 Xiangming Shi
Xiangming Shi Jing Zhao
Jing Zhao Yongchen Wang
Yongchen Wang Robert P. Mason
Robert P. Mason- 1Department of Marine Sciences, University of Connecticut, Groton, CT, United States
- 2Department of Chemistry, University of Connecticut, Storrs, CT, United States
Transformations of mercury (Hg) forms in the aquatic environment is a crucial aspect of Hg fate, transport and the bioaccumulation of methylmercury (CH3Hg; MeHg), which is the form that drives most human health concerns. Transformations between Hg forms on surfaces have been inadequately studied but here we report on the interaction of inorganic Hg (HgII) and MeHg with chalcogenide nanoparticles (NPs); specifically L-cysteine capped CdSe nanocrystals. The study sheds light on the transformation of the Hg species and the interaction mechanisms, by examining the product composition, reaction mass balance and the distribution between the liquid and solid phase. The results showed that the quenching of the photoluminescence (PL) of CdSe NPs was greater for HgII than MeHg, and that HgII caused significant PL quenching even when its concentration was in the nM range. Over 90% of HgII was found associated with the solid phase while most MeHg existed in the liquid phase in the experimental solutions. No dimethylmercury ((CH3)2Hg; DMeHg) was produced from the interaction of MeHg and the NPs, in contrast to findings with microparticles. However, a fast and complete MeHg transformation into HgII occurred when the MeHg + NPs mixture was exposed to light. A scheme for the MeHg degradation was derived and is presented, and it was concluded that the precipitation of HgSe accelerated the MeHg degradation. These results provide insight into the abiotic pathways for MeHg degradation in environmental waters in the presence of NPs.
1 Introduction
Mercury (Hg) is regarded as one of the most hazardous heavy metals in the environment. In its organic form of methylated mercury, primarily monomethylmercury (CH3Hg; MeHg), it is highly bioaccumulative and is present at concentrations harmful to human health in higher trophic level aquatic organisms consumed by humans and wildlife (Driscoll et al., 2013; Sunderland et al., 2018). Numerous studies have examined the formation of MeHg in aquatic environments and have concluded that its formation is primarily mediated by anaerobic microbes, such as sulfate and iron reducing anaerobic bacteria (Jensen and Jernelöv, 1969; Benoit et al., 2003; Warner et al., 2003; Kerin et al., 2006; Parks et al., 2013; Gilmour et al., 2018). However, although MeHg is relatively unstable in the environment, and its degradation has been shown in many studies, it is not clear whether biotic or abiotic processes are more important for its degradation (Craig and Bartlett, 1978; Khan and Wang, 2010; Susana et al., 2011). In surface waters, photochemical degradation has been shown to be important, but the mechanisms by which this degradation occurs has not been clearly elucidated (Seller et al., 1996; Hammerschmidt and Fitzgerald, 2006; Tai et al., 2014; DiMento and Mason, 2017; Du et al., 2019). Recent studies have also suggested that metal sulfide and selenide surfaces can enhance MeHg transformations (Bjorklund, 2015; Du et al., 2019; Jonsson et al., 2016; Zhang and Hsu-Kim, 2010; West et al., 2020). However, few studies have examined the role of nanomaterials in Hg transformation and MeHg degradation in the aquatic environment.
Both commercially produced nanoparticles (NPs), released by human activity and usage, and natural nanomaterials can be found in aquatic systems, as the mechanisms for their formation commercially and in the laboratory are replicated in the environment (Handy et al., 2008; Robinson, 2009; Nick et al., 2014). Specifically, metal-sulfide and metal-selenide NPs have been produced in the laboratory under typical environmental conditions (Borm et al., 2006; Park et al., 2010) and such NPs have also been found in natural waters, sediments and hydrothermal vents (Lee and Richards, 2004; Wigginton et al., 2007; Jeevanandam et al., 2018; Turan et al., 2019). As these chalcogen elements have been shown to mediate Hg reactions in the environment, the understanding on MeHg + NPs reactions is vital in ascertaining the role that natural or commercial NPs in the environment may have on MeHg cycling. Prior research has shown that inorganic Hg (HgII) can be reduced to elemental Hg in association with FeS microparticles (Coulibaly et al., 2021) and that MeHg can be converted to dimethylmercury ((CH3)2Hg; DMeHg), as well (Jonsson et al., 2016). Yet, as natural waters are generally enriched with dissolved organic matter (DOM), the NPs are likely coated with organic ligands, which also helps maintain the dispersion of NPs in aquatic systems. CdSe NPs are a common photocatalyst (Zhao et al., 2013; Edwards et al., 2020) which may induce MeHg photolysis under some conditions; and given that the sulphur-containing ligand L-cysteine is essential in organomercurial lyase (Li et al., 2010), its presence as a capping agent may also enhance MeHg degradation. Thus, we choose to use L-cysteine capped CdSe NPs to examine the influence of coated NPs on MeHg cycling, while it is likely that the capping agents in the natural environment are larger organic molecules. Using a well characterized ligand however allowed for the interactions and mechanisms to be studied. Although our recent study (Wang et al., 2019) indicated that the interaction between HgII and L-cysteine capped CdSe NPs only occurred at the surface as a ligand association, further studies are still needed to explore whether there is a species transformation occuring and how it may occur, especially for MeHg, and how it may occur as this may mediate the high toxicity and bioaccumulation of MeHg in the biosphere.
Here, we investigate the interactions of inorganic Hg and MeHg interactions with CdSe NPs by examining the reaction products. By quantifying a Hg mass balance in the various phases, and with a special focus on the potential volatile Hg species that might have been ignored in prior studies, we further elucidated the reaction mechanisms for these transformations. Considering that Hg exists at pico-to-nano molar levels in natural aqueous systems, in this study, the concentrations of Hg species in our manufactured system were decreased to a range of ∼1–150 nM compared to the prior work. We also deployed light and dark conditions when monitoring the change of MeHg in the presence of the CdSe NPs since the coordination between NPs and Hg species and the chemical/physical properties of NPs may be variable in natural environments with water depth (Aldana et al., 2001; Rivera et al., 2019). These experiments advance understanding of the mechanisms for the transformation of Hg species in the presence of NPs, and have elucidated the potential role of NPs in MeHg cycling in the environment even though experiments were done at higher concentrations, and with a simple capping ligand. The role of photochemical processes in these transformations also has implications for the cycling of MeHg in natural waters as it is known to be more readily degraded in the photic zone of aquatic systems (Hammerschimdt and Fitzgerald, 2006; DiMento and Mason, 2017).
2 Materials and Methods
2.1 Nanoparticle Synthesis, Purification and Characterization
The synthesis method, as well as the reaction conditions, followed the studies of Park et al. (2010); Park et al. (2011). The synthesized CdSe NPs are stable at room temperature for over 2 months. In detail, accurately weighted selenium powder (100 mg; 99.99%, Alfa Aesar) and sodium sulfite (478 mg; ≥ 98%, Alfa Aesar) were added into 25 mL of water in a flask to obtain the Se precursor (Na2SeSO3). The mixture was held in a water bath at 85°C with magnetic stirring. After >12 h of reaction, the mixture became clear and ready to use as the Se precursor.
When synthesizing CdSe NPs, 6.0 mL of sodium hydroxide solution (1 M), 169 mg of L-cysteine (≥ 99%, Alfa Aesar), 1.0 mL of cadmium sulfate solution (0.16 M) and 0.78 mL of hot Se precursor were added into 100 mL of degassed water strictly in order, with magnetic stirring at all times. In particular, a 10 min or longer time interval was needed between each chemical/solution addition, and the reaction was carried out in a N2-filled glove box. The CdSe NPs with L-cysteine ligand gradually formed and showed a bright yellow color after 5 days. Stirring was stopped on the 7th day and the aqueous NPs were stored in a dark and inert environment. Considering that some of the chemicals used are sensitive to oxygen, all reactants were made in degassed water, which was prepared by boiling deionized (DI) water (18.2 mΩ) under N2 bubbling for 20 min.
Purification of the NPs is necessary to discontinue the growth of particles and decrease the interference from the free L-cysteine on the mercury species. To separate the particulates from the solution, the aqueous NP solution was mixed with ethanol (≥ 99.5%) in a volume ratio of 3:2 (NP solution:ethanol). A yellow precipitate, namely L-cysteine capped CdSe NPs (designated as NPs below), was obtained after centrifuging the mixture at 4700 G-force for 10 min. Immediately after centrifugation, the supernatant was decanted and traces of water were further removed with a pipette. The remaining yellow solid was the purified NPs.
Earlier study had revealed that the temporal stability of CdSe NPs is greatly affected by the dispersing substrate and pH (Aldana et al., 2005; Baker et al., 2011). Thus, for long-term conservation, the purified NPs were dispersed in a basic Cd-cysteine complex solution, i.e., the synthesis mixture without the Se precursor (pH = 12.3). For optical characterization, the dispersed NPs was diluted by a factor of 3–4 with the same basic solution to avoid self-absorption. Then, an aliquot of 3 ml diluted NP solution was sealed in a 1 cm quartz cuvette with a screw cap under a N2 atmosphere. UV-Vis spectra were measured with a Hitachi U3010 UV-Vis spectrometer. Fluorescence spectra were collected with a Cary Eclipse Fluorescence Spectrophotometer (Agilent) with a 400 nm excitation wavelength.
2.2 Interactions between NPs and Hg Species
2.2.1 NPs Fluorescence Quenching with Hg Species
Firstly, the interactions of Hg species with NPs can be assessed from the quenching of the NP photoluminescence. The NP solution for quenching assays was prepared with the same procedure as described above, except that the basic Cd-cysteine complex solution was replaced with degassed DI water for NP redispersion to avoid L-cysteine complexation with Hg. 3 ml of diluted NPs was sealed in a quartz cuvette and the quencher solution was injected with a microvolume needle through the septa in the cuvette cap to prevent oxidation. Relatively low and high concentrations of methylmercury chloride (CH3HgCl) and mercury chloride (HgCl2) were used in this study. Specifically, 0.52 µM CH3HgCl and 0.58 µM HgCl2 stock solutions were utilized for the low-level quenching series; while for the high-level series, the concentrations of the solutions were 2.6 and 3.3 µM, respectively; i.e., the higher concentration solution was ∼5 times that of the lower solution. The quencher solution was added into the NP solution at a volume interval of 10–20 µL so the concentration of MeHg and HgII in the low series ranged from 0 to 40 nM and 0–26 nM, respectively; in contrast, the range for the high series was 0–115 nM for MeHg and 0–160 nM for HgII.
After the quenching experiments, the NPs with Hg quenchers were processed for solid-liquid separation to ascertain the phase distributions of MeHg and HgII, including the Hg concentrations of the bulk solution, liquid phase and solid phase. A volume of 30–100 µL of the NP-Hg mixture was firstly taken for the bulk MeHg/total Hg determination. Then the solution in the cuvette was transferred into a 15 mL tube, followed by the addition of an equal volume of 1 M NaCl solution. NaCl can potentially ionize the particle surfaces, making the NPs aggregate more easily. However, NaCl would not displace Hg from the binding sites on NPs or interfere with the Hg measurements. The solid phase was then separated from the solution by centrifugation (at 4700 G-force for 20 min). The final MeHg and total Hg concentration in the liquid phase was calibrated accounting for volume changes from NaCl dilution. The solid phase was only used for total Hg determination.
2.2.2 Reaction Assays with the MeHg + NPs Mixture
By examining the products derived from the interactions between NPs and MeHg, we explored whether the NPs can mediate a MeHg transformation. In practice, the MeHg solutions were added to the purified solid NPs directly. The mixture was shaken and ultra-sonicated to evenly disperse the NPs and to ensure the reaction was not limited by coagulation. According to the prior research showing that DMeHg formation was mediated by the presence of inorganic iron sulfide microparticles (Jonsson et al., 2016), the ratios of MeHg:NP were chosen accordingly, and ranged from 0.06–0.6 nmol μmol−1. To test for the potential of DMeHg formation, 10 ml of freshly-diluted MeHg solutions in concentrations of 1.9, 3.4 and 17.5 nM were prepared in duplicate. One group was kept in brown borosilicate vials for determining background concentrations. The other group was mixed with newly-purified NPs (solid) extracted from 0.5 ml of NP growth solution. The assay schematic is shown in Supplementary Figure S1A of the Supporting Information (SI). When examining the change of MeHg in the NP mixture, we kept the MeHg:NP ratio in the DMeHg test of its presence the same, and used a MeHg solution of ∼3.4 nM. Given the detection limit of the mercury analyzer, a higher concentration solution of MeHg was also used in comparison so that a solid phase concentration was detectable when MeHg associated with the NPs. A batch of 6 replicates of purified NPs (solid, from 1.0 ml of NP growth solution) were prepared: one group in duplicate was phase separated immediately after mixing; the two other groups were allowed to stay in dark/light for 2 days to monitor their composition change. 2 ml of MeHg solution of ∼3.4 and ∼74 nM was used for quantification for the low- and high-concentration group, respectively. A control group, 2 ml of the low-concentration MeHg solution mixed with 1 ml of the basic Cd-cysteine complex solution, was set up along with each of the abovementioned groups. These procedures are illustrated with SI Supplementary Figure S1B as two batches were processed at the same time.
2.3 Analysis of Mercury Species
2.3.1 Dimethylmercury
After mixing the NPs and MeHg solution, the test tube was wrapped with alumina foil to lower the influence of light. The tube cap was quickly replaced with a rubber stopper holding two clean tubules. One was connected with a carbon trap column (CarbotrapTM, Supelco), following a soda lime drier. The other one was connected to the gas tubing. After ensuring the system was gas tight, the solution was bubbled with N2 for 30 min (flow rate = 200 mL/min) and the volatile Hg species was carried out with the outflow and collected onto the carbon trap. Then the carbon trap was heated to 180°C for up to 45 s to release all Hg species. The vapor containing Hg species was subjected to isothermal gas chromatography separation and the constituents were separated based on their respective retention time, with the carrier gas Ar flow rate of 40 mL/min (Baya et al., 2013). Finally, all Hg species were decomposed to Hg0 by pyrolysis and detected with a cold vapor atomic fluorescence spectrophotometer (CVAFS, Tekran 2500). Under these conditions, the DMeHg peak appeared at 2 min, identified with the characteristic peak represented by the DMeHg standard (Jonsson et al., 2016). For safety considerations, DMeHg was routinely quantified based on the standard curve derived from the ethylated form of MeHg (methylethyl mercury; MeEtHg) determined with the Tekran 2500 since it is quantitatively produced from the ethylation of the MeHg standard (see Section 2.3.2).
2.3.2 Methylmercury in Bulk and Aqueous Solutions
The quantity of MeHg in the solution was determined using a Tekran 2700 with an automatic purging system, following the procedure of EPA method 1630 (Tseng et al., 2004). Briefly, the measurement matrix was prepared in a brown borosilicate vial with ∼30 mL DI water. After adding 2.5% ascorbic acid and buffering with 2 M acetate buffer to the suitable pH for derivitization, an aliquot of 10–100 µL of the samples was added into the matrix. As MeHg is very unstable in acidic NPs environments (discussed in Section 3.5), the prepared solution was immediately ethylated with NaTEB (1% sodium tetraethyl borate) to form volatile MeEtHg. The Tekran 2700 automated the sample purging, gas chromatographic separation and thermal decomposition of the various species (MeEtHg and diethylmercury (DEtHg)) for quantification of Hg0 by CVAFS. Specifically, from the size of DEtHg peak, one can also identify, qualitatively, the relative change of HgII in the solution over time. For the MeHg control group in the product testing assays, the measured aliquot was firstly acidified with 1% sulfuric acid overnight to eliminate the effect of complexation on the ethylation efficiency of Hg. Overall, the detection limit of the Tekran 2700 was 0.1 pM and the recovery of MeHg standard spike (Alfa Aesar) was over 90%.
2.3.3 Total Mercury in the Solid and Liquid Phases
After the phase separation, an aliquot of DI water (<0.5 mL) was added to assist the solid transfer from the tube to a sample boat of the direct thermal decomposition mercury analyzer (MA-3000, Nippon©). In the analyzer, mercury species were completely transformed into volatile Hg0 for determination. All Hg species within the solid were released when combusted at high temperature in oxygen and detected by cold vapor atomic absorption. Hence, the MA-3000 can only quantify the total amount of mercury in the samples without identifying any species. As such, the result of the solid phase derived from MeHg + NPs mixture represents the total Hg content. Similarly, the total Hg of the liquid phase was directly determined with a volume range of 0.1–0.5 mL. The detection limit of the MA-3000 was ∼0.5 ng, calculated based on three times the value of the background signal.
3 Results and Discussion
3.1 Properties of L-Cysteine Capped CdSe Nanoparticles
Figure 1A shows the optical spectra of the purified NPs in the basic Cd-cysteine complex solution (pH = 12.3). The first extinction peak at 419 nm of the UV-Vis spectrum is almost identical to that of the NPs reported by Park et al. (2010) The absorption spectra remained the same for 102 h after synthesis, showing that the L-cysteine capped CdSe NPs were stable for several days. The photoluminescence (PL) spectra exhibited a characteristic emission peak at ∼430 nm, consistent with the reference (Figure 2A in Park et al., 2010). In addition, there is a broad peak at longer wavelength around 530 nm. Based on the Stokes shift rule (Zhu et al., 2018), and the reference peak in Park et al. (2010), the narrow peak characterized the excitonic PL corresponding to the NP bandgap; and the broad peak likely resulted from the trap state emission. Figure 1B shows the influence of the dispersing matrices on the PL spectra. Compared with the unpurified NP solution, when the dispersion medium contained Cd-cysteine complexes (pH = 12.3), the PL spectrum showed higher intensities at both 430 and 530 nm. In contrast, in the DI-dispersed NP solution (pH = ∼8), the PL spectrum had a higher peak at 550 nm and a lower peak at 430 nm. The red shift and intensity increase of the 530 nm peak is coincident with the phenomena found by Park et al., that NPs synthesized in low L-cysteine solutions showed stronger surface trap PL (Park et al., 2011). This is also consistent with previous studies that the PL derived from the trap states was significantly affected by multiple factors including the precursor Cd/Se ratio, ligand concentration, size or the evolution time (Qu and Peng, 2002; Park et al., 2011; Ben Brahim et al., 2017). However, the peak at 430 nm remained at a consistent location in the different dispersing solutions (Figure 1B), implying that the emission from the band gap of the CdSe core was relatively independent of the surrounding environment.
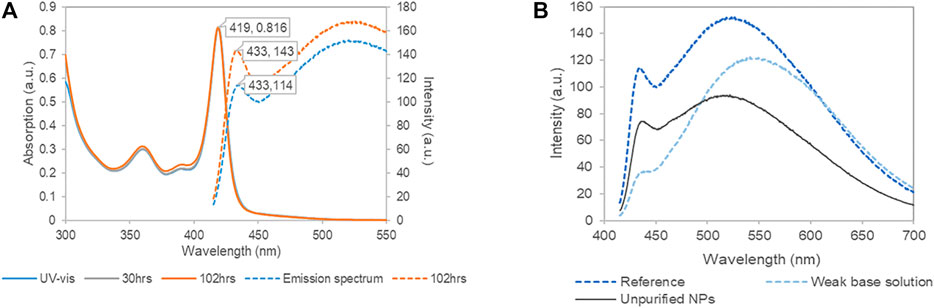
FIGURE 1. Optical characteristics of L-cysteine capped CdSe nanoparticles (NPs). (A) Temporal changes (4 day interval) of UV-vis absorption spectra (solid lines) and photoluminescence spectra with excitation of 400 nm (dashed lines) of the purified NP solution. (B) The photoluminescence spectra in different substrate. The dark blue refers to the same one in (A); the light blue refers to purified NP in DI-dispersing solution (pH = 8); the black refers to the diluted unpurified NPs. Each solution held the same amount of NPs.
3.2 Quenching by Mercury Species on Nanoparticles
The PL spectra of NPs quenched with different concentrations of Hg species are shown in Figure 2. The intensity ratio between the initial (F0) and quenched NPs (F0/F) at ∼530 nm (see Supplementary Section 2.1 for details) were calculated and fitted using the Stern-Volmer equation (Lakowicz, 2006). The HgII showed a stronger quenching effect than MeHg. The F0/F vs. HgII concentration showed a linear trend that reflects the conventional Stern-Volmer relationship. Interestingly, the quenching efficiency of the low-level HgII solutions was higher by a factor of 3 than that at the higher concentrations (based on the slope of the Stern-Volmer curves; Figures 2A,B), even though the concentration range was ∼5 times higher. Even with the lower level Hg quenching, the results showed that quenching was more effective when [HgII] was less than 15 nM. In fact, several studies (Zeng et al., 2013; Ke et al., 2014) on metal ion induced quenching have reported a similar effect where a lower level addition of the quencher resulted in a greater quenching efficiency. The non-linear quenching efficiency with concentration of the low vs. high level additions manifests as a Stern-Volmer plot curving non-linearly when the PL of the NPs was observed over an HgII concentration range of several orders of magnitude. One possibility causing these differences could be that some HgII in the solution is inaccessible to quench the NPs. As the earlier study found, Hg was bound with amine or carboxylic group of the ligand (Wang et al., 2019), and given that there was a large difference in the concentrations between L-cysteine and mercury species (milli vs. nano-molarity) in our experiments, some of the added HgII could bind to the L-cysteine on the outer layer of the NPs, which unlikely induces the PL quenching. This is likely more significant when higher concentrations of HgII are added, as found previously. Practically, if CdSe NPs were used as a HgII sensor, it is clear that the linear calibration range would be constrained by the differences in the quenching efficiency over the concentration range for <15 nM compared to higher concentrations.
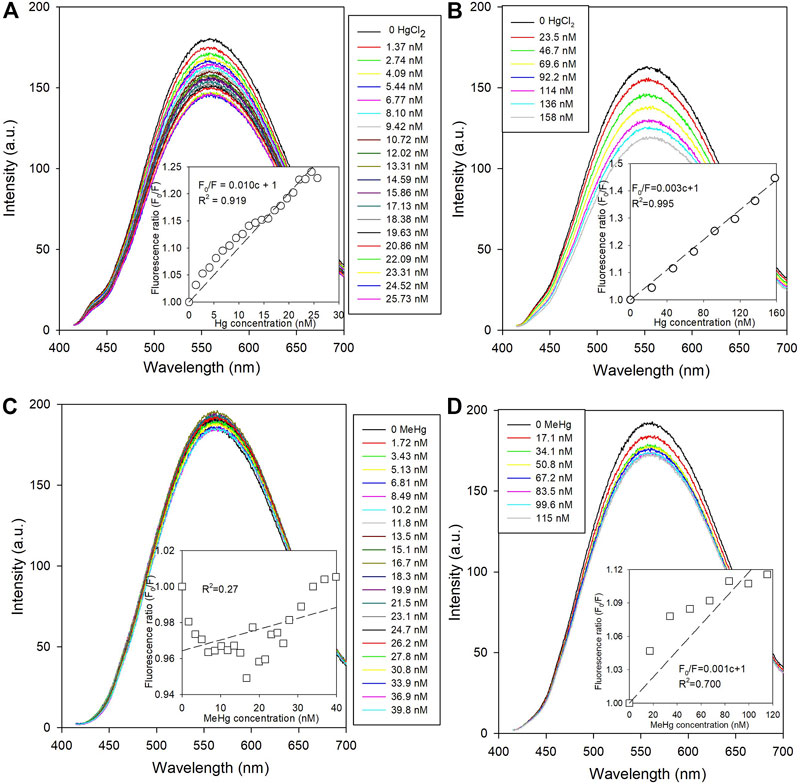
FIGURE 2. The change of fluorescence spectra of L-cysteine capped CdSe NPs with mercury species quenching (Ex = 400 nm). The inserted diagrams show the Stern-Volmer fittings plotted with the concentrations of quenchers and the fluorescence intensity ratios at ∼560 nm. (A) the low-level with HgCl2 concentration of 0–26 nM; (B) the high-level with HgCl2 concentration of 0–160 nM; (C) the low-level with MeHg concentration of 0–40 nM; and (D) the high-level with MeHg concentration of 0–115 nM.
In contrast, MeHg additions presented the opposite quenching trend. For the higher level MeHg addition, a small decrease in the fluorescence intensity of the NPs was observed but the F0/F ratio did not show a linear Stern-Volmer relationship with respect to MeHg concentration (Figure 2D). Moreover, the PL of the NPs with the lower level MeHg additions showed very little change even though the average mercury content was higher than that for the lower level HgII quenching solutions.
3.3 Mechanism for the Different Quenching Efficiencies Based on the Phase Distributions of Mercury Species
After the quenching assays, the concentration of Hg species in each quenched solution was determined and the results are presented in Table 1. Particularly, the Hg content in the solid phase is the sum of solids from the double phase separations (see Supplementary Section 2.2). The predicted concentration is the concentration of the mercury species calculated from the total amount of standard added and the final volume of the quenched solution. Clearly, we have identified that different Hg species have different distributions in the quenching systems: HgII was mostly bound onto the solid phase while MeHg mainly existed in the liquid phase. This observation coincides with the higher quenching efficiency of HgII compared to MeHg. Additionally, L-cysteine on the quenched NPs was determined to be ∼1.0 mM, comparable to that of the unquenched purified NP solution (see Supplementary Section 2.3). This implies that the HgII quenching of the NPs was not constrained by the presence of sulfhydryl groups, although Hg generally has a strong binding affinity with reduced S. One the other aspect, when an equivalent amount of EDTA was added to the HgII + NPs solution, there was no recovery of the PL of the NPs. This experiment suggests that the HgII was strongly interacting with the NPs, likely with the inorganic CdSe core. This conjecture is supported by the change in the energy of the XPS peak of Se (3 d) with the same NPs quenched solution in Wang et al. (2019). Notably, there was no significant shift in the PL spectra during the quenching in our experiments (see Figure 2), suggesting that HgII was not replacing Cd through a cation exchange reaction, but was simply deposited onto or associated with the surface (Boles et al., 2016). This assumption was also supported by the absence of Cd in the quenched solutions (Wang et al., 2019). Furthermore, we also examined HgII quenching on the purified CdSe NPs that were redispersed in a Cd-cysteine complex solution. When a similar amount of HgCl2 was added, significant PL quenching was observed even though there were abundant free L-cysteine molecules existing in solution (see Supplementary Figure S2). Both PL peaks at ∼430 and ∼530 nm decreased with HgII additions, indicating that HgII quenched both the band edge emission of the CdSe core and the surface state emission, and such quenching was not reduced by free L-cysteine which should have complexed the HgII. This result further confirms that Hg has a higher affinity to Se in the CdSe NPs than to thiol in the L-cysteine, which is expected given the higher binding constant of HgSe compared to HgS (Jackson, 1998).
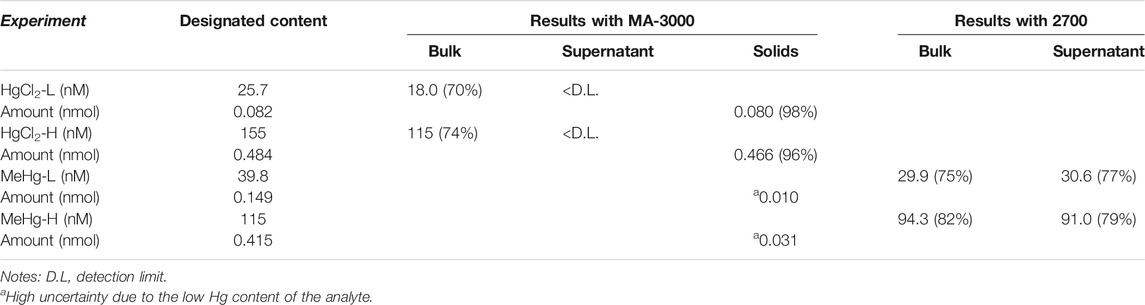
TABLE 1. Phase distributions of Hg species after quenching of the NPs. The percentage in the parentheses refers to the fraction relative to the designated content.
In the MeHg + NPs solutions, MeHg was primarily observed in the liquid phase (Table 1). Moreover, MeHg has a lower efficiency towards quenching the PL of the NPs. We presume that the methyl group prevents MeHg from approaching the NP surface. Since the CdSe core was passivated by the organic ligands, the steric hindrance may prevent MeHg from penetrating the ligand layer. To further test this hypothesis, we examined the MeHg distribution in solutions with ligand-free CdSe particles (see Supplementary Section 1.1). Briefly, MeHg solution was added to ligand-free CdSe particles synthesized and purified. The phase distribution of sample was then tested with the MA-3000 immediately. We observed that >90% of the Hg was detected in the solid phase, totally opposite of the phase distribution pattern in the L-cysteine capped NP solution. This result supports the assumption that L-cysteine prevented MeHg from associating with the solid phase of the NP solution. In summary, the PL of NPs is more sensitive to inorganic Hg. The ligand L-cysteine, as a stabilizer of CdSe NPs, likely acted as a barrier for MeHg.
3.4 Products from the Interactions of Nanoparticles and Mercury Species
3.4.1Dimethylmercury
Prior studies (Jonsson et al., 2016) found that DMeHg was formed when MeHg adsorbed to disordered machinawite (FeSm) particles. But in this study, we did not observe DMeHg formation in the MeHg + NPs mixtures. Only a small DMeHg peak was found in the MeHg control solution. Yet, DMeHg was often detected in pure MeHg solution (see Supplementary Section 2.4; Supplementary Figure S3). Therefore, we argue that any DMeHg observed in this study was likely sourced from the added MeHg solutions, and was not the product of the interaction of MeHg with the NPs. Studies have shown that MeHg can form di-complexes ((CH3Hg)2X, where X = OH−, S2-, RS−) and these have been shown to decompose into (CH3)2Hg and HgX, especially for the sulfide and thiol ligands (Craig and Bartlett, 1978; Loux, 2007; Asaduzzaman and Schreckenbach, 2011). This pathway was the proposed reaction for the formation of DMeHg in the presence of FeSm(s), for example (Jonsson et al., 2016). While we have no details for a potential reaction mechanism, such a disproportionation could lead to the formation of trace amounts of DMeHg in the concentrated MeHg solutions. In the presence of water, the equilibrium constant for the formation of (CH3Hg)2OH (logK = 6.1)(Stumm and Morgan, 1996) is low, suggesting that the dimer is not an important species in distilled water. However, the presence of traces of organic ligands or chloride in the concentrated MeHg solutions may enhance the potential for the reactions outlined above.
Overall, although our result is not consistent with the earlier study (Jonsson et al., 2016) that showed DMeHg formation on the reduced sulfur surfaces, other research has indicated DMeHg degradation in the presence of dissolved sulfide and FeSm in aquatic systems (West et al., 2020). The inconsistency suggests that there are still some issues in the DMeHg studies, and not all reaction pathways have been identified. Hence, one must be cautious when performing experiments examining DMeHg production due to the potential for artifactual formation. We suggest that the MeHg solution used should be freshly diluted or N2 purged to exclude the potential for the presence of background DMeHg. Additionally, the absence of DMeHg in the Cd-cysteine complex solutions (with free L-cysteine) or NPs (Supplementary Figure S4) implies that the ligand likely hindered the production of DMeHg, and the potential reaction with the CdSe core. Likewise, Jonsson et al. (2016) demonstrated that the fraction of MeHg being converted to DMeHg was much lower when organic thiol compounds were in solution compared with that in association with naked FeSm particles.
3.4.2 Methylmercury Change in the MeHg + NPs Mixture
Although the MeHg concentration determined was ∼20% lower compared with the predicted concentration in the quenched NP solutions (see Table 1), a HgII peak was apparent in the mercury fluorescence spectra when the bulk was measured using the Tekran 2700 (see Supplementary Figure S5). This implies that the lost MeHg was probably transformed into HgII. The change in both the low-level and high-level MeHg + NPs mixtures further confirmed this argument (Table 2). In the MeHg + NPs mixture, MeHg consistently remained in the liquid phase (the 3rd and 4th columns of Table 2). The dark experiments also presented a similar distribution pattern after mixing for 24 h (the 5th column of Table 2). On the contrary, the MeHg concentration rapidly decreased in the duplicate kept under room light, including both the bulk and supernatant concentrations (the 6th column of Table 2). Ultimately, most Hg in the illuminated group was detected in the solid phase (76% for the low-level and 91% for the high-level Hg additions; the last column of Table 2).
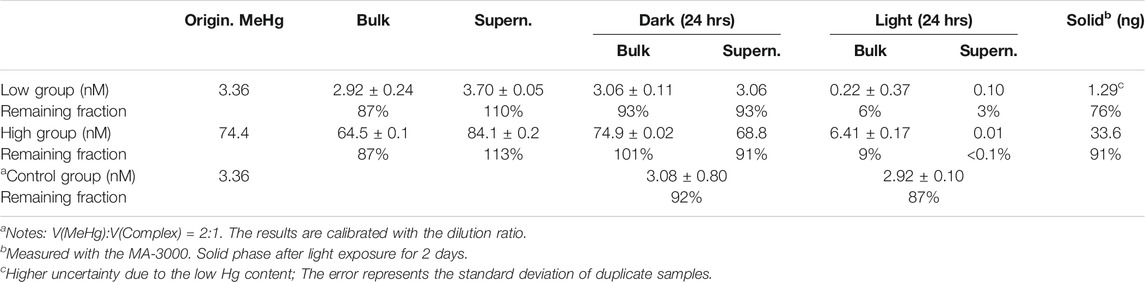
TABLE 2. Testing results of the reaction of MeHg and L-cysteine capped CdSe NPs. Most results refer to the MeHg concentrations derived from Tekran 2700.
If we compare the change in the Hg0 detector fluorescence spectra (from the Tekran 2700) of the bulk solutions in the dark and light (Figure 3), the decrease of the MeHg peak (area change from 3,350 to 110) is coincident to the increase of the HgII peak (area from 126 to 3,023), suggesting that a significant amount of MeHg was transformed into HgII when the solution was exposed to light. The produced HgII was bound onto the solid phase, in agreement with the previous observation of the HgII quenching of the NPs. The control group (no NPs), either in the dark or light, did not show MeHg loss as significant as the illuminated NPs group, indicating that self-degradation of MeHg was not the main factor accounting for the species transformation.
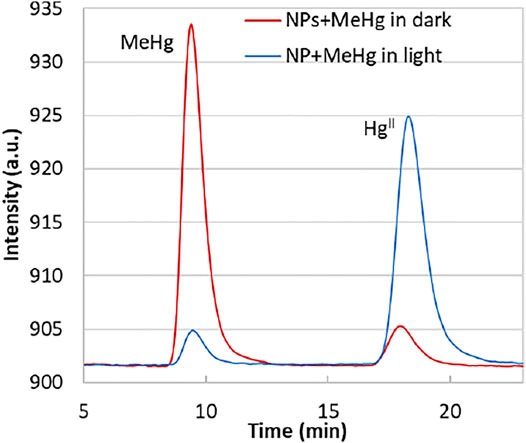
FIGURE 3. Mercury atomic fluorescence spectra from the Tekran 2700. The peak MeHg represents methylethyl mercury sourced from methylmercury by derivitization; HgII represents the diethyl mercury produced from HgII. The lines represent the spectra of bulk solutions from the higher concentration additions. For the exposure in the dark (red line), the peak area of MeHg = 3,550; HgII = 126; for the light exposure (blue line), MeHg = 110; HgII = 3,023.
The dominant MeHg degradation could be facilitated by CdSe NPs because they are known photocatalysts. Upon photoexcitation, bound electron-hole pairs (aka “excitons”) are formed in the quantum dots (QDs) (Boles et al., 2016), such as our NPs. When species that could accept or donate electrons are associated with the QDs or in the vicinity, electrons or holes could transfer from QDs to these species and vice versa. Since our NPs are passivated with L-cysteine, the L-cysteine molecules could be involved in photo-induced transformation and dissociate from the QD surface. To examine the assumption, the atomic composition of the NPs kept in the dark vs. in the light was determined by transmission electron microscopy (TEM, see Supplementary Section 1.2; Supplementary Figure S6). The results are shown in Figure 4 and in the inserted tables. Using the Se signal as a reference, there was significantly more S in the sample in the dark (25.5:1 S:Se) than in the light (2.0:1 S:Se), suggesting that the majority of the L-cysteine had dissociated from the NPs in the light. The absence of N in the illuminated NPs also supports this inference, and suggests the potential dissociation of L-cysteine. In addition, the Cd:Se ratio also decreased in the samples exposed to light, indicating some Cd on the surface may have been associated with the L-cysteine ligands.
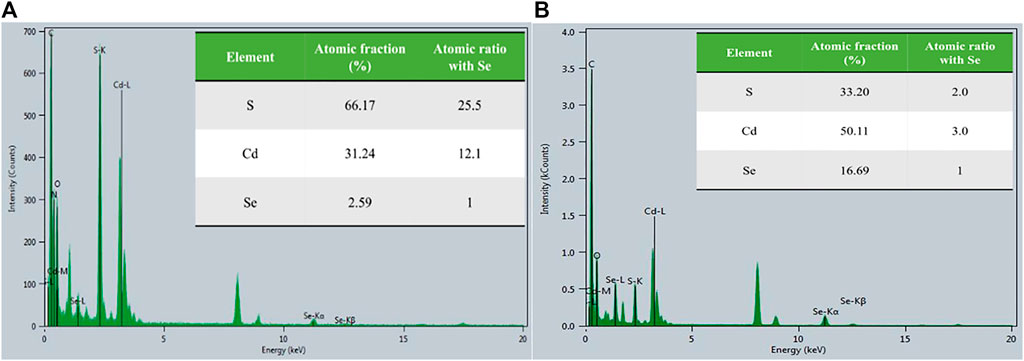
FIGURE 4. Atomic spectra for L-cysteine capped CdSe NP in a dark condition (A) and in the light condition (B). The inserted table shows the atomic fraction of scanned NP and the atomic ratios relative to Se.
When the MeHg + NPs mixture was exposed to the light, the dissociation of L-cysteine could expose more Se on the NP surface for Hg binding. As a result, MeHg could access the CdSe core and form a structure like Cd-Se-Hg-CH3 on the particle surface. Given the strong binding of Se with Hg, the bond between Hg-C would become less stable as the electron pair was pushed toward C. A mechanistic study on thiol dissociation revealed that the photoexcitation of CdSe NPs promoted electrons to the conduction band, resulting in proton reduction on the NP surface (Li et al., 2014).
Hence, we propose a mechanism of MeHg degradation based on the electron transfer mediated by the CdSe NP core and the binding of the resultant HgII to the NPs. The complexation of the HgII with the surface is energetically highly favorable, promoting the MeHg degradation. Whether methane is a product of this degradation or the methyl radical reacts with other constituents in solution could not be determined given the low concentrations involved, but the mass balance and Hg speciation measurements confirm that the reaction is occurring. The overall mechanism is similar to that proposed by others for the degradation of MeHg in the presence of metal sulfides. In the experiments of Jonsson et al. (2016) with micro FeSm(s), the higher concentration of MeHg on the surface allowed for a methyl transfer reaction between two MeHg molecules but also resulted in the precipitation of Hg on the solid surface.
In addition, the mechanism may explain the observed quenching of the NPs at the higher level MeHg additions (see Figure 2D). During fluorescence measurements, the NPs were irradiated with the instrument excitation light, which could have promoted photocatalysis. Therefore, the component quenching the NPs could be the HgII derived from the MeHg photodegradation, but only at the higher concentrations. The photodegradation could also explain the MeHg deficiency in the bulk solution (∼20%) after the characterization and quenching assays (Table 1). The Hg detected in the solid phase (Table 2) could have been HgII associated with the NPs from such reactions.
3.5 The Role of the Ligand in the MeHg + NPs Interaction
A time-dependent change of MeHg with the presence of CdSe NPs in dark was examined (see Supplementary Section 1.3) after the DMeHg test. The results are shown in Table 3. Compared with the estimated concentration, MeHg decreased by ∼20%, and this loss is consistent with the results of the MeHg quenching studies (Table 1). The MeHg content in the supernatant gradually decreased with time. The lost portion was likely relocated onto the particles as the Hg in the solid phase accounted for the difference between the original MeHg and the supernatant on the 6th day. Overall, Hg mass balance was found in both cases. These results reveal that MeHg can also degrade in the dark when mixed with NPs, but at a much slower rate. As the one-time phase separation that was used in our experiments cannot precipitate out the solids completely (see Supplementary Section 2.2), there were likely some NPs remaining in the supernatant. Since the NPs are not stable in DI water, some L-cysteine molecules could have dissociated from the DI-dispersed NPs (see Supplementary Section 2.6). These slowly exposed CdSe surfaces could therefore interact with MeHg, leading to degradation.
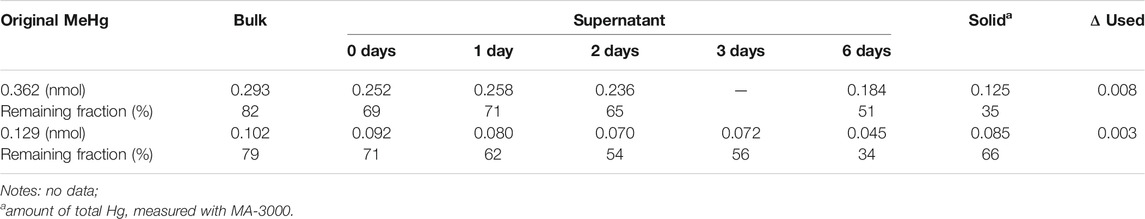
TABLE 3. The change of MeHg amounts after mixing MeHg with NPs in dark. ΔUsed refers to the portion transferred for the concentration determination.
Earlier studies have proposed several mechanisms for the transformation of MeHg; and the interaction with S/Se is an essential factor to mediate these reactions (Jonsson et al., 2016; Khan and Wang, 2010). Thus, we expected that the bare CdSe particles without a ligand coating (see Supplementary Section 1.1) should also trigger the transformation. In the assays where we studied the MeHg distribution on the ligand-free CdSe particles (see Supplementary Section 3.3), although most Hg was bound on the particles, a significant HgII peak was still observed when determining the MeHg concentration of the supernatant in both light and dark conditions. This implies that MeHg was degraded via the inorganic interaction. Other than reacting directly with the ligand-free CdSe, fast MeHg degradation was also observed in the acidified MeHg + NPs mixture (see Supplementary Section 2.5). Given that H+ can shift the metastable equilibrium of NPs (Aldana et al., 2005), and the pKa of the thiol in cysteine is 8.6 (Kyte, 2006), the acidification (pH < 5.5) could have promoted the dissociation of L-cysteine from the NP surface. This further indicates that mutual reactions between MeHg and the core are the vital factor that leads to the breaking of Hg-C bonds. In fact, based on the atomic composition (see Supplementary Figure S7), we found that HgSe dominated the particles when we mixed the ligand-free CdSe with the Hg solution. The precipitation of HgSe was likely the reaction that enhanced the degradation rate. Therefore, the L-cysteine coating on the NPs surface acted more like an isolation layer between MeHg and the CdSe NP core, protecting MeHg from degradation.
In natural waters, there are unlikely to be uncoated NPs as the levels of DOM are sufficient to coat these surfaces, as found for suspended particles. Therefore, MeHg degradation could be hindered although ligand degradation or dissociation should also occur with natural chalcogenide particles under environmental conditions when the change of their microenvironments significantly affects their stability, and depending on the level of light penetration into the water. The exposure of the chalcogenide core would then have a potential influence on the behavior of mercury species in their surroundings, such as the demethylation of MeHg and/or the association of HgII found in this study. Additionally, this may provide a hint on the mechanism for the change of the toxicity of Hg species when associated with reduced Se in biotic tissue if demethylation is promoted (Skerfving, 1978; Wang et al., 2011).
4 Conclusion
Compared to previous studies, we have added to the understanding of the interactions of Hg and MeHg with NPs through the study of the interaction of MeHg with L-cysteine capped CdSe NPs, and explored influences on the quenching efficiency and the mechanism on the reaction, and addressed the pathway of MeHg degradation. We have shown that the potential importance of photochemical enhancement of MeHg degradation.
We found that HgII was much more effective than MeHg in quenching the PL of the NPs. The phase-distribution assays showed that most of the HgII quickly associated with the solid phase of the NP solution while MeHg was mostly dispersed in the liquid phase. This observation indicates that the capability of Hg species to associate with the NPs determined their quenching effect. By comparing the quenching results at different concentration levels and different matrices, we deduced that under some conditions a portion of the added quencher was inaccessible to quench the PL.
By examining the products in the MeHg + NPs mixture, we found no evidence that DMeHg formed under our experimental conditions. However, significant HgII was detected when the MeHg + NPs mixture was illuminated, indicating that photodegradation of MeHg occurred. It was deduced that this degradation was triggered by the electron transfer mediated by the irradiated NPs, and the association (co-precipitation) of HgII with the NP core. We also observed the dissociation of L-cysteine from the NPs under some specific conditions, which exposed MeHg to the core structure and accelerated the reaction of MeHg with the CdSe, thus leading to MeHg transformation. Therefore, organic ligands are not only crucial to maintain the character of the NPs, but also in preventing MeHg degradation in the presence of the NPs. The presence and concentration of DOM in natural waters is therefore likely to be an important parameter determining the stability of MeHg in the presence of NPs as its coating not only stabilizes the NPs and lessens the MeHg degradation reaction, but also as the presence of DOM in solution can alter the degree of light penetration and therefore the abiotic photochemical degradation of MeHg.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
XS, RM and JZ designed the experiments and the overall study. XS completely most of the laboratory studies with the help of YW. All authors were involved in the synthesis and interpretation of the results and all authors were involved in the writing of the article and in its editing.
Funding
The project was funded by the United States National Science Foundation Environmental Chemical Science program through award #1607913 to RM and JZ.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Claudia Koerting for the use of and help with the UV-Vis spectrometer and fluorescence spectrophotometer. We are grateful to the reviewers for the constructive comments on this paper. XS thanks Xudong Wang for the guide in NP synthesis, and Gunnar Hansen, Wesley Huffman and Yipeng He for their help in the lab.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fenvc.2021.762052/full#supplementary-material
References
Aldana, J., Lavelle, N., Wang, Y., and Peng, X. (2005). Size-dependent Dissociation pH of Thiolate Ligands from Cadmium Chalcogenide Nanocrystals. J. Am. Chem. Soc. 127 (8), 2496–2504. doi:10.1021/ja047000+
Aldana, J., Wang, Y. A., and Peng, X. (2001). Photochemical Instability of CdSe Nanocrystals Coated by Hydrophilic Thiols. J. Am. Chem. Soc. 123 (36), 8844–8850. doi:10.1021/ja016424q
Asaduzzaman, A. M., and Schreckenbach, G. (2011). Degradation Mechanism of Methyl Mercury Selenoamino Acid Complexes: A Computational Study. Inorg. Chem. 50 (6), 2366–2372. doi:10.1021/ic1021406
Baker, J. S., Nevins, J. S., Coughlin, K. M., Colón, L. A., and Watson, D. F. (2011). Influence of Complex-Formation Equilibria on the Temporal Persistence of Cysteinate-Functionalized CdSe Nanocrystals in Water. Chem. Mater. 23 (15), 3546–3555. doi:10.1021/cm2013347
Baya, P. A., Hollinsworth, J. L., and Hintelmann, H. (2013). Evaluation and Optimization of Solid Adsorbents for the Sampling of Gaseous Methylated Mercury Species. Analytica Chim. Acta 786, 61–69. doi:10.1016/j.aca.2013.05.019
Ben Brahim, N., Bel Haj Mohamed, N., Poggi, M., Ben Chaâbane, R., Haouari, M., Ben Ouada, H., et al. (2017). Interaction of L-Cysteine Functionalized CdSe Quantum Dots with Metallic Cations and Selective Binding of Cobalt in Water Probed by Fluorescence. Sensors Actuators B: Chem. 243, 489–499. doi:10.1016/j.snb.2016.12.003
Benoit, J. M., Gilmour, C. C., Heyes, A., Mason, R. P., and Miller, C. L., (2003). Geochemical and Biological Controls over Methylmercury Production and Degradation in Aquatic Ecosystems. In: Y. Cai, and O. C. Braids (Editors), Biogeochemistry of Environmentally Important Trace Elements, Washington, D. C.: American Chemical Societypp. 262–297.
Bjorklund, G. (2016). Selenium as an antidote in the treatment of mercury intoxication. BioMetals 28 (4), 605–614.
Boles, M. A., Ling, D., Hyeon, T., and Talapin, D. V. (2016). The Surface Science of Nanocrystals. Nat. Mater 15 (2), 141–153. doi:10.1038/nmat4526
Borm, P. J., Robbins, D., Haubold, S., Kuhlbusch, T., Fissan, H., Donaldson, K., et al. (2006). The Potential Risks of Nanomaterials: a Review Carried Out for ECETOC. Part. Fibre Toxicol. 3 (1), 11. doi:10.1186/1743-8977-3-11
Coulibaly, M., Mazrui, N. M., Jonsson, S., and Mason, R. P. (2021). Abiotic Reduction of Mercury(II) in the Presence of Sulfidic Mineral Suspensions. Front. Environ. Chem. 2, 660058.
Craig, P. J., and Bartlett, P. D. (1978). The Role of Hydrogen Sulphide in Environmental Transport of Mercury. Nature 275 (5681), 635–637. doi:10.1038/275635a0
DiMento, B. P., and Mason, R. P. (2017). Factors Controlling the Photochemical Degradation of Methylmercury in Coastal and Oceanic Waters. Mar. Chem. 196, 116–125. doi:10.1016/j.marchem.2017.08.006
Driscoll, C. T., Mason, R. P., Chan, H. M., Jacob, D. J., and Pirrone, N. (2013). Mercury as a Global Pollutant: Sources, Pathways, and Effects. Environ. Sci. Technol. 47 (10), 4967–4983. doi:10.1021/es305071v
Du, H., Ma, M., Igarashi, Y., and Wang, D. (2019). Biotic and Abiotic Degradation of Methylmercury in Aquatic Ecosystems: A Review. Bull. Environ. Contam. Toxicol. 102 (5), 605–611. doi:10.1007/s00128-018-2530-2
Edwards, E. H., Fertig, A. A., McClelland, K. P., Meidenbauer, M. T., Chakraborty, S., Krauss, T. D., et al. (2020). Enhancing the Activity of Photocatalytic Hydrogen Evolution from CdSe Quantum Dots with a Polyoxovanadate Cluster. Chem. Commun. 56 (62), 8762–8765. doi:10.1039/d0cc03163a
Gilmour, C. C., Bullock, A. L., McBurney, A., Podar, M., and Elias, D. A. (2018). Robust Mercury Methylation across Diverse Methanogenic Archaea. mBio 9 (2), e02403–17. doi:10.1128/mBio.02403-17
Hammerschmidt, C. R., and Fitzgerald, W. F. (2006). Photodecomposition of Methylmercury in an Arctic Alaskan lake. Environ. Sci. Technol. 40 (4), 1212–1216. doi:10.1021/es0513234
Handy, R. D., Owen, R., and Valsami-Jones, E. (2008). The Ecotoxicology of Nanoparticles and Nanomaterials: Current Status, Knowledge Gaps, Challenges, and Future Needs. Ecotoxicology 17 (5), 315–325. doi:10.1007/s10646-008-0206-0
Jackson, T. A. (1998). “Mercury in Aquatic Ecosystems,” in Metal Metabolism in Aquatic Environments. Editors W. J. Langston, and M. J. Bebianno (Boston, MA: Springer US), 77–158. doi:10.1007/978-1-4757-2761-6_5
Jeevanandam, J., Barhoum, A., Chan, Y. S., Dufresne, A., and Danquah, M. K. (2018). Review on Nanoparticles and Nanostructured Materials: History, Sources, Toxicity and Regulations. Beilstein J. Nanotechnol. 9, 1050–1074. doi:10.3762/bjnano.9.98
Jensen, S., and Jernelöv, A. (1969). Biological Methylation of Mercury in Aquatic Organisms. Nature 223 (5207), 753–754. doi:10.1038/223753a0
Jonsson, S., Mazrui, N. M., and Mason, R. P. (2016). Dimethylmercury Formation Mediated by Inorganic and Organic Reduced Sulfur Surfaces. Sci. Rep. 6 (1), 27958. doi:10.1038/srep27958
Ke, J., Li, X., Zhao, Q., Hou, Y., and Chen, J. (2014). Ultrasensitive Quantum Dot Fluorescence Quenching Assay for Selective Detection of Mercury Ions in Drinking Water. Sci. Rep. 4 (1), 5624. doi:10.1038/srep05624
Kerin, E. J., Gilmour, C. C., Roden, E., Suzuki, M. T., Coates, J. D., and Mason, R. P. (2006). Mercury Methylation by Dissimilatory Iron-Reducing Bacteria. Appl. Environ. Microbiol. 72 (12), 7919–7921. doi:10.1128/aem.01602-06
Khan, M. A. K., and Wang, F. (2010). Chemical Demethylation of Methylmercury by Selenoamino Acids. Chem. Res. Toxicol. 23 (7), 1202–1206. doi:10.1021/tx100080s
Kyte, J. (2006). Structure in Protein Chemistry. New York, New York, United States: Garland Science.
Lakowicz, J. R. (2006). “Quenching of Fluorescence,” in Principles of Fluorescence Spectroscopy. Editor J. R. Lakowicz (Boston, MA: Springer US), 277–330.
Lee, S. H., and Richards, R. J. (2004). Montserrat Volcanic Ash Induces Lymph Node Granuloma and Delayed Lung Inflammation. Toxicology 195 (2), 155–165. doi:10.1016/j.tox.2003.09.013
Li, X.-B., Li, Z.-J., Gao, Y.-J., Meng, Q.-Y., Yu, S., Weiss, R. G., et al. (2014). Mechanistic Insights into the Interface-Directed Transformation of Thiols into Disulfides and Molecular Hydrogen by Visible-Light Irradiation of Quantum Dots. Angew. Chem. Int. Ed. 53 (8), 2085–2089. doi:10.1002/anie.201310249
Li, X., Liao, R.-Z., Zhou, W., and Chen, G. (2010). DFT Studies of the Degradation Mechanism of Methyl Mercury Activated by a Sulfur-Rich Ligand. Phys. Chem. Chem. Phys. 12 (16), 3961–3971. doi:10.1039/b918402c
Loux, N. T. (2007). An Assessment of Thermodynamic Reaction Constants for Simulating Aqueous Environmental Monomethylmercury Speciation. Chem. Speciation Bioavailability 19 (4), 183–196. doi:10.3184/095422907x255947
Nick, S. T., Bolandi, A., Samuels, T. A., and Obare, S. O. (2014). Advances in Understanding the Transformation of Engineered Nanoparticles in the Environment. Pure Appl. Chem. 86 (7), 1129–1140. doi:10.1515/pac-2014-0301
Park, Y.-S., Dmytruk, A., Dmitruk, I., Kasuya, A., Okamoto, Y., Kaji, N., et al. (2010). Aqueous Phase Synthesized CdSe Nanoparticles with Well-Defined Numbers of Constituent Atoms. J. Phys. Chem. C 114 (44), 18834–18840. doi:10.1021/jp107608b
Park, Y.-S., Okamoto, Y., Kaji, N., Tokeshi, M., and Baba, Y. (2011). Aqueous Phase-Synthesized Small CdSe Quantum Dots: Adsorption Layer Structure and strong Band-Edge and Surface Trap Emission. J. Nanopart Res. 13 (11), 5781–5798. doi:10.1007/s11051-011-0273-7
Parks, J. M., Johs, A., Podar, M., Bridou, R., Hurt, R. A., Smith, S. D., et al. (2013). The Genetic Basis for Bacterial Mercury Methylation. Science 339 (6125), 1332–1335. doi:10.1126/science.1230667
Qu, L., and Peng, X. (2002). Control of Photoluminescence Properties of CdSe Nanocrystals in Growth. J. Am. Chem. Soc. 124 (9), 2049–2055. doi:10.1021/ja017002j
Rivera, N. A., Bippus, P. M., and Hsu-Kim, H. (2019). Relative Reactivity and Bioavailability of Mercury Sorbed to or Coprecipitated with Aged Iron Sulfides. Environ. Sci. Technol. 53 (13), 7391–7399. doi:10.1021/acs.est.9b00768
Robinson, B. H. (2009). E-waste: An Assessment of Global Production and Environmental Impacts. Sci. Total Environ. 408 (2), 183–191. doi:10.1016/j.scitotenv.2009.09.044
Seller, P., Kelly, C. A., Rudd, J. W. M., and MacHutchon, A. R. (1996). Photodegradation of Methylmercury in Lakes. Nature 380 (6576), 694–697. doi:10.1038/380694a0
Skerfving, S. (1978). Interaction between Selenium and Methylmercury. Environ. Health Perspect. 25, 57–65. doi:10.1289/ehp.782557
Stumm, W., and Morgan, J. J. (1996). Aquatic Chemistry: Chemical Equilibria and Rates in Natural Waters. Hoboken, New Jersey, United States: Wiley.
Sunderland, E. M., Li, M., and Bullard, K. (2018). Decadal Changes in the Edible Supply of Seafood and Methylmercury Exposure in the United States. Environ. Health Perspect. 126 (1), 017006. doi:10.1289/ehp2644
Susana, S., Dias, T., and Ramalhosa, E. (2011). “Mercury Methylation versus Demethylation: Main Processes Involved,” in Methylmercury: Formation, Sources and Health Effects. Editor A. P. Clampet (Hauppauge, New York, United States: Nova Science Publishers), 123–166.
Tai, C., Li, Y., Yin, Y., Scinto, L. J., Jiang, G., and Cai, Y. (2014). Methylmercury Photodegradation in Surface Water of the Florida Everglades: Importance of Dissolved Organic Matter-Methylmercury Complexation. Environ. Sci. Technol. 48 (13), 7333–7340. doi:10.1021/es500316d
Tseng, C.-M., Hammerschmidt, C. R., and Fitzgerald, W. F. (2004). Determination of Methylmercury in Environmental Matrixes by On-Line Flow Injection and Atomic Fluorescence Spectrometry. Anal. Chem. 76 (23), 7131–7136. doi:10.1021/ac049118e
Turan, N. B., Erkan, H. S., Engin, G. O., and Bilgili, M. S. (2019). Nanoparticles in the Aquatic Environment: Usage, Properties, Transformation and Toxicity-A Review. Process Saf. Environ. Prot. 130, 238–249. doi:10.1016/j.psep.2019.08.014
Wang, F., Lemes, M., and Khan, M. A. K. (2011). “Metallomics of Mercury: Role of Thiol- and Selenol-Containing Biomolecules,” in Environmental Chemistry and Toxicology of Mercury. Editors G. Liu, Y. Cai, and O. D. Nelson, 517–544. doi:10.1002/9781118146644.ch16
Wang, X., Seelen, E. A., Mazrui, N. M., Kerns, P., Suib, S. L., Zhao, J., et al. (2019). The Interaction of Mercury and Methylmercury with Chalcogenide Nanoparticles. Environ. Pollut. 255, 113346. doi:10.1016/j.envpol.2019.113346
Warner, K. A., Roden, E. E., and Bonzongo, J.-C. (2003). Microbial Mercury Transformation in Anoxic Freshwater Sediments under Iron-Reducing and Other Electron-Accepting Conditions. Environ. Sci. Technol. 37 (10), 2159–2165. doi:10.1021/es0262939
West, J., Graham, A. M., Liem-Nguyen, V., and Jonsson, S. (2020). Dimethylmercury Degradation by Dissolved Sulfide and Mackinawite. Environ. Sci. Technol. 54 (21), 13731–13738. doi:10.1021/acs.est.0c04134
Wigginton, N. S., Haus, K. L., and Hochella Jr, M. F. (2007). Aquatic Environmental Nanoparticles. J. Environ. Monit. 9 (12), 1306–1316. doi:10.1039/b712709j
Zeng, T., Hu, Y., Wang, N., Xia, C., Li, S., Zu, Y., et al. (2013). Effects of Different Metal Ions on the Fluorescence of CdSe/ZnS Quantum Dots Capped with Various Thiolate Ligands. Phys. Chem. Chem. Phys. 15 (42), 18710–18715. doi:10.1039/c3cp52666f
Zhao, J., Holmes, M. A., and Osterloh, F. E. (2013). Quantum Confinement Controls Photocatalysis: A Free Energy Analysis for Photocatalytic Proton Reduction at CdSe Nanocrystals. ACS Nano 7 (5), 4316–4325. doi:10.1021/nn400826h
Zhang, T., Hsu-Kim, H., and Osterloh, F. E. (2010). Photolytic degradation of methylmercury enhanced by binding to natural organic ligands. Nature Geoscience 3 (7), 473–476.
Keywords: mercury, nanoparticles, methylmercury, cadmium selenide, methylmercury degradation, quenching effeciency
Citation: Shi X, Zhao J, Wang Y and Mason RP (2021) The Transformation of Inorganic and Methylmercury in the Presence of l-Cysteine Capped CdSe Nanoparticles. Front. Environ. Chem. 2:762052. doi: 10.3389/fenvc.2021.762052
Received: 20 August 2021; Accepted: 08 December 2021;
Published: 24 December 2021.
Edited by:
Marc Amyot, Université de Montréal, CanadaReviewed by:
Carl Mitchell, University of Toronto Scarborough, CanadaIgor Lehnherr, University of Toronto Mississauga, Canada
Copyright © 2021 Shi, Zhao, Wang and Mason. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert P. Mason, Um9iZXJ0Lm1hc29uQHVjb25uLmVkdQ==