 Lorenz Schwab1,2*
Lorenz Schwab1,2* Florian M. Rothe1
Florian M. Rothe1 David S. McLagan3,4,5Alexandra Alten3Stephan M. Kraemer1Harald Biester3
David S. McLagan3,4,5Alexandra Alten3Stephan M. Kraemer1Harald Biester3 Jan G. Wiederhold1,6
Jan G. Wiederhold1,6- 1Department for Environmental Geosciences, Centre for Microbiology and Environmental System Science, University of Vienna, Vienna, Austria
- 2Doctoral School in Microbiology and Environmental Science, University of Vienna, Vienna, Austria
- 3Institute of Geoecology, Technical University of Braunschweig, Braunschweig, Germany
- 4Department of Geological Sciences and and Geological Engineering, Queen’s University, Kingston, ON, Canada
- 5School of Environmental Studies, Queen’s University, Kingston, ON, Canada
- 6German Federal Institute of Hydrology (BfG), Qualitative Hydrology, Koblenz, Germany
Mercury (Hg) release from contaminated legacy sites is a large contributor to riverine ecosystems and can represent a significant local and regional environmental issue even long after the initial site contamination. Understanding processes of in-stream species transformation is therefore important to assess the fate and bioavailability of the released Hg. In this study, we investigated in-stream Hg transformation processes with analyses of Hg binding forms and Hg stable isotopes. Stream sediments were collected downstream of a former kyanization facility (Black Forest, SW Germany), where highly soluble Hg(II)-chloride (HgCl2) was used as an anti-fouling agent to treat timber. Exfiltration of partly anoxic, contaminated groundwater with Hg concentrations of up to 700 μg L−1 into the adjacent Gutach stream is the main source of Hg to sediments. Total Hg concentrations in the stream bottom sediments (<2 mm) ranged from background values of 6.3 µg kg−1 upstream of the contaminated site to 77 mg kg−1 near the location of exfiltration of contaminated groundwater. A five-step sequential extraction procedure and Hg pyrolytic thermal desorption (PTD) analyses indicated changes in Hg binding forms in the sediments along the flow path towards a higher proportion of organically bound Hg. A large shift towards negative δ202Hg values was observed downstream of the contaminated site (change of ≈2‰) along with a minor offset in mass-independent fractionation. Binary mixing models based on Hg isotope ratios using one industrial and different natural background endmembers were tested to estimate their respective contribution of Hg to the sediments but failed to produce plausible allocations. Based on the observed changes in isotopic composition, total Hg concentrations and Hg binding forms, we propose that the large extent of fractionation observed in downstream sediments is the result of a combination of kinetic isotope effects during sorption, redistribution of Hg within the sediment and the preferential transport of Hg associated with the sediment fine fraction. These results highlight the importance of transformation processes when assessing the sources and fate of Hg in environmental systems and show limitations of using simple mixing models based on Hg stable isotopes.
1 Introduction
Mercury (Hg) is a global pollutant emitted by natural processes and human activities (Selin, 2009; Streets et al., 2019; UN Environment, 2019). The use of Hg in industrial processes has created local Hg contamination hotspots in different ecosystems around the world (Eckley et al., 2020), which continue to represent a significant source of Hg to the atmosphere and aquatic systems (Kocman et al., 2013). This anthropogenic legacy Hg, released and deposited previously, is recycled through re-emission and links past human activities to current environmental Hg cycling (Cooke et al., 2020). Once released, the main concern with respect to human exposure is Hg transformation into methylmercury, which bioaccumulates in aquatic organisms (Ullrich et al., 2001; Branfireun et al., 2020; Bravo and Cosio, 2020) and presents the main Hg uptake pathway for humans through fish consumption (Lavoie et al., 2018). Rivers represent an important long-range transport pathway, connecting contaminated legacy sites to estuaries and coastal zones (Horvat et al., 1999; Araujo et al., 2017) and are the largest source of Hg to coastal oceans (Liu et al., 2021). Understanding Hg transport from the release of Hg in the stream along the flow path to the river mouth is of critical interest.
The fate of Hg in environmental compartments such as river systems largely depends on its chemical speciation (Liu et al., 2011a). The majority of Hg released to the environment is in the form of inorganic Hg(0) or Hg(II) compounds (Eckley et al., 2020). Transformation processes in aquatic systems include the association/dissociation with numerous organic and inorganic ligands, sorption/desorption, precipitation/dissolution, oxidation/reduction, methylation/demethylation and volatilization/deposition to/from the atmosphere (Liu et al., 2011b). In addition to these biogeochemical transformations, physical transport mechanisms are of special interest in the case of fluvial Hg transport, which in most systems occurs predominantly in particulate form (Amos et al., 2014), because discharge velocity and energy control particle entrainment, suspension and ultimately transport (Boszke et al., 2004; Kocman et al., 2011; Kelly and Rudd, 2018). The partitioning of Hg at the particle-solution interface also affects its availability for methylating microorganisms and biological uptake (Hsu-Kim et al., 2013; Zhang et al., 2019).
Despite all the recent advances in understanding Hg biogeochemical cycling in the environment, many questions regarding Hg transformation processes and mechanisms remain unanswered (Gustin et al., 2020). The determination of the exact speciation of Hg in environmental samples remains challenging, leaving the determination of operationally defined pools as the most common method to assess the mobility, bioavailability and changes in Hg binding forms. There are numerous sequential extraction protocols (SEP) aiming to separate Hg into specified pools (Issaro et al., 2009; Fernández-Martínez and Rucandio, 2013; Reis et al., 2016). Other than chemical extractions, pyrolytic thermal desorption (PTD) analyses use the release of different Hg compounds at specific temperatures and are an effective method for the identification of the presence of Hg(0) (released at low temperatures) and Hg2+ sulfides and sulfates released at high temperatures) (Biester and Scholz, 1997; Reis et al., 2015; Rumayor et al., 2016).
Mercury stable isotope ratios are a potential tracer for sources and pathways of Hg in the environment and have been applied as a tool in an increasing number of field studies for source attribution and transport processes in stream systems contaminated by different sources such as former Hg mines (e.g., Foucher et al., 2013; Smith et al., 2015a; Baptista-Salazar et al., 2018; Pribil et al., 2020), gold mining or processing activities (e.g., Donovan et al., 2016; Marshall et al., 2018; Goix et al., 2019), chlor-alkali or other industrial activities (e.g., Donovan et al., 2014; Washburn et al., 2017; Demers et al., 2018; Grigg et al., 2018; Reinfelder and Janssen, 2019; Crowther et al., 2021; Janssen et al., 2021). In addition to mass-dependent fractionation (MDF), Hg isotopes also exhibit mass-independent fractionation (MIF) related to the magnetic isotope effect (MIE) and the nuclear volume effect (NVE, also known as nuclear field shift effect) (Blum and Bergquist, 2007; Bergquist and Blum, 2009). Fewer processes have been demonstrated to induce MIF (e.g., photochemical redox transformations) and it is therefore considered to be a more conservative tracer than MDF and might be conserved during accumulation in soils or plants (Bergquist and Blum, 2009; Sonke, 2011).
Simple binary mixing models based on δ202Hg isotope signatures and inverse total Hg concentrations (1/THg) have been applied to estimate source contributions of two different sources or an industrial source versus background contribution to sediments and soils in numerous studies (Foucher et al., 2009; Feng et al., 2010; Feng et al., 2019; Ma et al., 2013; Gray et al., 2015; Guédron et al., 2016; Yin et al., 2016; Washburn et al., 2017; Washburn et al., 2018; Woerndle et al., 2018; Zhang et al., 2018; Zhang et al., 2020; Zhu et al., 2018; Goix et al., 2019; Reinfelder and Janssen, 2019; Pribil et al., 2020; Tsui et al., 2020). Mixing models were also extended to include the relationship between MDF vs. MIF instead of MDF vs. 1/THg (Liu et al., 2011c; Lepak et al., 2015; Chen et al., 2016; Meng et al., 2019; Janssen et al., 2021) and in some studies the two-endmember mixing models were extended to include three or more endmembers (e.g., Bartov et al., 2013; Yin et al., 2018; Jung et al., 2021; Jung et al., 2022; Chen et al., 2022) or additional parameters such as carbon stable isotopes (Campeau et al., 2022). Binary mixing models require the precise definition of two distinct endmembers (e.g., contaminant and natural background) with a sufficiently different isotopic composition (Foucher et al., 2009). These mixing models are based on a linear relationship between δ202Hg and 1/THg and assume that the isotopic composition of samples between the endmembers is the result of conservative mixing of these two distinct endmembers.
Compared to source tracing, the application of Hg isotopes for process tracing in rivers is more limited (Washburn et al., 2017; Demers et al., 2018; Crowther et al., 2021; McLagan et al., 2022a). Mercury transformation processes in river systems have also been investigated using stable Hg isotope signatures, for example, partitioning among dissolved and particulate phases (Washburn et al., 2017; Demers et al., 2018), methylation of inorganic Hg (Donovan et al., 2016) or photodegradation of methylmercury (Tsui et al., 2013).
Here we assessed Hg stable isotopes as a tool to trace Hg sources, understand Hg transport processes and identify changes in Hg binding forms in contaminated freshwater environments. For this purpose, we investigated stream sediments contaminated by continuous release of Hg from a former industrial site where timber was treated with a 0.67% solution of highly soluble mercuric chloride (HgCl2) for preservation in a process called kyanization (Troschel, 1916; Nowak, 1952; Kallipoliti, 2010). The combination of analyses of Hg binding forms employing a SEP and PTD with Hg stable isotope analysis is expected to provide additional information about the processes governing the biogeochemical changes and the transport mechanism in the stream sediments. With our study we address the question whether the highly mobile HgCl2 originally released at the contamination source changes its binding forms during the transport along the stream and will be retained in sediments. Further, we investigate if this change in binding forms is reflected in the different isotopic compositions of Hg in sediment samples and if differences in Hg isotope ratios in bulk sediments compared to the fine fraction can provide additional information about the dominant transport mechanisms and the changes in binding forms. The objective is to assess in-stream processes that can explain the observed shifts in Hg binding forms and isotopic composition. This contamination case further provides an excellent opportunity to investigate the potential fractionation of Hg isotopes in a stream system and an assessment of the suitability of Hg isotopes and endmember mixing models as a source tracing tool.
2 Materials and methods
2.1 Site description
All stream sediment and stream water samples were collected in the vicinity of a former wood impregnation facility located in the Black Forest (South-West Germany, 830 m a.s.l.; site A in McLagan et al., 2022b, site C in Richard et al., 2016b). From the late 19th century until the early 1960s, HgCl2 was used in the kyanization process as a wood preservation agent at this site. Extensive contamination of soil and groundwater resulted from spillages and drying of treated wood on unsealed ground (Heckmann, 2002; Eisele, 2004). The facility is divided into northern and southern parts by the Gutach stream (Figure 1), which was dammed by a small barrage in 1968/69, creating a small artificial pond (Okalsee) at the western end of the site (Eisele, 2004) (photos provided in Supplementary Section S1). This did not only affect the sediment transport from the upstream part of Gutach stream, which features a natural brownwater lake “Titisee” a few km upstream, but also the location of the streambed was slightly altered at that time. The northern part of the former industrial area has been developed into a commercial shopping area with stores and parking lots in 2002/03 (Eisele, 2004). The southern part is today occupied by several smaller company buildings, but also exhibits larger open areas with unsealed land surfaces. There are two vertically arranged aquifers with mostly south-eastern flow direction. The upper groundwater body is in exchange with the stream via exfiltration of Hg contaminated groundwater (Heckmann, 2002; Schuler, 2012).
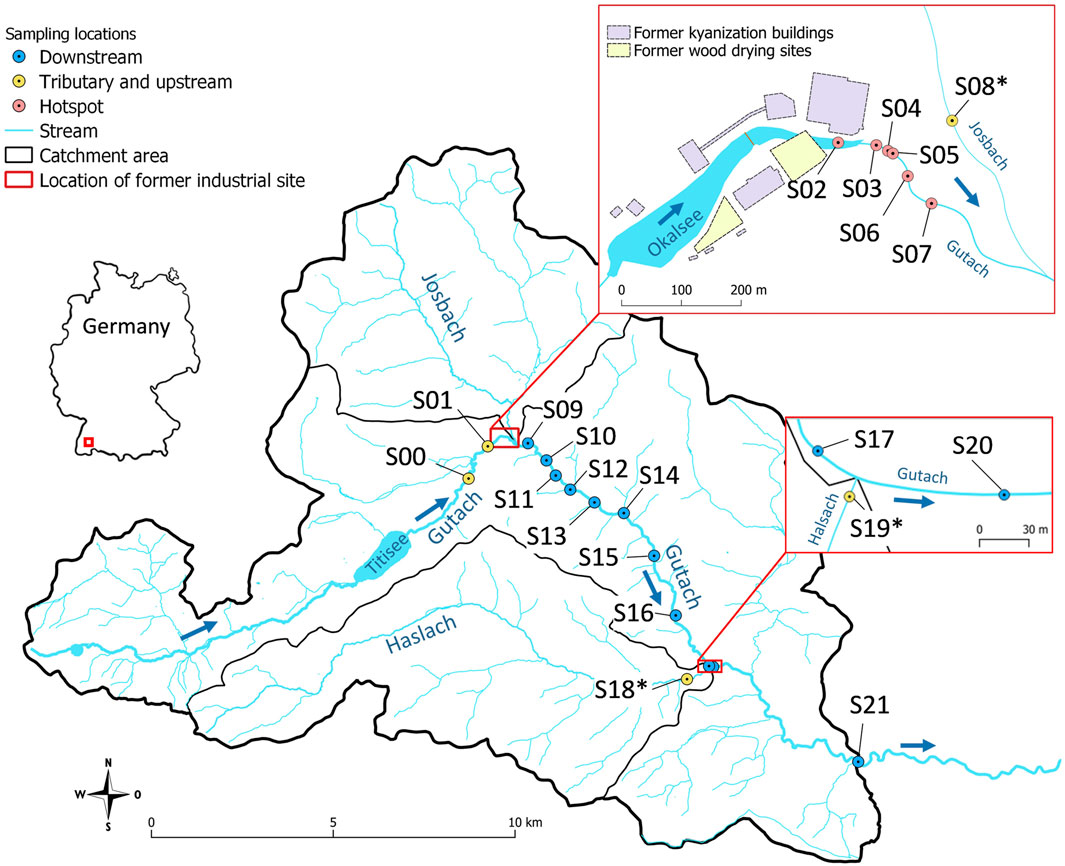
FIGURE 1. Location of the sampling sites. Samples from tributary creeks are denoted with an asterisk.
The (hydro)geology of the area is dominated by Quaternary fluvial sediments overlaying Paleozoic granite basement (LGRB, 2021). Between lake Titisee and the former kyanization site, there is only a small altitude gradient and Gutach stream is flowing at a slow velocity. Shortly after the confluence with the Josbach stream (see Figure 1) the terrain gets steeper and the flow velocities increase resulting in a streambed consisting of coarser gravel and stones with only little fine-grained particles settling (Gonser and Schwoerbel, 1985; Schuler, 2012). After the confluence with the Haslach stream the name changes to Wutach. The river Wutach is a tributary of the river Rhine, which ultimately drains to the North Sea. The mean discharge of the Wutach is 6 m3 s−1, but due to the close relation to rainfall and snowmelt events, it can increase to >100 m3 s−1 during high discharge events (see precipitation and discharge data in Supplementary Section S2), where large amounts of sediments can be mobilized (Ortlepp, 1997).
2.2 Sampling and sample preparation
Sediment samples from Gutach stream were collected at and downstream of the former industrial site (n = 17, see Figure 1) in 2018 and 2019. Two additional samples were collected upstream of the contaminated site (S00 and S01) to assess background Hg concentration and isotope signatures in the Gutach sediment. Additionally, sediment samples from the two main tributary streams Josbach (S08*) and Haslach (S18∗ and S19∗) were collected to assess their potential influence. The location of all sampling points is displayed in Figure 1. Sampling campaigns were carried out in months with comparably low precipitation and discharge (Supplementary Figures S2.1, S2.2). All sampling equipment was washed thoroughly and rinsed with stream water at the respective sampling location. Sediment samples were taken from the uppermost <5 cm with a hand shovel. Multiple grabs randomly distributed across the streambed were combined at each location to optimize representative sampling of the heterogeneous streambed and until a sufficient amount of sediment was collected (100–200 g, <2 mm). Samples were homogenized using a stainless-steel spatula and directly sieved to <2 mm in the field and filled into 180 ml polypropylene (PP) containers for transport to the laboratory. Stream water samples were filtered through 0.45 µm cellulose acetate (CA) filters into 50 ml PP vials and stabilized with 1% (v/v) BrCl (0.2 M BrCl in concentrated HCl, prepared according to Bloom et al. (2003). Electrical conductivity, pH and temperature of the stream water were measured in the field with respective electrodes (Supplementary Table S6). Sediment samples were dried at 60°C for 48 h. Samples used for PTD were not dried to avoid loss of Hg(0). An aliquot of the dried samples was dry-sieved to <125 µm to compare Hg concentrations and isotope ratios of the fine fraction with bulk sediments and assess its role for transport and transformation processes. Relative mass proportions were calculated using the mass of the <125 µm fraction in relation to the total mass of sieved aliquots. An aliquot of the <2 mm fraction was milled with a vibrating ball mill for 30 s at 30 vibrations per second (Retsch MM 400).
2.3 Analyses
Total carbon (C), hydrogen (H), nitrogen (N) and sulfur (S) concentrations were determined using an elemental analyzer (Elementar, VarioMacro). Organic and inorganic carbon contents of the sediment samples were determined using a multiphase carbon analyzer (LECO RC612, Leco Corporation). For this analysis, an aliquot of the milled <2 mm fraction was heated from 105°C to 1,000°C at a rate of 5°C min−1. A peak separator was set at 550°C to distinguish between organic and inorganic carbon. Because the Leco analyzer contains parts made of gold, which could be damaged by the introduction of samples with elevated Hg contents, the total C of samples with Hg concentrations >1 mg kg−1 were analyzed by combustion and infra-red detection using a DIMA 1000NT (Dimatec). The organic carbon content of these samples was determined after removing the inorganic carbon fraction with concentrated HCl by the “carbonate-bomb” method (Müller and Gastner, 1971). Characterization of organic carbon was performed by Fourier-transform infrared spectroscopy (FTIR; Cary 630 FTIR Spectrometer, Agilent Technologies) on selected samples (details see Supplementary Section S8). Major elements were analyzed by ICP-OES (ICP-OES 5110, Agilent Technologies) on aqua regia digests (HNO3:HCl (1:3)). Samples were digested on a hotplate at 85°C for 2 h after the addition of 12 ml aqua regia to 0.5 g of sediment in loosely capped 50 ml PP vials. Aqua regia was refilled to 12 ml and digested for another hour, then filled to 50 ml with ultrapure water and filtered through 0.45 µm CA filters. Elemental analysis was further conducted on selected samples by energy dispersive X-ray fluorescence (XRF) spectrometry on wax pellets of the milled <2 mm fraction. The mineralogical composition of the milled bulk stream sediments was examined using X-ray diffraction and the COD database (XRD, Rigaku MiniFlex 600).
For THg analysis aqua regia extracts with BrCl (8 ml of conc. HCl, 3 ml of conc. HNO3 and 1 ml of conc. BrCl) of both <2 mm and <125 µm sediment fractions were analyzed for total Hg and, if concentrations allowed, also for Hg stable isotopes. Extractions were carried out in 50 ml PP centrifuge vials covered with perforated parafilm. Digests were run overnight (20–24 h) on a lateral shaker at 170 rpm. Samples were then diluted with 36 ml of ultrapure water (18.2 MΩ cm), resuspended and centrifuged at 3,000 rcf for 20 min (Herolab HiCen 21°C). After centrifugation, the supernatants were passed through 0.45 µm CA filters.
A five-step sequential extraction protocol (SEP) was carried out on selected sediment samples (<2 mm, not milled) based on the method described in Bloom et al. (2003) (for details see Supplementary Section S3). This method separates Hg into operationally defined fractions using the following extraction agents: deionized water (F1), 0.01 M HCl/0.1 M CH3COOH (F2), 1 M KOH (F3), 12 M HNO3 (F4), aqua regia (F5). Major elements in extracts were analyzed by ICP-OES (Supplementary Figure S3.1). The pH of sediment samples was measured for selected samples in the F1 extract (Supplementary Table S7). Pyrolytic thermal desorption (PTD) analysis was applied to characterize Hg binding forms similar to the procedure described in Biester and Scholz (1997). Aliquots of the untreated sediment samples were continuously heated to 650°C at a rate of 1°C per second in an N2 gas flow and the Hg release was measured as a function of temperature using atomic absorption spectrometry. The obtained release curves were compared to the characteristic release curves of Hg reference compounds (for details see Supplementary Section S4). Although both methods provide valuable and often complementary information about the nature of the dominant Hg compounds and their binding to different matrix components in the investigated sediments, it is important to realize that no individual chemical Hg species can be separated and identified in most cases.
Total Hg concentrations were determined on a direct mercury analyzer (DMA-80, Milestone Inc.) using thermal decomposition, amalgamation and subsequent atomic absorption spectrometry on the milled sample aliquots following EPA method 7473 (EPA, 2000). High Hg concentration samples from the hotspot area (S05-S07) were not included in this analysis because impractically low sample masses would have been required to remain within the calibration range of the instrument. Mercury concentrations of aqua regia and SEP extracts were analyzed using cold vapor atomic absorption/fluorescence spectrometry (CV-AAS/AFS, DMA-80L, Milestone Inc.) after pre-reduction of the samples with hydroxylamine-hydrochloride solution (NH2OH·HCl) and online reduction of Hg2+ to Hg(0) by SnCl2 following EPA method 1631 (EPA, 2002). Certified reference material (ERM CC-018) was extracted in parallel with samples as quality control and yielded recoveries of 93.3% ± 8.6% (SD, n = 4) for aqua regia digests and 87.4% ± 8.4% (SD, n = 4) for the sum of the five extracts in the sequential extraction protocol. All reagent blanks were below the limit of quantification. Stream water samples were analyzed using cold vapor atomic fluorescence spectrometry (CV-AFS, Mercur Analytic Jena) except for additional samples collected at later sampling events which were analyzed using the DMA-80L. Water samples are labeled in the same order as sediments (W00-W21) and were collected at the same location.
For isotope analysis, sediment samples with low Hg concentrations were pre-concentrated using combustion and trap with the DMA-80 direct mercury analyzer previously tested and successfully applied in our laboratory (McLagan et al., 2022b; McLagan et al., 2022a). Samples were weighed into sample boats (0.2–17 g, max. 1 g per boat), combusted and the released Hg purged into an oxidizing trap consisting of 5 ml 40% (v/v) inverse aqua regia (HNO3:BrCl (3:1)) as described in Janssen et al. (2019). The efficacy of the method was tested by preparing different reference materials and calculating recoveries with 1SD variation (BCR-482: 103% ± 12%, n = 13; CC-141: 95% ± 4%, n = 16; NIST-3133: 102% ± 4%, n = 12). Mercury isotope ratios of aqua regia digests and selected sequential extracts were determined with a Nu Plasma II multi-collector inductively coupled plasma mass spectrometer (MC-ICP-MS, Nu Instruments) connected to an HGX-200 cold vapor system (Teledyne Cetac) for Hg introduction and an Aridus II desolvating nebulizer (Teledyne Cetac) for Tl introduction. Mass bias and instrument drift were corrected using NIST-3133 standard-sample bracketing and element doping with NIST-997 Tl standard. Measurement sessions were conducted at concentrations of either 2.5 µg L−1, 5 µg L−1 or 10 µg L−1 Hg with concentration-matched bracketing standards. Standards and samples were diluted in a 1% (v/v) BrCl matrix to an acid strength of <10% (v/v). Isotope data are reported as δ-values in permille relative to NIST-3133 calculated according to Eq. 1:
Mass-independent fractionation (MIF) is reported as deviation from the theoretically predicted mass-dependent fractionation (MDF) using the following Eq. 2:
The βxxx values of 199Hg, 200Hg, 201Hg and 204Hg isotopes are 0.2520, 0.5024, 0.7520, and 1.4930, respectively (Blum and Bergquist, 2007). Assessment of accuracy and precision of the isotope analysis was done by regular measurements of the secondary standard “ETH Fluka”. Results were consistent with previous analyses of different laboratories (Smith et al., 2015a; Jiskra et al., 2017; Obrist et al., 2017; Grigg et al., 2018; Brocza et al., 2019; Goix et al., 2019; McLagan et al., 2022b). The overall reproducibility (2SD, n = 69) was ±0.15‰ for δ202Hg and ±0.06‰ for Δ199Hg and Δ201Hg, but the standard deviations (2SD) reported for individual sample measurements correspond to the respective session reproducibility of “ETH Fluka” (overview in Supplementary Table S5.1).
3 Results
3.1 Stream water and sediment characterization
Background Hg concentrations, measured in Gutach stream before the contaminated area and the tributary streams, were below the detection limit except for W19* (0.6 ng L−1) consistent with previously reported low background Hg concentrations between 1.1 and 4.1 ng L−1 (Bischoff et al., 2015). Close to the former kyanization facility, between the artificial pond and the confluence with the Josbach stream (hereafter called “hotspot samples,” see Figure 1 inset), the water samples show elevated Hg concentrations reaching up to 835.4 ng L−1 at W05 (Supplementary Table S6), indicating exfiltration of contaminated groundwater (for details see Supplementary Section S6). This is supported by elevated THg concentrations in groundwater samples from a well close to the location where groundwater was exfiltrating ranging from 78 to 555 µg L−1 in five measurement campaigns (McLagan et al., 2022b; cf. sample WA10). “Hotspot” water samples were also characterized by elevated electrical conductivity (EC) and higher dissolved Fe and Mn concentrations than both upstream and downstream samples. After the confluence with Josbach stream (hereafter called “downstream samples”) the dissolved Hg concentrations remain elevated (14.8–50.8 ng L−1, Supplementary Table S6) up to and including the most distant sample (W21) approximately 16.5 km along the Gutach-Wutach channel. Stream water field parameters are summarized in Supplementary Table S6.
The pH of sediment samples ranged from 6.0 to 7.0 (Supplementary Table S7). The fine fraction (<125 µm) made up only a small mass proportion of 0.3% (S20) to 7.5% (S05) of the bulk sample mass (Supplementary Table S7). The peak identification of XRD patterns showed that the mineralogy is dominated by quartz and plagioclase feldspars such as albite and anorthite. There is some variation in the intensity of the major peaks and the occurrence of minor peaks between the sediment samples indicating a different relative composition and abundance of these minerals, but no clear distinction between the Gutach stream sediments and sediments from tributaries could be made based on mineralogical composition (XRD patterns are provided in Supplementary Section S9). Elemental composition results from ICP-OES and XRF measurements show that some of the samples from near the former industrial facility exhibit increased concentrations of contaminants related to former usage and release from the site (Pb, Cr, Zn) and higher concentrations of Fe, Mn and Ca (Supplementary Tables S10.1, S10.2). The XRF data also shows higher concentrations of Cl, S and P concentrations for the most contaminated samples. For Fe and Mn this is likely caused by the exfiltration of anoxic groundwater and subsequent oxidation and precipitation and settling of Fe and Mn. However, XRD patterns do not allow the identification of Fe or Mn minerals as a component of the sediment samples, either because of their poorly-crystalline structure or their low relative abundance. The FTIR spectra showed subtle differences in absorbance among the selected samples indicating a slightly higher proportion of organic matter in sediments S05 and S08* (Supplementary Figures S8.1, S8.2). The total carbon (TC) content of the bulk sediments in Gutach stream and the tributaries ranged from 0.16% to 1.02% (details see Supplementary Table S7 and Supplementary Figure S7). Most of the carbon was present as organic carbon (86% ± 8%) (Supplementary Table S7). Overall, the differences in the elemental and mineralogical composition were not very pronounced and did not allow an estimate of the sediment contribution from each tributary stream. An overview of the sediment characterization is presented in Supplementary Section S7.
3.2 Hg concentrations and binding forms
Total mercury (THg) concentrations in bulk sediment and fine fraction (<125 µm) samples varied strongly along the investigated section of Gutach stream. Bulk sediments from upstream of the former wood treatment facility (S00 and S01), which can be considered as natural background, had low THg concentrations in the bulk (6.32 µg kg−1 and 19.3 µg kg−1, respectively) and fine (115 µg kg−1 and 74.9 µg kg−1, respectively) fractions (Figure 2). The hotspot samples (S02-S07) are impacted by the contamination case and showed highly elevated THg concentrations ranging from 4.37 mg kg−1 to 77.0 mg kg−1 in bulk sediments (S02–S07). For these samples, the fine fraction also had much higher THg concentrations of up to 340 mg kg−1 (S05). The downstream samples showed a higher Hg concentration (range from 267 to 726 µg kg−1) than the background values of the Gutach or the tributaries indicating an influence of the Hg contamination case as far as 16.5 km downstream of the source. Both the Josbach and Haslach streams had similarly low Hg concentrations as the uncontaminated upstream Gutach stream in both fractions. One of the samples from the Haslach stream however had elevated THg concentrations [S19*: 296 µg kg−1 (<2 mm) and 1.19 mg kg−1 (<125 µm)]. Sample S19* was collected near the confluence with the Gutach stream and considering the differences in Hg concentrations between S18* and S19* an influence or mixture of contaminated sediments cannot be excluded, especially during high discharge events. The fine fraction had systematically higher Hg concentrations in all investigated samples by a factor of 4.4–21.7 in comparison to the respective coarse fraction (≥125 µm–<2 mm). Most of the total Hg was nonetheless associated with the coarse fraction (61%–97%) because the fine fraction made up only a small mass proportion of the bulk sample.
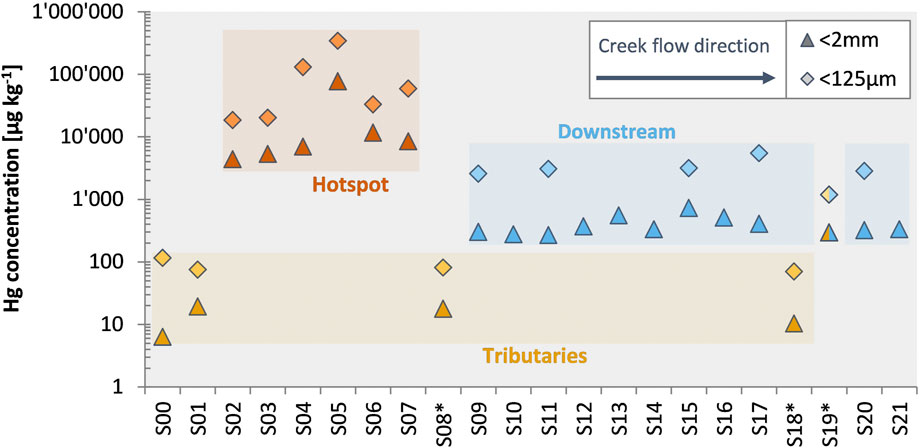
FIGURE 2. Total mercury (THg) concentrations in bulk (<2 mm) and fine (<125 µm) fractions of the sediment samples. Samples are displayed in the flow direction from S00 to S21. Tributary creeks are marked with an asterisk. Sample S19* was collected only about 10 m upstream of the confluence with the Gutach creek and might be affected by the Gutach during high discharge events. Error bars are smaller than data symbols. Accuracy and precision of THg measurements was assessed by replicate analysis of reference materials: NIST-3133 (recovery: 103% ± 3%, 1SD, n = 108) for DMA-80L; ERM CC-141 (recovery: 96% ± 6%, 1SD, n = 20) and NIST-2711 (recovery: 105% ± 7%, 1SD, n = 8) for DMA-80.
Both SEP and PTD of bulk sediments showed a distinct shift of the Hg binding form of hotspot sediments directly affected by exfiltrating groundwater compared to sediments from the tributaries and the downstream sediments. The PTD release curve of hotspot samples started at 180°C with a peak maximum of around 250°C while the release curve for downstream sediments shifted to higher temperatures with a start at 250°C and a peak maximum at 300°C (Figure 3B). None of the samples showed any Hg(0) as would be indicated by a low release temperature of around 100°C or lower. Although there are a large number of potential Hg2+ species with maximum release peaks between 200 and 300°C, including HgCl2, metacinnabar (β-HgS), Hg adsorbed to Fe(OH)3 as well as Hg bound to organic matter and humic acids (Biester and Scholz, 1997; Rumayor et al., 2016; McLagan et al., 2022b), the consistent shift in peak maxima gives further weight to the interpretation of a shift in binding forms in downstream sediments (Supplementary Figures S4.1–S4.3). The presence of metacinnabar should be indicated by a second peak shoulder or a peak at higher temperatures, which was not observed in the samples. There was a slight tailing in the release curve of S02 which could be attributed to sulfidic Hg, but slight differences in release curves can also be caused by differences in the sample matrix (e.g., grain size) and Hg concentrations (Biester and Scholz, 1997; Baptista-Salazar et al., 2017; Petranich et al., 2022). Generally, Hg associated with organic matter (humic acid in case of the standard) has a release temperature that is slightly higher than Hg associated with Fe or other “matrix-bound” Hg forms (around 300°C, see Supplementary Figure S4.3) and the observed shift can be interpreted as a change in Hg binding form towards a higher proportion of organic matter bound Hg in downstream sediments (Biester et al., 2000; Baptista-Salazar et al., 2018).
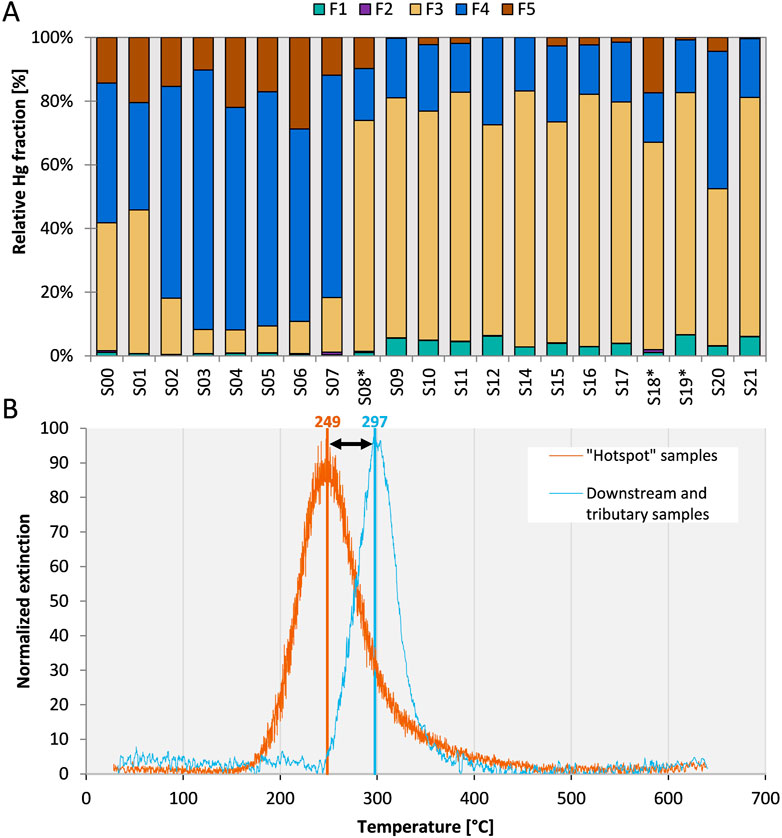
FIGURE 3. (A) Results of the SEP displayed as Hg pools normalized to the sum of extracts (sum of extracts = 100%). Samples from tributary creeks are marked with an asterisk. The performance of the SEP was assessed with repeated extractions of the reference material ERM CC-018 (F1: 0.13% ± 0.05%; F2: 0.02% ± 0.01%; F3: 3.9% ± 1.1%; F4: 77.9% ± 4.7%; F5: 3.6% ± 3.6%, Sum of extracts: 87.8% ± 8.4%. n = 5). (B) Moving average of PTD release peaks of sediments from “hotspot” section (S02-S07, peak centers ranging from 232 to 257°C) and downstream plus tributary creek sediments (peak centers ranging from 292 to 304°C). Signals were normalized to the maximum extinction of individual samples. See Supplementary Table S4 for peaks of individual samples.
For hotspot samples (S02–S07), most of the Hg was extracted in the F4 step (60%–80%) targeting strongly complexed Hg (Figure 3A) The relative proportion of the F5 pool (residual Hg, HgS) decreased from 10%–22% in hotspot samples to <5% after the confluence with Josbach stream, suggesting a minor role of sulfide-bound Hg in these sediments. For the hotspot samples, approximately 10% of Hg was extracted in the F3 fraction, which is associated with Hg bound to organic matter. This pool had a much higher relative abundance in the samples after the confluence with Josbach stream (66%–80%) and to a slightly lower degree in the uncontaminated Gutach. In absolute terms, the pool of Hg in the F3 pool was larger in the hotspot samples compared to the downstream samples, despite the lower relative abundance. Especially with samples that are not pure compounds but more complex mixtures, several issues can influence the results of SEP (Kim et al., 2003; Hall et al., 2005; Hall and Pelchat, 2005) and resorption of Hg can cause the release of more labile Hg in later extraction steps. In the case of the hotspot samples, the concentrations are high and incomplete removal of organically bound Hg or resorption of Hg during the F3 step causing a later extraction in step F4 cannot be completely ruled out. Even though the sediment composition has a large effect on Hg adsorption (Pelcová et al., 2010), the general sediment characterization (elemental and mineralogical composition) did not indicate clear shifts in the sediment composition between the sampling locations. Differences in organic matter content can also not explain the shift towards a higher proportion of organically bound Hg. There is only a weak relationship between TOC content and absolute Hg extracted in F3 (p = 0.06, R2 = 0.235) and no relationship between TOC content and the relative Hg fraction extracted in F3 (p = 0.42, R2 = 0.047) (Supplementary Figure S3.3). With general sediment characterization and TOC having no apparent relationship to Hg binding forms, we measured Hg isotope ratios to further investigate the reasons for the observed changes in Hg binding forms.
3.3 Hg isotope composition of bulk and fine sediment fractions
Along the investigated section of Gutach stream, large variations in the isotopic composition of Hg in sediments were observed in both bulk and fine fraction samples, with a notable shift of δ202Hg values towards more negative values in downstream sediments (Figure 4A). Gutach stream samples from upstream with very low Hg concentrations can be used to approximate the background Hg isotope signature of uncontaminated Gutach stream sediment. Both samples from upstream of the site had highly negative δ202Hg values of −2.27‰ ± 0.11‰ (S00) and −1.84‰ ± 0.11‰ (S01). The background sample from Josbach stream had a lighter isotopic composition (S08*; −2.91‰ ± 0.11‰) than Gutach upstream samples while Haslach stream had an isotopic composition that is distinct from the Gutach and Josbach streams. Like THg, the two samples of the Haslach stream were also isotopically different. The sample from further up the gorge (S18*) had a less negative δ202Hg value (−1.27‰ ± 0.11‰) compared to the sample close to the confluence (S19*; −3.12‰ ± 0.11‰). As mentioned earlier, S19* was likely influenced by contaminated Gutach sediments and water and the background of Haslach stream is better represented by S18*. The bulk isotope signature of the sediment samples from the hotspot section (S02-S07) had a δ202Hg value of −0.31‰ ± 0.37‰ (1SD, n = 6), which is close to values in the groundwater (McLagan et al., 2022b). Exfiltration of contaminated groundwater is expected to be the major source of Hg in Gutach stream and the analysis of groundwater from the most contaminated wells at the former kyanization site showed mean δ202Hg values of −0.20‰ ± 0.07‰ and Δ199Hg values of −0.07‰ ± 0.03‰ indicated little to no mass-independent fractionation in the aquifer [1SD, wells WA7 and WA10a in McLagan et al. (2022b)]. The isotopic compositions of contaminated groundwater and hotspot sediments were remarkably similar, indicating that exfiltration is likely the dominant source of Hg in these samples and an influence of contaminated surface runoff and erosion of contaminated soil from the site is of minor importance and likely restricted to heavy rainfall events. For the downstream sediments a shift towards remarkably light δ202Hg values of −2.35‰ ± 0.37‰ (1SD, n = 12) was observed. The isotopic composition of the fine fraction showed an offset with more positive δ202Hg values compared to the bulk fraction (in 13 out of 16 samples; Figure 4A). The offset was most pronounced in the hotspot and the tributary samples, whereas the downstream samples exhibited no clear δ202Hg difference between the size fractions. Apart from this offset, the isotopic composition of the fine fraction followed the same general trends and changes observed for the bulk sediments. The extent of MIF observed in the sediment samples was very low. No even-mass MIF beyond the analytical uncertainty was observed (Supplementary Table S5.2) suggesting no significant contribution of Hg to the sediments from atmospheric precipitation or dry deposition (Gratz et al., 2010; Chen et al., 2012). However, there was a minor shift in odd-mass MIF (Δ199Hg and Δ201Hg, see Figures 4B, C) towards higher values (∼0.1‰) in downstream samples compared to hotspot or tributary sediments.
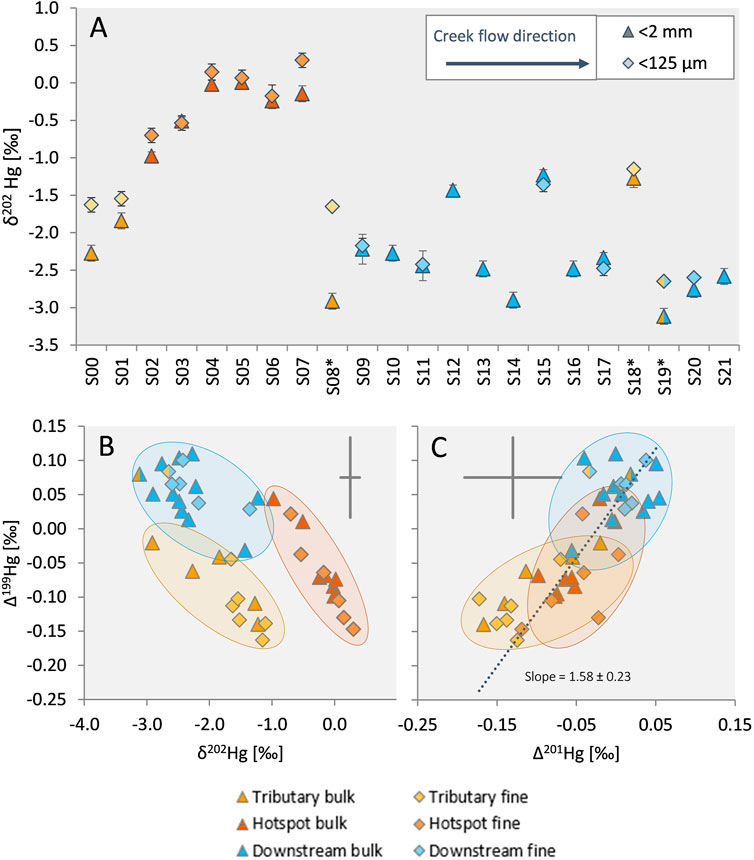
FIGURE 4. (A) δ202Hg (MDF) of bulk and fine sediment samples. Samples are displayed in flow direction from S00 to S21. Tributary creeks are marked with an asterisk. Hotspot and downstream section are indicated by colored symbols as in Figure 2. Error bars indicate 2SD based on the standard reproducibility of the measurement session, but are smaller than symbol size in some cases. (B) Relationship of MDF (δ202Hg) and MIF (Δ199Hg). (C) Relationship of Δ201Hg and Δ199Hg. The dotted line represents a York regression for combined bulk and fine sediment samples calculated using the IsoplotR package in R (Vermeesch, 2018). The grey crosses indicate the average 2SD for the respective parameter.
4 Discussion
4.1 Mixing model based on MDF and Hg concentrations
To test the hypothesis of conservative mixing of natural Hg and industry-derived mercury, a binary mixing model was applied according to eqs 3, 4:
In the model fnat and find represent the fractions of Hg originating from natural background and industrial contamination, respectively. The isotopic signature of the endmembers in the model are defined as δ202Hgind representing the industrial contamination source (exfiltrating groundwater) and δ202Hgnat of different natural background samples (Figure 5A). The exact isotopic signature of the industrial endmember is difficult to determine since there is no record of HgCl2 solutions that were used at the site and Hg exfiltrating from the groundwater has potentially already undergone transformation processes as it interacts with the aquifer material (McLagan et al., 2022b). Nonetheless, we consider δ202Hg values of highly contaminated groundwater wells to be the closest estimate of the industrial Hg isotope source signature for this study. The second endmember, the natural background signature is equally difficult to define. Five approaches were tested with different assumptions made about the regional background signature (details see Supplementary Section S11).
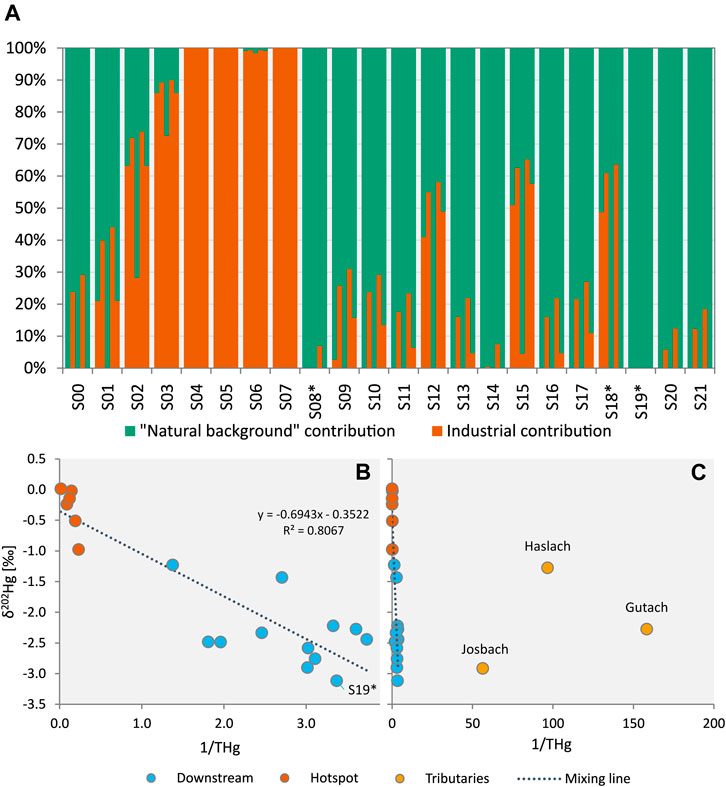
FIGURE 5. (A) Results of two endmember mixing models, each one using a different natural background endmember from left to right: Gutach upstream sample S00 (δ202Hg = −2.27‰), Josbach S08* (δ202Hg = −2.91‰), Haslach S18* (δ202Hg = −1.27‰), S19* (δ202Hg = −3.12‰) and an approach using average δ202Hg values of tributaries after each confluence (see Supplementary Section S11). (B) Plot of δ202Hg values of creek sediments versus 1/THg (mg kg−1 basis). Excluding the tributary and upstream sediments (Gutach S00, Josbach S08* and Haslach S18*) used as endmembers in the mixing models results in a good fit for the linear regression (mixing line) with a hypothetical endmember with a very low δ202Hg value and sufficiently high Hg concentrations (e.g. δ202Hg ≈ −3.0‰ and 0.25 mg kg−1 Hg). (C) This panel shows that δ202Hg values of creek sediments do not lie between the industrial endmember (δ202Hg ≈ −0.22‰) and the δ202Hg values of sediments of the tributary creeks and the Gutach upstream sample.
The bulk isotope signatures of the sediment samples from the hotspot section had a δ202Hg value of −0.31‰ ± 0.37‰ which is close to that of the groundwater (−0.20‰ ± 0.07‰). For the allocation of Hg in the hotspot samples, the mixing models predicted that most of the Hg (63%–100%) is derived from exfiltrating groundwater. The model using Haslach stream as natural endmember predicted a lower contribution (only 27% for S02) in the hotspot samples, which is very unlikely based on the remarkably high Hg concentrations in these samples and the low background concentration. For most of the downstream samples, only a small amount of Hg was allocated to the industrial source. The industrial contribution ranged between 0% and 3% (Gutach endmember), 0%–26% (Josbach endmember), 0%–31% (S19* endmember) and 0%–16% (mixed endmember). Only for the S19* endmember the downstream sediments plot along the mixing line (Figure 5B). However, for S19* an influence of contaminated Gutach sediment or stream water is likely and it is questionable if S19* can be regarded as “natural background.” The downstream sediments had Hg concentrations between 268 and 727 µg kg−1, which is more than 15-fold higher than the background samples (6.3–17.8 µg kg−1 Hg). There is no known other major Hg source in the area and based on this difference in Hg concentrations the contribution of industrial Hg is expected to be >90%. Irrespective of the chosen natural background endmember the contribution of industrial Hg is therefore largely underestimated in the mixing models. Due to the low background Hg concentrations, a significant contribution of Hg from other sources within the tributary catchment area (industrial or natural/geogenic) to the total Hg budget in Gutach stream sediments through conservative mixing seems unlikely, even for endmembers with a light isotope signature such as Josbach stream (Figure 5C). Extending the mixing model with MIF values (Δ199Hg) would be one option to add a more conservative dimension to the system. The extent of odd-mass MIF in our samples was however small. Plotting MDF and MIF values against each other reveals that the downstream sediments do not lie between the hotspot samples, with a similar isotopic composition as the industrial endmember, and the tributary sediments which can be regarded as natural background endmember (Figure 4B).
Both MDF and MIF suggest that the downstream samples are at least partially affected by processes causing isotope fractionation and not conservative transport and mixing of the industrial source (exfiltrating contaminated groundwater) with Hg in sediments derived from natural sources. The change in the binding form, as indicated by SEP and PTD, further implies transformation processes within the sediment or between the sediment and the liquid phase which in turn could lead to isotope fractionation.
4.2 Isotope fractionation mechanisms
The shift towards higher release temperatures in PTD and the higher proportion of Hg extractable in F3 in the downstream samples compared to hotspot samples both indicated a higher proportion of organically bound Hg. This observed shift can be the result of in-stream transformation (e.g., from F4 extractable Hg to an F3 extractable form) as well as preferential transport of different Hg pools. Many biotic and abiotic Hg species transformation processes can cause fractionation of Hg isotopes and the identification of the exact processes involved in the isotopic fractionation is challenging. Comparing the expected fractionation effects of different processes with the Hg isotope ratios in the stream sediments, provides the means to narrow down the drivers of the observed isotopic offset between hotspot and downstream samples. In the following section the potential influence of different processes on the Hg isotopic composition in Gutach stream after the exfiltration of contaminated groundwater are discussed. This allows to exclude certain processes and identify more probable mechanisms that explain the observed variability in isotope ratios.
Mercury is microbially methylated under anoxic conditions in aquatic environments (Hsu-Kim et al., 2013; Tang et al., 2020). However, the conditions in Gutach stream are predominantly oxic and potential methylation is presumably restricted to localized areas (e.g., still water areas with low flow velocities and floodplains). Methylmercury (MeHg) would be extracted in the F3 step of SEP (Bloom et al., 2003) which is found in a higher proportion in downstream sediments. Methylation was also found to cause mass-dependent isotope fractionation leading to an enrichment of light isotopes in the produced methylmercury (Janssen et al., 2016; Zhang et al., 2022) and is further isotopically fractionated throughout environmental reactions in the water column and internal cycling within biological tissue (Rosera et al., 2022). If methylation takes place in Gutach sediments, this could only change the isotopic composition of the bulk sediment to the observed negative δ202Hg values if MeHg is accumulating and the remaining Hg(II) is preferentially removed. Additionally, MeHg usually contributes only a minor part of total Hg in river sediments (Skyllberg, 2012) and Hg methylation is therefore not expected to result in a measurable shift of the overall isotopic signature of the sediments.
The minor offset in MIF observed in downstream sediments compared to hotspot sediments suggests an influence of in-stream fractionation affecting both the MDF and MIF signals of sediment samples, such as, for example, redox processes. Based on experimental studies the ratio of Δ199Hg and Δ201Hg has been proposed to be indicative of specific fractionation processes. These experiments include photoreduction of Hg(II) (slope = 1.00 ± 0.02) (Bergquist and Blum, 2007), photoreduction of Hg(II) at different Hg/DOC ratios (slopes ranging from 1.19 ± 0.02 to 1.31 ± 0.14) (Zheng and Hintelmann, 2009) and photoreduction of MeHg (slope = 1.36 ± 0.02) (Bergquist and Blum, 2007). Experiments investigating reactions influenced by the nuclear volume effect (NVE) showed different characteristic slopes of 1.61 ± 0.06 for abiotic reduction of Hg(II) by DOM (Zheng and Hintelmann, 2010b) and 1.54 ± 0.22 for equilibrium fractionation between dissolved Hg(II) species and thiol-bound Hg (Wiederhold et al., 2010) which is close to the theoretically predicted value of 1.65 for NVE (Wiederhold et al., 2010). Applying this slope as diagnostic tool in stream sediments, Washburn et al. (2019) suggested that Hg in the investigated rivers was influenced by Hg photoreduction (observed Δ199Hg/Δ201Hg slopes of 1.57 ± 0.49 and 1.40 ± 0.27). In our study the slope of the orthogonal regression (York et al., 2004) of the relationship between Δ199Hg and Δ201Hg of all sediment samples (fine and bulk) is 1.58 ± 0.23 (Figure 4C). The slope of separate regressions for the bulk sediments only (1.57 ± 0.31) and the fine fraction only (1.62 ± 0.34) showed similar trends. For hotspot and downstream samples this slope indicates an influence of processes involving isotope fractionation influenced by NVE rather than photoreduction. The slope of the regression for the tributary samples only however is 0.95 ± 0.70, closer to values reported for Hg affected by photochemical reduction (Bergquist and Blum, 2007; Zheng and Hintelmann, 2009). Microbial reduction of Hg(II) to Hg(0) (Kritee et al., 2008, 2013), photochemical reduction by organic compounds (Zheng and Hintelmann, 2010a; Motta et al., 2020) and dark abiotic reduction (Zheng and Hintelmann, 2010b) are known to impart MDF as well as MIF. However, the PTD results do not indicate the presence of Hg(0) in the sediment samples. The fact that Hg(0) is not present in the sediments does not entirely exclude an influence of reduction processes because Hg(0) could easily volatilize to the atmosphere. The effects of reduction processes would nonetheless be detectable in the remaining Hg pool in sediments by causing a shift towards heavier δ202Hg values along the flow path as reduction of Hg(II) leads to an enrichment of light isotopes in the produced Hg(0) fraction. This is opposite to the observed shift towards lighter isotopic composition in downstream samples.
Formation of Hg(0) in the aquifer of this site has been demonstrated (McLagan et al., 2022b) and in certain groundwater wells at the contamination site Hg(0) has been detected (Richard et al., 2016b) which could potentially exfiltrate to the stream despite the dominant species being inorganic Hg(II). Data on isotope fractionation during Hg(0) oxidation in aqueous systems is still limited, but dark abiotic oxidation of dissolved Hg(0) has been shown to enrich heavier isotopes in the oxidized Hg(II) and shift Δ199Hg to more negative values (Zheng et al., 2019). Exfiltration of Hg(0) and subsequent oxidation is therefore an unlikely cause of the observed negative δ202Hg values in downstream sediment samples and the minor offset in Δ199Hg towards more positive values.
The groundwater at the investigated site showed changing redox states, and the importance of hydrous ferric oxides for the retention of Hg in parts of the adjacent groundwater aquifer has been demonstrated by Richard et al. (2016a). If dissolved Fe(II) exfiltrates with anoxic groundwater it will rapidly precipitate as ferrihydrite and other poorly-crystalline hydrous ferric oxides with a large reactive surface when it enters the well-mixed oxic stream water. These Fe-phases can function as a temporary sink for Hg through sorption and co-precipitation. Hg(II) sorption to goethite as a model compound for Fe-oxides under equilibrium conditions was reported to cause an overall δ202Hg fractionation of ∼−0.4‰ (Jiskra et al., 2012). In experiments, the precipitation of metacinnabar (β-HgS) and montroydite (HgO) caused an enrichment of light isotopes in the precipitates relative to the dissolved fraction by a factor of up to −0.63‰ (Smith et al., 2015b). Immediate precipitation of a small fraction of Hg exfiltrating from groundwater as HgO or β-HgS could also cause hotspot sediments to have a slightly lighter isotopic composition than the exfiltrating groundwater, shifting the isotope ratios in the observed direction. However, the release curves of the PTD do not indicate the presence of HgO or β-HgS in the sediment samples (except for S02 to a minimal extent). If Hg precipitated, then more likely as co-precipitates with hydrous ferric oxides. A large fraction of Hg in the hotspot sediments is extracted in steps F4 and F5, the steps in which the majority of Fe is extracted, and thus in which Hg associated with Fe-oxides is expected to be released (Supplementary Figure S3.1). The determined equilibrium fractionation factors are however not large enough to explain the large extent of fractionation observed in the downstream sediments solely based on equilibrium sorption to Fe oxides or precipitation of Hg as HgO or β-HgS. Precipitation of Hg with mineral phases and subsequent sorption on these mineral phases is rather expected directly after the exfiltration of contaminated groundwater in the hotspot area immobilizing Hg in sediments in the hotspot section without causing a large shift in isotopic composition.
Organic matter is one of the primary parameters controlling Hg sorption and mobility in natural aquatic systems due to the formation of extremely strong ionic bonding between Hg and reduced sulfur (S) or thiol groups (-SH) commonly present in aquatic organic matter (Ravichandran, 2004; Skyllberg et al., 2006; Wang et al., 2022). Mercury exfiltrating as inorganic Hg(II) with contaminated groundwater is expected to rapidly bind to organic sorption sites in the bottom sediments as well as algae and biofilms. However, it has been suggested that Hg in highly contaminated areas is also adsorbed onto mineral fractions of sediments because high-affinity organic matter sorption sites are rapidly saturated by high Hg concentrations (Hissler and Probst, 2006). Similarly, high-affinity sites associated with colloidal DOM may quickly become saturated after initial exposure and the remaining Hg may become associated with lower affinity sites and larger particulates (Washburn et al., 2017). The high THg concentrations in hotspot sediments suggest rapid removal of Hg from the water phase by sorption of Hg to the bottom sediments. Relative proportions in SEP indicate the formation of poorly soluble high-concentration Hg pools (released in SEP steps F4 and F5) in that section of the stream, but the absolute pool size of F3 is also larger in the hotspot sediments compared to downstream sediments highlighting the importance of organic matter in the retention of Hg in the hotspot section. Sorption of Hg to organic matter is expected to cause isotope fractionation as shown by equilibrium sorption experiments of dissolved Hg(II) on thiols which is associated with an enrichment of light isotopes (δ202Hg) in the sorbed fraction in the range of −0.53‰ (for HgCl2) and −0.62‰ (for Hg(OH)2) (Wiederhold et al., 2010). Like sorption to goethite, the enrichment factors for equilibrium sorption to functional groups (thiol, carboxyl) of organic matter alone are not large enough to explain the negative δ202Hg values observed in downstream sediments. Exchange experiments between dissolved Hg(II) species and solid bound Hg(II) on carboxyl- and thiol-resins or goethite indicated that kinetic effects may play an important role in metal isotope fractionation during sorption (Jiskra et al., 2014). The timeframe to reach equilibrium in these experiments was days to several months depending on the properties of the dissolved ligand phase. In a dynamic, open system such as Gutach stream, the contact time of dissolved Hg(II) and sorption sites in the bottom sediment is likely not sufficiently long to reach complete equilibrium conditions. Thus, an influence of initial kinetic effects on isotope fractionation during sorption and transport of Hg in the water phase and suspended particles needs to be considered.
Dissolved Hg concentrations in Gutach stream remain elevated at downstream sampling locations (14.8–50.8 ng L−1; Supplementary Table S6). There are two possible explanations for these elevated dissolved Hg concentrations: 1.) The majority of Hg remains in the liquid phase after the exfiltration of contaminated groundwater in soluble forms or associated with high affinity sorption sites of DOM and is transported in the stream water over long distances. 2.) The contaminated bottom sediments constantly release Hg and sustain elevated Hg concentrations in stream water. In the first case most of the released Hg remains in the liquid phase and only a small fraction of Hg is sorbed to the bottom sediments. The observed shift towards negative δ202Hg values would result from preferential sorption of light isotopes. The large extent of fractionation could be explained by the short contact time which doesn’t allow the establishment of equilibrium conditions (Jiskra et al., 2014). An initial kinetic effect with minor re-equilibration can lead to a larger extent of fractionation compared to equilibrium sorption experiments. The removal of only a small pool of Hg enriched in light isotopes would cause a minimal (negligible) change in the isotope signature of the large remaining dissolved Hg pool that cannot be analytically resolved in downstream samples.
In the second case, the Hg pool size in bottom sediments needs to be large enough to constantly release Hg and sustain elevated Hg concentrations in stream water. Our results showed that the largest amount of Hg in Gutach stream sediments was associated with the coarse fraction (61%–97%), similar to results reported by Xu et al. (2021), Hissler and Probst (2006) and Smith et al. (2015a). The proportion of Hg released from bulk sediments in the water-soluble pool in downstream samples was higher compared to hotspot samples, but still relatively small (4.3% ± 1.3%). The overall pool size of this labile Hg fraction in sediments can nonetheless be large considering the overall mass of the sediments. Crowther et al. (2021) hypothesized that abundant Hg in strongly-bound fractions (F4 and F5 steps in SEP) may continuously be remobilized, contributing small amounts of Hg to both the weakly-bound Hg pools in sediments (F1 step in SEP) and replenishing dissolved Hg in the stream water. A similar redistribution of Hg in Gutach stream is plausible, however the transformations of Hg binding forms observed in PTD and SEP suggest that Hg is re-adsorbed to organic matter (F3 in SEP) rather than the water-soluble pool. The Hg pool mobilized from contaminated bulk sediments is expected to be isotopically heavier to an extent similar to that expected from equilibrium sorption experiments (δ202Hg difference 0.3‰ to 0.6‰) (Wiederhold et al., 2010; Jiskra et al., 2012). Leaching experiments also showed that the labile Hg pools mobilized from soils or mine waste (Stetson et al., 2009; Wiederhold et al., 2013; Yin et al., 2013) and aquifer material from drill cores contaminated with HgCl2 (Brocza et al., 2019; McLagan et al., 2022b) were consistently enriched in heavier isotopes compared to bulk samples. Water extracts of contaminated sediments in Grigg et al. (2018) and Crowther et al. (2021) however were not significantly different from bulk sediments showing that the extent of Hg fractionation during leaching is site-specific, depending on Hg speciation, geochemical conditions and spatial heterogeneity. The limited data of isotope ratios in SEP extracts in this study showed that both the F1 and F3 extracts of the most contaminated sediment samples (S05 and S02) had a heavier δ202Hg value compared to the bulk sample (Supplementary Figure S3.3) suggesting that Hg mobilized and re-adsorbed is indeed isotopically heavier and the Hg associated with the bulk sediments isotopically lighter. Complementary to MDF, sorption of Hg in equilibrium experiments was also associated with a small extent of odd-mass MIF related to NVE, resulting in a Δ199Hg/Δ201Hg slope of 1.54 (Wiederhold et al., 2010). This is consistent with the results of sediment samples in this study. The extent of mass dependent enrichment of heavy isotopes is however again smaller (<1‰ difference) than the differences observed in the stream sediments and can only explain the observed variation when particle size and transport effects are included.
The redistribution of Hg to organic matter is likely also a redistribution to the finer sediment fractions due to the increase in specific reactive surface area with decreasing grain size (Kelly and Rudd, 2018; Dickson et al., 2019; Xu et al., 2021). The sediment fine fraction has often been observed to contain higher THg concentrations than bulk sediments (Kelly and Rudd, 2018). Previous studies also found slight differences in Hg isotopic composition between sediment size fractions, with the fine fraction exhibiting less negative δ202Hg values than the coarse fraction (Donovan et al., 2014; Smith et al., 2015a). This difference was attributed to the input of Hg from isotopically distinct sources (contaminated streambanks and soils) that contribute to sediment size fractions to a different extent. While this may also be the case for sediments at our study site, including sediments from tributary streams not affected by direct industrial contamination that also exhibit a large offset between δ202Hg values of the size fractions, we propose that the in-situ redistribution of Hg from the bulk sediments to organic matter in the fine fractions also leads to the isotopic difference between the size fractions which was observed in some samples (Figure 4). The differences between the size fractions are however not as pronounced and consistent as could be expected from this process because of preferential mobilization and removal of the fine fraction. It has been demonstrated that hydrological conditions can have an effect on Hg speciation in suspended matter of rivers (Lin et al., 2011; Baptista-Salazar et al., 2017; Yu et al., 2022). Particles of different size and composition exhibit different settling and resuspension behaviors, with the finer particles remaining suspended in water for much longer and being transported further (Kelly and Rudd, 2018). This can cause a separation of different Hg binding forms associated with these particles (Riscassi et al., 2016; Baptista-Salazar and Biester, 2019; Nair et al., 2022). Similar to the findings of Demers et al. (2018) that the suspended sediment load is “spiraling” downstream as it is deposited and resuspended from streambed biofilm, the sediment fine fraction is expected to act as a transport vector for Hg in Gutach stream. The observed difference between the hotspot section and the downstream sediments can also be attributed to the particle transport regime, which changes shortly after the Gutach stream and the Josbach stream converge. The hotspot section is relatively flat and right after the weir of the artificial Okalsee pond which will reduce the mobilization of coarse sediment in this section compared to the downstream section. The coarse fraction, and with it the largest Hg pool in Gutach stream, is expected to only be transported in the hotspot section during high discharge periods [∼10 times per year (Ortlepp, 1997)]. Mercury associated with finer particles however can be more easily resuspended and transported during the prevalent low to moderate flow conditions. This difference in transport velocities between the sediment size fractions leads to a separation of these pools with an isotopic fractionation behavior that is different from what would be expected in a closed system batch experiment without transport. The continuous replenishment of fine sediment and suspended particulate matter during regular flow conditions in the downstream section and redistribution of Hg to organic matter in this sediment fraction removes the product of the partitioning process. In comparison to equilibrium fractionation, the extent of fractionation in systems with continuous removal of the product can be much larger. For such systems with removal of the reaction product a Rayleigh fractionation model describes the extent of fractionation more accurately and using the fractionation factors determined for equilibrium sorption processes in a Rayleigh model shows that the observed offset of about −2‰ in the remaining pool is possible if a very large relative fraction of Hg (∼95%) has been redistributed and removed. Considering the much lower THg concentrations in downstream samples compared to hotspot samples, the removal of a large proportion of Hg from the sediments by partitioning processes, redistribution and preferential transport of remobilized Hg from sediment components which were previously affected by isotope fractionation between different components of the sediment is another possible explanation for the observed changes in Hg binding forms and isotope values (a conceptualization of this redistribution and fractionation mechanism is presented in Figure 6).
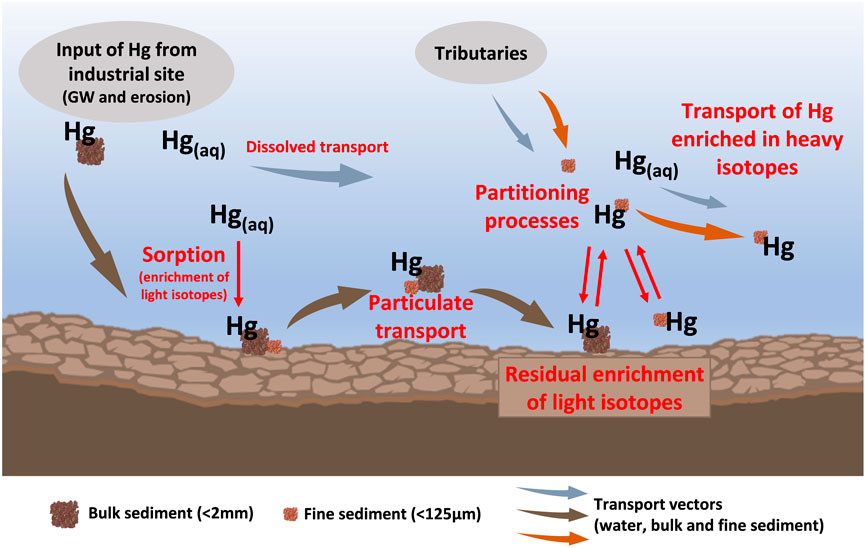
FIGURE 6. Conceptual model of the proposed mechanisms leading to the observed light δ202Hg values in downstream sediments after the exfiltration of contaminated groundwater (GW) into the stream. Briefly, the processes involved include rapid sorption to the solid phase (including lower affinity sorption sites) after exfiltration of contaminated groundwater to the stream. Mercury is then mainly transported with particulate matter during high discharge events. Partitioning processes lead to a redistribution of Hg during the further transport, mobilizing Hg from the particulate phase and re-adsorbing to high affinity sites such as organic matter or fine fraction. The partitioning of Hg between the solid and liquid phase leads to isotopic fractionation and an enrichment of light isotopes in the residual phase. Hg re-adsorbed from the heavier pool is preferentially transported within the stream, leading to a large extent of Hg isotope fractionation in the residual Hg pool. Please note that this is a simplified visualization and not all potentially relevant components and processes are depicted.
The observed change in Hg binding forms during the transport of Hg in Gutach stream may have implications for the methylation potential of Hg transported further downstream of the former HgCl2 source. Methylation rates and bioavailability potentials are known to span over a large range depending on the binding form of Hg in sediments and local geochemical conditions (Jonsson et al., 2012; Zhang et al., 2019; Xu et al., 2021) and the amount of Hg removed in the F3 fraction has been reported to be correlated with Hg methylation potential for aquatic sediments (Bloom et al., 2003; Brooks et al., 2017). The redistribution of Hg to organically bound Hg in downstream samples may therefore indicate a greater potential for Hg methylation and subsequent accumulation in the aquatic food web.
5 Conclusion
This study demonstrates that Hg released as highly soluble HgCl2 from a contaminated site is rapidly sorbed to stream sediments and can be retained long-term followed by particulate transport and redistribution within the sediment. The observed large shift to negative δ202Hg values in Gutach sediments downstream of the contaminated site stream could not be explained by conservative binary mixing models using industrially derived Hg from the former kyanization facility and natural background endmembers. All tested scenarios resulted in a large underestimation of the industrial contribution. More likely, transformation of Hg binding forms as indicated by the analysis of Hg binding forms together with transport processes have caused the large extent of isotopic fractionation. Based on the observed MDF and MIF signatures, we were able to conclude that several potentially relevant in-stream processes, which would lead to a fractionation causing an enrichment of heavy isotopes in the sediments such as methylation, precipitation or (photo-)reduction and evaporative losses to the atmosphere, were not able to influence the Hg budget in the studied stream system to a significant extent. The rapid sorption of Hg to the sediments is expected to retain a large fraction of the released Hg in the bulk sediments which is only transported during high discharge events, while Hg mobilized and re-adsorbed to organic matter in fine sediment fractions is expected to be removed also under the prevalent low to moderate flow conditions. This redistribution of Hg in the sediments results in an offset of δ202Hg between bulk and fine sediments. The large extent of isotopic fractionation between hotspot and contaminated downstream sediments is proposed to be a combination of kinetic effects during the redistribution with the preferential transport of the fine fraction and the removal of the redistributed fraction preventing re-equilibration. These findings highlight the importance of processes governing the isotopic composition of environmental samples such as stream sediments impacted by a single point source of contamination and the apparent limitations of the application of conservative mixing models using Hg isotopes. Furthermore, the observed change in Hg binding form has implications for the potential of Hg to become available for methylation and subsequent accumulation in the aquatic food web. The combination of methods used in this study allowed to narrow down the processes leading to the observed large extent of Hg isotope fractionation. Given the complexity of the system and the influence of many different (biogeo)chemical and physical processes occurring simultaneously in a natural stream environment the exact allocation of governing processes is challenging and the proposed mechanisms still need to be verified and confirmed in future studies. Ideally, the sampling and isotopic analysis should target multiple sample matrices such as stream water, suspended particulate matter, and potentially biota, in addition to sediments. Although stream sediments may potentially provide an integrated record of past Hg releases from industrial sites, the interpretation of sediment Hg isotope data is complicated because the age (time of deposition) of a collected sediment sample is usually unknown as well as due to the difficulty of establishing mass balance estimates of Hg pool sizes and fluxes in such heterogeneous natural systems where transport is often dominated by hydrological extreme events. Concerning the interpretation of Hg isotope data, a more comprehensive understanding of the Hg isotope effects of certain processes is still lacking [e.g., (de)sorption and complexation under kinetically controlled conditions] and experiments under environmentally relevant conditions are needed to link field observations to laboratory studies. While SEP and PTD provide a useful general picture of dominant Hg binding forms, advances in spectroscopic techniques may allow obtaining more detailed information of the Hg speciation in environmental samples (e.g., Poulin et al., 2016; Xu et al., 2021; Nehzati et al., 2022) which in turn could potentially provide further insights into the mechanisms and processes involved. Moreover, recent advancements in analytical methods and sample preparation enable the analysis of Hg isotope ratios in many different matrices (e.g., water, suspended particulate matter, biota), also at very low Hg concentrations (e.g., Janssen et al., 2019; Yang et al., 2022) as well as species specific Hg isotope analysis (e.g., Rosera et al., 2022; Bouchet et al., 2022). We therefore recommend that the design of future process-oriented studies on the behavior and fate of Hg at contaminated sites and surrounding environmental systems should take advantage of these recent analytical advances and include the investigation of multiple sample matrices and the combination of different methods approaches to elucidate and disentangle the intricate mechanisms and processes controlling environmental Hg cycling.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author contributions
LS: Writing of original draft, fieldwork, sample analysis, data evaluation and visualization. FR: Fieldwork, sample analysis, data evaluation. AA: FTIR analysis. DM: Fieldwork, sample analysis, manuscript reviews. SK: Manuscript reviews. HB: Study design and concept, manuscript reviews. JW: Study design and concept, fieldwork, sample analysis, data evaluation, manuscript reviews.
Funding
This research was funded by the Austrian Science Fund (FWF) grant I-3489-N28 and the German Research Foundation (DFG) grant BI 734/17-1.
Acknowledgments
The authors acknowledge Herwig Lenitz, Kurt Barmettler, Petra Schmidt, and Adelina Calean for analytical assistance and thank the Environmental Geochemistry Group at the University of Vienna for inspiring discussion. We thank the three reviewers for helpful comments.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fenvc.2022.1058890/full#supplementary-material
References
Amos, H. M., Jacob, D. J., Kocman, D., Horowitz, H. M., Zhang, Y., Dutkiewicz, S., et al. (2014). Global biogeochemical implications of mercury discharges from rivers and sediment burial. Environ. Sci. Technol. 48, 9514–9522. doi:10.1021/es502134t
Araujo, B. F., Hintelmann, H., Dimock, B., Almeida, M. G., and Rezende, C. E. (2017). Concentrations and isotope ratios of mercury in sediments from shelf and continental slope at Campos Basin near Rio de Janeiro, Brazil. Chemosphere 178, 42–50. doi:10.1016/j.chemosphere.2017.03.056
Baptista-Salazar, C., and Biester, H. (2019). The role of hydrological conditions for riverine Hg species transport in the Idrija mining area. Environ. Pollut. 247, 716–724. doi:10.1016/j.envpol.2019.01.109
Baptista-Salazar, C., Hintelmann, H., and Biester, H. (2018). Distribution of mercury species and mercury isotope ratios in soils and river suspended matter of a mercury mining area. Environ. Sci. Process. Impacts 20, 621–631. doi:10.1039/c7em00443e
Baptista-Salazar, C., Richard, J. H., Horf, M., Rejc, M., Gosar, M., and Biester, H. (2017). Grain-size dependence of mercury speciation in river suspended matter, sediments and soils in a mercury mining area at varying hydrological conditions. Appl. Geochem. 81, 132–142. doi:10.1016/j.apgeochem.2017.04.006
Bartov, G., Deonarine, A., Johnson, T. M., Ruhl, L., Vengosh, A., and Hsu-Kim, H. (2013). Environmental impacts of the Tennessee Valley Authority Kingston coal ash Spill. 1. Source apportionment using mercury stable isotopes. Environ. Sci. Technol. 47, 2092–2099. doi:10.1021/es303111p
Bergquist, B. A., and Blum, J. D. (2007). Mass-dependent and -independent fractionation of Hg isotopes by photoreduction in aquatic systems. Science 318, 417–420. doi:10.1126/science.1148050
Bergquist, B. A., and Blum, J. D. (2009). The odds and evens of mercury isotopes: Applications of mass-dependent and mass-independent isotope fractionation. Elements 5, 353–357. doi:10.2113/gselements.5.6.353
Biester, H., Gosar, M., and Covelli, S. (2000). Mercury speciation in sediments affected by dumped mining residues in the drainage area of the Idrija mercury mine, Slovenia. Environ. Sci. Technol. 34, 3330–3336. doi:10.1021/es991334v
Biester, H., and Scholz, C. (1997). Determination of mercury binding forms in contaminated soils: Mercury pyrolysis versus sequential extractions. Environ. Sci. Technol. 31, 233–239. doi:10.1021/es960369h
Bischoff, C., Richard, J., and Biester, H. (2015). Abschlussbericht Änderungen der Quecksilber, Arsen und Chromkonzentrationen durch Grundwasserexfiltration in die Gutach im Bereich der ehemaligen Kyanisieranlage Okal-Werk. Braunschweig, Germany: Technical University of Braunschweig (unpublished).
Bloom, N. S., Preus, E., Katon, J., and Hiltner, M. (2003). Selective extractions to assess the biogeochemically relevant fractionation of inorganic mercury in sediments and soils. Anal. Chim. Acta X. 479, 233–248. doi:10.1016/S0003-2670(02)01550-7
Blum, J. D., and Bergquist, B. A. (2007). Reporting of variations in the natural isotopic composition of mercury. Anal. Bioanal. Chem. 388, 353–359. doi:10.1007/s00216-007-1236-9
Boszke, L., Kowalski, A., and Siepak, J. (2004). Grain size partitioning of mercury in sediments of the middle Odra River (Germany/Poland). Water Air Soil Pollut. 159, 125–138. doi:10.1023/B:WATE.0000049171.22781.bd
Bouchet, S., Tessier, E., Masbou, J., Point, D., Lazzaro, X., Monperrus, M., et al. (2022). In situ photochemical transformation of Hg species and associated isotopic fractionation in the water column of high-altitude lakes from the Bolivian altiplano. Environ. Sci. Technol. 56, 2258–2268. doi:10.1021/acs.est.1c04704
Branfireun, B. A., Cosio, C., Poulain, A. J., Riise, G., and Bravo, A. G. (2020). Mercury cycling in freshwater systems - an updated conceptual model. Sci. Total Environ. 745, 140906. doi:10.1016/j.scitotenv.2020.140906
Bravo, A. G., and Cosio, C. (2020). Biotic formation of methylmercury: A bio–physico–chemical conundrum. Limnol. Oceanogr. 65, 1010–1027. doi:10.1002/lno.11366
Brocza, F. M., Biester, H., Richard, J.-H., Kraemer, S. M., and Wiederhold, J. G. (2019). Mercury isotope fractionation in the subsurface of a Hg(II) chloride-contaminated industrial legacy site. Environ. Sci. Technol. 53, 7296–7305. doi:10.1021/acs.est.9b00619
Brooks, S., Eller, V., Dickson, J., Earles, J., Lowe, K., Mehlhorn, T., et al. (2017). Mercury content of sediments in east fork poplar creek: Current assessment and past trends. Available at: http://www.osti.gov/scitech/(Accessed August 8, 2022).
Campeau, A., Eklöf, K., Soerensen, A. L., Åkerblom, S., Yuan, S., Hintelmann, H., et al. (2022). Sources of riverine mercury across the Mackenzie River Basin; inferences from a combined Hg–C isotopes and optical properties approach. Sci. Total Environ. 806, 150808. doi:10.1016/j.scitotenv.2021.150808
Chen, J. Bin, Hintelmann, H., Feng, X. Bin, and Dimock, B. (2012). Unusual fractionation of both odd and even mercury isotopes in precipitation from Peterborough, ON, Canada. Geochim. Cosmochim. Acta 90, 33–46. doi:10.1016/j.gca.2012.05.005
Chen, J., Hintelmann, H., Zheng, W., Feng, X., Cai, H., Wang, Z. Z. Z., et al. (2016). Isotopic evidence for distinct sources of mercury in lake waters and sediments. Chem. Geol. 426, 33–44. doi:10.1016/j.chemgeo.2016.01.030
Chen, X., Zheng, L., Sun, R., Liu, S., Li, C., Chen, Y., et al. (2022). Mercury in sediment reflecting the intensive coal mining activities: Evidence from stable mercury isotopes and Bayesian mixing model analysis. Ecotoxicol. Environ. Saf. 234, 113392. doi:10.1016/j.ecoenv.2022.113392
Cooke, C. A., Martínez-Cortizas, A., Bindler, R., and Sexauer Gustin, M. (2020). Environmental archives of atmospheric Hg deposition – a review. Sci. Total Environ. 709, 134800. doi:10.1016/j.scitotenv.2019.134800
Crowther, E. R., Demers, J. D., Blum, J. D., Brooks, S. C., and Johnson, M. W. (2021). Use of sequential extraction and mercury stable isotope analysis to assess remobilization of sediment-bound legacy mercury. Environ. Sci. Process. Impacts 23, 756–775. doi:10.1039/d1em00019e
Demers, J. D., Blum, J. D., Brooks, S. C., Donovan, P. M., Riscassi, A. L., Miller, C. L., et al. (2018). Hg isotopes reveal in-stream processing and legacy inputs in east fork poplar creek, oak ridge, Tennessee, USA. Environ. Sci. Process. Impacts 20, 686–707. doi:10.1039/c7em00538e
Dickson, J. O., Mayes, M. A., Brooks, S. C., Mehlhorn, T. L., Lowe, K. A., Earles, J. K., et al. (2019). Source relationships between streambank soils and streambed sediments in a mercury-contaminated stream. J. Soils Sediments 19, 2007–2019. doi:10.1007/s11368-018-2183-0
Donovan, P. M., Blum, J. D., Demers, J. D., Gu, B., Brooks, S. C., and Peryam, J. (2014). Identification of multiple mercury sources to stream sediments near Oak Ridge, TN, USA. Environ. Sci. Technol. 48, 3666–3674. doi:10.1021/es4046549
Donovan, P. M., Blum, J. D., Singer, M. B., Marvin-Dipasquale, M., and Tsui, M. T. K. (2016). Isotopic composition of inorganic mercury and methylmercury downstream of a historical gold mining region. Environ. Sci. Technol. 50, 1691–1702. doi:10.1021/acs.est.5b04413
Eckley, C. S., Gilmour, C. C., Janssen, S., Luxton, T. P., Randall, P. M., Whalin, L., et al. (2020). The assessment and remediation of mercury contaminated sites: A review of current approaches. Sci. Total Environ. 707, 136031. doi:10.1016/j.scitotenv.2019.136031
Eisele, G. (2004). Absicherbarkeit von Risiken beim Flächenrecycling, Forschungsbericht FZKA-BWPLUS. Available at: https://pudi.lubw.de/detailseite/-/publication/99447-Arbeitshilfe_Absicherbarkeit_von_Risiken_beim_Flächenrecycling.pdf.
EPA (2002). Method 1631, revision E: Mercury in water by oxidation, purge and trap, and cold vapor atomic fluorescence spectrometry. Available at: https://www.nemi.gov/methods/method_summary/9628/ (Accessed September 2, 2021).
EPA (2000). Method 7473, Mercury in solids and solutions by thermal decomposition, amalgamation, and atomic absorption spectrophotometry. Available at: https://www.epa.gov/esam/epa-method-7473-sw-846-mercury-solids-and-solutions-thermal-decomposition-amalgamation (Accessed December 9, 2022).
Feng, C., Pedrero, Z., Lima, L., Olivares, S., de la Rosa, D., Berail, S., et al. (2019). Assessment of Hg contamination by a Chlor-Alkali Plant in riverine and coastal sites combining Hg speciation and isotopic signature (Sagua la Grande River, Cuba). J. Hazard. Mat. 371, 558–565. doi:10.1016/j.jhazmat.2019.02.092
Feng, X., Foucher, D., Hintelmann, H., Yan, H., He, T., and Qiu, G. (2010). Tracing mercury contamination sources in sediments using mercury isotope compositions. Environ. Sci. Technol. 44, 3363–3368. doi:10.1021/es9039488
Fernández-Martínez, R., and Rucandio, I. (2013). Assessment of a sequential extraction method to evaluate mercury mobility and geochemistry in solid environmental samples. Ecotoxicol. Environ. Saf. 97, 196–203. doi:10.1016/j.ecoenv.2013.07.013
Foucher, D., Hintelmann, H., Al, T. A., and MacQuarrie, K. T. (2013). Mercury isotope fractionation in waters and sediments of the Murray Brook mine watershed (New Brunswick, Canada): Tracing mercury contamination and transformation. Chem. Geol. 336, 87–95. doi:10.1016/j.chemgeo.2012.04.014
Foucher, D., Ogrinc, N., and Hintelmann, H. (2009). Tracing mercury contamination from the idrija mining region (Slovenia) to the gulf of trieste using Hg isotope ratio measurements. Environ. Sci. Technol. 43, 33–39. doi:10.1021/es801772b
Goix, S., Maurice, L., Laffont, L., Rinaldo, R., Lagane, C., Chmeleff, J., et al. (2019). Quantifying the impacts of artisanal gold mining on a tropical river system using mercury isotopes. Chemosphere 219, 684–694. doi:10.1016/j.chemosphere.2018.12.036
Gonser, T., and Schwoerbel, J. (1985). Chemische und biologische Untersuchung des Gutach-Wutach-Flusssystems zwischen Neustadt und Weizener Steg. Beih. Veröffentlichungen für Naturschutz Landschaftspfl. Baden-württemb 44, 9–112. Available at: http://www.ephemeroptera-galactica.com/pubs/pub_g/pubgonsert1985p9.pdf (Accessed December 9, 2022).
Gratz, L. E., Keeler, G. J., Blum, J. D., and Sherman, L. S. (2010). Isotopic composition and fractionation of mercury in Great Lakes precipitation and ambient air. Environ. Sci. Technol. 44, 7764–7770. doi:10.1021/es100383w
Gray, J. E., Van Metre, P. C., Pribil, M. J., and Horowitz, A. J. (2015). Tracing historical trends of Hg in the Mississippi River using Hg concentrations and Hg isotopic compositions in a lake sediment core, Lake Whittington, Mississippi, USA. Chem. Geol. 395, 80–87. doi:10.1016/j.chemgeo.2014.12.005
Grigg, A. R. C., Kretzschmar, R., Gilli, R. S., and Wiederhold, J. G. (2018). Mercury isotope signatures of digests and sequential extracts from industrially contaminated soils and sediments. Sci. Total Environ. 636, 1344–1354. doi:10.1016/j.scitotenv.2018.04.261
Guédron, S., Amouroux, D., Sabatier, P., Desplanque, C., Develle, A. L., Barre, J., et al. (2016). A hundred year record of industrial and urban development in French Alps combining Hg accumulation rates and isotope composition in sediment archives from Lake Luitel. Chem. Geol. 431, 10–19. doi:10.1016/j.chemgeo.2016.03.016
Gustin, M. S., Bank, M. S., Bishop, K., Bowman, K., Branfireun, B., Chételat, J., et al. (2020). Mercury biogeochemical cycling: A synthesis of recent scientific advances. Sci. Total Environ. 737, 139619. doi:10.1016/j.scitotenv.2020.139619
Hall, G. E. M., Pelchat, P., and Percival, J. B. (2005). The design and application of sequential extractions for mercury, part 1. Optimization of HNO3 extraction for all non-sulphide forms of Hg. Geochem. Explor. Environ. Anal. 5, 107–113. doi:10.1144/1467-7873/03-061
Hall, G. E. M., and Pelchat, P. (2005). The design and application of sequential extractions for mercury, part 2. Resorption of mercury onto the sample during leaching. Geochem. Explor. Environ. Anal. 5, 115–121. doi:10.1144/1467-7873/03-062
Heckmann, G. (2002). Sanierungsplan ehemaliges Betriebsgelände der Fa. Himmelsbach (“OKAL-Gelände”) an der Freiburgerstraße in 79822 Titisee-Neustadt. Landkreis Breisgau-Hochschwarzwald, Germany: Institut für Sanierung und Umweltschutz Dr. Eisele GmbH.
Hissler, C., and Probst, J. L. (2006). Chlor-alkali industrial contamination and riverine transport of mercury: Distribution and partitioning of mercury between water, suspended matter, and bottom sediment of the Thur River, France. Appl. Geochem. 21, 1837–1854. doi:10.1016/j.apgeochem.2006.08.002
Horvat, M., Covelli, S., Faganeli, J., Logar, M., Mandić, V., Rajar, R., et al. (1999). Mercury in contaminated coastal environments; a case study: The Gulf of Trieste. Sci. Total Environ. 237–238, 43–56. doi:10.1016/S0048-9697(99)00123-0
Hsu-Kim, H., Kucharzyk, K. H., Zhang, T., and Deshusses, M. A. (2013). Mechanisms regulating mercury bioavailability for methylating microorganisms in the aquatic environment: A critical review. Environ. Sci. Technol. 47, 2441–2456. doi:10.1021/es304370g
Issaro, N., Abi-Ghanem, C., and Bermond, A. (2009). Fractionation studies of mercury in soils and sediments: A review of the chemical reagents used for mercury extraction. Anal. Chim. Acta X. 631, 1–12. doi:10.1016/j.aca.2008.10.020
Janssen, S. E., Lepak, R. F., Tate, M. T., Ogorek, J. M., DeWild, J. F., Babiarz, C. L., et al. (2019). Rapid pre-concentration of mercury in solids and water for isotopic analysis. Anal. Chim. Acta X. 1054, 95–103. doi:10.1016/j.aca.2018.12.026
Janssen, S. E., Schaefer, J. K., Barkay, T., and Reinfelder, J. R. (2016). Fractionation of mercury stable isotopes during microbial methylmercury production by iron- and sulfate-reducing bacteria. Environ. Sci. Technol. 50, 8077–8083. doi:10.1021/acs.est.6b00854
Janssen, S. E., Tate, M. T., Krabbenhoft, D. P., DeWild, J. F., Ogorek, J. M., Babiarz, C. L., et al. (2021). The influence of legacy contamination on the transport and bioaccumulation of mercury within the Mobile River Basin. J. Hazard. Mat. 404, 124097. doi:10.1016/j.jhazmat.2020.124097
Jiskra, M., Saile, D., Wiederhold, J. G., Bourdon, B., Björn, E., and Kretzschmar, R. (2014). Kinetics of Hg(II) exchange between organic ligands, goethite, and natural organic matter studied with an enriched stable isotope approach. Environ. Sci. Technol. 48, 13207–13217. doi:10.1021/es503483m
Jiskra, M., Wiederhold, J. G., Bourdon, B., and Kretzschmar, R. (2012). Solution speciation controls mercury isotope fractionation of Hg(II) sorption to goethite. Environ. Sci. Technol. 46, 6654–6662. doi:10.1021/es3008112
Jiskra, M., Wiederhold, J. G., Skyllberg, U., Kronberg, R.-M. M., and Kretzschmar, R. (2017). Source tracing of natural organic matter bound mercury in boreal forest runoff with mercury stable isotopes. Environ. Sci. Process. Impacts 19, 1235–1248. doi:10.1039/c7em00245a
Jonsson, S., Skyllberg, U., Nilsson, M. B., Westlund, P. O., Shchukarev, A., Lundberg, E., et al. (2012). Mercury methylation rates for geochemically relevant HgII species in sediments. Environ. Sci. Technol. 46, 11653–11659. doi:10.1021/es3015327
Jung, S., Kwon, S. Y., Hong, Y., Yin, R., and Motta, L. C. (2021). Isotope investigation of mercury sources in a creek impacted by multiple anthropogenic activities. Chemosphere 282, 130947. doi:10.1016/j.chemosphere.2021.130947
Jung, S., Kwon, S. Y., Li, M. L., Yin, R., and Park, J. (2022). Elucidating sources of mercury in the west coast of Korea and the Chinese marginal seas using mercury stable isotopes. Sci. Total Environ. 814, 152598. doi:10.1016/j.scitotenv.2021.152598
Kallipoliti, L. (2010). Dry rot: The chemical origins of British preservation. Future Anterior 7, 1–19. doi:10.1353/fta.2010.0000
Kelly, C. A., and Rudd, J. W. M. (2018). Transport of mercury on the finest particles results in high sediment concentrations in the absence of significant ongoing sources. Sci. Total Environ. 637–638, 1471–1479. doi:10.1016/j.scitotenv.2018.04.234
Kim, C. S., Bloom, N. S., Rytuba, J. J., and Brown, G. E. (2003). Mercury speciation by X-ray absorption fine structure spectroscopy and sequential chemical extractions: A comparison of speciation methods. Environ. Sci. Technol. 37, 5102–5108. doi:10.1021/es0341485
Kocman, D., Horvat, M., Pirrone, N., and Cinnirella, S. (2013). Contribution of contaminated sites to the global mercury budget. Environ. Res. 125, 160–170. doi:10.1016/j.envres.2012.12.011
Kocman, D., Kanduč, T., Ogrinc, N., and Horvat, M. (2011). Distribution and partitioning of mercury in a river catchment impacted by former mercury mining activity. Biogeochemistry 104, 183–201. doi:10.1007/s10533-010-9495-5
Kritee, K., Blum, J. D., and Barkay, T. (2008). Mercury stable isotope fractionation during reduction of Hg(II) by different microbial pathways. Environ. Sci. Technol. 42, 9171–9177. doi:10.1021/es801591k
Kritee, K., Blum, J. D., Reinfelder, J. R., and Barkay, T. (2013). Microbial stable isotope fractionation of mercury: A synthesis of present understanding and future directions. Chem. Geol. 336, 13–25. doi:10.1016/j.chemgeo.2012.08.017
Lavoie, R. A., Bouffard, A., Maranger, R., and Amyot, M. (2018). Mercury transport and human exposure from global marine fisheries. Sci. Rep. 8, 6705–6709. doi:10.1038/s41598-018-24938-3
Lepak, R. F., Yin, R., Krabbenhoft, D. P., Ogorek, J. M., Dewild, J. F., Holsen, T. M., et al. (2015). Use of stable isotope signatures to determine mercury sources in the great lakes. Environ. Sci. Technol. Lett. 2, 335–341. doi:10.1021/acs.estlett.5b00277
LGRB Landesamt für Geologie Rohstoffe und Bergbau (2021). Hydrogeologische Großeinheiten. LGRB-GeoPortal. Available at: https://maps.lgrb-bw.de/.
Lin, Y., Larssen, T., Vogt, R. D., Feng, X., and Zhang, H. (2011). Transport and fate of mercury under different hydrologie regimes in polluted stream in mining area. J. Environ. Sci. 23, 757–764. doi:10.1016/S1001-0742(10)60473-1
Liu, G., Cai, Y., and O’Driscoll, N. (2011a). Environmental Chemistry and toxicology of mercury. Hoboken, NJ, USA: John Wiley & Sons. doi:10.1002/9781118146644
Liu, G., Li, Y., and Cai, Y. (2011b). Adsorption of mercury on solids in the aquatic environment. Environ. Chem. Toxicol. Mercur., 367–387. doi:10.1002/9781118146644.ch11
Liu, J., Feng, X., Yin, R., Zhu, W., and Li, Z. (2011c). Mercury distributions and mercury isotope signatures in sediments of Dongjiang, the Pearl River Delta, China. Chem. Geol. 287, 81–89. doi:10.1016/j.chemgeo.2011.06.001
Liu, M., Zhang, Q., Maavara, T., Liu, S., Wang, X., and Raymond, P. A. (2021). Rivers as the largest source of mercury to coastal oceans worldwide. Nat. Geosci. 14, 672–677. doi:10.1038/s41561-021-00793-2
Ma, J., Hintelmann, H., Kirk, J. L., and Muir, D. C. G. (2013). Mercury concentrations and mercury isotope composition in lake sediment cores from the vicinity of a metal smelting facility in Flin Flon, Manitoba. Chem. Geol. 336, 96–102. doi:10.1016/j.chemgeo.2012.10.037
Marshall, B. G., Veiga, M. M., Kaplan, R. J., Adler Miserendino, R., Schudel, G., Bergquist, B. A., et al. (2018). Evidence of transboundary mercury and other pollutants in the Puyango-Tumbes River basin, Ecuador-Peru. Environ. Sci. Process. Impacts 20, 632–641. doi:10.1039/c7em00504k
McLagan, D. S., Biester, H., Navrátil, T., Kraemer, S. M., and Schwab, L. (2022a). Internal tree cycling and atmospheric archiving of mercury : Examination with concentration and stable isotope analyses. Biogeosciences 19, 4415–4429. doi:10.5194/bg-2022-124
McLagan, D. S., Schwab, L., Wiederhold, J. G., Chen, L., Pietrucha, J., Kraemer, S. M., et al. (2022b). Demystifying mercury geochemistry in contaminated soil–groundwater systems with complementary mercury stable isotope, concentration, and speciation analyses. Environ. Sci. Process. Impacts 24, 1406–1429. doi:10.1039/d1em00368b
Meng, M., Sun, R. Y., Liu, H. W., Yu, B., Yin, Y. G., Hu, L. G., et al. (2019). An integrated model for input and migration of mercury in Chinese coastal sediments. Environ. Sci. Technol. 53, 2460–2471. doi:10.1021/acs.est.8b06329
Motta, L. C., Kritee, K., Blum, J. D., Tsz-Ki Tsui, M., and Reinfelder, J. R. (2020). Mercury isotope fractionation during the photochemical reduction of Hg ( II ) coordinated with organic ligands. J. Phys. Chem. A 124, 2842–2853. doi:10.1021/acs.jpca.9b06308
Müller, G., and Gastner, M. (1971). The “Karbonat-Bombe”, a simple device for the determination of carbonate content in sediment, soils, and other materials. Neues Jahrb. für Mineral. - Monatsh. 10, 466–469. doi:10.013/epic.27884
Nair, S. S., DeRolph, C., Peterson, M. J., McManamay, R. A., and Mathews, T. (2022). Integrated watershed process model for evaluating mercury sources, transport, and future remediation scenarios in an industrially contaminated site. J. Hazard. Mat. 423, 127049. doi:10.1016/j.jhazmat.2021.127049
Nehzati, S., Dolgova, N. V., Young, C. G., James, A. K., Cotelesage, J. J. H., Sokaras, D., et al. (2022). Mercury Lα1 high energy resolution fluorescence detected X-ray absorption spectroscopy: A versatile speciation probe for mercury. Inorg. Chem. 61, 5201–5214. doi:10.1021/acs.inorgchem.1c03196
Nowak, A. (1952). Die Kyanisierung von Leitungsmasten. Holz als Roh- und Werkst 10, 12–15. doi:10.1007/BF02605401
Obrist, D., Agnan, Y., Jiskra, M., Olson, C. L., Colegrove, D. P., Hueber, J., et al. (2017). Tundra uptake of atmospheric elemental mercury drives Arctic mercury pollution. Nature 547, 201–204. doi:10.1038/nature22997
Ortlepp, J. (1997). Fischereirechtlicher Hegeplan Wutach. Available at: https://docplayer.org/32635320-Fischereilicher-hegeplan-wutach.html.
Pelcová, P., Margetínová, J., Vaculovič, T., Komárek, J., and Kubáň, V. (2010). Adsorption of mercury species on river sediments - effects of selected abiotic parameters. Open Chem. 8, 116–125. doi:10.2478/s11532-009-0128-6
Petranich, E., Predonzani, S., Acquavita, A., Mashyanov, N., and Covelli, S. (2022). Rapid thermoscanning technique for direct analysis of mercury species in contaminated sediments: From pure compounds to real sample application. Appl. Geochem. 143, 105393. doi:10.1016/j.apgeochem.2022.105393
Poulin, B. A., Aiken, G. R., Nagy, K. L., Manceau, A., Krabbenhoft, D. P., and Ryan, J. N. (2016). Mercury transformation and release differs with depth and time in a contaminated riparian soil during simulated flooding. Geochim. Cosmochim. Acta 176, 118–138. doi:10.1016/j.gca.2015.12.024
Pribil, M. J., Rimondi, V., Costagliola, P., Lattanzi, P., and Rutherford, D. L. (2020). Assessing mercury distribution using isotopic fractionation of mercury processes and sources adjacent and downstream of a legacy mine district in Tuscany, Italy. Appl. Geochem. 117, 104600. doi:10.1016/j.apgeochem.2020.104600
Ravichandran, M. (2004). Interactions between mercury and dissolved organic matter - a review. Chemosphere 55, 319–331. doi:10.1016/j.chemosphere.2003.11.011
Reinfelder, J. R., and Janssen, S. E. (2019). Tracking legacy mercury in the Hackensack River estuary using mercury stable isotopes. J. Hazard. Mat. 375, 121–129. doi:10.1016/j.jhazmat.2019.04.074
Reis, A. T., Coelho, J. P., Rucandio, I., Davidson, C. M., Duarte, A. C., and Pereira, E. (2015). Thermo-desorption: A valid tool for mercury speciation in soils and sediments? Geoderma 237, 98–104. doi:10.1016/j.geoderma.2014.08.019
Reis, A. T., Davidson, C. M., Vale, C., and Pereira, E. (2016). Overview and challenges of mercury fractionation and speciation in soils. TrAC Trends Anal. Chem. 82, 109–117. doi:10.1016/j.trac.2016.05.008
Richard, J. H., Bischoff, C., Ahrens, C. G. M., and Biester, H. (2016a). Mercury (II) reduction and co-precipitation of metallic mercury on hydrous ferric oxide in contaminated groundwater. Sci. Total Environ. 539, 36–44. doi:10.1016/j.scitotenv.2015.08.116
Richard, J. H., Bischoff, C., and Biester, H. (2016b). Comparing modeled and measured mercury speciation in contaminated groundwater: Importance of dissolved organic matter composition. Environ. Sci. Technol. 50, 7508–7516. doi:10.1021/acs.est.6b00500
Riscassi, A., Miller, C., and Brooks, S. (2016). Seasonal and flow-driven dynamics of particulate and dissolved mercury and methylmercury in a stream impacted by an industrial mercury source. Environ. Toxicol. Chem. 35, 1386–1400. doi:10.1002/etc.3310
Rosera, T. J., Janssen, S. E., Tate, M. T., Lepak, R. F., Ogorek, J. M., Dewild, J. F., et al. (2022). Methylmercury stable isotopes: New insights on assessing aquatic food web bioaccumulation in legacy impacted regions. ACS Environ. Sci. Technol. Water. 2, 701–709. doi:10.1021/acsestwater.1c00285
Rumayor, M., Lopez-Anton, M. A., Díaz-Somoano, M., Maroto-Valer, M. M., Richard, J. H., Biester, H., et al. (2016). A comparison of devices using thermal desorption for mercury speciation in solids. Talanta 150, 272–277. doi:10.1016/j.talanta.2015.12.058
Schuler, D. (2012). Bericht 2012-69 Gefahrenverdachtserkundung Gewässeruntersuchung Gutach. Germany Unpublished report by Solum Büro für Boden + Geologie, Freiburg i. Br.
Selin, N. E. (2009). Global biogeochemical cycling of mercury: A review. Annu. Rev. Environ. Resour. 34, 43–63. doi:10.1146/annurev.environ.051308.084314
Skyllberg, U., Bloom, P. R., Qian, J., Lin, C. M., and Bleam, W. F. (2006). Complexation of mercury(II) in soil organic matter: EXAFS evidence for linear two-coordination with reduced sulfur groups. Environ. Sci. Technol. 40, 4174–4180. doi:10.1021/es0600577
Skyllberg, U. (2012). Chemical speciation of mercury in soil and sediment. Environ. Chem. Toxicol. Mercur., 219–258. doi:10.1002/9781118146644.ch7
Smith, R. S., Wiederhold, J. G., Jew, A. D., Brown, G. E., Bourdon, B., and Kretzschmar, R. (2015a). Stable Hg isotope signatures in creek sediments impacted by a former Hg mine. Environ. Sci. Technol. 49, 767–776. doi:10.1021/es503442p
Smith, R. S., Wiederhold, J. G., and Kretzschmar, R. (2015b). Mercury isotope fractionation during precipitation of metacinnabar (β-HgS) and montroydite (HgO). Environ. Sci. Technol. 49, 4325–4334. doi:10.1021/acs.est.5b00409
Sonke, J. E. (2011). A global model of mass independent mercury stable isotope fractionation. Geochim. Cosmochim. Acta 75, 4577–4590. doi:10.1016/j.gca.2011.05.027
Stetson, S. J., Gray, J. E., Wanty, R. B., and Macalady, D. L. (2009). Isotopic variability of mercury in ore, mine-waste calcine, and leachates of mine-waste calcine from areas mined for mercury. Environ. Sci. Technol. 43, 7331–7336. doi:10.1021/es9006993
Streets, D. G., Horowitz, H. M., Lu, Z., Levin, L., Thackray, C. P., and Sunderland, E. M. (2019). Five hundred years of anthropogenic mercury: Spatial and temporal release profiles. Environ. Res. Lett. 14, 084004. doi:10.1088/1748-9326/ab281f
Tang, W. L., Liu, Y. R., Guan, W. Y., Zhong, H., Qu, X. M., and Zhang, T. (2020). Understanding mercury methylation in the changing environment: Recent advances in assessing microbial methylators and mercury bioavailability. Sci. Total Environ. 714, 136827. doi:10.1016/j.scitotenv.2020.136827
Troschel, E. (1916). Handbuch der Holzkonservierung. Berlin Heidelberg: Springer. doi:10.1007/978-3-662-42439-1
Tsui, M. T. K., Blum, J. D., Finlay, J. C., Balogh, S. J., Kwon, S. Y., and Nollet, Y. H. (2013). Photodegradation of methylmercury in stream ecosystems. Limnol. Oceanogr. 58, 13–22. doi:10.4319/lo.2013.58.1.0013
Tsui, M. T. K., Uzun, H., Ruecker, A., Majidzadeh, H., Ulus, Y., Zhang, H., et al. (2020). Concentration and isotopic composition of mercury in a blackwater river affected by extreme flooding events. Limnol. Oceanogr. 65, 2158–2169. doi:10.1002/lno.11445
Ullrich, S. M., Tanton, T. W., and Abdrashitova, S. A. (2001). Mercury in the aquatic environment: A review of factors affecting methylation. Crit. Rev. Environ. Sci. Technol. 31, 241–293. doi:10.1080/20016491089226
UN Environment, (2019). Global mercury assessment 2018. UN environment programme, chemicals. U. N. Environ. Program. Chem. heal. Branch Geneva 58. doi:10.1021/acs.est.8b01246
Vermeesch, P. (2018). IsoplotR: A free and open toolbox for geochronology. Geosci. Front. 9, 1479–1493. doi:10.1016/j.gsf.2018.04.001
Wang, Y., Liu, J., Liem-Nguyen, V., Tian, S., Zhang, S., Wang, D., et al. (2022). Binding strength of mercury (II) to different dissolved organic matter: The roles of DOM properties and sources. Sci. Total Environ. 807, 150979. doi:10.1016/j.scitotenv.2021.150979
Washburn, S. J., Blum, J. D., Demers, J. D., Kurz, A. Y., and Landis, R. C. (2017). Isotopic characterization of mercury downstream of historic industrial contamination in the south river, Virginia. Environ. Sci. Technol. 51, 10965–10973. doi:10.1021/acs.est.7b02577
Washburn, S. J., Blum, J. D., Donovan, P. M., and Singer, M. B. (2019). Isotopic evidence for mercury photoreduction and retention on particles in surface waters of Central California, USA. Sci. Total Environ. 674, 451–461. doi:10.1016/j.scitotenv.2019.04.145
Washburn, S. J., Blum, J. D., Kurz, A. Y., and Pizzuto, J. E. (2018). Spatial and temporal variation in the isotopic composition of mercury in the South River, VA. Chem. Geol. 494, 96–108. doi:10.1016/j.chemgeo.2018.07.023
Wiederhold, J. G., Cramer, C. J., Daniel, K., Infante, I., Bourdon, B., and Kretzschmar, R. (2010). Equilibrium mercury isotope fractionation between dissolved Hg(II) species and thiol-bound Hg. Environ. Sci. Technol. 44, 4191–4197. doi:10.1021/es100205t
Wiederhold, J. G., Smith, R. S., Siebner, H., Jew, A. D., Brown, G. E., Bourdon, B., et al. (2013). Mercury isotope signatures as tracers for Hg cycling at the new idria Hg mine. Environ. Sci. Technol. 47, 6137–6145. doi:10.1021/es305245z
Woerndle, G. E., Tsz-Ki Tsui, M., Sebestyen, S. D., Blum, J. D., Nie, X., and Kolka, R. K. (2018). New insights on ecosystem mercury cycling revealed by stable isotopes of mercury in water flowing from a headwater peatland catchment. Environ. Sci. Technol. 52. doi:10.1021/acs.est.7b04449
Xu, J., Bland, G. D., Gu, Y., Ziaei, H., Xiao, X., Deonarine, A., et al. (2021). Impacts of sediment particle grain size and mercury speciation on mercury bioavailability potential. Environ. Sci. Technol. 55, 12393–12402. doi:10.1021/acs.est.1c03572
Yang, L., Yu, B., Liu, H., Ji, X., Xiao, C., Liang, Y., et al. (2022). Online determination of mercury isotopic compositions at ultratrace levels by automated purge and trap coupled with multicollector inductively coupled plasma-mass spectrometry. J. Anal. At. Spectrom. 37, 2480–2489. doi:10.1039/d2ja00148a
Yin, R., Feng, X., Hurley, J. P., Krabbenhoft, D. P., Lepak, R. F., Hu, R., et al. (2016). Mercury isotopes as proxies to identify sources and environmental impacts of mercury in sphalerites. Sci. Rep. 6, 18686. doi:10.1038/srep18686
Yin, R., Feng, X., Wang, J., Bao, Z., Yu, B., and Chen, J. (2013). Mercury isotope variations between bioavailable mercury fractions and total mercury in mercury contaminated soil in Wanshan Mercury Mine, SW China. Chem. Geol. 336, 80–86. doi:10.1016/j.chemgeo.2012.04.017
Yin, R., Guo, Z., Hu, L., Liu, W., Hurley, J. P., Lepak, R. F., et al. (2018). Mercury inputs to Chinese marginal seas: Impact of industrialization and development of China. J. Geophys. Res. Oceans 123, 5599–5611. doi:10.1029/2017JC013691
York, D., Evensen, N. M., Martińez, M. L., and De Basabe Delgado, J. (2004). Unified equations for the slope, intercept, and standard errors of the best straight line. Am. J. Phys. 72, 367–375. doi:10.1119/1.1632486
Yu, C., Liu, M., Guo, J., Lin, H., Yan, Y., Zhang, Q., et al. (2022). Transport of mercury in a regulated high-sediment river and its input to marginal seas. Water Res. 214, 118211. doi:10.1016/j.watres.2022.118211
Zhang, L., Wu, S., Zhao, L., Lu, X., Pierce, E. M., and Gu, B. (2019). Mercury sorption and desorption on organo-mineral particulates as a source for microbial methylation. Environ. Sci. Technol. 53, 2426–2433. doi:10.1021/acs.est.8b06020
Zhang, L., Yin, Y., Li, Y., and Cai, Y. (2022). Mercury isotope fractionation during methylmercury transport and transformation: A review focusing on analytical method, fractionation characteristics, and its application. Sci. Total Environ. 841, 156558. doi:10.1016/j.scitotenv.2022.156558
Zhang, R., Russell, J., Xiao, X., Zhang, F., Li, T., Liu, Z., et al. (2018). Historical records, distributions and sources of mercury and zinc in sediments of East China sea: Implication from stable isotopic compositions. Chemosphere 205, 698–708. doi:10.1016/j.chemosphere.2018.04.100
Zhang, Y., Chen, J., Zheng, W., Sun, R., Yuan, S., Cai, H., et al. (2020). Mercury isotope compositions in large anthropogenically impacted Pearl River, South China. Ecotoxicol. Environ. Saf. 191, 110229. doi:10.1016/j.ecoenv.2020.110229
Zheng, W., and Hintelmann, H. (2009). Mercury isotope fractionation during photoreduction in natural water is controlled by its Hg/DOC ratio. Geochim. Cosmochim. Acta 73, 6704–6715. doi:10.1016/j.gca.2009.08.016
Zheng, W., Demers, J. D., Lu, X., Bergquist, B. A., Anbar, A. D., Blum, J. D., et al. (2019). Mercury stable isotope fractionation during abiotic dark oxidation in the presence of thiols and natural organic matter. Environ. Sci. Technol. 53, 1853–1862. doi:10.1021/acs.est.8b05047
Zheng, W., and Hintelmann, H. (2010a). Isotope fractionation of mercury during its photochemical reduction by low-molecular-weight organic compounds. J. Phys. Chem. A 114, 4246–4253. doi:10.1021/jp9111348
Zheng, W., and Hintelmann, H. (2010b). Nuclear field shift effect in isotope fractionation of mercury during abiotic reduction in the absence of light. J. Phys. Chem. A 114, 4238–4245. doi:10.1021/jp910353y
Keywords: mercury stable isotopes, stream sediment, source tracing, species transformation processes, contaminated legacy sites
Citation: Schwab L, Rothe FM, McLagan DS, Alten A, Kraemer SM, Biester H and Wiederhold JG (2022) Large extent of mercury stable isotope fractionation in contaminated stream sediments induced by changes of mercury binding forms. Front. Environ. Chem. 3:1058890. doi: 10.3389/fenvc.2022.1058890
Received: 30 September 2022; Accepted: 06 December 2022;
Published: 20 December 2022.
Edited by:
Robert Peter Mason, University of Connecticut, United StatesReviewed by:
Amrika Deonarine, Texas Tech University, United StatesBrett Poulin, University of California, Davis, United States
Daniel Steven Grégoire, Carleton University, Canada
Copyright © 2022 Schwab, Rothe, McLagan, Alten, Kraemer, Biester and Wiederhold. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lorenz Schwab, bG9yZW56LnNjaHdhYkB1bml2aWUuYWMuYXQ=