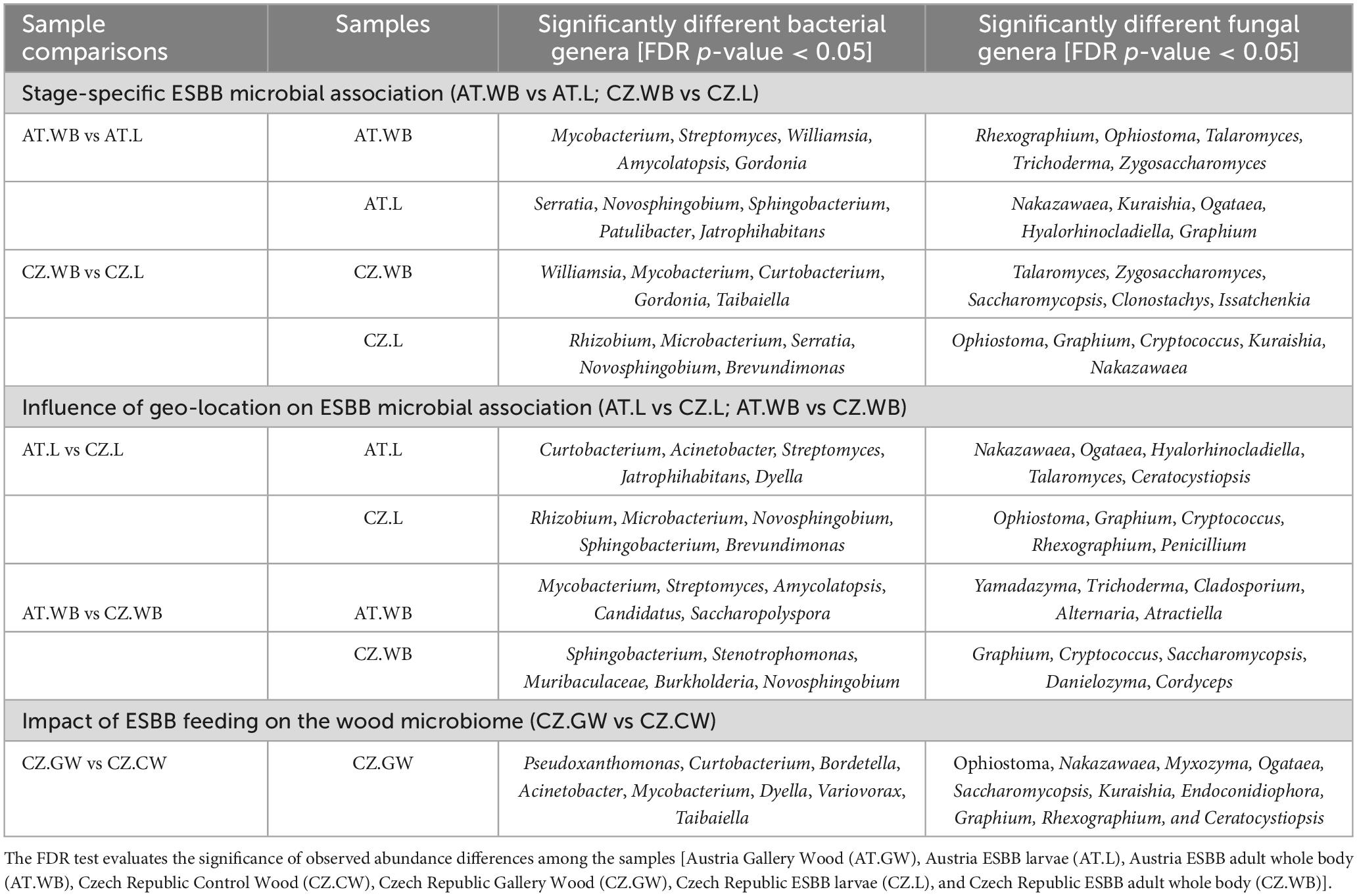
Table 3. Metastat analysis documenting the significant abundance of bacterial and fungal species within the two life stages of Ips typographus (larvae and adults) collected from Austria and Czech Republic, along with their control wood and gallery wood samples.
Similarly, 75 bacterial ASVs were common between CZ.WB and CZ.L, while 2101 and 1809 were unique ASVs, respectively (Figure 3C and Supplementary Material 6). MetaStat and t-test analyses demonstrated a significantly high abundance of Williamsia, Curtobacterium, Mycobacterium, Gordonia, and Taibaiella in CZ.WB, whereas Rhizobium, Microbacterium, Novosphingobium, Brevundimonas, Serratia, and Patulibacter were prevalent in CZ.L (Table 3 and Supplementary Table 2). Additionally, LEfSe analysis revealed the bacterial species belonging to Norcardiacea, Enterobacteriaceae, Entomoplasmatales, Corynebacteriales, Bacilli, Lachnospirales, and Clostridia as biomarkers for CZ.WB. While members of the class Alphaproteobacteria and order Rhizobiales were present as biomarkers in CZ.L (Figure 3D and Supplementary Figure 3B).
3.3.1.2. Fungal assemblage in larvae and adult ESBB
Like the ESBB bacteriome, the adult beetles and larvae from Austria comprised 65 common fungal ASVs, while 217 and 115 unique ASVs were observed, respectively (Figure 4A and Supplementary Material 6). The life stage-specific variation in the fungal communities demonstrated by MetaStat and t-test analyses (P < 0.05) revealed a significantly higher abundance of Rhexographium, Ophiostoma, Talaromyces, Cladosporium, Zygosaccharomyces, and Trichoderma in AT.WB, whereas Nakazawaea, Kuraishia, Ogataea, Graphium, and Hyalorhinocladiella were dominant in AT.L (Table 3 and Supplementary Table 2). Furthermore, LEfSe analysis demonstrated that the members of the classes, Eurotiomycetes and Sordariomycetes, including Aspergillus, Yamadazyma, Rhexographium, Ophiostoma, and Talaromyces, as fungal biomarkers in AT.WB (Figure 4B and Supplementary Figure 3C), while members of the class Saccharomycetes including yeasts like Ogataea, Yamadazyma, Kuraishia, and Nakazawaea, were biomarkers in AT.L.
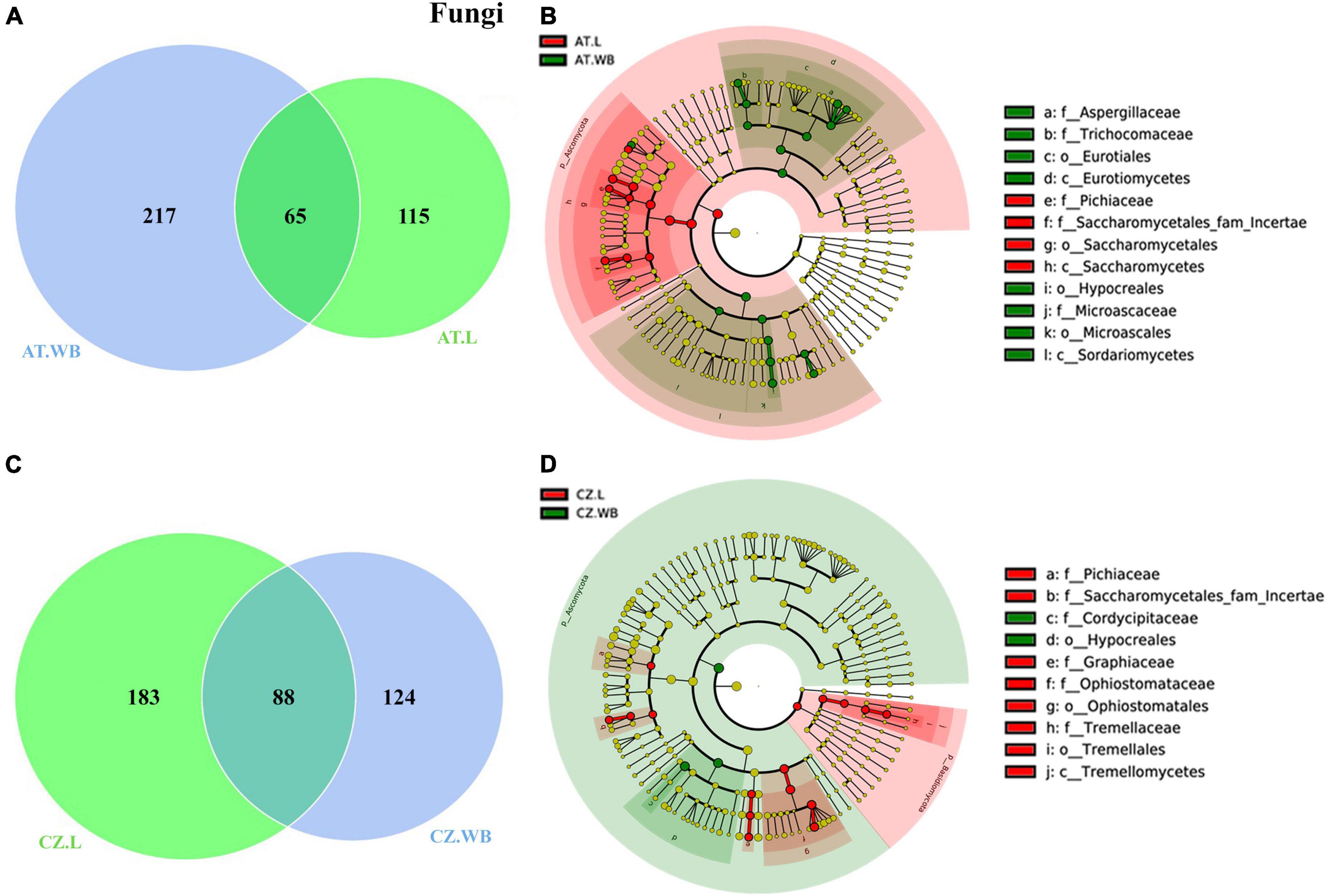
Figure 4. Life stage-specific fungal association in ESBB. Venn diagrams show fungal ASVs distribution between adult and larval samples from Austria (A) and Czech Republic (C). The number of shared or unique ASVs is denoted. LEfSe analysis displaying the significantly different fungal biomarkers between larvae and adults from Austria (B) and Czech Republic (D). Each circle represents a distinct taxon at the respective taxonomic level. The color of the fungal biomarkers corresponds with the color of the life stages. Yellowish-green circles denote non-significant fungal species. Red and green nodes resemble fungal taxon, highly contributing to the group. Letters above the circles represent fungal biomarkers [Austria Larva (AT.L), Austria Adult Whole Body (AT.WB), Czech Republic Larva (CZ.L), and Czech Republic Adult Whole Body (CZ.WB)].
Likewise, CZ.WB and CZ.L shared 88 common fungal ASVs, and 124 and 183 were unique ASVs, respectively (Figure 4C). The common fungal genera include Wickerhamomyces, Ophiostoma, Yamadazyma, Rhexographium, and Endoconidiophora (Supplementary Material 6). A high abundance of Ophiostoma, Graphium, Cryptococcus, Kuraishia, and Nakazawaea was observed in CZ.L, while Talaromyces was observed in CZ.WB (Table 3 and Supplementary Table 2). LEfSe analysis detected the members of the classes such as Saccharomycetes, Sordariomycetes, and Tremellomycetes, including fungal genera Kuraishia, Cryptococcus, Graphium, and Ophiostoma to be the biomarkers in CZ.L whereas class Sordariomycetes (order- Hypocreales) was abundant in CZ.WB (Figure 4D and Supplementary Figure 3D).
3.3.2. Geographic location impact on the microbial assemblage
3.3.2.1. Geographic location influencing bacterial assemblage
ESBB larvae from Austria and the Czech Republic (AT.L, CZ.L) comprised a consortium of 121 common bacterial ASVs (Figure 5A). The core bacterial community implies the bacterial population prevalent in time and space in or outside beetles, although their abundance may vary due to various factors. These shared ASVs belonged to 6 bacterial orders dominated by Enterobacterales and Xanthomonadales. The core bacteriome includes bacterial genera Pseudoxanthomonas, Pseudomonas, Serratia, Rhizobium, Brevundimonas, and Novosphingobium (Supplementary Material 7). Moreover, a total of 1283 and 1763 unique bacterial ASVs were observed in AT.L and CZ.L, respectively. MetaStat and t-test analyses revealed abundant (P < 0.05) bacterial genera, including Curtobacterium, Acinetobacter, and Streptomyces in AT.L, while Rhizobium, Microbacterium, Novosphingobium, and Sphingobacterium were prevalent in CZ.L (Table 3 and Supplementary Table 2). Furthermore, LEfSe analysis detected the class Alphaproteobacteria as a biomarker in CZ.L (Figure 5B and Supplementary Figure 4A).
Figure 5. Influence of geo-location on the bacterial association in ESBB. Venn diagrams showing the distribution of bacterial ASVs between larvae (A) and adult (C) ESBB from Austria and Czech Republic. The number of shared or unique ASVs is denoted. LEfSe analysis displaying the significantly different bacterial biomarkers between larvae (B) and adults (D) from Austria and Czech Republic. Each circle represents a distinct taxon at the respective taxonomic level. The color of the bacterial biomarkers corresponds with the color of the geographic locations. Yellowish-green circles denote non-significant bacterial species. Red and green nodes resemble bacterial taxon, highly contributing to the group. Letters above the circles represent bacterial biomarkers [Austria Larvae (AT.L), Austria Adult Whole Body (AT.WB), Czech Republic Larvae (CZ.L), and Czech Republic Adult Whole Body (CZ.WB)].
Comparably, adult beetles (AT.WB and CZ.WB) comprised 66 core bacterial ASVs with a high abundance of bacterial genera Cellulomonas, Mycobacterium, Pseudoxanthomonas, Gordonia, and Tyzerella (Figure 5C and Supplementary Material 7). Additionally, AT.WB and CZ.WB showed 2395 and 2110 unique ASVs, respectively. The significantly abundant bacterial genera Streptomyces, Amycolatopsis, Candidatus, and Saccharopolyspora were observed in AT.WB (Table 3 and Supplementary Table 2). While, Alphaproteobacteria, Actinobacteria, including members of Streptomycetaceae and Mycobacteriaceae were documented as the predominant biomarkers in AT.WB (Figure 5D and Supplementary Figure 4B). Similarly, Sphingobacterium, Serratia, and Muribaculaceae were prevalent in CZ.WB (Table 3 and Supplementary Table 2), with bacterial species belonging to Bacteroidia, Clostridia, Gammaproteobacteria, Lachnospiraceae, and Enterobacteriaceae as predominant biomarkers (Figure 5D and Supplementary Figure 4B).
The influence of geo-location on the gallery wood microbiome (AT.GW and CZ.GW) revealed the presence of 119 core bacterial ASVs, while 2536 and 1009 ASVs were unique to AT.GW and CZ.GW respectively (Supplementary Figure 5A and Supplementary Material 7). The order Xanthomonadales and the bacterial genus Pseudoxanthomonas dominated the core bacterial community. The bacterial biomarkers detected by LEfSe analysis include members from Acidobacteriae, Actinobacteria, Sphingobacteriaceae, Bacteroidia, Pseudomonadales in AT.GW, while Gammaproteobacteria, Enterobacterales, and Pseudoxanthomonas served as biomarkers of CZ.GW (Supplementary Figures 5B, C).
3.3.2.2. Geographic location influencing fungal assemblage
Comparing the mycobiome of ESBB larvae from both locations revealed 89 core ASVs, 91, and 182 unique ASVs to AT.L and CZ.L, respectively (Figure 6A). The core mycobiome explaining the conserved fungal communities consisted of 30 genera, including Wickerhamomyces, Ophiostoma, Yamadazyma, Kuraishia, and Nakazawaea (Supplementary Material 7). MetaStat analysis revealed a high abundance (p < 0.05) of Nakazawaea and Ogataea in AT.L, while Ophiostoma, Graphium, Cryptococcus, and Rhexographium were dominant in CZ.L (Table 3). Furthermore, the LDA score estimated in the LEfSe analysis reflected the presence of fungal biomarkers in CZ.WB belongs to Saccharomycetes, Nakazawaea, and Pichiaceae (Figure 6B and Supplementary Figure 4C). Several fungi belonging to the classes Sordariomycetes, and Tremellomycetes, including Ophiostoma, Graphium, Cryptococcus, Rhexographium, and Nectria served as biomarkers in CZ.L (Figure 6B and Supplementary Figure 4C).
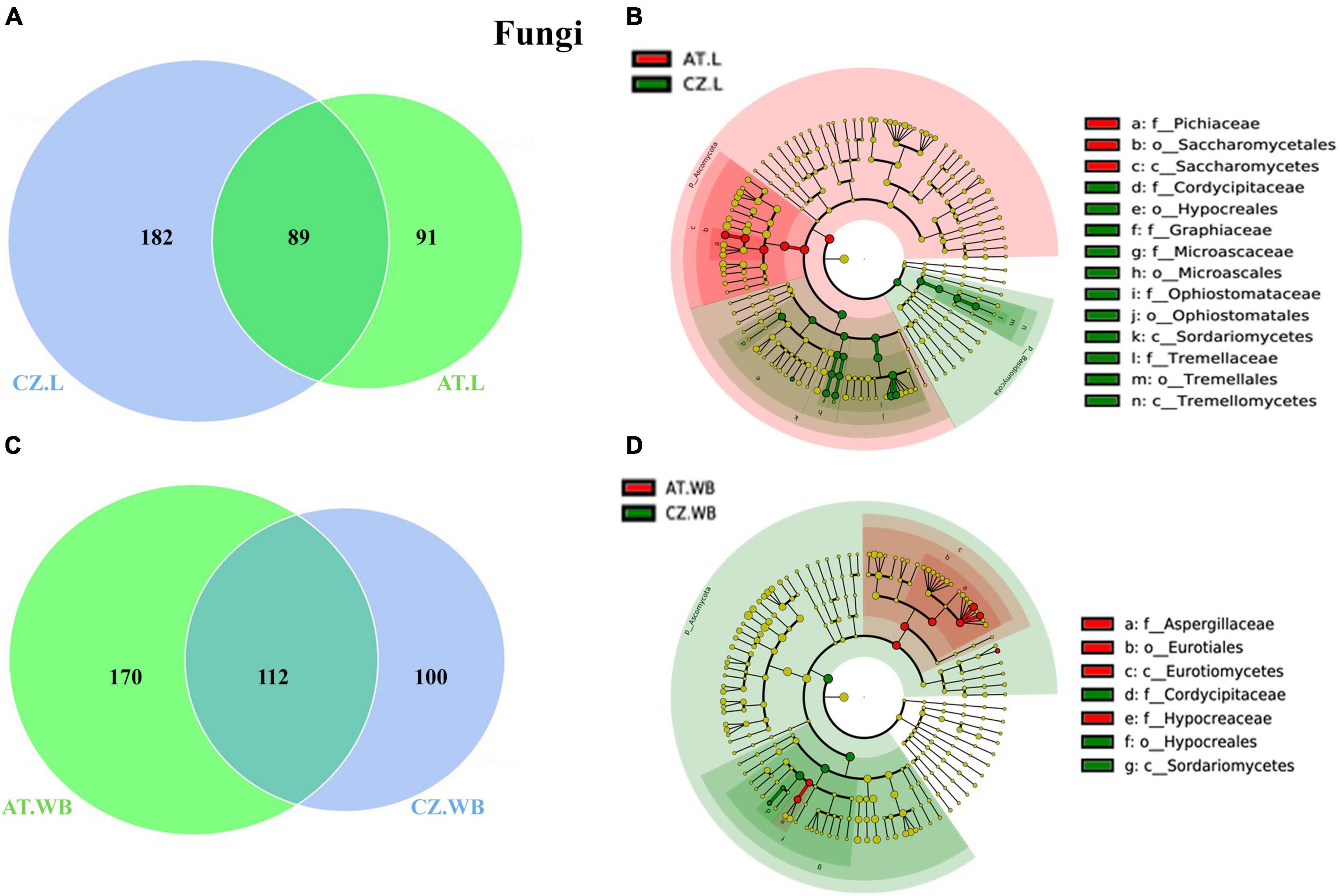
Figure 6. Influence of geo-location on the fungal association in ESBB. Venn diagrams showing the distribution of fungal ASVs between larvae (A) and adult (C) ESBB from Austria and Czech Republic. The number of shared or unique ASVs is denoted. LEfSe analysis displaying the significantly different fungal biomarkers between larvae (B) and adults (D) from Austria and Czech Republic. Each circle represents a distinct taxon at the respective taxonomic level. The color of the fungal biomarkers corresponds with the color of the geographic locations. Yellowish-green circles denote non-significant fungal species. Red and green nodes resemble fungal taxon, highly contributing to the group. Letters above the circles represent fungal biomarkers [Austria Larvae (AT.L), Austria Adult Whole Body (AT.WB), Czech Republic Larvae (CZ.L), and Czech Republic Adult Whole Body (CZ.WB)].
Similarly, adult beetles (AT.WB and CZ.WB) shared a consortium of 112 fungal ASVs, consisting of 24 families and 37 genera (Figure 6C and Supplementary Material 7), where Wickerhamomyces, Rhexographium, Aspergillus, Ophiostoma, Endoconidiophora, and Yamadazyma were dominant. Apart from the common ASVs, AT.WB and CZ.WB consisted of 170 and 100 unique fungal ASVs, respectively. The differentially abundant (p < 0.05) fungal genera included Yamadazyma, Trichoderma, Graphium, Cladosporium, Alternaria, Saccharomycopsis, Danielozyma, Cordyceps and Cryptococcus (Table 3). The LEfSe analysis detected the presence of Trichoderma, Alternaria, Aspergillus, and other members from Eurotiomycetes, Sordariomycetes, and Hypocreaceae, as biomarkers in AT.WB, while members of Sordariomycetes, Hypoocreales, and Cordycipitaceae served as biomarkers in CZ.WB (Figure 6D and Supplementary Figure 4D).
In addition, considering the gallery wood mycobiome, AT.GW and CZ.GW shared 87 fungal ASVs, whereas 183 and 87 unique ASVs, respectively (Supplementary Figure 5D and Supplementary Material 7). The conserved fungal communities consisted of 27 fungal families and 36 genera, including Ophiostoma, Nakazawaea, Cryptococcus, and Wickerhamomyces, as the most abundant fungal genera. LDA score determined Yamadazyma, Danielozyma, Nakazawaea, Kalmusia, and Cryptococcus as the biomarkers in AT.GW, whereas Ophiostoma, Nakazawaea, Ogataea, Saccharomycopsis, Kuraishia, and Cyberlindnera were the biomarkers in CZ.GW (Supplementary Figures 5E, F).
3.3.3. Host contribution to the microbial assemblage
3.3.3.1. Host contribution to bacterial assemblage
The gallery wood and the larval samples from Austria showed the presence of 83 core bacterial ASVs mainly predominated by Pseudoxanthomonas, Pseudomonas, Rhizobium, and Dyella genera (Figure 7A and Supplementary Material 8). MetaStat analysis revealed a higher abundance (p < 0.05) of Pseudomonas, Curtobacterium, Rhizobium, Terriglobus, and Dyella in AT.GW, whereas Novosphingobium, Streptomyces, Microbacterium, Jatrophihabitans, and Taibaiella were in higher abundance in AT.L samples (Table 3 and Supplementary Table 2). In comparison, adult beetles (AT.WB) shared 19 ASVs, with the gallery wood mostly belonging to Pseudoxanthomonas and Pseudomonas (Figure 7B and Supplementary Material 8). Mycobacterium, Streptomyces, Williamsia, Amycolatopsis, and Gordonia were more abundant in AT.WB samples in comparison to gallery wood (Table 3 and Supplementary Table 2). Similarly, CZ.L and CZ.GW shared a consortium of 171 ASVs dominated by bacterial genera belonging to Enterobacterales and Xanthomonadales (Figure 7A and Supplementary Material 8). MetaStat analysis (p < 0.05) showed a relatively higher abundance of Pseudoxanthomonas, Curtobacterium, and Acinetobacter in CZ.GW samples, whereas Rhizobium, Microbacterium, Novosphingobium, Sphingobacterium, and Brevundimonas were substantially higher in CZ.L samples (Table 3 and Supplementary Table 2). Similarly, CZ.WB and CZ.GW shared 70 bacterial ASVs (Figure 7A), whereas Pseudoxanthomonas, Curtobacterium, Serratia, Bordetella, and Sphingomonas were significantly higher in CZ.GW samples. In contrast, Williamsia, Mycobacterium, Sphingobacterium, Nocardioides, and Gordonia were more abundant in CZ.WB samples than CZ.GW (Table 3 and Supplementary Table 2).
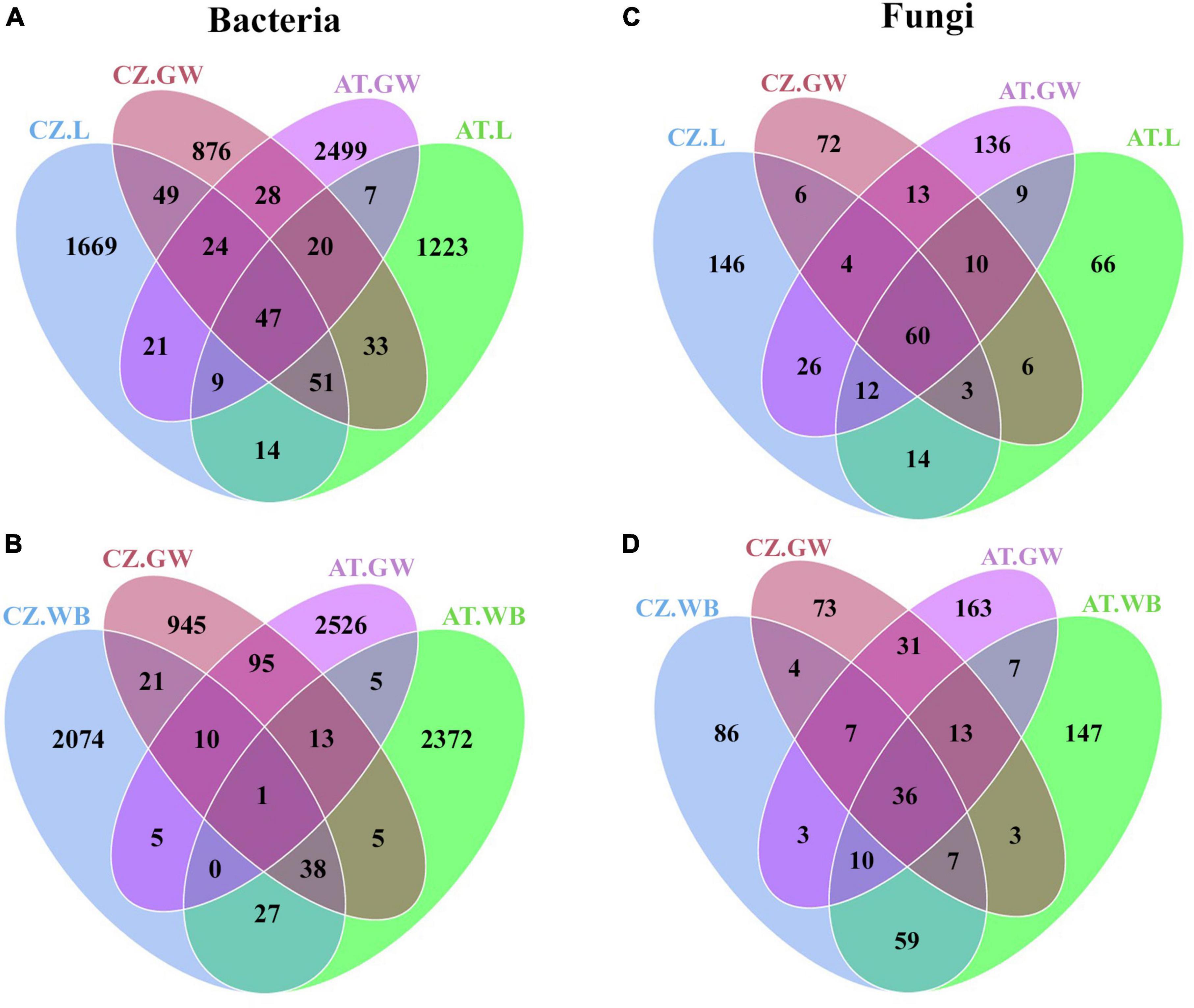
Figure 7. Impact of host contribution on ESBB microbial association. Venn diagrams showing the distribution of bacterial (A,B) and fungal (C,D) ASVs between gallery wood and larvae (A,C) and gallery wood and adult (B,D) samples from both Austria and Czech Republic locations. The number of shared or unique ASVs is denoted [Austria Larvae (AT.L), Austria Adult Whole Body (AT.WB), Austria Gallery Wood (AT.GW), Czech Republic Larvae (CZ.L), Czech Republic Adult Whole Body (CZ.WB), and Czech Republic Gallery Wood (CZ.GW)].
3.3.3.2. Host contribution to fungal assemblage
Similar to bacterial communities, AT.L and AT.WB shared 91 and 66 ASVs with AT.GW, respectively (Figures 7C, D and Supplementary Material 8). Furthermore, comparing AT.GW and AT.L in Metastat analysis revealed a significantly high abundance (p < 0.05) of Cryptococcus, Ophiostoma, Rhexographium, Eucasphaeria, and Ceratocystiopsis in AT.GW, whereas AT.L showed the prevalence of Nakazawaea, Wickerhamomyces, Kuraishia, Ogataea, and Hyalorhinocladiella (Table 3 and Supplementary Table 2). Similarly, AT.GW documented a higher abundance of Cryptococcus, Nakazawaea, Danielozyma, Eucasphaeria, Ceratocystiopsis compared to AT.WB, where Wickerhamomyces, Rhexographium, Talaromyces, Yamadazyma, and Trichoderma were significantly abundant (Table 3). Furthermore, CZ.L and CZ.WB showed the presence of 73 and 54 common ASVs with CZ.GW (Figures 7C, D). Additionally, Metastat analysis demonstrated a significantly higher abundance (p < 0.05) of Nakazawaea, Myxozyma, Ogataea, Saccharomycopsis, and Clonostachys in CZ.GW, compared to CZ.L, where Wickerhamomyces, Graphium, Cryptococcus, Yamadazyma, Rhexographium were abundant (Table 3). In comparison, CZ.GW revealed a high abundance of Ophiostoma, Nakazawaea, Myxozyma, Ogataea, and Saccharomycopsis, while Wickerhamomyces, Rhexographium, Yamadazyma, Talaromyces, and Penicillium were more abundant in CZ.WB (Table 3).
3.3.4. Impact of beetle feeding on the wood microbiome
3.3.4.1. Bacteria in control and gallery wood
Control wood (CZ.CW) shared 68 bacterial ASVs with the gallery wood (CZ.GW) from the Czech Republic. Apart from the common ASVs, there were 1256 and 1060 unique bacterial ASVs in control and gallery wood, respectively (Figure 8A and Supplementary Material 9). Furthermore, MetaStat analysis (p < 0.05) revealed a significantly higher abundance of Pseudoxanthomonas, Curtobacterium, and Acinetobacter in gallery wood. Additionally, LEfSe analysis identified several members of the classes Gammaproteobacteria and Actinobacteria, including genera like Pseudoxanthomonas, Curtobacterium, and Mycobacterium, as biomarkers of CZ.GW (Figure 8B and Supplementary Figure 6A).
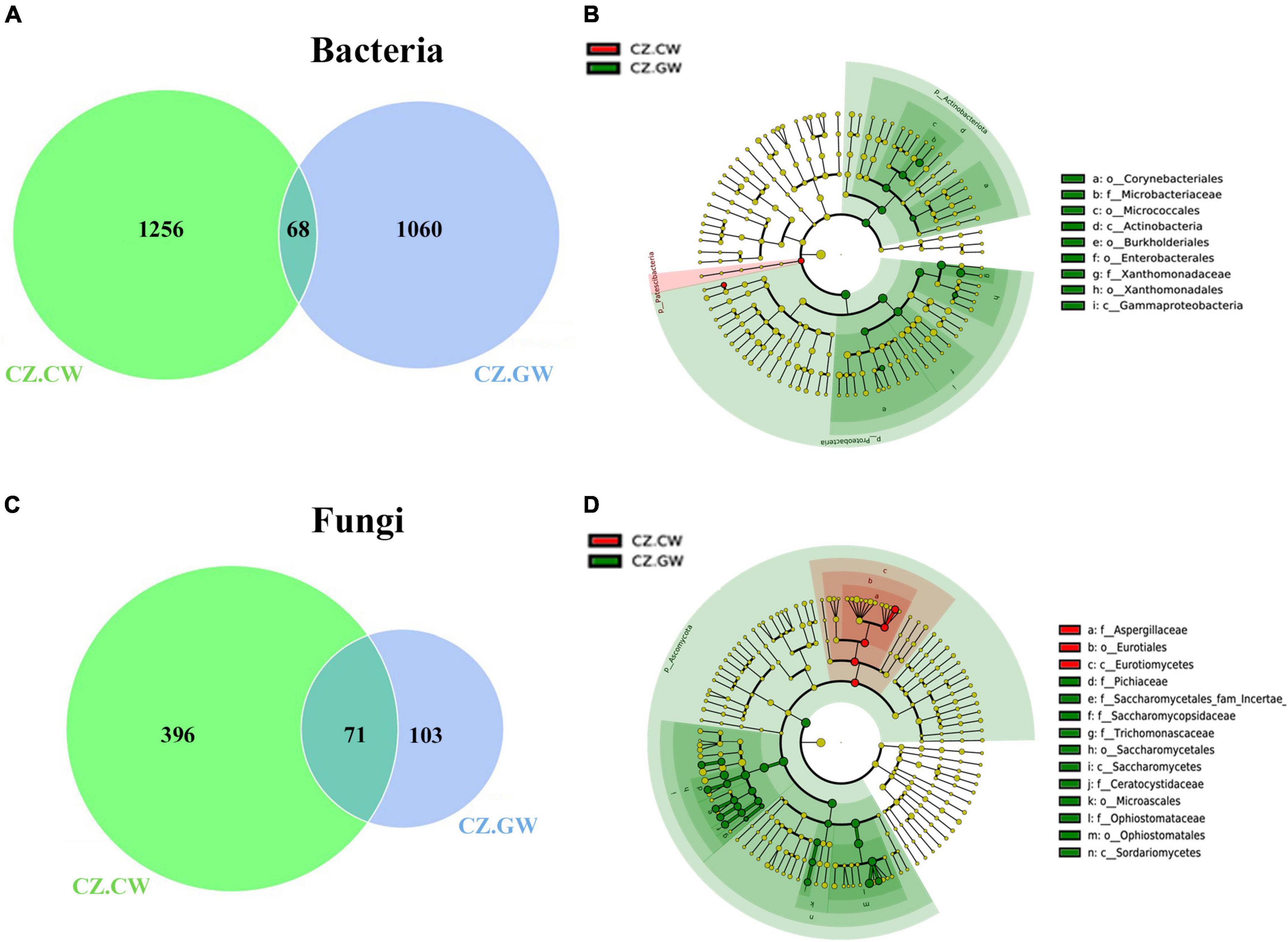
Figure 8. Impact of ESBB feeding on the microbial association in the wood samples from Czech Republic. Venn diagrams show the distribution of bacterial (A) and fungal (C) ASVs between control and gallery wood. The number of shared or unique ASVs is denoted. The unique ASVs in gallery wood represent the impact of feeding on microbial association. LEfSe analysis displays the significantly different bacterial (B) and fungal (D) biomarkers between the control and gallery wood. Each circle represents a distinct taxon at the respective taxonomic level. The color of the bacterial (B) and fungal (D) biomarkers correspond with the color of the wood samples. Yellowish-green circles denote non-significant bacterial (B) and fungal (D) species. Red and green nodes resemble bacterial (B) and fungal (D) taxon, highly contributing to the group. Letters above the circles represent fungal biomarkers [Czech Republic Control Wood (CZ.CW) and Czech Republic Gallery Wood (CZ.GW)].
3.3.4.2. Fungi in control and gallery wood
Nevertheless, the control and gallery wood from the Czech Republic (CZ.CW and CZ.GW) comprised 71 common fungal ASVs (Figure 8C and Supplementary Material 9). While 396 unique fungal ASVs were observed in control wood, and 103 unique ASVs were found in gallery wood. Among the core fungal genera, Ophiostoma, Nakazawaea, Myxozyma, and Ogataea showed significantly (p < 0.05) higher abundance in gallery wood (Table 2). Furthermore, LEfSe analysis suggested Ophiostoma, Nakazawaea, Myxozyma, Ogataea, Saccharomycopsis, Kuraishia, Cyberlindnera, Zygoascus, and Endoconidiophora genera as biomarkers in CZ.GW while Aspergillacaea, members of Eurotiomycetes as biomarkers in CZ.CW (Figure 8D and Supplementary Figure 6B).
3.4. Putative functional diversity profiling of bacteriome and mycobiome
Heatmaps demonstrated the putative functional diversity of the bacterial community in each sample depending on the contribution of each ASV to specific functions using PICRUSt2 (Figure 9 and Supplementary Figure 7). The top KEGG orthologs associated with transcription, replication and repair, chemotaxis, fatty acid biosynthesis, metabolism, and transport of amino acid, phosphate, sugar, iron complex, nickel/peptide, and phospholipid were the most abundant metabolic pathways in larvae, adult, and gallery wood samples (Figure 9A). Furthermore, Enzyme database (EC) documented a high abundance of alcohol dehydrogenase, fumarate reductase, enoyl-CoA hydratase, acetyl-CoA C-acetyltransferase, asparaginyl-tRNA synthase, glutaminyl-tRNA synthase, DNA-topoisomerase, serine/threonine protein kinase, and DNA-lyase in AT.WB samples (Figure 9B). Larvae and gallery wood samples from both locations had a high abundance of glutathione transferase.
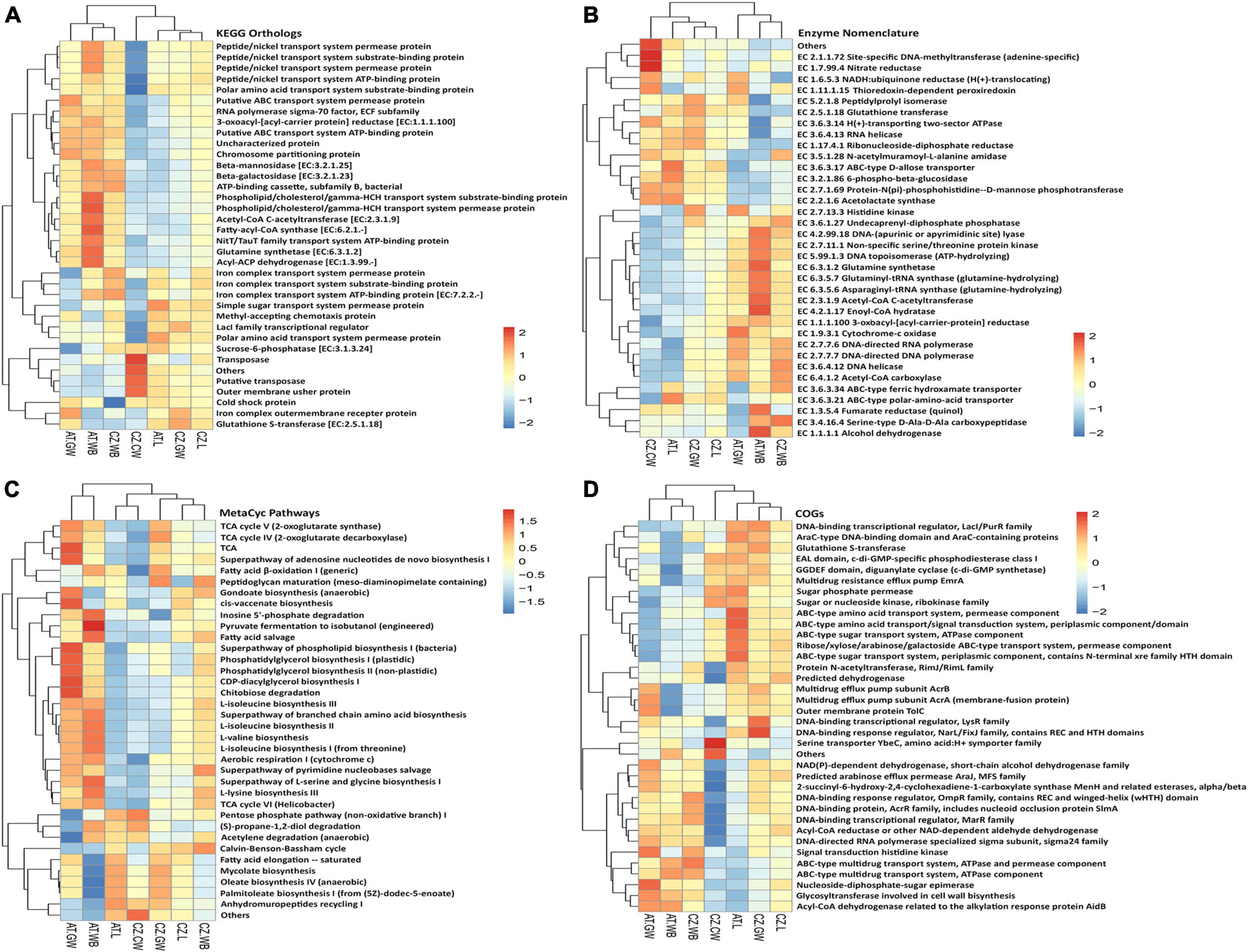
Figure 9. Functional prediction of the bacterial community using PICRUSt2. Heatmaps illustrate the functional profile inferred from significantly different (A) KEGG orthologs, (B) Enzymes, (C) MetaCyc Pathways, and (D) Cluster of orthologous genes (COGs), using PICRUSt2 analysis, representing the functional contribution of the bacterial community in control wood and Ips typographus larvae, adults, and gallery wood samples [Austria Larvae (AT.L), Austria Adult Whole Body (AT.WB), Austria Gallery Wood (AT.GW), Czech Republic Larvae (CZ.L), Czech Republic Adult Whole Body (CZ.WB), Czech Republic Gallery Wood (CZ.GW), and Czech Republic Control Wood (CZ.CW)].
Similarly, the MetaCyc database revealed the putative metabolic pathways related to fatty acid, phospholipids, sugar, amino acid, nucleotide biosynthesis, cell wall biosynthesis, fermentation of alcohols, aerobic respirations, fatty acid degradation, sugar derivative degradation, glycolysis, pentose phosphate pathway, TCA cycle, and Calvin cycle to be abundant. For instance, a higher abundance of pyruvate fermentation to isobutanol, inosine 5’-phosphate degradation, fatty acid salvage, and L-lysine biosynthesis pathways was documented in AT.WB samples (Figure 9C). Additionally, a comparison of ASVs in the COG database predicted several ABC-type amino acid and sugar transport system components to be highly abundant in AT.L samples (Figure 9D). Other databases used to predict functional diversity in the samples, including Pfam and TIGRFAM domain databases, documented different protein families to be involved, which might be associated with the bacterial communities present (Supplementary Figure 7). The putative functional diversity profile in the control wood differed from the gallery wood and the beetles (adults/larvae) (Figure 9 and Supplementary Figure 7).
Similarly, the putative potential roles of the fungal community in the samples from two different locations were predicted using FUNGuild (Figure 10 and Supplementary Figure 8). The main ecological guilds observed in all the samples include saprotrophs, symbiotrophs, and pathotrophs. In our study, the saprotrophs mainly consist of undefined saprotrophs, soil saprotrophs, and wood saprotrophs. The symbiotrophs include ectomycorrhizal, ericoid mycorrhizal, endophyte, and lichenized, while animal pathogens, fungal parasites, lichen parasites, and plant pathogens belong to the pathotrophs. The relative abundance of saprotrophs was significantly higher in AT.L (95%) compared to CZ.L (61%) (Supplementary Figure 8). The beetles (larvae/adults) have a high abundance of saprotrophic fungi that might facilitate the acquisition of nutrients from the dead and decaying wood (Figure 10 and Supplementary Figure 8). The influence of feeding might be explained by the abundance of saprotrophs in gallery wood compared to control uninfested wood. The pathotrophs were higher in CZ.L (30%) and CZ.GW (37%) compared to the rest of the samples. No remarkable difference in the relative abundance of symbiotrophs was observed in the beetle samples as well as the gallery wood samples (Figure 10). Among the beetle samples, pathotroph-symbiotrophs were highly abundant in larval samples (CZ.L, AT.L), while saprotroph-symbiotrophs were abundant in the beetle whole body samples (CZ.WB, AT.WB) and absent in larval samples. Comparing the beetle-infested gallery wood and the control uninfested wood, the saprotrophs and plant pathogens were highly abundant in gallery wood (Supplementary Figure 8).
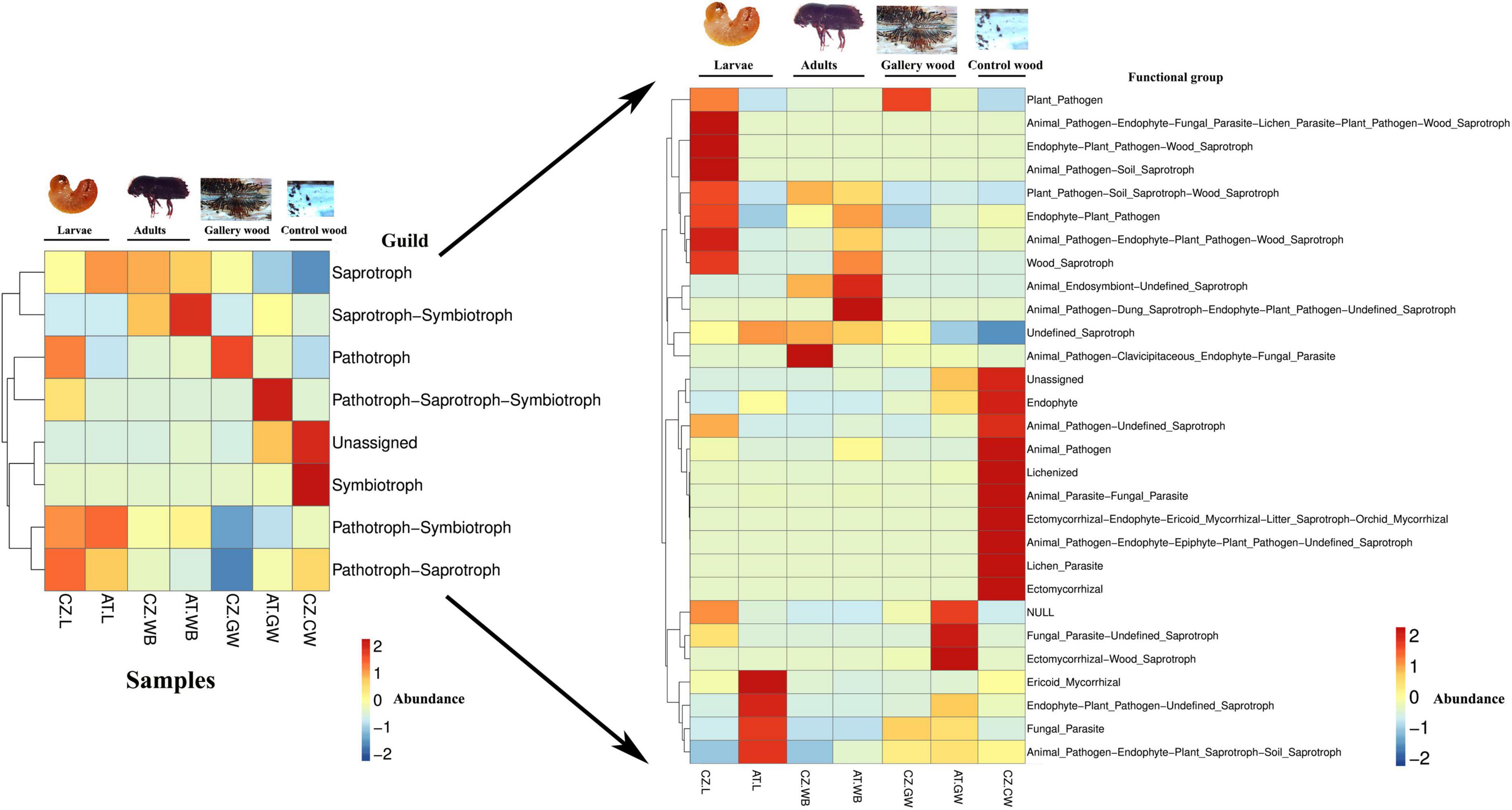
Figure 10. Functional prediction of the fungal community using FUNGuild. Heatmaps representing the predicted fungal functional groups in ESBB larvae, adults, and gallery wood from two different locations [Austria Larvae (AT.L), Austria Adult Whole Body (AT.WB), Austria Gallery Wood (AT.GW), Czech Republic Larvae (CZ.L), Czech Republic Adult Whole Body (CZ.WB), Czech Republic Gallery Wood (CZ.GW)] and Control Wood from Czech Republic (CZ.CW).
4. Discussion
The symbiosis with microbes paves the way for the ecological dominance of different pests, including tree-killing bark beetles (Frago et al., 2012; Fisher et al., 2017; Singh et al., 2021). The life stage, host tree, and environment also play a crucial role in fine-tuning such associations of ecological relevance (Hartmann et al., 2017; Hernández-García et al., 2018; Liu et al., 2019; Kaltenpoth, 2020). For instance, D. valens was documented to have a dynamic gut bacteriome, which differed among different beetle populations (Adams et al., 2010). Alternatively, D. ponderosae was reported to uphold relatively stable gut microbial communities resistant to perturbation (Adams et al., 2013). There are other independent observations with beetles or weevils supporting variable or stable microbial associations across wide geographic locations, making it difficult to generalize (Berasategui et al., 2016; Hernández-García et al., 2018; Moussa et al., 2023). Nevertheless, such variations in symbiont selection and maintenance throughout the life cycle among closely related bark beetles are fascinating in an ecological context and enhance our interest in bark beetles as holobionts. Unfortunately, there is still limited information on microbial associations in ESBB, the most aggressive tree-killing bark beetle in Europe. The present study documented the life stage and geo-location contribution in microbial assemblages in ESBB (Coleoptera: Curculionidae).
ESBB larvae spend their entire time under the bark, developing and acquiring a large part of their microbial associates during feeding and metamorphosis. Some of these microbes, primarily related to nutrition, detoxification, and other vital functions essential for development, might persist in other stages, suggesting their pivotal role in beetles (Peral-Aranega et al., 2023). Our study reported a high abundance of distinct microbial genera between the two developmental stages (larvae and adults) of ESBB. The larval stage is the gregarious feeding stage, where the associated microbial communities primarily function in nutrient acquisition and detoxification of plant terpenes (Peral-Aranega et al., 2020). The high abundance of Serratia, Novosphingobium, Rhizobium, Patulibacter, Microbacterium, and Brevundimonas in the larval population may be correlated with larval survival by performing important roles in detoxifying monoterpenes, inhibition of antagonistic fungi, degradation of cellulose and nitrogen fixation (Cardoza et al., 2006; Fröhlich et al., 2007; Boone et al., 2013; Xu et al., 2015, 2016; Zhou et al., 2016). Like bacterial communities, ESBB larvae are associated with fungi that may perform essential functions in beetle holobiont (Six, 2013). For instance, Ogataea, Cryptococcus, and Graphium were abundant in larvae that were mainly involved in producing volatile semiochemicals that inhibit antagonistic fungi growth (Leufvén and Nehls, 1986; Davis et al., 2011). Additionally, ESBB larval microbial communities might facilitate cold tolerance if larvae experience some dormancy during their life cycle. However, such possibilities need to be explored in future.
ESBB adults are involved in maturation feeding, host finding, new galleries, and reproduction. Such responsibilities in ESBB adults can also be associated with symbiont selection and maintenance (Kaltenpoth, 2020). Moreover, ESBB adults thrive in a saprophytic environment where the requirement of microbial assistance is obligatory. The presence of distinct bacterial genera in the adult beetles implies such possibilities. For instance, Cellulomonas and Curtobacterium are involved in cellulose and xylan degradation and thus aiding ESBB adult nutrition (Morales-Jiménez et al., 2009; Fabryová et al., 2018). Streptomyces and Amycolatopsis demonstrated antifungal activities, and their presence can be correlated with the inhibition of beetle-antagonistic fungi during and after new host colonization (Scott et al., 2008; Sen et al., 2009). Similarly, fungal genera Wickerhamomyces, Rhexographium, Aspergillus, Ophiostoma, Endoconidiophora, and Yamadazyma were dominant in the adult beetles that might be involved in anti-microbial activity to defend bark beetles against pathogens, degrade plant defense compounds, and contribute to beetle nutrition (Coda et al., 2011; Wadke et al., 2016; Parafati et al., 2017; Zhao et al., 2019b; Zaman et al., 2023). The core mycobiome, including Ophiostoma and Endoconidiophora can degrade plant phenolics and other defense compounds and utilize them as carbon sources (Wadke et al., 2016; Zhao et al., 2019b; Zaman et al., 2023). Yamadazyma and Kuraishia (=Hansenula) play an essential role in anti-aggregation pheromone production by converting cis and trans-verbenol to verbenone (Leufvén et al., 1984; Hunt and Borden, 1990). Moreover, ESBB shows overwintering behavior and undergoes diapause to endure low temperatures during winter, which also impacts their development, reproduction, and voltism (Schebeck et al., 2023). Diapause behavior may also influence the microbial community structure within the beetles and help in overwintering the chilling period (Moussa et al., 2023). Studies revealed a shift in the bacterial communities during overwintering and seasonal variation in Dendroctonus beetles suggesting the potential role of the microbiome in contributing to the success of host overwintering (Wang et al., 2017; Hou et al., 2021). In addition, change in temperature and photoperiod influences diapause plasticity and voltism in ESBB (Schroeder and Dalin, 2017). In the present study, the ESBB collected from the Czech Republic and Austria is typically bivoltine or multivoltine (Schebeck et al., 2017). The bivoltine or multivoltine nature of the species might impact the beetle microbiome, hence demands dedicated investigation.
Interestingly, the microbial richness between the larvae and adult ESBB did not differ significantly. Larvae and adults might acquire similar microbial associates during host feeding. This possibility corroborates with the recent findings where high bacterial diversity was observed in the ESBB larvae, which decreased in the pupal stage and further lowered in the teneral adults but recovered in the adults (Peral-Aranega et al., 2023). During metamorphosis, the microbial composition shrinks due to the structural changes in the host. The organogenesis and compartmentalization of the internal structures determine different habitat and physiological conditions, such as oxygen concentration, pH, and redox potential, influencing the distribution and survival of the microbial communities (Callegari et al., 2021). After metamorphosis, callow beetles (pre-emerged) acquire terminal and highly selected microbial communities, which are enriched again when the beetle starts feeding on a new host for colonization.
Predicting putative functional profiles of the microbial communities aids in understanding the role of microbes in ESBB holobiont. Our data indicated the putative functional potential of the bacterial and fungal communities from ESBB larvae, adults, and galleries (Figures 9, 10). Symbiotic bacteria and yeasts might contribute to the degradation of complex plant polymers, nitrogen fixation, and supplement essential amino acids and vitamins to the beetles (García-Fraile, 2018; Peral-Aranega et al., 2020). The presence of sugar derivative degradation, fatty acid degradation, and amino acid biosynthesis pathways in the samples might endorse the putative role of the bacterial community in nutritional supplementation. For instance, the core bacterial communities, including Pseudoxanthomonas, Pseudomonas, Cellulomonas, and Curtobacterium, might be involved in the cellulolytic activity and degradation of complex sugars (Hu et al., 2014; Fabryová et al., 2018). Furthermore, studies revealed that Acinetobacter is involved in the degradation of fatty acids and lipids (Briones-Roblero et al., 2017), while Pseudomonas function in essential amino acid supplementation to the bark beetles (Saati-Santamaría et al., 2021). The pathways related to the fermentation of alcohols observed in the study might endorse the bacterial role in pheromone production (Chiu and Bohlmann, 2022; Cao et al., 2023). For example, Pseudomonas and Serratia isolated from D. valens can convert cis-verbenol into verbenone, a common pheromone of bark beetles (Xu et al., 2015). Similarly, Glutathione S-transferase (GST) activity by the bacterial communities associated with the beetles plays a crucial role in detoxifying plant defense allelochemicals (Allocati et al., 2009; Gao et al., 2020). Pseudomonas and Sphingomonas have been documented to possess GST genes (Vuilleumier and Pagni, 2002) that might be beneficial in protecting against oxidative stress and detoxifying toxic metabolites (Gao et al., 2020).
FUNGuild helps to predict the putative functional groups of the fungal communities in the samples (Figure 10). The high abundance of saprotrophs in ESBB indicates their involvement in the acquisition of beetle nutrition in the saprophytic environment. Saprotrophic fungi can breakdown the dead and decaying organic matter into simpler forms that can be utilized for beetle nutrition (Crowther et al., 2012). For instance, Endoconidiophora, Graphium, and Rhexographium associated with bark beetles have been reported to endure tree defense, utilize the toxic defensive compounds as carbon sources, and boost beetle fitness (Hammerbacher et al., 2013; Cale et al., 2016; Wadke et al., 2016; Zhou et al., 2016). The high abundance of pathotrophs in ESBB larvae such as Ophiostoma depicts the role of fungi in enduring tree defensive compounds and damaging host tree cells for survival under the bark (Wingfield, 1995; Kirisits, 2007). However, it is important to note that PICRUSt 2 and FUNGuild are the software for predicting functional abundances based on marker genes; hence the putative functional diversity profiling of microbiome needs further validation using metatranscriptomic, metaproteomic or other culture-based approaches.
Geographic locations often influence microbial assemblage in bark beetles (Adams et al., 2010; Moussa et al., 2023). The ANOSIM analysis indicated a significant impact of geo-location on the ESBB microbial assemblage. The environmental factors, including temperature (Wang et al., 2020), landscape (Park et al., 2019), tree diversity (Cohen et al., 2020), and maturity of the trees, might influence the beetle microbiome in different geo-locations. Hence, the difference in the host microclimate of Austria and Czechia can also influence the ESBB microbial assemblages or the dominance of selective ones. For instance, a high abundance of Curtobacterium, Streptomyces, Acinetobacter were documented in larval samples from Austria that might be involved in cellulose degradation, antagonistic fungi inhibition and lipid degradation (Briones-Roblero et al., 2017; Engl et al., 2018; Fabryová et al., 2018). Consequently, larvae from the Czech Republic showed a dominance of Rhizobium, Microbacterium, Novosphingobium, and Sphingobacterium. These bacterial genera were reportedly involved in functions like nitrogen supplementation, antagonist inhibition, and plant defense compound degradation (Cardoza et al., 2006; Fröhlich et al., 2007; Cheng et al., 2018). Furthermore, the α-diversity indices did not differ when comparing the same stages of bark beetles across the two locations. Such observations explain a life-stage-specific conserved microbial community in ESBB (Hernández-García et al., 2018). However, other studies documented the impact of the environment on ESBB microbial assemblages while comparing beetles collected from more diverse geo-locations (Moussa et al., 2023), endorsing the requirements of ESBB sampling from vast geographic locations to capture more noticeable environmental impact. Such a sampling effort is challenging and must be a harmonized collaborative effort from different beetle collectors worldwide to minimize the other variabilities that can potentially disguise the actual environmental effect.
Microbial influence on plant-insect interaction is well documented (Chakraborty and Roy, 2021). Although plants and insects host microorganisms, how much their microbiomes overlap and how much they alter each other’s microbiomes is ambiguous (Pirttilä et al., 2023). How far the ESBB colonization influences the spruce holobiont merits investigation. The present study provides evidence for the enrichment of the spruce microbiome upon beetle feeding and vice versa. We presume that while feeding on wood, bark beetles transmit microbes through oral secretions, exoskeletons, and fecal deposition that structure the gallery wood microbiome (Popa et al., 2012; Cheng et al., 2018; Pessotti et al., 2021). Pseudoxanthomonas, Curtobacterium, and Acinetobacter were present in significantly higher abundance in galleries than in control wood samples. Curtobacterium and Acinetobacter were often observed in the bark beetle’s oral secretions and crushed bodies (Cardoza et al., 2009; Muratoğlu et al., 2011) and might be inoculated to the phloem during feeding aids in the initial cellulose and lipid degradations (Briones-Roblero et al., 2017; Fabryová et al., 2018). Nevertheless, many insects acquire symbionts via horizontal transmission from the environment (Kikuchi et al., 2007; Hartmann et al., 2017). The bacterial genera Pseudoxanthomonas, Pseudomonas, Rhizobium, and Dyella were abundant and shared between gallery wood and ESBB (Figure 7 and Supplementary Material 8). These bacteria are commonly found in the soil, water, or plants (Preston, 2004; Weller, 2007; Fierer, 2017; Mohapatra et al., 2018). There is a possibility that these bacteria are present in the microhabitat of bark beetles; hence were found in higher abundance in bark beetles and gallery wood (Du et al., 2015; Li et al., 2019). Notably, all these bacteria can degrade cellulose and other complex sugars and provide nitrogen, thereby essential for the bark beetles to survive in the saprophytic environment (Morales-Jiménez et al., 2013; Hu et al., 2014; Menendez et al., 2015). Similar to bacteria, the fungal genera Ophiostoma, Nakazawaea, Myxozyma, and Ogataea that were highly abundant in gallery wood were shown to be associated with bark beetle adult and larval samples (Rivera et al., 2009; Cheng et al., 2018; Chakraborty et al., 2020b). These fungi are primarily present in the soil, plant materials, and trees, including conifers (Durand et al., 2017; Jankowiak et al., 2019) that beetles might acquire during feeding or overwintering under the soil. These fungi often aid in nutrition, degradation of plant chemicals, and pheromone production in beetles (Davis et al., 2011). Such microbial exchange enriches the bark beetle microhabitat and can influence the fitness of hosts (i.e., spruce and beetles). However, dedicated investigations can unveil the true ecological relevance.
5. Conclusion
The present study reports the ESBB microbial community encompassing two life stages (adults and larvae) from two geographic locations (Czech Republic and Austria). The study documented the influence of beetle life stage and geo-location on the core and unique bacterial and fungal communities in ESBB. It also documented that beetle feeding modulates the gallery microbiome and thus enriches the microhabitat. The microbial communities at the feeding gallery need further attention to understand their ecological relevance to the complex life of the bark beetles. Furthermore, the present study provides insights into the influence of drivers, such as life stage and geo-location, in shaping ESBB microbial associations and their maintenance after metamorphosis. Comparing the impact of these two drivers revealed a more decisive influence of the developmental stages on the ESBB microbiome than their geo-location. Studying ESBB microbiomes from more diverse geo-locations can be more revealing. Nevertheless, current understanding enables the formulation of ecologically relevant testable hypotheses on bark beetle symbiosis for downstream functional studies and may facilitate the formulation of symbiont-mediated bark beetle management strategies (Joga et al., 2021).
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://dataview.ncbi.nlm.nih.gov/object/PRJNA855649, https://dataview.ncbi.nlm.nih.gov/object/PRJNA855680, PRJNA855680 (bacteriome) and PRJNA855649 (mycobiome).
Author contributions
AR and AC planned the research. RM and AK collected and maintained the bark beetles. AC, AP, and AK conducted the molecular experiments and data analysis and made biological interpretations of the results. AP, AC, and AR prepared the manuscript. All authors read and approved the manuscript.
Funding
The project was financed by “EXTEMIT – K,” No. CZ.02.1.01/0.0/0.0/15_003/0000433 financed by OP RDE. The financial support for AP, AC was provided by “EVA 4.0,” No. CZ.02.1.01/0.0/0.0/16_019/0000803 financed by OP RDE. The salary for AR, RM, was obtained from “EXTEMIT – K,” No. CZ.02.1.01/0.0/0.0/15_003/0000433 financed by OP RDE. AK was partially supported by Internal Grant Agency (IGA 2021-2022; project number- A_21_05) at the Faculty of Forestry and Wood Sciences, Czech University of Life Sciences, Prague, Czech Republic. AR and AC are also supported by “Excellent Team Grants” (2023-2024) from the Faculty of Forestry and Wood Sciences, Czech University of Life Sciences, Prague, Czech Republic.
Acknowledgments
We acknowledge Dr. Sigrid Netherer, University of Natural Resources and Life Sciences, BOKU, Austria for her contribution to the beetle collection from Austria and for gifting us for the microbiome study. Special thanks to Prof. Fredrik Schlyter for his constructive comments and encouragement. All beetle experiments complied with the ARRIVE guidelines and were carried out per (Scientific Procedures) Act, 1986 and associated guidelines, EU Directive 2010/63/EU for animal experiments. We also thank the handling editor and reviewers for their constructive comments on the earlier version of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/ffgc.2023.1176160/full#supplementary-material
Supplementary Figure 1 | Rarefaction Curves. Different colors and symbols denote different samples.
Supplementary Figure 2 | Boxplot showing microbial α-diversity indices among ESBB larvae, adults, gallery wood, and control wood samples. Bacterial (A–C) and Fungal (D–F) community richness are indicated by Chao1 analysis (A,D), and diversity is represented by the Shannon index (B,E) and Simpson index (C,F). The statistical analysis for the significant differences between groups is done by The Kruskal-Wallis test, where “*” denotes p < 0.05, “**” and denotes p < 0.01 [Austria Gallery Wood (AT.GW), Austria ESBB larvae (AT.L), Austria ESBB adult whole body (AT.WB), Czech Republic Control Wood (CZ.CW), Czech Republic Gallery Wood (CZ.GW), Czech Republic ESBB larvae (CZ.L), and Czech Republic ESBB adult whole body (CZ.WB)].
Supplementary Figure 3 | Life stage-specific microbial association in ESBB. Histograms of the LDA scores illustrate the presence of bacterial (A,B) and fungal (C,D) species (biomarkers) whose abundance is significantly different between larval and adult samples from Austria (A,C) and Czech Republic (B,D). The LDA score at log 10 > 4 is set as the threshold, and the length of each bin, i.e., the LDA score, represents the extent to which the microbial biomarker differs among the groups. The color of each bin corresponds with the bark beetle’s life stages [Austria ESBB larvae (AT.L), Austria ESBB adult whole body (AT.WB), Czech Republic ESBB larvae (CZ.L), and Czech Republic ESBB adult whole body (CZ.WB)].
Supplementary Figure 4 | Influence of geo-location on the microbial association in ESBB. Histograms of the LDA scores illustrate the presence of bacterial (A,B) and fungal (C,D) species (biomarkers) whose abundance is significantly different between larvae (A,C) and adults (B,D) from Austria and Czech Republic sites. The LDA score at log 10 > 4 is set as the threshold, and the length of each bin, i.e., the LDA score, represents the extent to which the microbial biomarker differs among the groups. The color of each bin corresponds with the geographical location [Austria ESBB larvae (AT.L), Austria ESBB adult whole body (AT.WB), Czech Republic ESBB larvae (CZ.L), and Czech Republic ESBB adult whole body (CZ.WB)].
Supplementary Figure 5 | Influence of geo-location on the microbial association in ESBB gallery wood samples. Venn diagrams showing the distribution of bacterial (A) and fungal (D) ASVs between gallery wood of both locations (Austria and Czech Republic). The number of shared or unique ASVs are denoted. LEfSe analysis displays the significantly different bacterial (B) and fungal (E) biomarkers between Austria and Czech Republic gallery wood samples. Each circle represents a distinct taxon at the respective taxonomic level. The color of the microbial biomarkers corresponds with the color of the geographic locations. Yellowish-green circles denote non-significant bacterial (B) and fungal (E) species. Red and green nodes resemble microbial taxon, highly contributing to the group. The letters above the circles represent microbial biomarkers. Histograms of the LDA scores illustrate the presence of bacterial (C) and fungal (F) species (biomarkers) whose abundance is significantly different between gallery wood samples from Austria and Czech Republic sites. The LDA score at log 10 > 4 is set as the threshold, and the length of each bin, i.e., the LDA score, represents the extent to which the microbial biomarker differs among the groups. The color of each bin corresponds with the geographical locations [Austria Gallery Wood (AT.GW) and Czech Republic Gallery Wood (CZ.GW)].
Supplementary Figure 6 | Impact of ESBB feeding on the microbial association in the wood samples from Czech Republic. Histograms of the LDA scores illustrate the presence of bacterial (A) and fungal (B) species (biomarkers) whose abundance is significantly different between control wood and gallery wood samples from Czech Republic site. The LDA score at log 10 > 4 is set as the threshold, and the length of each bin, i.e., the LDA score, represents the extent to which the microbial biomarker differs among the groups. The color of each bin corresponds with control wood and gallery wood [Czech Republic Gallery Wood (CZ.GW) and Czech Republic Control Wood (CZ.CW)].
Supplementary Figure 7 | Functional prediction of the bacterial community using PICRUSt2. Heatmaps illustrate the functional profile inferred from significantly different (A) Pfam domains and (B) TIGRFAM entries, using PICRUSt2 analysis, representing the functional contribution of the bacterial community in control wood and Ips typographus larvae, adults, and gallery wood samples 01 [Austria Gallery Wood (AT.GW), Austria ESBB larvae (AT.L), Austria ESBB adult whole body (AT.WB), Czech Republic Control Wood (CZ.CW), Czech Republic Gallery Wood (CZ.GW), Czech Republic ESBB larvae (CZ.L), and Czech Republic ESBB adult whole body (CZ.WB)].
Supplementary Figure 8 | t-test analysis illustrating the significant fungal guilds between the samples collected from two different geo-locations [Austria ESBB Larvae (AT.L), Austria ESBB Adult Whole Body (AT.WB), Austria ESBB Gallery Wood (AT.GW), Czech Republic ESBB Larvae (CZ.L), Czech Republic ESBB Adult Whole Body (CZ.WB), Czech Republic ESBB Gallery Wood (CZ.GW) and Czech Republic Control Wood (CZ.CW)]. The comparisons were made between ESBB life stages, geo-locations, ESBB vs infested phloem (gallery wood) and infested and uninfested phloem.
Supplementary Table 1 | ADONIS analysis based on Unweighted Unifrac representing the significant differences between the samples. Df denotes the degree of freedom; SS represents sums of squares of deviations, MS stands for SS/Df. F. Model represents F-test value. The R2 value illustrates the ratio of grouping variance and total variance. The p-value (<0.05) determines the significant variation between the bark beetles.
Supplementary Table 2 | t-test analysis illustrating significant bacterial and fungal species variation in ESBB larvae and adults from Austria [Austria ESBB larvae (AT.L), Austria ESBB adult whole body (AT.WB)] and Czech Republic [Czech Republic ESBB larvae (CZ.L), and Czech Republic ESBB adult whole body (CZ.WB)] with control wood and gallery wood [Czech Republic Gallery Wood (CZ.GW), Czech Republic Control Wood (CZ.CW) and Austria Gallery Wood (AT.GW)].
Supplementary Material 1 | QC statistics and qualified clean read counts.
Supplementary Material 2 | The ASV table representing the ASV abundance in ESBB (adults and larvae), gallery wood, and control wood samples (with biological replicates).
Supplementary Material 3 | Relative abundance of bacteriome and mycobiome (class level) present in ESBB (adults and larvae), gallery wood, and control wood samples (with biological replicates).
Supplementary Material 4 | Relative abundance of bacteriome and mycobiome (genus level) present in ESBB (adults and larvae), gallery wood, and control wood samples (with biological replicates).
Supplementary Material 5 | Significant differences between samples in bacterial and fungal richness (Chao1) and diversity (Shannon index and Simpson index) by Kruskal-Wallis-pairwise-group test.
Supplementary Material 6 | Life-stage specific microbial association; the shared and unique microbial communities when compared between ESBB life-stages (adult and larvae) from both the locations (Austria and Czech Republic).
Supplementary Material 7 | Impact of geo-location on the microbial association; the shared and unique microbial communities comparing similar life-stages (adult or larvae) in ESBB or gallery wood between different locations (Austria and Czech Republic).
Supplementary Material 8 | Host contribution to the microbial association; the shared microbial communities while compared between ESBB (adult or larvae) and the gallery wood from different locations (Austria and Czech Republic).
Supplementary Material 9 | Impact of ESBB feeding on the wood microbiome; the unique microbial communities in gallery wood due to beetle feeding compared to control wood from Czech Republic.
Footnotes
- ^ http://ccb.jhu.edu/software/FLASH/
- ^ http://www.arb-silva.de/
- ^ http://huttenhower.sph.harvard.edu/lefse/
- ^ http://funguild.org
References
Adams, A. S., Adams, S. M., Currie, C. R., Gillette, N. E., and Raffa, K. F. (2010). Geographic variation in bacterial communities associated with the red turpentine beetle (Coleoptera: Curculionidae). Environ. Entomol. 39, 406–414.
Adams, A. S., Aylward, F. O., Adams, S. M., Erbilgin, N., Aukema, B. H., Currie, C. R., et al. (2013). Mountain pine beetles colonizing historical and naive host trees are associated with a bacterial community highly enriched in genes contributing to terpene metabolism. Appl. Environ. Microbiol. 79, 3468–3475. doi: 10.1128/AEM.00068-13
Adams, A., Currie, C., Cardoza, Y., Klepzig, K., and Raffa, K. (2009). Effects of symbiotic bacteria and tree chemistry on the growth and reproduction of bark beetle fungal symbionts. Can. J. For. Res. 39, 1133–1147.
Allocati, N., Federici, L., Masulli, M., and Di Ilio, C. (2009). Glutathione transferases in bacteria. FEBS J. 276, 58–75.
Anderson, M. J. (2001). A new method for non-parametric multivariate analysis of variance. Austr. Ecol. 26, 32–46.
Berasategui, A., Axelsson, K., Nordlander, G., Schmidt, A., Borg-Karlson, A. K., Gershenzon, J., et al. (2016). The gut microbiota of the pine weevil is similar across Europe and resembles that of other conifer-feeding beetles. Mol. Ecol. 25, 4014–4031.
Berasategui, A., Salem, H., Paetz, C., Santoro, M., Gershenzon, J., Kaltenpoth, M., et al. (2017). Gut microbiota of the pine weevil degrades conifer diterpenes and increases insect fitness. Mol. Ecol. 26, 4099–4110. doi: 10.1111/mec.14186
Bokulich, N. A., Kaehler, B. D., Rideout, J. R., Dillon, M., Bolyen, E., Knight, R., et al. (2018). Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6:90. doi: 10.1186/s40168-018-0470-z
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857.
Boone, C. K., Keefover-Ring, K., Mapes, A. C., Adams, A. S., Bohlmann, J., and Raffa, K. F. (2013). Bacteria associated with a tree-killing insect reduce concentrations of plant defense compounds. J. Chem. Ecol. 39, 1003–1006. doi: 10.1007/s10886-013-0313-0
Briones-Roblero, C. I., Rodríguez-Díaz, R., Santiago-Cruz, J. A., Zúñiga, G., and Rivera-Orduña, F. N. (2017). Degradation capacities of bacteria and yeasts isolated from the gut of Dendroctonus rhizophagus (Curculionidae: Scolytinae). Folia Microbiol. 62, 1–9. doi: 10.1007/s12223-016-0469-4
Cale, J. A., Collignon, R. M., Klutsch, J. G., Kanekar, S. S., Hussain, A., and Erbilgin, N. (2016). Fungal volatiles can act as carbon sources and semiochemicals to mediate interspecific interactions among bark beetle-associated fungal symbionts. PLoS One 11:e0162197. doi: 10.1371/journal.pone.0162197
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Callegari, M., Crotti, E., Fusi, M., Marasco, R., Gonella, E., De Noni, I., et al. (2021). Compartmentalization of bacterial and fungal microbiomes in the gut of adult honeybees. NPJ Biofilms Microbiomes 7:42. doi: 10.1038/s41522-021-00212-9
Cao, Q., Koski, T. M., Li, H., Zhang, C., and Sun, J. (2023). The effect of inactivation of aldehyde dehydrogenase on pheromone production by a gut bacterium of an invasive bark beetle, Dendroctonus valens. Insect Sci. 30, 459–472. doi: 10.1111/1744-7917.13101
Cardoza, Y. J., Klepzig, K. D., and Raffa, K. F. (2006). Bacteria in oral secretions of an endophytic insect inhibit antagonistic fungi. Ecol. Entomol. 31, 636–645.
Cardoza, Y. J., Vasanthakumar, A., Suazo, A., and Raffa, K. F. (2009). Survey and phylogenetic analysis of culturable microbes in the oral secretions of three bark beetle species. Entomol. Exp. Appl. 131, 138–147.
Ceja-Navarro, J. A., Karaoz, U., Bill, M., Hao, Z., White, I. R. A., Arellano, A., et al. (2019). Gut anatomical properties and microbial functional assembly promote lignocellulose deconstruction and colony subsistence of a wood-feeding beetle. Nat. Microbiol. 4, 864–875. doi: 10.1038/s41564-019-0384-y
Chakraborty, A., and Roy, A. (2021). “Microbial influence on plant–insect interaction,” in Plant-pest interactions: from molecular mechanisms to chemical ecology: chemical ecology, eds K. Singh and A. Singh (Amsterdam: Springer), 337–363.
Chakraborty, A., Ashraf, M. Z., Modlinger, R., Synek, J., Schlyter, F., and Roy, A. (2020a). Unravelling the gut bacteriome of Ips (Coleoptera: Curculionidae: Scolytinae): identifying core bacterial assemblage and their ecological relevance. Sci. Rep. 10:18572. doi: 10.1038/s41598-020-75203-5
Chakraborty, A., Modlinger, R., Ashraf, M. Z., Synek, J., Schlyter, F., and Roy, A. (2020b). Core mycobiome and their ecological relevance in the gut of five Ips bark beetles (Coleoptera: Curculionidae: Scolytinae). Front. Microbiol. 11:568853. doi: 10.3389/fmicb.2020.568853
Chapman, M., and Underwood, A. (1999). Ecological patterns in multivariate assemblages: information and interpretation of negative values in ANOSIM tests. Mar. Ecol. Progr. Ser. 180, 257–265.
Cheng, C., Wickham, J. D., Chen, L., Xu, D., Lu, M., and Sun, J. (2018). Bacterial microbiota protect an invasive bark beetle from a pine defensive compound. Microbiome 6:132. doi: 10.1186/s40168-018-0518-0
Chiu, C. C., and Bohlmann, J. (2022). Mountain pine beetle epidemic: an interplay of terpenoids in host defense and insect pheromones. Annu. Rev. Plant Biol. 73, 475–494. doi: 10.1146/annurev-arplant-070921-103617
Clarke, K. R. (1993). Non-parametric multivariate analyses of changes in community structure. Austr. J. Ecol. 18, 117–143.
Coda, R., Cassone, A., Rizzello, C. G., Nionelli, L., Cardinali, G., and Gobbetti, M. (2011). Antifungal activity of Wickerhamomyces anomalus and Lactobacillus plantarum during sourdough fermentation: identification of novel compounds and long-term effect during storage of wheat bread. Appl. Environ. Microbiol. 77, 3484–3492. doi: 10.1128/AEM.02669-10
Cohen, H., McFrederick, Q. S., and Philpott, S. M. (2020). Environment shapes the microbiome of the blue orchard bee, Osmia lignaria: RRH: environmental drivers of bee microbiome. Microb. Ecol. 80, 897–907. doi: 10.1007/s00248-020-01549-y
Crowther, T. W., Boddy, L., and Jones, H. T. (2012). Functional and ecological consequences of saprotrophic fungus–grazer interactions. ISME J. 6, 1992–2001. doi: 10.1038/ismej.2012.53
D’Argenio, V., Casaburi, G., Precone, V., and Salvatore, F. (2014). Comparative metagenomic analysis of human gut microbiome composition using two different bioinformatic pipelines. Biomed Res. Int. 2014:325340. doi: 10.1155/2014/325340
Davis, T. S., Hofstetter, R. W., Foster, J. T., Foote, N. E., and Keim, P. (2011). Interactions between the yeast Ogataea pini and filamentous fungi associated with the western pine beetle. Microb. Ecol. 61, 626–634. doi: 10.1007/s00248-010-9773-8
Davis, T. S., Stewart, J. E., Mann, A., Bradley, C., and Hofstetter, R. W. (2019). Evidence for multiple ecological roles of Leptographium abietinum, a symbiotic fungus associated with the North American spruce beetle. Fungal Ecol. 38, 62–70.
Douglas, G. M., Maffei, V. J., Zaneveld, J. R., Yurgel, S. N., Brown, J. R., Taylor, C. M., et al. (2020). PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 38, 685–688.
Du, R., Yan, J., Li, S., Zhang, L., Zhang, S., Li, J., et al. (2015). Cellulosic ethanol production by natural bacterial consortia is enhanced by Pseudoxanthomonas taiwanensis. Biotechnol. Biofuels 8, 1–10. doi: 10.1186/s13068-014-0186-7
Durand, A., Maillard, F., Foulon, J., Gweon, H. S., Valot, B., and Chalot, M. (2017). Environmental metabarcoding reveals contrasting belowground and aboveground fungal communities from poplar at a Hg phytomanagement site. Microb. Ecol. 74, 795–809. doi: 10.1007/s00248-017-0984-0
Ebner, G. (2020). Significantly more damaged wood in 2019. Available online at: https://www.timber-online.net/log_wood/2020/02/significantly-more-damaged-wood-in-2019.html
Engl, T., Kroiss, J., Kai, M., Nechitaylo, T. Y., Svatoš, A., and Kaltenpoth, M. (2018). Evolutionary stability of antibiotic protection in a defensive symbiosis. Proc. Natl. Acad. Sci. U.S.A. 115, E2020–E2029. doi: 10.1073/pnas.1719797115
Fabryová, A., Kostovčík, M., Díez-Méndez, A., Jiménez-Gómez, A., Celador-Lera, L., Saati-Santamaría, Z., et al. (2018). On the bright side of a forest pest-the metabolic potential of bark beetles’ bacterial associates. Sci. Total Environ. 619, 9–17. doi: 10.1016/j.scitotenv.2017.11.074
Fierer, N. (2017). Embracing the unknown: disentangling the complexities of the soil microbiome. Nat. Rev. Microbiol. 15, 579–590. doi: 10.1038/nrmicro.2017.87
Fisher, R. M., Henry, L. M., Cornwallis, C. K., Kiers, E. T., and West, S. A. (2017). The evolution of host-symbiont dependence. Nat. Commun. 8:15973.
Frago, E., Dicke, M., and Godfray, H. C. J. (2012). Insect symbionts as hidden players in insect–plant interactions. Trends Ecol. Evol. 27, 705–711. doi: 10.1016/j.tree.2012.08.013
Franceschi, V. R., Krokene, P., Christiansen, E., and Krekling, T. (2005). Anatomical and chemical defenses of conifer bark against bark beetles and other pests. N. phytol. 167, 353–376.
Fröhlich, J., Koustiane, C., Kämpfer, P., Rosselló-Mora, R., Valens, M., Berchtold, M., et al. (2007). Occurrence of rhizobia in the gut of the higher termite Nasutitermes nigriceps. Syst. Appl. Microbiol. 30, 68–74. doi: 10.1016/j.syapm.2006.03.001
Gao, H., Dai, L., Fu, D., Sun, Y., and Chen, H. (2020). Isolation, expression profiling, and regulation via host allelochemicals of 16 Glutathione S-Transferases in the Chinese white pine beetle, Dendroctonus armandi. Front. Physiol. 11:546592. doi: 10.3389/fphys.2020.546592
García-Fraile, P. (2018). Roles of bacteria in the bark beetle holobiont–how do they shape this forest pest? Ann. Appl. Biol. 172, 111–125.
Good, I. J. (1953). The population frequencies of species and the estimation of population parameters. Biometrika 40, 237–264.
Hammerbacher, A., Schmidt, A., Wadke, N., Wright, L. P., Schneider, B., Bohlmann, J., et al. (2013). A common fungal associate of the spruce bark beetle metabolizes the stilbene defenses of Norway spruce. Plant physiol. 162, 1324–1336. doi: 10.1104/pp.113.218610
Hartmann, A. C., Baird, A. H., Knowlton, N., and Huang, D. (2017). The paradox of environmental symbiont acquisition in obligate mutualisms. Curr. Biol. 27, 3711–3716.e3. doi: 10.1016/j.cub.2017.10.036
Hernández-García, J. A., Gonzalez-Escobedo, R., Briones-Roblero, C. I., Cano-Ramírez, C., Rivera-Orduña, F. N., and Zúñiga, G. (2018). Gut bacterial communities of Dendroctonus valens and D. mexicanus (Curculionidae: Scolytinae): a metagenomic analysis across different geographical locations in Mexico. Int. J. Mol. Sci. 19, 2578. doi: 10.3390/ijms19092578
Hlásny, T., König, L., Krokene, P., Lindner, M., Montagné-Huck, C., Müller, J., et al. (2021). Bark beetle outbreaks in Europe: state of knowledge and ways forward for management. Curr. For. Rep. 7, 138–165.
Hlásny, T., Krokene, P., Liebhold, A., Montagné-Huck, C., Müller, J., Qin, H., et al. (2019). Living with bark beetles: impacts, outlook and management options. Joensuu: European Forest Institute.
Hou, Z., Dong, Y., Shi, F., Xu, Y., Ge, S., Tao, J., et al. (2021). Seasonal shifts in cold tolerance and the composition of the gut microbiome of Dendroctonus valens LeConte occur concurrently. Forests 12:888.
Hu, X., Yu, J., Wang, C., and Chen, H. (2014). Cellulolytic bacteria associated with the gut of Dendroctonus armandi larvae (Coleoptera: Curculionidae: Scolytinae). Forests 5, 455–465.
Hulcr, J., Gomez, D. F., and Johnson, A. J. (2022). Collecting and preserving bark and ambrosia beetles (Coleoptera: Curculionidae: Scolytinae & Platypodinae). PLoS One 17:e0265910. doi: 10.1371/journal.pone.0265910
Hunt, D., and Borden, J. (1990). Conversion of verbenols to verbenone by yeasts isolated from Dendroctonus ponderosae (Coleoptera: Scolytidae). J. Chem. Ecol. 16, 1385–1397. doi: 10.1007/BF01021034
Jankowiak, R., Bilañski, P., Strzałka, B., Linnakoski, R., Bosak, A., and Hausner, G. (2019). Four new Ophiostoma species associated with conifer-and hardwood-infesting bark and ambrosia beetles from the Czech Republic and Poland. Antonie Leeuwenhoek 112, 1501–1521. doi: 10.1007/s10482-019-01277-5
Joga, M. R., Mogilicherla, K., Smagghe, G., and Roy, A. (2021). RNA interference-based forest protection products (FPPs) against wood-boring coleopterans: hope or hype? Front. Plant Sci. 12:733608. doi: 10.3389/fpls.2021.733608
Kaltenpoth, M. (2020). An endosymbiont’s journey through metamorphosis of its insect host. Proc. Natl. Acad. Sci. U.S.A. 117, 20994–20996. doi: 10.1073/pnas.2014598117
Keeling, C. I., and Bohlmann, J. (2006). Genes, enzymes and chemicals of terpenoid diversity in the constitutive and induced defence of conifers against insects and pathogens. N. Phytol. 170, 657–675.
Kikuchi, Y., Hosokawa, T., and Fukatsu, T. (2007). Insect-microbe mutualism without vertical transmission: a stinkbug acquires a beneficial gut symbiont from the environment every generation. Appl. Environ. Microbiol. 73, 4308–4316. doi: 10.1128/AEM.00067-07
Kirisits, T. (2007). “Fungal associates of European bark beetles with special emphasis on the ophiostomatoid fungi,” in Bark and wood boring insects in living trees in Europe, a synthesis, eds F. Lieutier, K. R. Day, A. Battisti, J. Grégoire, and H. F. Evans (Dordrecht: Springer), 181–236.
Klindworth, A., Pruesse, E., Schweer, T., Peplies, J., Quast, C., Horn, M., et al. (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41:e1. doi: 10.1093/nar/gks808
Leufvén, A., and Nehls, L. (1986). Quantification of different yeasts associated with the bark beetle, Ips typographus, during its attack on a spruce tree. Microb. Ecol. 12, 237–243. doi: 10.1007/BF02011208
Leufvén, A., Bergström, G., and Falsen, E. (1984). Interconversion of verbenols and verbenone by identified yeasts isolated from the spruce bark beetle Ips typographus. J. Chem. Ecol. 10, 1349–1361. doi: 10.1007/BF00988116
Li, Q.-Q., Chen, X., Zhou, X.-K., Dong, L.-M., Xiao, M., Fang, B.-Z., et al. (2019). Dyella tabacisoli sp. nov., a bacterium isolated from an arable soil sample of Nicotiana tabacum L. Int. J. Syst. Evol. Microbiol. 69, 3338–3343. doi: 10.1099/ijsem.0.003138
Liu, F., Wickham, J. D., Cao, Q., Lu, M., and Sun, J. (2020). An invasive beetle–fungus complex is maintained by fungal nutritional-compensation mediated by bacterial volatiles. ISME J. 14, 2829–2842. doi: 10.1038/s41396-020-00740-w
Liu, F., Ye, F., Cheng, C., Kang, Z., Kou, H., and Sun, J. (2022). Symbiotic microbes aid host adaptation by metabolizing a deterrent host pine carbohydrate d-pinitol in a beetle-fungus invasive complex. Sci. Adv. 8:eadd5051. doi: 10.1126/sciadv.add5051
Liu, H., Macdonald, C. A., Cook, J., Anderson, I. C., and Singh, B. K. (2019). An ecological loop: host microbiomes across multitrophic interactions. Trends Ecol. Evol. 34, 1118–1130. doi: 10.1016/j.tree.2019.07.011
Lozupone, C., Lladser, M. E., Knights, D., Stombaugh, J., and Knight, R. (2011). UniFrac: an effective distance metric for microbial community comparison. ISME J. 5, 169–172.
Magoč, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Magurran, A. E. (1988). Ecological diversity and its measurement. Princeton, NJ: Princeton university press.
Menendez, E., Ramírez-Bahena, M. H., Fabryova, A., Igual, J. M., Benada, O., Mateos, P. F., et al. (2015). Pseudomonas coleopterorum sp. nov., a cellulase-producing bacterium isolated from the bark beetle Hylesinus fraxini. Int. J. Syst. Evol. Microbiol. 65(Pt. 9), 2852–2858. doi: 10.1099/ijs.0.000344
Mikkelson, K. M., Bearup, L. A., Maxwell, R. M., Stednick, J. D., McCray, J. E., and Sharp, J. O. (2013). Bark beetle infestation impacts on nutrient cycling, water quality and interdependent hydrological effects. Biogeochemistry 115, 1–21.
Mohapatra, B., Sar, P., Kazy, S. K., Maiti, M. K., and Satyanarayana, T. (2018). Taxonomy and physiology of Pseudoxanthomonas arseniciresistens sp. nov., an arsenate and nitrate-reducing novel gammaproteobacterium from arsenic contaminated groundwater, India. PLoS One 13:e0193718. doi: 10.1371/journal.pone.0193718
Morales-Jiménez, J., Vera-Ponce de León, A., García-Domínguez, A., Martínez-Romero, E., Zúñiga, G., and Hernández-Rodríguez, C. (2013). Nitrogen-fixing and uricolytic bacteria associated with the gut of Dendroctonus rhizophagus and Dendroctonus valens (Curculionidae: Scolytinae). Microb. Ecol. 66, 200–210. doi: 10.1007/s00248-013-0206-3
Morales-Jiménez, J., Zúñiga, G., Villa-Tanaca, L., and Hernández-Rodríguez, C. (2009). Bacterial community and nitrogen fixation in the red turpentine beetle, Dendroctonus valens LeConte (Coleoptera: Curculionidae: Scolytinae). Microb. Ecol. 58, 879–891. doi: 10.1007/s00248-009-9548-2
Moussa, A., Nones, S., Vannucchi, P. E., Shahzad, G.-R., Dittmer, J., Corretto, E., et al. (2023). The bacterial community of the European spruce bark beetle in space and time. bioRxiv [Prerpint]. doi: 10.1101/2023.04.28.538755
Muratoğlu, H., Sezen, K., and Demirbağ, Z. (2011). Determination and pathogenicity of the bacterial flora associated with the spruce bark beetle, Ips typographus (L.)(Coleoptera: Curculionidae: Scolytinae). Turk. J. Biol. 35, 9–20.
Netherer, S., Kandasamy, D., Jirosová, A., Kalinová, B., Schebeck, M., and Schlyter, F. (2021). Interactions among Norway spruce, the bark beetle Ips typographus and its fungal symbionts in times of drought. J. Pest Sci. 94, 591–614. doi: 10.1007/s10340-021-01341-y
Nguyen, N. H., Song, Z., Bates, S. T., Branco, S., Tedersoo, L., Menke, J., et al. (2016). FUNGuild: an open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 20, 241–248.
Nilsson, R. H., Larsson, K.-H., Taylor, A. F. S., Bengtsson-Palme, J., Jeppesen, T. S., Schigel, D., et al. (2019). The UNITE database for molecular identification of fungi: handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 47, D259–D264. doi: 10.1093/nar/gky1022
Nunberg, M. (1981). Klucze do rozpoznawania owadów Polski. Część XIX. Chrząszcze–Coleoptera, Korniki–Scolytidae, Wyrynniki–Platypodidae. Zeszyt, 99-100. Warsaw: PWN.
Parafati, L., Cirvilleri, G., Restuccia, C., and Wisniewski, M. (2017). Potential role of exoglucanase genes (WaEXG1 and WaEXG2) in the biocontrol activity of Wickerhamomyces anomalus. Microb. Ecol. 73, 876–884. doi: 10.1007/s00248-016-0887-5
Park, R., Dzialo, M. C., Spaepen, S., Nsabimana, D., Gielens, K., Devriese, H., et al. (2019). Microbial communities of the house fly Musca domestica vary with geographical location and habitat. Microbiome 7:147. doi: 10.1186/s40168-019-0748-9
Paulson, J. N., Pop, M., and Bravo, H. C. (2011). Metastats: an improved statistical method for analysis of metagenomic data. Genome Biol. 12:P17. doi: 10.1371/journal.pcbi.1000352
Peral-Aranega, E., Saati-Santamaría, Z., Ayuso-Calles, M., Kostovčík, M., Veselská, T., Švec, K., et al. (2023). New insight into the bark beetle Ips typographus bacteriome reveals unexplored diversity potentially beneficial to the host. Environ. Microbiome 18:53. doi: 10.1186/s40793-023-00510-z
Peral-Aranega, E., Saati-Santamaría, Z., Kolaøik, M., Rivas, R., and García-Fraile, P. (2020). Bacteria belonging to Pseudomonas typographi sp. nov. from the bark beetle Ips typographus have genomic potential to aid in the host ecology. Insects 11:593. doi: 10.3390/insects11090593
Pessotti, R. D. C., Hansen, B. L., Reaso, J. N., Ceja-Navarro, J. A., El-Hifnawi, L., Brodie, E. L., et al. (2021). Multiple lineages of Streptomyces produce antimicrobials within passalid beetle galleries across eastern North America. Elife 10:e65091. doi: 10.7554/eLife.65091
Pfeffer, A. (1955). Fauna čSR. Svazek 6: Kùrovci-Scolytoidea. Øád: Brouci-Coleoptera. Prague: Nakladatelství československé akadmie vìd.
Pirttilä, A., Brusila, V., Koskimäki, J., Wäli, P., Ruotsalainen, A., Mutanen, M., et al. (2023). Exchange of microbiomes in plant-insect herbivore interactions. Mbio 14, e03210–e03222. doi: 10.1128/mbio.03210-22
Popa, V., Déziel, E., Lavallée, R., Bauce, E., and Guertin, C. (2012). The complex symbiotic relationships of bark beetles with microorganisms: a potential practical approach for biological control in forestry. Pest Manag. Sci. 68, 963–975. doi: 10.1002/ps.3307
Powell, D., Groβe-Wilde, E., Krokene, P., Roy, A., Chakraborty, A., Löfstedt, C., et al. (2021). A highly-contiguous genome assembly of the Eurasian spruce bark beetle, Ips typographus, provides insight into a major forest pest. Commun. Biol. 4:1059. doi: 10.1038/s42003-021-02602-3
Preston, G. M. (2004). Plant perceptions of plant growth-promoting Pseudomonas. Philos. Trans. R. Soc. London Ser. B Biol. Sci. 359, 907–918.
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2012). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
R Core Team (2013). R: A language and environment for statistical computing (Version 2.15. 3). Vienna: R Foundation for Statistical Computing.
Raffa, K. F., Gregoire, J. C., and Lindgren, B. S. (2015). “Natural history and ecology of bark beetles,” in Bark beetles. Eds F. E. Vega and R. W. Hofstetter (Cambridge, MA: Academic Press), 1–40.
Rivera, F. N., Gonzalez, E., Gomez, Z., Lopez, N., Hernandez-Rodriguez, C., Berkov, A., et al. (2009). Gut-associated yeast in bark beetles of the genus Dendroctonus Erichson (Coleoptera: Curculionidae: Scolytinae). Biol. J. Linnean Soc. 98, 325–342.
Rognes, T., Flouri, T., Nichols, B., Quince, C., and Mahé, F. (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ 4:e2584. doi: 10.7717/peerj.2584
Saati-Santamaría, Z., López-Mondéjar, R., Jiménez-Gómez, A., Díez-Méndez, A., Vìtrovskı, T., Igual, J. M., et al. (2018). Discovery of phloeophagus beetles as a source of Pseudomonas strains that produce potentially new bioactive substances and description of Pseudomonas bohemica sp. nov. Front. Microbiol. 9:913. doi: 10.3389/fmicb.2018.00913
Saati-Santamaría, Z., Rivas, R., Kolaøik, M., and García-Fraile, P. (2021). A new perspective of Pseudomonas—host interactions: distribution and potential ecological functions of the genus Pseudomonas within the Bark Beetle Holobiont. Biology 10:164. doi: 10.3390/biology10020164
Sauvard, D. (2007). “General biology of bark beetles,” in Bark and wood boring insects in living trees in Europe, a synthesis, eds F. Lieutier, K. R. Day, A. Battisti, J. Grégoire, and H. F. Evans (Dordrecht: Springer), 63–88.
Schebeck, M., Hansen, E. M., Schopf, A., Ragland, G. J., Stauffer, C., and Bentz, B. J. (2017). Diapause and overwintering of two spruce bark beetle species. Physiol. Entomol. 42, 200–210. doi: 10.1111/phen.12200
Schebeck, M., Schopf, A., Ragland, G. J., Stauffer, C., and Biedermann, P. H. (2023). Evolutionary ecology of the bark beetles Ips typographus and Pityogenes chalcographus. Bull. Entomol. Res. 113, 1–10. doi: 10.1017/S0007485321000353
Schroeder, M., and Dalin, P. (2017). Differences in photoperiod-induced diapause plasticity among different populations of the bark beetle Ips typographus and its predator Thanasimus formicarius. Agric. For. Entomol. 19, 146–153.
Scott, J. J., Oh, D.-C., Yuceer, M. C., Klepzig, K. D., Clardy, J., and Currie, C. R. (2008). Bacterial protection of beetle-fungus mutualism. Science 322, 63–63.
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12:R60.
Seidl, R., Schelhaas, M.-J., Rammer, W., and Verkerk, P. J. (2014). Increasing forest disturbances in Europe and their impact on carbon storage. Nat. Climate Change 4, 806–810.
Sen, R., Ishak, H. D., Estrada, D., Dowd, S. E., Hong, E., and Mueller, U. G. (2009). Generalized antifungal activity and 454-screening of Pseudonocardia and Amycolatopsis bacteria in nests of fungus-growing ants. Proc. Natl. Acad. Sci. U.S.A. 106, 17805–17810. doi: 10.1073/pnas.0904827106
Singh, S., Singh, A., Baweja, V., Roy, A., Chakraborty, A., and Singh, I. K. (2021). Molecular rationale of insect-microbes symbiosis—from insect behaviour to mechanism. Microorganisms 9:2422. doi: 10.3390/microorganisms9122422
Six, D. L. (2013). The bark beetle holobiont: why microbes matter. J. Chem. Ecol. 39, 989–1002. doi: 10.1007/s10886-013-0318-8
Skrodenytė-Arbačiauskienė, V., Radžiutė, S., Stunžėnas, V., and Būda, V. (2012). Erwinia typographi sp. nov., isolated from bark beetle (Ips typographus) gut. Int. J. Syst. Evol. Microbiol. 62(Pt. 4), 942–948. doi: 10.1099/ijs.0.030304-0
Stat, M., Pochon, X., Franklin, E. C., Bruno, J. F., Casey, K. S., Selig, E. R., et al. (2013). The distribution of the thermally tolerant symbiont lineage (Symbiodinium clade D) in corals from Hawaii: correlations with host and the history of ocean thermal stress. Ecol. Evol. 3, 1317–1329. doi: 10.1002/ece3.556
Tanin, S. M., Kandasamy, D., and Krokene, P. (2021). Fungal interactions and host tree preferences in the spruce bark beetle Ips typographus. Front. Microbiol. 12:695167. doi: 10.3389/fmicb.2021.695167
Valiev, A., Ogel, Z. B., and Klepzig, K. D. (2009). Analysis of cellulase and polyphenol oxidase production by southern pine beetle associated fungi. Symbiosis 49, 37–42.
Veselská, T., Švec, K., Kostovčík, M., Peral-Aranega, E., Garcia-Fraile, P., Křížková, B., et al. (2023). Proportions of taxa belonging to the gut core microbiome change throughout the life cycle and season of the bark beetle Ips typographus. FEMS Microbiol. Ecol.
Vuilleumier, S., and Pagni, M. (2002). The elusive roles of bacterial glutathione S-transferases: new lessons from genomes. Appl. Microbiol. Biotechnol. 58, 138–146. doi: 10.1007/s00253-001-0836-0
Wadke, N., Kandasamy, D., Vogel, H., Lah, L., Wingfield, B. D., Paetz, C., et al. (2016). The bark-beetle-associated fungus, Endoconidiophora polonica, utilizes the phenolic defense compounds of its host as a carbon source. Plant Physiol. 171, 914–931. doi: 10.1104/pp.15.01916
Wang, J., Gao, G., Zhang, R., and Chen, H. (2017). Metabolism and cold tolerance of Chinese white pine beetle Dendroctonus armandi (Coleoptera: Curculionidae: Scolytinae) during the overwintering period. Agric. For. Entomol. 19, 10–22.
Wang, K., Miettinen, I., Jaber, E. H., and Asiegbu, F. O. (2023). “Anatomical, chemical, molecular, and genetic basis for tree defenses,” in Forest microbiology, eds F. Asiegbu and A. Kovalchuk (Amsterdam: Elsevier), 33–57.
Wang, Y., Kapun, M., Waidele, L., Kuenzel, S., Bergland, A. O., and Staubach, F. (2020). Common structuring principles of the Drosophila melanogaster microbiome on a continental scale and between host and substrate. Environ. Microbiol. Rep. 12, 220–228. doi: 10.1111/1758-2229.12826
Wang, Y., Lim, L., Madilao, L., Lah, L., Bohlmann, J., and Breuil, C. (2014). Gene discovery for enzymes involved in limonene modification or utilization by the mountain pine beetle-associated pathogen Grosmannia clavigera. Appl. Environ. Microbiol. 80, 4566–4576.
Weller, D. M. (2007). Pseudomonas biocontrol agents of soilborne pathogens: looking back over 30 years. Phytopathology 97, 250–256. doi: 10.1094/PHYTO-97-2-0250
White, T. J., Bruns, T., Lee, S., and Taylor, J. (1990). “Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics,” in PCR protocols: a guide to methods and applications, Vol. 18, eds M. Innis, D. Gelfand, J. Sninsky, and T. White (San Diego, CA: Academic Press), 315–322.
Wingfield, M. J. (1995). “Do conifer bark beetles require fungi to kill trees?” in Barkbeetles, blue-stain fungi, and conifer defence systems, ed. E. Christiansen (Norway: Aktuelt fra Skogforsk).
Xu, L. T., Lu, M., and Sun, J. H. (2016). Invasive bark beetle-associated microbes degrade a host defensive monoterpene. Insect Sci. 23, 183–190. doi: 10.1111/1744-7917.12255
Xu, L., Lou, Q., Cheng, C., Lu, M., and Sun, J. (2015). Gut-associated bacteria of Dendroctonus valens and their involvement in verbenone production. Microb. Ecol. 70, 1012–1023. doi: 10.1007/s00248-015-0625-4
Yun, J.-H., Roh, S. W., Whon, T. W., Jung, M.-J., Kim, M.-S., Park, D.-S., et al. (2014). Insect gut bacterial diversity determined by environmental habitat, diet, developmental stage, and phylogeny of host. Appl. Environ. Microbiol. 80, 5254–5264. doi: 10.1128/AEM.01226-14
Zaman, R., May, C., Ullah, A., and Erbilgin, N. (2023). Bark beetles utilize ophiostomatoid fungi to circumvent host tree defenses. Metabolites 13, 239. doi: 10.3390/metabo13020239
Zhao, T., Axelsson, K., Krokene, P., and Borg-Karlson, A.-K. (2015). Fungal symbionts of the spruce bark beetle synthesize the beetle aggregation pheromone 2-methyl-3-buten-2-ol. J. Chem. Ecol. 41, 848–852. doi: 10.1007/s10886-015-0617-3
Zhao, T., Ganji, S., Schiebe, C., Bohman, B., Weinstein, P., Krokene, P., et al. (2019a). Convergent evolution of semiochemicals across Kingdoms: bark beetles and their fungal symbionts. ISME J. 13, 1535–1545. doi: 10.1038/s41396-019-0370-7
Zhao, T., Kandasamy, D., Krokene, P., Chen, J., Gershenzon, J., and Hammerbacher, A. (2019b). Fungal associates of the tree-killing bark beetle, Ips typographus, vary in virulence, ability to degrade conifer phenolics and influence bark beetle tunneling behavior. Fungal ecol. 38, 71–79.
Keywords: IPS typographus, amplicon sequence variants (ASVs), symbiosis, holobiont, metagenomic analysis, microbial biomarkers, wood microbiome, spruce
Citation: Chakraborty A, Purohit A, Khara A, Modlinger R and Roy A (2023) Life-stage and geographic location determine the microbial assemblage in Eurasian spruce bark beetle, Ips typographus L. (Coleoptera: Curculionidae). Front. For. Glob. Change 6:1176160. doi: 10.3389/ffgc.2023.1176160
Received: 28 February 2023; Accepted: 29 June 2023;
Published: 13 July 2023.
Edited by:
Juan A. Martin, Polytechnic University of Madrid, SpainReviewed by:
Dezene Philip Wiebe Huber, University of Northern British Columbia, CanadaGerardo Zúñiga, National Polytechnic Institute, Mexico
Copyright © 2023 Chakraborty, Purohit, Khara, Modlinger and Roy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amit Roy, Um95QGZsZC5jenUuY3o=
†These authors have contributed equally to this work