 Sudipta Rakshit
Sudipta Rakshit Avedananda Ray1
Avedananda Ray1 Ali Taheri
Ali Taheri Michael E. Essington
Michael E. Essington
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Environ. Sci., 09 May 2022
Sec. Soil Processes
Volume 10 - 2022 | https://doi.org/10.3389/fenvs.2022.894581
Nucleic acid derived organic phosphorus (Po) is an important source of plant available P when degraded to inorganic phosphate (P(V)i). It is known that when nucleic acids or components are adsorbed on mineral surfaces, the enzymatic degradation is hindered or delayed. Thus, understanding adsorption/desorption mechanisms of nucleic acids and their derivatives are key to assess the biogeochemical pathways of Po cycling. Here we report adsorption mechanisms of adenosine-5′-monophosphate (AMP) on hematite, a common iron oxide mineral, under various solution properties using macroscopic and in situ attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectroscopic probes. The effects of citrate, mimicking organic acids from roots exudates, and the influence of P(V)i, representing inorganic fertilizer application, were also evaluated on the release of adsorbed AMP under various solution properties. The results suggested that AMP adsorbed with the hematite surface via the phosphate moiety, N7 atom and the π electron systems of the adenine moiety. The presence of citrate significantly decreased the AMP adsorption, which was also corroborated by the negative phosphate IR bands in the results of AMP and citrate competitive adsorption experiments monitored by in situ ATR-FTIR probe. Like citrate, P(V)i also reduced AMP adsorption on hematite. Our findings suggest a potential novel pathway of nucleic acid derived Po cycling in the soil environment.
It has been recognized that there are only finite reserves of inorganic phosphorus (Pi) in geologic rock phosphate deposits (Alewell et al., 2020). Thus, studies on cycling of organic forms of P and mineralization to inorganic phosphate (P(V)i) have been conducted extensively since 1900s (Potter and Benton, 1916; Bower, 1949; Greaves and Wilson, 1970; McLaren et al., 2015; Haygarth et al., 2018; McLaren et al., 2020). The amount of organic P (Po) in the soil ranges from about 29 to 65% of the total soil P (Harrison, 1987; Berg and Joern, 2006). Nucleic acids and derivatives constitute a significant portion of the soil Po and can be converted to plant available P(V)i (Bower, 1949; Greaves and Wilson, 1970; Bowman and Cole, 1978; Levy-Booth et al., 2007; Parikh et al., 2014).
Free or dissolved nucleic acids and their derivatives are rapidly degraded in soil to P(V)i (Greaves and Wilson, 1970; Levy-Booth et al., 2007). However, the general observation by many researchers revealed that when nucleic acid or its components were adsorbed on the soil minerals, the enzymatic degradation was hindered (Bower, 1949; Greaves and Wilson, 1970; Romanowski et al., 1991; Khanna and Stotzky, 1992; Cai et al., 2006; Pedreira-Segade et al., 2016). For example, Bower (1949) indicated that P(V)i production decreased from 90 to 16% when nucleic acid and nucleotides were adsorbed on soil minerals.
Thus, many researchers studied the retention mechanisms of nucleic acids and derivatives on soil minerals to improve the understandings on the biogeochemical pathways of Po cycling (Berg and Joern, 2006; Levy-Booth et al., 2007). In general, the macroscopic adsorption studies and in situ ATR-FTIR investigations indicated that various nucleic acids and derivatives have large affinities for the iron oxide minerals and the main modes of surface complexation were via phosphate moiety (Omoike and Chorover, 2004; Parikh and Chorover, 2006; Cleaves et al., 2011; Schmidt and Martinez, 2017; Sit et al., 2020). Iron oxides are known sinks for inorganic P (V) as well; therefore, a competitive adsorption scenario can be envisioned, but not evaluated in detail under environmentally relevant conditions. Recent study by Sit et al. (2020) evaluated the effects of adsorbed P(V)i on nucleotide adsorption on nanosized hematite and proposed that nucleotide adsorption could be greatly affected by the preadsorbed P(V)i. In addition, the authors noted minor release of preadsorbed P(V)i upon adsorption of deoxy-AMP (dAMP) on nano-hematite. However, the concentrations of P(V)i used in their study were in the range of 300 μM, relevant to groundwater conditions, but not suitable for soil pore water, in which soluble P(V)i concentration was thought to be less than 100 µM (Hinsinger, 2001; Barreto et al., 2020a). This is also supported by the thermodynamic calculation using MINTEQ (Gustafsson, 2014), in which a positive saturation index of P-mineral can be predicted at pH > 7.5 in the presence of other dissolved cations (i.e., dissolved Ca2+ as low as 20 µM) at dissolved P(V)i greater than or equal to 300 µM.
Citrate, a common organic acid detected in high concentrations in the root zone, is known to promote desorption of mineral bound P(V)i (Hinsinger, 2001; Johnson and Loeppert, 2006; Barreto et al., 2020a). In fact, the concentrations of citrate in the root exudates can rise upto mM level in some cases (Jones et al., 1996). Although a great many studies are conducted to decipher the surface complexation mechanisms of citrate promoted desorption of P(V)i adsorbed on soil minerals (Geelhoed et al., 1998; Geelhoed et al., 1999; Barrow et al., 2018; Barreto et al., 2020a), little is known about the effects of citrate on nucleic acid derived organic P desorption from the mineral surfaces. Thus, nucleotide adsorption on minerals and subsequent desorption by organic acids could potentially unravel a novel biogeochemical pathway of soil organic P cycling.
In order to evaluate the potential retention and release mechanisms of small nucleotides, here we propose to study the adsorption mechanisms of adenosine-5′-monophosphate (AMP) on a common soil iron oxide mineral, hematite, under various solution properties using macroscopic and in situ attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectroscopic experiments. In addition, we evaluated the effect of citrate on the AMP adsorption on hematite. To assess the effects of anthropogenic inorganic anions added with fertilizers, we also studied the effects of P(V)i on the adsorption mechanism of AMP on hematite. The importance of AMP is unique in the sense that not only does AMP constitute a fundamental building block of nucleic acids, but it is also an intermediate of biochemical pathways of ATP (adenosine-5′-triphosphate) production. In addition, a recently published study (Klein et al., 2019) indicated that the abiotic dephosphorylation mechanism was negligible for AMP. Thus, the retention and release mechanisms of AMP on hematite will reveal potential abiotic mechanisms of Po cycling.
Adenosine-5′-monophosphate (AMP) was purchased from Sigma-Aldrich and stored at −20°C. A fresh stock solution of AMP was always prepared. Milli-Q water (18.2 MΩ cm) was used for all the experiments. Commercial hematite was purchased from Fisher Scientific (catalog number 50-901-14645) and used as received. All other chemicals used were of analytical or high-performance liquid chromatography (HPLC) grade.
The in situ ATR-FTIR experimental system with flow cell attachment was explained in detail in the supporting documents (SI) and in our earlier studies (Rakshit et al., 2013; Rakshit et al., 2017; Anuo et al., 2021). The adsorption mechanism of IR active solutes at the solid mineral-water interface was successfully probed in the past using similar in situ ATR-FTIR experiments. The detailed principles can be found in the literature (Hug and Sulzberger, 1994; Hind et al., 2001; Lefèvre, 2004; Hinkle et al., 2015). For AMP time series spectra, a freshly prepared AMP stock solution was added to a 500 ml reaction vessel to achieve a 10 µM initial concentration. The pH was controlled at∼ 5.0 ± 0.04. The incremental IR bands were recorded until no enhancement of the IR signal was noticed (∼2 h). For comparing the IR bands of the hematite adsorbed AMP spectrum with that of aqueous AMP spectrum, a 100 µM AMP was dissolved in carbonate-free (Ar purged for 2 h) 0.01 M NaCl solution inside an anaerobic glove box (Coy laboratory products, Grass Lake, MI) at pH 5.0 ± 0.04. Then an aliquot of the aqueous AMP was spread on a Ge-HATR (horizontal ATR, Pike Technologies, Madison, WI) cell and a spectrum was collected. Before collection of this spectrum, carbonate-free 0.01 M NaCl solution at the same pH value was spread on the Ge-HATR cell to collect background spectrum. The sample chamber is Ar purged to avoid interference of CO2. The collected aqueous spectrum was normalized against the highest IR band in the range of 800–1,800 cm−1 to avoid the differences in relative intensity factors in the hematite adsorbed AMP spectrum collected at the similar solution properties. Owing to low detection of IR bands originated from aqueous AMP spectrum, some studies have used significantly greater concentrations (i.e., mM level) of aqueous AMP compared to the adsorption experiments (Sit et al., 2020). However, the intermolecular H-bonding and self-association of AMP could potentially complicate the interpretations while comparing with adsorbed spectrum. Thus, in our study only the 100 µM AMP was used.
For understanding the effects of pH on the adsorption mechanisms of AMP on hematite, initially a 10 µM AMP was equilibrated on the hematite deposit at pH 5.0 ± 0.04 until a stable IR band was obtained and no spectral changes were observed. Then the pH was gradually increased in the range of 5–9. For each incremental pH value, the IR band that resulted from the final equilibrated AMP was recorded. To identify the spectral change upon increase of pH value, raw spectra were subtracted from the pH five spectra and stacked as difference spectra.
For competitive adsorption experiments of AMP with citrate and P(V)i, 10 µM AMP was equilibrated with the hematite deposit at pH 5 (±0.04) and ionic strength 0.01 M NaCl until a final spectrum was obtained. After that, 100 µM citrate was added and the resultant IR bands due to citrate adsorption and AMP desorption were recorded. Two types of difference spectra were plotted to evaluate the desorption of AMP upon addition of citrate. Time series difference spectra were obtained by subtracting the spectrum at a particular time with the spectrum immediately before (i.e., t2-t1, where t2 is the second time point and t1 is the first time point). Negative IR bands associated with these spectra were analyzed for the evidence of desorption of AMP. Final difference spectra were obtained by subtracting the final equilibrated spectrum of AMP + citrate with AMP only. Similar competitive adsorption experiments with 1 µM AMP and 10 µM citrate were also conducted. For P (V), 10 μM P(V) was added to the final equilibrated reaction mixture of 10 µM AMP, then the spectra were collected until equilibrium was reached. Then a 100 μM P (V) was added to the reaction mixture and the final equilibrium AMP + P(V) spectrum was recorded. The difference spectrum was obtained by subtracting AMP + either 10 or 100 μM P final equilibrated spectrum with AMP only spectrum.
The pH envelope of AMP, AMP + citrate, and AMP + P(V) were conducted following the adsorption edge experimental methods described elsewhere (Essington and Stewart, 2016; 2018).Briefly, 1 g L−1 hematite was pre-hydrated in a 1 L reaction vessel using 0.01 M NaCl solution under anaerobic conditions in a glove box (Coy laboratory product, Grass Lake, MI) for 24 h. Next, the reaction vessel was placed on a magnetic stirrer outside the glove box and continuously purged with Ar. A pH electrode was inserted in the reaction vessel to monitor the pH changes. All solutions were Ar purged prior to use to remove CO2. A similar set up for control experiments without hematite was prepared. To begin the pH envelope experiment, first the pH was controlled at 5.0 ± 0.05 using small additions of 0.1 M NaOH or HCl. Then an aliquot of fresh stock solution of AMP was added to the reaction vessel to reach 10 µM. The suspension was allowed to equilibrate for 1 h after addition of AMP stock. Preliminary kinetic study indicated that the adsorption did not increase significantly after 24 h equilibration. After the equilibration period (1 h), the suspension pH was recorded and 10 ml of the suspension was removed, centrifuged, and filtered using a 0.2-µm syringe filter. The filtrate was analyzed for AMP using UV-VIS spectrophotometer (Lambda 650, Perkin Elmer, Shelton, CT) at 260 nm wavelength (Feuillie et al., 2013; Pedreira-Segade et al., 2016). A portion of the filtrate was also analyzed for total phosphorus in ICP-OES (iCAP 7,400, Thermo Electron, West Palm Beach, FL) to corroborate the UV-VIS measurements. Generally, a good agreement (∼10% variability) was obtained (data not shown). The pH value of the suspension was then raised to the next value and allowed to equilibrate for 1 h and the suspension was removed. This process was continued until pH 9. For competitive adsorption experiments, the stock solutions of AMP and citrate or P(V) were added together and a similar procedure as above was followed. The final concentrations of AMP and citrate or P (V) were 10, and 100 µM.
Adsorption of 10 µM AMP on a hematite coated ZnSe HATR cell at a fixed pH (5.0 ± 0.04) and ionic strength (I = 0.01 M NaCl) indicated rapid growth of IR bands at locations 1,648, 1,605, 1,579, 1,480, 1,425, 1,378, 1,337, 1,305, 1,250, 1,215, 1,121, 1,075, and 990 cm−1 (Table 1). This type of IR band enhancement is due to the surface interactions of AMP on the hematite coated ATR crystal and suggests that adsorption occurred either via the outer-sphere (OS) mode or via H-bonding (Müller and Lefèvre, 2011). For inner-sphere bonding, the IR bands would be shifted or indicate significant change of shape in the spectrum of hematite adsorbed AMP compared to the aqueous AMP spectrum under similar solution properties (Müller and Lefèvre, 2011). Alternatively, many researchers conducted the adsorption experiment at varying ionic strength and evaluated the changes in IR band absorbance for identifying the inner-sphere mode of binding (Müller and Lefèvre, 2011).
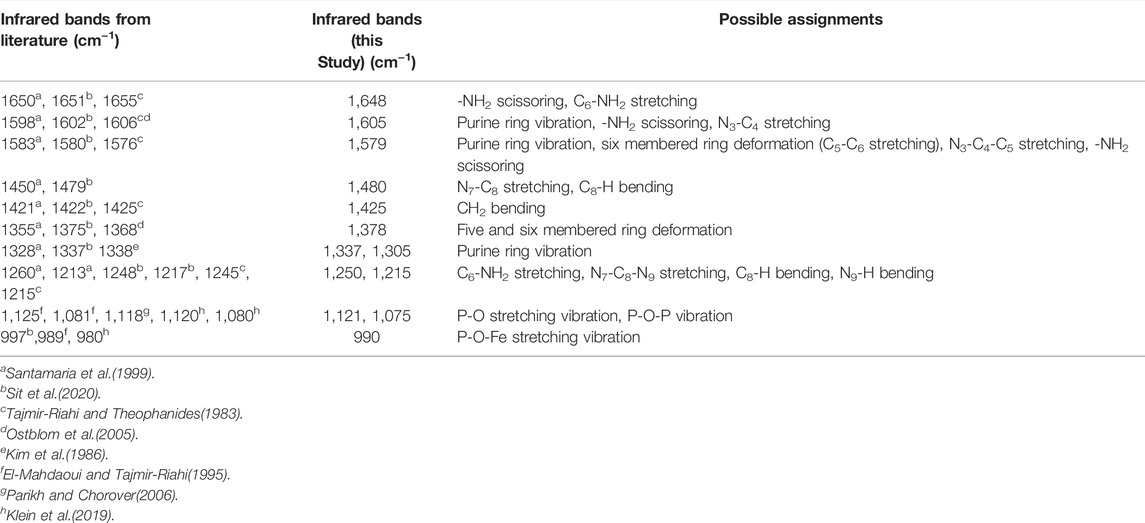
TABLE 1. Infrared bands for adenosine-5′-monopshophate from the literature and this study.
To understand the nature of the surface interaction of AMP with hematite, the final hematite adsorbed AMP spectrum was stacked on top of aqueous AMP spectrum collected at the similar solution properties (pH ∼5 and I = 0.01 M NaCl). The spectra were normalized in the 1800–800 cm−1 wavenumber range with respect to the highest IR band (Figure 1A). There are two main portions of AMP that interact with the hematite surface to cause IR band vibrations: i) the N-atoms and π electrons in the purine ring system and ii) the phosphate moiety (Tajmir-Riahi and Theophanides, 1983; Östblom et al., 2005; Kundu et al., 2009; Klein et al., 2019). Past IR spectroscopic studies with metal complexation by AMP indicated that when the N7 atom of the purine ring system had bound with metal ions, the IR bands originating from N7C8-H bending and N7-C8 stretching caused some shifts or intensity change in the 1,506 and 1,479 cm−1 IR bands (Tajmir-Riahi and Theophanides, 1983; Tajmir-Riahi et al., 1986). For complexation of ATP (adenosine-5′-triphopshate) with trivalent metal cation [Fe (III)], researchers (El-Mahdaoui and Tajmir-Riahi, 1995) suggested that Fe(III)-ATP coordination by N7 atom in the purine ring caused an intensity increase and shift of the 1,477 cm−1 IR band. In our study, the IR band at 1,465 cm−1 of aqueous AMP spectrum most likely shifted due to change in N7-C8 stretching in the hematite adsorbed AMP spectrum to 1,480 cm−1, suggesting strong surface interactions of AMP with hematite using the N7 atom (Table 1; Figures 1A,B). Many researchers indicated the possible binding of adenine and derivatives on solid surface through the exocyclic -NH2 group as well (Giese and McNaughton, 2002; McNutt et al., 2003; Yamada et al., 2004; Östblom et al., 2005). Generally, -NH2 scissoring modes in the purine ring of adenine and derivatives appear in the range 1,620–1,655 cm−1 (Tajmir-Riahi and Theophanides, 1983; El-Mahdaoui and Tajmir-Riahi, 1995; Giese and McNaughton, 2002; Kundu et al., 2009; Sit et al., 2020). A strong band at 1,648 cm−1 did appear in the IR spectrum of AMP adsorbed on hematite at pH 5 (Figure 1A). The change of this -NH2 scissoring band in the adsorbed AMP spectrum could not be assigned to surface complexation of AMP on hematite because the aqueous AMP (100 µM) spectrum indicated a large interference from carbonate or water band in this region. For divalent and trivalent metal binding to AMP and ATP, this -NH2 scissoring mode has not been assigned to metal-NH2 interaction, instead it was speculated as an indication of indirect cation-H2O–NH2 binding (Tajmir-Riahi and Theophanides, 1983; El-Mahdaoui and Tajmir-Riahi, 1995). Other IR bands (1,425, 1,378, 1,337, and 1,305 cm−1) in the hematite adsorbed AMP spectrum can be attributed largely due to the change in five and six membered ring deformations (Santamaria et al., 1999; Giese and McNaughton, 2002; Östblom et al., 2005). Surface complexation of AMP via N7 would cause a change in electron distribution on the N7-C8-N9 of the five membered ring, whereas binding via exocyclic -NH2 would cause a difference in electron delocalization in the six membered ring. Thus, these IR bands originating from various ring deformations are corroborating the surface complexation of AMP via the N7 atom of the five membered ring and exocyclic -NH2 group.
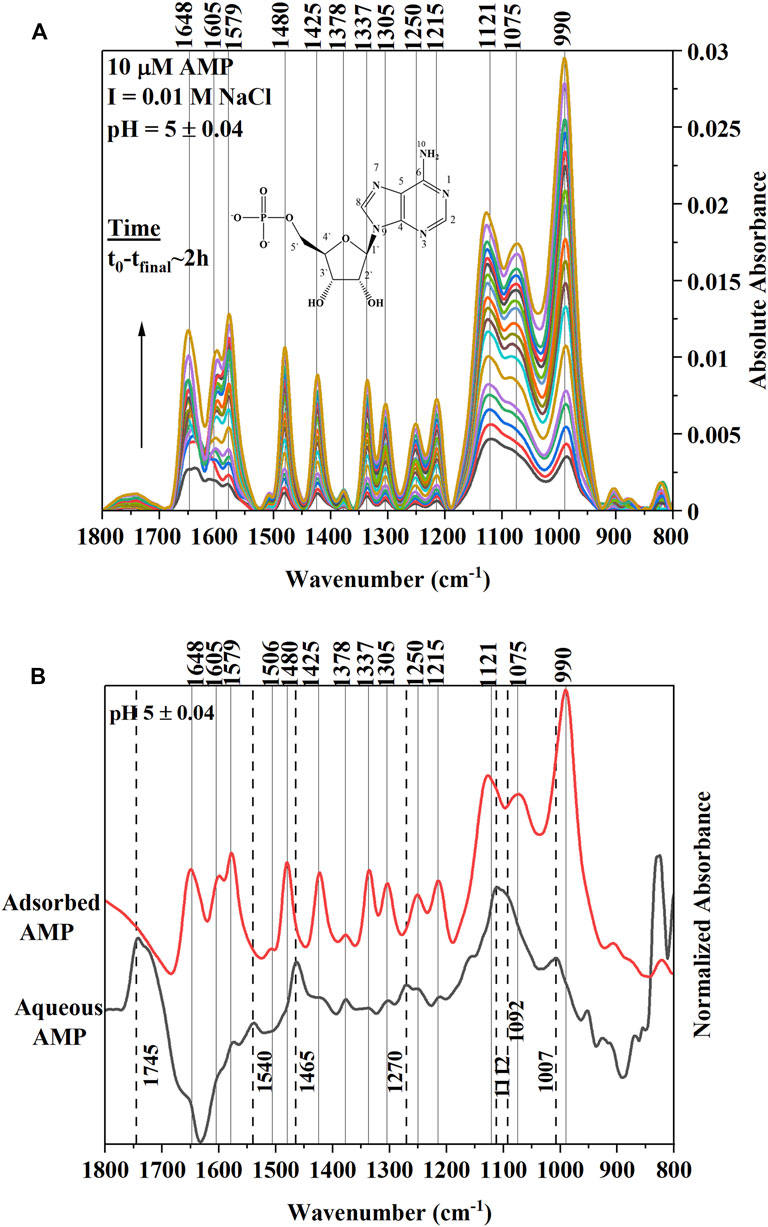
FIGURE 1. (A) (Upper Panel) Time series in situ ATR-FTIR spectra of 10 µM AMP adsorption on hematite at ionic strength of 0.01 M NaCl and pH 5.0 ± 0.04. The t0-tfinal indicates approximately the time interval of collecting first and the last IR spectra after addition of AMP in the reaction mixture. The Y axis is indicating the absolute value of the IR absorbance. The inset is a representative of chemical structure of AMP. (B) (Lower Panel) Final equilibrated spectrum of 10 µM AMP stacked on top of aqueous AMP spectrum collected at similar pH (5.0 ± 0.04) and ionic strength values. Both spectra were normalized with respect to the highest IR band in the 1,800–800 cm−1range to compare. Dotted lines are connecting the IR bands related to aqueous AMP spectrum.
Alternatively, many researchers found that adenine (or adenine moiety in AMP) can interact with a metal surface using the purine π electron system when the adenine orients in a flat conformation (Koglin et al., 1980; Kim et al., 1986; Suh and Moskovits, 1986; Yamada et al., 2004; Kundu et al., 2009). Indeed, the binding mode or orientation of adenine or the adenine portion of the AMP on a solid surface is controversial (Harroun, 2018). However, some inferences can be drawn based on the IR band change or enhancements in the hematite adsorbed AMP spectrum in our study. Researchers (Kim et al., 1986) observed in surface enhanced Raman scattering (SERS) study of adenine and AMP on silver (Ag) sol that relative intensity of the 1,338 cm−1 peak was enhanced when adenine and AMP oriented flat on the Ag surface. This 1,338 cm−1 band had been assigned to adenine ring vibrations in literature both in infrared and Raman spectroscopic studies. A parallel orientation of AMP adsorbed on ferrihydrite has been supported by a recent molecular modeling study as well (Klein et al., 2019). In Figure 1A, the enhancement of 1,337 cm−1 IR band in the hematite adsorbed AMP spectra was clear compared to the aqueous AMP band. Thus, it is likely that in our study at pH 5, AMP also interacted directly with hematite using the purine π electron system. However, a conclusive molecular orientation cannot be deduced based on only infrared results.
Due to the complexities of the interpretations of the surface interactions of the adenine portion of AMP, many published studies attributed the important surface interactions of nucleic acid molecules and derivatives with oxide minerals mainly to the phosphate moiety attached to the ribose sugar and nucleobases (Parikh and Chorover, 2006; Lü et al., 2017; Schmidt and Martinez, 2017; Klein et al., 2019; Sit et al., 2020). For surface complexation of AMP and derivatives on iron oxides (Parikh and Chorover, 2006; Hammami et al., 2016; Klein et al., 2019) or aqueous complexation with the di- and trivalent metals(Tajmir-Riahi and Theophanides, 1983; El-Mahdaoui and Tajmir-Riahi, 1995), the 1,150–900 cm−1 region in the IR spectroscopy is assigned to the vibrations of the phosphate group. In our study, a strong IR band at 990 cm−1 indicated growth in the time series spectra (Figure 1A). This IR band most likely shifted from 1,007 cm−1 IR peak in the aqueous AMP spectrum (Figure 1B). These findings are consistent with the appearance of an IR band in this region due to P-O-Fe stretching (ν) vibrations originating from surface complexation of AMP on iron oxides (Parikh and Chorover, 2006; Klein et al., 2019; Sit et al., 2020). The broad IR band at 1,121 cm−1 and the shoulder at 1,075 cm−1 in the adsorbed AMP spectrum can be assigned to P-O stretching vibrations (El-Mahdaoui and Tajmir-Riahi, 1995; Omoike and Chorover, 2004; Parikh and Chorover, 2006; Klein et al., 2019), which most likely shifted from 1,112 to 1,092 cm−1 in the aqueous AMP spectrum (Figures 1A,B).
The infrared (IR) spectra of hematite-adsorbed AMP at different pH values (5–9) were presented in Figure 2A. The raw spectra were stacked to identify the changes in IR band due to variations of pH values. Below this, a series of difference spectra were presented by subtracting the raw spectra of adsorbed AMP at pH 5 from the other pH values (6, 7, 8, and 9) (Figure 2B). These difference spectra would identify the changes in IR bands when pH was increased from 5 to 9. Since, in our macroscopic experiments, we have observed an overall decrease in AMP adsorption on hematite upon increasing pH, negative bands associated with difference spectra can suggest the change of surface interactions of AMP functional groups at varying pH values (Figure 2B).
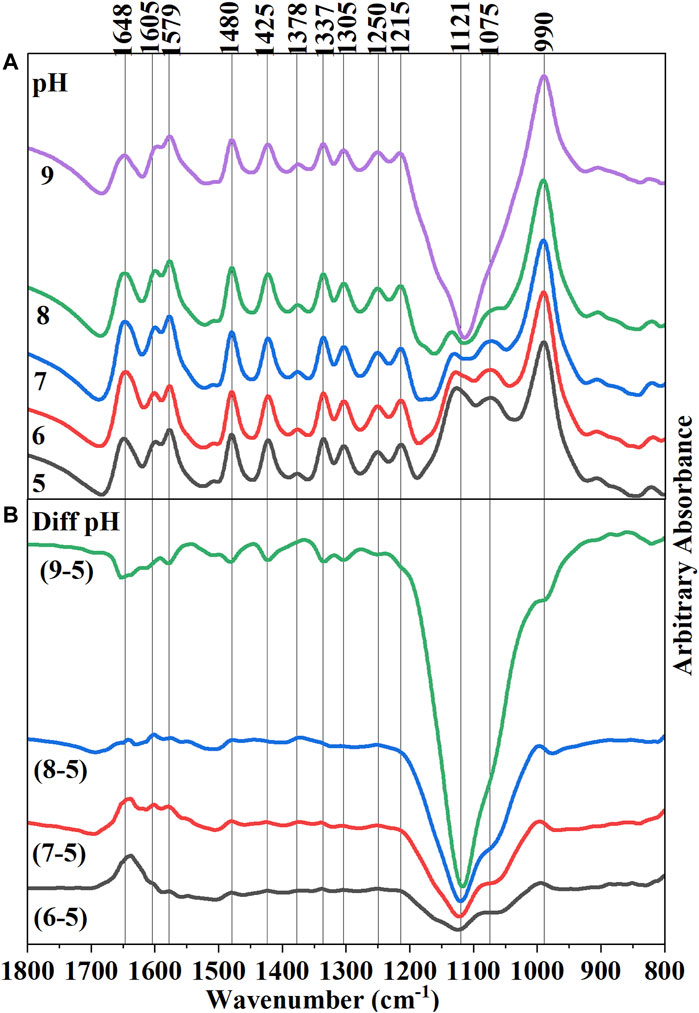
FIGURE 2. (A) (Upper Panel) Final equilibrated in situ ATR-FTIR spectra of 10 µM adsorbed AMP on hematite at pH 5–9. Raw spectra were stacked on each other to visually observe the differences in IR bands, including the absorbance value. (B) (Lower Panel) Difference spectra of subsequent pH increments obtained from subtracting pH 5 spectrum form all other spectra. Thus, Diff pH (9–5) means pH 5 spectrum was subtracted from the final equilibrated adsorbed AMP spectrum on hematite at pH 9.
A noticeable change can be identified with the IR band at 1,648 cm−1 for the subtracted spectra of pH 9 from pH 5, suggesting a change in -NH2 scissoring mode (Figure 2B). Interpretation of the change of the IR band at this location upon variations in pH is somewhat complicated. In addition, owing to the presence of overlapping IR bands of water and carbonate in this location, clear identification is difficult. However, some inference can be made about the change of the -NH2 scissoring mode: i) pKa of the N1H+ of AMP is approximately around 3.96 (Smith et al., 1991; Budnyak et al., 2021); however, there are many tautomeric forms possible in which N7H+ and N3H+ can also be protonated at that pH value (Fonseca Guerra et al., 2006; Verdolino et al., 2008). Thus, at pH < ∼ 4 the lone pair of electrons on exocyclic -NH2 would be less available due to delocalization in the N10-C6-N1H+ system (Ref. AMP structure in Figure 1A). When the pH is raised above 4, the lone pair of electrons on -NH2 would be delocalized in a different way. Thus, a change in -NH2 scissoring and hence the change in the IR band is expected.
ii) The change of -NH2 scissoring is also expected when the extent of surface complexation by this exocyclic -NH2 group would increase upon increasing pH. However, a straightforward interpretation may not be possible because the wet chemical data suggested that overall adsorption of AMP decreased with increasing pH (Figure 3). iii) the intra- and intermolecular H-bonding in AMP would vary as the pH increases above pKa ∼4.0.
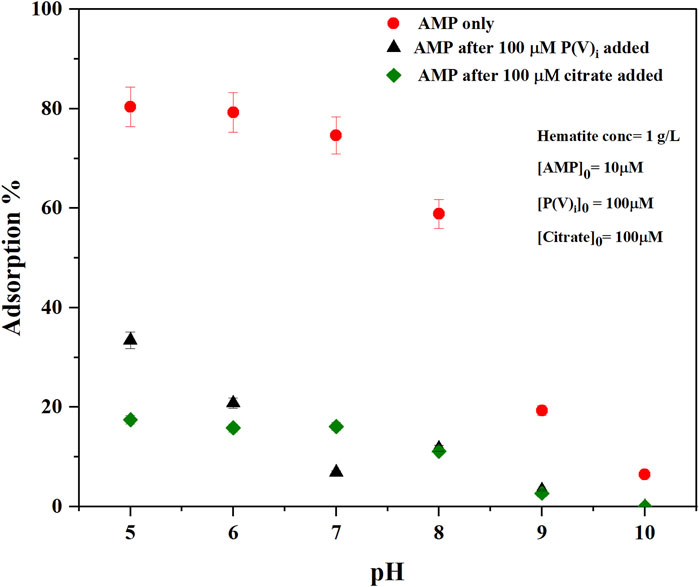
FIGURE 3. Macroscopic adsorption % of AMP on hematite as a function of pH at ionic strength (I = 0.01 M NaCl) without any other adsorbates (AMP only) or with competitive adsorption of either inorganic phosphate (P(V)i) (AMP after 100 μM P(V)i added) or citrate (AMP after 100 µM citrate added) [AMP]0 [Phosphate]0, and [Citrate]0 represent initial concentrations of added adsorbate in the reaction mixture. Error bars represent standard deviation of three replicates.
Increasing pH indicated changes in IR bands consistent with changes in electron delocalization in the five and six membered rings (1,425, 1,378, 1,337, and 1,305 cm−1), and change in the nucleophilic character of the N7 center (1,480 cm−1). Negative IR bands at these locations were most prominent in the pH (9-5) difference spectrum (Figure 2B), indicating at higher pH the purine ring vibrations have reduced. Interestingly, on analyzing the difference spectra of pH (9–5), a negative band at 1,337 cm−1 was observed (Figure 2B). In our earlier discussions on the mechanisms of AMP adsorption on hematite at pH 5, it was noted that a strong IR absorbance in this region was assigned to adenine skeletal ring vibration and a likely indication of molecular orientation of the adsorbed AMP. Thus, on increasing pH, the AMP molecule could have different orientation on the surface (i.e., flat at pH 5 and perpendicular at pH 9). However, this evaluation is confounded by the fact that there is desorption of AMP from the hematite surface when pH is raised (Figure 3). Thus, the decrease in the 1,337 cm−1 IR band can also be related to lower amount of AMP adsorption on hematite.
The pKa of the phosphate moiety in AMP is around 6.46 (Smith et al., 1991; Budnyak et al., 2021). Thus, increasing the pH above this pKa value would change the surface interactions of the phosphate moiety. Major changes in the IR bands due to P-O-Fe and P-O vibrations were noticed at locations of 990 and 1,121 cm−1, which drastically became negative above pH 7 (i.e., the difference spectra of pH (8–5) and pH (9–5)). This is consistent with our macroscopic adsorption data (Figure 3) and P(V) adsorption on iron oxide minerals, in which increasing pH decreases P(V) adsorption (Goldberg and Sposito, 1984; 1985). Due to the large and broad negative band in 1,121 cm−1 region in the difference spectrum, interpretations of the negative band at 990 cm−1 is somewhat complicated.
Drastic reduction of AMP adsorption on hematite was noticed at all experimental pH values (5–10 range) after the addition of citric acid (Figures 3, 4). About 60% of the AMP adsorption on hematite was decreased at pH 5- to 8 when citrate was present (Figure 3). These results are consistent with those studies that evaluated the effects of organic acids in influencing the desorption of P(V)i from oxide minerals (Geelhoed et al., 1998; Geelhoed et al., 1999; Hinsinger, 2001; Johnson and Loeppert, 2006; Barreto et al., 2020a). Release of AMP upon citrate addition was also corroborated by the in situ ATR-FTIR experiments, in which the time series difference spectra indicated the negative IR bands at locations of 1,121 and 990 cm−1 (Figure 4B). In our earlier discussions of AMP adsorption mechanisms on hematite at pH 5, these two IR bands were assigned to P-O and P-O-Fe stretching vibrations. Thus, negative peaks at these locations are indicative of desorption of phosphate moiety of AMP. A recent study (Barreto et al., 2020a) reported similar negative IR bands in the range 900–1,200 cm−1 corresponding to P(V) desorption upon accumulation of citrate on the hematite surface.
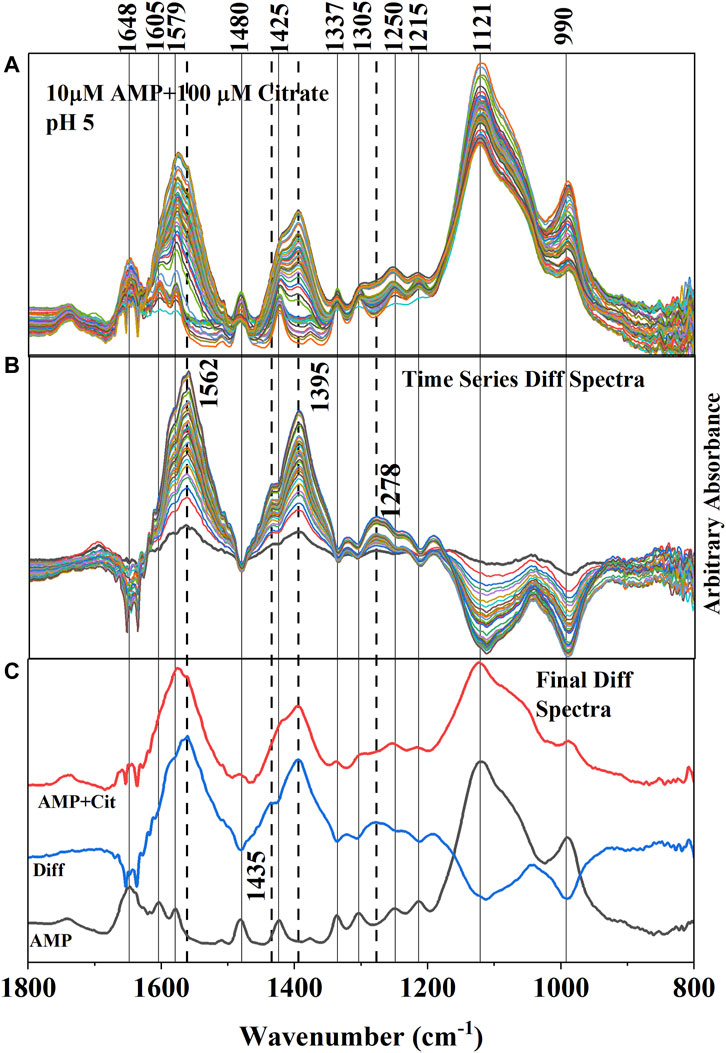
FIGURE 4. (A) (Upper Panel) Time series in situ ATR-FTIR spectra of adsorbed AMP and citrate at pH 5 ± 0.04. The time difference between first and last collected spectra is ∼ 2 h. (B) (Middle Panel) Difference IR spectra (obtained by subtracting the spectra collected after and before citrate addition) representing the resultant IR bands due to adsorption of citrate and concomitant desorption of AMP. The negative IR bands are indicating the weakening of the IR bands associated with adsorption of AMP. Dotted lines are connecting the new IR bands representing the adsorption of citrate on hematite. (C) (Lower Panel) Final equilibrated IR spectra of only adsorbed AMP and adsorbed (AMP + Cit) (when citrate added) were stacked in between the difference spectrum (Diff obtained from subtracting the AMP spectrum from the AMP + Cit spectrum).
Adsorption of citrate on the hematite surface in this work can also be seen by the IR band enhancements in the time series spectrum (Figures 4A–C). However, the interpretations are complicated by the overlapping IR bands of AMP in the same locations. It is unlikely that IR bands from AMP in the 1,800–1,200 cm−1 spectral region would increase upon addition of citrate in the system and is further supported by wet chemical data (Figure 3), in which a 60% reduction of adsorption was noticed at pH 5. Thus, the enhancement of the IR bands in the 1,800–1,200 cm−1 spectral region most likely originated from citrate adsorbing on hematite. In fact, the locations and shapes of the major IR bands (i.e., ∼ 1,570, 1,395, and 1,260 cm−1) resulting from adsorbed citrate on hematite in the absence of AMP (Supplementary Figures S1, S2) resembled the difference spectra (Figure 4C), further supporting citrate adsorption on hematite during the desorption of adsorbed AMP from hematite.
Aqueous citric acid has three pKa values in the range of 3.13, 4.76, and 6.40 (Lackovic et al., 2003; Yeasmin et al., 2014). Thus, at pH∼ 5, the majority of the citrate ions would be in the monoprotonated, divalent anionic form. Earlier studies investigating citrate aqueous speciation using IR spectroscopy indicated that fully deprotonated citrate (citrate3-) has prominent IR absorption bands at 1,570 and 1,390 cm−1, which were assigned to asymmetric (νas COO−) and symmetric (νs COO−) C-O stretching, respectively (Lackovic et al., 2003; Lindegren et al., 2009; Barreto et al., 2020a). The weak and broad feature ∼1,230–1,320 cm−1 in the solution spectra had been assigned to C-OH bending (∼1,280 cm−1) and C-H rocking of the citrate skeleton originating from CH2 groups (∼1,255 and 1,295 cm−1) (Noerpel and Lenhart, 2015; Barreto et al., 2020a) When citric acid is adsorbed on iron oxides, researchers observed that the asymmetric (νas COO−) stretching band of the solution spectrum at ∼1,570 cm−1 was shifted to a lower wavenumber (1,550 cm−1 for goethite and ferrihydrite; 1,562 cm−1 for hematite in this study) and became broader due to structural distortion of carboxylate groups resulting from surface interactions (Lackovic et al., 2003; Yeasmin et al., 2014; Barreto et al., 2020a). Similar to Barreto et al. (2020b) the symmetric (νs COO−) C-O stretching band appeared to split into 1,395 and 1,435 cm−1 in the hematite adsorbed citrate spectrum at pH 5 (Figures 4A–C and Supplementary Figure S1). However, the IR band at 1,435 cm−1 could be due to -CH2 bending mode as well (Mudunkotuwa and Grassian, 2010). Detailed evaluation of citrate binding mode on hematite is not a principal focus of this study and has already been published by many researchers. Here, in the discussion above it is demonstrated that the citrate strongly adsorbed on the hematite surface and concomitant desorption of AMP was evident from both wet chemical and in situ ATR-FTIR data (Figures 3, 4).
Since in the environment, the reported citrate concentrations from root exudates could be as low as 10–50 µM (Hinsinger, 2001), competitive in situ ATR-FTIR adsorption experiments using 10 µM citrate and 1 µM AMP were conducted as well. The results indicated a similar trend (Supplementary Figure S2) to that described above.
Similar to citrate, P(V)i significantly reduced the adsorption of AMP on hematite in the pH of 5–10 range (Figures 5A–C). Compared to 100 µM citrate, 100 μM P(V)i was less efficient in desorbing AMP at pH 5–6 (Figure 3). However, at higher pH values, P(V)i has either greater (pH ∼ 7) or similar effects compared to citrate. In situ ATR-FTIR data corroborated the desorption of AMP in the presence of P(V)i. The final difference spectrum indicated negative IR bands at locations 1,648, 1,605, 1,579, 1,480, 1,425, 1,337, and 1,305 cm−1 when 100 μM P(V) was added to the system (Figure 5C). These IR bands are representative of interactions of the adenine portion of the AMP molecule and negative IR bands are indicative of lack of interactions. Since the IR bands of AMP at locations 1,121–990 cm−1 has a considerable overlap with the IR bands from P(V)i, negative IR bands at these ranges could not be observed. However, the shapes of the IR bands in the difference spectrum (Figure 5C) at the 1,121–990 cm−1 range resembled the IR bands originating from P (V) adsorption on hematite (Elzinga and Sparks, 2007). Upon further visual inspection of the difference spectrum of 10 μM P(V)i, it became clear that the low intensity broad IR band in the 1,121–990 cm−1 range most likely originated from P(V)i. Interestingly, the negative IR bands in the 1,640–1300 cm−1 range were not observed for 10 μM P(V) additions, indicating the interactions of the adenine portion of the AMP molecule possibly could not be hindered at this level of P(V)i.
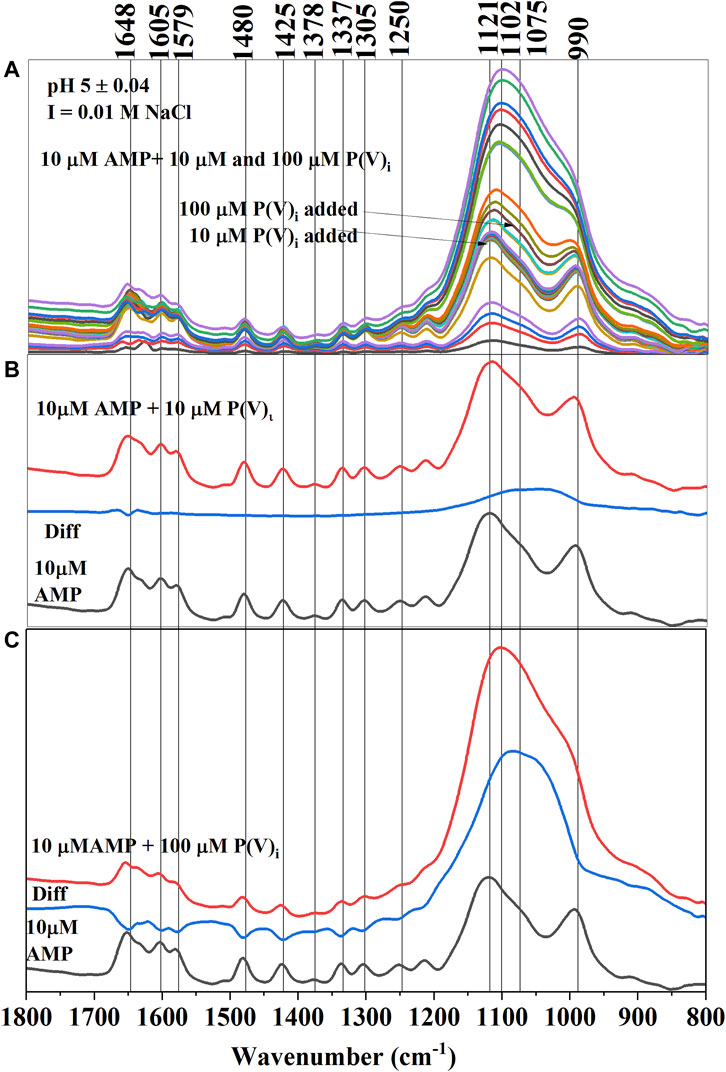
FIGURE 5. (A) (Upper Panel) Time series in situ ATR-FTIR spectra of adsorbed AMP and inorganic phosphate (P(V)i) at pH 5 ± 0.04. (B) (Middle Panel) Final equilibrated IR spectra of only adsorbed AMP and adsorbed (AMP+10 μM P(V)i) (when 10 μM P(V)i was added) were stacked in between the difference spectrum (Diff obtained from subtracting the AMP spectrum from the AMP+ 10 μM P(V)i spectrum). (C) (Lower Panel) Final equilibrated IR spectra of only adsorbed AMP and adsorbed (AMP+100 μM P(V)i) (when 100 μM P(V)i was added) were stacked in between the difference spectrum (Diff obtained from subtracting the AMP final spectrum from the AMP+ 100 μM P(V)i final spectrum).
The results presented here contribute significantly to the understanding of nucleic acid based organic P cycling in the environment in the presence of P(V)i (anthropogenic source) and citrate from root exudates. Knowledge of this mechanistic pathway will not only be helpful to understand plant nutrient cycling, but also to model the fate or P(V)i as a pollutant because the desorbed AMP can be degraded easily by microbes to P(V)i, and then either be absorbed by the plant roots or mobilize to sensitive aquatic environments potentially causing eutrophication. In situ ATR-FTIR spectroscopic data presented here supported the mechanisms of novel biogeochemical pathways of small nucleotide desorption from the iron oxide rich soil environment in the presence of co-adsorbing inorganic (P(V)i) and organic (citrate) anions.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
National Institute of Food and Agriculture, Evans Allen program, Project Number TENX-2129-CCAP. Project Accession Number 1025021.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fenvs.2022.894581/full#supplementary-material
Alewell, C., Ringeval, B., Ballabio, C., Robinson, D. A., Panagos, P., and Borrelli, P. (2020). Global Phosphorus Shortage Will Be Aggravated by Soil Erosion. Nat. Commun. 11 (1), 4546. doi:10.1038/s41467-020-18326-7
Anuo, C. O., Rakshit, S., and Essington, M. E. (2021). Influence of Oxytetracycline on Boron Adsorption at the Hematite-Water Interface: A Macroscopic and In Situ ATR-FTIR Study. Soil Sci. Soc. Am. J. 85 (3), 606–618. doi:10.1002/saj2.20235
Barreto, M. S. C., Elzinga, E. J., and Alleoni, L. R. F. (2020a). Attenuated Total Reflectance-Fourier Transform Infrared Study of the Effects of Citrate on the Adsorption of Phosphate at the Hematite Surface. Soil Sci. Soc. Am. J. 84 (1), 57–67. doi:10.1002/saj2.20005
Barreto, M. S. C., Elzinga, E. J., and Alleoni, L. R. F. (2020b). The Molecular Insights into Protein Adsorption on Hematite Surface Disclosed by In-Situ ATR-FTIR/2D-COS Study. Sci. Rep. 10 (1), 13441. doi:10.1038/s41598-020-70201-z
Barrow, N. J., Debnath, A., and Sen, A. (2018). Mechanisms by Which Citric Acid Increases Phosphate Availability. Plant Soil 423 (1), 193–204. doi:10.1007/s11104-017-3490-8
Berg, A. S., and Joern, B. C. (2006). Sorption Dynamics of Organic and Inorganic Phosphorus Compounds in Soil. J. Environ. Qual. 35 (5), 1855–1862. doi:10.2134/jeq2005.0420
Bower, C. A. (1949). Studies on the Forms and Availability of Soil Organic Phosphorus. Iowa Agric. Home Econ. Exp. Stn. Res. Bull. 28 (362), 1.
Bowman, R. A., and Cole, C. V. (1978). Transformations of Organic Phosphorus Substrates in Soils as Evaluated by NaHCO3 Extraction. Soil Sci. 125, 49–54. doi:10.1097/00010694-197801000-00008
Budnyak, T. M., Vlasova, N. N., Golovkova, L. P., Markitan, O., Baryshnikov, G., Ågren, H., et al. (2021). Nucleotide Interaction with a Chitosan Layer on a Silica Surface: Establishing the Mechanism at the Molecular Level. Langmuir 37 (4), 1511–1520. doi:10.1021/acs.langmuir.0c03050
Cai, P., Huang, Q.-Y., and Zhang, X.-W. (2006). Interactions of DNA with Clay Minerals and Soil Colloidal Particles and Protection against Degradation by DNase. Environ. Sci. Technol. 40 (9), 2971–2976. doi:10.1021/es0522985
Cleaves, H. J., Crapster-Pregont, E., Jonsson, C. M., Jonsson, C. L., Sverjensky, D. A., and Hazen, R. A. (2011). The Adsorption of Short Single-Stranded DNA Oligomers to Mineral Surfaces. Chemosphere 83 (11), 1560–1567. doi:10.1016/j.chemosphere.2011.01.023
El-Mahdaoui, L., and Tajmir-Riahi, H. A. (1995). A Comparative Study of ATP and GTP Complexation with Trivalent Al, Ga and Fe Cations. Determination of Cation Binding Site and Nucleotide Conformation by FTIR Difference Spectroscopy. J. Biomol. Struct. Dyn. 13 (1), 69–86. doi:10.1080/07391102.1995.10508822
Elzinga, E. J., and Sparks, D. L. (2007). Phosphate Adsorption onto Hematite: an In Situ ATR-FTIR Investigation of the Effects of pH and Loading Level on the Mode of Phosphate Surface Complexation. J. Colloid Interface Sci. 308 (1), 53–70. doi:10.1016/j.jcis.2006.12.061
Essington, M. E., and Stewart, M. A. (2016). Adsorption of Antimonate by Gibbsite: Reversibility and the Competitive Effects of Phosphate and Sulfate. Soil Sci. Soc. Am. J. 80 (5), 1197–1207. doi:10.2136/sssaj2016.04.0129
Essington, M. E., and Stewart, M. A. (2018). Adsorption of Antimonate, Sulfate, and Phosphate by Goethite: Reversibility and Competitive Effects. Soil Sci. Soc. Am. J. 82 (4), 803–814. doi:10.2136/sssaj2018.01.0003
Feuillie, C., Daniel, I., Michot, L. J., and Pedreira-Segade, U. (2013). Adsorption of Nucleotides onto Fe-Mg-Al Rich Swelling Clays. Geochim. Cosmochim. Acta 120, 97–108. doi:10.1016/j.gca.2013.06.021
Fonseca Guerra, C., Bickelhaupt, F. M., Saha, S., and Wang, F. (2006). Adenine Tautomers: Relative Stabilities, Ionization Energies, and Mismatch with Cytosine. J. Phys. Chem. A 110 (11), 4012–4020. doi:10.1021/jp057275r
Geelhoed, J. S., Hiemstra, T., and Van Riemsdijk, W. H. (1998). Competitive Interaction between Phosphate and Citrate on Goethite. Environ. Sci. Technol. 32 (14), 2119–2123. doi:10.1021/es970908y
Geelhoed, J. S., Van Riemsdijk, W. H., and Findenegg, G. R. (1999). Simulation of the Effect of Citrate Exudation from Roots on the Plant Availability of Phosphate Adsorbed on Goethite. Eur. J. Soil Sci. 50 (3), 379–390. doi:10.1046/j.1365-2389.1999.00251.x
Giese, B., and McNaughton, D. (2002). Surface-Enhanced Raman Spectroscopic and Density Functional Theory Study of Adenine Adsorption to Silver Surfaces. J. Phys. Chem. B 106 (1), 101–112. doi:10.1021/jp010789f
Goldberg, S., and Sposito, G. (1984). A Chemical Model of Phosphate Adsorption by Soils: I. Reference Oxide Minerals. Soil Sci. Soc. Am. J. 48 (4), 772–778. doi:10.2136/sssaj1984.03615995004800040015x
Goldberg, S., and Sposito, G. (1985). On the Mechanism of Specific Phosphate Adsorption by Hydroxylated Mineral Surfaces: A Review. Commun. Soil Sci. Plant Anal. 16 (8), 801–821. doi:10.1080/00103628509367646
Greaves, M. P., and Wilson, M. J. (1970). The Degradation of Nucleic Acids and Montmorillonite-Nucleic-Acid Complexes by Soil Microorganisms. Soil Biol. Biochem. 2 (4), 257–268. doi:10.1016/0038-0717(70)90032-5
Gustafsson, J. P. (2014). Visual Minteq 3.1. Stockholm: KTH, Department of Land and Water Resources Engineering.
Hammami, K., El-Feki, H., Marsan, O., and Drouet, C. (2016). Adsorption of Nucleotides on Biomimetic Apatite: The Case of Adenosine-5`-Triphosphate (ATP). Appl. Surf. Sci. 360, 979–988. doi:10.1016/j.apsusc.2015.11.100
Harrison, A. F. (1987). Soil Organic Phosphorus : A Review of World Literature. Wallingford: CAB International.
Harroun, S. G. (2018). The Controversial Orientation of Adenine on Gold and Silver. Chemphyschem 19 (9), 1003–1015. doi:10.1002/cphc.201701223
Haygarth, P. M., Harrison, A. F., and Turner, B. L. (2018). On the History and Future of Soil Organic Phosphorus Research: a Critique across Three Generations. Eur. J. Soil Sci. 69 (1), 86–94. doi:10.1111/ejss.12517
Hind, A. R., Bhargava, S. K., and McKinnon, A. (2001). At the Solid/liquid Interface: FTIR/ATR-the Tool of Choice. Adv. Colloid Interface Sci. 93 (1), 91–114. doi:10.1016/S0001-8686(00)00079-8
Hinkle, M. A. G., Wang, Z., Giammar, D. E., and Catalano, J. G. (2015). Interaction of Fe(II) with Phosphate and Sulfate on Iron Oxide Surfaces. Geochim. Cosmochim. Acta 158, 130–146. doi:10.1016/j.gca.2015.02.030
Hinsinger, P. (2001). Bioavailability of Soil Inorganic P in the Rhizosphere as Affected by Root-Induced Chemical Changes: a Review. Plant Soil 237 (2), 173–195. doi:10.1023/A:1013351617532
Hug, S. J., and Sulzberger, B. (1994). In Situ Fourier Transform Infrared Spectroscopic Evidence for the Formation of Several Different Surface Complexes of Oxalate on TiO2 in the Aqueous Phase. Langmuir 10 (10), 3587–3597. doi:10.1021/la00022a036
Johnson, S. E., and Loeppert, R. H. (2006). Role of Organic Acids in Phosphate Mobilization from Iron Oxide. Soil Sci. Soc. Am. J. 70 (1), 222–234. doi:10.2136/sssaj2005.0012
Jones, D. L., Darah, P. R., and Kochian, L. V. (1996). Critical Evaluation of Organic Acid Mediated Iron Dissolution in the Rhizosphere and its Potential Role in Root Iron Uptake. Plant Soil 180 (1), 57–66. doi:10.1007/BF00015411
Khanna, M., and Stotzky, G. (1992). Transformation of Bacillus Subtilis by DNA Bound on Montmorillonite and Effect of DNase on the Transforming Ability of Bound DNA. Appl. Environ. Microbiol. 58 (6), 1930–1939. doi:10.1128/aem.58.6.1930-1939.1992
Kim, S. K., Joo, T. H., Suh, S. W., and Kim, M. S. (1986). Surface-enhanced Raman Scattering (SERS) of Nucleic Acid Components in Silver Sol: Adenine Series. J. Raman Spectrosc. 17 (5), 381–386. doi:10.1002/jrs.1250170503
Klein, A. R., Bone, S. E., Bakker, E., Chang, Z., and Aristilde, L. (2019). Abiotic Phosphorus Recycling from Adsorbed Ribonucleotides on a Ferrihydrite-Type Mineral: Probing Solution and Surface Species. J. Colloid Interface Sci. 547, 171–182. doi:10.1016/j.jcis.2019.03.086
Koglin, E., Sequaris, J. M., and Valenta, P. (1980). Surface Raman Spectra of Nucleic Acid Components Adsorbed at a Silver Electrode. J. Mol. Struct. 60, 421–425. doi:10.1016/0022-2860(80)80102-5
Kundu, J., Neumann, O., Janesko, B. G., Zhang, D., Lal, S., Barhoumi, A., et al. (2009). Adenine− and Adenosine Monophosphate (AMP)−Gold Binding Interactions Studied by Surface-Enhanced Raman and Infrared Spectroscopies. J. Phys. Chem. C 113 (32), 14390–14397. doi:10.1021/jp903126f
Lackovic, K., Johnson, B. B., Angove, M. J., and Wells, J. D. (2003). Modeling the Adsorption of Citric Acid onto Muloorina Illite and Related Clay Minerals. J. Colloid Interface Sci. 267 (1), 49–59. doi:10.1016/S0021-9797(03)00693-3
Lefèvre, G. (2004). In Situ Fourier-transform Infrared Spectroscopy Studies of Inorganic Ions Adsorption on Metal Oxides and Hydroxides. Adv. Colloid Interface Sci. 107 (2-3), 109–123. doi:10.1016/j.cis.2003.11.002
Levy-Booth, D. J., Campbell, R. G., Gulden, R. H., Hart, M. M., Powell, J. R., Klironomos, J. N., et al. (2007). Cycling of Extracellular DNA in the Soil Environment. Soil Biol. Biochem. 39 (12), 2977–2991. doi:10.1016/j.soilbio.2007.06.020
Lindegren, M., Loring, J. S., and Persson, P. (2009). Molecular Structures of Citrate and Tricarballylate Adsorbed on α-FeOOH Particles in Aqueous Suspensions. Langmuir 25 (18), 10639–10647. doi:10.1021/la900852p
Lü, C., Yan, D., He, J., Zhou, B., Li, L., and Zheng, Q. (2017). Environmental Geochemistry Significance of Organic Phosphorus: An Insight from its Adsorption on Iron Oxides. Appl. Geochem. 84, 52–60. doi:10.1016/j.apgeochem.2017.05.026
McLaren, T. I., Smernik, R. J., McLaughlin, M. J., McBeath, T. M., Kirby, J. K., Simpson, R. J., et al. (2015). Complex Forms of Soil Organic Phosphorus-A Major Component of Soil Phosphorus. Environ. Sci. Technol. 49 (22), 13238–13245. doi:10.1021/acs.est.5b02948
McLaren, T. I., Smernik, R. J., McLaughlin, M. J., Doolette, A. L., Richardson, A. E., and Frossard, E. (2020). The Chemical Nature of Soil Organic Phosphorus: A Critical Review and Global Compilation of Quantitative Data. Adv. Agron. 160, 51–124. doi:10.1016/bs.agron.2019.10.001
McNutt, A., Haq, S., and Raval, R. (2003). RAIRS Investigations on the Orientation and Intermolecular Interactions of Adenine on Cu(110). Surf. Sci. 531 (2), 131–144. doi:10.1016/s0039-6028(03)00444-8
Müller, K., and Lefèvre, G. (2011). Vibrational Characteristics of Outer-Sphere Surface Complexes: Example of Sulfate Ions Adsorbed onto Metal (Hydr)oxides. Langmuir 27 (11), 6830–6835. doi:10.1021/la200514z
Mudunkotuwa, I. A., and Grassian, V. H. (2010). Citric Acid Adsorption on TiO2 Nanoparticles in Aqueous Suspensions at Acidic and Circumneutral pH: Surface Coverage, Surface Speciation, and its Impact on Nanoparticle-Nanoparticle Interactions. J. Am. Chem. Soc. 132 (42), 14986–14994. doi:10.1021/ja106091q
Noerpel, M. R., and Lenhart, J. J. (2015). The Impact of Particle Size on the Adsorption of Citrate to Hematite. J. Colloid Interface Sci. 460, 36–46. doi:10.1016/j.jcis.2015.08.028
Omoike, A., and Chorover, J. (2004). Spectroscopic Study of Extracellular Polymeric Substances from Bacillus Subtilis: Aqueous Chemistry and Adsorption Effects. Biomacromolecules 5, 1219–1230. doi:10.1021/bm034461z
Östblom, M., Liedberg, B., Demers, L. M., and Mirkin, C. A. (2005). On the Structure and Desorption Dynamics of DNA Bases Adsorbed on Gold: A Temperature-Programmed Study. J. Phys. Chem. B 109 (31), 15150–15160. doi:10.1021/jp051617b
Parikh, S. J., and Chorover, J. (2006). ATR-FTIR Spectroscopy Reveals Bond Formation during Bacterial Adhesion to Iron Oxide. Langmuir 22 (20), 8492–8500. doi:10.1021/la061359p
Parikh, S. J., Mukome, F. N. D., and Zhang, X. (2014). ATR-FTIR Spectroscopic Evidence for Biomolecular Phosphorus and Carboxyl Groups Facilitating Bacterial Adhesion to Iron Oxides. Colloids Surf. B. 119, 38–46. doi:10.1016/j.colsurfb.2014.04.022
Pedreira-Segade, U., Feuillie, C., Pelletier, M., Michot, L. J., and Daniel, I. (2016). Adsorption of Nucleotides onto Ferromagnesian Phyllosilicates: Significance for the Origin of Life. Geochim. Cosmochim. Acta 176, 81–95. doi:10.1016/j.gca.2015.12.025
Potter, R. S., and Benton, T. H. (1916). The Organic Phosphorus of Soil. Soil Sci. 2 (3), 291–298. doi:10.1097/00010694-191609000-00004
Rakshit, S., Elzinga, E. J., Datta, R., and Sarkar, D. (2013). In Situ attenuated Total Reflectance Fourier-Transform Infrared Study of Oxytetracycline Sorption on Magnetite. J. Environ. Qual. 42 (3), 822–827. doi:10.2134/jeq2012.0412
Rakshit, S., Sallman, B., Davantés, A., and Lefèvre, G. (2017). Tungstate (VI) Sorption on Hematite: An In Situ ATR-FTIR Probe on the Mechanism. Chemosphere 168, 685–691. doi:10.1016/j.chemosphere.2016.11.007
Romanowski, G., Lorenz, M. G., and Wackernagel, W. (1991). Adsorption of Plasmid DNA to Mineral Surfaces and Protection against DNase I. Appl. Environ. Microbiol. 57 (4), 1057–1061. doi:10.1128/aem.57.4.1057-1061.1991
Santamaria, R., Charro, E., Zacarías, A., and Castro, M. (1999). Vibrational Spectra of Nucleic Acid Bases and Their Watson-Crick Pair Complexes. J. Comput. Chem. 20 (5), 511–530. doi:10.1002/(sici)1096-987x(19990415)20:5<511::aid-jcc4>3.0.co;2-8
Schmidt, M. P., and Martínez, C. E. (2017). Ironing Out Genes in the Environment: An Experimental Study of the DNA-Goethite Interface. Langmuir 33 (34), 8525–8532. doi:10.1021/acs.langmuir.7b01911
Sit, I., Sagisaka, S., and Grassian, V. H. (2020). Nucleotide Adsorption on Iron(III) Oxide Nanoparticle Surfaces: Insights into Nano-Geo-Bio Interactions through Vibrational Spectroscopy. Langmuir 36 (51), 15501–15513. doi:10.1021/acs.langmuir.0c02633
Smith, R. M., Martell, A. E., and Chen, Y. (1991). Critical Evaluation of Stability Constants for Nucleotide Complexes with Protons and Metal Ions and the Accompanying Enthalpy Changes. Pure Appl. Chem. 63 (7), 1015–1080. doi:10.1351/pac199163071015
Suh, J. S., and Moskovits, M. (1986). Surface-enhanced Raman Spectroscopy of Amino Acids and Nucleotide Bases Adsorbed on Silver. J. Am. Chem. Soc. 108 (16), 4711–4718. doi:10.1021/ja00276a005
Tajmir-Riahi, H. A., and Theophanides, T. (1983). Adenosine-5′-monophosphate Complexes of Pt(II) and Mg(II) Metal Ions. Synthesis, FTIR Spectra and Structural Studies. Inorg. Chim. Acta 80, 183–190. doi:10.1016/S0020-1693(00)91280-5
Tajmir-Riahi, H. A., Bertrand, M. J., and Theophanides, T. (1986). Synthesis, Structure, Proton-Nuclear Magnetic Resonance, and Fourier Transform Infrared Spectroscopy of Several Transition and Nontransition Metal-Adenosine-5-Triphosphate Complexes. Can. J. Chem. 64 (5), 960–966. doi:10.1139/v86-160
Verdolino, V., Cammi, R., Munk, B. H., and Schlegel, H. B. (2008). Calculation of pKa Values of Nucleobases and the Guanine Oxidation Products Guanidinohydantoin and Spiroiminodihydantoin Using Density Functional Theory and a Polarizable Continuum Model. J. Phys. Chem. B 112 (51), 16860–16873. doi:10.1021/jp8068877
Yamada, T., Shirasaka, K., Takano, A., and Kawai, M. (2004). Adsorption of Cytosine, Thymine, Guanine and Adenine on Cu(110) Studied by Infrared Reflection Absorption Spectroscopy. Surf. Sci. 561 (2), 233–247. doi:10.1016/j.susc.2004.05.095
Yeasmin, S., Singh, B., Kookana, R. S., Farrell, M., Sparks, D. L., and Johnston, C. T. (2014). Influence of Mineral Characteristics on the Retention of Low Molecular Weight Organic Compounds: A Batch Sorption-Desorption and ATR-FTIR Study. J. Colloid Interface Sci. 432, 246–257. doi:10.1016/j.jcis.2014.06.036
Keywords: nucleotide, organic phosphate, adsorption, adenosine-5′monophosphate, citrate
Citation: Rakshit S, Ray A, Taheri A and Essington ME (2022) Influence of Citrate and Phosphate on the Adsorption of Adenosine-5′-Monophosphate at the Hematite Water Interface. Front. Environ. Sci. 10:894581. doi: 10.3389/fenvs.2022.894581
Received: 11 March 2022; Accepted: 18 April 2022;
Published: 09 May 2022.
Edited by:
Jun Zhou, University of Massachusetts Lowell, United StatesReviewed by:
Zhi-Neng Hong, Institute of Soil Science (CAS), ChinaCopyright © 2022 Rakshit, Ray, Taheri and Essington. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sudipta Rakshit, c3Jha3NoaXRAdG5zdGF0ZS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.