 Teresa Einzmann
Teresa Einzmann Moritz Schroll
Moritz Schroll Jan F. Kleint
Jan F. Kleint Markus Greule
Markus Greule Frank Keppler
Frank Keppler- 1Institute of Earth Sciences, Heidelberg University, Heidelberg, Germany
- 2Department of Environmental Sciences, University of Basel, Basel, Switzerland
- 3Department of Geosciences, MARUM – Center for Marine Environmental Sciences, University of Bremen, Bremen, Germany
- 4Heidelberg Center for the Environment (HCE), Heidelberg University, Heidelberg, Germany
Methane (CH4) emissions from aquatic systems have recently been comprised to account for up to 50% of global CH4 emissions, with lakes representing one of the largest CH4 sources within this pool. However, there is large uncertainty associated with CH4 emissions from freshwater environments to the atmosphere, because of a lack of understanding in the spatial and temporal dynamics of CH4 sources and sinks, as well as underlying mechanisms and processes. In this study, we investigated the concentrations and stable carbon (δ13C-CH4) and hydrogen (δ2H-CH4) isotope composition of CH4 in a small eutrophic lake (Lake Willersinnweiher) with seasonal stratification and its spatial and temporal variation. We found that while supersaturation of CH4 in the entire water column was present throughout the whole year, the isotopic composition of CH4 in sediment and water column varied depending on lake stratification, physiochemical conditions, and lake depth. During the stratification period, isotopic characteristics of pelagic surface water CH4 differed from littoral and sedimentary CH4, suggesting likely mixing of CH4 from different sources including vertical and lateral input as well as groundwater input and potentially oxic methane production in the mixed surface water layer. Aerobic CH4 oxidation indicated by a strong increase in both δ13C-CH4 and δ2H-CH4 values at the bottom of the oxycline was found to significantly reduce upward migrating CH4 released at the sediment-water interface. In the sediment, stable isotope characteristics of CH4 showed an increasing dominance of the acetoclastic CH4 formation pathway from the pelagic towards the littoral area. Furthermore, the occurrence of sulfate-dependent anaerobic methane oxidation in the sediment was suggested by an increase in δ13C-CH4 and δ2H-CH4 values. During the mixing period, the isotopic CH4 composition of the water column was distinctively less negative than during the stratification period potentially resulting from a greater impact of groundwater CH4 input compared to the stratification period. Our findings implicate that the application of concentrations and dual isotope measurements of CH4 is a promising approach for constraining CH4 sinks and sources in Lake Willersinnweiher and potentially other small lakes to clearly disentangle the complex CH4 dynamics in lakes both spatially and seasonally.
Introduction
Lakes and other freshwater systems cover only a small area of the earth’s continental land surface (>3%; Downing et al., 2006), but play an important role in the global carbon cycle and greenhouse gas emissions to the atmosphere (Oswald et al., 2015). Amongst aquatic systems, lakes constitute one of the largest sources of CH4 emission, releasing 23–142 Tg CH4 yr−1 to the atmosphere (Rosentreter et al., 2021). Thus, CH4 cycles in lakes and their fluxes to the atmosphere have been investigated extensively in recent years (e.g. Casper et al., 2000; Bastviken et al., 2004; Natchimuthu et al., 2016; Donis et al., 2017; Peeters et al., 2019) and global lake CH4 emissions were found to be dominated by small lakes (surface area <1 km2) (Thottathil et al., 2022). Nevertheless, many key factors regarding CH4 sources and sinks in lakes and their spatial and temporal variability remain unknown (Duc et al., 2010; Loken et al., 2019). Methane dynamics in lacustrine environments are complex and dependent on various biological production and consumption mechanisms as well as different transport pathways, impacting the distribution and accumulation of CH4 in the water column (Günthel et al., 2019) (Figure 1). In particular, the source of CH4 supersaturation in the surface water layer has been intensely debated in recent years. Whilst some research groups support lateral (riverine, littoral) and vertical CH4 input from anoxic sources as the main mechanisms behind this observation (Fernández et al., 2016; Peeters et al., 2019; Peeters and Hofmann, 2021), others provide evidence for CH4 production under oxic conditions as a potentially important process in the surface water layer of lakes (Grossart et al., 2011; Tang et al., 2016; Donis et al., 2017; Günthel et al., 2019; Hartmann et al., 2020; Thottathil et al., 2022). However, the large discrepancies in estimations of the contribution of oxic CH4 production to surface water CH4 emissions, varying from negligible contributions (Peeters and Hofmann, 2021) to a contribution of oxic methanogenesis to surface water emissions of up to 90% (Donis et al., 2017), reveal a lack of understanding of the processes contributing to surface supersaturation and their spatial and temporal variability. Although the determination of dissolved CH4 concentrations and CH4 fluxes is helpful to establish the magnitude of emissions, it cannot resolve the responsible pathways and production mechanisms (Cadieux et al., 2016). Measurement of stable carbon and hydrogen isotopes of CH4 (expressed as δ13C-CH4 and δ2H-CH4 values) are known to assist with identifying sources and sinks of CH4 (e.g., Waldron et al., 1999; Whiticar, 2020; Douglas et al., 2021).
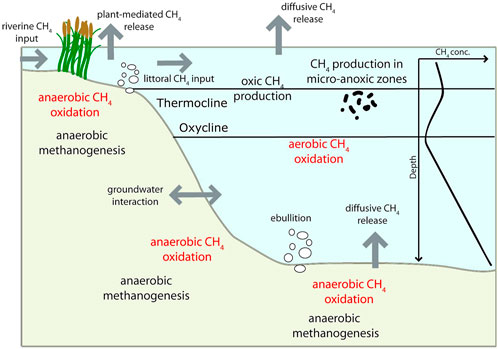
FIGURE 1. Overview of sources and sinks of CH4 as well as transport processes (transport direction indicated by grey arrows) and a typical concentration profile of CH4 in the lacustrine environment during thermal stratification. Modified after Tang et al. (2016).
In aquatic systems, biogenic CH4 production has until recently been assumed to be solely performed by anaerobic methanogenic archaea occurring in the anoxic sediment and water column (e.g. Lessner, 2009). Microbial methanogenesis leads to the formation of 13C and 2H depleted CH4. In anoxic sediment, methanogenesis typically involves two metabolic pathways using competitive substrates: Carbon dioxide (CO2) reduction via H2 (hydrogenotrophic) and acetate fermentation (acetoclastic). Methane production through CO2 reduction generates δ13C-CH4 values ranging from −110 to −60‰ and δ2H-CH4 values from −250 to −150‰, whereas acetoclastic methanogenesis generates δ13C-CH4 values varying from −60 to −40‰ and δ2H-CH4 values from −400 to −250‰ (Whiticar, 1999; Belle et al., 2015). However, the δ2H-CH4 values were found not be exclusively controlled by the methanogenic formation pathway, but other variables such as CH4 oxidation and δ2H-H2O values also have significant impact on the stable hydrogen isotope composition of CH4 (e.g., Waldron et al., 1999; Douglas et al., 2021).
The observation of CH4 oversaturation in the oxic surface mixed water layer of aquatic systems challenges the view of methanogenesis exclusively occurring under anoxic conditions (Donis et al., 2017; Bižić et al., 2020a; Hartmann et al., 2020). Recent research has shown that CH4 production is possible under oxic conditions, both in terrestrial environments, e.g. plants and fungi (Keppler et al., 2006; Lenhart et al., 2012), as well as in aquatic environments, e.g. phytoplankton and cyanobacteria (Lenhart et al., 2015; Bižić et al., 2020b). Recently, Ernst et al. (2022) proposed a reaction mechanism for CH4 formation potentially occurring on a cellular level across all living organisms through the interaction of reactive oxygen species with free iron and methylated sulfur and nitrogen compounds in living cells. Furthermore, the authors found that increased levels of oxidative stress enhanced CH4 production in all of the investigated organisms, providing a possible explanation not only for CH4 emissions under oxic conditions but also for the large variability of emission rates observed for many organisms in aquatic and terrestrial environments. However, the related δ13C and δ2H isotopic patterns of CH4 produced in the oxic environment remain yet to be clarified.
Aerobic CH4 oxidation (MOx) as a counteracting mechanism to CH4 production is usually observed mainly at the oxic-anoxic interface in aquatic systems where high CH4 concentrations and dissolved oxygen are present. MOx is generally characterized by an increase in δ13C-CH4 and δ2H-CH4 values, since methanotrophic bacteria prefer to oxidize the light carbon and hydrogen stable isotopes, leading to a relative enrichment of 13C and 2H in the CH4 pool (Barker and Fritz, 1981; Whiticar, 1999). In the anoxic sediment, anaerobic oxidation of CH4 (AOM) through electron acceptors other than oxygen can be observed leading to an enrichment in 13C and 2H of CH4, however other isotopic effects discussed later might obscure this typical isotope enrichment. AOM coupled to sulfate reduction is a common and widely described process in the ocean, oxidizing >90% of CH4 produced in oceanic sediment (Knittel and Boetius, 2009). Due to usually low sulfate concentrations in freshwater systems, sulfate-dependent AOM only occurs in some specific lake environments.
Tsunogai et al. (2020) suggested that the relation between the original carbon and hydrogen isotopic composition of CH4 can be inferred from the relation between δ13C-CH4 and δ2H-CH4 values of residual CH4, irrespective of the isotopic fractionation caused by CH4 oxidation. Therefore, they introduced a novel stable isotope indicator ∆(2,13), which corrects for the kinetic isotope fractionation effect associated to oxidation by using the ratio of hydrogen to carbon stable isotopes during microbial oxidation and thus can help in characterizing sources of CH4 in a system. Through the application of dual stable isotopes and flux measurements, model predictions can be complemented and provide a better comprehension of processes occurring in the system and disentangling production and consumption mechanisms.
So far, application of dual isotope and concentration measurements has only found limited use in aquatic environments and an overview of dual stable isotope characterization of limnic CH4 processes is missing. Therefore, we analyzed the δ13C-CH4 and δ2H-CH4 values as well as CH4 concentrations in Lake Willersinnweiher - a seasonally stratified, eutrophic lake in southwestern Germany - and furthermore applied the novel ∆(2,13) indicator. Detailed profiles of the water column and pore water of the sediments were collected during the stratified and non-stratified lake periods in order to gain a better understanding of the CH4 sources and sinks and the diffusive and ebullitive processes involved in CH4 cycling of the lake. We aim to isotopically characterize different sources of CH4 to the lake and thus to disentangle the contribution of these sources to the complex CH4 cycling at Lake Willersinnweiher both spatially and temporally.
Material and methods
Site description and geochemical characterization
Lake Willersinnweiher is situated in the plain of the Upper Rhine Graben (Germany), northwest of Ludwigshafen (49.499950 °N; 8.397138 °E) and covers an area of 17 ha (Figure 2). It is one of four gravel pit lakes and has neither a surface inflow nor outflow, thereby making inflowing groundwater its main source of water and solutes (Wollschläger et al., 2007). Lake Willersinnweiher is composed of a shallower northeastern basin with a maximum water depth of 14 m and a deeper southwestern basin with a maximum water depth of 20 m. The average water depth in the lake is 8 m (Sandler, 2000).
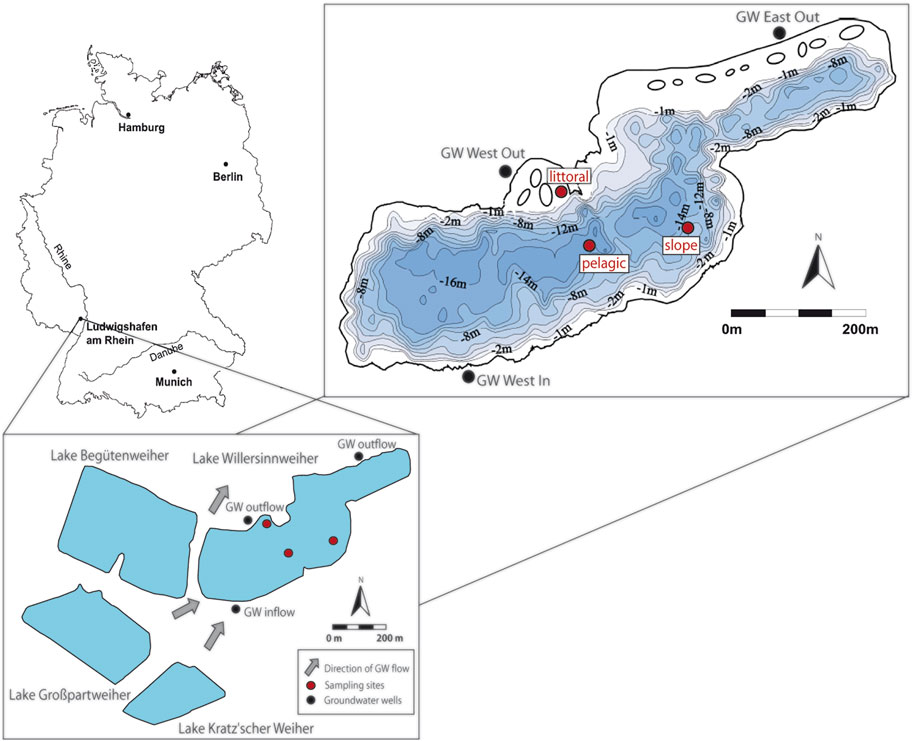
FIGURE 2. Location of Lake Willersinnweiher and the three adjacent lakes in Germany modified after Kleint et al. (2021). Lake sampling sites (pelagic, slope and littoral) and groundwater sampling sites (GW West In, GW West Out, GW East Out) are shown as red and black dots, respectively. Groundwater flow direction was modified after Wollschläger et al. (2007).
Geochemically, Lake Willersinnweiher can be characterized as an eutrophic hardwater lake with an average water residence time of 3.7 years and experiences seasonal water stratification during summer (stratification period) (Wollschläger et al., 2007). From November/December to March/April, the water column of Lake Willersinnweiher is fully mixed (mixing period). This leads to fully oxic conditions during the mixing period in the lake with O2 reaching the upper few millimeters of the sediment (Schröder, 2004). With warming temperatures in spring, thermal stratification starts to build up and reaches a maximum in late summer, leading to the formation of a warm, fully oxygenated epilimnion and the anoxic hypolimnion separated by a thermocline at the upper boundary of the metalimnion and a chemocline/oxycline at the lower boundary of the metalimnion. Chlorophyll-a measurements show a peak in algal activity at the thermocline and at the transition from metalimnion to hypolimnion. As thermal stratification weakens in October due to decreasing temperatures, characteristics of mixing are observed throughout the entire water column reflecting the transition to whole lake circulation prevailing during winter.
Sulfate (SO42−) concentrations in the lake are unusually high for a freshwater environment (∼2 mmol L−1) and are the result of sulfate-rich groundwater inflow at the southwestern shore of the lake (Schröder, 2004). Hence, due to high SO42− concentrations in Lake Willersinnweiher, the potential for sulfate-dependent AOM is high compared to most other limnic environments. The main process of organic matter turnover in Lake Willersinnweiher is degradation via sulfate reduction (Kleint et al., 2021). This results in the production of sulfide (S2−) in the lake sediment, causing diffusive release into the bottom water and consequently leading to euxinic conditions in the hypolimnion during the stratification period. Pyrite oxidation in the Quaternary river sediments of the Rhine cause high SO42− concentrations in the catchment upstream of the lake (Schröder, 2004; Isenbeck-Schröter et al., 2016). A detailed review of geochemical processes in the lake water and sediment of Lake Willersinnweiher can be found in a recent study by Kleint et al. (2021).
Eutrophic conditions in the lake are caused by agriculture and land use in the vicinity of the lake, resulting in the release of nutrients into the groundwater, which ultimately reach the lake (Laukenmann, 2002). Groundwater flow in the area of Lake Willersinnweiher is generally directed from southwest to northeast with low flow velocities due to a nearly horizontal groundwater table. Inflowing groundwater at the southwestern shore has already passed at least one of the adjacent smaller lakes upstream of Lake Willersinnweiher (Wollschläger et al., 2007).
Field methods
Sampling of the lake water and sediment was performed at three different sites in the southwestern basin. The pelagic sampling site in the center of the lake has a water depth of 16 m, the slope site is 9 m and the littoral site about 1.5 m deep. Groundwater sampling was carried out at three wells surrounding the lake (see Figure 2). The lake water, groundwater and sediment were sampled in July 2020 during the stratification period and in March 2021 during the mixing period.
Changes in lake water parameters (temperature, pH, dissolved oxygen and chlorophyll-a) with depth were recorded using an Exo1 multiparameter probe (Xylem Analytics, Norway). The probe was calibrated for each of the monitored parameters prior to each sampling session.
Samples of the water column were taken at different depth levels using a submersible pump (COMET-Pumpen Systemtechnik GmbH & Co. KG, Germany) and prepared for measurement of CH4 concentration and isotopic composition using a headspace technique (Kampbell et al., 1989). Samples collected for dissolved ion and dissolved inorganic carbon (DIC) analysis were filtered through a 0.2 μm filter into falcon tubes (Corning, United States). Samples for cation analysis were acidified with 150 μl of 6 M nitric acid.
For pore-water analysis, two sediment cores (core length ∼21–29 cm) were taken at each site with a manually operated gravity corer. Pore-water was extracted from one of the cores immediately after sampling in defined intervals using rhizons (Rhizosphere Research Products, Netherlands) with a pore-size of 0.15 μm. For measurement of sedimentary CH4 concentration and its isotopic composition, the sediment of the second core was subsampled in the same intervals as the pore-water by transferring 3 ml of lake sediment with a cut-off plastic syringe into glass vials. The sediment was treated with 5 ml of sodium hydroxide (1 M NaOH) and subsequently the vials were crimped with a lid containing a butyl rubber septum and shaken vigorously to impede any further microbial activity. In the laboratory, the sediment samples were shaken for ∼10 min to equilibrate porewater CH4 and the gas headspace. Afterwards, the headspace was extracted for further analysis. The sediment samples were dried in an oven at 105°C for several days to determine their water content and porosity.
Groundwater wells were sampled using a submersible pump (MP1, Grundfos GMBH, Germany) after pumping until groundwater parameters showed steady values (∼30 min). Groundwater samples were prepared in the same way as lake water samples.
Ebullitive CH4 was sampled for its isotopic composition in November 2020 and September 2021 by deploying an in-house built bubble trap consisting of inverted plastic funnels. A gravity corer was dropped into the sediment in order to release gas bubbles trapped in the sediment. Released gas bubbles were collected with the bubble traps directly under the lake surface and the accumulated gas was analyzed for CH4 concentration as well as its carbon and hydrogen isotopic composition. The collection of ebullitive CH4 via this method was only performed in November 2020 and September 2021 at the littoral and slope sites, whereas at the pelagic site, it was not possible to collect gas bubbles at the surface after dropping the weight into the sediment.
Gas samples for measurement of δ13C-CH4 and δ2H-CH4 values released to the atmosphere via diffusion were taken between March and September 2021 using a floating chamber. The floating chamber consisted of a plastic body with a volume of 8.6 L, two tubes equipped with three-way valves in order to take samples and a floatable ring made from polyethylene that kept the chamber afloat and the edges of the chamber in the water at a depth between 2 and 3 cm. Samples were collected when the chamber was placed in the water and after 15 and 30 min. All gas samples collected from the water column, sediment, groundwater and via the floating chamber were analyzed for their CH4 concentrations, δ13C-CH4 and δ2H-CH4 values in the laboratory.
Laboratory methods
All laboratory analyses were performed at the Institute of Earth Sciences at Heidelberg University, Germany.
δ13C and δ2H stable isotope analysis of CH4 and DIC
The natural stable isotopic composition of CH4 is expressed in the conventional δ-notation in permil (‰) versus Vienna Peedee Blemnite (V-PDB) for carbon and Vienna Standard Mean Ocean Water (V-SMOW) for hydrogen. The δ-notation is defined as the relative difference of isotope ratios of a sample compared to the standard substance, hence the 13C/12C ratio of a sample compared to V-PDB (δ13C value) and the 2H/1H ratio of a sample compared to V-SMOW (δ2H value) (Eq. 1):
A DeltaPLUS XL IRMS (Thermo Fisher Scientific, Bremen, Germany) was used to analyze δ13C-CH4, δ2H-CH4 and δ13C-DIC values in the headspace samples. For 13C-DIC analysis, the water sample was first acidified with a few drops of hydrochloric acid (≥25% HCl) in order to transform all DIC to CO2 prior to sampling the headspace gas. The IRMS was coupled to a HP 6890N GC (Agilent Technologies, United States) via a GC Combustion III Interface (GC-C; ThermoFisher Scientific, United States) with an oxidation reactor at 960°C and a thermo conversion reactor (GC-TC) at 1450°C for carbon and hydrogen stable isotopic analysis, respectively. The GC was equipped with a CP-PoraPLOT Q capillary column (length: 27.5 m; inner diameter 0.25 mm; film thickness: 8 μm; Varian, United States). For δ13C-CH4 and δ2H-CH4 measurements, the GC-C/TC-IRMS was linked to a cryogenic pre-concentration unit, whereas for δ13C-DIC an A200S autosampler was applied (CTC Analytics, Switzerland).
For CH4 measurements with the pre-concentration unit, the headspace gas samples were transferred to an evacuated 40 ml sample loop. Methane was trapped on HayeSep D at −125°C, separated from other remaining compounds by GC, and then introduced into the IRMS system via the interface described above. All δ13C and δ2H values were corrected using two CH4 reference standards (Isometric instruments, Canada) with δ13C-CH4 values of −42.32‰ and −66.35‰ and δ2H-CH4 values of −190.6‰ and −149.9‰, respectively, that were calibrated against International Atomic Energy Agency (IAEA) and National Institute of Standards and Technology (NIST) reference substances. Sample values were normalized according to Paul et al. (2007).
Analysis of CH4 via gas chromatography
Pore-water, water column and groundwater samples were analyzed using a gas chromatograph (14B GC-FID, Shimadzu, Japan) equipped with a flame ionization detector for CH4 concentrations ranging from 100 ppbv to 50 ppmv and a gas chromatograph (GC-2010 BID, Shimadzu, Japan) equipped with a barrier discharge ionization detector for CH4 concentrations higher than 50 ppmv. The 14B GC-FID was fitted with a stainless-steel column (length: 2 m; inner diameter: 1/8 inches) filled with a molecular sieve 5A (60–80 mesh; pore-size: 5 Å diameter). The GC oven temperature was isothermal at 125°C. Two reference standards (2.192 ppmv and 9.655 ppmv) were also analyzed for quality control. The GC-2010 BID was fitted with a stainless-steel ShinCarbon ST packed column (80/100 mesh; length: 2 m; diameter: 0.53 mm). The GC oven temperature was programmed to hold at 30°C for 6.5 min, rise to 75°C at a rate of 10°C/min and then to 180°C at a rate of 30°C/min. The GC-2010 BID was calibrated using several standards ranging from 50 ppmv to 97% CH4. A 1000 ppmv CH4 standard was analyzed for quality control.
DIC, SO42− and S2− analysis
For determination of the dissolved inorganic carbon (DIC) concentration, a TOC-V CPH (Shimadzu, Japan) was used. The instrument was calibrated by repeated analysis of an in-house standard solution prior to each measurement. The SO42− concentration was analyzed by a Dionex™ ICS-1100 Ion Chromatography System (ThermoFisher Scientific, United States). The measurement precision for each element was <3% and derived from long-term repeated analysis of reference material SPS-NUTR-WW1. The S2− concentration within the water column, pore-water and groundwater samples was determined photometrically (DREL 2800, Hach, United States) immediately after returning to the laboratory from sampling in the field. The samples were prepared using the Spectroquant® Sulfide Reagent Test (Merck, Germany) and measured at a wavelength of 665 nm. The concentrations of cations in the water samples were determined using an Inductively Coupled Plasma Optical Emission Spectrometer (ICP-OES 720, Agilent Technologies, United States). For quality control, the reference material SPS-SW2 was analyzed along with the samples with a measurement precision for each element <2%.
The ionic balance was determined by the sum of major cation and anion concentrations in order to qualitatively control the results of total dissolved ion composition analysis. Results of ionic balance calculations showing a deviation less than 5% displayed good quality control of the measurements.
Application of stable isotopes of methane
Methane formation and consumption processes are associated with a kinetic isotope effect leading to a change in its isotopic composition. The magnitude of this kinetic isotope effect is expressed in the isotope fractionation factor α. Processes of CH4 formation can be distinguished by determining the apparent carbon isotopic fractionation factor αCH4-CO2 between CH4 and CO2 according to Eq. 2:
In order to calculate the δ13C-CO2 values from δ13C-DIC values the methodology as described by Gonzalez Moguel et al. (2021) was used. Shortly, in a first step the DIC concentrations as well as the pH values in the respective depth were used to calculate the distribution of CO32−, HCO3− and dissolved CO2. Using this species distribution and the isotope fractionation factors between gaseous CO2 and dissolved CO2, HCO3− and CO32− as reported by Zhang et al. (1995), dissolved δ13C-CO2 values were calculated. An αCH4-CO2 between 1.050 and 1.060 indicates acetoclastic methanogenesis whilst between 1.060 and 1.090 shows predominance of hydrogenotrophic methanogenesis (Thottathil and Prairie, 2021).
Isotopic fractionation factors 13α for carbon and 2α for hydrogen during CH4 oxidation were determined using the Rayleigh model for closed systems according to Bastviken et al. (2002) (Eq. 3):
where f is the fraction of CH4 being oxidized,
The Keeling plot method was used to determine the δ13C-CH4 and δ2H-CH4 values of CH4 released from the water column to the atmosphere (Keeling, 1958). For a detailed description of the Keeling plot method, we refer the reader to Pataki et al. (2003) and Keppler et al. (2016). Briefly, a graphical approach based on the Keeling plot method was used to estimate stable isotope source values of CH4. Here, the inverse CH4 mixing ratios of the three individual measurements during sampling of diffusion (x-axis) are plotted against their respective δ13C-CH4 or δ2H-CH4 values measured via GC-C-IRMS (y-axis). Then a linear regression was employed and the intercept of this linear regression with the y-axis reflects the δ13C-CH4 or δ2H-CH4 source value. Reported errors are based on the standard error of the linear regression.
A novel stable isotope indicator
The
To calculate the
Results
Dissolved CH4 concentrations and isotopic composition of CH4 in the sediment
Sediment porewater profiles of CH4 concentrations at the pelagic site showed an increase with greater sediment depth during the stratification period with maximum concentrations ranging around 1 mmol L−1, which were accompanied by δ13C-CH4 values of −80 to −75‰ and δ2H-CH4 values of −314 to −328‰ (Figure 3D). A brief shift towards more positive isotopic values of CH4 within a few centimeters in the upper sediment occurred, where δ13C-CH4 values showed a maximum increase of 11‰, however δ2H-CH4 data is missing for this depth interval due to problems in collecting samples. The upper sediment core at the pelagic site was characterized by decreasing CH4 concentrations towards the sediment surface. Sulfate concentrations decreased with depth in the upper few centimeters (3–5 cm) of the sediment while simultaneously a rise in S2− concentrations was recorded in the same depth interval (Figure 3E). Also, DIC concentrations in the pore-water increased with depth and showed lowest δ13C-DIC values of around −14‰ in the upper part and an enrichment in 13C with increasing sediment depth to −9‰ (Figure 3F).
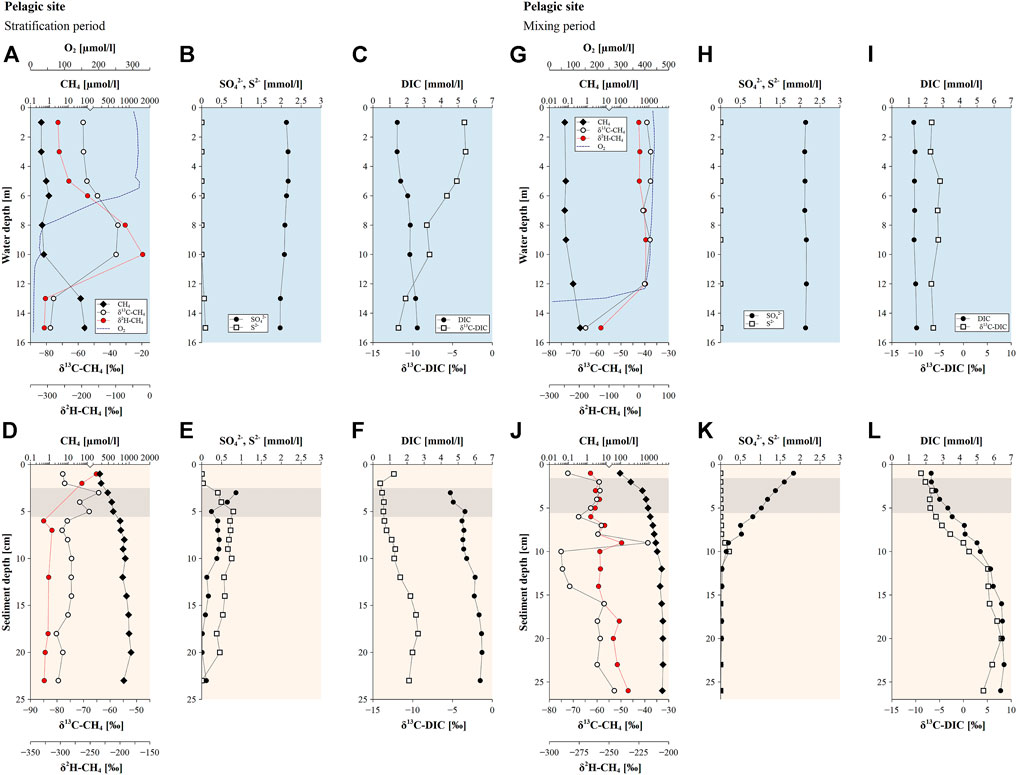
FIGURE 3. Methane (CH4) concentrations, δ13C-CH4 and δ2H-CH4 values along with sulfate, sulfide, dissolved organic carbon (DIC) concentrations and δ13C-DIC values observed at the pelagic site during (A–F) the stratification period (July 2020) and (G–L) the mixing period (March 2021). Blue graph background colors indicate profiles in the water column, orange background colors show profiles of sediment cores and grey highlighted sediment depths indicate the occurrence of the SMTZ.
At the slope site an increase in CH4 concentrations with depth was observed accompanied by δ13C-CH4 values varying from −77 to −69‰ and δ2H-CH4 values ranging from −338 to −271‰ (Figure 4D). Similarly to the pelagic site, a decrease in SO42− and an increase in S2− concentration with depth was recorded (Figure 4E), whereas DIC concentrations increased towards the bottom of the sediment core coinciding with an overall decrease in δ13C-DIC values (Figure 4F).
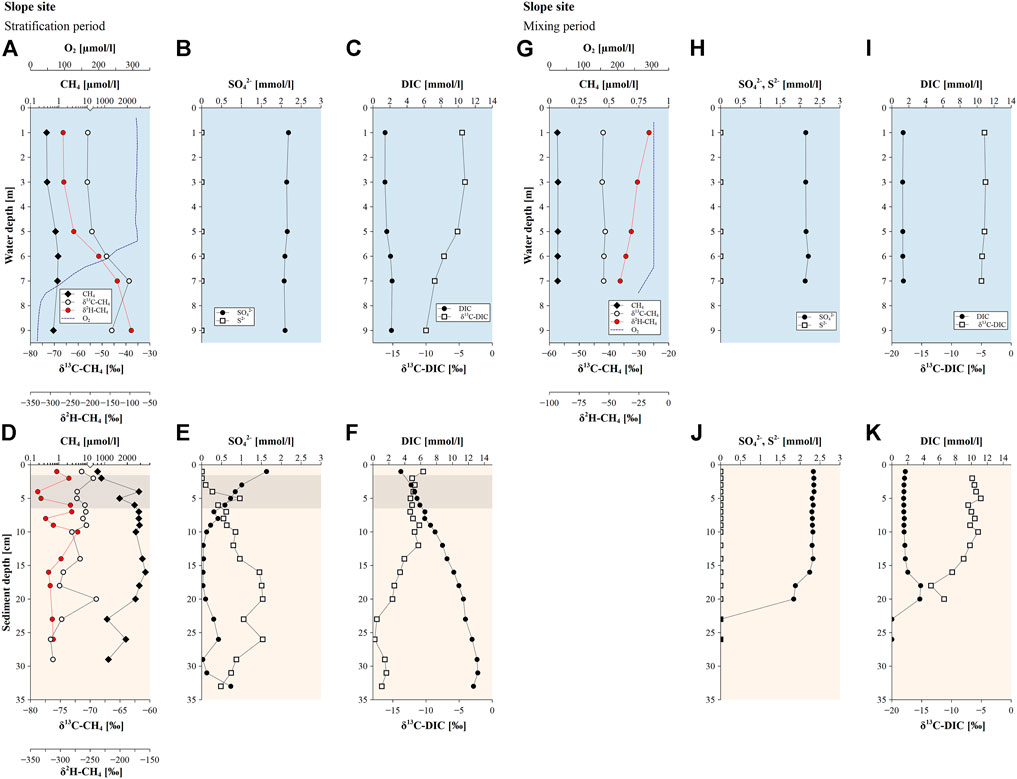
FIGURE 4. Methane (CH4) concentrations, δ13C-CH4 and δ2H-CH4 values along with sulfate, sulfide, dissolved organic carbon (DIC) concentrations and δ13C-DIC values observed at the slope site during (A–F) the stratification period (July 2020) and (G–K) the mixing period (March 2021). No sediment CH4 data available for March 2021 at the slope site. Blue background colors indicate profiles in the water column, orange background colors show profiles of sediment cores and grey highlighted sediment depths indicate the occurrence of the SMTZ.
At the littoral site, profiles of the sediment porewater showed CH4 concentrations of up to 1.3 mmol L−1 (Figure 5D). 13C-enriched CH4 values compared to the pelagic and slope sites around −58 to −52‰ were found during the stratification period along with δ2H-CH4 values ranging from −333 to −256‰ (Figure 5D). Similar to the deeper sites, a shift towards a more positive isotopic signal particularly in δ2H-CH4 values was recorded in the upper sediment layers, whereas δ13C-CH4 values showed a gradual increase from −57‰ at 3 cm to −51‰ at 9 cm sediment depth.
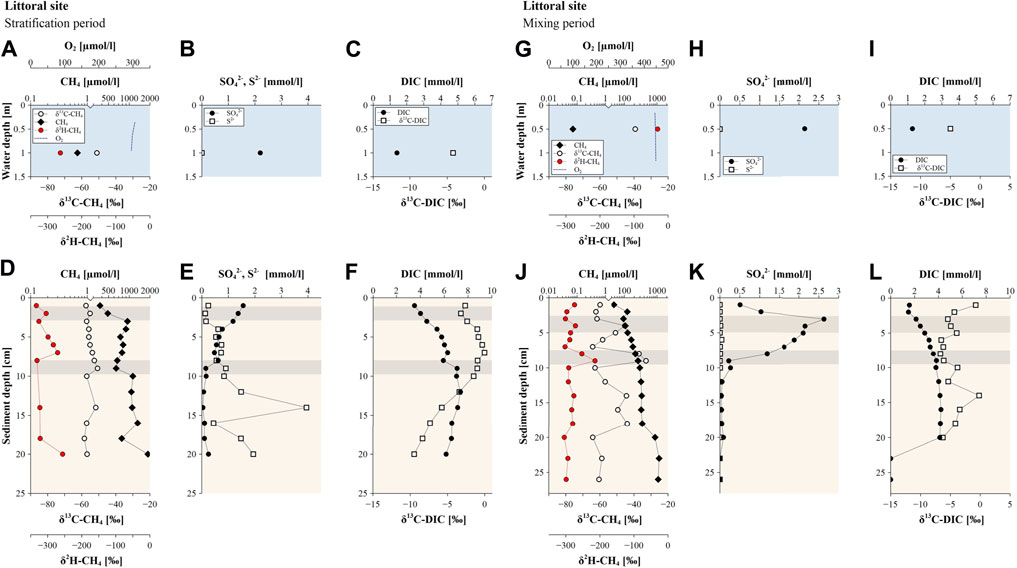
FIGURE 5. Methane (CH4) concentrations, δ13C-CH4 and δ2H-CH4 values along with sulfate, sulfide, dissolved organic carbon (DIC) concentrations and δ13C-DIC values observed at the slope site during (A–F) the stratification period (July 2020) and (G–L) the mixing period (March 2021). Blue background colors indicate profiles in the water column, orange background colors show profiles of sediment cores and grey highlighted sediment depths indicate the occurrence of the SMTZ.
During the mixing period, porewater CH4 at the pelagic site showed highly variable δ13C-CH4 values throughout the entire sediment core varying from −39 to −75‰, whereas δ2H-CH4 showed a rather uniform distribution ranging between −234 and −266‰, respectively (Figures 3J–L). Methane concentrations were at a much higher level than during the stratification period at the pelagic site, increasing to 3.1 mmol L−1 in the lower part of the core. In contrast, the littoral sediment recorded significantly lower CH4 concentrations around 0.3 mmol L−1 during the mixing period (Figure 5J). For the slope site no CH4 data is available for the mixing period. Similar to the stratification period, SO42− concentrations decreased in the upper part of the pelagic sediment and stayed at very low levels in the lower part of the core (Figure 3K). No increase in S2− concentration was observed in the sediment of the three investigated sites during the mixing period. DIC concentrations showed a similar trend as during the stratification period and increased with sediment depth accompanied by increasing δ13C-DIC values (Figure 3L).
Dissolved CH4 concentrations and isotopic composition of CH4 in the water column
In the lake water column, CH4 concentrations showed a decrease from the hypolimnion towards the surface water accompanied by an enrichment in 13C-CH4 and 2H-CH4 during the stratification period (Figure 3A). Methane in the bottom water of Lake Willersinnweiher showed δ13C-CH4 and δ2H-CH4 values of −77‰ and −309‰, respectively, and increased to δ13C-CH4 values of −56‰ and −269‰, respectively, in the surface water layer during the stratification period. Methane concentrations in the surface water ranged from 0.37 to 0.67 µmol L−1 with highest concentrations found at the littoral site (Figure 5A). DIC concentrations rose from 1.4 mmol L−1 in the surface water to 2.6 mmol L−1 in the bottom water, while δ13C-DIC showed a decrease from −4 to −12‰ throughout the water column from top to bottom (Figure 3C).
A pronounced zone of low CH4 concentrations was observed at the pelagic site during the stratification period in the lower metalimnion, showing a maximum decline in CH4 of 0.52 µmol L−1 from 6 to 8 m water depth (Figure 3A). This minimum in CH4 concentrations coincided with maximum δ13C-CH4 and δ2H-CH4 values in the water column.
A local peak in CH4 concentrations in the oxic upper water column was found at a water depth of 6 m during the stratification period at the pelagic site, showing a maximum increase of 0.56 µmol L−1 compared to surface water CH4 concentrations during the stratification period (Figure 3A). This was accompanied by an increase in the δ13C-CH4 and δ2H-CH4 values of about 9‰ and 83‰, respectively, compared to surface water values, resulting in δ13C-CH4 values of −48‰ and δ2H-CH4 values of −83‰ at 6 m water depth.
The slope site showed similar surface water CH4 concentrations (0.37–0.39 µmol L−1) and isotopic compositions (δ13C-CH4 of -56‰ and δ2H-CH4 of -268‰) as the pelagic site and recorded an increase in the concentration with depth up to 0.94 µmol L−1, which was accompanied by an increase in δ13C-CH4 and δ2H-CH4 values of 17‰ and 136‰, respectively (Figure 4A). The littoral site displayed CH4 enriched in 13C-CH4 and 2H-CH4 (δ13C-CH4 and δ2H-CH4 values of −51 and −263‰) in the surface water (Figure 5A) compared to the pelagic and slope sites.
During the mixing period, distinctively lower CH4 concentrations around 0.07 µmol L−1 at the pelagic site throughout almost the entire water column were characterized by δ13C-CH4 values of −39‰ and +11‰ for δ2H-CH4 values (Figure 3G). The bottom water showing CH4 concentrations of 0.4 µmol L−1 recorded more negative isotopic values of −65‰ and −127‰. The concentration of DIC in the water column stayed at the same level throughout the water column and the carbon isotopic pattern recorded a minor shift towards slightly more positive values from −7 to −5‰ at intermediate water depths (Figure 3I). While CH4 concentrations and δ13C-CH4 values at the slope and littoral site were similar to those found at the pelagic site, δ2H-CH4 values were more negative varying around −30‰ (Figures 4G, 5G).
Isotopic fractionation factors in the sediment and water column
The apparent carbon isotopic fractionation factor αCH4-CO2 between CH4 and CO2 (Eq. 2) in the sediment was determined for sediment depths where CH4 concentrations were highest, therefore most likely indicating the occurrence of methanogenesis in these depths. The αCH4-CO2 was found to be lowest at the littoral site (1.047) and rose with increasing lake depth (1.058 at the slope site and 1.066 at the pelagic site) during the stratification period (Supplementary Table S1). During the mixing period, the pelagic and littoral site exhibited a αCH4-CO2 of 1.070 and 1.047, respectively.
Isotopic fractionation factors associated with CH4 oxidation (Eq. 3) were determined in the upper sediment layers of the three sampled locations where a zone of decreasing CH4 was observed. During the stratification period, isotopic fractionation in these zones was found to range from 1.005 to 1.031 for 13α and from 1.042 to 1.124 for 2α (Table 1). During the mixing period, 13α varied from 1.051 to 1.087 and 2α from 1.015 to 1.169. In the water column, an isotopic fractionation factor was determined for the zone between the upper and the lower metalimnion at the pelagic site in which a decrease in CH4 concentrations was apparent for the stratification period, where an 13α of 1.009 and an 2α of 1.057 was calculated. An isotopic fractionation factor for the mixing period could not be calculated since no changes in the isotopic composition of CH4 was observed in the water column and similar CH4 concentrations throughout the entire water column were recorded (Figure 3G).
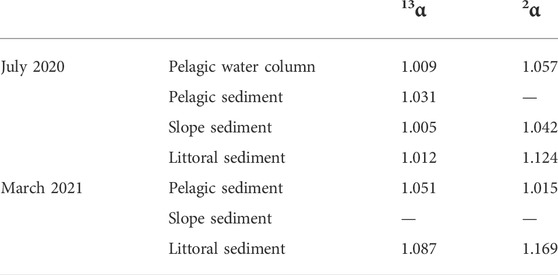
TABLE 1. Isotopic fractionation factors for carbon (13α) and hydrogen (2α) calculated for the for zones of decreasing CH4 concentrations in the water column (bottom water to lower metalimnion) and in the upper sediment during the stratification period (July 2020) and for the upper sediment during the mixing period (March 2021).
Concentration and isotopic composition of diffusive and ebullitive CH4
Methane released via diffusion from the lake surface water into the atmosphere during the stratification period (May to September) recorded similar δ13C-CH4 values at each of the three sampled sites, varying between −52 and −58‰ (Table 2). δ2H-CH4 values were more variable ranging from −195 to −310‰ at the different sites, whereby the pelagic and slope site showed in July the most positive values during the stratification period (−208 ± 3 and −205 ± 14‰, respectively) and the littoral site in August (−195 ± 22‰). During the mixing period, CH4 emitted from the lake was considerably enriched in the heavier isotopes at all investigated sites compared to the stratification period. δ13C-CH4 values ranged from −36 to −42‰ and δ2H-CH4 values from 1 to 26‰.

TABLE 2.
The concentrations and isotopic composition of ebullitive CH4 were analyzed for the littoral and slope site of the lake in November 2020 and September 2021 (Supplementary Table S2). Ebullitive CH4 was not collected for analysis in July 2020 and March 2021 due to logistical reasons. Nevertheless, the stratified lake period is represented by the data from September 2021, whereas the mixing period is represented by the data from November 2020.
In September during thermal stratification, CH4 concentrations in gas bubbles in the littoral area reached 80.8 ± 4.5%. In November, CH4 concentrations of ebullitive CH4 were in the range of 62.1 ± 7.4% and 66.8 ± 3.2% at the littoral and slope site, respectively. δ13C-CH4 values at the littoral site were more negative in September compared to November with −58 ± 1‰ and −50 ± 12‰, respectively. In contrast, δ2H-CH4 values in September and November at the littoral site were very similar with −323 ± 3‰ and −326 ± 4‰. At the slope site, δ13C-CH4 values were more negative compared to the littoral site with −64 ± 11‰, but δ2H-CH4 values were in a similar range compared to the littoral site with values of −318 ± 1‰.
Groundwater methane
Groundwater in the area of Lake Willersinnweiher showed highly variable CH4 concentrations, δ13C-CH4 and δ2H-CH4 values and was strongly enriched in 2H-CH4 and 13C-CH4 (Table 3). During the stratification period, inflowing groundwater yielded a CH4 concentration of 1.01 µmol L−1 with an isotopic composition enriched in 13C and 2H (δ13C-CH4 value of −24‰ and δ2H-CH4 value of +106‰). Outflowing groundwater of the southwestern basin showed slightly lower CH4 concentrations of 0.95 µmol L−1 along with δ13C-CH4 and δ2H-CH4 values of +18‰ and +582‰, respectively. CH4 concentrations of groundwater flowing out of the northeastern basin were 0.77 µmol L−1 during the stratification period accompanied by δ13C-CH4 values of −50‰ and δ2H-CH4 values of −187‰. Between the two outflowing groundwater wells in the western and eastern part of Lake Willersinnweiher a disparity in CH4 concentrations and stable isotope values is worth noticing. This observation is probably caused as outflowing groundwater in the western part consists of a mixture of outflowing groundwater from the lake and groundwater with properties similar to inflowing groundwater, that also passed subjacent lakes, due to its location and groundwater flow direction (see Figure 2). During the mixing period, groundwater at the three wells showed CH4 concentrations of 1.97 and 2.71 µmol L−1 for inflowing and outflowing groundwater of the southwestern basin, respectively, and even 30.89 µmol L−1 for groundwater flowing out of the northeastern basin. Groundwater outflow of the southwestern basin showed a δ13C-CH4 value of −37‰ and a δ2H-CH4 value of +24‰. The isotopic composition of groundwater at the other two groundwater wells was in the range of values reported during the stratification period (Table 3).
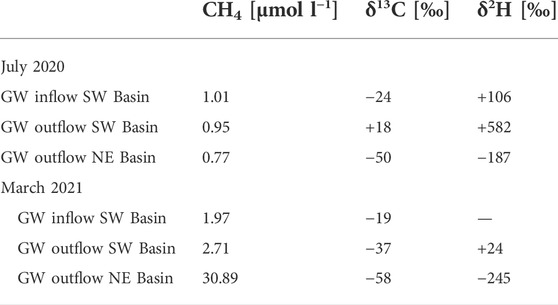
TABLE 3. Methane concentration and its stable isotopic composition of the three different groundwater wells during the stratification period (July 2020) and the mixing period (March 2021).
Discussion
Methanogenesis in the sediment
Methanogenesis is the last step in the microbial degradation of organic matter and is performed by methanogenic archaea in the anaerobic environment (Conrad, 2005). However, in presence of other electron donors (such as SO42−), organic matter degradation via sulfate reduction is favored over methanogenesis. This is most likely the case for Lake Willersinnweiher, where sulfate-reducing bacteria are believed to thermodynamically outcompete methanogenic archaea for available carbon compounds and H2 in the upper sediment due to high SO42− concentrations in the lake water (Kleint et al., 2021). In aquatic systems with high SO42− concentrations, anaerobic methanogenesis is therefore restricted to greater sediment depths, where SO42− concentrations are low (Holmer and Storkholm, 2001; Reeburgh, 2007; Schubert et al., 2011). Methanogenesis in Lake Willersinnweiher is suggested to occur in zones of the deeper sediment where CH4 in the sediment accumulated (Figures 3D, 4D, 5D). Hydrogenotrophic methanogenesis as the dominant process of CH4 production in the sediment during the stratification period was indicated by relatively negative δ13C-CH4 values (ranging from around −80 to −75‰) observed in the lower sediment of the pelagic and slope site, where maximum CH4 concentrations were recorded (Figure 3D). Whereas the associated δ2H-CH4 values of around −321 to −246‰ were rather negative for this type of methanogenesis (Whiticar, 1999). However, δ2H-CH4 values in this environment are likely mostly linked to the δ2H value of the source water (Waldron et al., 1999; Douglas et al., 2021) and seem to be less indicative of the methanogenic pathway compared to δ13C-CH4 values as δ2H-CH4 values are less studied and influenced by more factors. Above that a mixing of the hydrogenotrophic and acetoclastic pathway is possible (Sugimoto and Wada, 1995; Walter et al., 2008). During methanogenesis, hydrogen originating from ambient water is incorporated into the CH4 molecule, thus δ2H-CH4 values are mostly dependent on the hydrogen isotopic composition of the water (Sugimoto and Wada, 1995). Plotting sedimentary δ13C-CH4 values against δ2H-CH4 values and classifying them after Whiticar (2020) indicated an overlap of the two different methanogenic pathways at the pelagic and slope site (Figure 6). For the littoral site, a dominance of acetoclastic methanogenesis seems to be implied due to less negative δ13C-CH4 values compared to the pelagic and slope site.
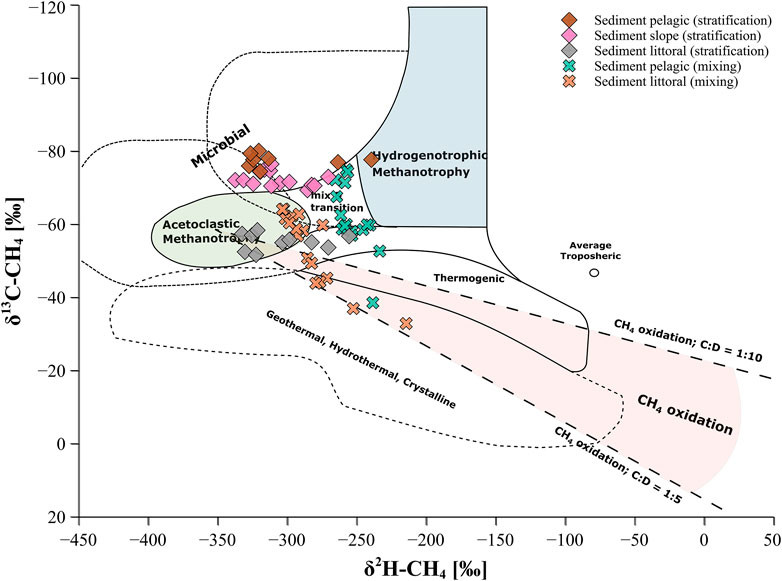
FIGURE 6. 2-dimensional isotope plot of sedimentary CH4 data from the stratification and mixing period implemented into the classification of methanogenic pathways modified after (Whiticar, (2020). The dotted lines represent the range of ratios of 13C and 2H (C:D) enrichment during anaerobic CH4 oxidation as reported by Martens et al. (1999), Holler et al. (2009) and Rasigraf et al. (2012).
Furthermore, based on the carbon isotopic shift between CH4 and CO2, we determined apparent carbon isotopic fractionation factors associated to anaerobic methanogenesis, which is an indicator to distinguish between the hydrogenotrophic and acetoclastic pathway. At the pelagic site, an αCH4-CO2 of 1.066 supports the dominance of hydrogenotrophic methanogenesis during the stratification period (Supplementary Table S1). The αCH4-CO2 decreased towards the littoral site, indicating an increasing contribution of the acetoclastic pathway. Overlapping characteristics were hence especially visible at the slope site, where δ13C-CH4 values around −75‰ point to hydrogenotrophic methanogenesis whereas the calculated αCH4-CO2 of 1.058 is representative for acetoclastic CH4 formation. The proposed increasing dominance of acetoclastic methanogenesis from the pelagic towards the littoral area is based on the isotopic values of sedimentary CH4 observed at Lake Willersinnweiher and might be coupled to changes in sediment temperatures and differences in organic matter availability with varying lake depth (Thottathil and Prairie, 2021). However, further investigations are needed to gain more insight into the occurrence and distribution of the different methanogenic pathways in the sediment of Lake Willersinnweiher.
During the mixing period, the observed αCH4-CO2 of 1.070 for the pelagic site (Supplementary Table S1) was higher than during the stratification period suggesting a higher dominance of hydrogenotrophic CH4 formation. This might possibly reflect the limited availability of fresh organic material in the lake. However, higher sedimentary CH4 concentrations found at the pelagic site during the mixing period compared to the stratification period (Figures 3D,J) seemed to contradict the expected decrease of CH4 concentrations due to lower primary production in winter and therefore reduced organic matter availability. This indicated that processes other than microbial activity in the sediment must have contributed to the high observed sedimentary CH4 concentrations and isotopic pattern in the sediment at the pelagic site and highlights the complex interaction of the occurring processes influencing the sedimentary CH4 cycle. However, we suggest that infiltrating groundwater as a potential CH4 source might also play an important role at Lake Willersinnweiher since it yielded very high CH4 concentrations characterized by extremely positive isotopic δ13C-CH4 and δ2H-CH4 values both for inflowing and outflowing groundwater (Table 3). Mixing of groundwater and lake sediment CH4 might potentially skew the calculated fractionation factors for the mixing period, hence these have to be considered with caution and can only be used as a preliminary indicator for the methanogenic pathway.
Anaerobic methane oxidation in the sediment
The shift to more positive δ13C-CH4 and δ2H-CH4 values partially observed in the upper sediment layers during the stratification period was most likely caused by the anaerobic oxidation of CH4 coupled to SO42−reduction. The occurrence of sulfate-dependent AOM in Lake Willersinnweiher was described in a recent study by Kleint et al. (2021) and we therefore refer the reader to this study for more detailed discussion on this process at Lake Willersinnweiher. Here we want to give a brief summary on sulfate-dependent AOM at Lake Willersinnweiher and discuss the associated isotopic effects of this process. In the sediment, upward rising CH4 and downward diffusing SO42− meet in the so-called sulfate-methane transition zone (SMTZ), where anaerobic methanotrophic archaea in syntrophic association with sulfate-reducing bacteria are able to reverse methanogenesis and oxidize CH4 using SO42− (Boetius et al., 2000). In most freshwater environments, low SO42− concentrations usually preclude sulfate-dependent AOM, whereas in the oceans, sulfate-dependent CH4 oxidation is a widely described and common process due to high SO42− concentrations in the ocean water (Knittel and Boetius, 2009). Since Lake Willersinnweiher shows high SO42− concentrations, compared to other lakes, as a result of sulfate-rich groundwater input, sulfate-dependent AOM is possible.
Indicative of AOM might furthermore be the observation of DIC enriched in 12C in the upper lake sediment compared to the lower sediment during the stratification period since 12C-CH4 is preferably oxidized by the microorganisms, thereby providing HCO3− relatively enriched in 12C to the sedimentary DIC pool (Schubert et al., 2011). With increasing sediment depth, microbial hydrogenotrophic CH4 production caused a depletion in isotopically light DIC, thus leading to DIC enriched in 13C-DIC in the sediment pore-water.
Zones in the lake sediment where sulfate-dependent AOM was presumed to take place were not always characterized by a clear shift towards less negative δ13C-CH4 and δ2H-CH4 values (Figures 3J, 5D), as one might typically expect for CH4 oxidation or restrained to a few centimeters within the upper sediment at the investigated sites of Lake Willersinnweiher. Plotting sedimentary δ13C-CH4 values against δ2H-CH4 values shows a progression of the littoral sediment data of the mixing period within a predicted CH4 oxidation line between a ratio of C:D of 1:5 and 1:10 (e.g., Wang et al., 2016; Whiticar, 2020), referring to an enrichment of CH4 in hydrogen isotopes 5–10 times more than in carbon isotopes during AOM (Figure 6). This observation is in good accordance with previously reported C:D ratios during AOM ranging between 6.4 and 10 (Martens et al., 1999; Holler et al., 2009; Feisthauer et al., 2011; Rasigraf et al., 2012). However, this trend was less pronounced at the other two sites, limiting the use of carbon and hydrogen stable isotopes of CH4 to characterize AOM in the lake sediment. Nevertheless, carbon and hydrogen isotopic fractionation factors 13α and 2α were also determined to identify the occurrence of potential AOM in the respective zones of the sediment. The calculated carbon isotopic fractionation factors 13α values for the observed enrichment in 13C-CH4 and 2H-CH4 and associated with AOM in the sediment (1.002–1.019, Table 1) were on the lower end of 13α values determined for AOM in marine and brackish environments and in laboratory studies (1.009–1.039, Whiticar and Faber, 1986; Holler et al., 2009; Knittel and Boetius, 2009). The hydrogen isotopic fractionation factors 2α (1.033–1.057) determined for Lake Willersinnweiher were substantially lower than 2α modelled for aquatic environments and found in in vitro experiments (1.109–1.315, Whiticar and Faber, 1986; Alperin et al., 1988; Holler et al., 2009). It is important to mention that natural systems might differ significantly from modelled conditions and in vitro experiments, thus leading to discrepancies in isotopic fraction factors. Furthermore, it must be noted that isotopic fractionation factors of AOM determined from in situ isotopic signatures, as in the case of Lake Willersinnweiher, have to be considered with caution since AOM and methanogenesis might overlap in the sediment and carbon isotope equilibrium effects caused by forward and backward AOM influence the isotopic composition of the CH4 pool in opposite ways (Holler et al., 2009; Yoshinaga et al., 2014; Chang et al., 2019). A study by Wegener et al. (2021) furthermore showed, that the accumulation of 13C and 2H-depleted CH4 in zones of AOM was driven under low sulfate conditions (similar to SO42− concentrations found in lake Willersinnweiher) because of intracellular reactions associated with the enzymatic AOM mechanism. They found that the reactions in the enzymatic AOM pathway became more reversible and thus closer to equilibrium at low SO42− conditions driving the formation of 13C and 2H depleted CH4. The lack of a clear enrichment in the heavier isotopes of CH4 in sediment zones where AOM is presumed to occur in Lake Willersinnweiher might therefore be resulting from the above-mentioned isotopic effects.
Due to AOM in the lake sediment, the diffusive release of CH4 at the sediment-water interface is reduced in Lake Willersinnweiher (Kleint et al., 2021). Our concentration depth profiles of CH4 in the sediment show a decreasing gradient towards the sediment water interface, caused by CH4 diffusion into the bottom water and most likely AOM. Significant decreases in sedimentary CH4 concentrations observed particularly at the slope and littoral site were suggested to be related to AOM as they coincided with an enrichment in 13C-CH4 and 2H-CH4 (Figures 4D, 5D). Methane diffusion from the sediment into the bottom water significantly elevated hypolimnic CH4 concentrations (up to 74 µmol L−1 at the pelagic site). The negative isotope values of hypolimnic CH4 of −77‰ and −309‰ for δ13C-CH4 and δ2H-CH4 values, respectively, during the stratification period (Figure 3A) fell well within the range of δ13C-CH4 and δ2H-CH4 values found in sediment depths where anaerobic methanogenesis is presumed to occur, hence suggesting anaerobic CH4 production in the sediment as its origin.
During the mixing period, shifts towards more positive δ13C-CH4 and δ2H-CH4 values in the upper sediment at the pelagic and littoral site suggested the occurence of AOM in the sediment (Figures 3J, 5J). Furthermore, decreasing CH4 concentrations and increasing SO42− concentrations towards the sediment surface might be indicative of the occurrence of sulfate-dependent AOM (Figures 3J,K, 5J,K). The apparent lack of S2− accumulation in the sediment might be caused by re-oxidation of sulfide to sulfate via manganese oxides in the sediment pore-water as described by Kleint et al. (2021). The determined fractionation factors associated to AOM during the mixing period (1.051–1.087 for 13α and 1.015–1.169 for 2α, Table 1) do not reflect typical fractionation factors for AOM, however as discussed above, these fractionation factors have to be considered cautiously as different isotopic effects can influence the isotopic composition of CH4 in the SMTZ. Furthermore, a mixture with other CH4 sources, e.g., groundwater CH4 input, might affect the carbon and hydrogen isotopic composition of sedimentary CH4 since mixing of groundwater and sedimentary CH4 might lead to an increase in sedimentary δ13C-CH4 and δ2H-CH4 values due to the very enriched 13C and 2H isotopic composition of groundwater CH4. AOM most likely reduced the release of CH4 at the sediment-water interface, however diffusion from the uppermost sediment layers into the bottom water layer caused elevated CH4 concentrations in the bottom water.
Aerobic methane oxidation in the water column
During the stratification period, a strong increase in the isotopic composition of CH4 (δ13C-CH4 values of −35‰ and δ2H-CH4 values of −72‰, Figure 3A) along with a decrease in CH4 concentrations at the bottom of the oxycline in the water column compared to surface water CH4 suggested MOx of sedimentary released CH4. The enrichment in the δ13C-CH4 and δ2H-CH4 signal is caused by the preference of methanotrophic organisms to metabolize light atoms 12C and 1H of CH4, leading to an enrichment in 13C-CH4 and 2H-CH4 of the remaining CH4 (Silverman and Oyama, 1968; Barker and Fritz, 1981; Coleman et al., 1981). From the 2-dimensional isotope plot it can be inferred that MOx occurs in the metalimnion during the stratification period since the metalimnic stable isotope data are distributed along a gradient showing a stronger enrichment of 2H compared to 13C during MOx with a C:D ratio of 9.5 (Figure 7). This observation fits well within similar reported C:D ratios during MOx ranging between 5.9 and 14.9 (Coleman et al., 1988; Kinnaman et al., 2007; Powelson et al., 2007; Feisthauer et al., 2011; Wang et al., 2016). In comparison, epilimnic water samples plot did not show this progression, hence implying that δ13C-CH4 and δ2H-CH4 values there are most likely not or less affected by MOx.
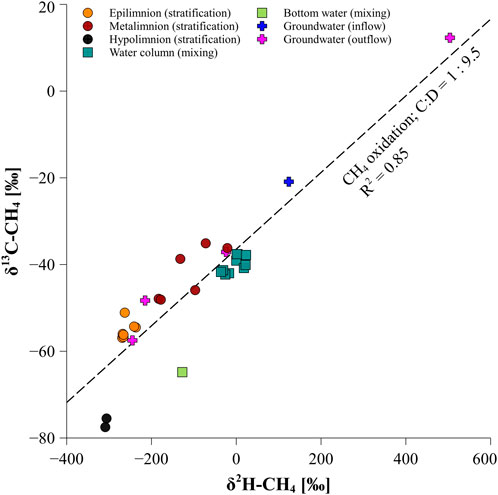
FIGURE 7. 2-dimensional isotope plot of water column and groundwater CH4 data from the stratification and mixing period. The dotted line represents the ratio of 13C to 2H (C:D) enrichment during CH4 oxidation in the water column and groundwater at lake Willersinnweiher.
The carbon isotopic fractionation factor 13α of 1.009 determined for the observed enrichment in Lake Willersinnweiher furthermore suggested the occurrence of MOx at this depth as it is in the range of 13α determined for MOx in experimental studies (1.003–1.039; Templeton et al., 2006). The estimated 2α of 1.057 on the other hand is substantially lower than values determined for MOx in closed cultures (1.103–1.325; Alperin et al., 1988). However, a higher 2α than 13α (factor of ∼9.5) at Lake Willersinnweiher confirms the larger isotopic fractionation generally observed for stable hydrogen isotopes compared to stable carbon isotopes during microbial oxidation. The isotopic partitioning between hydrogen isotopes is greater than between carbon isotopes because of a larger mass difference between 2H and 1H than between 13C and 12C (e.g., Wang et al., 2016). This leads to a stronger enrichment in hydrogen isotopes than in carbon isotopes during oxidation.
The zone of MOx at the oxic-anoxic interface in Lake Willersinnweiher during the stratification period acts as a barrier and effectively limits the release of sedimentary produced CH4 into the surface mixed water layer to minimum concentrations (Kleint et al., 2021). The occurrence of MOx at the bottom of the oxycline indicated by maximum δ13C-CH4 and δ2H-CH4 values suggests that CH4 oxidizing bacteria are most active at low O2 levels and their presence is restricted as O2 concentrations rise in the upper water body (Schubert et al., 2010; Thottathil et al., 2019). At the littoral site, the isotopic composition of the water column agrees well with sedimentary δ13C-CH4 and δ2H-CH4 values, suggesting that MOx did not occur here. The apparent absence of MOx at the littoral sampling site might be caused by fully oxygenated conditions in the shallow water column. Another controlling factor of MOx might be the availability of light in the photic zone of the water column. Murase and Sugimoto (2005) showed that MOx was inhibited under light conditions, whereas a linkage between a light driven supply of O2 through photosynthesis and MOx was found in a study by Oswald et al. (2015), implying that MOx is dependent on light availability.
MOx leads to the formation of CO2, which is converted to HCO3− according to the bicarbonate buffer system due to a prevailing pH of 8.5–7.3 in the water column of the lake. This contributed to an increase in DIC concentrations in the metalimnion, which was accompanied by decreasing δ13C-DIC values (Figure 3C) caused by CH4 oxidizing organisms and their preference for 12CH4 consumption leading to the production of 12C enriched DIC. However, increased DIC concentrations might also originate from the degradation of organic matter. Organic material is generally characterized by relatively negative δ13C values, consequently causing the trend to more negative δ13C-DIC values with depth as degradation proceeds and inorganic carbon is released from the sediment into the water column.
During the mixing period, constant CH4 concentrations as well as similar δ13C-CH4 and δ2H-CH4 values in the water column, except between a depth of 12 and 15 m, where stable isotope values of CH4 became more enriched, did not suggest MOx (Figure 3G). Since almost the entire water column was fully oxygenated, this might have prevented efficient MOx as methane oxidizing bacteria were found to be sensitive to high O2 concentrations (Thottathil et al., 2019). However, MOx could still have occurred in the lake water column in the absence of O2, δ13C-CH4 and δ2H-CH4 gradients due to mixing and thus might contribute to CH4 enriched in 13C and 2H during the mixing period compared to the stratification period. Plotting δ13C-CH4 values against δ2H-CH4 values further suggests the occurrence of MOx during the mixing period as the data points show a distribution along the gradient of isotope enrichment (Figure 7). Beyond this the enrichment in 13C and 2H of lake water CH4 during the mixing period (δ13C-CH4 values of −39‰ and δ2H-CH4 values of +11‰) compared to lake water values from the stratification period, the enrichement might also have originated from the input of groundwater CH4 since inflowing and outflowing groundwater recorded extremely positive δ13C-CH4 and δ2H-CH4 values (Table 3). Groundwater CH4 might therefore have constituted an important source to lake water CH4 during the mixing period and contributed to the shift towards more positive δ13C-CH4 and δ2H-CH4 values of lake water during the mixing period, when CH4 concentrations in the water column were much lower compared to the stratification period and the relative importance of groundwater on lake water CH4 thus increases.
Methane supersaturation in the surface water layer
During the stratification period, fully oxygenated conditions in the epilimnion of Lake Willersinnweiher likely prevented efficient MOx in the upper water column, thus leading to the accumulation of CH4 in the surface mixed water layer. The surface water of Lake Willersinnweiher with CH4 concentrations ranging from 0.3 to 0.6 µmol L−1 during the stratification period was oversaturated compared to the atmosphere, since water in equilibrium with the atmosphere shows CH4 concentrations of ∼3 nmol L−1 (Wiesenburg and Guinasso, 1979).
During the stratification period, the δ13C-CH4 and δ2H-CH4 values of CH4 in the pelagic surface water were around −56‰ and −270‰, respectively. These values were significantly different from hypolimnic δ13C-CH4 values of −77‰ and δ2H-CH4 values −323‰. This difference raises the question for the source of CH4 supersaturation in the surface water layer (Figure 8A). The isotopic signatures of surface water CH4 is likely altered by the mixing of different CH4 pools, e.g., through littoral and vertical input, ebullition, groundwater interactions and internal oxic CH4 production (e.g., Tang et al., 2016; Hartmann et al., 2020).
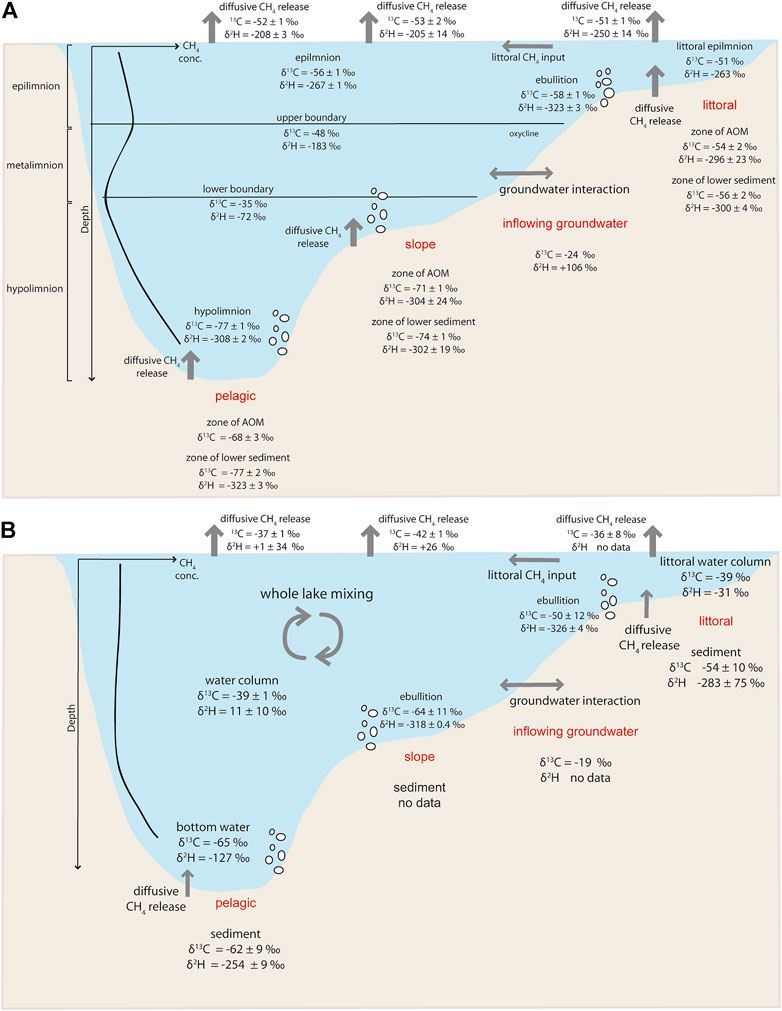
FIGURE 8. Methane cycling in Lake Willersinnweiher showing CH4 sources and sinks with δ13C-CH4 and δ2H-CH4 values (where available) as well as transport mechanisms (grey) of different water layers (black) observed at the three investigated sites during (A) the stratification period (July 2020, for diffusive release at water-air interface data from July 2021 is shown and for ebullition data from September 2021) and (B) the mixing period (March 2021 and for ebullition data from November 2020 is shown).
Vertical input of CH4 from the sediment is strongly reduced in the lower metalimnion through MOx (Figure 3A). However, sedimentary CH4 might still migrate into the upper water layer and become enriched in 13C and 2H through MOx, resulting in less negative surface water δ13C-CH4 and δ2H-CH4 values compared to hypolimnic values. Transport of CH4 from the littoral sediment to pelagic lake areas comprises an important source especially in small lakes with organic rich littoral zones, where CH4 deriving from the sediment can be distributed in the surface water layer of the entire lake through turbulences, e.g., induced by wind activity (Tang et al., 2014). As thermal stratification builds up in the Lake Willersinnweiher, around 0.3 µmol L−1 higher CH4 concentrations can be observed in the littoral area of Lake Willersinnweiher than in the epilimnion of the pelagic and slope sites during the stratification period. Sediments in the littoral zone are usually not isolated through thermal stratification from the overlying water column compared to pelagic sites, thus often resulting in higher CH4 concentrations in littoral waters (Loken et al., 2019). Higher sediment temperatures and disruption by waves fuel CH4 production and release in the littoral zone during summer (Hofmann et al., 2010). A similar pattern of spatiotemporal CH4 distribution was observed in other lakes and highlights the importance of the littoral area considering CH4 emission to the atmosphere (Wang et al., 2006; Tsunogai et al., 2020). During maximum stratification, the isotopic CH4 pattern of littoral surface water showed a δ13C-CH4 value of −51‰ which is different from the values observed for the pelagic and slope sites (δ13C-CH4 value of −56‰), whereas δ2H-CH4 values were rather similar around −265‰, suggesting that sources of CH4 in the surface water might differ between the littoral and deeper sites (Figure 8A). However, it is possible that mixing of littoral and vertical CH4 input caused the observed more negative isotopic values of surface water CH4 in the pelagic zone of the lake compared to the littoral area. A further potential source of CH4 in the surface water layer of aquatic system is oxic CH4 production despite the long-standing paradigm defining methanogenesis as a process occurring only under anoxic conditions (Grossart et al., 2011; Tang et al., 2014, 2016; Donis et al., 2017; Günthel et al., 2019; Hartmann et al., 2020; Thottathil et al., 2022). In Lake Willersinnweiher, CH4 in the oxic upper water column peaked at a water depth of 6 m during the stratification period, which was accompanied by an increase in the δ13C-CH4 and δ2H-CH4 values to −48‰ and −183‰, respectively, compared to surface water δ13C-CH4 values of −57‰ and δ2H-CH4 values of −269‰ (Figure 3A). Although the isotopic signature of CH4 produced in the oxic water column is hitherto unknown, Hartmann et al. (2020) proposed that oxic CH4 production might cause an enrichment in 13C in the CH4 pool in the lake surface layer. More recently, δ13C mass balance estimates by Thottathil et al. (2022) supported this finding by stating that CH4 produced from oxic sources is relatively enriched in 13C compared to anoxic sources with δ13C-CH4 values ranging between −64 and −38‰. More positive δ13C-CH4 values in the surface mixed layer compared to sedimentary δ13C-CH4 values have also been found in other oxic lakes such as Lake Stechlin (−50‰; Tang et al., 2014), Lake Cromwell (−40‰; Bogard et al., 2014) and Lake Lugano (−55‰; Blees et al., 2014). These isotopic values are in line with the observed increase in δ13C-CH4 values to around −48‰ in the upper metalimnion during the stratification period in Lake Willersinnweiher. The slight enrichment in 13C might be due to different precursors and pathways of in situ oxic CH4 production compared to anoxic methanogenesis and/or an interplay of co-occurring MOx. (Donis et al., 2017). Beyond δ13C-CH4 values, better constraints on δ2H-CH4 values of oxic CH4 production would assist in the characterization of in situ produced CH4 in the oxic environment as well as the possible influence of MOx in the observation of enriched stable isotope values of CH4.
The exact pathways of CH4 formation in the oxic water column are not fully understood to date. However recently, Ernst et al. (2022) suggested a reaction mechanism for CH4 formation associated to the interaction of reaction oxygen species with free iron and methylated sulfur and nitrogen compounds occurring on a cellular level across all living organisms and that increasing the level of oxidative stress enhanced the production of CH4. Moreover, it has been discovered that cyanobacteria produce CH4 (Bižić et al., 2020b) and that light as well as temperature yield a significant control over oxic CH4 production rates by influencing phytoplankton communities (Klintzsch et al., 2020). Furthermore, the role of submerged macrophytes for oxic methane production has so far been ignored, but might also contribute significantly (Hilt et al., 2022). Hence, oxic CH4 production is most likely closely coupled to primary production, potentially contributing to an increase in CH4 concentrations in the surface water layer during the stratification period as it was also observed at Lake Willersinnweiher. A peak in chlorophyll-α co-occurring with maximum CH4 concentrations in the upper metalimnion at Lake Willersinnweiher might support a linkage between CH4 production and algal activity as proposed already in other studies (Grossart et al., 2011; Bogard et al., 2014; Tang et al., 2016; Hartmann et al., 2020; Thottathil et al., 2022). Nevertheless, Donis et al. (2017) suggested that the accumulation of CH4 at the thermocline can be caused by physical processes in the water column and the highest production rates of CH4 can be found in the surface water layer.
Beyond the 2-dimensional stable isotope approach, we applied a novel stable isotopic indicator
Infiltrating groundwater might further contribute to CH4 supersaturation in the surface water layer of Lake Willersinnweiher and most likely influenced lake water CH4 during the entire year due to the lake’s direct accessibility to the upper aquifer and high groundwater CH4 concentrations around 1 µmol L−1 (Wollschläger et al., 2007). Possibly, groundwater CH4 yielding less negative δ13C-CH4 and δ2H-CH4 values could also have contributed to the observed enrichment in 13C and 2H at the upper thermocline. So far the origin of the groundwater CH4 remains unclear and different possibilities have been proposed to explain the source of CH4 in the inflowing groundwater and its unusually positive isotopic signature including CH4 production in a contamination site through oxidative biodegradation of hydrocarbons such as volatile chlorinated organic compounds, in organic-rich sediments from old branches of the Rhine river or in aquifers located upstream of Lake Willersinnweiher (Wollschläger et al., 2007; Kleint et al., 2021). Only limited data about groundwater CH4 concentrations and δ13C-CH4 values are available to date and groundwater that enriched in 13C-CH4 and 2H-CH4 seems to be only a rare occurrence (Schloemer et al., 2016). The highly enriched isotopic values of infiltrating groundwater might point to intense AOM since the groundwater is suboxic. The oxidation might therefore be coupled to the reduction of other electron acceptors such as nitrate, nitrite, manganese and iron (Kleint et al., 2021). Methane oxidation causing the highly enriched isotopic composition of groundwater CH4 might also be supported by the 2-dimensional isotope plot, revealing a distribution of inflowing and outflowing groundwater along the gradient of CH4 oxidation at Lake Willersinnweiher (Figure 7).
Mixing of lake and groundwater might lead to a change towards more positive isotope values in the lake water column, however the impact of groundwater CH4 on lake water CH4 is hard to estimate based on the current dataset and more detailed research concerning groundwater CH4 in the area of Lake Willersinnweiher is desirable. Due to lower primary productivity and lower CH4 concentrations in the lake during the mixing period we suggest that groundwater input might be an important source of CH4 to the lake especially during the mixing period. This might also contribute to the less negative isotopic composition of lake water CH4 observed during the mixing period (Figures 3G, 4G, 5G).
Supersaturation of CH4 in the surface water leads to CH4 release into the atmosphere during the stratification period. Additionally, the surface water was also oversaturated with CH4 during the mixing period (70 nmol L−1, Figures 3G, 4G, 5G), highlighting that Lake Willersinnweiher emits CH4 all year round. Methane can be emitted via plant-mediated processes, diffusion or ebullitive transport (Bastviken et al., 2004), however since plant-coverage at Lake Willersinnweiher is negligible, plant-mediated release of CH4 to the atmosphere is considered to be of minor importance. Methane emitted via diffusion from the surface water into the atmosphere showed a great range in the isotopic composition over 1 year with most positive values found at the three investigated sites during the mixing period (δ13C-CH4 of −42 to −37‰ and δ2H-CH4 of +1 to +26‰) and distinctively lower values observed during the stratification period (δ13C-CH4 of −59 to −51‰ and δ2H-CH4 of −310 to −195‰, Table 2). These isotopic values reflect the isotopic composition of lake surface water, which is enriched in the heavier isotopes during the mixing period. Especially during the mixing period, 13C-CH4 and 2H-CH4 were much more enriched compared to previously reported values (e.g., from Thottathil and Prairie, 2021 ranging from −58.8‰ to −43.5‰), highlighting the potential impact and importance of diffusive CH4 emissions from lakes with similar properties, such as high sulfate concentrations, for regional and/or global isotope source apportionment studies.
We found that in Lake Willersinnweiher, δ13C-CH4 and δ2H-CH4 values of CH4 released via ebullition displayed temporal and spatial disparities (Figure 8). At the deeper sampling site, CH4 transported via ebullition yielded more negative δ13C-CH4 values (−64 ± 11‰) when compared to δ13C-CH4 values at the littoral site (−50 ± 12‰ during the mixing period in November 2021). These differences in the isotopic composition were most likely induced by CH4 formation taking place mainly via the hydrogenotrophic and acetoclastic pathways at the slope and littoral sites, respectively, as discussed above. Ebullition plays a particularly important role in the littoral area of aquatic systems since the potential for CH4 transported via gas bubbles rises to reach the surface water with decreasing water depth. In particular, CH4 release to the atmosphere via ebullition likely represents an important pathway and has been found to vary significantly spatially and temporally (West et al., 2016; Thottathil and Prairie, 2021). Interestingly, δ2H-CH4 values only show small differences between the littoral and slope site at Lake Willersinnweiher (326 ± 4‰ for the littoral site and 318 ± 1‰ for the slope site, Figure 8B). In previous studies, changes in the δ2H-CH4 values of CH4 emitted via ebullition were attributed to changes in the δ2H values of water (e.g. due to evaporation in shallow lakes), which is a hydrogen source for methanogens (Wik et al., 2020). As Lake Willersinnweiher is a distinctively deeper lake (average depth of 8 m), this effect might only play a minor role and therefore have a smaller influence on δ2H-CH4 values. However, Douglas et al. (2021) showed that even though the δ2H-H2O values highly influenced δ2H-CH4 values, other processes such as methane oxidation and the type of the methanogenic pathway might also contribute to the isotopic composition of CH4. Furthermore, sulfate-dependent AOM in the sediments of lake Willersinnweiher might play an important role regarding the isotopic composition of CH4 released via ebullition and therefore might affect δ13C-CH4 values and to a lesser extent δ2H-CH4 values (Wegener et al., 2021). However, the influence of AOM is hard to estimate as AOM most probably takes place heterogeneously throughout the sediment. In November, when there is less organic matter input than in September (which is also indicated by the lower CH4 concentrations within the gas bubbles, Supplementary Table S2), pockets of CH4 within the sediment might be subject to heterogeneously occurring AOM, which might contribute to the relatively high variability of δ13C-CH4 values. During the uprise of gas bubbles in the water column, CH4 partly dissolves into the water (e.g. McGinnis et al., 2006). However, isotope fractionation associated with dissolution of CH4 was found to be small and most likely lower than the variability of the measured isotope values (Fuex, 1980; Bergamaschi, 1997). Additionally, the surface water layer showed isotopic values enriched in 13C and 2H compared to the gas bubbles (δ13C-CH4 of −56‰ and δ2H-CH4 of −270‰ for lake surface water and δ13C-CH4 of −64 ± 11‰ and δ2H-CH4 of −318 ± 1‰ for ebullitive CH4). We therefore conclude that ebullition is not the main source of surface water CH4 supersaturation in the pelagic area of the lake.
Conclusion
We investigated the sources and sinks of CH4 in a seasonally stratified freshwater lake using concentration measurements of CH4 and dual isotope analysis. Our main findings are presented in Figures 8A,B for the stratification period and the mixing period:
• δ13C-CH4 and δ2H-CH4 values of sedimentary CH4, anaerobic methanogenesis at the three investigated sites indicated a mix of the hydrogenotrophic and acetoclastic pathway at the pelagic and slope site and an increasing dominance of the acetoclastic methanogenesis towards the shallower littoral site. During the mixing period, the dominance of hydrogenotrophic methanogenesis potentially increased at the pelagic and slope sites. Throughout the whole year, sulfate-dependent AOM caused shifts towards heavier δ13C-CH4 and δ2H-CH4 values in the upper sediment and reduced the diffusive release of CH4 at the sediment-water interface.
• MOx efficiently consumed upward migrating CH4 in the water column during thermal stratification of the lake, indicated by a strong enrichment of 13C-CH4 and 2H-CH4 values.
• Supersaturation of CH4 in the surface water was present both during the stratification period and the mixing period, whereas supersaturation during the stratification period was significantly higher than supersaturation during the mixing period. Supersaturation of CH4 in the pelagic area of the lake possibly originated from the vertical and lateral input of sedimentary and littoral CH4, however oxic CH4 production might also supply CH4 to the surface water layer of the lake and might contribute to a local peak in concentrations at the upper thermocline during the stratification period. On the contrary, CH4 input from groundwater with unusually positive δ13C-CH4 and δ2H-CH4 values and CH4 diffusion from the littoral sediment were most likely the main source of lake water CH4 during the mixing period, leading to rather positive isotopic δ13C-CH4 and δ2H-CH4 values in the water column.
• Methane released from the water into the atmosphere via diffusion covered a wide range from −59 to −36‰ for δ13C-CH4 and −310 to +26‰ for δ2H-CH4 values during the stratification period and mixing of the water column during the mixing period, highlighting the potential importance of diffusive CH4 emissions from lakes with similar properties for regional and/or global isotope source apportionment studies.
Our study provides an overview of the isotopic characteristics of CH4 sources and sinks in a seasonally stratified lake and gives new insight into the sources contributing to CH4 supersaturation in the surface water layer of lacustrine systems. It furthermore outlines the potential for disentangling different sources and sinks both spatially and temporally based on their isotopic signature, especially considering the still highly debated contribution of oxic CH4 production to lake water CH4 concentrations. This clearly highlights the need for a multiparameter approach including the investigation of stable isotopes when researching the complex CH4 dynamics in an aquatic system since only then can a complete understanding of the involved processes be achieved.
Data availability statement
The experimental data used in this study are available from heiDATA, which is an institutional repository for research data of the Heidelberg University (https://doi.org/10.11588/data/KLFDVF). Requests to access the datasets should be directed to dGVyZXNhLmVpbnptYW5uQHVuaWJhcy5jaA==.
Author contributions
FK, TE, and MS conceived the study. TE and MS performed the field measurements and experiments under supervision of FK. TE and MS analyzed the data and visualized the results. MG validated stable isotope measurements of methane and analyzed stable carbon isotope measurements of dissolved inorganic carbon. TE, MS, FK, and JK discussed the results. The manuscript was written under the lead of TE and MS, with contribution of all authors.
Funding
This study was supported by DFG (German Research Foundation, project KE 884/16-2).
Acknowledgments
We thank Stefan Rheinberger, Silvia Rheinberger and Christian Scholz for analytical support. We are grateful to Bernd Knape for technical support in the laboratory and field. We thank Kai Ernst, Hannah Geisinger, Martin Lienenlüke, Iva Ralenekova and Timo Schreiter for help with field work and support in data analysis.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fenvs.2022.865862/full#supplementary-material
References
Alperin, M. J., Reeburgh, W. S., and Whiticar, M. J. (1988). Carbon and hydrogen isotope fractionation resulting from anaerobic methane oxidation. Glob. Biogeochem. Cycles 2, 279–288. doi:10.1029/GB002i003p00279
Barker, J. F., and Fritz, P. (1981). Carbon isotope fractionation during microbial methane oxidation. Nature 293, 289–291. doi:10.1038/293289a0
Bastviken, D., Cole, J., Pace, M., and Tranvik, L. (2004). Methane emissions from lakes: Dependence of lake characteristics, two regional assessments, and a global estimate. Glob. Biogeochem. Cycles 18, 1–12. doi:10.1029/2004GB002238
Bastviken, D., Ejlertsson, J., and Tranvik, L. (2002). Measurement of methane oxidation in lakes: A comparison of methods. Environ. Sci. Technol. 36, 3354–3361. doi:10.1021/es010311p
Belle, S., Verneaux, V., Millet, L., Parent, C., and Magny, M. (2015). A case study of the past CH4 cycle in lakes by the combined use of dual isotopes (carbon and hydrogen) and ancient DNA of methane-oxidizing bacteria: Rearing experiment and application to lake remoray (eastern France). Aquat. Ecol. 49, 279–291. doi:10.1007/s10452-015-9523-6
Bergamaschi, P. (1997). Seasonal variations of stable hydrogen and carbon isotope ratios in methane from a Chinese rice paddy. J. Geophys. Res. 102, 25383–25393. doi:10.1029/97jd01664
Bižić, M., Grossart, H., and Ionescu, D. (2020a). “Methane paradox,” in eLS (Chichester: John Wiley & Sons). doi:10.1002/9780470015902.a0028892
Bižić, M., Klintzsch, T., Ionescu, D., Hindiyeh, M. Y., Günthel, M., Muro-Pastor, A. M., et al. (2020b). Aquatic and terrestrial cyanobacteria produce methane. Sci. Adv. 6, eaax5343–10. doi:10.1126/sciadv.aax5343
Blees, J., Niemann, H., Wenk, C. B., Zopfi, J., Schubert, C. J., Kirf, M. K., et al. (2014). Micro-aerobic bacterial methane oxidation in the chemocline and anoxic water column of deep south-Alpine Lake Lugano (Switzerland). Limnol. Oceanogr. 59, 311–324. doi:10.4319/lo.2014.59.2.0311
Boetius, A., Ravenschlag, K., Schubert, C. J., Rickert, D., Widdel, F., Gieseke, A., et al. (2000). A marine microbial consortium apparently mediating anaerobic oxidation of methane. Nature 407, 623–626. doi:10.1038/35036572
Bogard, M. J., del Giorgio, P. A., Boutet, L., Chaves, M. C. G., Prairie, Y. T., Merante, A., et al. (2014). Oxic water column methanogenesis as a major component of aquatic CH4 fluxes. Nat. Commun. 5, 5350. doi:10.1038/ncomms6350
Cadieux, S. B., White, J. R., Sauer, P. E., Peng, Y., Goldman, A. E., and Pratt, L. M. (2016). Large fractionations of C and H isotopes related to methane oxidation in Arctic lakes. Geochimica Cosmochimica Acta 187, 141–155. doi:10.1016/j.gca.2016.05.004
Casper, P., Maberly, S. C., Hall, G. H., and Finlay, B. J. (2000). Fluxes of methane and carbon dioxide from a small productive lake to the atmosphere. Biogeochemistry 49, 1–19. doi:10.1023/a:1006269900174
Chang, P.-C., Yang, T. F., Wallmann, K., Matsumoto, R., Hu, C.-Y., Chen, H. W., et al. (2019). Carbon isotope exchange during anaerobic oxidation of methane (AOM) in sediments of the northeastern South China Sea. Geochimica Cosmochimica Acta 246, 138–155. doi:10.1016/j.gca.2018.11.003
Coleman, D. D., Risatti, J. B., and Schoell, M. (1981). Fractionation of carbon and hydrogen isotopes by methane-oxidizing bacteria. Geochimica Cosmochimica Acta 45, 1033–1037. doi:10.1016/0016-7037(81)90129-0
Conrad, R. (2005). Quantification of methanogenic pathways using stable carbon isotopic signatures: A review and a proposal. Org. Geochem. 36, 739–752. doi:10.1016/j.orggeochem.2004.09.006
Donis, D., Flury, S., Stöckli, A., Spangenberg, J. E., Vachon, D., and McGinnis, D. F. (2017). Full-scale evaluation of methane production under oxic conditions in a mesotrophic lake. Nat. Commun. 8, 1661. doi:10.1038/s41467-017-01648-4
Douglas, P. M. J., Stratigopoulos, E., Park, S., and Phan, D. (2021). Geographic variability in freshwater methane hydrogen isotope ratios and its implications for global isotopic source signatures. Biogeosciences 18, 3505–3527. doi:10.5194/bg-18-3505-2021
Downing, J. A., Prairie, Y. T., Cole, J. J., Duarte, C. M., Tranvik, L. J., Striegl, R. G., et al. (2006). The global abundance and size distribution of lakes, ponds and impoundments. Limnol. Oceanogr. 51, 2388–2397. doi:10.4319/lo.2006.51.5.2388
Duc, N. T., Crill, P., and Bastviken, D. (2010). Implications of temperature and sediment characteristics on methane formation and oxidation in lake sediments. Biogeochemistry 100, 185–196. doi:10.1007/s10533-010-9415-8
Ernst, L., Steinfeld, B., Barayeu, U., Klintzsch, T., Kurth, M., Grimm, D., et al. 2022, methane formation driven by reactive oxygen species across all living organisms Nature, 603, 482, doi:10.1038/s41586-022-04511-9
Feisthauer, S., Vogt, C., Modrzynski, J., Szlenkier, M., Krüger, M., Siegert, M., et al. (2011). Different types of methane monooxygenases produce similar carbon and hydrogen isotope fractionation patterns during methane oxidation. Geochimica Cosmochimica Acta 75, 1173–1184. doi:10.1016/j.gca.2010.12.006
Fernández, J. E., Peeters, F., and Hofmann, H. (2016). On the methane paradox: Transport from shallow water zones rather than in situ methanogenesis is the major source of CH4 in the open surface water of lakes. J. Geophys. Res. Biogeosci. 121, 2717–2726. doi:10.1002/2016JG003586
Fuex, A. N. (1980). Experimental evidence against an appreciable isotopic fractionation of methane during migration. Phys. Chem. Earth 12, 725–732. doi:10.1016/0079-1946(79)90153-8
Gonzalez Moguel, R., Bass, A. M., Garnett, M. H., Pilote, M., Keenan, B., Matveev, A., et al. (2021). Radiocarbon data reveal contrasting sources for carbon fractions in thermokarst lakes and rivers of eastern Canada (nunavik, quebec). J. Geophys. Res. Biogeosci. 126, 1–16. doi:10.1029/2020JG005938
Grossart, H.-P., Frindte, K., Dziallas, C., Eckert, W., and Tang, K. W. (2011). Microbial methane production in oxygenated water column of an oligotrophic lake. Proc. Natl. Acad. Sci. U. S. A. 108, 19657–19661. doi:10.1073/pnas.1110716108
Günthel, M., Donis, D., Kirillin, G., Ionescu, D., Bižić, M., McGinnis, D. F., et al. (2019). Contribution of oxic methane production to surface methane emission in lakes and its global importance. Nat. Commun. 10, 5497. doi:10.1038/s41467-019-13320-0
Hartmann, J. F., Günthel, M., Klintzsch, T., Kirillin, G., Grossart, H.-P., Keppler, F., et al. (2020). High spatiotemporal dynamics of methane production and emission in oxic surface water. Environ. Sci. Technol. 54, 1451–1463. doi:10.1021/acs.est.9b03182
Hilt, S., Grossart, H. P., McGinnis, D. F., and Keppler, F. (2022). Potential role of submerged macrophytes for oxic methane production in aquatic ecosystems. Limnol. Oceanogr. doi:10.1002/lno.12095
Hofmann, H., Federwisch, L., and Peeters, F. (2010). Wave-induced release of methane: Littoral zones as source of methane in lakes. Limnol. Oceanogr. 55, 1990–2000. doi:10.4319/lo.2010.55.5.1990
Holler, T., Wegener, G., Knittel, K., Boetius, A., Brunner, B., Kuypers, M. M. M., et al. (2009). Substantial 13C/12C and D/H fractionation during anaerobic oxidation of methane by marine consortia enriched in vitro. Environ. Microbiol. Rep. 1, 370–376. doi:10.1111/j.1758-2229.2009.00074.x
Holmer, M., and Storkholm, P. (2001). Sulphate reduction and sulphur cycling in Lake sediments: A review. Freshw. Biol. 46, 431–451. doi:10.1046/j.1365-2427.2001.00687.x
Isenbeck-Schröter, M., Aeschbach, W., Al Najem, S., Freundt, F., and Schmidt, G. (2016). Trace - TiefenReservoir-Analyse und Charakterisierung von der Erdoberfläche: Geochemisch-isotopisches Multimethoden-Konzenpt zur Charakterisierung tiefer Aquifere: Abschlussbericht TRACE Teil B. Tech. Inf. Hann. 95. doi:10.2314/GBV:871282461
Kampbell, D. H., Wilson, J. T., and Vandegrift, S. A. (1989). Dissolved oxygen and methane in water by a GC headspace equilibration technique. Int. J. Environ. Anal. Chem. 36, 249–257. doi:10.1080/03067318908026878
Keeling, C. D. (1958). The concentration and isotopic abundances of atmospheric carbon dioxide in rural areas. Geochimica Cosmochimica Acta 13, 322–334. doi:10.1016/0016-7037(58)90033-4
Keppler, F., Hamilton, J. T. G., Braß, M., and Röckmann, T. (2006). Methane emissions from terrestrial plants under aerobic conditions. Nature 439, 187–191. doi:10.1038/nature04420
Keppler, F., Schiller, A., Ehehalt, R., Greule, M., Hartmann, J., and Polag, D. (2016). Stable isotope and high precision concentration measurements confirm that all humans produce and exhale methane. J. Breath. Res. 10, 016003. doi:10.1088/1752-7155/10/1/016003
Kinnaman, F. S., Valentine, D. L., and Tyler, S. C. (2007). Carbon and hydrogen isotope fractionation associated with the aerobic microbial oxidation of methane, ethane, propane and butane. Geochimica Cosmochimica Acta 71, 271–283. doi:10.1016/j.gca.2006.09.007
Kleint, J. F., Wellach, Y., Schroll, M., Keppler, F., and Isenbeck-Schröter, M. (2021). The impact of seasonal sulfate–methane transition zones on methane cycling in a sulfate-enriched freshwater environment: Limnology and Oceanography, 1–19. doi:10.1002/lno.11754
Klintzsch, T., Langer, G., Wieland, A., Geisinger, H., Lenhart, K., Nehrke, G., et al. (2020). Effects of temperature and light on methane production of widespread marine phytoplankton. J. Geophys. Res. Biogeosci. 125. doi:10.1029/2020jg005793
Knittel, K., and Boetius, A. (2009). Anaerobic oxidation of methane: Progress with an unknown process. Annu. Rev. Microbiol. 63, 311–334. doi:10.1146/annurev.micro.61.080706.093130
Laukenmann, S. (2002). Transport und Austausch redoxsensitiver Elemente zwischen Freiwasser und Sediment in einem eutrophen Hartwassersee (Willersinnweiher/Ludwigshafen), unter besonderer Berücksichtigung des geochemischen Verhaltens von Uran. Heidelberg, BW: Ruprecht-Karls-Universität Heidelberg, 125. doi:10.11588/heidok.00002919
Lenhart, K., Bunge, M., Ratering, S., Neu, T. R., Schuttmann, I., Greule, M., et al. (2012). Evidence for methane production by saprotrophic fungi. Nat. Commun. 3, 1046–1048. doi:10.1038/ncomms2049
Lenhart, K., Klintzsch, T., Langer, G., Nehrke, G., Bunge, M., Schnell, S., et al. (2015). Evidence for methane production by marine algae ( Emiliana huxleyi ) and its implication for the methane paradox in oxic waters. Biogeosciences Discuss. 12, 20323–20360. doi:10.5194/bgd-12-20323-2015
Lessner, D. J. (2009). “Methanogenesis biochemistry,” in eLS. doi:10.1002/9780470015902.a0000573.pub2Chichester
Loken, L. C., Crawford, J. T., Schramm, P. J., Stadler, P., Desai, A. R., and Stanley, E. H. (2019). Large spatial and temporal variability of carbon dioxide and methane in a eutrophic lake. J. Geophys. Res. Biogeosci. 124, 2248–2266. doi:10.1029/2019JG005186
Martens, C. S., Albert, D. B., and Alperin, M. J. (1999). Stable isotope tracing of anaerobic methane oxidation in the gassy sediments of Eckernfoerde Bay, German Baltic Sea. Am. J. Sci. 299, 589–610. doi:10.2475/ajs.299.7-9.589
McGinnis, D. F., Greinert, J., Artemov, Y., Beaubien, S. E., and Wüest, A. (2006). Fate of rising methane bubbles in stratified waters: How much methane reaches the atmosphere? J. Geophys. Res. 111, C09007. doi:10.1029/2005JC003183
Murase, J., and Sugimoto, A. (2005). Inhibitory effect of light on methane oxidation in the pelagic water column of a mesotrophic lake (Lake Biwa, Japan). Limnol. Oceanogr. 50, 1339–1343. doi:10.4319/lo.2005.50.4.1339
Natchimuthu, S., Sundgren, I., Gålfalk, M., Klemedtsson, L., Crill, P., Danielsson, Å., et al. (2016). Spatio-temporal variability of lake CH4 fluxes and its influence on annual whole lake emission estimates. Limnol. Oceanogr. 61, S13–S26. doi:10.1002/lno.10222
Oswald, K., Milucka, J., Brand, A., Littmann, S., Wehrli, B., Kuypers, M. M. M., et al. (2015). Light-dependent aerobic methane oxidation reduces methane emissions from seasonally stratified lakes. PLoS ONE 10, e0132574. doi:10.1371/journal.pone.0132574
Pataki, D. E., Ehleringer, J. R., Flanagan, L. B., Yakir, D., Bowling, D. R., Still, C. J., et al. (2003). The application and interpretation of Keeling plots in terrestrial carbon cycle research. Glob. Biogeochem. Cycles 17, 1022. doi:10.1029/2001gb001850
Peeters, F., Fernandez, J. E., and Hofmann, H. (2019). Sediment fluxes rather than oxic methanogenesis explain diffusive CH4 emissions from lakes and reservoirs. Sci. Rep. 9, 243. doi:10.1038/s41598-018-36530-w
Peeters, F., and Hofmann, H. (2021). Oxic methanogenesis is only a minor source of lake-wide diffusive CH4 emissions from lakes. Nat. Commun. 12, 1206. doi:10.1038/s41467-021-21215-2
Powelson, D. K., Chanton, J. P., and Abichou, T. (2007). Methane oxidation in biofilters measured by mass-balance and stable isotope methods. Environ. Sci. Technol. 41, 620–625. doi:10.1021/es061656g
Rasigraf, O., Vogt, C., Richnow, H. H., Jetten, M. S., and Ettwig, K. F. (2012). Carbon and hydrogen isotope fractionation during nitrite-dependent anaerobic methane oxidation by Methylomirabilis oxyfera. Geochimica Cosmochimica Acta 89, 256–264. doi:10.1016/j.gca.2012.04.054
Reeburgh, W. S. (2007). Oceanic methane biogeochemistry. Chem. Rev. 107, 486–513. doi:10.1021/cr050362v
Rosentreter, J. A., Borges, A. V., Deemer, B. R., Holgerson, M. A., Liu, S., Song, C., et al. (2021). Half of global methane emissions come from highly variable aquatic ecosystem sources. Nat. Geosci. 14, 225–230. doi:10.1038/s41561-021-00715-2
Sandler, B. (2000). Die Wirkung von Sanierungs- und Restaurierungsmaßnahmen auf die Nährstoffströme und die biotische Dynamik eines anthropogenen Gewässers am Beispiel des Willersinnweihers/Ludwigshafen. Heidelberg, BW: Ruprecht-Karls-Universität Heidelberg, 190.
Schloemer, S., Elbracht, J., Blumenberg, M., and Illing, C. J. (2016)., 67. Germany, 118–132. doi:10.1016/J.APGEOCHEM.2016.02.005Distribution and origin of dissolved methane, ethane and propane in shallow groundwater of Lower Saxony, GermanyAppl. Geochem.
Schröder, H. (2004). Saisonale Redoxfronten im Kopplungsbereich zwischen Schwefel- Eisen- und Mangankreislauf im System Seewasser – sediment – Grundwasser des Willersinnweihers. Heidelberg, BW: Ruprecht-Karls-Universität Heidelberg, 122. doi:10.11588/heidok.00004894
Schubert, C. J., Lucas, F. S., Durisch-Kaiser, E., Stierli, R., Diem, T., Scheidegger, O., et al. (2010). Oxidation and emission of methane in a monomictic lake (Rotsee, Switzerland). Aquat. Sci. 72, 455–466. doi:10.1007/s00027-010-0148-5
Schubert, C. J., Vazquez, F., Lösekann-Behrens, T., Knittel, K., Tonolla, M., and Boetius, A. (2011). Evidence for anaerobic oxidation of methane in sediments of a freshwater system (Lago di Cadagno). FEMS Microbiol. Ecol. 76, 26–38. doi:10.1111/j.1574-6941.2010.01036.x
Silverman, M. P., and Oyama, V. I. (1968). Automatic apparatus for sampling and preparing gases for mass spectral analysis in studies of carbon isotope fractionation during methane metabolism. Anal. Chem. 40, 1833–1837. doi:10.1021/ac60268a001
Sugimoto, A., and Wada, E. (1995). Hydrogen isotopic composition of bacterial methane: CO2/H2 reduction and acetate fermentation. Geochimica Cosmochimica Acta 59, 1329–1337. doi:10.1016/0016-7037(95)00047-4
Tang, K. W., McGinnis, D. F., Frindte, K., Brüchert, V., and Grossart, H.-P. (2014). Paradox reconsidered: Methane oversaturation in well-oxygenated lake waters. Limnol. Oceanogr. 59, 275–284. doi:10.4319/lo.2014.59.1.0275
Tang, K. W., McGinnis, D. F., Ionescu, D., and Grossart, H.-P. (2016). Methane production in oxic lake waters potentially increases aquatic methane flux to air. Environ. Sci. Technol. Lett. 3, 227–233. doi:10.1021/acs.estlett.6b00150
Thottathil, S. D., and Prairie, Y. T. (2021). Coupling of stable carbon isotopic signature of methane and ebullitive fluxes in northern temperate lakes. Sci. Total Environ. 777, 146117. doi:10.1016/j.scitotenv.2021.146117
Thottathil, S. D., Reis, P. C. J., and Prairie, Y. T., 2022, Magnitude and drivers of oxic methane production in small temperate lakes, doi:10.1021/acs.est.2c01730
Thottathil, S. D., Reis, P. C. J., and Prairie, Y. T. (2019). Methane oxidation kinetics in northern freshwater lakes. Biogeochemistry 143, 105–116. doi:10.1007/s10533-019-00552-x
Tsunogai, U., Miyoshi, Y., Matsushita, T., Komatsu, D. D., Ito, M., Sukigara, C., et al. (2020). Dual stable isotope characterization of excess methane in oxic waters of a mesotrophic lake. Limnol. Oceanogr. 65, 2937–2952. doi:10.1002/lno.11566
Waldron, S., Lansdown, J. M., Scott, E. M., Fallick, A. E., and Hall, A. J. (1999). The global influence of the hydrogen isotope composition of water on that of bacteriogenic methane from shallow freshwater environments. Geochimica Cosmochimica Acta 63, 2237–2245. doi:10.1016/S0016-7037(99)00192-1
Walter, K. M., Chanton, J. P., Chapin, F. S., Schuur, E. A. G., and Zimov, S. A. (2008). Methane production and bubble emissions from arctic lakes: Isotopic implications for source pathways and ages. J. Geophys. Res. 113, G00A08. doi:10.1029/2007JG000569
Wang, D. T., Welander, P. V., and Ono, S. (2016). Fractionation of the methane isotopologues 13CH4, 12CH3D, and 13CH3D during aerobic oxidation of methane by Methylococcus capsulatus (Bath). Geochimica Cosmochimica Acta 192, 186–202. doi:10.1016/j.gca.2016.07.031
Wang, H., Lu, J., Wang, W., Yang, L., and Yin, C. (2006). Methane fluxes from the littoral zone of hypereutrophic Taihu Lake, China. J. Geophys. Res. 111, 17109. doi:10.1029/2005JD006864
Wegener, G., Gropp, J., Taubner, H., Halevy, I., and Elvert, M. (2021). Sulfate-dependent reversibility of intracellular reactions explains the opposing isotope effects in the anaerobic oxidation of methane. Sci. Adv. 7, eabe4939. doi:10.1126/sciadv.abe4939
West, W. E., Creamer, K. P., and Jones, S. E. (2016). Productivity and depth regulate lake contributions to atmospheric methane. Limnol. Oceanogr. 61, S51–S61. doi:10.1002/lno.10247
Whiticar, M. J. (1999). Carbon and hydrogen isotope systematics of bacterial formation and oxidation of methane. Chem. Geol. 161, 291–314. doi:10.1016/S0009-2541(99)00092-3
Whiticar, M. J., and Faber, E. (1986). Methane oxidation in sediment and water column environments - isotope evidence. Org. Geochem. 10, 759–768. doi:10.1016/S0146-6380(86)80013-4
Whiticar, M. J. (2020). “The biogeochemical methane cycle,” in Hydrocarbons, oils and lipids: Diversity, origin, chemistry and fate. Editor H. Wilkes (Cham: Springer International Publishing), 669–746. doi:10.1007/978-3-319-90569-3_5
Wiesenburg, D. A., and Guinasso, N. L. (1979). Equilibrium solubilities of methane, carbon monoxide, and hydrogen in water and sea water. J. Chem. Eng. Data 24, 356–360. doi:10.1021/je60083a006
Wik, M., Thornton, B. F., Varner, R. K., McCalley, C., and Crill, P. M. (2020). Stable methane isotopologues from northern lakes suggest that ebullition is dominated by sub‐lake scale processes. J. Geophys. Res. Biogeosci. 125. doi:10.1029/2019JG005601
Wollschläger, U., Ilmberger, J., Isenbeck-Schröter, M., Kreuzer, A. M., von Rohden, C., Roth, K., et al. (2007). Coupling of groundwater and surface water at Lake Willersinnweiher: Groundwater modeling and tracer studies. Aquat. Sci. 69, 138–152. doi:10.1007/s00027-006-0825-6
Yoshinaga, M. Y., Holler, T., Goldhammer, T., Wegener, G., Pohlman, J. W., Brunner, B., et al. (2014). Carbon isotope equilibration during sulphate-limited anaerobic oxidation of methane. Nat. Geosci. 7, 190–194. doi:10.1038/NGEO2069
Keywords: δ13C-CH4 values, δ2H-CH4 values, methane sources, methane sinks, lake, stable isotopes, stratification
Citation: Einzmann T, Schroll M, Kleint JF, Greule M and Keppler F (2022) Application of concentration and 2-dimensional stable isotope measurements of methane to constrain sources and sinks in a seasonally stratified freshwater lake. Front. Environ. Sci. 10:865862. doi: 10.3389/fenvs.2022.865862
Received: 30 January 2022; Accepted: 12 October 2022;
Published: 03 November 2022.
Edited by:
Tonya DelSontro, University of Waterloo, CanadaReviewed by:
Sarah Beth Cadieux, Rensselaer Polytechnic Institute, United StatesPeter Douglas, McGill University, Canada
Regina Gonzalez Moguel, McGill University, Canada
Copyright © 2022 Einzmann, Schroll, Kleint, Greule and Keppler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Teresa Einzmann, dGVyZXNhLmVpbnptYW5uQHVuaWJhcy5jaA==; Moritz Schroll, bW9yaXR6LnNjaHJvbGxAZ2Vvdy51bmktaGVpZGVsYmVyZy5kZQ==
†These authors have contributed equally to this work and share first authorship