 Wenhui Xu
Wenhui Xu Yanlu Qiao1,2*
Yanlu Qiao1,2* Qing Jiang
Qing Jiang Jianliang Xue
Jianliang Xue
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Environ. Sci., 02 May 2022
Sec. Toxicology, Pollution and the Environment
Volume 10 - 2022 | https://doi.org/10.3389/fenvs.2022.865636
This article is part of the Research TopicWaste Management of Petroleum Hydrocarbons in Marine EnvironmentView all 9 articles
Marine oil spill pollution was one of the factors affecting the marine ecology of the northeastern South China Sea (nSCS). The submarine oil produced after the oil spill had a long-term impact on the microbial community in the sediment. In this study, 16S rRNA genes high-throughput sequencing and quantitative PCR were used to study the composition and distribution of bacterial communities in deep-sea sediments; meanwhile, petroleum hydrocarbon degrading bacteria were isolated, of which activity were detected. Proteobacteria and Planctomycetota were the main bacterial phyla found in the samples studied in this study. 29 bacterial strains capable of degrading petroleum hydrocarbons were isolated from S02 and S39 sediment samples, belonging to genus Stenotrophomonas, Pseudidiomarina, Sulfitobacter, Pseudomonas, Halomonas and so on. Strains from Stenotrophomonas degraded petroleum hydrocarbons efficiently. This research provided new insights into distribution pattern of benthic microbial community in the nSCS, and validated the degradation potential of petroleum hydrocarbons by indigenous bacteria.
Deep-sea sediments were the largest ecosystem on Earth and contained rich biological resources (Cinzia, 2015). The environment of marine sediments was characterized by low temperature, high pressure, and lack of oxygen, and there were also special areas such as high pH and high salinity (Louvado et al., 2015; Danovaro et al., 2016; Perez Calderon et al., 2019). The source of organic matter in the deep sea mainly included surface water with organics and died marine organisms (Biddle, 2012; Azpiroz-Zabala et al., 2017). These factors resulted in significant differences among community composition of deep-sea sediments (Kirkpatrick et al., 2019). Extensive investigation of this difference was essential for further research on the distribution of marine microorganisms (Webb et al., 2010).
The extraction and transportation of offshore oil resulted in the release of large quantities of petroleum hydrocarbons into the environment (Duran and Cravo-Laureau, 2016). Oil spills would not only have a toxic effect on organisms in water, but would also form submarine oil under the action of particle aggregation, etc., which would be toxic to benthic organisms and affect the benthic ecosystem for a long time (Jacketti et al., 2020; Xue et al., 2021; Yu et al., 2021). After the oil spill in the Gulf of Mexico, the detriment and treatment of deep-sea oil spills and submarine oil aroused people’s attention (Kostka et al., 2011; Kimes et al., 2013). The remediation of oil pollution in the deep sea was still a difficult problem, and the isolation of high-efficiency deep-sea petroleum hydrocarbon degrading bacteria was the key to remediate polluted environment in deep-sea (Agarwal and Liu, 2015). Moreover, the low temperature in deep-sea would significantly affect the degradation performance of petroleum hydrocarbon degrading bacteria (Ferguson et al., 2017; Fasca et al., 2018). Recent studies demonstrated there were plenty of hydrocarbon degrading bacteria in the Mariana Trench (Liu J. et al., 2019). Although several petroleum hydrocarbon degrading bacteria had been isolated from deep-sea sediments (Gao et al., 2015; Tomasino et al., 2021), the current research on their degradation potential was still limited.
The northeastern South China Sea (nSCS) was surrounded by the coastal sedimentary belt of the Pearl River, the southwest side of Taiwan Island, and the west side of the Philippines Island (Luan et al., 2012). The nSCS water was affected by the Kuroshio Current and the deep cold water of the Pacific flowing from the Luzon Strait (Wang et al., 2018; Zhao, 2020). The change of microbial community in this area was greatly affected by exogenous pollutants (Graw et al., 2018). At the same time, this sea area was major for oil exploration and transportation. The distribution of petroleum hydrocarbons was affected by ocean dynamics and sedimentation (Yang et al., 2015). In the investigation of the distribution and source tracing of petroleum hydrocarbons in this area, Gong et al. (2020) found that the northeastern part of the SCS might have been polluted by oil for a long time. Therefore, studying the microbial community structure and diversity in the nSCS as well as isolating potential native petroleum hydrocarbon degrading bacteria were of great significance for the assessment and remediation of environmental pollution.
In this study, according to the complex environmental characteristics of the nSCS, a large-scale sediment sampling method was adopted. After that, high-throughput sequencing and quantitative PCR were used to study bacterial communities in sediments at different regions and water depths. In this non-culturing approach, the microbial diversity of indigenous deep-sea sediments was explored. Two sampling sites with the largest difference in water depths were selected for the isolation of low-temperature petroleum hydrocarbon degrading bacteria, and the petroleum hydrocarbon degradation potential of isolated bacteria were investigated.
Samples were collected by the R/V Xiang Yang Hong 18 during a cruise in the SCS from August to September, 2020. The sediment samples were collected with a box corer, then 0.5 cm thick surface layer of sediment was scraped with a sterile spatula and placed in steriled tubs. Sediment samples were stored at 4°C (for cultivation) and −80°C (for DNA extraction). The distribution of sampling sites in the study area was shown in Figure 1, and details of sampling sites was listed in Supplementary Table S1.
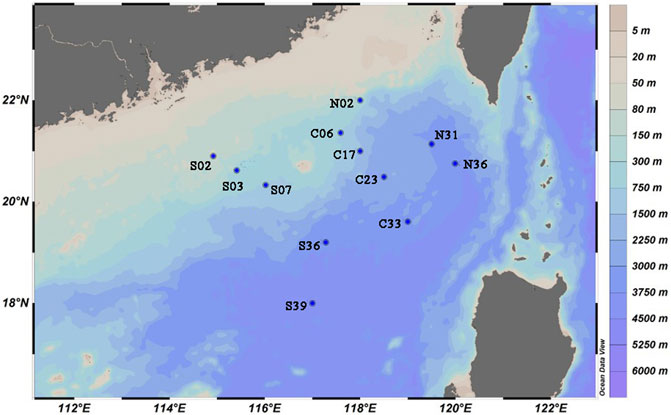
FIGURE 1. Water depths (unit: m) and sampling points in the northeastern South China Sea.
Diesel was selected as target material for microbial degradation of petroleum hydrocarbon pollutants in this study. Diesel (0#) was obtained by PetroChina and filtered with a 0.22 μm filter membrane. All chemicals, if not mentioned, were of analytical grade.
One aliquot of the sediment was sent to Shanghai Meiji Biomedical Technology Co., Ltd., for DNA extraction, high-throughput sequencing and quantitative PCR. Primers and PCR cycling conditions were shown in Table 1. Microbial community structure and diversity were analyzed using the online platform of Majorbio I-Sanger cloud platform (www.i-sanger.com). The sequencing data had been uploaded to NCBI SRA database (PRJNA802797) (https://www.ncbi.nlm.nih.gov/sra/PRJNA802797).
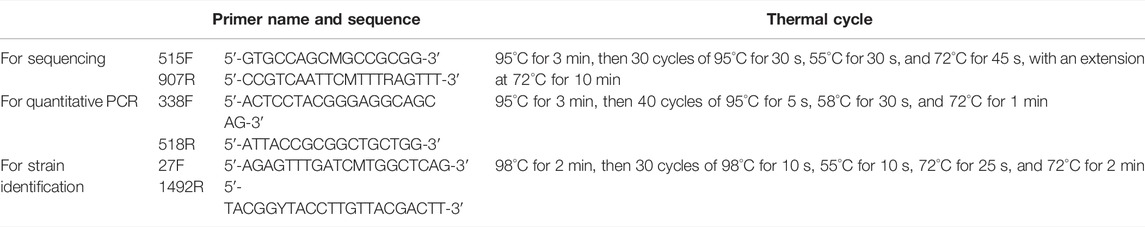
TABLE 1. Primers and PCR cycling conditions.
Four culture media were used: Artificial seawater (AS) media, Mineral salt medium (MSM), simplified 2216E medium and 1/50 2216E medium. Formulas are shown in Supplementary Table S2.
Concerned on the water depths and geographical differences of sampling points, the sediments of S02 (S) and S39 (Q) sites were selected for bacterial isolation and culture in order to explore the abundance and ability of cultivable petroleum hydrocarbon degrading bacteria in deep and shallow sediments.
Cultivable bacteria were measured using three methods. Y method was inoculating 1 g sediment directly into AS medium containing 1% diesel (v/v) and incubating at 10°C, 140 rpm for 30 days. The bacteria suspension was coated on the 2216E agar plate. C method was to dilute the sediment and to spread supernatant on 2216E agar plate coated with 200 μl diesel. G method was to dilute the sediment and to spread supernatant on 1/50 2216E agar plate.
Bacteria in these samples were cultured at 10°C. Individual colonies were purified by repeated streaking on 2216E agar plate, and biodegradability test was conducted with purified isolates.
Biodegradability test used the 2,6-Dichloroindophenol and sodium salt hydrate (DCPIP) redox indicator. Isolated strains were pre-cultured in 5 ml of 2216E (28°C, 140 rpm) until the optical density at 600 nm became >1.0. After centrifugation at 4000 rpm for 10 min, samples were washed with AS, and then the cell density was adjusted to 1.0 according to the optical density at 600 nm. 4 ml sterilized AS medium, 200 μl bacterial suspension, 400 μl 1 g/L DCPIP, 1% (v/v) diesel and 0.05% Tween 80 were added into the test tube. The control experiment was prepared without inoculum (Kubota et al., 2008; Montagnolli et al., 2015). The above system was divided into two groups and incubated at 28 and 10°C in shaker at 140 rpm for 5 days, respectively. The experiment was observed daily, and color changed from blue to colorless gradually. After 5 days, 2 ml of liquid was taken from each tube into an EP tube and centrifuged at 8000 rpm for 15 min. The supernatant was then analysed at 600 nm using the ultra violet-visible (UV–VIS) spectrophotometer (Obi et al., 2016). According to the equation, the decolorization rate was calculated as follows:
Where D is the decolorization rate (%), A0 is the absorbance of control and Ai is the absorbance of treated sample.
The degradation effect of diesel was evaluated according to the decolorization rate. The strains with decolorization ability were selected for further identification.
After DNA extraction from purified target strains, PCR was performed using the MiniAmp thermal cycler (Applied Biosystems, United States). The PCR amplification system was to mix 20 μl T3 Super PCR Mix, 1 μl Primer 27F, 1 μl Primer 1492R, and 3 μl DNA templates. The PCR products were sent to the Qingke Biotechnology Co., Ltd., (Qingdao, China) for sequencing. The 16S rRNA genes were performed by comparing with EzBioCloud databases (Yoon et al., 2017) (http://www.ezbiocloud.net/). The phylogenetic co-relation of each isolate was analyzed by using MEGA 7.0 (Hall, 2013).
The biodegradation experiments were conducted in 100 ml Erlenmeyer flasks with 50 ml of sterilized MSM supplemented and 1% (v/v) diesel. Then, 1% (v/v) of the single bacteria suspension was added into the Erlenmeyer flasks and incubated at 28 and 10°C on a shaker at 140 rpm for 5 days.
After 5 days, the remaining diesel was examined through UV spectrophotometry (Liu et al., 2018). The remaining diesel at 10°C for control and experimental groups on day 5 was detected by gas chromatography-mass spectrometry (GC-MS) (Agilent 19091S-433, United States) (Shi et al., 2019).
Community composition was based on the data table in the tax_summary_a folder, using R programming (v.3.3.1) tools to map. Sequences were clustered into distance-based (97% similarity) operational taxonomic units (OTUs) using mothur (version v.1.30.2). Microbial diversity was assessed using alpha and beta diversity using QIIME. Samples were clustered based on beta diversity, using Bray-curtis as a distance measure and hierarchical UPGMA as a clustering method. Principal co-ordinates analysis (PCoA) was to use R language PCoA statistical analysis and mapping. The difference in species abundance between the two samples was compared with the chi-square test. Analysis of similarities (ANOSIM) was performed using the anosim function from the R package “vegan” with the Bray-Curtis metric. Differences in species abundance between the two samples were compared between arms using Fisher exact test and 95% exact CIs. PICRUSt was used to infer gene representation using taxanomic information from 16S rRNA genes sequencing (Langille et al., 2013). Surfer v17 (Golden Software) was used to visualize the nSCS geographic distribution of the species diversity and functional pathways.
To estimate the community richness and diversity of the communities, the α-diversity indices were analyzed at the OTU level. The coverage index was greater than 97% (Supplementary Table S3), which indicated that the sequencing results in this run could represent the real situation of microorganisms in the sample. The Shannon curve tended to be flat, indicating that the amount of sequencing data could reflect the vast majority of microbial diversity information in a sample (Supplementary Figure S1). In total, 64 phyla were observed in the 12 sediment samples. Proteobacteria was the most dominant phylum, of which the abundances of γ-proteobacteria and α-proteobacteria was 16.45 and 13.05%, respectively. This was followed by abundance of Planctomycetota (15.26%), Acidobacteriota (11.26%), Actinobacteriota (9.61%), Chloroflexi (7.14%), NB1-j (5.05%) and Bacteroidota (4.01%). The differences in bacterial communities of sediments were mostly related to factors such as organic matter deposition (Giovannelli et al., 2013), temperature, salinity, and oxygen content (Conte et al., 2018). Combined with the hydrological characteristics of this sea area, this difference was speculated to be mainly related to the influence of water depths and exogenous organic matter (Graw et al., 2018).
The sample similarity analysis based on the Bray-Curtis dissimilarity at the OTU level showed that the community composition of the samples was associated with water depths (Figure 2). Sampling sites with similar water depths showed stronger clustering relationships. All samples were clustered by water depths (shallow:≤ 1000 m, deep:> 1000 m) (ANOSIM, r = 0.4138, p = 0.006). Similarly, PCoA based on unweighted UniFrac distances showed that samples could be clustered into two categories according to the water depths (Figure 3). The above results could clearly show that water depths was an important factor affecting the composition of bacterial communities in this study. This conclusion was different from the study of the nSCS sediments by Zhang et al. (2019). The reason of the difference might be the deeper sampling water depths of this study.
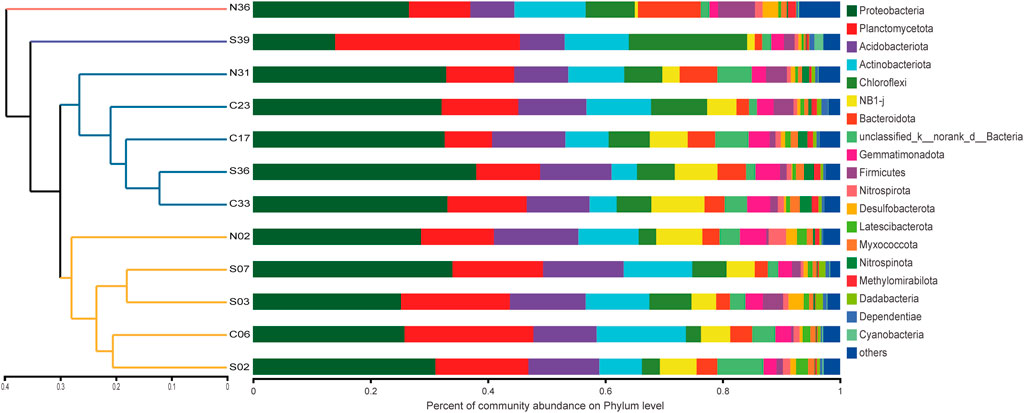
FIGURE 2. Bacterial community similarity and composition in 12 samples. Dendrogram (left) based on Bray-Curtis similarity of bacterial communities on OTU level. Bar charts (right) represented the relative abundance of bacterial phyla in each sample.
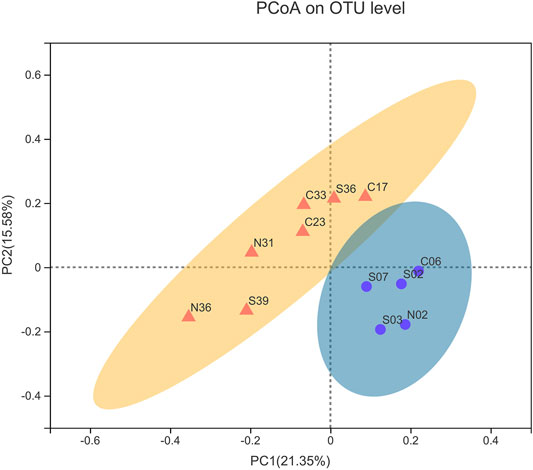
FIGURE 3. Principal coordinate analysis (PCoA) based on unweighted UniFrac. (Yellow represents >1000 m group, blue represents ≤1000 m group).
From Figure 4A, it could be seen that both richness and diversity were higher in sites near shore than sites far from shore. This phenomenon could be explained by the source of exogenous pollutants. S02 and S03 were affected by the terrigenous organic matter transported by the Pearl River (Wang et al., 2021), resulting in higher microbial diversity there. The study by Yuan et al. (2015) confirmed the impact of petroleum hydrocarbons input from the Pearl River on surface sediments. Liu X. et al. (2019) also confirmed that the microbial community at the estuary was largely affected by terrigenous organic matter. The higher bacterial diversity of N31 and N36 was influenced by various factors. They were located near the port on the southwest side of Taiwan, existing obvious submarine landslides (Zhang et al., 2021) and being close to the production site of natural gas hydrate (Han et al., 2021). The deep Pacific ocean current flowing in from the Luzon Strait passed through the area first which might carry organic pollutants from the east side of Taiwan Island (Wang et al., 2018),. These factors contributed to the complex diversity of exogenous organic matter here, resulting in high bacterial diversity. Compared with other sites, N36 and N31 had a higher abundance of Bacteroidota (10.67%) (Figure 2). Bacteroidota also showed high abundance in studies of the Arctic and deep waters of the Indian Ocean (Gao et al., 2021).
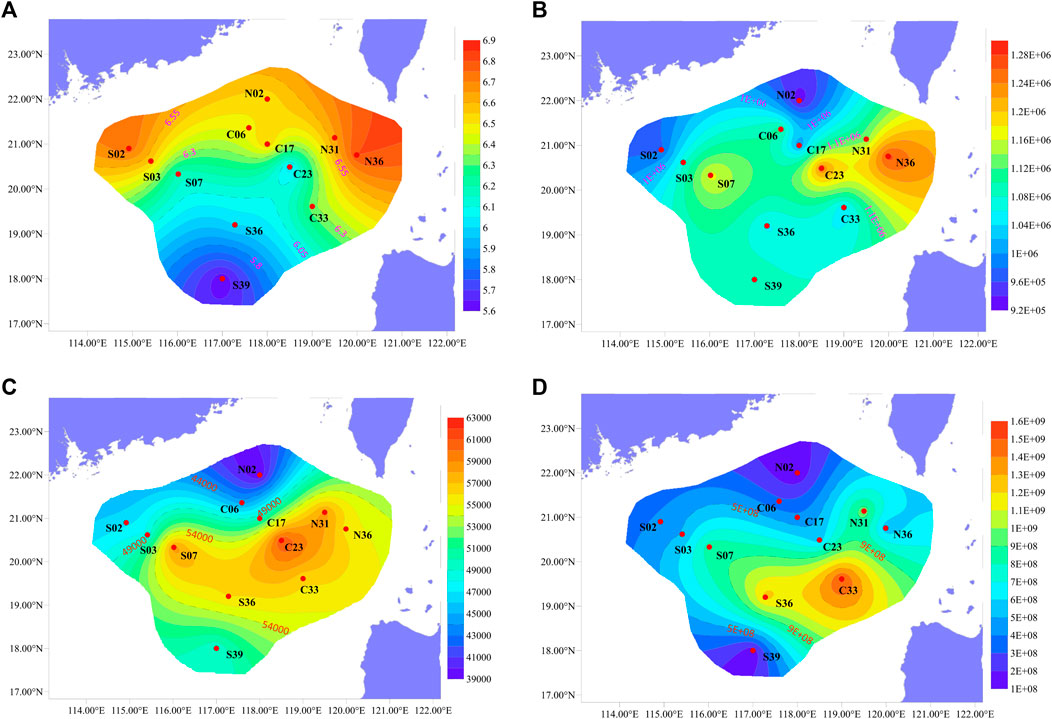
FIGURE 4. Regional distribution of (A) Shannon index, (B) Xenobiotics Biodegradation and Metabolism (at second level), (C) Metabolism of xenobiotics by cytochrome P450 (at third level), (D) 16S gene.
At the deepest site in this study, S39, which was also the furthest offshore site, different community abundance compositions was observed. In site S39, the Planctomycetota (31.48%) was the dominant phyla, and the second dominant phylum was Chloroflexi (20.16%) (Figure 2). Planctomycetota was widely present in marine sediments, and was able to utilize a variety of organic and inorganic substances, which played an important role in the global nitrogen and sulfur cycles (Anantharaman et al., 2018). Chloroflexi was highly diverse in sediments, utilizing hydrocarbons through anaerobic and aerobic pathways and effectively promoting carbon cycling in sediments (Hug et al., 2013; Vuillemin et al., 2020). In the study of nearby locations by Kong et al. (2021), Chloroflexi was also found to have high abundance. S39 located in a deep-sea basin, where sediments were transported here due to geological processes (Cao et al., 2015), resulting in changes of bacterial communities.
When the 16S gene abundance was studied by quantitative PCR, it was found that the gene abundance was higher in the basins deeper than 1000 m, and the overall performance was high in the east and low in the west (Figure 4D). Functional pathways were inferred from OTUs using PICRUSt and annotated with KEGG database. It was found that “Xenobiotics Biodegradation and Metabolism” and “Metabolism of xenobiotics by cytochrome P450” also showed similar distribution (Figures 4B,C). Kaiser et al. (2018) found that the content of petroleum hydrocarbons in the sediments of this sea area was extremely low, but it also showed high in the east and low in the west. Petroleum hydrocarbons could be deposited on the surface of particles such as sediment in the ocean, and the migration or resuspension of these sediments would significantly affect the distribution of petroleum hydrocarbons (Jia et al., 2019). The organic matter would be brought by the deep Pacific currents flowing into the Luzon Strait, and might be influenced by the internal currents of the SCS. Luna et al. (2016) proposed that ocean currents near the continental shelf could significantly influence on microbial function and biogeochemical cycles. Therefore, the impact of ocean currents on microbial communities in deep-sea sediments in the nSCS was speculated to be greater than former thought (Lo et al., 2014; Sun et al., 2021).
In subsequent study, S02 and S39 were selected to compare the diversity of potential low temperature petroleum hydrocarbon degrading bacteria in sediments at different water depths. The species composition of two samples, S02 and S39, differed significantly (Figure 5). In this study, three cultivation methods with different characteristics were used. Method Y was used to isolate strains that only relied on petroleum hydrocarbons as carbon source for growth. Moreover, Method C was to observe bacteria that can grow under oil pollution in the presence of other carbon sources, whereas Method G was to explore the ability of strains to grow under oligotrophic conditions, and to study whether it could degrade petroleum hydrocarbons.
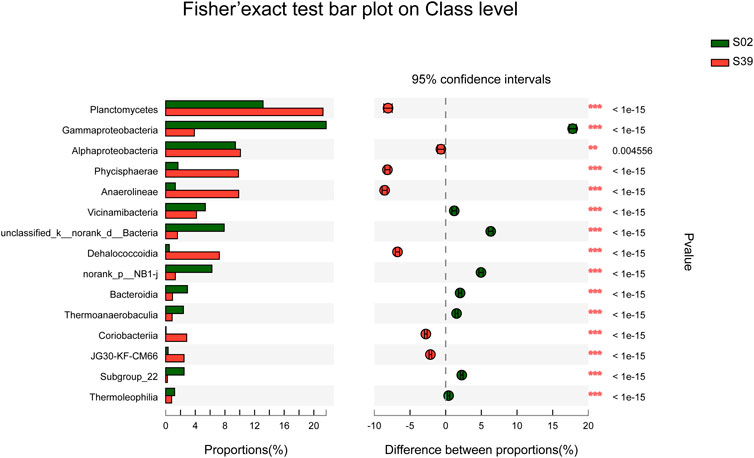
FIGURE 5. Comparison of S02 and S39 community abundance at the class level. (Fisher’s exact test (two-tailed), *p ≤ 0.05,**p ≤ 0.01,***p ≤ 0.001).
Total 47 strains were isolated by three cultivation methods. In order to efficiently screen out the strains with the ability to degrade petroleum hydrocarbons, the DCPIP method was used for preliminary screening. This method had been verified by Kubota et al. (2008) and Montagnolli et al. (2015), and could screen out potential petroleum hydrocarbon degrading bacteria quickly and accurately. According to the screening results of DCPIP evaluation method, the strains with decolorization rate over 60% could be considered to have degradation effect, and 29 strains were obtained that still had the degradation performance of petroleum hydrocarbons at low temperature (Figure 6).
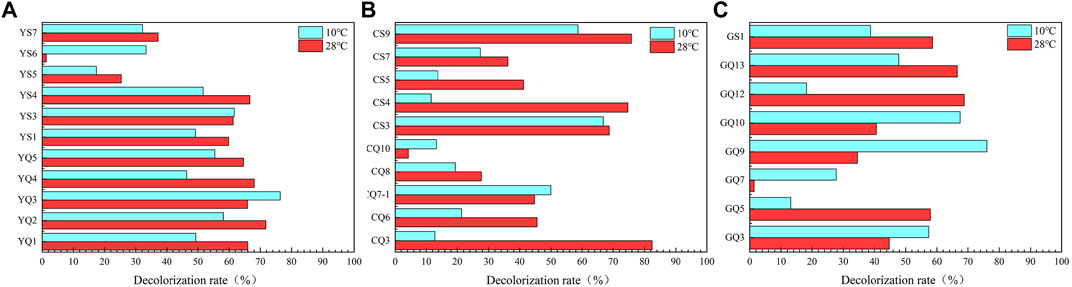
FIGURE 6. Decolorization rate of DCPIP, which indicating potential of diesel degradation ability. (A-method Y; B-method C; C-method G).
Most of the 29 petroleum hydrocarbon degrading strains belonged to Proteobacteria (Supplementary Figure S2). Among them, the diversity of γ-Proteobacteria was relatively high, which was also the main bacteria responsible for petroleum degradation in previous studies (Kostka et al., 2011; Mason et al., 2012). The petroleum hydrocarbon degrading bacterial strains isolated in this study mainly belonged to genus Stenotrophomonas, Pseudidiomarina, Sulfitobacter, Pseudomonas, and Halomonas, which all had been widely isolated in the study of petroleum hydrocarbon degradation (Varjani, 2017). In this study, Stenotrophomonas was more abundant, which was able to grow in harsh environments and use a variety of substances (Ryan et al., 2009). There were more low temperature petroleum hydrocarbon degrading strains isolated from S39 than from S02. Hydrocarbon degrading microorganisms were more abundant in the deep sea basin due to the accumulation of organic matter and low temperature (Tomasino et al., 2021).
According to the results of decolorization rate and identification results, the following 10 strains (YQ2, YQ3, YS3, YS4, CQ3, CS3, CS4, CS9, GQ10, GQ13) were selected as low-temperature petroleum hydrocarbon degrading strains in the comprehensive site for diesel degradation rate experiments.
As shown in Figure 7, the screened strains all had some degradation ability under both 10 and 28°C conditions. The degradation rate of CQ3, GQ13, YQ3 and YS3 was higher at 28°C, but they were significantly inhibited at 10°C. However, YQ2, YS4, CS3 and GQ10, showed paradoxical increase in degradation rate at 10°C, and also showed more than 30% degradation rate at 28°C. Among them, GQ10 had the highest degradation rate (51.42% at 10°C), and shared 99.8% similarity with Pseudomonas zhaodongensis. This species had been isolated in Heilongjiang Province, China (Zhang et al., 2015) and the South Shetland Trench sediment in Antarctica (Abdel-Mageed et al., 2020), and been proved to have high low temperature growth activity. In this study, the petroleum hydrocarbon degradation ability of Pseudomonas zhaodongensis was unambiguously determined for the first time. To further analyze the differences in the degradation of specific petroleum hydrocarbons by the strains, GC-MS analysis was used to study the degradation ability of 10 strains to diesel components at 10°C. The main alkane components of diesel used in this experiment are C10-C21. After alkanes were released into the environment, it could be degraded by microorganisms fastest (Varjani, 2017). For full GC-MS spectra, see Supplementary Figure S3. Figure 8 showed the peak heights of these alkanes in the GC-MS spectrum, ordered from lowest to highest. It could be seen that YQ2, CS3, YS4, GQ10 and CQ3 had obvious degradation to the petroleum alkane components in diesel, which matched the degradation rate measurement results. C13 remained the most abundant after degradation. Compared with other n-alkane, the degradation degree of C13 by the strain was low, but the difference was not obvious. The reason might be that C13 was the most abundant in diesel, and the degradation of the strains was relatively average for the degradation of different alkane, and did not show specific degradation of a certain n-alkane. Specifically, YQ2 had the strongest degradation ability (>60%) to long and medium-chain alkanes after 5 days at 10°C, followed by YS4 and CS3. They were all belonged to Stenotrophomonas. It also had degradation ability to polycyclic aromatic hydrocarbons (PAHs) (Arulazhagan et al., 2017). Stenotrophomonas was widely used in environmental remediation due to good ability to degrade petroleum hydrocarbons (Gargouri et al., 2011; Elufisan et al., 2020). So, our study clearly shows the potential of native microbial communities present in deep-sea sediments in the nSCS for applications in petroleum hydrocarbon pollution remediation.
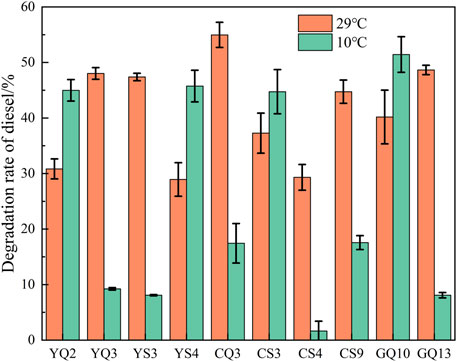
FIGURE 7. Diesel degradation rate over 5 days by 10 strains.
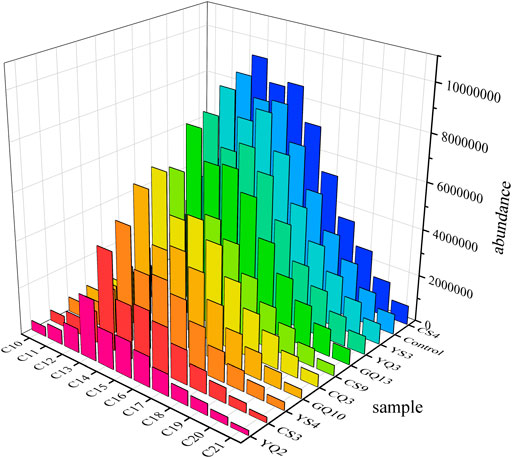
FIGURE 8. GC-MS of residual diesel. (Peak heights of n-alkanes C10-C21 in GC-MS spectrum).
There were obvious regional differences in bacterial community composition and diversity in deep-sea sediments in the nSCS, which were related to the influence of water depths and exogenous organic matter. Proteobacteria and Planctomycetota were the main bacterial phyla found in the samples in this paper. Strains of γ-proteobacteria (Stenotrophomonas, Pseudidiomarina, Sulfitobacter, Pseudomonas, and Halomonas) were isolated as the key degrading bacteria of petroleum hydrocarbons in two sediments (S02, S39). More potential petroleum hydrocarbon degrading bacteria were present in deep-sea sediments. Under low temperature conditions, Stenotrophomonas showed a stronger ability to degrade petroleum hydrocarbons.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI (accession: PRJNA802797).
WX: Writing-original draft, Methodology. YQ: Conceptualization, Formal analysis, Funding acquisition. JW: Data curation. QJ: Project administration. JX: Writing - review.
This work was supported by the scientific research fund project of the National Natural Science Foundation of China (grant numbers 42106151); the Natural Science Foundation of Shandong Province (grant numbers ZR2021QD103); the Weifang Science and Technology Development Plan Project (2021GX048).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank all of the scientists and crew members on the R/V Xiang Yang Hong 18 during the expedition for their great efforts and help in sample collection.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fenvs.2022.865636/full#supplementary-material
Abdel-Mageed, W. M., Lehri, B., Jarmusch, S. A., Miranda, K., Al-Wahaibi, L. H., Stewart, H. A., et al. (2020). Whole Genome Sequencing of Four Bacterial Strains from South Shetland Trench Revealing Biosynthetic and Environmental Adaptation Gene Clusters. Mar. Genomics 54, 100782. doi:10.1016/j.margen.2020.100782
Agarwal, A., and Liu, Y. (2015). Remediation Technologies for Oil-Contaminated Sediments. Mar. Pollut. Bull. 101 (2), 483–490. doi:10.1016/j.marpolbul.2015.09.010
Anantharaman, K., Hausmann, B., Jungbluth, S. P., Kantor, R. S., Lavy, A., Warren, L. A., et al. (2018). Expanded Diversity of Microbial Groups that Shape the Dissimilatory Sulfur Cycle. Isme J. 12 (7), 1715–1728. doi:10.1038/s41396-018-0078-0
Arulazhagan, P., Al-Shekri, K., Huda, Q., Godon, J. J., Basahi, J. M., and Jeyakumar, D. (2017). Biodegradation of Polycyclic Aromatic Hydrocarbons by an Acidophilic Stenotrophomonas Maltophilia Strain Ajh1 Isolated from a Mineral Mining Site in saudi arabia. Extremophiles 21 (1), 163–174. doi:10.1007/s00792-016-0892-0
Azpiroz-Zabala, M., Cartigny, M., Talling, P. J., Parsons, D. R., Sumner, E. J., and Clare, M. A. (2017). Newly Recognized Turbidity Current Structure Can Explain Prolonged Flushing of Submarine Canyons. Sci. Adv. 3 (10), e1700200. doi:10.1126/sciadv.1700200
Biddle, J. F. (2012). Prospects for the Study of Evolution in the Deep Biosphere. Front. Microbiol. 2. doi:10.3389/fmicb.2011.00285
Cao, L., Jiang, T., Wang, Z., Zhang, Y., and Sun, H. (2015). Provenance of Upper Miocene Sediments in the Yinggehai and Qiongdongnan Basins, Northwestern South China Sea: Evidence from REE, Heavy Minerals and Zircon U-Pb Ages. Mar. Geol. 361, 136–146. doi:10.1016/j.margeo.2015.01.007
Conte, A., Papale, M., Amalfitano, S., Mikkonen, A., Rizzo, C., De Domenico, E., et al. (2018). Bacterial Community Structure along the Subtidal Sandy Sediment Belt of a High Arctic Fjord (Kongsfjorden, Svalbard Islands). Sci. Total Environ. 619-620, 203–211. doi:10.1016/j.scitotenv.2017.11.077
Cinzia, C. (2015). New Perspectives in Benthic Deep-Sea Microbial Ecology. Front. Mar. Sci. 2 (17), 17. doi:10.3389/fmars.2015.00017
Danovaro, R., Molari, M., Corinaldesi, C., and Dell’Anno, A. (2016). Macroecological Drivers of Archaea and Bacteria in Benthic Deep-Sea Ecosystems. Sci. Adv. 2 (4), e1500961. doi:10.1126/sciadv.1500961
Duran, R., and Cravo-Laureau, C. (2016). Role of Environmental Factors and Microorganisms in Determining the Fate of Polycyclic Aromatic Hydrocarbons in the Marine Environment. FEMS Microbiol. Rev. 40 (6), 814–830. doi:10.1093/femsre/fuw031
Elufisan, T. O., Rodríguez-Luna, I. C., Oyedara, O. O., Sánchez-Varela, A., Hernández-Mendoza, A., Dantán Gonzalez, E., et al. (2020). The Polycyclic Aromatic Hydrocarbon (Pah) Degradation Activities and Genome Analysis of a Novel Strainstenotrophomonas Sp. Pemsol Isolated from mexico. PeerJ 8, e8102. doi:10.7717/peerj.8102
Fasca, H., de Castilho, L. V. A., de Castilho, J. F. M., Pasqualino, I. P., Alvarez, V. M., de Azevedo Jurelevicius, D., et al. (2018). Response of Marine Bacteria to Oil Contamination and to High Pressure and Low Temperature Deep Sea Conditions. MicrobiologyOpen 7 (2), e00550. doi:10.1002/mbo3.550
Ferguson, R. M. W., Gontikaki, E., Anderson, J. A., and Witte, U. (2017). The Variable Influence of Dispersant on Degradation of Oil Hydrocarbons in Subarctic Deep-Sea Sediments at Low Temperatures (0-5 °C). Sci. Rep. 7 (1), 2253. doi:10.1038/s41598-017-02475-9
Gao, P., Qu, L., Du, G., Wei, Q., Zhang, X., and Yang, G. (2021). Bacterial and Archaeal Communities in Deep Sea Waters Near the Ninetyeast Ridge in Indian Ocean. J. Ocean. Limnol. 39 (2), 582–597. doi:10.1007/s00343-020-9343-y
Gao, X., Gao, W., Cui, Z., Han, B., Yang, P., Sun, C., et al. (2015). Biodiversity and Degradation Potential of Oil-Degrading Bacteria Isolated from Deep-Sea Sediments of South Mid-atlantic Ridge. Mar. Pollut. Bull. 97 (1-2), 373–380. doi:10.1016/j.marpolbul.2015.05.065
Gargouri, B., Karray, F., Mhiri, N., Aloui, F., and Sayadi, S. (2011). Application of a Continuously Stirred Tank Bioreactor (Cstr) for Bioremediation of Hydrocarbon-Rich Industrial Wastewater Effluents. J. Hazard. Mater. 189 (1-2), 427–434. doi:10.1016/j.jhazmat.2011.02.057
Giovannelli, D., Molari, M., d’Errico, G., Baldrighi, E., Pala, C., and Manini, E. (2013). Large-scale Distribution and Activity of Prokaryotes in Deep-Sea Surface Sediments of the Mediterranean Sea and the Adjacent Atlantic Ocean. PLoS One 8 (8), e72996. doi:10.1371/journal.pone.0072996
Gong, S., Liu, W., Li, Y., Zhang, J., Chen, C., and Fu, J. (2020). Distribution Characteristics and Source Tracing of Petroleum Hydrocarbons in the Northeastern South china Sea. Chin. Chem. Lett. 31 (10), 2854–2858. doi:10.1016/j.cclet.2020.06.020
Graw, M. F., D'Angelo, G., Borchers, M., Thurber, A. R., Johnson, J. E., Zhang, C., et al. (2018). Energy Gradients Structure Microbial Communities across Sediment Horizons in Deep Marine Sediments of the South china Sea. Front. Microbiol. 9, 729. doi:10.3389/fmicb.2018.00729
Hall, B. G. (2013). Building Phylogenetic Trees from Molecular Data with Mega. Mol. Biol. Evol. 30 (5), 1229–1235. doi:10.1093/molbev/mst012
Han, W.-C., Chen, L., and Liu, C.-S. (2021). Distribution and Characteristics of Gas Chimneys in the Passive Margin Offshore Sw Taiwan. Mar. Geophys. Res. 42 (3), 25. doi:10.1007/s11001-021-09447-9
Hug, L. A., Castelle, C. J., Wrighton, K. C., Thomas, B. C., Sharon, I., Frischkorn, K. R., et al. (2013). Community Genomic Analyses Constrain the Distribution of Metabolic Traits across the Chloroflexi Phylum and Indicate Roles in Sediment Carbon Cycling. Microbiome 1 (1), 22. doi:10.1186/2049-2618-1-22
Jacketti, M., Beegle-Krause, C. J., and Englehardt, J. D. (2020). A Review on the Sinking Mechanisms for Oil and Successful Response Technologies. Mar. Pollut. Bull. 160, 111626. doi:10.1016/j.marpolbul.2020.111626
Jia, Y., Tian, Z., Shi, X., Liu, J. P., Chen, J., Liu, X., et al. (2019). Deep-sea Sediment Resuspension by Internal Solitary Waves in the Northern South china Sea. Sci. Rep. 9 (1), 12137–12138. doi:10.1038/s41598-019-47886-y
Kaiser, D., Schulz-Bull, D. E., and Waniek, J. J. (2018). Polycyclic and Organochlorine Hydrocarbons in Sediments of the Northern South china Sea. Mar. Pollut. Bull. 137, 668–676. doi:10.1016/j.marpolbul.2018.10.039
Kimes, N. E., Callaghan, A. V., Aktas, D. F., Smith, W. L., Sunner, J., Golding, B., et al. (2013). Metagenomic Analysis and Metabolite Profiling of Deep-Sea Sediments from the Gulf of Mexico Following the Deepwater Horizon Oil Spill. Front. Microbiol. 4, 50. doi:10.3389/fmicb.2013.00050
Kirkpatrick, J. B., Walsh, E. A., and D’Hondt, S. (2019). Microbial Selection and Survival in Subseafloor Sediment. Front. Microbiol. 10, 956. doi:10.3389/fmicb.2019.00956
Kong, Y., Lei, H., Zhang, Z., Cheng, W., Wang, B., Pan, F., et al. (2021). Depth Profiles of Geochemical Features, Geochemical Activities and Biodiversity of Microbial Communities in Marine Sediments from the Shenhu Area, the Northern South china Sea. Sci. Total Environ. 779, 146233. doi:10.1016/j.scitotenv.2021.146233
Kostka, J. E., Prakash, O., Overholt, W. A., Green, S. J., Freyer, G., Canion, A., et al. (2011). Hydrocarbon-degrading Bacteria and the Bacterial Community Response in Gulf of mexico Beach Sands Impacted by the Deepwater Horizon Oil Spill. Appl. Environ. Microbiol. 77 (22), 7962–7974. doi:10.1128/AEM.05402-11
Kubota, K., Koma, D., Matsumiya, Y., Chung, S.-Y., and Kubo, M. (2008). Phylogenetic Analysis of Long-Chain Hydrocarbon-Degrading Bacteria and Evaluation of Their Hydrocarbon-Degradation by the 2,6-dcpip Assay. Biodegradation 19 (5), 749–757. doi:10.1007/s10532-008-9179-1
Langille, M. G. I., Zaneveld, J., Caporaso, J. G., McDonald, D., Knights, D., Reyes, J. A., et al. (2013). Predictive Functional Profiling of Microbial Communities Using 16s Rrna Marker Gene Sequences. Nat. Biotechnol. 31 (9), 814–821. doi:10.1038/nbt.2676
Liu, J., Zheng, Y., Lin, H., Wang, X., Li, M., Liu, Y., et al. (2019). Proliferation of Hydrocarbon-Degrading Microbes at the Bottom of the Mariana Trench. Microbiome 7 (1), 47. doi:10.1186/s40168-019-0652-3
Liu, X., Sen, B., Zhao, Y., Bai, M., He, Y., Xie, Y., et al. (2019). Gradients of Three Coastal Environments off the South china Sea and Their Impacts on the Dynamics of Heterotrophic Microbial Communities. Sci. Total Environ. 659, 499–506. doi:10.1016/j.scitotenv.2018.12.405
Liu, Z., Xue, J., Shi, K., Wu, Y., Gao, Y., Xiao, X., et al. (2018). Performance and Community Structure Change of Immobilized Marine Petroleum-Degrading Bacteria in the Degradation Process. dwt 130, 117–123. doi:10.5004/dwt.2018.22964
Lo, W.-T., Dahms, H.-U., and Hwang, J.-S. (2014). Water Mass Transport through the Northern Bashi Channel in the Northeastern South china Sea Affects Copepod Assemblages of the Luzon Strait. Zool. Stud. 53 (1), 1–9. doi:10.1186/s40555-014-0066-7
Louvado, A., Gomes, N. C. M., Simões, M. M. Q., Almeida, A., Cleary, D. F. R., and Cunha, A. (2015). Polycyclic Aromatic Hydrocarbons in Deep Sea Sediments: Microbe-Pollutant Interactions in a Remote Environment. Sci. Total Environ. 526, 312–328. doi:10.1016/j.scitotenv.2015.04.048
Luan, X., Zhang, L., and Peng, X. (2012). Dongsha Erosive Channel on Northern South china Sea Shelf and its Induced Kuroshio South china Sea Branch. Sci. China Earth Sci. 55 (1), 149–158. doi:10.1007/s11430-011-4322-y
Luna, G. M., Chiggiato, J., Quero, G. M., Schroeder, K., Bongiorni, L., Kalenitchenko, D., et al. (2016). Dense Water Plumes Modulate Richness and Productivity of Deep Sea Microbes. Environ. Microbiol. 18 (12), 4537–4548. doi:10.1111/1462-2920.13510
Mason, O. U., Hazen, T. C., Borglin, S., Chain, P. S. G., Dubinsky, E. A., Fortney, J. L., et al. (2012). Metagenome, Metatranscriptome and Single-Cell Sequencing Reveal Microbial Response to Deepwater Horizon Oil Spill. ISME J. 6 (9), 1715–1727. doi:10.1038/ismej.2012.59
Montagnolli, R. N., Lopes, P. R. M., and Bidoia, E. D. (2015). Screening the Toxicity and Biodegradability of Petroleum Hydrocarbons by a Rapid Colorimetric Method. Arch. Environ. Contam. Toxicol. 68 (2), 342–353. doi:10.1007/s00244-014-0112-9
Obi, L. U., Atagana, H. I., and Adeleke, R. A. (2016). Isolation and Characterisation of Crude Oil Sludge Degrading Bacteria. SpringerPlus 5 (1), 1946. doi:10.1186/s40064-016-3617-z
Perez Calderon, L. J., Gontikaki, E., Potts, L. D., Shaw, S., Gallego, A., Anderson, J. A., et al. (2019). Pressure and Temperature Effects on Deep-Sea Hydrocarbon-Degrading Microbial Communities in Subarctic Sediments. MicrobiologyOpen 8 (6), e00768. doi:10.1002/mbo3.768
Ryan, R. P., Monchy, S., Cardinale, M., Taghavi, S., Crossman, L., Avison, M. B., et al. (2009). The Versatility and Adaptation of Bacteria from the Genus Stenotrophomonas. Nat. Rev. Microbiol. 7 (7), 514–525. doi:10.1038/nrmicro2163
Shi, K., Xue, J., Xiao, X., Qiao, Y., Wu, Y., and Gao, Y. (2019). Mechanism of Degrading Petroleum Hydrocarbons by Compound Marine Petroleum-Degrading Bacteria: Surface Adsorption, Cell Uptake, and Biodegradation. Energy fuels. 33 (11), 11373–11379. doi:10.1021/acs.energyfuels.9b02306
Sun, F.-l., Wang, Y.-S., Wu, M.-L., Sun, C.-C., Jiang, Z.-Y., Cheng, H., et al. (2021). Bacterial Community Variations in the South china Sea Driven by Different Chemical Conditions. Ecotoxicology 30 (9), 1808–1815. doi:10.1007/s10646-021-02455-w
Tomasino, M. P., Aparício, M., Ribeiro, I., Santos, F., Caetano, M., Almeida, C. M. R., et al. (2021). Diversity and Hydrocarbon-Degrading Potential of Deep-Sea Microbial Community from the Mid-atlantic Ridge, South of the Azores (North Atlantic Ocean). Microorganisms 9 (11), 2389. doi:10.3390/microorganisms9112389
Varjani, S. J. (2017). Microbial Degradation of Petroleum Hydrocarbons. Bioresour. Technol. 223, 277–286. doi:10.1016/j.biortech.2016.10.037
Vuillemin, A., Kerrigan, Z., D'Hondt, S., and Orsi, W. D. (2020). Exploring the Abundance, Metabolic Potential and Gene Expression of Subseafloor Chloroflexi in Million-Year-Old Oxic and Anoxic Abyssal Clay. FEMS Microbiol. Ecol. 96 (12), fiaa223. doi:10.1093/femsec/fiaa223
Wang, A., Du, Y., Peng, S., Liu, K., and Huang, R. X. (2018). Deep Water Characteristics and Circulation in the South china Sea. Deep Sea Res. Part I Oceanogr. Res. Pap. 134, 55–63. doi:10.1016/j.dsr.2018.02.003
Wang, Y., Lin, C., Zhang, Z., Zhang, B., and Liu, H. (2021). Sedimentary Evolution and Controlling Factors of Early-Mid Miocene Deltaic Systems in the Northern Pearl River Mouth Basin, South china Sea. Sci. Rep. 11 (1), 6134. doi:10.1038/s41598-021-85369-1
Webb, T. J., Vanden Berghe, E., and O'Dor, R. (2010). Biodiversity's Big Wet Secret: the Global Distribution of Marine Biological Records Reveals Chronic Under-exploration of the Deep Pelagic Ocean. PLoS One 5 (8), e10223. doi:10.1371/journal.pone.0010223
Xue, J., Shi, K., Chen, C., Bai, Y., Cui, Q., Li, N., et al. (2021). Evaluation of Response of Dynamics Change in Bioaugmentation Process in Diesel-Polluted Seawater via High-Throughput Sequencing: Degradation Characteristic, Community Structure, Functional Genes. J. Hazard. Mater. 403, 123569. doi:10.1016/j.jhazmat.2020.123569
Yang, X., Yuan, X., Zhang, A., Mao, Y., Li, Q., Zong, H., et al. (2015). Spatial Distribution and Sources of Heavy Metals and Petroleum Hydrocarbon in the Sand Flats of Shuangtaizi Estuary, Bohai Sea of china. Mar. Pollut. Bull. 95 (1), 503–512. doi:10.1016/j.marpolbul.2015.02.042
Yoon, S.-H., Ha, S.-M., Kwon, S., Lim, J., Kim, Y., Seo, H., et al. (2017). Introducing Ezbiocloud: a Taxonomically United Database of 16s Rrna Gene Sequences and Whole-Genome Assemblies. Int. J. Syst. Evol. Micr. 67 (5), 1613–1617. doi:10.1099/ijsem.0.001755
Yu, Y., Qi, Z., Xiong, D., Sun, R., Fu, S., and Li, W. (2021). Oil Dispersion and Aggregation with Suspended Particles in a Wave Tank. J. Environ. Manag. 278, 111572. doi:10.1016/j.jenvman.2020.111572
Yuan, K., Wang, X., Lin, L., Zou, S., Li, Y., Yang, Q., et al. (2015). Characterizing the Parent and Alkyl Polycyclic Aromatic Hydrocarbons in the Pearl River Estuary, Daya Bay and Northern South china Sea: Influence of Riverine Input. Environ. Pollut. 199, 66–72. doi:10.1016/j.envpol.2015.01.017
Zhang, J., Chen, M., Huang, J., Guo, X., Zhang, Y., Liu, D., et al. (2019). Diversity of the Microbial Community and Cultivable Protease-Producing Bacteria in the Sediments of the Bohai Sea, Yellow Sea and South china Sea. PLoS One 14 (4), e0215328. doi:10.1371/journal.pone.0215328
Zhang, K., Song, H., Sun, S., and Gao, J. (2021). Distribution and Genesis of Submarine Landslides in the Northeastern South china Sea. Geol. J. 56 (3), 1187–1201. doi:10.1002/gj.3996
Zhang, L., Pan, Y., Wang, K., Zhang, X., Zhang, C., Zhang, S., et al. (2015). Pseudomonas Zhaodongensis Sp. Nov., Isolated from Saline and Alkaline Soils. Int. J. Syst. Evol. Micr. 65 (Pt_3), 1022–1030. doi:10.1099/ijs.0.000057
Keywords: deep-sea sediments, the northeastern South China Sea, bacterial communities, petroleum hydrocarbon degrading bacteria, low temperature
Citation: Xu W, Qiao Y, Wei J, Jiang Q and Xue J (2022) Bacterial Communities and Culturable Petroleum Hydrocarbon Degrading Bacteria in Marine Sediments in the Northeastern South China Sea. Front. Environ. Sci. 10:865636. doi: 10.3389/fenvs.2022.865636
Received: 30 January 2022; Accepted: 15 April 2022;
Published: 02 May 2022.
Edited by:
Shan Zhao, Shandong University, ChinaReviewed by:
Xiaoshou Liu, Ocean University of China, ChinaCopyright © 2022 Xu, Qiao, Wei, Jiang and Xue. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanlu Qiao, cWlhb3lhbmx1b3VjQHNpbmEuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.