 Emily Rose Lancaster
Emily Rose Lancaster Christopher Lee Jerde
Christopher Lee Jerde Andrew Robert Mahon
Andrew Robert Mahon Erin Katherine Grey
Erin Katherine Grey- 1University of Maine, School of Marine Sciences and School of Ecology and Environmental Sciences, Orono, ME, United States
- 2University of New England, School of Marine and Environmental Programs, Biddeford, ME, United States
- 3University of Santa Barbara, Marine Science Institute, Santa Barbara, CA, United States
- 4Central Michigan University, Department of Biology, Mount Pleasant, MI, United States
1 Introduction
Human-mediated environmental change is impacting every corner of our planet (Pereira et al., 2010). The 2022 Intergovernmental Panel on Climate Change (IPCC) reported that climate change will cause increased extreme weather events, water and food insecurity, species loss, and declines in human health (IPCC 2022). Apart from climate impacts, anthropogenic effects on ecosystems also include moving species from one area to another either unintentionally (i.e., ballast or fouling) or intentionally (i.e., aquarium trade, aquaculture, biocontrol, or assisted migration). If the organism is released into the new environment, it has the potential to become a damaging invasive species. Not all invasive species are damaging, but the lack of evolutionary history between the invader and recipient community creates novel interactions with the potential to be antagonistic (Gilman et al., 2010). Invasive species can be found in all ecosystems and are documented on every continent (Simberloff, 2013; Bergstrom, 2022). If they do have an impact, they could affect food supply, fisheries and agriculture, human health, and ecosystem functioning (Charles and Dukes, 2008). For example, European green crab, Carcinus maenas, has negatively affected the softshell clam fishery as well as native eelgrass beds which help prevent coastal erosion (Garbary et al., 2014). Elsewhere, the domestic cat, Felis catus, decimates local bird and other small prey populations (Baker et al., 2005). In some cases, invasive species have benefit, including recreational value, such as introduced deer or salmonids, or regulatory services that help clean up waterways (Sax et al., 2022). Despite perceived benefits, ecological and cultural effects must be considered across ecosystems and temporal scales.
In 2014, the Invasive Species Specialist Group (ISSG) of the International Union for Conservation of Nature (IUCN) created a list of “100 of the World’s Worst Invasive Alien Species.” While invasive species not included in the list are also impactful, these 101 species were selected due to their relatively large impacts on humans and ecology (Global Invasive Species Database, 2005). This list has been useful for increasing global awareness and galvanizing research and mitigation efforts but is not perfect. For example, one recent study looked at the impact of marine invasive species and found that the listed five marine species (Caulerpa taxifolia, Mytilus galloprovincialis, Carcinus maenas, Undaria pinnatifida, Pterois spp., and Mnemiopsis leidyi) had little to no ecological impact on their invaded environments and a short-lived media impact (Geraldi et al., 2019). Furthermore, only one species of a given genus was chosen for the “Worst” list, where in some instances several congeneric species have invaded regions (e.g., North American freshwater systems by Dreissena spp.). The worst invasive species list includes fungi, plants, invertebrates, and vertebrates affecting six of seven continents. As of yet, none of these listed invasive species have reached Antarctica, though several species may have the physiological capacity to survive if introduced (Tepolt and Somero, 2014). While the World’s 100 Worst Invasive Alien Species List ought to be updated to better reflect the impacts and taxonomic breadth of invasive species, it remains widely cited (more than 2,600 times as of August, 2018) in the scientific literature and its species serve as models for invasion issues (Geraldi et al., 2019).
As invasive species continue to spread and threaten native biodiversity, detection and management are critical to protecting ecosystems, fisheries, and human wellbeing (Pimentel et al., 2004). Our ability to combat invasive species is hindered by lack of genetic information (Matheson and McGaughran, 2022), highlighted by the fact that 52 of the World’s Worst species do not have a fully sequenced reference genome listed in NCBI’s Genbank. Having a full genome for these species would augment early detection through environmental DNA or ploidy testing, and allow for investigation into population genetics, transcriptomics, genomics, and control methods such as genetically modifying organisms. Here, we advocate for full genome sequencing for the remaining World’s Worst species, outline a plan for collecting the remaining specimens, and calculate the cost of completing the remaining sequencing. Our ability to manage invasive species using genomics-based research is within reach (Sepulveda et al., 2020).
2 Why?
Full genome sequencing has a variety of applications, from early species detection to population control, with several applications highly relevant for invasive species management (Figure 1). Environmental DNA (eDNA) methods have been used for the early detection of species in aquatic ecosystems (Ficetola et al., 2008; Sepulveda et al., 2020). Environmental DNA is shed into the environment (air, soil, ice, and water) as an organism undergoes its normal activities; respiring, reproducing, and naturally exuding fluids and cells (Deiner et al., 2017; Taberlet et al., 2012). This eDNA can be extracted from a sample and used in molecular techniques to detect single species (e.g., quantitative polymerase chain reaction [qPCR]), the presence of males (e.g., via PCR targeting Y-linked genes like mammalian SRY), intraspecific genetic diversity (e.g., microsatellites and single-nucleotide polymorphisms), or a community of species (e.g., metabarcoding). Due to the sensitivity of these methods, eDNA techniques can be used without direct capture of an organism, allowing for early detection of invasive or cryptic species. Indeed, several studies have found eDNA methods are to be a valuable part of early detection strategies for invasive species (reviewed in Fonseca et al., 2023). Whole genome sequencing for the remaining World’s Worst Invasive Species would assist in eDNA invasive species detection by broadening the loci available for metabarcoding and qPCR assays to ensure species specificity. For example, when developing eDNA assays to detect a certain species, often looking outside of the typically used barcode genes provides increased opportunity to ensure species specificity (Lim and Thompson, 2021). Further, whole genome sequencing would open the door for broader use of molecular tools for population genetics applications that can require dozens to thousands of informative loci (Sigsgaard et al., 2016; Wheat et al., 2016; Adams et al., 2019, Adams et al., 2022; Andres et al., 2021, Andres et al., 2023).
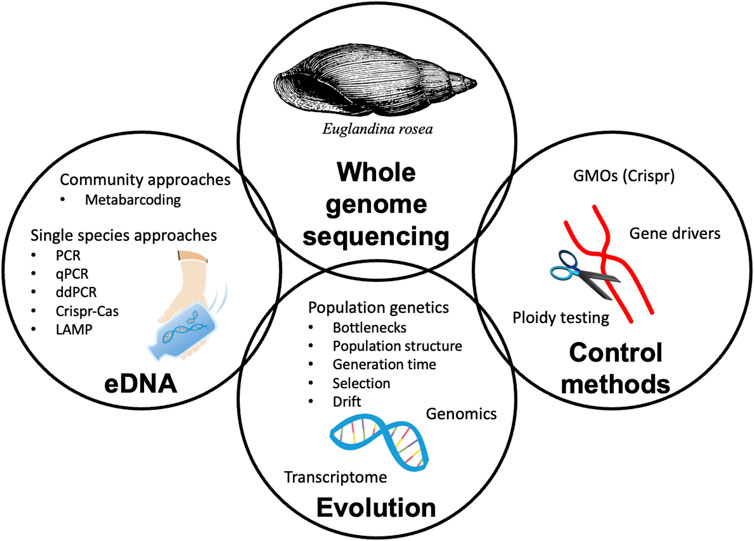
FIGURE 1. Conceptual diagram outlining benefits of whole genome sequencing for the study of invasive species.
While early detection with eDNA is one use of whole genome sequencing, it is but one of many potential applications for the use of these data. Whole genomes will bolster population genetic/genomic studies by providing a map for low-coverage genome sequencing. Population level research focusing on invasive species can help researchers gather information on the amount of introduction events (propagule pressure), genetic bottlenecks, generation time, natural selection, and genetic drift of the population in the recipient community. Additionally, these data can be used to investigate gene expression (transcriptomics) for the invading and native species and can then be used to identify novel genes that are under selection and up/downregulation. Furthermore, by comparing intraspecific and interspecific similarities in the genomes of invasive species, there is potential to predict future invasions through genomics informed species distribution modeling (Hamelin and Roe, 2020; Hudson et al., 2021). For example, genetic mapping of Ophiostoma novo-ulmi helped identify the gene transfer that causes Dutch elm disease to be incredibly invasive (Paoletti et al., 2006).
Lastly, whole genome sequences can assist in control methods for invasive species. These applications include genetically modifying organisms to decrease fecundity or otherwise decrease population levels using molecular techniques such as CRISPR/Cas9 (Li and Scott, 2016). Ploidy testing can be useful for assessing harms, as polyploidy in plants has been shown to increase invasiveness (i.e., Beest et al., 2012), and natural or synthetic ploidy may decrease the success of an invasive species. Transgene-based gene drives use RNA or polypeptides to modify an organism’s effect on the recipient community. Gene drive methods have been investigated for invasive mosquitoes to decrease disease transmission and have been considered to control reproduction of mice populations on islands (Gierus et al., 2022; Harvey-Samuel et al., 2019). Indeed, with any use of genome editing, ethical and biological considerations must be taken to mitigate any potential negative effects.
3 Estimated cost of sequencing and sample collection
Currently, genomic sequencing costs are conservatively estimated to be approximately $5,000 per Gb of the genome, based on externally sourced prices from a number of commercial facilities (A. Mahon, unpublished data). Genome sizes for each of the species lacking a sequenced genome were estimated by comparing data from closely related species (Leitch et al., 2019; Gregory, 2022). Other genome sizes have been estimated through real-time PCR or flow cytometry (Wilhelm et al., 2003; Johnston et al., 2019; Leitch et al., 2019; Gregory, 2022). To obtain the best estimates for genome sizes, if many related species had their genomes sequenced, the estimated genome size represents an average of those species (see Supplementary Table S1 for estimates). If between two and five related species had their genomes sequenced, the estimate reflects the largest genome size. For these 52 species, total sequencing cost is estimated to be less than $600,000. One species, Pinus pinaster, has a large, estimated genome size, which may be inflating the cost to sequence and should be investigated further.
The true global costs of invasive species are largely unknown and unquantified. However, Cuthbert et al. (2022) estimated the cost, with 60 of the 100 worst invasive species for which estimates are reliably known, to be at least $148.9 billion (USD), or approximately 248 times greater than the total cost ($0.6 million USD) of sequencing the 52 un-sequenced invasive species. The economic benefits of the 100 worst invasive species, such as Oreochromis mossambicus as a food source, are also largely unknown (but see Gu et al., 2019) for a discussion of a cost-benefit trade-off for tilapia). However, if the invasive species is a net benefit overall, there are emerging conservation and management benefits to also having the whole genome sequencing information available (Shafer et al., 2015).
We strongly recommend that sample collection and sequencing occur in the native range of the organism in question if possible. Sequencing from the native region ensures that any loss in diversity resulting from genetic bottlenecks, which are common in invasive species populations if few organisms are introduced, will not skew the genomic data. This recommendation only stands if the organism is not endangered or hybridized in its native range; to our understanding, none of these 52 species fall into this category, however Oreochromis mossambicus is considered “near threatened” in its native range due to hybridization (Firmat et al., 2013). We also recommend that the samples be processed in the native region, if possible, for both logistical and equity reasons. First, given complications arising from the Nagoya protocol and restriction on shipping samples in ethanol, sequencing nearby the point of collection would be easier and cheaper. Further, sequencing locally would direct scientific funds more equitably across the world, helping to build and strengthen genomics capacities in underserved areas that would improve research and public health both locally and globally (Blasiak et al., 2020; Knyazev et al., 2022). If local sequencing is not possible, we recommend using the Nagoya protocol for data sharing to ensure there is an equal benefit to all participating groups, with consent and cooperation with indigenous groups and communities (Convention on Biological Diversity, 2011). Invasive species genomes may benefit these groups by decreasing the risk of invasive species impacts on local ecosystems and food security via enhanced early detection and mitigation tools, so we believe sharing this knowledge with them quickly and accurately is extremely important.
4 Discussion
The World’s 100 Worst Invasive Species list aims to unite researchers and managers around the world to mitigate the spread and harms of biological invasions. Genomics can play a key role in this aim, but is currently hampered by lack of reference genomes, even though obtaining such genomes is easier and more affordable than ever. We see a great opportunity for collaborative efforts to obtain these missing genomes and increase genomic research capacity throughout the world. To realize this opportunity, researchers and funders must recognize that the benefits of reference genomes extend further than individual projects and value these contributions accordingly. In this way, we can propel invasion genomics research forward in a way that benefits the world equitably.
A significant number of the World’s Worst Invasive Species do not have their genome sequenced. As we have highlighted here, genome sequencing will help with the early detection of these invaders and increase our general knowledge of invasion biology and population ecology. Ultimately, filling this genetic knowledge gap may allow scientists to work towards molecular strategies leading to the control or eradication of pests. Sequencing costs are continuing to drop. As calculated here, the remaining sequencing could be done for less than $1 million dollars. We hope that by calculating the cost and suggesting a way forward, this message will motivate groups of researchers to complete the required sequencing of the World’s Worst and carry on with other more costly invasive species (Cuthbert et al., 2022).
Author contributions
EL: Conceptualization, Data curation, Investigation, Visualization, Writing–original draft, Writing–review and editing. CJ: Conceptualization, Data curation, Investigation, Visualization, Writing–review and editing. AM: Conceptualization, Data curation, Investigation, Visualization, Writing–review and editing. EG: Conceptualization, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. EL and EG were supported by National Science Foundation award #OIA-1849227 to Maine EPSCoR at the University of Maine. Jerde was funded by the Gordon and Betty Moore Foundation’s support of the California Biodiversity Network.
Acknowledgments
Thank you to Maine-EPSCOR and the Gordon and Betty Moore Foundation for supporting this work.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fenvs.2023.1258880/full#supplementary-material
References
Adams, C. I., Hepburn, C., Jeunen, G. J., Cross, H., Taylor, H. R., Gemmell, N. J., et al. (2022). Environmental DNA reflects common haplotypic variation. Environmental DNA.
Adams, C. I., Knapp, M., Gemmell, N. J., Jeunen, G. J., Bunce, M., Lamare, M. D., et al. (2019). Beyond biodiversity: can environmental DNA (eDNA) cut it as a population genetics tool? Genes 10 (3), 192. doi:10.3390/genes10030192
Andres, K. J., Lodge, D. M., and Andrés, J. (2023). Environmental DNA reveals the genetic diversity and population structure of an invasive species in the Laurentian Great Lakes. Proc. Natl. Acad. Sci. 120 (37), e2307345120. doi:10.1073/pnas.2307345120
Andres, K. J., Sethi, S. A., Lodge, D. M., and Andrés, J. (2021). Nuclear eDNA estimates population allele frequencies and abundance in experimental mesocosms and field samples. Mol. Ecol. 30 (3), 685–697. doi:10.1111/mec.15765
Baker, P. J., Bentley, A. J., Ansell, R. J., and Harris, S. (2005). Impact of predation by domestic cats Felis catus in an urban area. Mammal. Rev. 35, 302–312. doi:10.1111/j.1365-2907.2005.00071.x
Bergstrom, D. M. (2022). Maintaining Antarctica’s isolation from non-native species. Trends Ecol. Evol. 37 (1), 5–9. doi:10.1016/j.tree.2021.10.002
Blasiak, R., Wynberg, R., Grorud-Colvert, K., Thambisetty, S., Bandarra, N. M., Canario, A. V. M., et al. (2020). The Ocean genome: conservation and the fair, equitable and sustainable use of marine genetic resources. Washington, DC: World Resources Institute. online Available at: www.oceanpanel.org/blue-papers/ocean-genome-conservation-andfair-equitable-and-sustainable-use-marine-genetic.
Charles, H., and Dukes, J. S. (2008). “Impacts of invasive species on ecosystem services,” in Biological invasions (Berlin, Heidelberg: Springer), 217–237.
Convention on Biological Diversity (2011). Nagoya protocol access to genetic resources and the fair and equitable sharing of benefits arising from their utilization to the convention on biological diversity. Available at: https://www.cbd.int/abs/doc/protocol/nagoya-protocol-en.pdf.
Cuthbert, R. N., Diagne, C., Haubrock, P. J., Turbelin, A. J., and Courchamp, F. (2022). Are the “100 of the world’s worst” invasive species also the costliest? Biol. Invasions 24 (7), 1895–1904. doi:10.1007/s10530-021-02568-7
Deiner, K., Bik, H. M., Mächler, E., Seymour, M., Lacoursière-Roussel, A., Altermatt, F., et al. (2017). Environmental DNA metabarcoding: transforming how we survey animal and plant communities. Mol. Ecol. 26 (21), 5872–5895. doi:10.1111/mec.14350
Ficetola, G. F., Miaud, C., Pompanon, F., and Taberlet, P. (2008). Species detection using environmental DNA from water samples. Biol. Lett. 4 (4), 423–425. doi:10.1098/rsbl.2008.0118
Firmat, C., Alibert, P., le Losseau, M., Baroiller, J.-F., and Schliewen, U. K. (2013). Successive invasion-mediated interspecific hybridizations and population structure in the endangered cichlid Oreochromis mossambicus. PLOS One 8 (5), e63880. doi:10.1371/journal.pone.0063880
Fonseca, V. G., Davison, P. I., Creach, V., Stone, D., Bass, D., and Tidbury, H. J. (2023). The application of eDNA for monitoring aquatic non-indigenous species: practical and policy considerations. Diversity 15 (5), 631. doi:10.3390/d15050631
Garbary, D. J., Miller, A. G., Williams, J., and Seymour, N. R. (2014). Drastic decline of an extensive eelgrass bed in Nova Scotia due to the activity of the invasive green crab (Carcinus maenas). Mar. Biol. 161 (1), 3–15. doi:10.1007/s00227-013-2323-4
Gierus, L., Birand, A., Bunting, M. D., Godahewa, G. I., Piltz, S. G., Oh, K. P., et al. (2022). Leveraging a natural murine meiotic drive to suppress invasive populations. Proc. Natl. Acad. Sci. 119 (46), e2213308119. doi:10.1073/pnas.2213308119
Gilman, S. E., Urban, M. C., Tewksbury, J., Gilchrist, G. W., and Holt, R. D. (2010). A framework for community interactions under climate change. Trends Ecol. Evol. 25 (6), 325–331. doi:10.1016/J.TREE.2010.03.002
Global Invasive Species Database (2005). Global invasive species database. Available from: http://www.issg.org/database/species/ecology.asp?si=19&fr=1&sts=sss (Accessed September 1, 2005).
Gregory, T. R. (2022). Animal genome size database. Available at: http://www.genomesize.com.
Gu, D. E., Yu, F. D., Yang, Y. X., Xu, M., Wei, H., Luo, D., et al. (2019). Tilapia fisheries in Guangdong Province, China: socio-economic benefits, and threats on native ecosystems and economics. Fish. Manag. Ecol. 26 (2), 97–107. doi:10.1111/fme.12330
Hamelin, R. C., and Roe, A. D. (2020). Genomic biosurveillance of forest invasive alien enemies: a story written in code. Evol. Appl. 13 (1), 95–115. doi:10.1111/eva.12853
Harvey-Samuel, T., Campbell, K. J., Edgington, M., and Alphey, L. (2019). Trialling gene drives to control invasive species: what, where and how? Occas. Pap. SSC (62), 618–627. doi:10.2305/IUCN.CH.2019.SSC-OP.62.en
Hudson, J., Castilla, J. C., Teske, P. R., Beheregaray, L. B., Haigh, I. D., McQuaid, C. D., et al. (2021). Genomics-informed models reveal extensive stretches of coastline under threat by an ecologically dominant invasive species. Proc. Natl. Acad. Sci. 118 (23), e2022169118. doi:10.1073/pnas.2022169118
Johnston, J. S., Bernardini, A., and Hjelmen, C. E. (2019). Genome size estimation and quantitative cytogenetics in insects. Methods Mol. Biol. 1858, 15–26. doi:10.1007/978-1-4939-8775-7_2
Knyazev, S., Chhugani, K., Sarwal, V., Ayyala, R., Singh, H., Karthikeyan, S., et al. (2022). Unlocking capacities of genomics for the COVID-19 response and future pandemics. Nat. Methods 19 (4), 374–380. doi:10.1038/s41592-022-01444-z
Leitch, I., Johnston, E., Pellicer, J., Hidalgo, O., and Bennett, M. (2019). Plant DNA C-values database. Plant DNA C-values Database. Available from: https://cvalues.science.kew.org/ (Accessed December 2, 2022).
Li, F., and Scott, M. J. (2016). CRISPR/Cas9-mediated mutagenesis of the white and Sex lethal loci in the invasive pest, Drosophila suzukii. Biochem. Biophysical Res. Commun. 469 (4), 911–916. doi:10.1016/J.BBRC.2015.12.081
Lim, S. J., and Thompson, L. R. (2021). Mitohelper: a mitochondrial reference sequence analysis tool for fish eDNA studies. Environ. DNA 3 (4), 706–715. doi:10.1002/edn3.187
Lowe, S., Browne, M., Boudjelas, S., and De Poorter, M. (2000). 00 of the world's worst invasive alien species: a selection from the global invasive species database (Vol. 12). in 1. Auckland: Invasive Species Specialist Group.
Matheson, P., and McGaughran, A. (2022). Genomic data is missing for many highly invasive species, restricting our preparedness for escalating incursion rates. Sci. Rep. 12 (1), 13987–13988. doi:10.1038/s41598-022-17937-y
Paoletti, M., Buck, K. W., and Brasier, C. M. (2006). Selective acquisition of novel mating type and vegetative incompatibility genes via interspecies gene transfer in the globally invading eukaryote Ophiostoma novo-ulmi. Mol. Ecol. 15 (1), 249–262. doi:10.1111/j.1365-294x.2005.02728.x
Pereira, H. M., Leadley, P. W., Proença, V., Alkemade, R., Scharlemann, J. P., Fernandez-Manjarrés, J. F., et al. (2010). Scenarios for global biodiversity in the 21st century. Science 330 (6010), 1496–1501. doi:10.1126/science.1196624
Pimentel, D., Zuniga, R., and Morrison, D. (2004). Update on the environmental and economic costs associated with alien-invasive species in the United States. doi:10.1016/j.ecolecon.2004.10.002
Sepulveda, A. J., Nelson, N. M., Jerde, C. L., and Luikart, G. (2020). Are environmental DNA methods ready for aquatic invasive species management? Trends Ecol. Evol. 35 (8), 668–678. doi:10.1016/j.tree.2020.03.011
Shafer, A. B., Wolf, J. B., Alves, P. C., Bergström, L., Bruford, M. W., Brännström, I., et al. (2015). Genomics and the challenging translation into conservation practice. Trends Ecol. Evol. 30 (2), 78–87. doi:10.1016/j.tree.2014.11.009
Sigsgaard, E. E., Nielsen, I. B., Bach, S. S., Lorenzen, E. D., Robinson, D. P., Knudsen, S. W., et al. (2016). Population characteristics of a large whale shark aggregation inferred from seawater environmental DNA. Nat. Ecol. Evol. 1 (1), 0004. doi:10.1038/s41559-016-0004
Simberloff, D. (2013). Invasive species: what everyone needs to know. Oxford: Oxford University Press.
Taberlet, P., Coissac, E., Hajibabei, M., and Riesberg, L. H. (2012). Environmental DNA. Mol. Ecol. 21 (8), 1789–1793. doi:10.1111/j.1365-294X.2012.05542.x
Te Beest, M., Le Roux, J. J., Richardson, D. M., Brysting, A. K., Suda, J., Kubešová, M., et al. (2012). The more the better? The role of polyploidy in facilitating plant invasions. Ann. Bot. 109, 19–45. doi:10.1093/aob/mcr277
Tepolt, C. K., and Somero, G. N. (2014). Master of all trades: thermal acclimation and adaptation of cardiac function in a broadly distributed marine invasive species, the European green crab, Carcinus maenas. Carcinus maenas. J. Exp. Biol. 217 (7), 1129–1138. doi:10.1242/jeb.093849
Wheat, R. E., Allen, J. M., Miller, S. D., Wilmers, C. C., and Levi, T. (2016). Environmental DNA from residual saliva for efficient noninvasive genetic monitoring of brown bears (Ursus arctos). PLoS One 11 (11), e0165259. doi:10.1371/journal.pone.0165259
Keywords: conservation, environmental DNA, genomics, invasive species, population genetics, reference genome, whole genome sequencing, world’s worst alien invasive species
Citation: Lancaster ER, Lee Jerde C, Mahon AR and Grey EK (2023) Time to invest in the worst: a call for full genome sequencing of the 100 worst invasive species. Front. Environ. Sci. 11:1258880. doi: 10.3389/fenvs.2023.1258880
Received: 14 July 2023; Accepted: 17 October 2023;
Published: 30 October 2023.
Edited by:
Antoinette J. Piaggio, Animal and Plant Health Inspection Service (USDA), United StatesReviewed by:
Rafael Miranda, University of Navarra, SpainCopyright © 2023 Lancaster, Lee Jerde, Mahon and Grey. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emily Rose Lancaster, emily.rose.pierce@maine.edu