 Joana F. Marques
Joana F. Marques Inis Winde1†
Inis Winde1† Anna Maria Jönsson
Anna Maria Jönsson Olle Anderbrant
Olle Anderbrant- 1Department of Biology, Lund University, Lund, Sweden
- 2Department of Physical Geography and Ecosystem Science, Lund University, Lund, Sweden
Introduction: Scale insects (Hemiptera: Coccomorpha) feed on a wide variety of agricultural crops and forest and ornamental trees worldwide. These pest insects damage plants not only by causing sap loss but also by reducing the plant's photosynthetic activity. This is because the honeydew they produce acts as a substrate for mold, which covers leaf surfaces. In the last decades, several outbreaks of Physokermes spp. (soft-scale insects) have occurred throughout Europe and have partly been attributed to unusual weather conditions or climate change, as some species seem to be expanding their distribution range. However, the small size of these insects and their large intraspecific morphological variation have hindered the identification of the species responsible for outbreaks.
Methods: In this study, mitochondrial DNA (cytochrome c oxidase subunit I, COI), ribosomal RNA (28S), and nuclear (elongation factor 1α, EF1α) DNA markers were used to reconstruct the phylogenetic relationships of four Physokermes species sampled throughout Europe in 2013–2015.
Results and discussion: The results allowed us to clearly distinguish P. hellenicus and P. inopinatus from each other, as these appeared in well-supported clades in the phylogenetic trees and from P. piceae and P. hemicryphys. However, P. hemicryphus appeared in a single clade in trees based on 28S and EF1α but among P. piceae in the COI tree. Further investigations are therefore required to determine the taxonomic status of P. piceae and P. hemicryphys, which seem to comprise a species complex.
1. Introduction
Scale insects (Hemiptera: Coccomorpha) are important plant pests feeding on a wide range of agricultural crops as well as on forest and ornamental trees worldwide (Park et al., 2011). In addition to directly affecting host plants through sap loss, the honeydew produced by scale insects covers leaf surfaces, allowing mold development and thus reducing photosynthetic activity (Kozár et al., 2012). Because they are small and frequently found living within the plant, scale insects are often unseen. This contributes to their invasive potential and presents a huge challenge for plant quarantine (Andersen et al., 2010) and a major phytosanitary problem. The genus Physokermes comprises an important group of soft-scale insects infesting coniferous plants in the Holarctic region. In total, 13 species are currently recognized within Physokermes (Papanastasiou et al., 2021), but only four are known to occur in Europe: the widespread P. hemicryphus (Dalman) and P. piceae (Schrank), the more easterly distributed P. inopinatus (Danzig & Kozár), and P. hellenicus, (Kozár & Gounari) described from Abies cephalonica (Pinaceae) in Greece (Kozár et al., 2012). Although commonly known as the Hungarian spruce scale, P. inopinatus caused a mass infestation in Sweden in 2010 (McCarthy and Skovsgaard, 2011), suggesting a considerable northward expansion of this species' distribution, possibly linked to climate change or the unrestricted transport of plants within Europe (Kozár et al., 2012). Indeed, the attack by P. inopinatus was triggered by a sequence of droughts that contributed to population expansions in the relatively dry summers of 2008–2009, leading to the severe mass infestation of 2010 (McCarthy and Skovsgaard, 2011; Olsson et al., 2012; Gertsson and Isacsson, 2014; Winde et al., 2018).
However, the identity of the species causing the 2010 outbreak in Sweden was not immediately determined. This was because morphological identification of scale insects requires a high level of expertise and is time consuming. It mostly relies on characters present only in adult females, which is difficult when the morphological variation observed between females of closely related species corresponds to natural intraspecific variation instead of interspecific variation (Malausa et al., 2011; Park et al., 2011). The limitations of this methodology have also raised concern about the possible non-identification of cryptic species complexes (Andersen et al., 2010), and thus, several DNA markers have been developed to distinguish between closely related species of scale insects, mainly mealybugs and armored scales (Kondo et al., 2008; Andersen et al., 2010; Gwiazdowski et al., 2010; Malausa et al., 2011 and references therein; Park et al., 2010; 2011; Abd-Rabou et al., 2012; Beltrà et al., 2012; Deng et al., 2012; Sethusa et al., 2014). Such markers include a set of gene regions providing reliable and rapid identification of scale insect species, even when only immature and male specimens are available. Although DNA barcoding based on cytochrome c oxidase subunit I (COI) sequence variation has become a popular tool for species delimitation in several insect groups (Hajibabaei et al., 2006), and therefore an ideal candidate for identifying scale insects (e.g., Kondo et al., 2008; Malausa et al., 2011; Park et al., 2011; Abd-Rabou et al., 2012; Beltrà et al., 2012; Deng et al., 2012; Sethusa et al., 2014), only 2,172 COI sequences of Coccidae are deposited in GenBank (http://www.ncbi.nlm.nih.gov; accessed November 8, 2022). Gene 28S has also been used to identify insect species (Campbell et al., 1994; Monaghan et al., 2006; Smith et al., 2008). Although lacking sufficient variation to delimitate some species (Park et al., 2011; Deng et al., 2012), gene 28S has been proposed as a complementary marker to COI in scale insects (Sethusa et al., 2014). This supports van Nieukerken et al. (2012) suggestion of including at least one nuclear marker in addition to COI in studies concerning cryptic lineages. Thus, in this study, three genes from three independent loci were amplified: COI (mitochondrial DNA), 28S (D2 and D3 regions, ribosomal RNA gene), and elongation factor 1alpha (EF1α, nuclear DNA). These genes represent a range of different rates of evolution: COI is a relatively fast-evolving mitochondrial gene (Simon et al., 1994), 28S is relatively conserved and represented in the nuclear genome as numerous tandem repeats (Hoy, 1994), and EF1α is a low-copy number nuclear protein-coding gene with conserved exons and variable introns (Hoy, 1994).
The DNA sequence analysis performed in this study for several Physokermes spp. populations across Europe aimed to (i) scan for cryptic species within taxonomically recognized Physokermes species; (ii) study the phylogenetic relationships between the identified taxa; and (iii) obtain a series of molecular markers suitable for the reliable identification of closely related Physokermes spp. in Europe, especially those with invasive pest status, thus contributing to more efficient pest management and quarantine procedures. Finding cryptic species will also provide the necessary background and molecular evidence to conduct thorough morphological studies on Physokermes specimens representative of several species. This will allow laying the foundations for a contemporary taxonomic revision and for the recognition of morphological characters important to species differentiation, similar to what has been done for Chionaspis spp. by Vea et al. (2012) following the work of Gwiazdowski et al. (2010). To date, only one molecular study of Physokermes spp. has been published, and it included COI sequence analysis of P. hellenicus from different Greek populations (Papanastasiou et al., 2018).
2. Materials and methods
2.1. Taxon sampling
In total, 203 P. inopinatus, P. piceae, P. hemicryphus, and P. hellenicus individuals were collected across their distribution ranges in Europe, i.e., from Turkey, in the southeast, to Sweden, in the northwest (Table 1). Sampling occurred in 2013–2015, and insects were immediately placed in 95% ethanol and kept at −20°C for DNA preservation. Taxonomic identity was determined by experienced scale insect specialists (see Acknowledgments) based on discriminating morphological characters published in reference studies (e.g., Kosztarab and Kozár, 1988; Kozár et al., 2012).
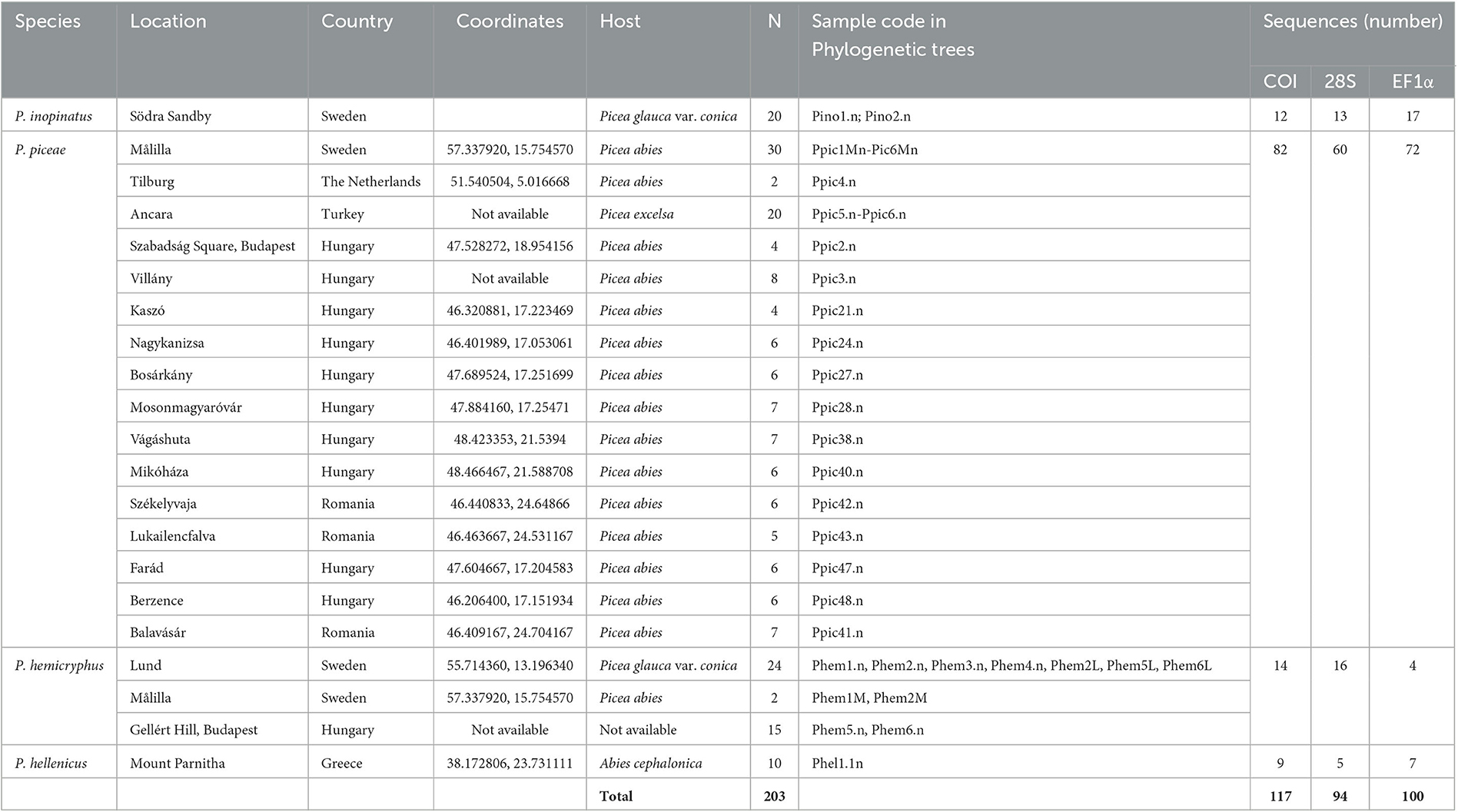
Table 1. Collection sites and the number of Physokermes spp. individuals examined in the present study, and the number of cytochrome oxidase I (COI), 28S, and elongation factor 1 α (EF1α) sequences obtained.
2.2. DNA extraction, amplification, and sequencing
Total genomic DNA was isolated from the 203 individuals using the QIAamp? DNA Mini Kit (QIAGEN, Hilden, Germany), following the manufacturer's protocol for the purification of total DNA from insects. The nucleotide sequences including the barcoding region of COI, 28S, and EF1α were amplified via PCR using the 5′-tailed primers and profiles described in Table 2. Amplification reaction mixtures (total volume = 20 μL) contained 10 μl DreamTaqTM PCR Master Mix (FermentasTM, Waltham, MA, USA), 1 μl each primer, 6 μl DNA-free water, and 2 μl template DNA. To check for contamination, negative controls (sterile water instead of template DNA) were included in each reaction array. The resulting PCR products were then visualized under UV light in 1% agarose gels stained with SYBRTM Safe DNA Gel Stain (ThermoFisher Scientific, Waltham, MA, USA). Those representing successful reactions were prepared for sequencing by digesting 5 μl of the PCR mixture with 0.5 μl Exonuclease I, and 1 μl FastAPTM Thermosensitive Alkaline Phosphatase (both from FermentasTM) during a thermal cycle consisting of 15 min at 37°C and 15 min at 85°C. PCR products were then sequenced in both directions, to ensure high-quality reads, using PCR primers (Table 2) and the BigDye Terminator Cycle Reaction Kit (Applied Biosystems, Foster City, CA, USA).
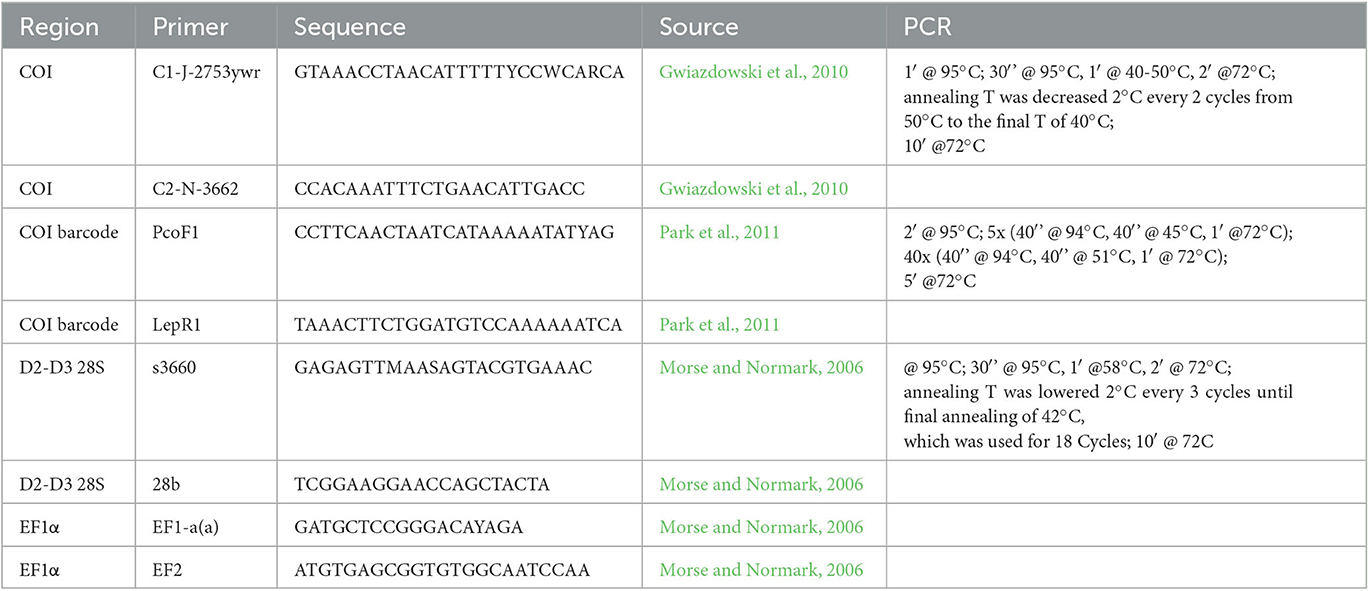
Table 2. Primers and PCR profiles used to amplify cytochrome oxidase I (COI), 28S, and elongation factor 1 α (EF1α) gene fragments in Physokermes spp.
2.3. Phylogenetic analyses
Contiguous sequences were assembled in Geneious® 8.1.15 (Biomatters, Auckland, New Zealand), aligned using the default parameters and cost matrix of the Geneious® alignment algorithm as included in Geneious® 8.1.15 and edited by eye to maximize blocks of sequence identity. Sequences were checked for homology by comparison with those deposited in GenBank, using the Basic Local Alignment Search Tool (BLAST) available on the NCBI website (NCBI, http://www.ncbi.nlm.nih.gov/Genbank). Translation into amino acids was also performed to confirm data correctness.
Using PartitionFinder 2 (Lanfear et al., 2012), the best partitioning scheme and model of nucleotide substitution were selected for each dataset based on the Bayesian information criterion. All possible combinations of codon positions were considered as candidate partitions (i.e., from a single partition for the entire sequence to each codon position treated as a partition), but only the nucleotide substitution models supported by MrBayes 3.2.6 (Ronquist and Huelsenbeck, 2003) were included in the candidate model pool. This procedure was repeated five times to ensure correct estimation. The resulting models and partition schemes were used in phylogenetic reconstructions based on maximum likelihood (ML) and Bayesian Inference (BI), performed in Garli 2.1 (Zwickl, 2006) and MrBayes 3.2.6, respectively. Because considerable statistical inconsistency can be found when using the maximum parsimony method of phylogenetic reconstruction on this type of data (different rates of evolution), it was not performed in this study.
In Garli, clade stability was evaluated by non-parametric bootstrapping (five independent search replicates of 10,000 generations each). In MrBayes, and starting from a random tree, alternative tree topologies were evaluated through the estimation of posterior probabilities by Markov chain Monte Carlo re-sampling. These were performed in two runs of four chains each (with the default settings implemented in MrBayes) for 10 × 106 generations with a sampling frequency of 100 (average standard deviation of split frequencies <0.01). These samples were then used to generate 50% majority-rule consensus trees. Commands “sumt” and “burnin = 5,000” were set to discard trees that did not reach the likelihood score detected with the “sump” command. Only bootstrap and posterior probabilities values >80% were considered to support clades.
The species delimitation plugin implemented in Geneious® was used to evaluate different scenarios of lineage divergence within Physokermes spp. based on a neighbor-joining (NJ) tree constructed for COI sequences. The distance-based species divergence criterion relies on the postulate that intraspecific genetic variation is small relative to interspecific variation. In this study, two clusters containing a pair of nearest neighbors with >2% distance from each other were considered different species, as this threshold has been proven efficient to discriminate other insect groups (Foottit et al., 2009; Zhu et al., 2017). Only monophyletic clades showing bootstrap support >80% were tested.
3. Results and discussion
The BLAST of all sequences against the NCBI database revealed that some (most of them corresponding to 28S sequences) matched those of internal parasites, parasitoids, or microorganisms infecting Physokermes spp.; hence, these sequences were excluded from further analysis. This higher-than-expected ratio of non-specific amplification might be due to the binding of one or both PCR primers to a sequence other than the target one, which is frequently a problem in the amplification of conserved sequences, such as ribosomal DNA. The 117 COI sequences (total length = 712 bp) and the 94 28S sequences (total length = 842 bp) presented the highest sequence homology to Physokermes jezoensis Siraiwa isolate S2, whereas the 100 EF1α sequences (total length = 1,056 bp) presented the highest sequence homology to Paralecanium sp. (Coccomorpha) isolate D0764B. These BLAST hits are probably the result of the very low number of Physokermes species nucleotide sequences deposited in GenBank (n = 22, Available online at: http://www.ncbi.nlm.nih.gov; accessed November 8, 2022). In addition, the handling of specimens for morphological identification might have damaged the DNA quality leading to amplified fragments shorter than ideal for BLAST hits. Nevertheless, the most similar sequences were used as outgroups in the subsequent phylogenetic analyses. The sequences obtained and analyzed in the present study are deposited in GenBank under accession numbers OQ433782–898 (COI), OQ470427–526 (EF1α), and OQ657360–453 (28S).
According to PartitionFinder, the best nucleotide substitution model for Physokermes spp. COI evolution was a generalized time reversible (GTR) with a gamma-distributed rate variation (G) among sites, whereas 28S evolved according to the Hasegawa–Kishino–Yano (HKY) model with a proportion of invariable sites (I). A partitioned model of evolution was found for EF1α sequences, with the first two positions evolving according to Felsenstein's 1981 model (F81) with G and the third according to HKY. The ML and BI analyses performed for all datasets (Figure 1) revealed similar results, although BI usually presented higher support values, and therefore, these are the trees presented here. One clade comprising P. hellenicus and another comprising P. inopinatus were clearly evidenced and well-supported in all trees. However, only 28S and EF1α BI trees evidenced clades comprising P. hemicryphus alone (Figures 1B, C, respectively), with this species appearing mostly among P. piceae and P. inopinatus. In addition, P. hemicryphus samples from Hungary (Phem5.n and Phem6.n) appeared in a different clade from P. hemicryphus collected in Sweden (all other Phem codes). These populations do not seem to constitute host races, because they were both sampled from Picea abies, but might have been isolated by distance. Thus, the 28S nuclear gene is complementary to COI in Physokermes spp. identification, as proposed by Sethusa et al. (2014).
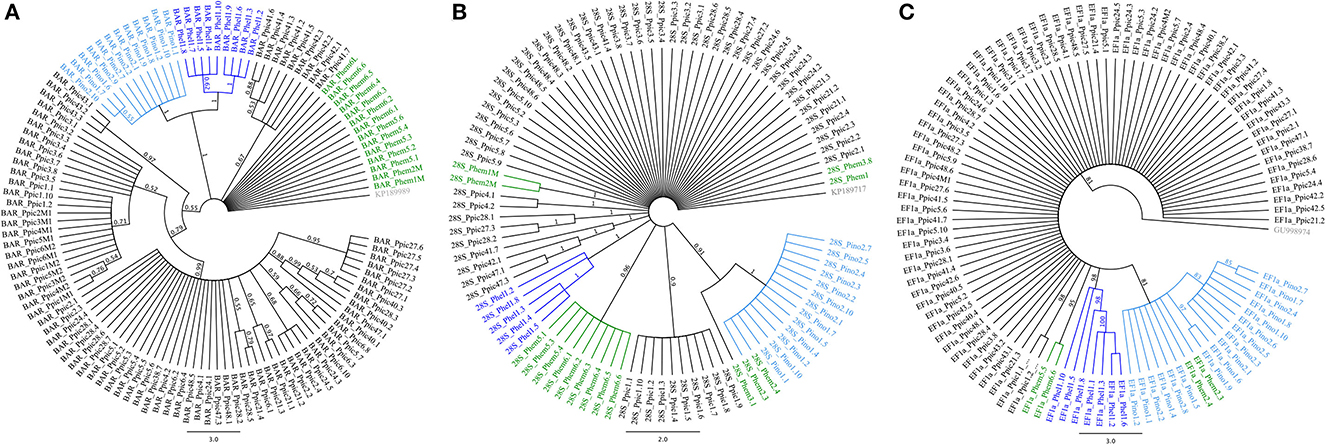
Figure 1. Bayesian inference phylogenetic trees constructed for Physokermes inopinatus (Pino), P. hellenicus (Phel), P. piceae (Ppic), and P. hemicryphus (Phem), based on cytochrome oxidase I [COI, (A)], 28S (B), and elongation factor 1 α [EF1α, (C)] sequences, using the generalized time reversible (GTR) model with a gamma-distributed rate variation (G) among sites, the Hasegawa–Kishino–Yano (HKY) model with a proportion of invariable sites (I), and a partitioned model in which the first two positions evolved according to Felsenstein's 1981 model (F81) with G and the third according to HKY, respectively. Posterior probabilities (PP) are shown above branches; only clades with PP > 0.80 were considered well-supported. Numbers (and letters) next to species codes correspond to sample codes; numbers (and letters) before the dot to population codes; and the numbers (and letters) after the dot to individuals (see Table 1).
The NJ tree produced for the COI sequences (Figure 2) also evidenced well-supported clades containing P. hellenicus and P. inopinatus, and three other clades corresponding to P. piceae from Kaszó in Hungary (Ppic21.1 and Ppic21.2) and from Balavásár (Ppic41.2 and Ppic41.3) and Lukailencfalva (P.pic43.1, Ppic43.2, and Ppic43.3) in Romania. All these clades were monophyletic and their distance to the nearest neighbor varied from 3.5% (between P. piceae clades from Romania) to 6.3% (between P. inopinatus and P. hellenicus). These results support the distinction between P. inopinatus and P. hellenicus, as indicated in the ML and BI analyses. Moreover, the genetic distance found is similar to the congeneric 5.8% average mitochondrial DNA divergence (Pons et al., 2006) and within the 0.0–7.95% range (Hebert et al., 2004) reported for insects. The clade support and genetic distances found within P. piceae from Hungary and Romania (>2%) suggest the existence of cryptic species. Similar to other scale insects, Physokermes spp. females are flightless, males have a short lifespan, and both sexes have low dispersal capacity, all contributing to population differentiation in different areas and/or host species and promoting cryptic speciation, as observed in Aspidiotus nerii in Australia (Andersen et al., 2010) and in Chionaspis heterophyllae and C. pinifoliae in North America (Gwiazdowski et al., 2010). Because all the genetically differentiated P. piceae were sampled from P. abies, host species does not appear to be the factor contributing to such differentiation. Although explanations for the recent outbreaks remain elusive, particularly given the limited dispersal ability of Physokermes spp., changes in climate or weather conditions inducing rapid reproduction of endemic (and thus escaping detection) populations are more likely than sudden range expansions.
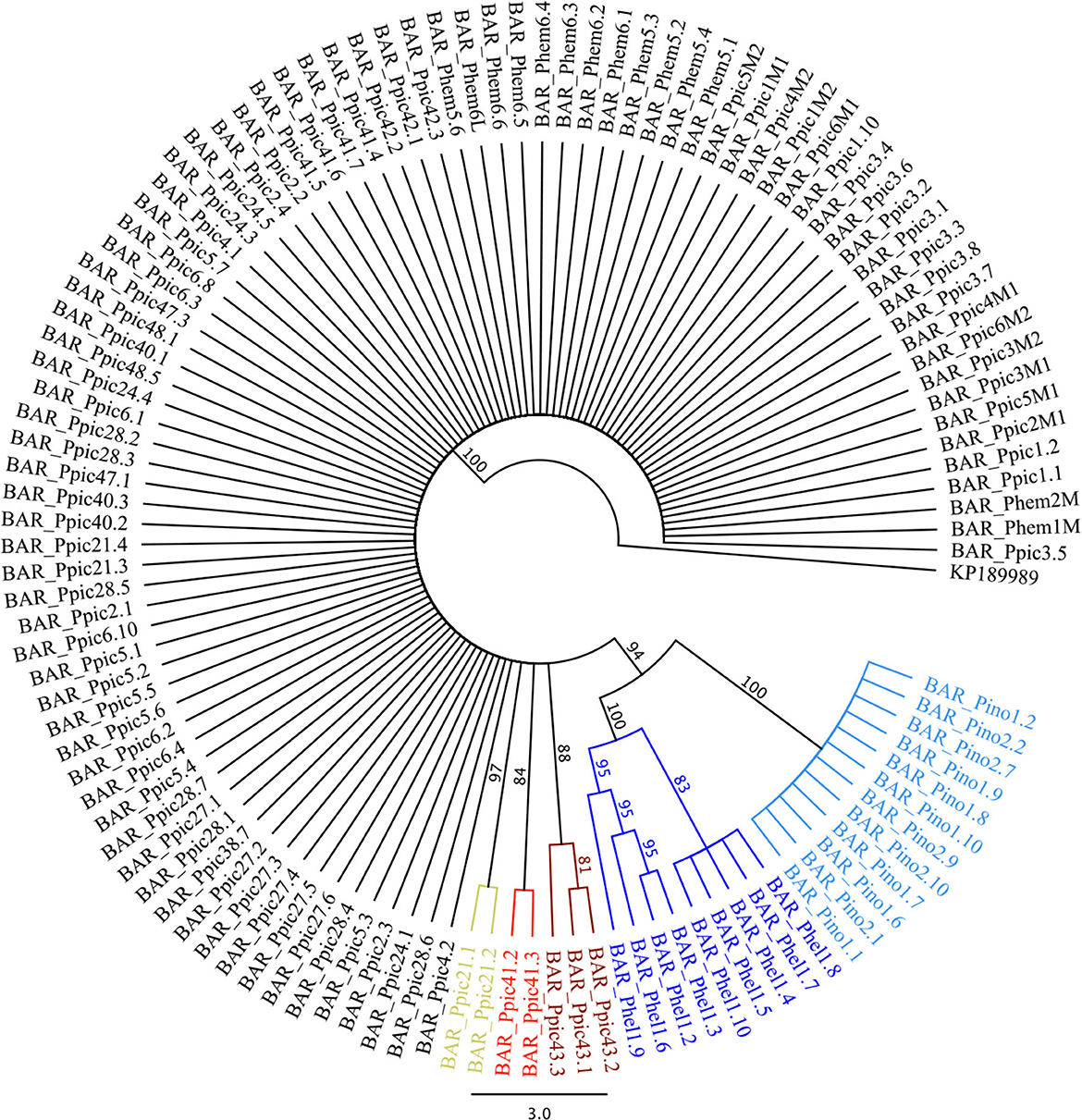
Figure 2. Neighbor-joining tree produced for the cytochrome oxidase I (COI) sequences obtained for Physokermes inopinatus (Pino), P. hellenicus (Phel), P. piceae (Ppic), and P. hemicryphus (Phem) to test different scenarios of species divergence, using the species delimitation plugin implemented in Geneious® 8.1.15. Clades with bootstrap support >80% were tested (presented in different colors). Numbers following species codes correspond to population codes (see Table 1). Physokermes jezoensis (GenBank accession KP189989) was used as an outgroup.
Overall, the results obtained here (i) clearly distinguished the closely related P. hellenicus, P. inopinatus, and (P. piceae + P. hemicryphus); (ii) revealed the existence of intraspecific variation and, possibly, of unknown complexes of species; and (iii) confirmed the value of using 28S in addition to COI for identifying scale insects. However, the differentiation of P. hemicryphus suggested in the nuclear data (particularly in 28S) analyses was not supported in the species delimitation analysis, as P. hemicryphus was not genetically distant from P. piceae. However, the sample size of P. hemicryphus and its geographic range was much smaller than that of P. piceae, and thus, further studies are needed to confirm the taxonomic status of P. hemicryphus.
Data availability statement
The gene sequences for this study can be found in GeneBank at https://www.ncbi.nlm.nih.gov/nuccore/?term=OQ433782:OQ433898[accn] for COI; https://www.ncbi.nlm.nih.gov/nuccore/?term=OQ4704273:OQ470526[accn] for EF1α; https://www.ncbi.nlm.nih.gov/nuccore/?term=OQ657360:OQ657453[accn] for 28S.
Author contributions
OA, AJ, and IW secured funding. IW ensured access to specimens. JM performed the genetic analyses and wrote the first draft of the manuscript. All authors conceived the study, contributed to the manuscript, and approved the submitted version.
Funding
The study is a contribution to the Strategic Research Area Biodiversity and Ecosystem Services in a Changing Climate (BECC), and it was supported by the Carl Trygger Foundation for Scientific Research (CTS 14:17), the Tranemåla Foundation, and the Swedish Research Council Formas (Svenska Forskningsrådet Formas) (2010-822).
Acknowledgments
We would like to thank Éva Szita, Selma Ülgentük, Maurica Jansen, Gyuri Csóka, and Iosif Papanastasiou for generously supplying the insect samples, making our investigation possible and Carl-Axel Gertsson for additional species identity verification and general support to our work.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abd-Rabou, S., Shalaby, H., Germain, J. F., Ris, N., Kreiter, P., and Malausa, T. (2012). Identification of mealybug pest species (Hemiptera: Pseudococcidae) in Egypt and France, using a DNA barcoding approach. Bull. Entomol. Res. 102, 515–523. doi: 10.1017/S0007485312000041
Andersen, J. C., Normark, B. B., Morse, G. E., and Gruwell, M. E. (2010). Cryptic diversity in the Aspidiotus nerii complex in Australia. Ann. Entomol. Soc. Am. 103, 844–854. doi: 10.1603/AN10060
Beltrà, A., Soto, A., and Malausa, T. (2012). Molecular and morphological characterisation of Pseudococcidae surveyed on crops and ornamental plants in Spain. Bull. Entomol. Res. 102, 165–172. doi: 10.1017/S0007485311000514
Campbell, B. C., Steffen-Campbell, J. D., and Werren, J. H. (1994). Phylogeny of the Nasonia species complex (Hymenoptera: Pteromalidae) inferred from an internal transcribed spacer (ITS2) and 28S rDNA sequences. Insect Mol. Biol. 2, 225–237. doi: 10.1111/j.1365-2583.1994.tb00142.x
Deng, J., Yu, F., Zhang, T. X., Hu, H. Y., Zhu, CD, Wu, S.-A, et al. (2012). DNA barcoding of six Ceroplastes species (Hemiptera: Coccoidea: Coccidae) from China. Mol. Ecol. Resour. 12, 791–796. doi: 10.1111/j.1755-0998.2012.03152.x
Foottit, R. G., Maw, H. E. L., and Pike, K. S. (2009). DNA barcodes to explore diversity in aphids (Hemiptera: Aphididae and Adelgidae). Redia 92, 87–91.
Gertsson, C.-A., and Isacsson, G. (2014). The Hungarian spruce scale, Physokermes inopinatus Danzig and Kozár (Hemiptera: Coccoidea: Coccidae) in Sweden. Acta Zool. Bulg. Suppl. 6, 83–86.
Gwiazdowski, R. A., Vea, I. M., Andersen, J., and Normark, B. B. (2010). Discovery of cryptic species among North American pine-feeding Chionaspis scale insects (Hemiptera: Diaspididae). Biol. J. Linn. Soc. 104, 47–62. doi: 10.1111/j.1095-8312.2011.01716.x
Hajibabaei, M., Janzen, D. H., Burns, J. M., Hallwachs, W., and Hebert, P. D. N. (2006). DNA barcodes distinguish species of tropical Lepidoptera. Proc. Natl. Acad. Sci. U.S.A. 103, 968–971. doi: 10.1073/pnas.0510466103
Hebert, P. D., Stoeckle, M. Y., Zemlak, T. S., and Francis, C. M. (2004). Identification of birds through DNA barcodes. PLoS Biol. 2, e312. doi: 10.1371/journal.pbio.0020312
Hoy, M. A. (1994). “Insect molecular systematics and evolution,” in Insect Molecular Genetics: An Introduction to Principles and Applications, ed M. A. Hoy (San Diego, CA: Academic Press) 337–387. doi: 10.1016/B978-0-12-357490-9.50016-X
Kondo, T., Gullan, P. J., and Williams, D. J. (2008). Coccidology. The study of scale insects (Hemiptera: Sternorrhyncha: Coccoidea). Revista Corpoica–Cienc. y Tecnol. Agropecu. 9, 55–61. doi: 10.21930/rcta.vol9_num2_art:118
Kosztarab, M., and Kozár, F. (1988). Scale Insects of Central Europe. Dr W. Junk, Budapest, Hungary. doi: 10.1007/978-94-009-4045-1
Kozár, F., Gounari, S., Hodgson, C. J., Fetyk, K., and Goras, G. (2012). A new species of Physokermes Targioni Tozzetti (Hemiptera: Coccoidea: Coccidae) from Greece. Zootaxa 3566, 23–38. doi: 10.11646/zootaxa.3566.1.3
Lanfear, R., Calcott, B., Ho, S. Y. W., and Guindon, S. (2012). Partitionfinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 29, 1695–1701. doi: 10.1093/molbev/mss020
Malausa, T., Fenis, A., Warot, S., Germain, J. F., Ris, N., Prado, E., et al. (2011). DNA markers to disentangle complexes of cryptic taxa in mealybugs (Hemiptera: Pseudococcidae). J. Appl. Entomol. 135, 142–155. doi: 10.1111/j.1439-0418.2009.01495.x
McCarthy, R., and Skovsgaard, J. P. (2011). Hungarian spruce scale on Norway spruce in southern Sweden: Correlation with climate, site and stand factors. Southern Swedish Forest Research Centre, SLU.
Monaghan, M. T., Balke, M., Pons, J., and Vogler, A. P. (2006). Beyond barcodes: complex DNA taxonomy of a south pacific island radiation. Proc. Royal Soc. B 273, 887–893. doi: 10.1098/rspb.2005.3391
Morse, E. M., and Normark, B. B. (2006). A molecular phylogenetic study of armoured scale insects (Hemiptera: Diaspididae). Syst. Entomol. 31, 338–349. doi: 10.1111/j.1365-3113.2005.00316.x
Olsson, P.-O., Jönsson, A. M., and Eklundh, L. (2012). A new invasive insect in Sweden – Physokermes inopinatus: Tracing forest damage with satellite based remote sensing. For. Ecol. Manage. 285, 29–37. doi: 10.1016/j.foreco.2012.08.003
Papanastasiou, I., Evangelou, V., Papoutsis, L., Bouga, M., and Emmanouil, N. (2018). Molecular taxonomy of the genus Physokermes (Hemiptera: Coccidae) species in Greece, based on mtDNA sequencing data. J. Apic. Res. 57, 479–483. doi: 10.1080/00218839.2018.1494886
Papanastasiou, I., Kavallieratos, N. G., Papadoulis, G. T., Emmanouli, C., and Emmanouel, N. G. (2021). Geographical distribution and long-term monitoring of Physokermes hellenicus (Hemiptera: Coccomorpha: Coccidae) on Abies spp. (Pinales: Pinaceae) in Greece. Insects 12, 1001. doi: 10.3390/insects12111001
Park, D.-S., Suh, S.-J., Hebert, P. D. N., Oh, H.-W., and Hong, K.-J. (2011). DNA barcodes for two scale insect families, mealybugs (Hemiptera: Pseudococcidae) and armoured scales (Hemiptera: Diaspididae). Bull. Entomol. Res. 101, 429–434. doi: 10.1017/S0007485310000714
Park, D.-S., Suh, S.-J., Oh, H.-W., and Hebert, P. D. N. (2010). Recovery of the mitochondrial COI barcode region in diverse Hexapoda through tRNA-based primers. BMC Genomics 11, 423. doi: 10.1186/1471-2164-11-423
Pons, J., Barraclough, T. G., Gomez-Zurita, J., Cardoso, A., Duran, D. P., Hazell, S., et al. (2006). Sequence-based species delimitation for the DNA taxonomy of undescribed insects. Syst. Biol. 55, 595–609. doi: 10.1080/10635150600852011
Ronquist, F., and Huelsenbeck, J. P. (2003). Mrbayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. doi: 10.1093/bioinformatics/btg180
Sethusa, M. T., Yessoufou, K., Van der Bank, M., Millar, I. M., and Jacobs, A. (2014). DNA barcode efficacy for the identification of economically important scale insects (Hemiptera: Coccoidea) in South Africa. Afr. Entomol. 22, 257–266. doi: 10.4001/003.022.0218
Simon, C., Frati, F., Beckenbach, A., Crespi, B., Liu, H., and Flook, P. (1994). Evolution, weighting and phylogenetic utility of mitochondrial gene sequences and a compilation conserved polymerase chain reaction primers. Ann. Entomol. Soc. Am. 87, 651–701. doi: 10.1093/aesa/87.6.651
Smith, M. A., Rodriguez, J. J., Whitfield, J. B., Deans, A. R., Janzen, D. H., Hallwachs, W., et al. (2008). Extreme diversity of tropical parasitoid wasps exposed by iterative integration of natural history, DNA barcoding, morphology, and collections. Proc. Natl. Acad. Sci. U.S.A. 105, 12359–12364. doi: 10.1073/pnas.0805319105
van Nieukerken, E. J., Doorenweerd, C., Stokvis, F. R., and Groenenberg, D. S. J. (2012). DNA barcoding of the leaf-mining moth subgenus Ectoedemia s. str. (Lepidoptera: Nepticulidae) with COI and EF1-α: two are better than one in recognizing cryptic species. Contrib. Zool. 81, 1–24. doi: 10.1163/18759866-08101001
Vea, I. M., Gwiazdowski, R. A., and Normark, B. B. (2012). Corroborating molecular species discovery: Four new pine-feeding species of Chionaspis (Hemiptera, Diaspididae). ZooKeys 270, 37–58. doi: 10.3897/zookeys.270.2910
Winde, I., Anderbrant, O., and Jönsson, A. M. (2018). Tree recovery during the aftermath of an outbreak episode of the Hungarian spruce scale in southern Sweden. Scand. J. For. Res. 33, 313–319. doi: 10.1080/02827581.2018.1424932
Zhu, X.-C., Chen, J., Chen, R., Jiang, L.-Y., and Qiao, G.-X. (2017). DNA barcoding and species delimitation of Chaitophorinae (Hemiptera, Aphididae). ZooKeys 656, 25–50. doi: 10.3897/zookeys.656.11440
Keywords: Hungarian spruce scale, phylogeny, invasive species, forest pest, climate change
Citation: Marques JF, Winde I, Jönsson AM and Anderbrant O (2023) Taxonomic relationship among four European Physokermes species (Hemiptera: Coccomorpha) based on nuclear and mitochondrial DNA. Front. For. Glob. Change 6:1167541. doi: 10.3389/ffgc.2023.1167541
Received: 16 February 2023; Accepted: 11 May 2023;
Published: 09 June 2023.
Edited by:
Quan Lu, Chinese Academy of Forestry, ChinaReviewed by:
San-an Wu, Beijing Forestry University, ChinaZhuhong Wang, Fujian Agriculture and Forestry University, China
Copyright © 2023 Marques, Winde, Jönsson and Anderbrant. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Olle Anderbrant, b2xsZS5hbmRlcmJyYW50QGJpb2wubHUuc2U=
†Present address: Inis Winde, Department of Aquatic Sciences and Assessment, Uppsala, Sweden