
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Environ. Sci. , 09 May 2016
Sec. Marine Pollution
Volume 4 - 2016 | https://doi.org/10.3389/fenvs.2016.00033
 Stephen M. Techtmann1*
Stephen M. Techtmann1* Kathleen S. Fitzgerald2,3Savannah C. Stelling2,3Dominique C. Joyner2,3Sagar M. Uttukar4,5Austin P. Harris2,3Noor K. Alshibli2,3
Kathleen S. Fitzgerald2,3Savannah C. Stelling2,3Dominique C. Joyner2,3Sagar M. Uttukar4,5Austin P. Harris2,3Noor K. Alshibli2,3 Steven D. Brown4,5
Steven D. Brown4,5 Terry C. Hazen2,3,6*
Terry C. Hazen2,3,6*Many studies have shown that microbes, which share nearly identical 16S rRNA genes, can have highly divergent genomes. Microbes from distinct parts of the ocean also exhibit biogeographic patterning. Here we seek to better understand how certain microbes from the same species have adapted for growth under local conditions. The phenotypic and genomic heterogeneity of three strains of Colwellia psychrerythraea was investigated in order to understand adaptions to local environments. Colwellia are psychrophilic heterotrophic marine bacteria ubiquitous in cold marine ecosystems. We have recently isolated two Colwellia strains: ND2E from the Eastern Mediterranean and GAB14E from the Great Australian Bight. The 16S rRNA sequence of these two strains were greater than 98.2% identical to the well-characterized C. psychrerythraea 34H, which was isolated from arctic sediments. Salt tolerance, and carbon source utilization profiles for these strains were determined using Biolog Phenotype MicoArrays. These strains exhibited distinct salt tolerance, which was not associated with the salinity of sites of isolation. The carbon source utilization profiles were distinct with less than half of the tested carbon sources being metabolized by all three strains. Whole genome sequencing revealed that the genomes of these three strains were quite diverse with some genomes having up to 1600 strain-specific genes. Many genes involved in degrading strain-specific carbon sources were identified. There appears to be a link between carbon source utilization and location of isolation with distinctions observed between the Colwellia isolate recovered from sediment compared to water column isolates.
Understanding the diversity and geographic distribution of microbes in the environment is an important line of investigation in microbial ecology. Many recent studies have sought to understand the extent of geographic constraints on microbial communities (Martiny et al., 2006; Brown et al., 2012; Hanson et al., 2012; Malmstrom et al., 2013). It is believed that microbes are not limited by dispersal and therefore should be ubiquitously distributed. A recent study used 16S rRNA deep sequencing to demonstrate that in the oceans there is a persistent seed bank of ubiquitously distributed microbial taxa (Gibbons et al., 2013). These taxa are found in most marine samples and their abundance is determined by local factors and environmental conditions. While similar microbial taxa may be present throughout the world's oceans, biogeographic patterning may be seen in the genomic diversity of these globally distributed taxa. As suggested by Becking (1934), the environment may select for not only the relative abundance of taxa but also for particular phenotypes able to thrive under local conditions. There is a possibility that natural selective pressure may drive microbial biogeography on a genomic level. Therefore, genomic analysis may provide key insights into the potential for microbial biogeography.
In addition to natural selection, neutral evolution may also play a role in determining the local diversity of microbial populations. A recent study modeled the role of neutral evolution in dictating biogeographic patterns in microbes found in the surface oceans (Hellweger et al., 2014). The authors of this study concluded that biogeographic patterns do exist in marine systems and suggested that microbes evolve faster than the ocean can disperse them (Hellweger et al., 2014). This conclusion would indicate that the same species from different parts of the ocean ought to have distinct genomic content. In this modeling study, natural selection was ignored and the authors were able to demonstrate that neutral evolution was sufficient to result in biogeographic patterns. A number of studies have previously described the fact that members of the same microbial species show dramatic genotypic diversity (Konstantinidis and Tiedje, 2005; Hunt et al., 2008; Luo et al., 2011; Caro-Quintero and Konstantinidis, 2012; Shapiro et al., 2012). This diversity is believed to confer enhanced survival under distinct environmental conditions and thus is the result of natural selection (Cordero and Polz, 2014).
We were interested in identifying the extent to which genomic differences among closely related strains could be related to adaptation to their environment. To this end, we sought to identify phenotypic and genotypic adaptations that might confer enhanced survival under distinct conditions found in the ecosystems studied. If the genomic differences observed in these strains confer phenotypic differences that are related to distinctions in environmental conditions, it may be that these mutations arose as a result of environmental selection for a particular phenotype. However, if the majority of differences do not relate to the distinct environmental conditions, it may be that their genomes have been predominantly shaped by neutral evolution. To test this hypothesis, we investigated the genomic diversity of Colwellia species isolated from distinct marine locations.
Colwellia species are psychrophilic heterotrophs found in many cold marine environments including sea ice, polar sediments, deep-sea trenches, and as symbionts of marine animals (Nogi et al., 2004; Methé et al., 2005; Jung et al., 2006; Zhang et al., 2008; Choi et al., 2010; Yu et al., 2011; Kim et al., 2013). Colwellia have also been shown to degrade hydrocarbons and were present at high abundance in the microbial community that responded to the Deepwater Horizon oil spill (Baelum et al., 2012; Redmond and Valentine, 2012; Dubinsky et al., 2013; Gutierrez et al., 2013; Mason et al., 2014). Sequences from Colwellia sp. have also been recovered from marine metagenomes (Kennedy et al., 2008). In the current study, we isolated representatives of Colwellia psychrerythraea from two deep-sea basins: Eastern Mediterranean and the Great Australian Bight. These strains were compared to a well-characterized strain of C. psychrerythraea—strain 34H—previously isolated from arctic sediments (Huston et al., 2000; Methé et al., 2005).
C. psychrerythraea is a model psychrophilic heterotroph and much of our understanding of the adaptations for microbial growth in cold environments comes from studies performed on C. psychrerythraea 34H (Junge et al., 2003; Methé et al., 2005; Casanueva et al., 2010; Yamauchi et al., 2012). In the present study, phenotypic comparison was performed using the Biolog high-throughput Phenotype MicroArray system to assess carbon source utilization and salt tolerance. We also sequenced the genomes of the two recently isolated strains and compared them with the genome of strain 34H. A better understanding of phenotypic and genomic heterogeneity of these ubiquitous psychrophiles and the sources of heterogeneity will add to our understanding of how genetic changes can impact diversity of psychrophilic microbes and lead to biogeographic patterning. An understanding of biogeographic patterning will help to clarify the drivers of microbial biodiversity in oceans. Furthermore, Colwellia spp. are known to be responders to various oil spills. Therefore, differences in phenotype and genotypes of closely related Colwellia strains from different deep sea basins may have implications in terms of their response to potential oil spills.
C. psychrerythraea 34H was previously isolated from Arctic marine sediments (Huston et al., 2000). In this study, strain 34H was routinely cultured in marine broth at temperatures between 4 and 14°C. C. psychrerythraea ND2E was isolated from a water sample collected from the Eastern Mediterranean Sea at a depth of 495 m and a temperature of 13.8°C. C. psychrerythraea GAB14E was isolated from a water sample from the Great Australian Bight collected at a depth of 1472 m and a temperature of 2.7°C. Isolates were obtained by plating raw seawater on ONR7a (Dyksterhouse et al., 1995) agar plates supplemented with peptone (1 g/L) and 100 ppm of local crude oil. Colonies were observed after a week of incubation at near in situ temperatures (ND2E at 14°C and GAB14E at 4°C). Isolated colonies were struck onto the same medium and transferred into liquid ONR7a supplemented with peptone and 100 ppm of oil. Following isolation, cultures were routinely grown in marine broth at 14°C.
DNA was extracted from strains ND2E and GAB14E collected at mid log phase using the UltraClean Microbial DNA Isolation Kit (MO BIO Laboratories, Carlsbad, CA). The 16S rRNA gene was amplified using bacterial primers 27f and 1492r. Amplicon were sequenced using an ABI 3730. The taxonomy of isolates was determined using the RDP classifier (Wang et al., 2007) on the nearly full-length 16S rRNA gene sequences.
Carbon source utilization profiles were generated by the Biolog Phenotype MicroArray (PM) technology. Cells were grown to late log phase in marine broth, and resuspended in minimal medium (ONR7a) lacking carbon source (Dyksterhouse et al., 1995). Resuspended cells were inoculated into ONR7a lacking a carbon source at a 10% inoculum concentration and inoculated onto PM01A and PM02A panels with 1X dye H (Biolog, Hayward, CA). Plates were incubated aerobically in a humidified chamber at 14°C for 14 days in duplicate. PM 09 panel was prepared as above in ONR7a amended with 1 g/L peptone. Positive carbon source metabolism was hand scored based a redox reaction where the development of a purple color indicates reduction of a tetrazolium salt named dye H to its formazan endproduct. Dye reduction therefore, represents positive metabolism. No color development was considered the inability to metabolize the substrate. Color development for the PM09 panel was scored by measuring absorbance at 560 nm.
Genome libraries were prepared using the Nextera XT DNA Preparation Kit (Illumina, San Diego, CA) following the standard workflow. Genomes were sequenced on the Illumina MiSeqTM using a 600-cycle v3 Reagent Kit with 300 bp paired-end reads. Genome sequencing of C. psychrerythraea ND2E and GAB14E generated 4,796,093 and 4,469,340 paired-end reads for ND2E and GAB14E respectively. Quality-based trimming was performed using Trimmomatic with the following parameters: SLIDINGWINDOW:4:15 MINLEN:36 (Bolger et al., 2014). After quality filtering 4,152,069 and 4,029,602 paired-end reads remained for C. psychrerythraea ND2E and GAB14E respectively resulting in 3.35 and 3.36 Gbp of sequence data for strain ND2E and GAB14E respectively. The average read length after quality filtering was 218 and 229 for ND2E and GAB14E respectively. We applied several assembly methods as described previously (Utturkar et al., 2014) and assembly with optimal statistics were selected as the best draft genome sequences. The genome of C. psychrerythraea ND2E was assembled using SPAdes version 3.1 (Nurk et al., 2013) into 57 large (≥ 500 bp) contigs, with a total genome size of 5.2 Mb. The N50 contig size for strain ND2E was 297,116 bp with the largest contig being 643,864 bp. The genome of C. psychrerythraea GAB14E was assembled using ABySS version 1.5.1 (Simpson et al., 2009) into 77 large (> 500 bp) contigs, with a total genome size of 5.7 Mb. The N50 contig size for strain GAB14E was 218,121 bp with the largest contig being 489,615 bp. The genome of C. psychrerythraea 34H was previously sequenced and assembled into one contig (Methé et al., 2005).
Genes were identified using the Prodigal algorithm (Hyatt et al., 2010) as part of the Oak Ridge National Laboratory genome annotation pipeline. The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) nonredundant database, UniProt, TIGRFam, Pfam, PRIAM, KEGG, COG, and InterPro databases. Non-coding genes and other features were predicted using tRNAscan-SE (Lowe and Eddy, 1997), AND RNAMMer (Lagesen et al., 2007).
Average nucleotide identity (ANI) was determined using Jspecies (Richter and Rossello-Mora, 2009). The genes shared between taxa were determined using an all vs. all BLASTP. Proteins that were greater than 50% identity over the length of the gene were considered to be homologs. Pan-genome trees were constructed using CMG biotools (Vesth et al., 2013). Strain-specific gene families were identified using the subset function in CMG biotools. The strain-specific genes were classified in the KEGG categories using the BlastKOALA (Kanehisa et al., 2015). BLAST atlas were constructed using CIRCOS (Krzywinski et al., 2009).
The genomes of ND2E and GAB14E were deposited in Genbank. ND2E Accession number JQED00000000. The accession number for GAB14E is JQEC00000000. Raw sequence data is available through NCBI Sequence Read Archive (SRA) database under accession SRP045527 (ND2E) and SRP045528 (GAB14E).
Three Colwellia strains were isolated from distant oceanographic basins (Figure 1). C. psychrerythraea 34H was previously isolated from sediment collected in the Arctic (Huston et al., 2000). Strain ND2E was isolated as part of this study from deep-sea water from the Mediterranean Sea. GAB14E was isolated as part of this study from deep-sea water from the Indian Ocean in the Great Australian Bight. The environmental conditions of the locations from which these C. psychrerythraea strains were isolated were distinct (Table 1). The temperature and salinity of these waters are all very different. The temperature in the sediments from which C. psychrerythraea 34H was isolated was 0.7°C. Alternately, strain GAB14E was isolated from deep-sea water that was 2.7°C, and ND2E was isolated from deep-sea water that was 13.8°C. Salinity was also very different at each of these sampling locations. The salinity at the site of sampling for strain 34H was not reported, but the salinity of the seawater from which GAB14E was isolated was 35.1 psu, whereas ND2E was isolated from waters with a salinity of 38.9 psu. While a number of environmental variables were measured at the sampling locations for GAB14E and ND2E (Supplemental Table 1), only temperature was reported for the sampling location from which 34H was derived. These differences in environmental conditions may select for distinct populations capable of surviving in those conditions.
Figure 1. Map of sampling locations for strains 34H, ND2E, and GAB14E. Locations are shown as circles and the strain that originated from that location is indicated by text.
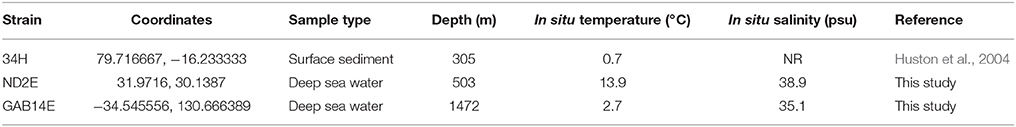
Table 1. Environmental conditions for sampling locations for three C. psychrerythraea.
In addition to differences in temperature and salinity these environments are distinct in terms of the types of available carbon. Strain 34H was isolated from arctic sediments typically with high organic carbon content as is the case in many sediment environments (Jiao et al., 2010). GAB14E and ND2E were both derived from deep water environments. Strain ND2E was isolated from a sample of ultraoligotrophic Eastern Mediterranean Deep water from a depth of 495 m directly above the North Alexandria Mud Volcano (Feseker et al., 2010). Mud volcanoes are capable of injecting both methane as well as other carbon sources into the overlaying seawater (Loncke et al., 2004; Mastalerz et al., 2009). GAB14E was derived from a sample collected at 1472 m, which should contain carbon that is quite difficult for microbes to degrade (recalcitrant) (Jiao et al., 2010). The available carbon at this depth has been subjected to microbial degradation through the water column resulting in the most labile carbon being degrader at more shallow depths. These differences in available carbon sources may have selected for distinct profiles of carbon source utilization for each strain.
The microbial community was profiled in these samples using 16S rRNA sequencing (Eastern Mediterranean, Techtmann et al., 2015 and GAB, unpublished data). These Colwellia isolates were present in both samples, albeit at low levels. ND2E was 0.005% of recovered reads from the sample from the Eastern Mediterranean and GAB14E was present at 0.02% of the community from the Great Australian Bight sample. No community analysis was done at the sampling location of 34H, it is therefore not possible to know the abundance of 34H in the environment. These abundances indicate that while these strains are present in these three environments, they are minor components of the community under the ambient conditions.
In an effort to understand differential adaptation to local conditions, the physiological response of these strains was measured under different temperatures, salt concentrations, and carbon sources. All three strains showed a temperature optimum of 8°C (Figure 2), which is in line with the previously reported growth parameters for 34H (Methé et al., 2005). Despite the common optimal temperature, the growth parameters were significantly different between these strains. For example, there were significant differences in growth rate at 4°C for each strain (Figure 2, Supplemental Table 2). ND2E showed the highest growth rate at 4°C followed by GAB14E, with 34H having the slowest growth rate. This is contrary to expectation that strains isolated from environments with colder temperatures would have faster growth rates under colder temperatures. Instead, what is observed is that strain ND2E is able to grow better under a broad range of temperatures whereas 34H is more restricted in temperature range.
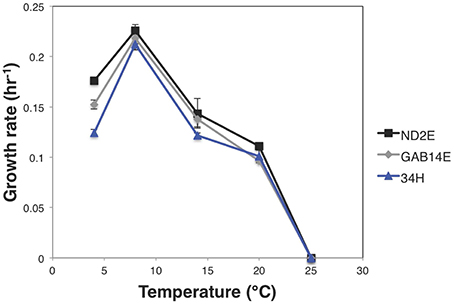
Figure 2. Growth rate vs. temperature. Strains were grown in marine broth at different temperatures. Growth rate was calculated for each temperature and strain and growth rate was plotted as function of temperature for each strain. Error bars represent the standard error for three biological replicates. Strain 34H is shown in blue, ND2E is shown in black, and GAB14E is shown in gray.
The salinity of the environments from which these strains were isolated also varied with ND2E coming from the high salinity Mediterranean whereas GAB14E and 34H were derived from environments with salinities that were similar to other oceanic contexts. To test the hypothesis that strains isolated from high saline environments would have an increased tolerance for salt, we used the PM panel PM09 to test a variety of salt stressors. These strains exhibited differential response to salt stress (Figure 3A). For example, strain GAB14E was able to tolerate up to 1% higher concentration of sodium chloride compared to the other two strains. Additionally, 34H was able to grow at 4% urea, whereas GAB14E was only able to grow at 3% urea and ND2E was inhibited above 2% urea. The phenotypes resulting from the salt tolerance screen do not correlate to what was expected based on the organisms native environment. It is possible that the differences in the bulk salinity of the sampling locations are at much finer scale than the 1% differences tested in these experiments.
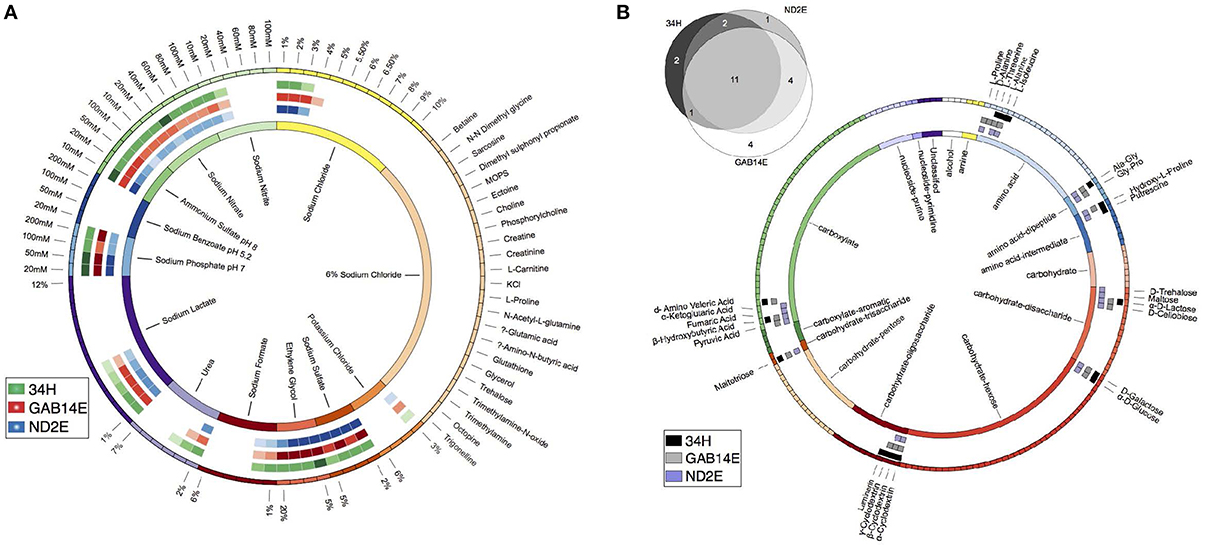
Figure 3. Phenotypic characterization of three C. psychrerythraea isolates. (A) Salt tolerance. The boxes on the outer circle represent each of the 90 salt concentrations tested. Colors represent groups of salts. Boxes in circles two, three, and four represent salt concentrations that allowed for measurable metabolism (OD560 > 0.5). Colors in circles two, three and four correspond to the isolate. Green corresponds to 34H, blue corresponds to ND2E and red corresponds to GAB14E. The inner circle represents the salt groupings with labels. (B) Venn Diagram showing the number of shared carbon sources between the isolates. Detailed description of the carbon sources able to support metabolism of each isolate. The boxes on the outer circle represent each of the 190 carbon sources tested. Colors represent carbon source class and are detailed in the inner circle. Boxes in circles two, three, and four represent a carbon source that could support growth of an isolate. Colors in circles two, three, and four correspond to the isolate. Black corresponds to 34H, gray corresponds to ND2E and purple corresponds to GAB14E. The inner circle represents the carbon source groupings with labels.
To investigate the carbon source utilization profiles for each of these strains, PM panels PM01A and PM02A were used to test metabolism of 190 carbon-based substrates. These three strains showed positive results indicative of metabolism and possible growth on 25 of the 190 carbon sources tested (Figure 3B). 34H was able to grow on 16 of the 190 carbon sources. ND2E was able to grow on 18 of the 190 substrates. GAB14E was able to grow on 20 of the 190 substrates. Eleven of the carbon sources could be utilized by all three strains. The eleven shared substrates were spread between the three major groups of compounds tested—amino acids (four substrates), carbohydrates (six substrates), and carboxylates (one substrate). ND2E and GAB14E had 15 substrates in common, whereas ND2E had only 13 common substrates with 34H. 34H and GAB14E shared 12 common substrates. GAB14E was able to metabolize four substrates that the other two strains could not. Strain 34H had two strain-specific substrates, and ND2E only had one strain-specific substrate.
The 25 substrates that were metabolized were spread between amino acids (nine substrates), carbohydrates (11 substrates) and carboxylates (five substrates) (Figure 3B). Within these categories, there was strain-to-strain variation. For example, GAB14E was able to grow using the widest range of disaccharides, whereas 34H could only metabolize one of the disaccharides tested. Conversely, 34H could metabolize four of the 12 oligosaccharides tested, and GAB14E could only metabolize two of the 12 oligosaccharides. Additionally, 34H was only able to grow on two of the carboxylates tested (β-Hydroxybutyric Acid and d-Amino Valeric Acid).
Only 11 substrates were shared between the three isolates, indicating that the carbon source utilization profiles are quite divergent. An understanding of the common substrates sheds light onto the set of compounds that many Colwellia spp. are able to metabolize. Many of these shared substrates are also used by other species within the Colwellia, which are able to use a large number of polymers including many oligosaccharides (Choi et al., 2010). While the majority of the common substrates are carbohydrates, a number of amino acids could be metabolized. These amino acids may serve as both carbon and nitrogen sources for Colwellia. These finding further expands our understanding of Colwellia species by confirming that all known Colwellia species are heterotrophs able to metabolize an array of carbohydrates and amino acids. There is however, a large differences in the carbon sources able to be utilized by each strain.
Since the relative concentration of each of the tested carbon sources in these environments is not known, it is difficult to extrapolate the selective advantage that metabolism of a particular carbon source confers. However, these isolates are derived from distinct environments with differing carbon qualities. Previous studies have shown that enzymes involved in polysaccharide metabolism are more active and able to degrade a broader range of substrates in sediments compared to the water column (Teske et al., 2011). ND2E was isolated from water directly above the active North Alex Mud Volcano, which releases gas and fluids into the overlying seawater (Feseker et al., 2010). This process may expose the microbial community at this site to carbon sources found in the sediments and sub-sea floor. The microbial community from this same sample was highly enriched in an unclassified group of Flavobacteria (Techtmann et al., 2015) that have been shown to be involved in polymer degradation and growth off of high molecular weight organic matter (Fernandez-Gomez et al., 2013). This could indicate that the microbes in this environment have been exposed to high levels of polysaccharides typically found in sediments. It is these findings that would suggest that 34H would be more adept at degrading oligosaccharides and ND2E would have the next highest potential. This is what is observed with the phenotypes, as 34H is able to utilize four different oligosaccharides, followed by ND2E, which can metabolize three distinct oligosaccharides. GAB14E is only able to use two oligosaccharides.
Genome analysis is able to provide key insights into the genotypic diversity and biogeographic patterns of microbes. The genomes of these three species were sequenced to better understand the genetic diversity of Colwellia and clarify the core and accessory genomes of these strains. The original genome assembly of strain 34H was closed into a single contig. ND2E was assembled into 57 large contigs (> 500 bp). The genome of GAB14E was assembled into 77 contigs (Supplemental Table 3). The three genomes were of different sizes with ND2E being the smallest at 5.2 Mb, followed by 34H at 5.4 Mb. GAB14E was the largest at 5.7 Mb (Supplemental Table 3). The G+C content was very similar between the three strains at 38% ± 0.05. The number of predicted protein-coding genes follows the genome size with GAB14E having the most (4691) followed by 34H (4510) and ND2E (4381). Between 69.1 and 72.1% of the genes in these three genomes were annotated as having a function. More than two thirds of the predicted genes in these genomes were assigned to a cluster of orthologous genes (COG).
Despite the differences observed in phenotypes between these three strains, they did not exhibit a trend in terms of phenotypes presumed to confer selective advantage under the in situ conditions. Comparison of these genomes was performed in order to understand how divergent these genomes were. To examine genomic difference among these strains on a whole genome level, the average nucleotide identity (ANI) for each was compared to C. psychrerythraea 34H (Supplemental Table 3). ND2E had an ANI of 84.5% compared to 34H, whereas GAB14E had an ANI of 79.8% compared to 34H. While all three isolates have 16S rRNA genes that are greater than 98.2% identical, the average nucleotide identity is quite different. Interestingly, the two strains with the highest identity on the 16S rRNA gene level are the most distant when comparing the whole genomes; the 16S rRNA gene identity for 34H and GAB14E is 99.2%, but the average nucleotide identity for these two strains is 79.8%.
The three strains shared 2595 gene families (Figures 4A,B). Strains ND2E and 34H were most closely related, sharing 3296 gene families. Strains 34H and GAB14E have 2723 gene families in common. ND2E and GAB14E have 2709 gene families shared between the two strains. GAB14E had the largest accessory genome, with 1671 gene families only found in GAB14E. 34H had 1249 strain-specific gene families and ND2E has 814 strain-specific gene families.
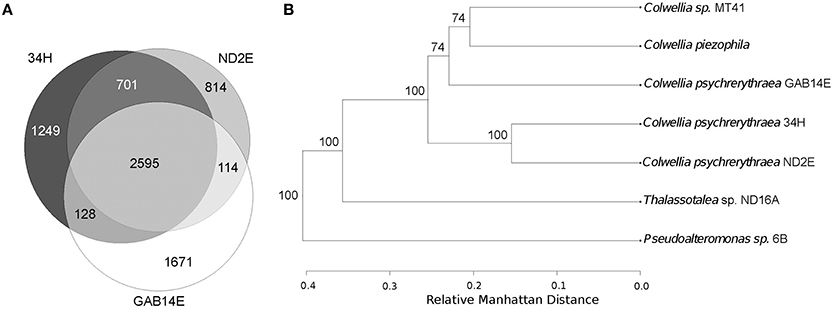
Figure 4. Genomic Comparison. (A) Venn Diagram showing shared protein families between the three strains. (B) Whole genome tree based on relative manhattan distances of similarities of genes families. Bootstrap values of greater than 50% are shown at the nodes of the branches.
To understand the functional significance of these large accessory genomes, representatives of these strain-specific gene families were categorized using KEGG categories (Table 2). The majority of these strain-specific genes were unclassified and were annotated as hypothetical proteins. The strain-specific genes that could be assigned to a KEGG category were spread across different categories. For example, strain 34H has 24 strain-specific genes involved in carbohydrate metabolism, 24 strain-specific genes involved in amino acid metabolism, and to 18 genes involved in membrane transport. Strain ND2E has eight strain-specific genes involved in replication and repair compared to only three and two in 34H and GAB14E, respectively. Furthermore, strain GAB14E has 19 strain-specific genes involved in signal transduction and nine strain-specific genes involved in cell motility. One notable difference is in the number of strain-specific genes classified as involved in amino acid metabolism. GAB14E had 39 strain-specific genes classified as involved in amino acid metabolism compared to 11 and 24 in ND2E and 34H respectively. For the most part, these strain-specific gene families were classified within similar KEGG categories. This could indicate that these strain-specific genes might encode for similar functional capacity and thus fulfill similar roles.
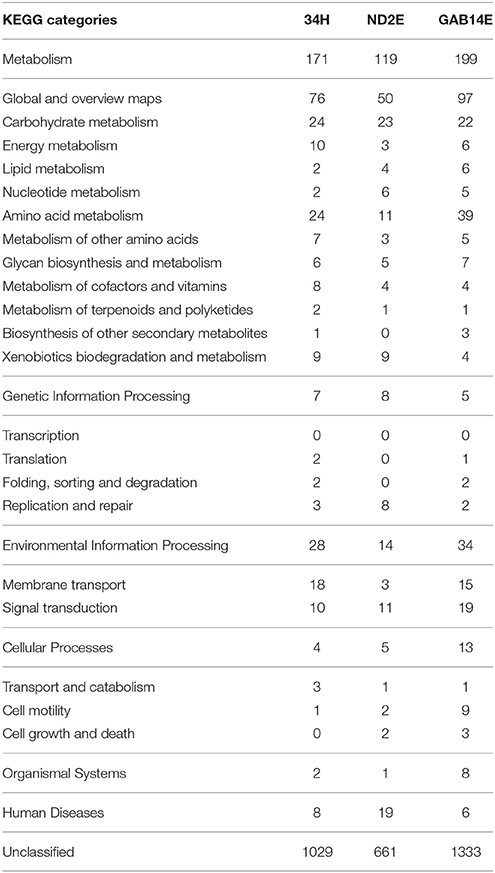
Table 2. KEGG categories for strain-specific genes from each of the three isolates.
Despite the fact that many of the gene families that are specific to one organism and fall into similar categories, differences within these categories may in part explain some of the phenotypic differences. For example, both 34H and GAB14E are able to grow using putrescene as a sole carbon source, whereas ND2E is not. Putrescene has been shown to be a ubiquitous chemical in the marine environment and can be used by marine microbes as both a carbon and nitrogen source (Höfle, 1984). Spermidine/Putrescine transporters are encoded in the accessory genomes of the 34H and GAB14E and are absent from the ND2E genome. This finding would indicate that the distinctions in the accessory genome contribute to the phenotypic diversity. This finding also suggests that the different gene complement can in part explain the functional differences observed. Furthermore, GAB14E is able to utilize both trehalose and maltose, whereas 34H and ND2E are not. The accessory genome of GAB14E encodes a maltose binding protein and a trehalose transporter. These differences in transporters appear to be important for trehalose and maltose metabolism. Trehalose has the potential to be a cryoprotectant for cells and biomolecules (Kikawada et al., 2007; De Maayer et al., 2014). Therefore, the presence of trehalose transport functionality in GAB14E may not only confer enhanced carbon utilization but may also allow GAB14E to use trehalose to cope with cold conditions encountered in the deep ocean.
To understand the mechanisms behind the observed genomic heterogeneity, every gene from strains ND2E and GAB14E were examined by BLAST analysis using the 34H genome as a reference. Homologous genes (>50% identity) were plotted against the 34H chromosome to identify regions of the 34H chromosome without homologous genes in either of the genomes of the two new isolates (Figure 5). Several large stretches of the 34H genome were shown to have no homologous genes in the other two strains. In some cases the lack of homology is due to gaps in the draft genomes. Some of these regions with gaps in homology between the three strains are found in the middle of the contigs in the draft sequences and flanked by regions with high homology.
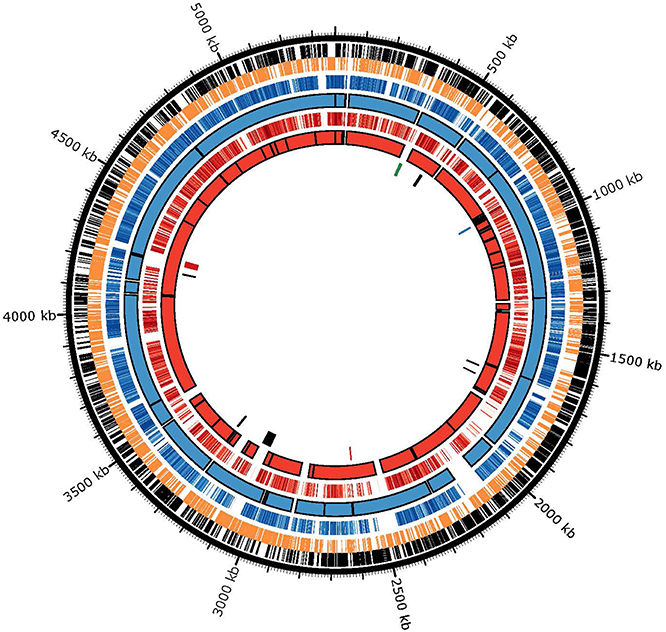
Figure 5. BLAST atlas comparing homologous genes. (Outer ring) 34H positive strand genes are shown in black. (Second ring) 34H negative strand genes are shown in orange (Third ring) Genes in ND2E with greater than 50% nucleotide identity to a gene in 34H are shown as blue lines. Darker blue lines are genes with higher identity to the gene in 34H and lighter blue lines have are less identical to the gene in 34H. (Fourth ring) ND2E Contigs are depicted as blue blocks. (Fifth ring) Genes in GAB14E with greater than 50% nucleotide identity to a gene in 34H are shown as red lines. Darker red lines are genes with higher identity to the gene in 34H and lighter red lines have are less identical to the gene in 34H. (Sixth ring) GAB14E Contigs are depicted as blue blocks. (Seventh ring) Putative genomic islands as predicted by island viewer. Genomic islands shown in red have been previously described (Collins and Deming, 2013). The genomic islands shown in blue corresponds to one of the two previously described filamentous phage (Methé et al., 2005). One of the previously described filamentous phage that was not predicted to be a genomic island is shown in green.
To determine if these gaps in homology are due to horizontal gene transfers (HGT), we used Island Viewer to identify putative genomic islands in strain 34H. Genomic islands are regions of the genome whose sequence composition is divergent from the overall genome averages. These genomic islands may be the result of horizontal transfer of genes from unrelated organisms. Island Viewer predicted nine genomic islands in the 34H genome. Many of these genomic islands include a number of transposases as well as some phage-related genes. These genomic islands vary in size from 4 to 50 kb. A couple of the regions with few homologous genes correspond to some of the predicted genomic islands, suggesting that these gaps in homology correspond to regions putatively obtained via horizontal gene transfer after 34H diverged from ND2E and GAB14E. These findings indicate that horizontal gene transfer has contributed to the genomic diversity of these strains. While it has been suggested that horizontal transfer can be the result of neutral evolution (Gogarten and Townsend, 2005), recent studies have concluded that neutral evolution is not sufficient to explain the frequency of HGT events in many genomes (Soucy et al., 2015).
These genomes show large differences in genomic and phenotypic diversity. This can be traced back to large segments of the genome that appear to be acquired by horizontal gene transfer. While there is some evidence that genes have been acquired and confer increased functionality and in turn potential selective advantage, the majority of differences do not appear to be related to adaptation to different environmental lifestyles. This suggests that a mixture of natural selection and neutral evolution have contributed to the divergence of these organisms and the great genetic and phenotypic diversity present within this species. This study examined one isolate of a Colwellia sp. recovered from three different locations. Further work involving analysis of many Colwellia isolates recovered from the same location is required to better understand Colwellia populations in the world's oceans in order to better quantify the core genome. This would further identify how distinct populations have adapted for growth under differeing environmental conditions.
ST performed the experiments, analyzed data and wrote the manuscript. KF, SS, and DJ performed the phenotypic analysis and genome sequencing. AH, NA were involved in isolations. SU, SB were involved in analysis of genome sequences. TH was involved in analysis of data and manuscript preparation and was the PI on the overall project.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This research was supported by contract A13-0119-001 Deep Sea Basin Microbiology between the University of Tennessee and BP. The authors are grateful to BP and its partners for support in the sampling effort.
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fenvs.2016.00033
Baelum, J., Borglin, S., Chakraborty, R., Fortney, J. L., Lamendella, R., Mason, O. U., et al. (2012). Deep-sea bacteria enriched by oil and dispersant from the Deepwater Horizon spill. Environ. Microbiol. 14, 2405–2416. doi: 10.1111/J.1462-2920.2012.02780.X
Becking, L. B. (1934). Geobiologie of Inleiding Tot de Milieukunde [Geobiology or Introduction to the Science of the Environment]. Den Haag: W. P. Van Stockum & Zoon.
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Brown, M. V., Lauro, F. M., DeMaere, M. Z., Muir, L., Wilkins, D., Thomas, T., et al. (2012). Global biogeography of SAR11 marine bacteria. Mol. Syst. Biol. 8:595. doi: 10.1038/msb.2012.28
Caro-Quintero, A., and Konstantinidis, K. T. (2012). Bacterial species may exist, metagenomics reveal. Environ. Microbiol. 14, 347–355. doi: 10.1111/J.1462-2920.2011.02668.X
Casanueva, A., Tuffin, M., Cary, C., and Cowan, D. A. (2010). Molecular adaptations to psychrophily: the impact of ‘omic’ technologies. Trends Microbiol. 18, 374–381. doi: 10.1016/J.Tim.2010.05.002
Choi, F. J., Kwon, H. C., Koh, H. Y., Kim, Y. S., and Yang, H. O. (2010). Colwellia asteriadis sp nov., a marine bacterium isolated from the starfish Asterias amurensis. Int. J. Syst. Evol. Microbiol. 60, 1952–1957. doi: 10.1099/Ijs.0.016055-0
Collins, R. E., and Deming, J. W. (2013). An inter-order horizontal gene transfer event enables the catabolism of compatible solutes by Colwellia psychrerythraea 34H. Extremophiles 17, 601–610. doi: 10.1007/S00792-013-0543-7
Cordero, O. X., and Polz, M. F. (2014). Explaining microbial genomic diversity in light of evolutionary ecology. Nat. Rev. Microbiol. 12, 263–273. doi: 10.1038/nrmicro3218
De Maayer, P., Anderson, D., Cary, C., and Cowan, D. A. (2014). Some like it cold: understanding the survival strategies of psychrophiles. EMBO Rep. 15, 508–517. doi: 10.1002/embr.201338170
Dubinsky, E. A., Conrad, M. E., Chakraborty, R., Bill, M., Borglin, S. E., Hollibaugh, J. T., et al. (2013). Succession of Hydrocarbon-degrading bacteria in the aftermath of the Deepwater Horizon oil spill in the Gulf of Mexico. Environ. Sci. Technol. 47, 10860–10867. doi: 10.1021/es401676y
Dyksterhouse, S. E., Gray, J. P., Herwig, R. P., Lara, J. C., and Staley, J. T. (1995). Cycloclasticus Pugetii gen-nov, sp-nov, an Aromatic Hydrocarbon-degrading bacterium from Marine-Sediments. Int. J. Syst. Bacteriol. 45, 116–123.
Fernandez-Gómez, B., Richter, M., Schuler, M., Pinhassi, J., Acinas, S. G., González, J. M., et al. (2013). Ecology of marine Bacteroidetes: a comparative genomics approach. ISME J. 7, 1026–1037. doi: 10.1038/Ismej.2012.169
Feseker, T., Brown, K. R., Blanchet, C., Scholz, F., Nuzzo, M., Reitz, A., et al. (2010). Active mud volcanoes on the upper slope of the western Nile deep-sea fan-first results from the P362/2 cruise of R/V Poseidon. Geo Marine Lett. 30, 169–186. doi: 10.1007/S00367-010-0192-0
Gibbons, S. M., Caporaso, J. G., Pirrung, M., Field, D., Knight, R., and Gilbert, J. A. (2013). Evidence for a persistent microbial seed bank throughout the global ocean. Proc. Natl. Acad. Sci. U.S.A. 110, 4651–4655. doi: 10.1073/pnas.1217767110
Gogarten, J. P., and Townsend, J. P. (2005). Horizontal gene transfer, genome innovation and evolution. Nat. Rev. Microbiol. 3, 679–687. doi: 10.1038/nrmicro1204
Gutierrez, T., Singleton, D. R., Berry, D., Yang, T. T., Aitken, M. D., and Teske, A. (2013). Hydrocarbon-degrading bacteria enriched by the Deepwater Horizon oil spill identified by cultivation and DNA-SIP. ISME J.l 7, 2091–2104. doi: 10.1038/Ismej.2013.98
Hanson, C. A., Fuhrman, J. A., Horner-Devine, M. C., and Martiny, J. B. H. (2012). Beyond biogeographic patterns: processes shaping the microbial landscape. Nat. Rev. Microbiol. 10, 497–506. doi: 10.1038/nrmicro2795
Hellweger, F. L., van Sebille, E., and Fredrick, N. D. (2014). Biogeographic patterns in ocean microbes emerge in a neutral agent-based model. Science 345, 1346–1349. doi: 10.1126/science.1254421
Höfle, M. G. (1984). Degradation of putrescine and cadaverine in seawater cultures by Marine-Bacteria. Appl. Environ. Microbiol. 47, 843–849.
Hunt, D. E., David, L. A., Gevers, D., Preheim, S. P., Alm, E. J., and Polz, M. F. (2008). Resource partitioning and sympatric differentiation among closely related bacterioplankton. Science 320, 1081–1085. doi: 10.1126/Science.1157890
Huston, A. L., Krieger-Brockett, B. B., and Deming, J. W. (2000). Remarkably low temperature optima for extracellular enzyme activity from Arctic bacteria and sea ice. Environ. Microbiol. 2, 383–388. doi: 10.1046/J.1462-2920.2000.00118.X
Huston, A. L., Methe, B., and Deming, J. W. (2004). Purification, characterization, and sequencing of an extracellular cold-active aminopeptidase produced by marine psychrophile Colwellia psychrerythraea strain 34H. Appl. Environ. Microbiol. 70, 3321–3328. doi: 10.1128/AEM.70.6.3321-3328.2004
Hyatt, D., Chen, G.-L., LoCascio, P., Land, M., Larimer, F., and Hauser, L. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformat. 11:119. doi: 10.1186/1471-2105-11-119
Jiao, N., Herndl, G. J., Hansell, D. A., Benner, R., Kattner, G., Wilhelm, S. W., et al. (2010). Microbial production of recalcitrant dissolved organic matter: long-term carbon storage in the global ocean. Nat. Rev. Microbiol. 8, 593–599. doi: 10.1038/nrmicro2386
Jung, S. Y., Oh, T. K., and Yoon, J. H. (2006). Colwellia aestuarii sp nov., isolated from a tidal flat sediment in Korea. Int. J. Syst. Evol. Microbiol. 56, 33–37. doi: 10.1099/Ijs.0.63920-0
Junge, K., Eicken, H., and Deming, J. W. (2003). Motility of Colwellia psychrerythraea strain 34H at subzero temperatures. Appl. Environ. Microbiol. 69, 4282–4284. doi: 10.1128/Aem.69.7.4282-4284.2003
Kanehisa, M., Sato, Y., and Morishima, K. (2015). BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731. doi: 10.1016/j.jmb.2015.11.006
Kennedy, J., Marchesi, J. R., and Dobson, A. D. W. (2008). Marine metagenomics: strategies for the discovery of novel enzymes with biotechnological applications from marine environments. Microb. Cell Fact. 7:27. doi: 10.1186/1475-2859-7-27
Kikawada, T., Saito, A., Kanamori, Y., Nakahara, Y., Iwata, K. I., Tanaka, D., et al. (2007). Trehalose transporter 1, a facilitated and high-capacity trehalose transporter, allows exogenous trehalose uptake into cells. Proc. Natl. Acad. Sci. U.S.A. 104, 11585–11590. doi: 10.1073/pnas.0702538104
Kim, Y. O., Park, S., Nam, B. H., Jung, Y. T., Kim, D. G., and Yoon, J. H. (2013). Colwellia meonggei sp nov., a novel gammaproteobacterium isolated from sea squirt Halocynthia roretzi. Antonie Van Leeuwenhoek Int. J. Gen. Mol. Microbiol. 104, 1021–1027. doi: 10.1007/S10482-013-0022-2
Konstantinidis, K. T., and Tiedje, J. M. (2005). Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. U.S.A. 102, 2567–2572. doi: 10.1073/Pnas.0409727102
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/Gr.092759.109
Lagesen, K., Hallin, P., Rødland, E. A., Stærfeldt, H.-H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Loncke, L., Mascle, J., and Parties, F. S. (2004). Mud volcanoes, gas chimneys, pockmarks and mounds in the Nile deep-sea fan (Eastern Mediterranean): geophysical evidences. Marine Petrol. Geol. 21, 669–689. doi: 10.1016/J.Marpetgeo.2004.02.004
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 0955–0964. doi: 10.1093/nar/25.5.0955
Luo, C. W., Walk, S. T., Gordon, D. M., Feldgarden, M., Tiedje, J. M., and Konstantinidis, K. T. (2011). Genome sequencing of environmental Escherichia coli expands understanding of the ecology and speciation of the model bacterial species. Proc. Natl. Acad. Sci. U.S.A. 108, 7200–7205. doi: 10.1073/Pnas.1015622108
Malmstrom, R. R., Rodrigue, S., Huang, K. H., Kelly, L., Kern, S. E., Thompson, A., et al. (2013). Ecology of uncultured Prochlorococcus clades revealed through single-cell genomics and biogeographic analysis. ISME J. 7, 184–198. doi: 10.1038/ismej.2012.89
Martiny, J. B. H., Bohannan, B. J. M., Brown, J. H., Colwell, R. K., Fuhrman, J. A., Green, J. L., et al. (2006). Microbial biogeography: putting microorganisms on the map. Nat. Rev. Microbiol. 4, 102–112. doi: 10.1038/nrmicro1341
Mason, O., Han, J., Woyke, T., and Jansson, J. (2014). Single-cell genomics reveals features of a Colwellia species that was dominant during the Deepwater Horizon oil spill. Front. Microbiol. 5:332. doi: 10.3389/fmicb.2014.00332
Mastalerz, V., de Lange, G. J., and Dahlmann, A. (2009). Differential aerobic and anaerobic oxidation of hydrocarbon gases discharged at mud volcanoes in the Nile deep-sea fan. Geochim. Cosmochim. Acta 73, 3849–3863. doi: 10.1016/J.Gca.2008.12.030
Methé, B. A., Nelson, K. E., Deming, J. W., Momen, B., Melamud, E., Zhang, X. J., et al. (2005). The psychrophilic lifestyle as revealed by the genome sequence of Colwellia psychrerythraea 34H through genomic and proteomic analyses. Proc. Natl. Acad. Sci. U.S.A. 102, 10913–10918. doi: 10.1073/Pnas.0504766102
Nogi, Y., Hosoya, S., Kato, C., and Horikoshi, K. (2004). Colwellia piezophila sp nov., a novel piezophilic species from deep-sea sediments of the Japan Trench. Int. J. Syst. Evol. Microbiol. 54, 1627–1631. doi: 10.1099/Ijs.0.03049-0
Nurk, S., Bankevich, A., Antipov, D., Gurevich, A., Korobeynikov, A., Lapidus, A., et al. (2013). Assembling Genomes and Mini-metagenomes from Highly Chimeric Reads, in Research in Computational Molecular Biology, eds M. Deng, R. Jiang, F. Sun, and X. Zhang (Berlin;Heidelberg: Springer), 158–170.
Redmond, M. C., and Valentine, D. L. (2012). Natural gas and temperature structured a microbial community response to the Deepwater Horizon oil spill. Proc. Natl. Acad. Sci. U.S.A. 109, 20292–20297. doi: 10.1073/Pnas.1108756108
Richter, M., and Rossello-Mora, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U.S.A. 106, 19126–19131. doi: 10.1073/Pnas.0906412106
Shapiro, B. J., Friedman, J., Cordero, O. X., Preheim, S. P., Timberlake, S. C., Szabó, G., et al. (2012). Population genomics of early events in the ecological differentiation of Bacteria. Science 336, 48–51. doi: 10.1126/Science.1218198
Simpson, J. T., Wong, K., Jackman, S. D., Schein, J. E., Jones, S. J. M., and Birol, I. (2009). ABySS: a parallel assembler for short read sequence data. Genome Res. 19, 1117–1123. doi: 10.1101/Gr.089532.108
Soucy, S. M., Huang, J. L., and Gogarten, J. P. (2015). Horizontal gene transfer: building the web of life. Nat. Rev. Genet. 16, 472–482. doi: 10.1038/nrg3962
Techtmann, S. M., Fortney, J. L., Ayers, K. A., Joyner, D. C., Linley, T. D., Pfiffner, S. M., et al. (2015). The Unique Chemistry of Eastern mediterranean water masses selects for distinct microbial communities by depth. PLoS ONE 10:e0120605. doi: 10.1371/journal.pone.0120605
Teske, A., Durbin, A., Ziervogel, K., Cox, C., and Arnosti, C. (2011). Microbial community composition and function in permanently cold seawater and sediments from an Arctic Fjord of Svalbard. Appl. Environ. Microbiol. 77, 2008–2018. doi: 10.1128/Aem.01507-10
Utturkar, S. M., Klingeman, D. M., Land, M. L., Schadt, C. W., Doktycz, M. J., Pelletier, D. A., et al. (2014). Evaluation and validation of de novo and hybrid assembly techniques to derive high quality genome sequences. Bioinformatics 30, 2709–2716. doi: 10.1093/bioinformatics/btu391
Vesth, T., Lagesen, K., Acar, O., and Ussery, D. (2013). CMG-Biotools, a Free Workbench for basic comparative microbial genomics. PLoS ONE 8:e60120. doi: 10.1371/journal.pone.0060120
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Yamauchi, S., Ueda, Y., Matsumoto, M., Inoue, U., and Hayashi, H. (2012). Distinct features of protein folding by the GroEL system from a psychrophilic bacterium, Colwellia psychrerythraea 34H. Extremophiles 16, 871–882. doi: 10.1007/S00792-012-0483-7
Yu, Y., Li, H. R., and Zeng, Y. X. (2011). Colwellia chukchiensis sp. nov., a psychrotolerant bacterium isolated from the Arctic Ocean. Int. J. Syst. Evol. Microbiol. 61, 850–853. doi: 10.1099/Ijs.0.022111-0
Keywords: genomic diversity, comparative genomics, psychrophile, phenotype, evolution, marine ecology
Citation: Techtmann SM, Fitzgerald KS, Stelling SC, Joyner DC, Uttukar SM, Harris AP, Alshibli NK, Brown SD and Hazen TC (2016) Colwellia psychrerythraea Strains from Distant Deep Sea Basins Show Adaptation to Local Conditions. Front. Environ. Sci. 4:33. doi: 10.3389/fenvs.2016.00033
Received: 04 March 2016; Accepted: 25 April 2016;
Published: 09 May 2016.
Edited by:
Naser A. Anjum, University of Aveiro, PortugalReviewed by:
Bernardo Duarte, Marine and Environmental Sciences Centre, PortugalCopyright © 2016 Techtmann, Fitzgerald, Stelling, Joyner, Uttukar, Harris, Alshibli, Brown and Hazen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephen M. Techtmann, c210ZWNodG1AbXR1LmVkdQ==;
Terry C. Hazen, dGNoYXplbkB1dGsuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.