 Mary Ann Cielo V. Relucio-San Diego
Mary Ann Cielo V. Relucio-San Diego Paul Christian T. Gloria
Paul Christian T. Gloria Marie Christine M. Obusan
Marie Christine M. Obusan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
DATA REPORT article
Front. Environ. Sci., 28 May 2024
Sec. Water and Wastewater Management
Volume 12 - 2024 | https://doi.org/10.3389/fenvs.2024.1390323
This article is part of the Research TopicBiological Contaminants of Concern in Water and Wastewater: An Environmental Health PerspectiveView all 15 articles
Wastewaters are loaded with many types of microorganisms but are predominantly composed of bacteria with different roles in the water treatment system (Kumar et al., 2022). Some belong to functional genera, which are mainly responsible for the removal of pollutants in wastewater while others are potentially pathogenic bacteria, which can pose threats to public health and the environment (Fan et al., 2018). The functional bacterial groups reported to be found in wastewater treatment plants (WWTPs) include ammonia-oxidizing bacteria (AOB), nitrite-oxidizing bacteria (NOB), polyphosphate-accumulating organisms (PAOs), glycogen-accumulating organisms (GAOs), acetogens, methanogens, and sulfur-reducing bacteria (Gallert and Winter, 2005; López-Vazquez et al., 2007; Dueholm et al., 2022). These types of bacteria are very favorable for plant operations because of their high pollutant removal rates, hence, ensuring their maintenance and sustainability in the systems is critical for the continuous efficacy of wastewater treatment operations (Gude, 2015). WWTPs are also known reservoirs of pathogenic microorganisms (Azli et al., 2022). Current microbial monitoring for contamination is limited to a few indicators such as coliforms, Salmonella sp., Shigella sp., Vibrio sp., Legionella sp., and fecal streptococci and enterococci (Naidoo and Olaniran, 2014; Garrido-Cardenas et al., 2017). However, the microbial indicators may mask the concentration of pathogens due to their lower concentration than the microbial indicators, hence a more sensitive approach is needed for pathogen detection (Zhang et al., 2021). Understanding the composition of bacterial communities is thus crucial in providing the basis for optimizing treatment conditions favoring the functional bacteria and detecting the presence of pathogenic bacteria. With the advent of new sequencing technologies, 16S metabarcoding may be utilized to explore and characterize wastewater microbiomes without the need for conventional culture methods. For these reasons, we sought to profile the bacterial communities in a poultry WWTP to serve as reference and baseline information on potentially functional and pathogenic bacteria present in wastewater, and to infer possible implications to wastewater treatment and management.
Here, we report data on the taxonomic composition of four treatment stages, i.e., Influent Tank, two aeration tanks where activated sludges were used (Aeration Tank 3 and Aeration Tank 1), and the Effluent flow, in a poultry wastewater treatment plant. The metabarcoding data offer insights into the bacterial diversity through ecological indices and may be used for further analyses relevant to the role of bacteria in wastewater treatment.
The samples were collected from a poultry dressing plant with a wastewater treatment facility in Cavite, Region IV-A, Philippines on 1 August 2022. Short interviews were conducted with the plant engineers to determine the operation protocols of the plant. The following consequent sampling points were chosen: (1) Influent Tank, where raw wastewater from the dressing plant was kept before entering the treatment system, (2) Aeration Tank 3, where the raw wastewater entered the system and where activated sludge was used, (3) Aeration Tank 1, where activated sludge was also used after treatment in the anaerobic digester, and the (4) Effluent, where it was free-flowing towards the nearest creek.
Triplicate 1L samples were collected using properly labeled sterile bottles at four sampling areas: Influent Tank, Aeration Tank 3, Aeration Tank 1, and Effluent flow. For the Influent Tank, only the top portion of the tank was collected due to the limitations of the sampler and the small opening of the tank. For the Aeration Tanks, the sampling protocol of the Global Water Microbiome Consortium (GWMC) was followed with modifications (Wu et al., 2019), wherein the sampling points depended only on the accessible areas of the tanks instead of having three different positions per zone. Hence, only one sampling point at a time was accessed with a time interval of 10 min per sampling. For the Effluent, since this was free-flowing, the samples were collected directly from the effluent water.
The twelve samples collected were immediately processed for sub-sampling. Approximately 300 mg of sludge/precipitate was collected from each sampling container and transferred into 1.5 mL cryovials. Triplicate samples from each treatment stage were processed. DNA was extracted following the manufacturer’s protocol using Qiagen DNeasy PowerSoil Pro (QIAGEN, United States). DNA concentration was measured using a Denovix DS-11 fluorometer (Denovix, United States) and samples that met sequencing standards were sent to Macrogen, South Korea for 16S V1-V3 metabarcoding employing 27F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 534R (5′-ATTACCGCGGCTGCTGG-3′) primers (Dueholm et al., 2022). The sequencing platform used was an Illumina MiSeq platform, 2 × 300 bp, 100,000 reads per sample, using Nextera XT-prepared libraries. Raw reads were deposited in the National Center for Biotechnology Information (NCBI) Short Read Archive (SRA) (Supplementary Table S1).
An end-to-end workflow for 16S metabarcoding was employed using QIIME2-2022.8 (Hall and Beiko, 2018). Raw reads were quality-checked, primer trimmed at 0% error rate, and merged using 70 as the optimal --p-minovlen parameter. All merged reads were quality-filtered at Phred 30, dereplicated, and clustered at 97% using an open reference OTU picking technique through VSEARCH (Rognes et al., 2016). Subsequently, singletons and chimeras were removed before taxonomy assignment using a Naive-Bayes trained classifier from SILVA 138-99 (Quast et al., 2012). The resulting taxonomy assignments were filtered to retain only bacterial sequences. Samples were rarefied to a depth of 8,900 features and subsequently, ecological indices along with statistical comparisons were done through the QIIME2 --diversity plugin. The taxonomy bar plot showing relative frequency was generated in R (R Core Team, 2021) using the package ggplot2 (Wickham, 2016) while other figures were directly lifted from QIIME2-generated tools.
Resulting raw reads for all samples yielded a total of 1029761 sequences which was reduced to 806085 after primer trimming. Merging at a minimum overlap length of 70bp yielded 513586 merged reads and subsequent quality trimming at Q30, dereplication, and open-reference-based OTU clustering at 97% using SILVA 138-99 produced 306092 features. Filtering of features to include only bacterial sequences yielded 180324 features. Figure 1 shows the taxonomy bar plots of bacterial sequences present in each sample and treatment stage.
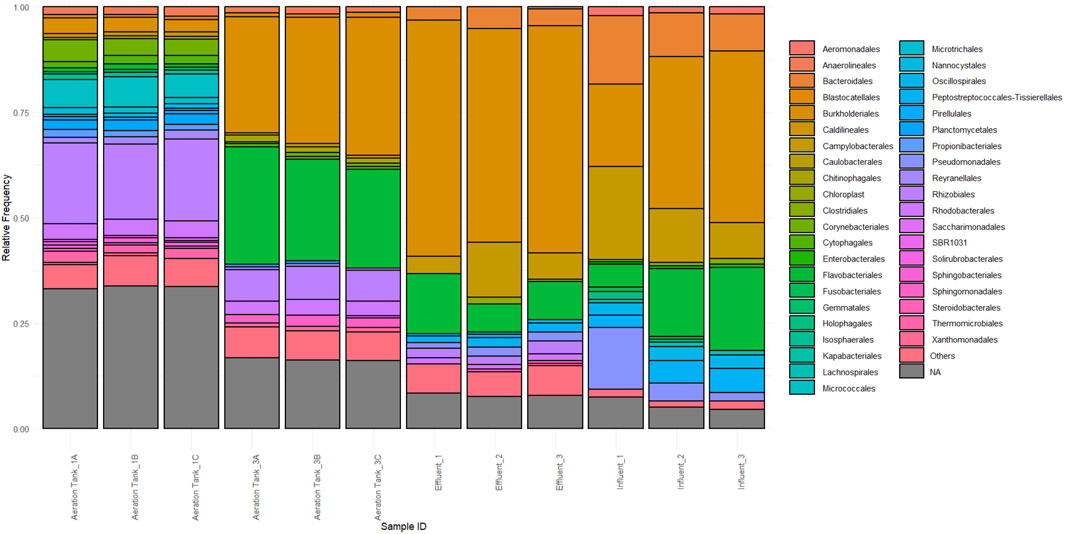
Figure 1. Taxonomy bar plot at the order level of triplicated wastewater samples at various treatment stages.
Effluent samples are dominated by Delftia sp. which comprises approximately 40% of each replicate. This genus has displayed remarkable biotechnological potential in wastewater applications. Delftia tsuruhatensis SDU2 showed excellent ammonium removal in high-strength nitrogen wastewater (Chen et al., 2023). Other studies demonstrated the ability of Delftia to metabolize organic pollutants (Custodio et al., 2022), inhibit biofilm formation in sludge through quorum quenching (Xu et al., 2023), and assimilate terephthalate from sludge (Shigematsu et al., 2003). Flavobacterium sp. is the next dominant member, with a relative frequency of 4.093%–7.283%. Other bacterial community members with a relative frequency of more than 1% are as follows: unclassified and uncultured bacteria (3.224%–4.497%), a bacterium under family Comamonadaceae (2.735%–5.153%), a bacterium under family Rhodocyclaceae (1.186%–1.938%), a bacterium under order Burkholderiales (1.832%–2.655%), a bacterium under order Rhizobiales (1.195%–1.389%), a bacterium under order Campylobacterales (2.126%–9.594%), a bacterium under family Arcobacteraceae (1.730%–3.234%), and an uncultured Tepidibacter sp. (1.616%–2.050%).
Influent samples show a high frequency of family Comamonadaceae, family Arcobacteriaceae members, and Aquaspirillum uncultured bacterium with relative frequency ranges of 7.247%–19.041%, 6.547%–7.799%, and 4.777%–10.197%, respectively. Family Comamonadaceae members were previously reported in influent raw wastewater samples collected from Canadian Arctic ponds (Huang et al., 2021). However, their role in other stages such as in activated sludge is more documented (Sadaie et al., 2007; Ge et al., 2015). Ge et al. (2015) have reported a novel PAO clade Comamonadaceae as integral in the removal of biological phosphorus from abattoir wastewater while its domination in sludge microbiome was seen as a result of sludge reduction due to reduced oxygen supply in a food-processing wastewater treatment plant (Sadaie et al., 2007). Family Arcobacteriaceae was found to be the dominant influent and “clean” effluent microbiome member in 14 Danish wastewater treatment plants (Kristensen et al., 2020). In addition, Arcobacter species have been isolated and characterized to harbor various multi-antibiotic resistance genes and phenotypes from poultry in Ghana and Tunisia (Jribi et al., 2020; Zautner et al., 2023). Thus, it is not surprising that this was also dominant in our Influent samples. Lastly, Aquaspirilum has been consistently found to be a major denitrifier in various reactors in WWTPs in Korea (Lee et al., 2008).
Some highly abundant members in the Effluent were also found in the Influent, namely, Flavobacterium sp. (1.956%–4.881%), a family Rhodocyclaceae member (1.179%–1.392%), an order Burkholderiales member (2.553%–3.306%), and an order Campylobacterales member (1.498%–14.929%). Other detected bacteria with high relative frequnecy are a class Bacteroidia member at 1.784%–3.634%, a family Prevotellaceae member at 2.200%–2.668%, a family Weeksellaceae member at 1.328%–2.842%, Bacteroides luti at 1.324%–1.616%, and Aeromonas E1-54 at 1.403%–1.997%.
Aeration Tank 3 samples are dominated by Flavobacterium sp. at 18.578%–19.575%, followed by the family Rhodocyclaceae at 12.960%–19.813%, and order Burkholderiales members at 7.266%–10.564%. Flavobacterium has been previously reported as an abundant member in wastewater effluent in springtime from a site in Korea (Chen et al., 2019); Flavobacterium has also been extensively documented in wastewater (Ryu et al., 2007; Fujii et al., 2014; Liu et al., 2017). Rhodocyclaceae is also a dominant member of wastewater microbiomes such as in poultry, cattle, and pigs WWTPs (Boukerb et al., 2021). In addition, they were pointed out as important denitrifiers in treatment plants as indicated by metagenomic and phylogenomic analyses (Wang et al., 2020). Order Burkholderiales, another dominant Aeration Tank 3 taxon, has been well documented in wastewater samples such as in the fermentation tank of a piggery WWTP (Shi et al., 2021), in a microbial fuel cell-treated wastewater input with high antibiotic load (Chen et al., 2022), and effluent input into riverine systems (Ruprecht et al., 2021). The following bacteria are detected in this treatment stage with their relative frequency ranges: the same unclassified and uncultured bacteria detected on the Effluent at 8.883%–9.320%, a family Comamonadaceae member at 2.961%–3.319%, a class Actinobacteria member at 1.459%–1.788%, an order Rhizobiales member at 4.394%–4.606%, a family Rhizobiaceae member at 1.235%–1.569%, Flavobacterium cucumis at 3.295%–7.102%, a family Rhodobacteraceae member at 1.981%–2.442%, a class Gammaproteobacteria member at 1.412%–1.774%, a class Alphaproteobacteria member at 1.119%–1.409%, and a family Sphingomonadaceae member at 1.995%–2.635%.
Lastly, Aeration Tank 1 samples show a high frequency of class Actinobacteria (12.824%–14.199%), the same uncultured bacteria seen on the Effluent and Aeration Tank 3 (8.835%–10.582%), an order Rhizobiales member (8.285%–8.680%), a family Rhizobiaceae member (5.472%–6.126%), and Lapilicoccus sp. (5.260%–6.918%). A family Comamonadaceae member (1.384%–1.892%), a class Bacteroidia member (1.286%–1.663%), a family Rhodobacteraceae member (1.648%–1.857%), a class Gammaproteobacteria member (2.081%–2.140%), a class Planctomycetes member (3.068%–3.358%), a class Alphaproteobacteria member (1.491%–1.589%), Mycobacterium sp. (2.215%–3.132%), Gemmobacter sp. (1.157%–1.715%), Reyranella sp. (1.350%–2.197%), an order Thermomicrobiales member (1.851%–2.691%), a family Rhizobiales incertae sedis member (1.468%–1.857%), and an order Corynebacteriales member (1.301%–1.791%) were also present in this treatment stage. A class Actinobacteria member showed high predominance in Aeration Tank 1 microbiomes. Such has been reported of high predominance in effluent microbiomes in a full-scale plant (Kaevska et al., 2016). Other studies show the potential of Actinobacterial members as bioflocculants that aid the treatment process (Agunbiade et al., 2016). Meanwhile, order Rhizobiales and class Rhizobiaceae members have increased in frequency in Aeration Tank 1 which is recapitulative of what is reported by Bia et al. (2021) where a switch of anoxic to oxic conditions permitted its sudden increase. Another group has also reported increased counts of Rhizobiales in activated sludge (Fredriksson et al., 2013). Lapilicoccus also showed high counts in this treatment stage. Interestingly, to our knowledge, there is no report of this genus in other wastewater microbiomes. To date, only two published studies are available: one which discusses its isolation from a botanical garden in Indonesia (Ratnakomala et al., 2016) and another that showed its presence in zinc-contaminated soil microbiome (Zaborowska et al., 2022).
In the case of the functional bacteria in WWTPs, their frequency in the wastewater treatment plant is quite low. Nirosomonas sp., a common NOB, is only present in Aeration Tank 3, with an average of 0.001%. Another bacterium, Thauera sp., which was reported to have denitrification and phosphorus removal capabilities (Ren et al., 2021), is more abundant in Aeration Tank 3, with an average relative frequency of 0.468%, than Aeration Tank 1 with an average relative frequency of 0.086%. A sulfate-reducing bacterium, Ochrobactrum sp. (Yang et al., 2021) was also detected in both Aeration Tanks 3 and 1 with mean relative frequencies of 0.260% and 0.215%, respectively. Two Nitrospira spp., which are AOB, were also detected on both Aeration Tanks with mean relative frequencies of 0.238% and 0.064% in Aeration Tank 3 and 0.051% and 0.115% in Aeration Tank 1. These may not reflect the true frequency of functional bacteria as there are many unidentified bacteria from the samples.
The use of metabarcoding for microbial monitoring revealed some pathogenic bacteria present in the poultry wastewater. Here, we report some of the potential pathogens found in the poultry wastewater treatment plant. Mycobacterium sp., a possible pathogen, has been detected in all treatment stages, with averages of 0.020%, 0.676%, 2.640%, and 0.157% in Influent, Aeration Tank 3, Aeration Tank 1, and Effluent, respectively. Another pathogen, Aeromonas sp., was also detected on the influent, with a relative frequency range of 1.403%–1.997%, in Aeration Tank 3 at 0.014%–0.034%, and in the Effluent at 0.151%–0.520%. Bacteroides spp. are clinically important pathogens where three different species, aside from B. luti, were detected in the Influent and Effluent with a total mean relative frequency of approximately 1.00% per sample. Pseudomonas sp., a ubiquitous and opportunistic pathogen, was present in all sites with the highest frequency in the Influent and the lowest at Aeration Tank 1. Leptospira sp. Was also detected with mean relative frequency of 0.040% in the Effluent and 0.094% in Aeration Tank 3.
The water flow in the treatment plant was as follows: Influent→ Aeration Tank 3 → Aeration Tank 1 → Effluent (Supplementary Figure S1). Aeration Tank 1 had the highest Shannon entropy values ranging from 8.42 to 8.45, which suggests the highest species diversity among all samples, while effluent had the lowest (Figure 2A). Also, Faith’s Phylogenetic Diversity of bacterial composition increased along the treatment stage except for the Effluent where there was a decline in bacterial diversity relative to Aeration Tank 1 (Figure 2B).
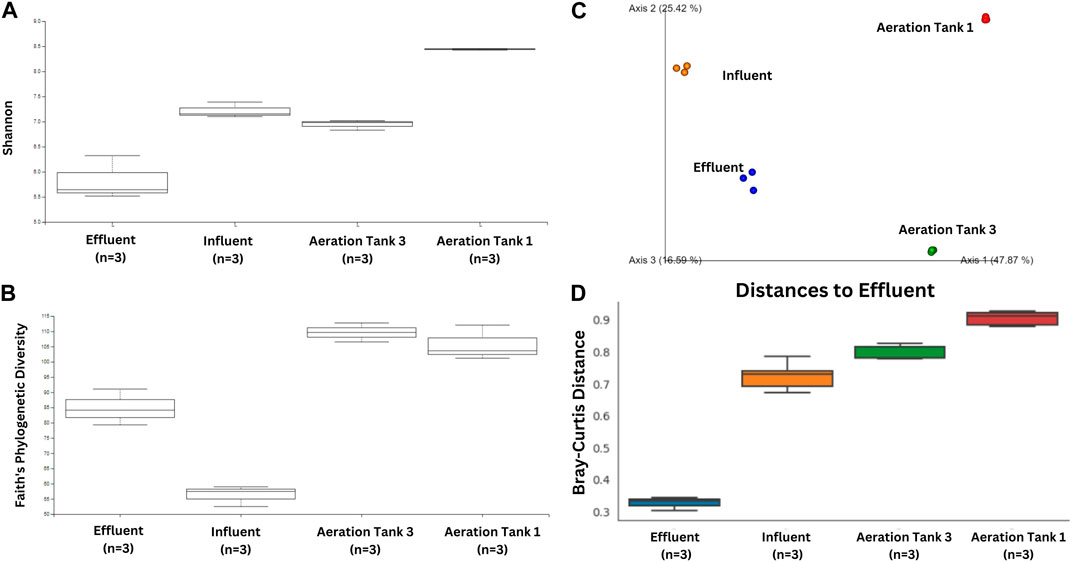
Figure 2. Ecological indices and microbiome composition comparisons. Box-and-whisker plots showing the alpha diversity indices of each wastewater treatment stage according to (A) Shannon’s and (B) Faith’s Phylogenetic Diversity (C) Principal Coordinates Analysis plot of the microbiome for each wastewater treatment stage. (D) PERMANOVA-subjected Box-and-whisker plot for Bray Curtis distances showing the beta diversity index of each treatment stage relative to the effluent bacterial community.
As for bacterial composition, distinct bacterial communities across treatment stages suggested significant alterations in communities as they passed through different tanks (Figure 2C). This observation was corroborated by Bray-Curtis distances relative to Effluent’s bacterial composition where a significant difference in beta diversity was observed. Shifts in each treatment stage’s bacterial community were observed (Figure 2D). In a microbial ecosystem, stochastic and deterministic processes determine community assembly (Dottorini et al., 2021). In the case of WWTPs, bacterial communities are affected by the plant’s capacity, which are composed of equipment capacity, volume of reactors, characteristics of the influent, sludge retention time, and hydraulic retention time (Kim et al., 2019). Hence, these processes may have contributed to the bacterial community shifts in the poultry WWTP system.
The bacterial community profiles of a poultry farm wastewater treatment plant in the Philippines showed that different bacteria dominated the different phases. In the Influent, the most prominent are members of the family Comamonadaceae and of family Arcobacteriaceae, and an uncultured Aquaspirillum bacterium while Delftia sp. and Flavobacterium sp. were dominant in the Effluent. Despite both having activated sludges, the bacterial composition of Aeration Tanks 3 and 1 were also different. For Aeration Tank 3, members of the family Rhodocyclaceae and order Burkholderiales, and Flavobacterium sp. had the highest frequencies while members of class Actinobacteria, order Rhizobiales, family Rhizobiaceae, and Lapilicoccus sp. were the highest for Aeration Tank 1. Common nitrifiers such as Nitrosomonas and Nitrospira were found at a low frequency. Mycobacterium sp., Aeromonas sp., Bacteroides spp., Pseudomonas sp., and Leptospira sp. were the potential pathogens detected in the system that may be disseminated in the environment. This will serve as baseline information on the microbial dimensions in poultry wastewater treatment plants and will allow other researchers to use it as a reference. The bacterial taxonomic information can guide the development of wastewater-related policies and guidelines for improved treatment processes and biotechnological explorations in the future.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
MR-S: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Writing–original draft, Writing–review and editing. PG: Data curation, Formal Analysis, Investigation, Methodology, Validation, Visualization, Writing–original draft, Writing–review and editing. MO: Conceptualization, Funding acquisition, Methodology, Resources, Supervision, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was funded by the University of the Philippines System Enhanced Creative Work and Research Grant (ECRWG-2021-2-13R). The study also received partial funding from the Department of Science and Technology through “Project 2. Microbial Community Composition and Function for the Removal of Excess Nitrogen and Endocrine Disruptors in Wastewater Treatment,” which is monitored by the Philippine Council for Industry, Energy and Emerging Technology Research and Development (PCIEERD).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fenvs.2024.1390323/full#supplementary-material
Agunbiade, M. O., Pohl, C. H., and Ashafa, A. O. (2016). A review of the application of biofloccualnts in wastewater treatment. Pol. J. Environ. Stud. 25 (4), 1381–1389. doi:10.15244/pjoes/61063
Azli, B., Razak, M. N., Omar, A. R., Mohd Zain, N. A., Abdul Razak, F., and Nurulfiza, I. (2022). Metagenomics insights into the microbial diversity and microbiome network analysis on the heterogeneity of influent to effluent water. Front. Microbiol. 13, 779196. doi:10.3389/fmicb.2022.779196
Bia, Y., Hanb, Z., Fengb, S., Wangb, X., Xud, Z., Zhange, Y., et al. (2021). Microbial community changes in a full-scale wastewater treatment system with a rotating biological contactor integrated into anaerobic-anoxic-oxic-oxic processes. Desalination Water Treat. 228, 165–175. doi:10.5004/dwt.2021.27346
Boukerb, A. M., Noël, C., Quenot, E., Cadiou, B., Chevé, J., Quintric, L., et al. (2021). Comparative analysis of fecal microbiomes from wild waterbirds to poultry, cattle, pigs, and wastewater treatment plants for a microbial source tracking approach. Front. Microbiol. 12, 697553. doi:10.3389/fmicb.2021.697553
Chen, J., Guo, J., Zhang, K., Wang, T., Luo, H., Chen, W., et al. (2022). Reduction and control of antibiotic-resistance genes and mobile genetic elements in tetracycline livestock wastewater treated by microbial fuel cell. J. Environ. Eng. 148 (8), 04022039. doi:10.1061/(asce)ee.1943-7870.0002028
Chen, L. F., Chen, L. X., Pan, D., Ren, Y. L., Zhang, J., Zhou, B., et al. (2023). Ammonium removal characteristics of Delftia tsuruhatensis SDU2 with potential application in ammonium-rich wastewater treatment. Int. J. Environ. Sci. Technol. 20 (4), 3911–3926. doi:10.1007/s13762-022-04219-3
Chen, X., Lang, X. L., Xu, A. L., Song, Z. W., Yang, J., and Guo, M. Y. (2019). Seasonal variability in the microbial community and pathogens in wastewater final effluents. Water 11 (12), 2586. doi:10.3390/w11122586
Custodio, M., Peñaloza, R., Espinoza, C., Espinoza, W., and Mezarina, J. (2022). Treatment of dairy industry wastewater using bacterial biomass isolated from eutrophic lake sediments for the production of agricultural water. Bioresour. Technol. Rep. 17, 100891. doi:10.1016/j.biteb.2021.100891
Dottorini, G., Michaelsen, T. Y., Kucheryavskiy, S., Andersen, K. S., Kristensen, J. M., Peces, M., et al. (2021). Mass-immigration determines the assembly of activated sludge microbial communities. Proc. Natl. Acad. Sci. 118 (27), e2021589118. doi:10.1073/pnas.2021589118
Dueholm, M. K. D., Nierychlo, M., Andersen, K. S., Rudkjøbing, V., Knutsson, S., Albertsen, M., et al. (2022). MiDAS 4: a global catalogue of full-length 16S rRNA gene sequences and taxonomy for studies of bacterial communities in wastewater treatment plants. Nat. Commun. 13 (1), 1908. doi:10.1038/s41467-022-29438-7
Fan, X. Y., Gao, J. F., Pan, K. L., Li, D. C., Dai, H. H., and Li, X. (2018). Functional genera, potential pathogens and predicted antibiotic resistance genes in 16 full-scale wastewater treatment plants treating different types of wastewater. Bioresour. Technol. 268, 97–106. doi:10.1016/j.biortech.2018.07.118
Fredriksson, N. J., Hermansson, M., and Wilén, B. M. (2013). The choice of PCR primers has great impact on assessments of bacterial community diversity and dynamics in a wastewater treatment plant. PloS one 8 (10), e76431. doi:10.1371/journal.pone.0076431
Fujii, D., Nagai, F., Watanabe, Y., and Shirasawa, Y. (2014). Flavobacterium longum sp. nov. and Flavobacterium urocaniciphilum sp. nov., isolated from a wastewater treatment plant, and emended descriptions of Flavobacterium caeni and Flavobacterium terrigena. Int. J. Syst. Evol. Microbiol. 64 (Pt_5), 1488–1494. doi:10.1099/ijs.0.054312-0
Gallert, C., and Winter, J. (2005). “Bacterial metabolism in wastewater treatment systems,” in Environmental Biotechnology. Concepts and applications (Hoboken, New Jersey, United States: Wiley).
Garrido-Cardenas, J. A., Polo-López, M. I., and Oller-Alberola, I. (2017). Advanced microbial analysis for wastewater quality monitoring: metagenomics trend. Appl. Microbiol. Biotechnol. 101, 7445–7458. doi:10.1007/s00253-017-8490-3
Ge, H., Batstone, D. J., and Keller, J. (2015). Biological phosphorus removal from abattoir wastewater at very short sludge ages mediated by novel PAO clade Comamonadaceae. Water Res. 69, 173–182. doi:10.1016/j.watres.2014.11.026
Gude, V. G. (2015). A new perspective on microbiome and resource management in wastewater systems. J. Biotechnol. Biomater. 5 (184), 2. doi:10.4172/2155-952x.1000184
Hall, M., and Beiko, R. G. (2018). 16S rRNA gene analysis with QIIME2. Microbiome analysis methods Protoc. 1849, 113–129. doi:10.1007/978-1-4939-8728-3_8
Huang, Y., Ragush, C. M., Johnston, L. H., Hall, M. W., Beiko, R. G., Jamieson, R. C., et al. (2021). Changes in bacterial communities during treatment of municipal wastewater in arctic wastewater stabilization ponds. Front. Water 3, 710853. doi:10.3389/frwa.2021.710853
Jribi, H., Sellami, H., Amor, S. B., Ducournau, A., Sifre, E., Benejat, L., et al. (2020). Occurrence and antibiotic resistance of arcobacter species isolates from poultry in Tunisia. J. Food Prot. 83 (12), 2080–2086. doi:10.4315/jfp-20-056
Kaevska, M., Videnska, P., and Vasickova, P. (2016). Changes in microbial composition of wastewater during treatment in a full-scale plant. Curr. Microbiol. 72, 128–132. doi:10.1007/s00284-015-0924-5
Kim, Y. K., Yoo, K., Kim, M. S., Han, I., Lee, M., Kang, B. R., et al. (2019). The capacity of wastewater treatment plants drives bacterial community structure and its assembly. Sci. Rep. 9 (1), 14809. doi:10.1038/s41598-019-50952-0
Kristensen, J. M., Nierychlo, M., Albertsen, M., and Nielsen, P. H. (2020). Bacteria from the genus Arcobacter are abundant in effluent from wastewater treatment plants. Appl. Environ. Microbiol. 86 (9), 030444-19–e3119. doi:10.1128/aem.03044-19
Kumar, A., Chalotra, T., Pathak, A. K., and Raina, N. (2022). Role of microbes in wastewater treatment and Energy generation potentials: a sustainable approach. Microb. Biotechnol. Role Ecol. Sustain. Res. 265–311. doi:10.1002/9781119834489.ch15
Lee, H. W., Park, Y. K., Choi, E. S., and Lee, J. W. (2008). Bacterial community and biological nitrate removal: comparisons of autotrophic and heterotrophic reactors for denitrification with raw sewage. J. Microbiol. Biotechnol. 18 (11), 1826–1835. doi:10.4014/jmb.0800.276
Liu, H., Lu, P., and Zhu, G. (2017). Flavobacterium cloacae sp. nov., isolated from waste water. Int. J. Syst. Evol. Microbiol. 67 (3), 659–663. doi:10.1099/ijsem.0.001684
López-Vasquez, C. M., Hooijmans, C. M., Brdjanovic, D., Gijzen, H. J., and van Loosdrecht, M. C. (2007). “A practical method for quantification of PAO and GAO populations in activated sludge systems,” in Nutrient removal and recovery symposium 2007 (St, Alexandria, VA, United States: Water Environment Federation), 39–63.
Naidoo, S., and Olaniran, A. O. (2014). Treated wastewater effluent as a source of microbial pollution of surface water resources. Int. J. Environ. Res. Public Health 11 (1), 249–270. doi:10.3390/ijerph110100249
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2012). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic acids Res. 41 (D1), D590–D596. doi:10.1093/nar/gks1219
Ratnakomala, S., Lisdiyanti, P., Prayitno, N. R., Triana, E., Lestari, Y., Hastuti, R. D., et al. (2016). Diversity of actinomycetes Eka Karya Botanical Garden, Bali. BIOTROPIA-The Southeast Asian J. Trop. Biol. 23 (1), 42–51. doi:10.11598/btb.2016.23.1.504
R Core Team (2021) R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing.
Ren, T., Chi, Y., Wang, Y., Shi, X., Jin, X., and Jin, P. (2021). Diversified metabolism makes novel Thauera strain highly competitive in low carbon wastewater treatment. Water Res. 206, 117742.
Rognes, T., Flouri, T., Nichols, B., Quince, C., and Mahé, F. (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ 4, e2584. doi:10.7717/peerj.2584
Ruprecht, J. E., Birrer, S. C., Dafforn, K. A., Mitrovic, S. M., Crane, S. L., Johnston, E. L., et al. (2021). Wastewater effluents cause microbial community shifts and change trophic status. Water Res. 200, 117206.
Ryu, S. H., Park, M., Jeon, Y., Lee, J. R., Park, W., and Jeon, C. O. (2007). Flavobacterium filum sp. nov., isolated from a wastewater treatment plant in Korea. Int. J. Syst. Evol. Microbiol. 57 (9), 2026–2030. doi:10.1099/ijs.0.65138-0
Sadaie, T., Sadaie, A., Takada, M., Hamano, K., Ohnishi, J., Ohta, N., et al. (2007). Reducing sludge production and the domination of Comamonadaceae by reducing the oxygen supply in the wastewater treatment procedure of a food-processing factory. Biosci. Biotechnol. Biochem. 71 (3), 791–799. doi:10.1271/bbb.60632
Shi, L., Liu, N., Liu, G., and Fang, J. (2021). Bacterial community structure and dynamic changes in different functional areas of a piggery wastewater treatment system. Microorganisms 9 (10), 2134. doi:10.3390/microorganisms9102134
Shigematsu, T., Yumihara, K., Ueda, Y., Numaguchi, M., Morimura, S., and Kida, K. (2003). Delftia tsuruhatensis sp. nov., a terephthalate-assimilating bacterium isolated from activated sludge. Int. J. Syst. Evol. Microbiol. 53 (5), 1479–1483.
Wu, L., Ning, D., Zhang, B., Li, Y., Zhang, P., Shan, X., et al. (2019). Global diversity and biogeography of bacterial communities in wastewater treatment plants. Nat. Microbiol. 4 (7), 1183–1195.
Xu, F., Liao, J., Hu, J., Feng, Y., Huang, Y., Feng, X., et al. (2023). Biofouling mitigation and microbial community dynamics in the membrane bioreactor by the indigenous quorum quenching bacterium Delftia sp. JL5. Bioresour. Technol. 388, 129753.
Yang, Z., Liu, Z., Dabrowska, M., Debiec-Andrzejewska, K., Stasiuk, R., Yin, H., et al. (2021). Biostimulation of sulfate-reducing bacteria used for treatment of hydrometallurgical waste by secondary metabolites of urea decomposition by Ochrobactrum sp. POC9: from genome to microbiome analysis. Chemosphere 282, 131064.
Zaborowska, M., Wyszkowska, J., Borowik, A., and Kucharski, J. (2022). Effect of separate and combined toxicity of bisphenol A and zinc on the soil microbiome. Int. J. Mol. Sci. 23 (11), 5937. doi:10.3390/ijms23115937
Zautner, A. E., Riedel, T., Bunk, B., Spröer, C., Boahen, K. G., Akenten, C. W., et al. (2023). Molecular characterization of Arcobacter butzleri isolates from poultry in rural Ghana. Front. Cell. Infect. Microbiol. 13, 1094067. doi:10.3389/fcimb.2023.1094067
Keywords: metabarcoding, bacteria, wastewater, poultry, treatment
Citation: Relucio-San Diego MACV, Gloria PCT and Obusan MCM (2024) 16S metabarcoding of the bacterial community of a poultry wastewater treatment plant in the Philippines. Front. Environ. Sci. 12:1390323. doi: 10.3389/fenvs.2024.1390323
Received: 23 February 2024; Accepted: 13 May 2024;
Published: 28 May 2024.
Edited by:
Visva Bharati Barua, University of North Carolina at Charlotte, United StatesReviewed by:
Yabing Li, Michigan State University, United StatesCopyright © 2024 Relucio-San Diego, Gloria and Obusan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mary Ann Cielo V. Relucio-San Diego, bXJzYW5kaWVnb0B1cC5lZHUucGg=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.