 Jee-Eun Choi1
Jee-Eun Choi1 Inwha Baek2,3,4*
Inwha Baek2,3,4*- 1Department of Pathology, School of Clinical Medicine, The University of Hong Kong, Pokfulam, Hong Kong SAR, China
- 2College of Pharmacy, Kyung Hee University, Seoul, Republic of Korea
- 3Department of Regulatory Science, Graduate School, Kyung Hee University, Seoul, Republic of Korea
- 4Institute of Regulatory Innovation through Science (IRIS), Kyung Hee University, Seoul, Republic of Korea
Tumors consist of cancer cells with different genetic, epigenetic, and phenotypic properties. Cancer stem cells are an important subpopulation of heterogeneous cancer cells and are capable of initiating and propagating tumors. The term cancer stem cells has become broader in efforts to understand their phenotypic plasticity to switch fates between self-renewal and differentiation. Cancer stem cell plasticity is significantly associated with the initiation of metastasis, resistance to therapy, and tumor recurrence. With our broadened knowledge of epigenetic regulation and metabolic reprogramming as key elements enabling such capabilities, an expansive body of literature has demonstrated the functional importance of each element in contributing to cancer stem cell characteristics. Recently, the direct interplay between epigenetic regulation and metabolic reprogramming has begun to be appreciated in the context of cancer stem cells with growing interest. In this review, we discuss the mechanisms by which cancer stem cells orchestrate the reciprocal regulation of cellular metabolism and epigenetic alterations. In the discussion, compelling, unanswered questions on this topic have been elaborated for the interest of the research community and how recent technological developments help tackle such research ideas. A comprehensive understanding of cancer stem cell attributes that are largely governed by epigenetic and metabolic reprogramming would enable the advancement of precise therapeutic options and the prediction of better responses to drugs, holding great promise in cancer treatment and cure.
1 Introduction
Tumor heterogeneity arises from multiple dimensions, including genetic and non-genetic levels with spatial and temporal differences. Our increasing understanding of tumor heterogeneity sheds light on cancer stem cells (CSCs), a subpopulation of cancer cells that demonstrate a higher capacity for tumor initiation, progression, metastasis, epithelial-mesenchymal plasticity, and drug resistance (Kreso and Dick, 2014; Nassar and Blanpain, 2016; Oskarsson et al., 2014). In addition to genetic heterogeneity within the tumor mass (Dagogo-Jack and Shaw, 2018), different non-genetic events can play functional roles in such capabilities in CSCs (Easwaran et al., 2014; Kim and DeBerardinis, 2019). Metabolic and epigenetic states are the two key non-genetic elements that contribute to CSC characteristics.
The interplay between metabolism and epigenetics in a pool of cancer cells has been extensively studied over the past decade at the population-averaged level (Izzo et al., 2021). In this review, we focus on the direct interplay between metabolic and epigenetic regulations in CSCs, which is a growing area of interest. Potential future research questions that are yet to be answered will be discussed to highlight the significance of this research area, which directly affects the therapeutic response and recurrence rate of patients with cancer.
2 CSCs: a minor but functionally critical subpopulation of cancer cells
Tumors are heterogeneous, as they continuously evolve at multiple layers, including genetic and epigenetic alterations and metabolic reprogramming (Meacham and Morrison, 2013). Cell-intrinsic and cell-extrinsic models to explain intratumor heterogeneity strongly suggest the presence of a tumor subpopulation with a unique ability to initiate and propagate tumors, metastasize with epithelial-mesenchymal plasticity, relapse post-therapy, and develop insensitivity to anti-cancer drugs, now known as CSCs (Kreso and Dick, 2014; Nassar and Blanpain, 2016; Oskarsson et al., 2014) (Figure 1). They are termed as such because of their functional resemblance to normal stem cells, which are characterized by self-renewal and differentiation potentials. Notably, CSCs were first identified in human acute myeloid leukemia (AML) by Bonnet and Dick based on their ability to propagate primary tumors and recreate phenotypic heterogeneity (Bonnet and Dick, 1997; Lapidot et al., 1994). Since pioneering studies, an increasing body of evidence has supported the existence of CSCs in various cancer types, such as breast cancer (Al-Hajj et al., 2003; Ginestier et al., 2007), colorectal cancer (O'Brien et al., 2007; Ricci-Vitiani et al., 2007), and glioblastoma (Singh et al., 2004).
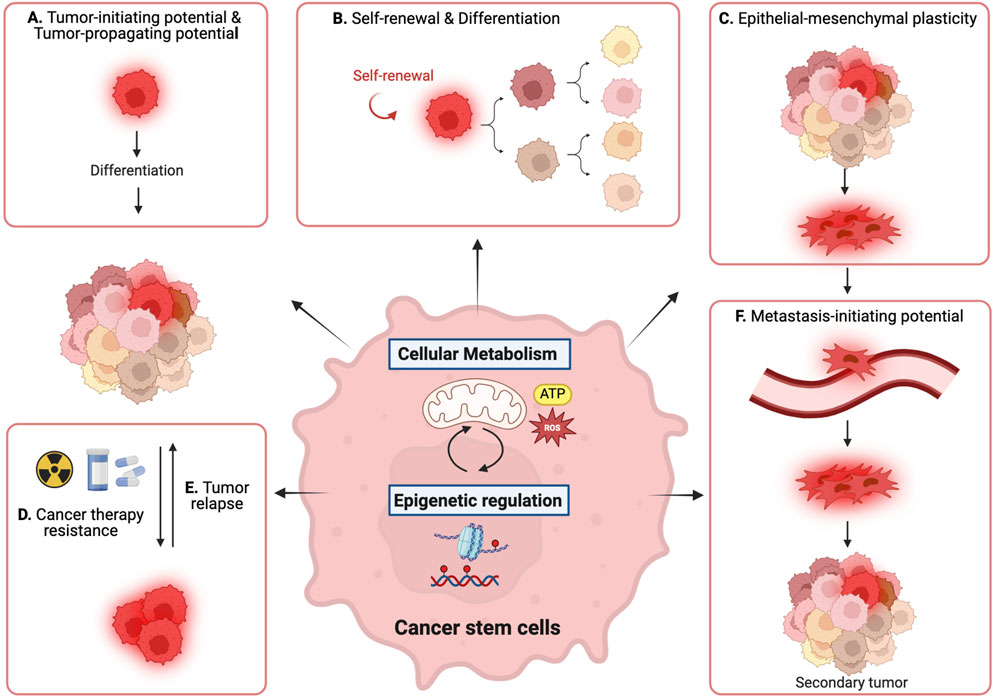
Figure 1. Reciprocal interaction between metabolic pathways and epigenetic regulation governs cancer stem cell (CSC) properties. (A) tumor-initiating and -propagating potential, (B) self-renewal and differentiation, (C) epithelial-mesenchymal plasticity, (D) cancer therapy resistance, (E) tumor relapse, and (F) metastasis-initiating potential.
Furthermore, CSCs are critical targets for anti-cancer therapeutics because of their distinctive roles in metastasis, therapy resistance, and tumor relapse. These abilities are mainly attributed to their phenotypic plasticity, including dedifferentiation and transdifferentiation (Pérez-González et al., 2023). In addition to cell-autonomous dysregulation of cell fate, CSCs can change their fate in response to environmental cues, similar to normal tissue stem cells (Batlle and Clevers, 2017). This dynamic cell fate transition involves the reprogramming of metabolic and epigenetic processes, which have been recognized as cancer hallmarks (Hanahan, 2022). This metabolic transition is thought to be partially achieved through epigenetic regulation. Conversely, metabolic alterations can affect epigenetic states. Metabolism and epigenetic regulation are not distinct but rather intricately connected as important reciprocal regulators of CSC plasticity, orchestrating CSC identity and characteristics. A comprehensive understanding of these two regulatory circuits in CSCs would enable the eradication of aggressive tumor cells and enhance tumor-free survival.
3 Metabolism controls epigenetics in CSCs
Metabolic rewiring is a well-known adaptation of cancer cells that provides survival and growth advantages in response to a tumor microenvironment with limited nutrients and oxygen and with stress (Faubert et al., 2020). Metabolic reprogramming during carcinogenesis and metastasis has been studied extensively over the last two decades (Pavlova and Thompson, 2016). Recently, metabolic heterogeneity has begun to be appreciated within the cancer cell population, along with the discovery of different cell surface markers and the development of metabolite measurement technologies for cellular resolution (Demicco et al., 2024). Remarkably, the metabolic phenotypes of CSCs differ from those of non-CSCs, raising exciting research questions regarding the functional roles of distinct metabolic pathways in CSCs.
Metabolic intermediates can serve as direct substrates or cofactors for the modifications of epigenetic marks (Wellen et al., 2009; Shyh-Chang et al., 2013; Carey et al., 2015). As epigenetic marks can be dynamically regulated during embryonic stem cell (ESC) differentiation, partly under the influence of metabolic changes, the metabolic state is a crucial determinant of cell lineage and identity (Ryall et al., 2015; Moussaieff et al., 2015; Sperber et al., 2015; Etchegaray and Mostoslavsky, 2016). During carcinogenesis, cell fate changes, partly through chromatin rearrangements and DNA/RNA modifications, favoring oncogenic reprogramming. As direct substrates or cofactors of enzymes responsible for epigenetic modifications, metabolites can regulate such phenotypes via epigenetic alterations, affecting the tumor-initiating, tumor-propagating, or metastatic potential. In this section, the key metabolites are discussed individually, focusing on their roles in writing and erasing epigenetic marks in CSCs.
3.1 Metabolites that generate epigenetic marks in CSC regulation
Acetyl-CoA and S-adenosyl methionine (SAM) are the most well-studied substrates for donating acetyl and methyl groups, respectively, to create new epigenetic modifications (Figure 2) (Boon et al., 2020). Acetyl-CoA is mainly produced during glucose metabolism and fatty acid oxidation (FAO). An increase in glycolysis and FAO results in abundant acetyl-CoA, which augments levels of global histone acetylation (Sivanand et al., 2018). Conversely, a decrease in the glycolysis-mediated generation of acetyl-CoA leads to a reduction in histone acetylation and thus affects the differentiation of ESCs, which emphasizes that the levels of intracellular metabolites feeding into epigenetic modifications can govern the cell differentiation state (Moussaieff et al., 2015). Hypoxia-induced upregulation of genes involved in glucose metabolism, such as glucose transporter 1 (GLUT1), pyruvate kinase isozyme M2, and pyruvate dehydrogenase A, enhances glucose influx and pyruvate production in breast cancer (Yang D. et al., 2020). This results in acetyl-CoA accumulation, an increase in histone H4 acetylation, and subsequent gene upregulation associated with cancer stemness, affecting the tumor-initiating capacity (Yang D. et al., 2020). An FAO-mediated increase in acetyl-CoA can epigenetically regulate epithelial-mesenchymal transition (EMT) gene expression, thus affecting EMT-driven metastasis in breast cancer (Loo et al., 2021). The balance between FAO and fatty acid storage regulates intracellular acetyl-CoA availability, a critical epigenetic module affecting epithelial-mesenchymal plasticity. These results suggest that the cell metabolic state can affect gene expression by providing surplus materials for writing epigenetic marks, thereby promoting the CSC potential of cellular plasticity, such as epithelial and mesenchymal states. Moreover, dietary palmitic acid can establish a stable epigenetic memory, particularly mediated by Set1A-dependent H3K4me3 deposition, critical for metastasis initiation and development of the pro-metastatic niche (Pascual et al., 2021). In addition to histone modifications, post-translational acetylation of non-histone proteins, such as signaling proteins and transcription factors (TFs) can also regulate the metastatic potential of breast cancer cells. For example, the exogenous supply of saturated fatty acid, palmitate, a rich source of acetyl-CoA, acetylates nuclear factor-kappaB p65 subunit, leading to the promotion of metastasis-initiating potential of breast cancer to lung and liver (Altea-Manzano et al., 2023).
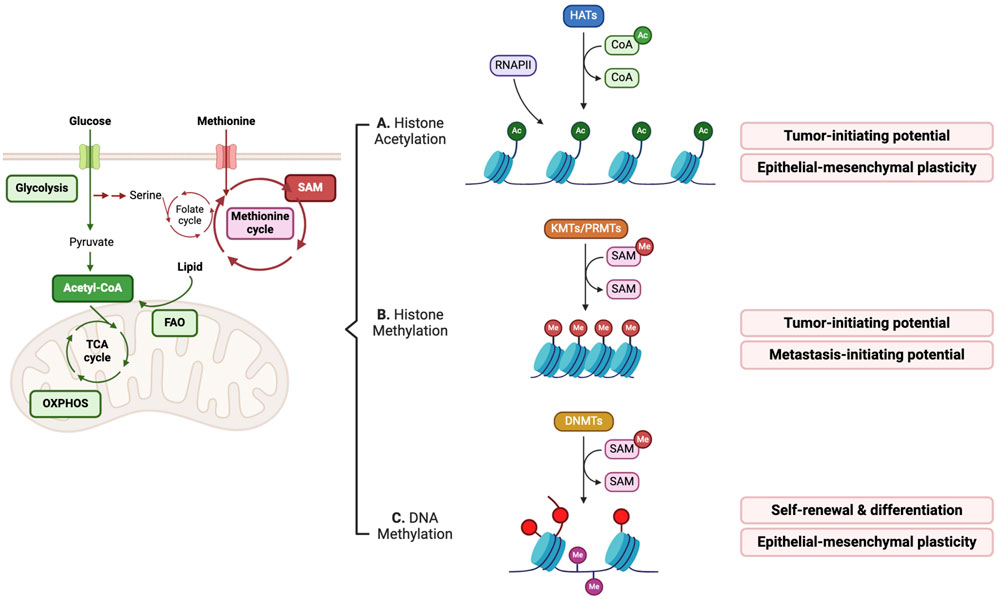
Figure 2. Metabolites serve as key substrates in producing epigenetic marks, thus affecting CSC properties. (A) Intracellular acetyl-CoA metabolism affects histone acetylation levels, in general promoting gene transcription associated with epithelial-mesenchymal transition (EMT) and enhancing tumor-initiating potential. (B,C) Intracellular S-Adenosyl methionine (SAM) abundance controls histone and DNA methylation states, which influences CSC features. RNAPII; RNA polymerase II.
As a methyl donor, SAM is involved in the creation of methylation marks on histone proteins, DNA and RNA. Aberrant regulations of glucose, glutamine, and serine/glycine metabolic pathways, and folate and methionine cycles have indirect and direct effects on the availability of intracellular SAM, thereby affecting the epigenetic modification of methyl group acceptors (Figure 2) (Mentch et al., 2015). Threonine and methionine, which are SAM-producing amino acids, can modulate histone methylation, thereby influencing ESC differentiation ability (Shyh-Chang et al., 2013; Shiraki et al., 2014). Histone methylation of lysine and arginine residues is mediated by lysine methyltransferases (KMTs) and protein arginine methyltransferases (PRMTs), respectively, which provide docking sites for epigenetic regulators (Stallcup, 2001). The number of covalent methyl groups in histones generates mono-, di-, and tri-methylation states and functions as regulatory signals that affect gene activation or repression, depending on the histone modifications at specific loci. In non-small cell lung cancer (NSCLC), tumor-initiating cells exhibit a unique metabolic dependence on methionine because of their demands for SAM. The key methionine cycle enzyme, methionine adenosyltransferase II alpha (MAT2A), plays a critical role in providing SAM for global increases in histone H3 methylation, such as H3K4me3, H3K9me3, H3K27me3, and H3K36me2/3, suggesting that the upregulation of histone methylation, regardless of active or repressive marks, can positively influence the tumor-initiating potential of NSCLC (Wang et al., 2019). In triple-negative breast cancer, restriction of SAM availability by MAT2A inhibition causes failure to maintain active H3K4me3 marks on the SOX9 gene locus, which is associated with CSC enrichment and tumor- and metastasis-initiating potential (Strekalova et al., 2019). In basal-like breast cancer, nicotinamide N-methyltransferase (NNMT), which negatively regulates SAM availability, can decrease H3K9me3 repressive marks and DNA methylation of the pro-metastatic genes, such as PR/SET domain-5, promoting basal-type cancer plasticity and metastasis-initiating potential by regulating H3K9me3 and DNA methylation (Couto et al., 2023).
DNA methylation occuring mainly at cytosine (5-methylcytosine [5-mC]) in cytosine-guanine (CpG) dinucleotides, is mediated by DNA methyltransferases (DNMTs), and regulates gene activation or repression by hypo or hypermethylation, respectively (Lyko, 2018). In glioblastoma stem cells (GSCs), increased NNMT expression depletes intracellular SAM levels, resulting in global DNA hypomethylation with a mesenchymal phenotype and the self-renewal of GSCs (Jung et al., 2017). The dependence of leukemia stem cells (LSCs) on oxidative phosphorylation (OXPHOS) in mitochondria links intracellular SAM availability to DNA methylation (Singh et al., 2020). An increase in mitochondrial copper under the inhibition of the mitochondria copper chaperone COX17 inhibits the enzymatic activity of S-adenosylhomocysteine hydrolase, thereby decreasing intracellular SAM levels (Singh et al., 2020). This specifically downregulates global DNA methylation but not histone methylation, which is essential for LSC viability and differentiation.
3.2 Metabolites that remove or regulate the removal of epigenetic marks in CSC regulation
Mitochondrial intermediates such as
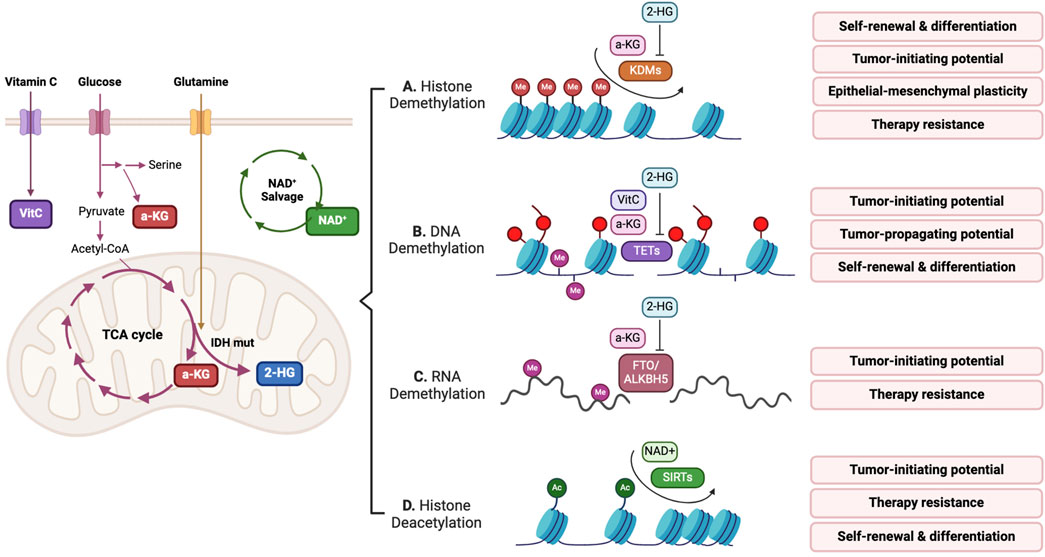
Figure 3. Metabolites take part in erasing epigenetic marks, which regulates CSC potential. (A–C)
Lysine demethylases (KDMs) catalyze the removal of methyl groups from histone lysine residues. Except for KDM1 (lysine-specific demethylase 1 [LSD1]), which utilizes flavin adenine dinucleotide (FAD) as a cofactor, the other KDMs (KDM2–7, JmjC domain-containing histone demethylases) are
Ten eleven translocation (TET) DNA demethylases belong to the
RNA methylation of N6-methyladenosine (m6A) is a recently recognized epitranscriptomic mark of mRNA that affects the transcription, translation, alternative splicing, and stability of mRNA (Wang and Tang, 2023). Demethylation of m6A can be mediated by
An oxidized form of nicotinamide adenine dinucleotide, NAD+, functions as an essential cofactor in histone deacetylation and deacylation mediated by Sirtuins (SIRTs). Notably, SIRTs are a part of the histone deacetylase family and uniquely utilize NAD+, translating cellular metabolic and redox conditions into histone modifications, thereby affecting gene expression (Choi and Mostoslavsky, 2014). Furthermore, NAD+ serves as a substrate in reactions catalyzed by ADP-ribosyltransferases, such as poly (ADP-ribose) polymerase (PARP). In colorectal cancer, the NAD salvage pathway enzyme nicotinamide phosphoribosyl transferase (NAMPT) increases stemness and EMT-related gene expression, resulting in the enrichment of tumor-initiating cells through SIRT1 and PARP (Lucena-Cacace et al., 2018). In glioblastoma, NAMPT can regulate the NAD+-dependent transcriptional program targeted by E2F2-ID (inhibitor of differentiation) axis (Gujar et al., 2016). Intracellular NAD+ levels sustained by the NAMPT-mediated salvage pathway are key to GSC self-renewal and therapy resistance to therapy.
4 Epigenetics controls metabolism in CSCs
Epigenetic reprogramming has emerged as a cancer hallmark due to its role in initiating and sustaining malignant tumors (Hanahan, 2022). Exploiting epigenetic reprogramming is beneficial for CSCs, as it enables them to adapt to both extrinsic and intrinsic changes (Wainwright and Scaffidi, 2017; Easwaran et al., 2014). For example, transcriptional reprogramming governed by epigenetic regulation confers plasticity to phenotypic switching. Metabolic adaptation is a critical alteration that enables better survival and growth, and is partly mediated by epigenetic regulation (Miranda-Goncalves et al., 2018).
Epigenetic regulation occurs at various levels ranging from DNA methylation to histone modification and chromatin rearrangement. Epigenetic regulators can be grouped into cis-regulatory elements and trans-regulatory elements (Maston et al., 2006). Cis-regulatory elements are specialized DNA regions capable of regulating the expression of target genes, and include promoters, enhancers, silencers, and insulators (Preissl et al., 2023; Rivera and Ren, 2013). They typically serve as binding platforms for trans-regulatory elements. Trans-regulatory elements include TFs along with a broader range of DNA-binding proteins that contribute to gene expression regulation. Notably, TFs function by recruiting coactivators and enzymes that modulate the chromatin structure, thereby regulating the accessibility of specific DNA regions to the transcription machinery (Ferrie et al., 2022). These enzymes include histone modifiers, chromatin remodellers, and DNMTs. In this section, we focus on how the aforementioned epigenetic regulators orchestrate tumor metabolism to form and maintain CSC potential.
4.1 Transcription factors
Stem cell pluripotency and EMT TFs are often upregulated in CSCs (Yang L. et al., 2020; Chaudhary et al., 2023). They help CSCs maintain stem cell-like characteristics, partly by changing their metabolic state, preferentially shifting from OXPHOS to glycolysis and FAO (Figure 4) (Lisowski et al., 2018). In basal-like breast cancer, Snail (encoded by SNAI1), a critical EMT TF, forms a complex with a G9a histone methyltransferase and DNMT. The Snail-G9a-DNMT complex directly represses the expression of fructose-1,6-biphosphatase, a rate-limiting enzyme in gluconeogenesis. This results in increased glycolysis and reduced reactive oxygen species (ROS) production, conferring stem-like features to breast cancer cells (Dong et al., 2013). Tumor-initiating stem-like cells in hepatocellular carcinoma (HCC) promote self-renewal and support drug resistance through NANOG-mediated expression of FAO gene and concomitant repression of OXPHOS genes (Chen et al., 2016). c-myc is an essential TF in the thrombopoietin (TPO)-responsive colorectal CD110+ CSCs that promote liver metastasis. Notably, TPO-induced c-myc recruits the histone acetyltransferase HBO1, which increases the H3K14 acetylation of metabolic genes involved in lysine catabolism and fatty acid synthesis (Wu et al., 2015).
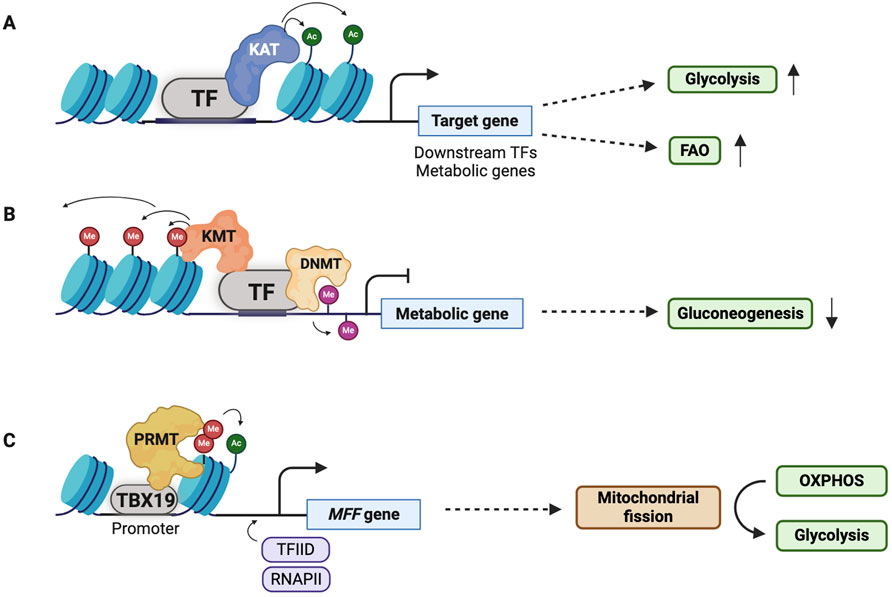
Figure 4. Transcription factors (TFs) can modulate metabolic adaptation of CSCs. (A,B) Stem cell pluripotency TFs, EMT TFs, and hypoxia-inducible factor-1 (HIF-1) activate or repress expression of metabolic genes through epigenetic modifications in CSCs. Typically, metabolic genes involved in glycolysis and fatty acid oxidation (FAO) or downstream TFs regulating metabolic adaptation are activated while metabolic genes involved in gluconeogenesis and oxidative phosphorylation (OXPHOS) are repressed. (C) T-box transcription factor 19 (TBX19), a TF, leads to a metabolic transition from OXPHOS to glycolysis via mitochondrial fission activated by expression of MFF. RNAPII; RNA polymerase II.
The tumor microenvironment influences CSC stemness, in part, through the reprogramming of tumor cell metabolism (Zheng et al., 2021; Elia and Haigis, 2021). Hypoxia, a common characteristic of the tumor microenvironment, epigenetically controls metabolic pathways primarily by activating the hypoxia-inducible factor 1 (HIF-1) (Chen et al., 2023). HIF-1 is a heterodimeric TF consisting of HIF-1α and HIF-1β. Upon activation, HIF-1α interacts with p300/CBP (CREB-binding protein), which in turn acetylates histones and recruits other TFs for gene activation (Gu et al., 2001; Lu et al., 2021). Recruited TFs such as OCT3/4, NANOG, SOX2, Snail, and TWIST contribute to the metabolic adaptation of CSCs by upregulating the expression of metabolic genes and/or their regulators (Zhang et al., 2020; Wang Y. et al., 2013; Jeter et al., 2015). Furthermore, HIF-1α can directly upregulate several glycolytic genes to meet high metabolic demands of CSCs, including GLUT1/2, hexokinases, aldolases, phosphoglycerate kinase 1, pyruvate kinase M, and enolases (Taylor and Scholz, 2022; Semenza, 2003).
Another interesting mechanism that strengthens the self-renewal capacity of CSCs is via T-box transcription factor 19 (TBX19)-mediated metabolic adaptation (Figure 4). TBX19 epigenetically regulates mitochondrial fission-mediated metabolic adaptation in liver CSCs (Tang et al., 2021). Specifically, TBX19 interacts with PRMT1, which increases the H4R3me2a/H3K9ac-mediated transcription of the mitochondrial fission factor (MFF) gene. The TBX19/MFF axis induces mitochondrial fission, which promotes the metabolic switch from OXPHOS to glycolysis. This metabolic switch, which involves mitochondrial fission, prevents ROS-mediated OCT4 degradation, thereby maintaining the stemness of liver CSCs.
4.2 Histone modification
Histone modifications can induce chromatin structural rearrangements and recruit chromatin factors, thereby affecting gene expression (Zentner and Henikoff, 2013). A wide array of histone modifications has been identified (Bannister and Kouzarides, 2011). Among these, histone acetylation/deacetylation and histone methylation/demethylation are the most well-characterized. We focus on how these two histone modification modules contribute to CSC metabolic adaptation.
4.2.1 Histone acetylation/deacetylation
Histone acetyltransferases (HATs) and histone deacetylases (HDACs) catalyze the addition or removal, respectively, of acetyl groups from the lysine residues of target proteins (Yang and Seto, 2007). HATs are also known as lysine acetyltransferases. HATs have six families and 17 subtypes in humans, and 18 HDACs are grouped into two families and four classes (Seto and Yoshida, 2014; Wapenaar and Dekker, 2016). Classes I and II HDACs require Zn2+ for deacetylation. Class III HDACs belong to the silent information regulator 2 (Sir2)-related protein family (Sirtuins, SIRTs) that contains SIRT1–7. The sirtuin family HDACs use NAD+ as the cofactor. Class IV HDAC includes HDAC11.
Acetylation and deacetylation of the ε-amino group of the lysine residue of histones directly affect the transcription of metabolic genes (Figure 5) (Verdone et al., 2006). In HCC, HDAC11 contributes to the maintenance of cancer stemness by regulating glycolysis (Bi et al., 2021). HDAC11 deacetylates H3K9ac in the serine threonine kinase 11 (STK11) promoter region, leading to its suppression. Since STK11 activates the AMP-activated protein kinase signaling pathway, which negatively regulates glucose uptake and glycolysis, HDAC11-mediated downregulation of STK11 promotes glycolysis, thereby maintaining the stemness of HCC CSCs. SIRT6 can deacetylate H3K9ac and H3K56ac, which negatively regulates the transcription of HIF-1α-dependent glycolytic genes, including GLUT1, pyruvate dehydrogenase kinase 1, and lactate dehydrogenase A (Zhong et al., 2010). In a colorectal cancer model with an Apc mutation, an increase in glycolysis due to the loss of Sirt6 can drive colorectal cancer initiation via aberrant histone acetylation (Sebastián et al., 2012). Furthermore, Sebastian et al. identified a subset of cancer cells with high glycolytic activity as quiescent tumor-initiating cells with better defense against oxidative stress (Sebastian et al., 2022). In line with these findings, in head and neck squamous cell carcinoma, CD34+ CSCs with Sirt6 loss display a high glycolytic phenotype, antioxidant protection, and nucleotide synthesis that collectively confers stemness and malignancy (Choi et al., 2021).
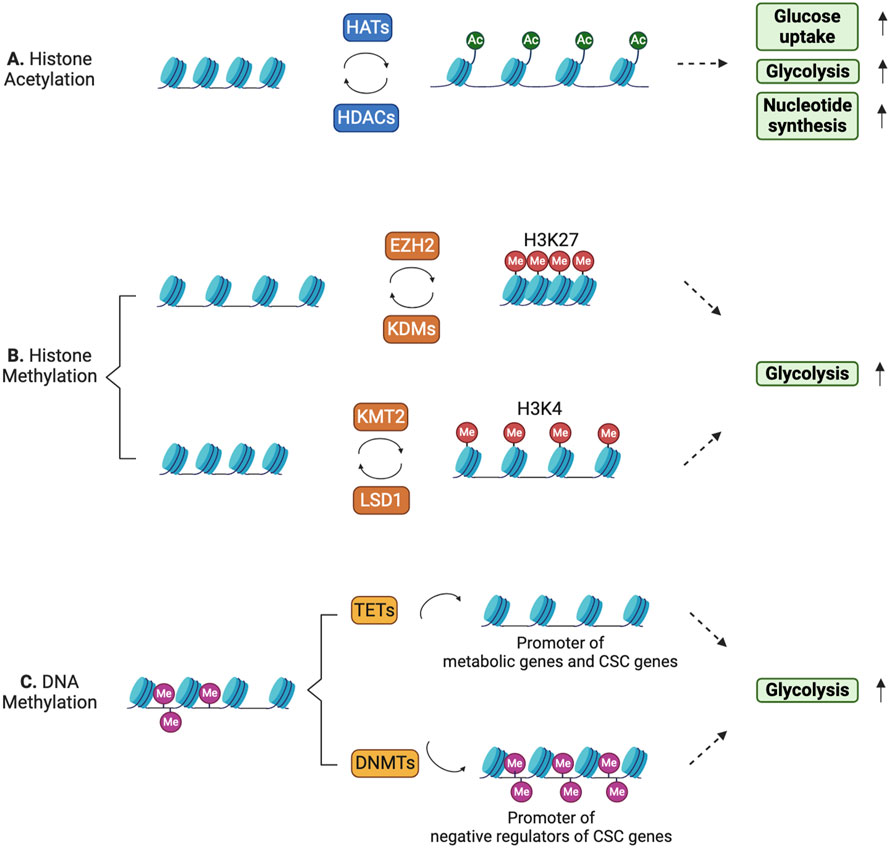
Figure 5. Histone and DNA modifications can dynamically control metabolic reprogramming in CSCs. (A) Histone acetylation/deacetylation patterns dynamically regulate gene expression in CSCs to meet their high energy demands. (B) Histone methylation leads to either gene repression or activation depending on the histone residues that are methylated. (C) DNA methylation at the promoter regions of metabolic genes and CSC-related genes is dynamically regulated.
Some HDACs have non-histone targets (Bannister and Kouzarides, 2011). CSCs leverage this noncanonical activity of HDACs for metabolic rewiring. For example, SIRT1 can deacetylate transcription coactivator PGC-1α, leading to increased expression of mitochondrial genes in chronic myeloid leukemia (CML) stem cells (Abraham et al., 2019). Upregulation of mitochondrial gene expression enhances mitochondrial activity and OXPHOS in response to the energy demands of CML stem cells. These findings underscore the multiple levels of metabolic control by epigenetic regulators.
4.2.2 Histone methylation/demethylation
Histone lysine residues are methylated and demethylated by KMTs and KDMs, respectively (Dimitrova et al., 2015; Husmann and Gozani, 2019). Up to three methyl groups can be added to the ε-nitrogen of lysine residues. Canonical lysine methylation sites were found in the N-terminal tails of histones H3 and H4. Dynamic regulation of histone methylation patterns and residues is critical for the epigenetic regulation of transcription (Hyun et al., 2017).
Enhancer of zeste homolog 2 (EZH2), a catalytic subunit of polycomb repressive complex 2, mediates the trimethylation of H3K27 (H3K27me3) (Duan et al., 2020). This histone modification is associated with the repression of gene expression. Additionally, EZH2 plays an essential role in stem cell fate decisions during development (Lee et al., 2022). The aberrant upregulation of EZH2 expression has been observed in CSCs across various cancer types (Wen et al., 2017), suggesting its involvement in CSC features (Figure 5). A recent study suggested that EZH2 epigenetically promotes c-Myc-mediated glycolysis and thus tumor cell proliferation, invasion, and stemness in pancreatic cancer (Zhai et al., 2021). Specifically, EZH2 silences the expression of long non-coding RNA LINC00261, which subsequently releases IGF2BP1 from its sequestration and thus stabilizing c-Myc RNA. Upregulation of c-Myc activates target genes that regulate CSC identity.
H3K4 methylation is associated with active transcription and is catalyzed by the KMT2 protein family (Van et al., 2024). H3K4 di-methylation (H3K4me2) and H3K4 tri-methylation (H3K4me3) are enriched in leukemia stem cells (LSCs) expressing c-Kit (Wong et al., 2015). This H3K4 methylation pattern was positively correlated with the expression of metabolic genes crucial for maintaining LSC potential (Somervaille et al., 2009; Wong et al., 2015), suggesting that the H3K4 methylation pattern is associated with the transcriptional program of metabolic genes that potentially govern CSC maintenance (Figure 5).
Epigenetic regulators that affect methionine metabolism can alter the H3K4 methylation states, and thus induce the transcription of CSC-related genes. In triple-negative breast cancer, aldehyde dehydrogenase+ CSCs express higher levels of EMSY, a proto-oncogene, than do non-CSCs (Liu et al., 2024). Overexpression of EMSY correlates with poor prognosis, tumorigenesis, and CSC self-renewal. EMSY promotes methionine metabolism and increases SAM and S-adenosylhomocysteine levels. Elevated SAM levels fuel H3K4me2 and H3K4me3, thereby driving the upregulation of CSC-related genes such as NANOG, POU5F1, and SOX2. Additionally, EMSY-mediated H3K4me2 and H3K4me3 are maintained by the competitive binding of EMSY to the JmjC domain of KDM5B, demethylating H3K4 methylation (Liu et al., 2024).
4.3 DNA methylation
DNA methylation typically refers to the methylation of 5-carbon cytosine (5-mC) in CpG dinucleotide repeats found in the promoter regions of most mammalian genes (Moore et al., 2013). DNA methylation is catalyzed by DNMTs (Lyko, 2018), of which DNMT1, DNMT3A, and DNMT3B are canonical DNMTs responsible for 5-mC. DNA methylation is reversed by TET enzymes that convert 5-mC to 5-hydroxymethylcytosine (Lyko, 2018). The counteractive actions of DNMTs and TETs dynamically shape DNA methylation patterns. A recent study has shown that TET3 is overexpressed in LSCs in AML and promotes the expression of glycolytic genes and stem cell signature genes (Pulikkottil et al., 2022). The list includes hexokinase 1/2 and enolase 2, which are involved in glucose metabolism, and HES1 and CCND1, which are associated with early myeloid progenitor gene signatures. Overexpression of TET3 in normal human hematopoietic stem and progenitors disrupts normal hematopoietic differentiation, suggesting that TET3 maintains distinct features of LSCs in AML (Pulikkottil et al., 2022). Zinc finger DHHC-type containing 1 (ZDHHC1, ZNF377) plays a tumor-suppressive function by downregulating stemness-related TFs and providing hostile metabolic conditions for tumor growth (Le et al., 2020). Multiple cancers downregulate ZDHHC1 expression by methylating its promoter region (Le et al., 2020). The expression of stem cell pluripotency TFs and metabolic genes that maintain CSC potential are dynamically regulated at the DNA methylation level (Figure 5).
5 Discussion
Understanding the mechanisms underlying CSC emergence and their ability to self-renew and differentiate has clinical implications and therapeutic potential, given their association with metastasis, therapy resistance, and tumor relapse. Over the past decade, studies have demonstrated an intricate interplay between metabolic pathways and the epigenetic regulation of CSCs. Alterations in metabolic pathways enable CSCs to generate metabolites that reshape the epigenetic landscape and drive transcriptional reprogramming, which is essential for sustaining CSC features. Conversely, epigenetic regulators target metabolic genes and regulators of metabolic genes, preferentially redirecting metabolic pathways toward glycolysis and FAO from OXPHOS. In this section, we discuss the outstanding questions to be answered in this field with the current technological advances.
Recently, acyl-CoA, in addition to acetyl-CoA, has been shown to serve as a direct substrate for generating epigenetic marks that involve distinct functions in gene expression (Simithy et al., 2017). A repertoire of acyl-CoA includes propionyl, butyryl,
The influence of metabolism on chromatin accessibility and chromatin topology remains another compelling area yet to be investigated in CSCs. The key components of the nucleosome remodeler SWI/SNF (BAF) complex are dependent on the supply of ATP to perform their enzymatic functions (Wilson and Roberts, 2011), which raises questions about how ATP availability affects the activity of the SWI/SNF complex and therefore CSC properties. The seminal paper by Bernstein and colleagues found that DNA hypermethylation in IDH mutant gliomas reduces CCCTC-binding factor (CTCF) binding and presents an associated decrease in insulator function, leading to an aberrant activation of PDGFRA oncogene (Flavahan et al., 2016). This paper highlights how chromosomal topology can be dysregulated in cancer under the influence of the oncometabolite. In line with it, poly (ADP-ribose) ylation (PARylation), mediated by PARP with NAD+ as a cofactor, is a well-known protein modification in the DNA damage response. In addition, PARP1 can regulate transcription, DNA methylation, and the function of CTCF insulator protein via PARylation (Beneke, 2012). Recently, PARylation of CTCF has been shown to affect its insulator function, thereby directly affecting the proper expression of tumor-suppressor genes such as CDH1, p16, and p19ARF (Farrar et al., 2010; Witcher and Emerson, 2009). To date, comprehensive understanding of how the functional interactions between PARP1, PAR, and CTCF can modulate the key characteristics of CSCs due to the dysregulation of NAD+ metabolism is lacking. Notably, in succinate dehydrogenase deficient gastrointestinal stromal tumors, global DNA hypermethylation-mediated, defective CTCF binding results in alterations in chromatin topology. The altered genome structure allows the activation of the FGF4 oncogene by interacting with enhancers (Flavahan et al., 2019). This will be another exciting area that needs future investigation in the context of CSCs, as deficiency in key metabolic enzymes affects the chromatin topology that can drive tumorigenesis.
Furthermore, m6A modification of RNA can influence gene expression (Jiang et al., 2021). This RNA modification process involves m6A addition and removal by m6A methyltransferases and demethylases, respectively. Key m6A methyltransferases include METTL3/14/16, RBM15/15B, ZC3H3, and VIRMA, whereas demethylases include FTO and ALKBH5. METTL3 directly interacts with and modifies the transcription of metabolic genes, including HK2 and GLUT1 (Shen et al., 2020). The m6A modification of these transcripts is recognized by readers such as IGF2BP2 or IGFBP2/3, triggering the activation of the glycolysis pathway. This activated glycolytic pathway provides essential sustenance for tumor cell growth. As the relationship between RNA methylation/demethylation and metabolic gene regulation has recently been established, future studies to understand how their interplay drives CSC characteristics should stimulate and expand this research area.
Considering the phenotypic plasticity of CSCs, the interplay between metabolic pathways and epigenetic regulation is dynamically orchestrated, and shapes their characteristics. This interplay can be controlled by external signals received by CSCs, such as the Notch, Hedgehog, and Wnt signaling pathways (Yuan et al., 2024; Takebe et al., 2015). Therefore, investigating how external signals modulate the interaction between metabolism and epigenetic regulation in CSCs will be interesting, albeit challenging. Understanding the distinctions between these signaling pathways in CSCs and normal stem cells and their effect on metabolic enzymes and epigenetic regulators constitutes crucial research questions.
Single-cell-based techniques, such as single-cell RNA-sequencing, single-cell epigenomic profiling, and single-cell metabolomics, in addition to spatially resolved metabolite analyses with cellular resolution (e.g., matrix-assisted laser desorption/ionization-mass spectrometry imaging), offer unprecedented resolution for dissecting the heterogeneity within CSC populations (Vegliante et al., 2022). The identification of metabolically and epigenetically distinct CSC subpopulations can enable us to uncover bona fide CSCs associated with cancer stemness (Yuan et al., 2022; Choi et al., 2021). Additionally, single-cell techniques can provide insights into how different cell types within CSC niches affect the metabolic states and epigenetic landscapes of CSCs. Technical advancements that enable the simultaneous capture of metabolic and epigenetic states will further deepen our understanding of the interplay between metabolism and epigenetic regulation in CSC biology.
Notably, CSCs possess the unique ability to initiate and propagate malignant tumors. Their pivotal role in tumors stems from their cellular plasticity. Furthermore, CSCs are capable of self-renewal and differentiation and can dynamically change their fate in response to intrinsic and extrinsic changes, similar to their normal stem cell counterparts. Cellular plasticity is partially achieved via metabolic and epigenetic reprogramming. Markedly, both engage in a complex interplay and fine-tune cellular states. Although various methods of communication between metabolic pathways and epigenetic regulation have been identified in CSCs, further studies are needed to comprehensively understand their interplay in CSCs. Such studies could explore how metabolites, including acyl-CoA and FAD, that are yet to be examined, contribute to epigenetic alterations. Such studies can also address the effects of metabolic reprogramming on spatiotemporal genome organization and identify new pathways of epigenetic influence on metabolic regulation, such as RNA modifications. Another pivotal research direction can be to examine how CSC-associated signaling pathways govern the interplay between metabolic and epigenetic reprogramming.
Intratumor heterogeneity has led to a model in which a tumor subpopulation, now considered CSCs, is responsible for metastasis, drug resistance, and tumor relapse. However, CSCs have been identified based on their functional ability to drive tumor initiation and progression or the expression of CSC biomarkers, such as CD44, CD90, and CD133. Recent advances in single-cell-based approaches for metabolic and epigenomic profiling can enable the identification of bona fide CSCs, presenting a promising opportunity for complete cancer remission.
Author contributions
J-EC: Conceptualization, Visualization, Writing–original draft, Writing–review and editing. IB: Conceptualization, Funding acquisition, Visualization, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. J-EC is a recipient of the RGC postdoctoral fellowship in Hong Kong SAR (PDFS2223-7S04). This work was supported by a grant from the National Research Foundation of Korea (NRF) funded by the Korea government (MSIT) (RS-2023-00278378) to IB and a grant from Kyung Hee University in 2023 (KHU-20233256) to IB.
Acknowledgments
The authors extend apologies to those colleagues whose important contributions could not be included in this article due to space limitations.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abraham, A., Qiu, S., Chacko, B. K., Li, H., Paterson, A., He, J., et al. (2019). SIRT1 regulates metabolism and leukemogenic potential in CML stem cells. J. Clin. Investigation 129, 2685–2701. doi:10.1172/jci127080
Agathocleous, M., Meacham, C. E., Burgess, R. J., Piskounova, E., Zhao, Z., Crane, G. M., et al. (2017). Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 549, 476–481. doi:10.1038/nature23876
Al-Hajj, M., Wicha, M. S., Benito-Hernandez, A., Morrison, S. J., and Clarke, M. F. (2003). Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. U. S. A. 100, 3983–3988. doi:10.1073/pnas.0530291100
Altea-Manzano, P., Doglioni, G., Liu, Y., Cuadros, A. M., Nolan, E., Fernández-García, J., et al. (2023). A palmitate-rich metastatic niche enables metastasis growth via p65 acetylation resulting in pro-metastatic NF-κB signaling. Nat. Cancer 4, 344–364. doi:10.1038/s43018-023-00513-2
Baksh, S. C., Todorova, P. K., Gur-Cohen, S., Hurwitz, B., Ge, Y., Novak, J. S. S., et al. (2020). Extracellular serine controls epidermal stem cell fate and tumour initiation. Nat. Cell Biol. 22, 779–790. doi:10.1038/s41556-020-0525-9
Bannister, A. J., and Kouzarides, T. (2011). Regulation of chromatin by histone modifications. Cell Res. 21, 381–395. doi:10.1038/cr.2011.22
Batlle, E., and Clevers, H. (2017). Cancer stem cells revisited. Nat. Med. 23, 1124–1134. doi:10.1038/nm.4409
Beneke, S. (2012). Regulation of chromatin structure by poly(ADP-ribosyl)ation. Front. Genet. 3, 169. doi:10.3389/fgene.2012.00169
Bi, L., Ren, Y., Feng, M., Meng, P., Wang, Q., Chen, W., et al. (2021). HDAC11 regulates glycolysis through the LKB1/AMPK signaling pathway to maintain hepatocellular carcinoma stemness. Cancer Res. 81, 2015–2028. doi:10.1158/0008-5472.can-20-3044
Bonnet, D., and Dick, J. E. (1997). Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 3, 730–737. doi:10.1038/nm0797-730
Boon, R., Silveira, G. G., and Mostoslavsky, R. (2020). Nuclear metabolism and the regulation of the epigenome. Nat. Metab. 2, 1190–1203. doi:10.1038/s42255-020-00285-4
Carey, B. W., Finley, L. W., Cross, J. R., Allis, C. D., and Thompson, C. B. (2015). Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 518, 413–416. doi:10.1038/nature13981
Chaudhary, A., Raza, S. S., and Haque, R. (2023). Transcriptional factors targeting in cancer stem cells for tumor modulation. Seminars Cancer Biol. 88, 123–137. doi:10.1016/j.semcancer.2022.12.010
Chen, C. L., Uthaya Kumar, D. B., Punj, V., Xu, J., Sher, L., Tahara, S. M., et al. (2016). NANOG metabolically reprograms tumor-initiating stem-like cells through tumorigenic changes in oxidative phosphorylation and fatty acid metabolism. Cell Metab. 23, 206–219. doi:10.1016/j.cmet.2015.12.004
Chen, Z., Han, F., Du, Y., Shi, H., and Zhou, W. (2023). Hypoxic microenvironment in cancer: molecular mechanisms and therapeutic interventions. Signal Transduct. Target. Ther. 8, 70. doi:10.1038/s41392-023-01332-8
Choi, J. E., and Mostoslavsky, R. (2014). Sirtuins, metabolism, and DNA repair. Curr. Opin. Genet. and Dev. 26, 24–32. doi:10.1016/j.gde.2014.05.005
Choi, J. E., Sebastian, C., Ferrer, C. M., Lewis, C. A., Sade-Feldman, M., Lasalle, T., et al. (2021). A unique subset of glycolytic tumour-propagating cells drives squamous cell carcinoma. Nat. Metab. 3, 182–195. doi:10.1038/s42255-021-00350-6
Chowdhury, R., Yeoh, K. K., Tian, Y. M., Hillringhaus, L., Bagg, E. A., Rose, N. R., et al. (2011). The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 12, 463–469. doi:10.1038/embor.2011.43
Cimmino, L., Dolgalev, I., Wang, Y., Yoshimi, A., Martin, G. H., Wang, J., et al. (2017). Restoration of TET2 function blocks aberrant self-renewal and leukemia progression. Cell 170, 1079–1095.e20. doi:10.1016/j.cell.2017.07.032
Couto, J. P., Vulin, M., Jehanno, C., Coissieux, M. M., Hamelin, B., Schmidt, A., et al. (2023). Nicotinamide N-methyltransferase sustains a core epigenetic program that promotes metastatic colonization in breast cancer. EMBO J. 42, e112559. doi:10.15252/embj.2022112559
Dagogo-Jack, I., and Shaw, A. T. (2018). Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 15, 81–94. doi:10.1038/nrclinonc.2017.166
Dang, L., White, D. W., Gross, S., Bennett, B. D., Bittinger, M. A., Driggers, E. M., et al. (2009). Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739–744. doi:10.1038/nature08617
Demicco, M., Liu, X. Z., Leithner, K., and Fendt, S. M. (2024). Metabolic heterogeneity in cancer. Nat. Metab. 6, 18–38. doi:10.1038/s42255-023-00963-z
Dimitrova, E., Turberfield, A. H., and Klose, R. J. (2015). Histone demethylases in chromatin biology and beyond. EMBO Rep. 16, 1620–1639. doi:10.15252/embr.201541113
Dong, C., Yuan, T., Wu, Y., Wang, Y., Fan, T. W., Miriyala, S., et al. (2013). Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell 23, 316–331. doi:10.1016/j.ccr.2013.01.022
Duan, R., Du, W., and Guo, W. (2020). EZH2: a novel target for cancer treatment. J. Hematol. Oncol. 13, 104. doi:10.1186/s13045-020-00937-8
Easwaran, H., Tsai, H. C., and Baylin, S. B. (2014). Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol. Cell 54, 716–727. doi:10.1016/j.molcel.2014.05.015
Elia, I., and Haigis, M. C. (2021). Metabolites and the tumour microenvironment: from cellular mechanisms to systemic metabolism. Nat. Metab. 3, 21–32. doi:10.1038/s42255-020-00317-z
Etchegaray, J. P., and Mostoslavsky, R. (2016). Interplay between metabolism and epigenetics: a nuclear adaptation to environmental changes. Mol. Cell 62, 695–711. doi:10.1016/j.molcel.2016.05.029
Farrar, D., Rai, S., Chernukhin, I., Jagodic, M., Ito, Y., Yammine, S., et al. (2010). Mutational analysis of the poly(ADP-ribosyl)ation sites of the transcription factor CTCF provides an insight into the mechanism of its regulation by poly(ADP-ribosyl)ation. Mol. Cell. Biol. 30, 1199–1216. doi:10.1128/mcb.00827-09
Faubert, B., Solmonson, A., and Deberardinis, R. J. (2020). Metabolic reprogramming and cancer progression. Science 368, eaaw5473. doi:10.1126/science.aaw5473
Ferrie, J. J., Karr, J. P., Tjian, R., and Darzacq, X. (2022). “Structure”-function relationships in eukaryotic transcription factors: the role of intrinsically disordered regions in gene regulation. Mol. Cell 82, 3970–3984. doi:10.1016/j.molcel.2022.09.021
Figueroa, M. E., Abdel-Wahab, O., Lu, C., Ward, P. S., Patel, J., Shih, A., et al. (2010). Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18, 553–567. doi:10.1016/j.ccr.2010.11.015
Flavahan, W. A., Drier, Y., Johnstone, S. E., Hemming, M. L., Tarjan, D. R., Hegazi, E., et al. (2019). Altered chromosomal topology drives oncogenic programs in SDH-deficient GISTs. Nature 575, 229–233. doi:10.1038/s41586-019-1668-3
Flavahan, W. A., Drier, Y., Liau, B. B., Gillespie, S. M., Venteicher, A. S., Stemmer-Rachamimov, A. O., et al. (2016). Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529, 110–114. doi:10.1038/nature16490
Ginestier, C., Hur, M. H., Charafe-Jauffret, E., Monville, F., Dutcher, J., Brown, M., et al. (2007). ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 1, 555–567. doi:10.1016/j.stem.2007.08.014
Gu, J., Milligan, J., and Huang, L. E. (2001). Molecular mechanism of hypoxia-inducible factor 1α-p300 interaction. J. Biol. Chem. 276, 3550–3554. doi:10.1074/jbc.m009522200
Gujar, A. D., Le, S., Mao, D. D., Dadey, D. Y., Turski, A., Sasaki, Y., et al. (2016). An NAD+-dependent transcriptional program governs self-renewal and radiation resistance in glioblastoma. Proc. Natl. Acad. Sci. U. S. A. 113, E8247–E8256. doi:10.1073/pnas.1610921114
Hanahan, D. (2022). Hallmarks of cancer: new dimensions. Cancer Discov. 12, 31–46. doi:10.1158/2159-8290.cd-21-1059
Husmann, D., and Gozani, O. (2019). Histone lysine methyltransferases in biology and disease. Nat. Struct. Mol. Biol. 26, 880–889. doi:10.1038/s41594-019-0298-7
Hyun, K., Jeon, J., Park, K., and Kim, J. (2017). Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 49, e324. doi:10.1038/emm.2017.11
Izzo, L. T., Affronti, H. C., and Wellen, K. E. (2021). The bidirectional relationship between cancer epigenetics and metabolism. Annu. Rev. Cancer Biol. 5, 235–257. doi:10.1146/annurev-cancerbio-070820-035832
Jeter, C. R., Yang, T., Wang, J., Chao, H. P., and Tang, D. G. (2015). Concise review: NANOG in cancer stem cells and tumor development: an update and outstanding questions. Stem Cells 33, 2381–2390. doi:10.1002/stem.2007
Jiang, X., Liu, B., Nie, Z., Duan, L., Xiong, Q., Jin, Z., et al. (2021). The role of m6A modification in the biological functions and diseases. Signal Transduct. Target. Ther. 6, 74. doi:10.1038/s41392-020-00450-x
Jung, J., Kim, L. J., Wang, X., Wu, Q., Sanvoranart, T., Hubert, C. G., et al. (2017). Nicotinamide metabolism regulates glioblastoma stem cell maintenance. JCI Insight 2, e90019. doi:10.1172/jci.insight.90019
Killian, J. K., Kim, S. Y., Miettinen, M., Smith, C., Merino, M., Tsokos, M., et al. (2013). Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov. 3, 648–657. doi:10.1158/2159-8290.cd-13-0092
Kim, J., and Deberardinis, R. J. (2019). Mechanisms and implications of metabolic heterogeneity in cancer. Cell Metab. 30, 434–446. doi:10.1016/j.cmet.2019.08.013
Klose, R. J., Kallin, E. M., and Zhang, Y. (2006). JmjC-domain-containing proteins and histone demethylation. Nat. Rev. Genet. 7, 715–727. doi:10.1038/nrg1945
Kooistra, S. M., and Helin, K. (2012). Molecular mechanisms and potential functions of histone demethylases. Nat. Rev. Mol. Cell Biol. 13, 297–311. doi:10.1038/nrm3327
Kreso, A., and Dick, J. E. (2014). Evolution of the cancer stem cell model. Cell Stem Cell 14, 275–291. doi:10.1016/j.stem.2014.02.006
Lapidot, T., Sirard, C., Vormoor, J., Murdoch, B., Hoang, T., Caceres-Cortes, J., et al. (1994). A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 367, 645–648. doi:10.1038/367645a0
Le, X., Mu, J., Peng, W., Tang, J., Xiang, Q., Tian, S., et al. (2020). DNA methylation downregulated ZDHHC1 suppresses tumor growth by altering cellular metabolism and inducing oxidative/ER stress-mediated apoptosis and pyroptosis. Theranostics 10, 9495–9511. doi:10.7150/thno.45631
Lee, S. H., Li, Y., Kim, H., Eum, S., Park, K., and Lee, C. H. (2022). The role of EZH1 and EZH2 in development and cancer. BMB Rep. 55, 595–601. doi:10.5483/BMBRep.2022.55.12.174
Letouzé, E., Martinelli, C., Loriot, C., Burnichon, N., Abermil, N., Ottolenghi, C., et al. (2013). SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell 23, 739–752. doi:10.1016/j.ccr.2013.04.018
Lisowski, P., Kannan, P., Mlody, B., and Prigione, A. (2018). Mitochondria and the dynamic control of stem cell homeostasis. EMBO Rep. 19, e45432. doi:10.15252/embr.201745432
Liu, C. C., Chen, L., Cai, Y. W., Chen, Y. F., Liu, Y. M., Zhou, Y. J., et al. (2024). Targeting EMSY-mediated methionine metabolism is a potential therapeutic strategy for triple-negative breast cancer. Cell Rep. Med. 5, 101396. doi:10.1016/j.xcrm.2024.101396
Loo, S. Y., Toh, L. P., Xie, W. H., Pathak, E., Tan, W., Ma, S., et al. (2021). Fatty acid oxidation is a druggable gateway regulating cellular plasticity for driving metastasis in breast cancer. Sci. Adv. 7, eabh2443. doi:10.1126/sciadv.abh2443
Losman, J. A., Koivunen, P., and Kaelin, W. G. (2020). 2-Oxoglutarate-dependent dioxygenases in cancer. Nat. Rev. Cancer 20, 710–726. doi:10.1038/s41568-020-00303-3
Losman, J. A., Looper, R. E., Koivunen, P., Lee, S., Schneider, R. K., Mcmahon, C., et al. (2013). (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 339, 1621–1625. doi:10.1126/science.1231677
Lu, C., Ward, P. S., Kapoor, G. S., Rohle, D., Turcan, S., Abdel-Wahab, O., et al. (2012). IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483, 474–478. doi:10.1038/nature10860
Lu, H., Lyu, Y., Tran, L., Lan, J., Xie, Y., Yang, Y., et al. (2021). HIF-1 recruits NANOG as a coactivator for TERT gene transcription in hypoxic breast cancer stem cells. Cell Rep. 36, 109757. doi:10.1016/j.celrep.2021.109757
Lucena-Cacace, A., Otero-Albiol, D., Jiménez-García, M. P., Muñoz-Galvan, S., and Carnero, A. (2018). NAMPT is a potent oncogene in colon cancer progression that modulates cancer stem cell properties and resistance to therapy through Sirt1 and PARP. Clin. Cancer Res. 24, 1202–1215. doi:10.1158/1078-0432.ccr-17-2575
Lyko, F. (2018). The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 19, 81–92. doi:10.1038/nrg.2017.80
Maston, G. A., Evans, S. K., and Green, M. R. (2006). Transcriptional regulatory elements in the human genome. Annu. Rev. Genomics Hum. Genet. 7, 29–59. doi:10.1146/annurev.genom.7.080505.115623
Meacham, C. E., and Morrison, S. J. (2013). Tumour heterogeneity and cancer cell plasticity. Nature 501, 328–337. doi:10.1038/nature12624
Mentch, S. J., Mehrmohamadi, M., Huang, L., Liu, X., Gupta, D., Mattocks, D., et al. (2015). Histone methylation dynamics and gene regulation occur through the sensing of one-carbon metabolism. Cell Metab. 22, 861–873. doi:10.1016/j.cmet.2015.08.024
Miranda-Goncalves, V., Lameirinhas, A., Henrique, R., and Jeronimo, C. (2018). Metabolism and epigenetic interplay in cancer: regulation and putative therapeutic targets. Front. Genet. 9, 427. doi:10.3389/fgene.2018.00427
Moore, L. D., Le, T., and Fan, G. (2013). DNA methylation and its basic function. Neuropsychopharmacology 38, 23–38. doi:10.1038/npp.2012.112
Moussaieff, A., Rouleau, M., Kitsberg, D., Cohen, M., Levy, G., Barasch, D., et al. (2015). Glycolysis-mediated changes in acetyl-CoA and histone acetylation control the early differentiation of embryonic stem cells. Cell Metab. 21, 392–402. doi:10.1016/j.cmet.2015.02.002
Nassar, D., and Blanpain, C. (2016). Cancer stem cells: basic concepts and therapeutic implications. Annu. Rev. Pathology Mech. Dis. 11, 47–76. doi:10.1146/annurev-pathol-012615-044438
O'brien, C. A., Pollett, A., Gallinger, S., and Dick, J. E. (2007). A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 445, 106–110. doi:10.1038/nature05372
Oskarsson, T., Batlle, E., and Massagué, J. (2014). Metastatic stem cells: sources, niches, and vital pathways. Cell Stem Cell 14, 306–321. doi:10.1016/j.stem.2014.02.002
Pan, M., Reid, M. A., Lowman, X. H., Kulkarni, R. P., Tran, T. Q., Liu, X., et al. (2016). Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat. Cell Biol. 18, 1090–1101. doi:10.1038/ncb3410
Pascual, G., Dominguez, D., Elosua-Bayes, M., Beckedorff, F., Laudanna, C., Bigas, C., et al. (2021). Dietary palmitic acid promotes a prometastatic memory via Schwann cells. Nature 599, 485–490. doi:10.1038/s41586-021-04075-0
Pavlova, N. N., and Thompson, C. B. (2016). The emerging hallmarks of cancer metabolism. Cell Metab. 23, 27–47. doi:10.1016/j.cmet.2015.12.006
Pérez-González, A., Bévant, K., and Blanpain, C. (2023). Cancer cell plasticity during tumor progression, metastasis and response to therapy. Nat. Cancer 4, 1063–1082. doi:10.1038/s43018-023-00595-y
Preissl, S., Gaulton, K. J., and Ren, B. (2023). Characterizing cis-regulatory elements using single-cell epigenomics. Nat. Rev. Genet. 24, 21–43. doi:10.1038/s41576-022-00509-1
Pulikkottil, A. J., Bamezai, S., Ammer, T., Mohr, F., Feder, K., Vegi, N. M., et al. (2022). TET3 promotes AML growth and epigenetically regulates glucose metabolism and leukemic stem cell associated pathways. Leukemia 36, 416–425. doi:10.1038/s41375-021-01390-3
Raffel, S., Falcone, M., Kneisel, N., Hansson, J., Wang, W., Lutz, C., et al. (2017). BCAT1 restricts αKG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature 551, 384–388. doi:10.1038/nature24294
Ricci-Vitiani, L., Lombardi, D. G., Pilozzi, E., Biffoni, M., Todaro, M., Peschle, C., et al. (2007). Identification and expansion of human colon-cancer-initiating cells. Nature 445, 111–115. doi:10.1038/nature05384
Rivera, C. M., and Ren, B. (2013). Mapping human epigenomes. Cell 155, 39–55. doi:10.1016/j.cell.2013.09.011
Rohle, D., Popovici-Muller, J., Palaskas, N., Turcan, S., Grommes, C., Campos, C., et al. (2013). An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 340, 626–630. doi:10.1126/science.1236062
Ryall, J. G., Cliff, T., Dalton, S., and Sartorelli, V. (2015). Metabolic reprogramming of stem cell epigenetics. Cell Stem Cell 17, 651–662. doi:10.1016/j.stem.2015.11.012
Sabari, B. R., Zhang, D., Allis, C. D., and Zhao, Y. (2017). Metabolic regulation of gene expression through histone acylations. Nat. Rev. Mol. Cell Biol. 18, 90–101. doi:10.1038/nrm.2016.140
Saggese, P., Pandey, A., Alcaraz, M., Fung, E., Hall, A., Yanagawa, J., et al. (2024). Glucose deprivation promotes pseudohypoxia and dedifferentiation in lung adenocarcinoma. Cancer Res. 84, 305–327. doi:10.1158/0008-5472.can-23-1148
Sebastian, C., Ferrer, C., Serra, M., Choi, J. E., Ducano, N., Mira, A., et al. (2022). A non-dividing cell population with high pyruvate dehydrogenase kinase activity regulates metabolic heterogeneity and tumorigenesis in the intestine. Nat. Commun. 13, 1503. doi:10.1038/s41467-022-29085-y
Sebastián, C., Zwaans, B. M., Silberman, D. M., Gymrek, M., Goren, A., Zhong, L., et al. (2012). The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell 151, 1185–1199. doi:10.1016/j.cell.2012.10.047
Semenza, G. L. (2003). Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 3, 721–732. doi:10.1038/nrc1187
Seto, E., and Yoshida, M. (2014). Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 6, a018713. doi:10.1101/cshperspect.a018713
Shen, C., Xuan, B., Yan, T., Ma, Y., Xu, P., Tian, X., et al. (2020). m(6)A-dependent glycolysis enhances colorectal cancer progression. Mol. Cancer 19, 72. doi:10.1186/s12943-020-01190-w
Shiraki, N., Shiraki, Y., Tsuyama, T., Obata, F., Miura, M., Nagae, G., et al. (2014). Methionine metabolism regulates maintenance and differentiation of human pluripotent stem cells. Cell Metab. 19, 780–794. doi:10.1016/j.cmet.2014.03.017
Shyh-Chang, N., Locasale, J. W., Lyssiotis, C. A., Zheng, Y., Teo, R. Y., Ratanasirintrawoot, S., et al. (2013). Influence of threonine metabolism on S-adenosylmethionine and histone methylation. Science 339, 222–226. doi:10.1126/science.1226603
Simithy, J., Sidoli, S., Yuan, Z. F., Coradin, M., Bhanu, N. V., Marchione, D. M., et al. (2017). Characterization of histone acylations links chromatin modifications with metabolism. Nat. Commun. 8, 1141. doi:10.1038/s41467-017-01384-9
Singh, R. P., Jeyaraju, D. V., Voisin, V., Hurren, R., Xu, C., Hawley, J. R., et al. (2020). Disrupting mitochondrial copper distribution inhibits leukemic stem cell self-renewal. Cell Stem Cell 26, 926–937.e10. doi:10.1016/j.stem.2020.04.010
Singh, S. K., Hawkins, C., Clarke, I. D., Squire, J. A., Bayani, J., Hide, T., et al. (2004). Identification of human brain tumour initiating cells. Nature 432, 396–401. doi:10.1038/nature03128
Sivanand, S., Viney, I., and Wellen, K. E. (2018). Spatiotemporal control of acetyl-CoA metabolism in chromatin regulation. Trends Biochem. Sci. 43, 61–74. doi:10.1016/j.tibs.2017.11.004
Somervaille, T. C., Matheny, C. J., Spencer, G. J., Iwasaki, M., Rinn, J. L., Witten, D. M., et al. (2009). Hierarchical maintenance of MLL myeloid leukemia stem cells employs a transcriptional program shared with embryonic rather than adult stem cells. Cell Stem Cell 4, 129–140. doi:10.1016/j.stem.2008.11.015
Sperber, H., Mathieu, J., Wang, Y., Ferreccio, A., Hesson, J., Xu, Z., et al. (2015). The metabolome regulates the epigenetic landscape during naive-to-primed human embryonic stem cell transition. Nat. Cell Biol. 17, 1523–1535. doi:10.1038/ncb3264
Stallcup, M. R. (2001). Role of protein methylation in chromatin remodeling and transcriptional regulation. Oncogene 20, 3014–3020. doi:10.1038/sj.onc.1204325
Strekalova, E., Malin, D., Weisenhorn, E. M. M., Russell, J. D., Hoelper, D., Jain, A., et al. (2019). S-adenosylmethionine biosynthesis is a targetable metabolic vulnerability of cancer stem cells. Breast Cancer Res. Treat. 175, 39–50. doi:10.1007/s10549-019-05146-7
Su, R., Dong, L., Li, C., Nachtergaele, S., Wunderlich, M., Qing, Y., et al. (2018). R-2HG exhibits anti-tumor activity by targeting FTO/m6A/MYC/CEBPA signaling. Cell 172, 90–105.e23. doi:10.1016/j.cell.2017.11.031
Takebe, N., Miele, L., Harris, P. J., Jeong, W., Bando, H., Kahn, M., et al. (2015). Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat. Rev. Clin. Oncol. 12, 445–464. doi:10.1038/nrclinonc.2015.61
Tang, M., Yang, M., Wu, G., Mo, S., Wu, X., Zhang, S., et al. (2021). Epigenetic induction of mitochondrial fission is required for maintenance of liver cancer-initiating cells. Cancer Res. 81, 3835–3848. doi:10.1158/0008-5472.can-21-0436
Taylor, C. T., and Scholz, C. C. (2022). The effect of HIF on metabolism and immunity. Nat. Rev. Nephrol. 18, 573–587. doi:10.1038/s41581-022-00587-8
Van, H. T., Xie, G., Dong, P., Liu, Z., and Ge, K. (2024). KMT2 family of H3K4 methyltransferases: enzymatic activity-dependent and -independent functions. J. Mol. Biol. 436, 168453. doi:10.1016/j.jmb.2024.168453
Vegliante, R., Pastushenko, I., and Blanpain, C. (2022). Deciphering functional tumor states at single-cell resolution. EMBO J. 41, e109221. doi:10.15252/embj.2021109221
Verdone, L., Agricola, E., Caserta, M., and Di Mauro, E. (2006). Histone acetylation in gene regulation. Brief. Funct. Genomic Proteomic 5, 209–221. doi:10.1093/bfgp/ell028
Wainwright, E. N., and Scaffidi, P. (2017). Epigenetics and cancer stem cells: unleashing, hijacking, and restricting cellular plasticity. Trends Cancer 3, 372–386. doi:10.1016/j.trecan.2017.04.004
Wang, F., Travins, J., Delabarre, B., Penard-Lacronique, V., Schalm, S., Hansen, E., et al. (2013a). Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science 340, 622–626. doi:10.1126/science.1234769
Wang, L., and Tang, Y. (2023). N6-methyladenosine (m6A) in cancer stem cell: from molecular mechanisms to therapeutic implications. Biomed. and Pharmacother. 163, 114846. doi:10.1016/j.biopha.2023.114846
Wang, Y., Shi, J., Chai, K., Ying, X., and Zhou, B. P. (2013b). The role of Snail in EMT and tumorigenesis. Curr. Cancer Drug Targets 13, 963–972. doi:10.2174/15680096113136660102
Wang, Z., Yip, L. Y., Lee, J. H. J., Wu, Z., Chew, H. Y., Chong, P. K. W., et al. (2019). Methionine is a metabolic dependency of tumor-initiating cells. Nat. Med. 25, 825–837. doi:10.1038/s41591-019-0423-5
Wapenaar, H., and Dekker, F. J. (2016). Histone acetyltransferases: challenges in targeting bi-substrate enzymes. Clin. Epigenetics 8, 59. doi:10.1186/s13148-016-0225-2
Wellen, K. E., Hatzivassiliou, G., Sachdeva, U. M., Bui, T. V., Cross, J. R., and Thompson, C. B. (2009). ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080. doi:10.1126/science.1164097
Wen, Y., Cai, J., Hou, Y., Huang, Z., and Wang, Z. (2017). Role of EZH2 in cancer stem cells: from biological insight to a therapeutic target. Oncotarget 8, 37974–37990. doi:10.18632/oncotarget.16467
Wilson, B. G., and Roberts, C. W. (2011). SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer 11, 481–492. doi:10.1038/nrc3068
Witcher, M., and Emerson, B. M. (2009). Epigenetic silencing of the p16 tumor suppressor is associated with loss of CTCF binding and a chromatin boundary. Mol. Cell 34, 271–284. doi:10.1016/j.molcel.2009.04.001
Wong, S. H., Goode, D. L., Iwasaki, M., Wei, M. C., Kuo, H. P., Zhu, L., et al. (2015). The H3K4-methyl epigenome regulates leukemia stem cell oncogenic potential. Cancer Cell 28, 198–209. doi:10.1016/j.ccell.2015.06.003
Wu, Z., Wei, D., Gao, W., Xu, Y., Hu, Z., Ma, Z., et al. (2015). TPO-induced metabolic reprogramming drives liver metastasis of colorectal cancer CD110+ tumor-initiating cells. Cell Stem Cell 17, 47–59. doi:10.1016/j.stem.2015.05.016
Xiao, M., Yang, H., Xu, W., Ma, S., Lin, H., Zhu, H., et al. (2012). Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 26, 1326–1338. doi:10.1101/gad.191056.112
Xu, W., Yang, H., Liu, Y., Yang, Y., Wang, P., Kim, S. H., et al. (2011). Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 19, 17–30. doi:10.1016/j.ccr.2010.12.014
Yang, D., Peng, M., Hou, Y., Qin, Y., Wan, X., Zhu, P., et al. (2020a). Oxidized ATM promotes breast cancer stem cell enrichment through energy metabolism reprogram-mediated acetyl-CoA accumulation. Cell Death Dis. 11, 508. doi:10.1038/s41419-020-2714-7
Yang, L., Shi, P., Zhao, G., Xu, J., Peng, W., Zhang, J., et al. (2020b). Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target. Ther. 5, 8. doi:10.1038/s41392-020-0110-5
Yang, X. J., and Seto, E. (2007). HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene 26, 5310–5318. doi:10.1038/sj.onc.1210599
Yuan, S., Almagro, J., and Fuchs, E. (2024). Beyond genetics: driving cancer with the tumour microenvironment behind the wheel. Nat. Rev. Cancer 24, 274–286. doi:10.1038/s41568-023-00660-9
Yuan, S., Stewart, K. S., Yang, Y., Abdusselamoglu, M. D., Parigi, S. M., Feinberg, T. Y., et al. (2022). Ras drives malignancy through stem cell crosstalk with the microenvironment. Nature 612, 555–563. doi:10.1038/s41586-022-05475-6
Zentner, G. E., and Henikoff, S. (2013). Regulation of nucleosome dynamics by histone modifications. Nat. Struct. Mol. Biol. 20, 259–266. doi:10.1038/nsmb.2470
Zhai, S., Xu, Z., Xie, J., Zhang, J., Wang, X., Peng, C., et al. (2021). Epigenetic silencing of LncRNA LINC00261 promotes c-myc-mediated aerobic glycolysis by regulating miR-222-3p/HIPK2/ERK axis and sequestering IGF2BP1. Oncogene 40, 277–291. doi:10.1038/s41388-020-01525-3
Zhang, C., Du, Z., Gao, Y., Lim, K. S., Zhou, W., Huang, H., et al. (2024). Methionine secreted by tumor-associated pericytes supports cancer stem cells in clear cell renal carcinoma. Cell Metab. 36, 778–792.e10. doi:10.1016/j.cmet.2024.01.018
Zhang, C., Samanta, D., Lu, H., Bullen, J. W., Zhang, H., Chen, I., et al. (2016). Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m 6 A-demethylation of NANOG mRNA. Proc. Natl. Acad. Sci. U. S. A. 113, E2047–E2056. doi:10.1073/pnas.1602883113
Zhang, Q., Han, Z., Zhu, Y., Chen, J., and Li, W. (2020). Role of hypoxia inducible factor-1 in cancer stem cells (Review). Mol. Med. Rep. 23, 1. doi:10.3892/mmr.2020.11655
Zheng, X., Yu, C., and Xu, M. (2021). Linking tumor microenvironment to plasticity of cancer stem cells: mechanisms and application in cancer therapy. Front. Oncol. 11, 678333. doi:10.3389/fonc.2021.678333
Keywords: epigenetics, cancer stem cell (CSC), cancer metabolism, oncogenic reprogramming, cellular plasticity, intratumor heterogeneity
Citation: Choi J-E and Baek I (2024) Interplay between epigenetics and metabolism controls cancer stem cell plasticity. Front. Epigenet. Epigenom. 2:1424163. doi: 10.3389/freae.2024.1424163
Received: 27 April 2024; Accepted: 30 August 2024;
Published: 11 September 2024.
Edited by:
Raul Mostoslavsky, Massachusetts General Hospital Cancer Center, United StatesReviewed by:
Eric Hanse, University of California, Irvine, United StatesBriana Prager, Massachusetts General Hospital and Harvard Medical School, United States
Copyright © 2024 Choi and Baek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Inwha Baek, aWJhZWtAa2h1LmFjLmty